


Abstract
DNA lesions or other barriers frequently compromise replisome progress. The SF2 helicase RecG is a key enzyme in the processing of postreplication gaps or regressed forks in Escherichia coli. A deletion of the recG gene renders cells highly sensitive to a range of DNA damaging agents. Here, we demonstrate that RecG function is at least partially complemented by another SF2 helicase, RadD. A ΔrecGΔradD double mutant exhibits an almost complete growth defect, even in the absence of stress. Suppressors appear quickly, primarily mutations that compromise priA helicase function or recA promoter mutations that reduce recA expression. Deletions of uup (encoding the UvrA-like ABC system Uup), recO, or recF also suppress the ΔrecGΔradD growth phenotype. RadD and RecG appear to avoid toxic situations in DNA metabolism, either resolving or preventing the appearance of DNA repair intermediates produced by RecA or RecA-independent template switching at stalled forks or postreplication gaps. Barriers to replisome progress that require intervention by RadD or RecG occur in virtually every replication cycle. The results highlight the importance of the RadD protein for general chromosome maintenance and repair. They also implicate Uup as a new modulator of RecG function.
INTRODUCTION
The replication of genomic DNA is an essential process that is carried out by a highly complex and regulated assembly of proteins called the replisome. As replication proceeds, the replisome encounters impediments. Exogenous damage from the environment, protein–DNA complexes, reactive oxygen species (ROS), and genotoxic agents can cause replisome stalling and fork collapse. If improperly repaired, lesions and breaks can produce mutagenesis. Mutagenesis in turn can give rise to human disease. In any organism, replication rarely, if ever, completes uninterrupted (1–3). The potential biological consequences and frequency of replication conflicts underscores the importance of understanding DNA repair and replication enzymes.
In most bacteria, replication initiates from a single origin called oriC. From there the two replication forks move bi-directionally on the circular chromosome until meeting at the terminus opposite of the origin. When a lesion is encountered by a replisome, repair can take many forms (Figure 1). Polymerase switching is a well-documented process in vitro that allows for lesion bypass by translesion DNA synthesis (4,5). In contrast, some lesions can be passed over by lesion skipping on either the leading or lagging strand. This consists of re-priming the replicative polymerase downstream of a roadblock for continued DNA synthesis (6–8), leaving the lesion behind in a gap. The postreplication gap left behind is filled by RecA in RecFOR mediated gap repair or RecA-independent template switching (9,10).
Figure 1.

Possible fates of abandoned replication forks. (A) The formation of post replication gaps by lesion skipping. Gaps generated by lesion skipping can be filled either by RecA-mediated homologous recombination or RecA-independent template switching. Synthesis is initiated from an undamaged template. Resolution of this intermediate can yield either crossover or non-crossover products. (B) The process of replication fork reversal and Holliday junction formation. The lesion can be re-incorporated into the parental duplex to grant other repair pathway access. The lesion can also be bypassed by nascent strand template switching on the free end of the Holliday junction.
If the replisome is unable to bypass a lesion it can disassociate, leaving behind an abandoned fork. Repair enzymes can then access the fork and re-anneal the parental duplex creating a Holliday Junction (HJ) in a process known as fork reversal or fork regression. Fork reversal is a frequent process, occurring in 25–40% of cells treated with a Topoisomerase I inhibitor (11). An important feature of fork reversal is the re-incorporation of the lesion back into the parental duplex. This allows Mismatch Repair (MMR), Base Excision Repair (BER), or Nucleotide Excision Repair (NER) enzymes to remove any lesion in question (Figure 1B). As an alternative, synthesis can occur on the free end of the reversed junction if the lagging strand has been replicated further than the leading strand. Both processes require branch migration back to a suitable fork substrate for PriA-mediated restart. Exonucleases may also digest the protruding arm on junctions to restore a fork (12). However, this would negate any template switch synthesis that has occurred. In bacteria, RecG, RecQ, RuvAB and RecA are all capable of or implicated in reversing replication forks (13–17). In humans, SMARCAL1, HLTF, RAD51 and ZRANB3 are enzymes involved in fork reversal and branch migration (10,11,18).
In Escherichia coli, the SF2 helicase RecG has emerged as a key player in this process (19–25). In vitro, RecG can reverse forks and, alternatively, branch migrate the resulting Holliday junction back to a fork structure. In vivo, a recG null strain is still capable of fork reversal, suggesting little or no involvement of RecG in the initial fork processing. However, the products of fork reversal, Holliday junctions, accumulate at sites of replisome stalling (17). This suggests that an important role of RecG in fork repair is to remodel Holliday junctions back to fork structures after repair.
Cells lacking recG function grow normally but are sensitive to many DNA damaging agents. When a ΔrecG strain is treated with UV, suppressor mutations arise in priA that may function by altering the PriA helicase activity without compromising the capacity of PriA to load DnaB for replication restart (12,26–31). The toxicity of a fully helicase-competent PriA in UV treated ΔrecG cells has yet to be fully explained. In vitro, some suppressor mutations render PriA incapable of unwinding the nascent lagging strand at a fork without a leading strand present. The emerging model is that, after repair, RecG restores a replication fork that has a nascent leading strand end in proximity to the junction. An end thus positioned correctly orients PriA to facilitate DnaB helicase loading and replication restart (3,15,19,26,28). Without a leading strand, PriA can incorrectly unwind the parental duplex. The presence of the single stranded DNA binding protein, SSB, allows for bypass of this leading strand requirement (30). High concentrations of SSB and its presumed presence at an abandoned replication fork suggest that toxicity is not from parental duplex unwinding (32).
We have identified an apparently complementary relationship between another SF helicase, RadD and RecG. RadD shares significant homology to the E. coli SF2 helicases RecQ and RecG. Previous work has shown RadD suppresses crossovers that can occur in postreplication gaps and can bind forked DNA structures (33). It is important for survival during tobramycin and ionizing radiation treatment. RadD also has a functional interaction with the C-terminal tail of the SSB (34–36), as does RecG and RecQ (37,38). This interaction places the primary function of RadD at the fork or single strand gaps. We also identified several suppressors of this phenotype that have given us more insight into the functions of RadD, PriA, Uup, RecA and RecG.
MATERIALS AND METHODS
Strain construction
Strains used in this report are in Table 1. A modification of the method by Datsenko & Wanner (39) was used to construct chromosomal gene knockouts and point mutations. The plasmid pEAW507 contains a kanamycin (Kan) cassette flanked by FRT recognition sites for the FLP recombinase (pJFS42 mutant FRT-KanR-wt FRT) was the template for gene deletions. PCR amplification across this region was carried out using primers with (a) 21 nucleotides of sequence complementary to one end of the cassette, and (b) an additional 50 nucleotide complementary sequence to regions flanking the gene of interest. Gel-purified PCR product was electroporated into cells containing pDK46, which expresses the lambda red recombinase. Recombinase expression was induced by the addition of l-arabinose. Kanamycin resistant colonies were screened for ampicillin sensitivity and used as a template for colony PCR confirmation. The KanR cassette was removed by transforming strains with a plasmid that harbors the FLP recombinase (pLH29). For strains containing multiple deletions, P1 transduction was used to introduce multiple alleles. The process of P1 transduction consisted of plating the initial transductants on LB + antibiotic. Resulting transductans were then streaked again on LB + antibiotic to ensure resistance. All strain constructions were confirmed by PCR amplification across all relevant deletion sites and/or direct sequencing.
Table 1.
Strains used in this study
| Strain | Genotype | Parent strain | Source/technique |
|---|---|---|---|
| MG1655 | uup+ radD+ recG+ | - | (65,66) |
| EAW9 | ΔrecA recG- | MG1655 | △recA to recG- 133FRT#1 from pEAW324 template |
| EAW114 | ΔrecO | MG1655 | Lambda RED recombination |
| EAW232 | Founder Δe14ΔradD | MG1655 | Lambda RED recombination |
| EAW242 | Founder Δe14Δuup | MG1655 | Lamda RED recombination |
| EAW368 | founderΔe14 ΔradD recG- | MG1655 | EAW232 transduced to recG- with P1 grown on EAW9 KanR |
| EAW401 | founderΔe14 ΔruvB | MG1655 | Lambda RED recombination |
| EAW408 | △lacIYZA | MG1655 | Lambda RED recombination |
| EAW505 | ΔrecG | MG1655 | Lambda RED recombination |
| EAW526 | ΔradD | MG1655 | Transduction of MG1655 with P1 grown on EAW232 KanR |
| EAW531 | ΔrecGΔradD | MG1655 | Transduction of EAW505 with P1 grown on EAW526 KanR |
| EAW552 | founderΔe14 ΔradD recG-, priA S278A-Tet | MG1655 | EAW368 suppressor#1 with wtFRT-TetR-wt FRT after priA |
| EAW553 | founderΔe14 ΔradD recG-, priA A520P-Tet | MG1655 | EAW368 suppressor#5 with wtFRT-TetR-wt FRT after priA |
| EAW629 | ΔrecF | MG1655 | Lambda RED recombination |
| EAW1073 | PrecA A → G | MG1655 | Lambda RED recombination |
| EAW1075 | ΔradD PrecA A → G | MG1655 | Transduction of EAW526 with P1 grown on EAW1073 KanR |
| EAW 1087 | ΔradD PrecA A → G ΔrecG | MG1655 | Transduction of EAW1075 with P1 grown on EAW505 KanR |
| EAW1097 | MG1655 ΔruvB | MG1655 | Transduction of MG1655 with P1 grown on EAW401 KanR |
| EAW1100 | ΔradD Δlac IZYA | EAW526 | Transduction of EAW526 with P1 grown on EAW408 KanR |
| EAW1102 | MG1655 ΔrecGΔlac IZYA | MG1655 | Transduction of EAW505 with P1 grown on EAW408 KanR |
| EAW1104 | MG1655 ΔradDΔuupΔlac IZYA | MG1655 | Transduction of ZJR04 with P1 from EAW408 KanR |
| EAW1132 | ΔradDΔlac IZYAΔuup | EAW1100 | Transduction of EAW1100 with P1 grown on EAW242 KanR |
| ZJR01 | Δuup° | MG1655 | Transduction of MG1655 with P1 grown on EAW242 KanR |
| ZJR04 | ΔuupΔradD | MG1655 | Transduction of ZJR01 with P1 grown on EAW232 KanR |
| ZJR10 | ΔuupΔrecG | MG1655 | Transduction of ZJR01 with P1 grown on EAW505 KanR |
| ZJR17 | ΔuupΔrecGΔradD | MG1655 | Transduction of ZJR10 with P1 grown on EAW 232 KanR |
| ZJR20 | ΔuupΔruvB | MG1655 | Transduction of ZJR01 with P1 grown on EAW 401 KanR |
| ZJR 22 | ΔradDΔLacIZYA PrecA A → G | MG1655 | Transduction of EAW1100 with P1 from EAW 1073 KanR |
| ZJR29 | ΔradD priA S278A | MG1655 | Transduction of EAW526 with P1 grown on EAW552 TetR |
| ZJR31 | ΔradDΔlacIZYA priA S278A | MG1655 | Transduction of EAW 1100 with P1 grown on EAW552 TetR |
| ZJR32 | ΔradDΔlacIZYA priA A520P | MG1655 | Transduction of EAW 1100 with P1 grown on EAW553 TetRR |
| ZJR34 | ΔrecG priAS278A | MG1655 | Transduction of EAW505 with P1 grown on EAW522 TetR |
| ZJR35 | ΔradD priA S278A △recG | MG1655 | Transduction of ZJR29 with P1 grown on EAW505 KanR |
| ZJR36 | ΔradDΔlacIZYA priA S278A ΔrecG | MG1655 | Transduction of ZJR31 with P1 grown on EAW 505 KanR |
| ZJR37 | ΔradDΔlacIZYA priA A520P ΔrecG | MG1655 | Transduction of ZJR32 with P1 grown on EAW505 KanR |
| ZJR40 | ΔradDΔLacIZYA PrecA A → G ΔrecG | MG1655 | Lambda RED recombination |
| ZJR41 | ΔrecG priA A520P | MG1655 | Transduction of EAW505 with P1 grown on EAW553 TetR |
| ZJR42 | ΔradD priA A520P ΔrecG | MG1655 | Transduction of ZJR41 with P1 grown on EAW232 KanR |
| ZJR49 | ΔradDΔlacIYZAΔrecO | MG1655 | Transduction of EAW1100 with P1 grown on EAW114 KanR |
| ZJR50 | ΔradDΔrecGΔlacIYZAΔrecO | MG1655 | Transduction of ZJR49 with P1 grown on EAW505 KanR |
| ZJR51 | △radD△recF△lacIYZA | MG1655 | Transduction of EAW1100 with EAW629 KanR |
| ZJR52 | ΔradDΔrecFΔlacIYZAΔrecG | MG1655 | Transduction of ZJR51 with P1 from EAW505 KanR |
| ZJR54 | ΔuupΔrnhA | MG1655 | Transduction of ZJR01 with P1 grown on JW0204 from Keio collection (67) KanR |
| ZJR55 | dnaA(46)ts | MG1655 | Transduction of MG1655 with P1 grown on MG1655 dnaA(46)ts (68) |
| ZJR56 | Δuup dnaA(46)ts | Transduction of ZJR01 with P1 grown on ZJR55 | |
| ZJR57 | dnaA(46)ts + ΔrnhA | Transduction of ZJR55 with P1 grown on JW0204 | |
| ZJR58 | ΔuupΔrnhA dnaA(46)ts | MG1655 | Transduction of ZJR54 with P1 grown on ZJR55 (68). |
To construct the ΔradDΔrecG strain, the radD deletion was introduced into the recG deletion strain by P1 transduction. Very small colonies appeared after growth overnight on LB plates containing Kanamycin. Multiple cultures from transductant colonies were grown overnight. Turbidity at this point was minimal but detectable. Several minimally turbid cultures were spun down, resuspended in 1 ml LB, and frozen. The presence of the two deletions was confirmed both by PCR amplification and by direct sequencing of the PCR product. This was the stock used for all experiments. Unless stated otherwise, all subsequent growth curves and spot plates were initiated by inoculating a fresh tube of media from the same frozen aliquot of ΔradDΔrecG cells.
Growth curves and SOS induction assays
In order to minimize growth before testing all growth curves had to be initiated from freezer stocks. 3 ml of LB was inoculated to a minimum OD600 of 0.01. Each culture was then diluted to give a starting OD600 of 0.005 and 100 μl of each culture was added to a 96-well plate. Growth was monitored at 37°C while shaking in a H1 Synergy Biotek plate reader. Optical density readings were taken every 10 min for 24 h.
For SOS induction assays, we utilized a plasmid containing SuperGlo GFP under control of the recN promoter (pEAW903). Each strain was transformed with pEAW903 and cultures were diluted to an initial OD600 of 0.005. SOS induction was monitored by measuring GFP fluorescence every 10 min for 24 h along with OD600 readings at 37°C while shaking in a H1 Synergy Biotek plate reader. Data was exported and data graphed using GraphPad Prism Software. Statistical analysis was based on at least three replicates in all experiments.
Mini-F pRC-7 plasmid assay
The pRC7 plasmid is a lac+ mini-F low copy derivative of pFZY1 (40). pJJ100 that harbors recG was a generous gift from Christian Rudolph and constructed as described previously (41). All strains were transformed with pJJ100 before adding the final mutation to be tested. For example, a ΔradD strain was transformed with pJJ100 and plated on 0.5× (Amp50) ampicillin. With plasmid selection present recG was deleted using P1 transduction and by plating on Kan 40 and Amp 50. This ensured that recG was always present and removed the chance of suppressor mutations arising. Once constructed, 3 ml overnights of each strain with the pJJ100 plasmid were set and allowed to grow overnight for 16 h. The following day 5 ml fresh LB was inoculated with 50 μl overnight; at this point antibiotic was withheld. Cultures were grown to an OD600 of 0.2 and placed on ice for a minimum of 5 min, serially diluted in 1× PBS Buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM CaCl2 and 0.5 mM MgCl2) and spread on X-gal IPTG plates. Plates were allowed to grow for a strict 16 h for initial blue and white colony counting. Plates were then allowed to grow for an addition 8 hours and colonies were recounted. All experiments were repeated at least three times with comparable results.
Sensitivity assays
All strains were grown in 3 ml LB culture overnight at 37°C while shaking. The following day 50 μl of overnight was used to inoculate 5 ml LB and grown to an OD600 of 0.2 while shaking at 37°C. Cultures were serially diluted in 10× steps to 10−6 in 1× PBS buffer in a 96-well plate. LB agar plates were made the day of the assay and kept in dark to prevent break down of DNA damaging agents. A total of 10 μl of each dilution was plated for all strains and the plates were photographed after growth at 37°C overnight. All experiments were repeated at least three times with comparable results.
Bright-field microscopy
For all measurements of cell filamentation, wide-field microscopy was conducted on an inverted microscope (IX-81, Olympus with a 1.49 NA 100× objective). Bright-field images were collected on a 512 × 512 pixel EM-CCD camera (C9100-13, Hamamatsu). For imaging of all strains we used glass coverslips functionalized with 3-amino-propyl-triethoxysilane (APTES, Alfa Aeser) to immobilize cells on the coverslip surface.
Coverslips were first sonicated for 30 min in 5M KOH to clean and activate the surface of the coverslip. Coverslips were then rinsed thoroughly with MilliQ water, then treated with 1 ml 5% (v/v) of APTES in MilliQ water for 10 min. Subsequently, coverslips were rinsed with ethanol twice and sonicated in ethanol for a further 20 s. Finally, functionalized coverslips were rinsed with MilliQ water and dried in a jet of N2 and stored under vacuum prior to use.
Live-cell imaging
For all imaging experiments, cells were grown overnight at 37°C with shaking in EZ rich defined medium (Teknova) that contained 0.2% (w/v) glucose. Overnight, saturated cultures were reset 1 in 1000 μl EZ glucose and grown out for 3 h before imaging. To initiate imaging, 20 μl of cells were loaded onto an APTES functionalized coverslip, sandwiched with a KOH cleaned coverslip and allowed to associate with the surface before being imaged. A single bright-field image (34 ms exposure) was taken at multiple fields of view to determine cell lengths and filamentation.
Analysis of cell filamentation
Bright-field images of all strains were imported into MicrobeTracker 0.937 (42), a MATLAB script, was used to create cell outlines as regions of interest (ROI’s). Cell outlines were manually created and designated via MicrobeTracker to ensure accuracy and that only non-overlapping, in-focus cells were selected for analysis. ROI’s were then exported Microsoft Excel to define cell parameters including cell length.
RESULTS
Cells lacking both RadD and RecG exhibit a severe growth defect
A radD deletion does not confer a significant growth defect on the host cell. In order to gain insight into the role of RadD in vivo, we have begun to explore its relationship with other cellular DNA helicases. We previously showed that removing both RadD and RadA function did not affect growth under standard conditions. However, when treated with Ciprofloxacin or UV, the ΔradAΔradD strain exhibited a significant loss in viability (34). Cells lacking both RadD and RecG function exhibited a more serious loss of viability, but that phenotype was not extensively characterized (34). The properties of the ΔradDΔrecG strain provided the starting point for the current study.
We began by measuring the growth rate of a ΔradDΔrecG strain and the related single deletion strains. All samples were normalized to an initial OD600 of 0.005 before being set for monitored growth to ensure that the initial number of cells per culture was comparable. Wild type, ΔradD, and ΔrecG strains grow unhindered as expected (Figure 2A), although deleting recG produces a slight lag in growth and reduction in growth rate. Deleting both radD and recG creates an extended lag phase lasting up to 8 h or more. Once growth begins, the culture approached saturation at a rate similar to a ΔrecG strain. Isolates from the saturated ΔradDΔrecG culture exhibited the same colony size as wild type cells when grown on plates from single cells overnight. These same isolates consistently did not exhibit the long growth lag of the original strain. This suggested the presence of suppressor mutations.
Figure 2.

Growth curves and SOS induction of recG and radD single and double mutants. (A) A minimum of 3 Log scale OD600 versus time traces of each strain shown in comparison to a wild type control. Wild type is always shown in black with mutants appearing in red. (B) SOS traces of recG and radD single and double mutants over time. The y-axis is the fluorescent signal divided by the optical density of the corresponding replicate. Experimental setup is further described in methods. The ΔradDΔrecG strain is designated ΔradDΔrecGsupp to highlight the presence of a suppressor.
The induction of SOS in the absence of damage was also measured for each strain. To monitor SOS induction, we used a plasmid harboring an early SOS-sensitive recN promoter that controls GFP expression. One caveat of this assay is the reporter plasmid carriers a pMB1 origin that might affect plasmid stability. It is important to note, all the strains were grown to saturation overnight. Each strain was diluted in fresh media to give a starting OD600 of 0.005. This additional growth prior to measurement will cause the ΔradDΔrecG strain to accumulate suppressors and grow much faster than observed in Figure 2A. Deleting radD produced no increased SOS induction in the absence of stress compared to wildtype (Figure 2B). A ΔrecG strain exhibited substantial SOS induction, again in the absence of stress. The signal halts after ∼500 min because the GFP signal saturates the capacity of the plate reader. The ΔradDΔrecG strain exhibited a higher induction of SOS before saturating our plate reader 3 h faster than a recG deletion alone. This signal is coming from a strain that has accumulated a suppressor, as detailed later in this study. The ΔradDΔrecG strain is thus designated ΔradDΔrecGsupp to highlight this status. These results support the idea that in the absence of either RadD or RecG, the requirement for the other activity is increased.
To further characterize this genetic relationship, we utilized a pRC7 synthetic lethality assay. The pRC7 plasmid is a mini-F derivative that contains the lac operon. The unstable nature of the mini-F element allows it to be rapidly cured by growing cells in media without selection (40). Placing an essential or conditionally essential gene on the plasmid will act as a form of selection in the absence of antibiotic. The resulting colonies, when plated on X-gal and IPTG, will be blue if the cells retain the plasmid or white if they have lost it (Figure 3A). We used a previously reported pRC7 construct called pJJ100, featuring an expressed wild type copy of recG (41). Note that plasmid stability may be affected when deleting multiple DNA repair genes. We use this assay only to underscore the importance of retaining the RecG-encoding plasmid for growth in the ΔradDΔrecG background. We do not draw conclusions from subtle differences in the retention % of the pRC7 plasmid. Cells lacking either radD or recG alone lost the plasmid expressing RecG and produced white colonies at a frequency of 43% and 35%, respectively, of the total after 16 hours of growth (Figure 3B, C). In contrast, when the ΔradDΔrecG strain was plated, white colonies represented only 4% of the total after 16 h of growth. In addition, the white colonies were much smaller than the blue colonies, again suggesting the appearance of suppressors. To monitor suppressor appearance, we took colony counts at both 16 and 24 h. At 24 h of growth, the ΔradDΔrecG plates accumulated additional white colonies. When five of these colonies were cultured again, each reproducibly displayed a restored growth phenotype. Both the radD and recG single deletion strains produced the same colony counts at 16 and 24 h, with no evident distinction in colony size between white and blue colonies.
Figure 3.
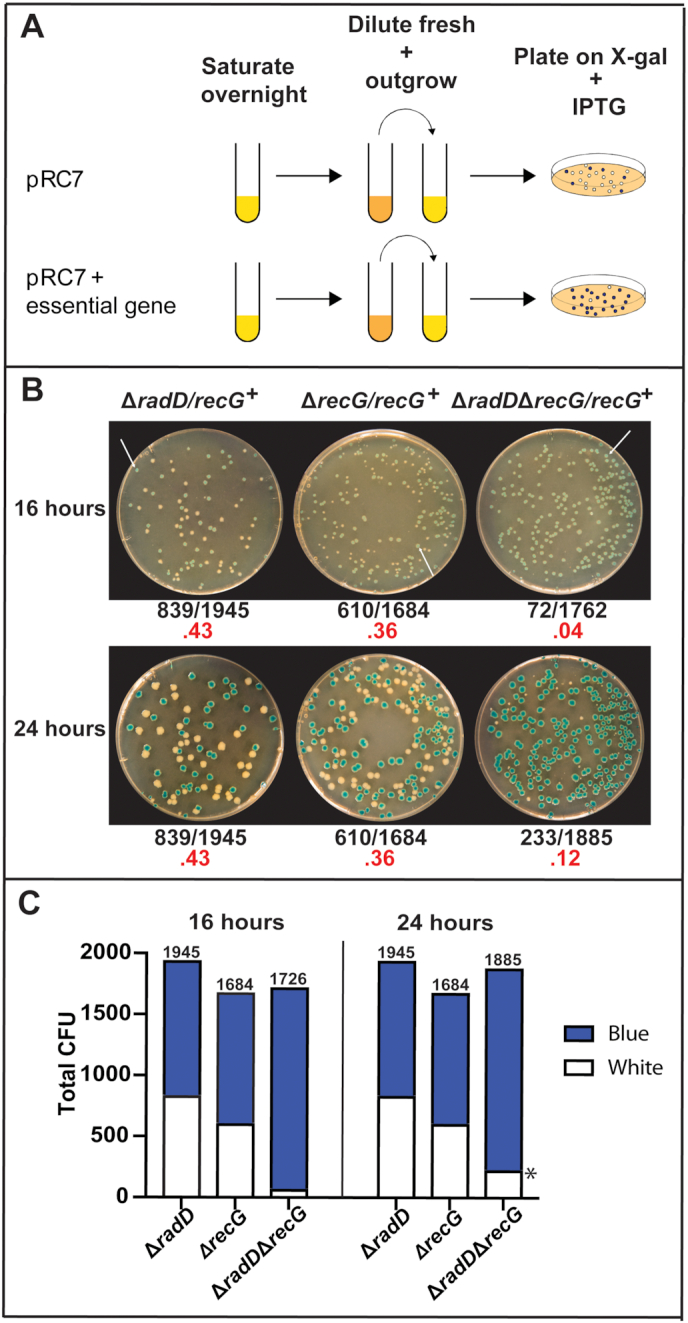
(A) Schematic of pRC7 synthetic lethality assay. (B) Images of results from the recG and radD single and double mutant pRC7 assay. Images of plates were taken at both 16 and 24 h to show the accumulation of white colonies after significant time in a radD recG double mutant. The white arrows at 16 h incubation point to blue colonies. Frequency of white colonies is highlighted in red underneath each image. (C) Stacked bar graph showing the total colony counts from each strain and their distribution of either white or blue colonies. The * denotes the appearance of suppressors that came up after 16 h colony counts.
Viability in ΔradDΔrecG is restored by suppressor mutations in priA and the recA promoter (PrecA)
The larger colony size in ΔradDΔrecG after the extended lag phase strongly suggested the presence of suppressor mutations. Eleven of the putative suppressor colonies were isolated. Mutations in priA suppress the DNA damage sensitivity of recG mutants (12,26–30). Based on this precedent, we first sequenced the priA gene in each isolate and found priA gene mutations in 10 of the 11 (Figure 4A). This suggested that priA mutations are the most common suppressors of ΔradDΔrecG (as confirmed below). Unlike the suppression observed in recG mutants alone, the suppression observed here occurs in the absence of elevated levels of DNA damage. We chose to do further studies on PriA S278A and PriA A520P. The latter suppressor has been observed in a previous study involving recG mutant suppression (31).
Figure 4.

(A) Domain layout of PriA protein and location of the 10 suppressor mutations isolated. The layout of the PriA protein is adapted from Bhattacharyya et al. (B) Layout of the RecA promoter and position of the PrecA suppressor mutation in the –10 region. Abbreviations are as follows: 3′ BD = 3′ binding domain, WH = winged helix domain, HL1 = helicase lobe 1, CRR = cysteine rich region, HL2 = helicase lobe 2, CTD = C-terminal domain.
One of the spontaneous suppressor isolates failed to produce a priA mutation when that gene was sequenced. This isolate was subjected to genomic sequencing that revealed a mutation in the recA promoter (PrecA). The base change is a T to C in the first position of the six-nucleotide Pribnow box sequence (Figure 4B). Mutations in this position will result in reduced expression of recA (43,44). RecA is the central recombinase in E. coli that facilitates homologous pairing in double strand break and daughter strand gap repair (45,46). The involvement of RecA in abandoned fork processing has been documented (13,41). Additional mutations found in the genomic sequencing of this suppressor strain are listed in Supplementary Table S1.
Validation of spontaneous suppressor mutations
To confirm the spontaneous suppressors identified, three of the mutations were separately introduced into a ΔradDΔrecG strain (adding the suppressor mutation prior to the introduction of one of the two helicase deletion mutations; with ΔradD usually added last). The PriA S278A, PriA A520P and PrecA mutations were all able to eliminate the growth defect, completely abrogating the extended lag phase of a ΔradDΔrecG strain (Figure 5A). This demonstrates that the identified mutations are responsible for the observed suppression. Each suppressor mutant was also tested for complementation by monitoring the appearance of white colonies when the pRC7-recG plasmid was introduced into the triple mutant strains. Strains in which the ΔradDΔrecG was suppressed by PriA S278A, PriA A520P or PrecA, lost the plasmid (indicating no requirement to retain RecG function) at a frequency equal to or greater than wild type strains (Figure 5B, C). Total CFU are reported after 24 h. Counts were taken at 16 and 24 h. White versus blue colony numbers stayed the same across both time points. Each of these three mutations, on their own, thus reproduce the suppression effect in its entirety.
Figure 5.

(A) A minimum of 3 Log scale OD600 versus time traces of ΔradDΔrecG strain with either priA S278A, priA A520P or PrecA suppressor mutations (red) in comparison to a wild type control (black). (B) Stacked bar graph quantifying results of pRC7 assay of ΔradDΔrecG with priA S278A, priA A520P or PrecA mutations. (C) Images of plates after 24 h for ΔradDΔrecG with priA S278A, priA A520P or PrecA suppressor mutations to show loss of plasmid increase. Frequency of white colonies is shown highlighted in red underneath each image.
The appearance of a suppressor in the recA promoter suggested to us that additional avenues of suppression of the ΔradDΔrecG growth defect might exist. We reasoned that the concentration of suppressors in the priA gene might simply reflect a multitude of SNP mutational paths to suitable functional priA suppressors, while alternative suppressors might require a more unlikely mutational change or complete inactivation of a gene or genes. To explore this idea, we abandoned the screen of spontaneous and random suppressor generation and tried a more directed approach. We made a series of triple mutants in which a candidate gene was deleted and combined with ΔradDΔrecG (in each case adding the radD deletion last) in a lac− background and tested them for suppression. Triple mutants combining deletions of the rep, ruvB, or rarA genes with ΔradDΔrecG failed to elicit suppression, with the strains very difficult to construct or maintain. However, good suppression was obtained when recF, recO, or uup deletions were introduced, as described in the next two sections.
Deleting recO or recF suppresses the ΔradDΔrecG growth defect
If reducing the concentration of RecA in the cell could suppress (the PrecA mutation), we wondered if blocking RecA loading could also suppress. The RecFOR proteins are implicated in loading RecA protein primarily in postreplication gaps (13,45–49). We made the triple ΔradDΔrecGΔrecO and ΔradDΔrecGΔrecF strains and tested them for suppression. Both strains were able to rescue the ΔradDΔrecG growth defect and to restore the appearance of white colonies to a ratio of 0.44 when the pRC7-recG plasmid was introduced (Figure 6A, B). This supports the idea that the deleterious ΔradDΔrecG phenotype involves an inability to resolve recombination intermediates being generated at a stalled replication fork or postreplication gap.
Figure 6.

(A) Stacked bar graph quantifying results of pRC7 assay of ΔradDΔrecG with either recF or recO deleted. (B) Images of plates after 24 h of results for ΔradDΔrecG with recF or recO deleted.
A full gene deletion of Uup suppresses the ΔradDΔrecG growth defect
Uup is a UvrA-like Class II ABC system that binds Holliday junctions. RadD and Uup help define at least two pathways for resolution of branched DNA intermediates during template switching in post replication gaps (33). Uup and RadD are responsible for the stabilization of tandem repeats that are susceptible to deletion (33). These deletion events mimic the RecA-mediated gap repair pathway. However, they are RecA-independent and can be mutagenic. Due to the ubiquitous nature of Holliday junctions in other pathways, we hypothesized that RadD and Uup may be involved in other repair processes. And as RecG and RadD appear to complement each other, we wondered if Uup and RecG might be involved in the same pathway. We thus decided to test if deleting uup suppresses the defect seen in the ΔradDΔrecG mutant strain.
As seen with the suppressors already described, deleting uup rescued colony size and suppressed the growth defect of ΔradDΔrecG (Figure 7). The triple ΔradDΔrecGΔuup mutant did produce a growth lag, similar to that observed in the ΔrecG single mutant. Combining Δuup with ΔradDΔrecG was also examined in mini-F plasmid assay. Here, loss of Uup function restored the appearance of white colonies to levels comparable to that seen for wild type strains. Thus, a deletion of uup appears to be effective in suppressing the growth defect of the ΔradDΔrecG double deletion mutant strain.
Figure 7.
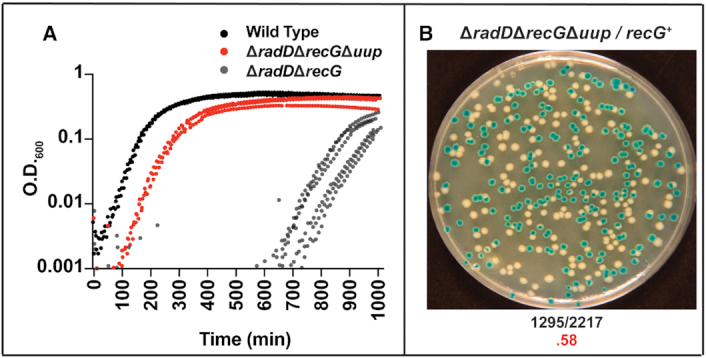
(A) A minimum of 3 Log scale OD600 versus time traces of ΔradDΔrecGΔuup (red) compared to wild type (black) and ΔradDΔrecG (gray). (B) Images of plates after 24 h of pRC7 results of ΔradDΔrecGΔuup strain with frequency of white colony formation highlighted below in red.
Effects of deleting combinations of uup, radD, and recG on sensitivity to DNA damaging agents
We wished to explore the potential connection between uup and recG further, determining whether the effects of suppression by deleting uup could be extended to conditions of stress. We treated all possible radD, recG and uup gene deletion combinations with various DNA damaging agents (Figure 8). The dose used was tailored to the high sensitivity of the ΔrecG and ΔradDΔrecG strains to DNA damaging agents. Multiple survival patterns were observed, varying not only with the mutants employed but also with the different DNA damaging agents. The latter effects presumably reflect variations in the pathways with which particular types of DNA lesions are normally resolved.
Figure 8.

(A) Sensitivity assays with all possible radD, recG and uup deletion combinations. Spot plates indicate compound, dose, and dilution above each plate. LB, Cipro, NFZ, HU, MMC, Trim and UV are Luria Broth, ciprofloxacin, nitrofurazone, hydroxyurea, mitomycin C, trimethoprim and ultraviolet light, respectively. The ΔradDΔrecG strain used in Figure 8 has undergone an extra overnight growth period so that its treatment is consistent with that of the other strains. Its facile growth on some of these plates demonstrates that it has picked up a suppressor. It is included for the sake of completion but is designated ΔradDΔrecGsupp to highlight this status. (B) Sensitivity assays with uup recG and ruvB gene deletion combinations. Damaging agent, dose and dilution are listed above each plate.
Pattern 1
Loss of Uup or RadD function alone had no significant effects on their own with any DNA damaging agent (Figure 8A). Loss of Uup and RadD together also had minimal effects except in the cases of Mitomycin C (MMC) and Trimethoprim (Trim) (increased sensitivity of the double deletion mutant has also been noted for Ciprofloxacin (Cipro) at levels higher than used here (33)). These results are consistent with earlier observations and provide one rationale for why Uup and RadD were largely overlooked until recently.
Pattern 2
Deletion of uup in combination with recG strongly suppressed the high sensitivity of ΔrecG strains to Cipro, Nitrofurazone (NFZ), Hydroxyurea (HU), and MMC. This effect is not seen for either Trimethoprim or UV irradiation. Survival on MMC wasn’t as greatly enhanced as the other three agents. At higher doses of NFZ and Cipro, the ΔuupΔrecG began to exhibit some sensitivity when compared to wild type (Supplemental Figure S2). This result in general suggests that many of the deleterious effects of a recG deletion (but not all) are dependent on the presence of a functional Uup protein. In the Discussion, we offer a hypothesis for a functional relationship between Uup and RecG that can explain these observations. The result also indicates that RadD can make a substantial contribution to survival when both RecG and Uup are missing.
Pattern 3
The addition of a radD deletion to construct the ΔradDΔrecGΔuup triple mutant generally eliminates the suppressive effect of a uup deletion on the DNA damage sensitivity of a ΔrecG strain. In some cases (Trim, MMC), the sensitivity of the triple mutant is somewhat greater than that seen with ΔrecG alone. This result again speaks to the existence of multiple, partially redundant pathways for repair, with a key alternative path blocked when radD is eliminated. Thus, although growth rates are restored under normal conditions with the triple mutant, it remains highly sensitive to elevated levels of DNA damage.
Pattern 4 (Figure 8B)
The suppression that a uup deletion confers on a ΔrecG phenotype does not extend to ruvB. RuvB is part of the resolvasome that is responsible for the resolution of HJs and replication fork processing (50,51). RuvB is also involved in replication fork reversal (3,52–54). There are no conditions in our trials where Δuup increases the survival of a ΔruvB strain, and one condition (MMC) where the sensitivity to DNA damage is exacerbated. The suppressive effects of a uup deletion are thus specific to recG.
We note that the ΔradDΔrecG strain used in Figure 8 has undergone an extra overnight growth period so that its treatment is consistent with that of the other strains. Its facile growth on some of these plates demonstrates that it has picked up a suppressor. It is included for the sake of completion but is designated ΔradDΔrecGsupp to highlight this status.
The various DNA-damaging agents utilized in Figure 8 function in different ways to inflict damage and affect replisome progress. Ciprofloxacin is a quinolone that inhibits DNA Gyrase. Inhibition of DNA Gyrase leads to a replication roadblock. Replisome stalling occurs ∼10 bases upstream of the halted gyrase cleavage site (55). Nitrofurazone at low doses induces base lesions in the form of N2-alkyl deoxyguanosine that relies on the nucleotide excision repair machinery to repair (56,57). Hydroxyurea is an inhibitor of ribonucleotide reductase and will deplete the nucleotide pool leading to replication stalling and disassociation (58). All three of these compounds have the potential to trigger formation of a reversed fork intermediate. Mitomycin C creates protein and DNA crosslinks that can pose stalling risks to the replisome machinery. Trimethoprim triggers rapid thymine depletion which then cascades to further DNA damage (59).
Uup suppresses the growth defect but not cell filamentation in the ΔradDΔrecG strain
We also wished to determine the status of the cells when the ΔradDΔrecG strain is suppressed by Δuup. We had previously observed that strains lacking Uup function filament rather extensively under normal growth conditions. As seen in Figure 9, strains lacking RecG did not alleviate the filamentation, but rather exacerbated it. The ΔradDΔrecGΔuup cells filamented extensively with the average cell length of these cells exceeding 19 μm. Thus, even if the growth defect of cells lacking RadD and RecG is suppressed by deleting Uup, deficiencies in replication, repair, and cell division are still abundantly evident.
Figure 9.
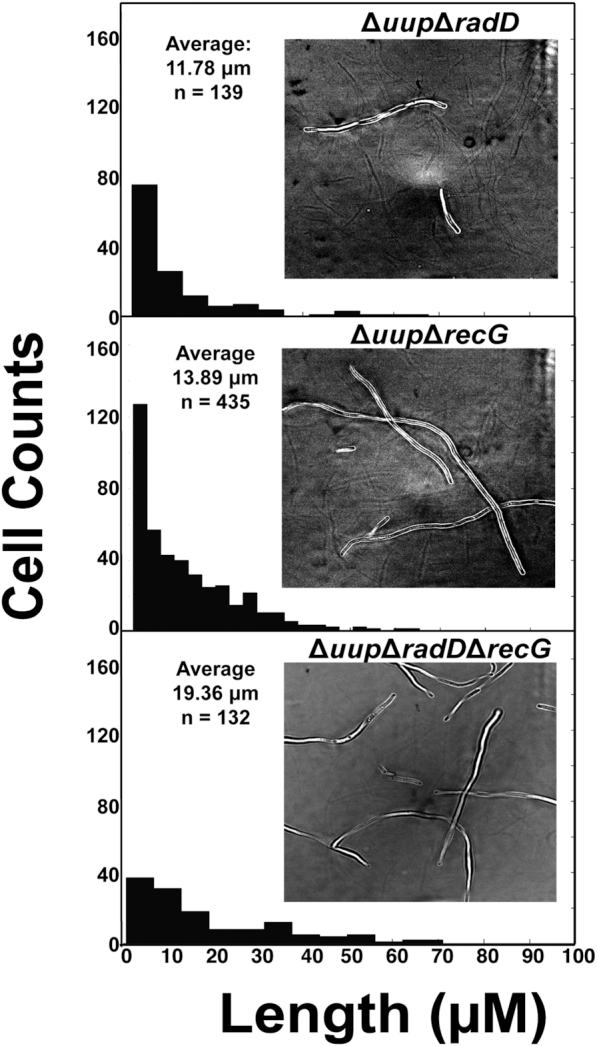
Cell filamentation measurements of ΔuupΔradD, ΔuupΔrecG and ΔuupΔradDΔrecG strains. Average length, number of cells and a bright field image are displayed for each strain.
Uup is required for SDR-dependent growth in rnhA dnaAts mutants
To further investigate the relationship between Uup and RecG, we explored a process with which RecG is closely associated, stable DNA replication or SDR. SDR is origin-independent replication, initiating at readily detectable levels in cells lacking the function of RnaseH or RecG (60–62). In E. coli rnhA mutants, SDR supports cell growth in the absence of oriC function, presumably via replication initiation at unprocessed R-loops scattered about the genome (61). In E. coli recG mutants, SDR is largely restricted to the terminus region where over-replication is initiated when RecG is unable to resolve structures created by fork collisions (62). Cell growth in the absence of oriC is not supported in a ΔrecG strain unless additional mutations in tus (to allow forks to escape the terminus region) and rpoB (to relieve replication-transcription conflicts) are also introduced (62). An rnhA recG double mutant is inviable and cannot be constructed, (63) presumably because RecG is needed to process the fork collisions that occur when oriC-independent replication is initiated in the absence of RnaseH. We reasoned that if Uup acted upstream of RecG, an absence of Uup function might also affect growth in a strain lacking RnaseH when oriC function was compromised.
Results are presented in Figure 10. We began with a dnaA(46) mutation which supports normal oriC-dependent replication at 30°C but not at 42°C (64). To this we added a ΔrnhA mutation, a Δuup mutation, or both. We also tested a ΔrnhAΔuup double mutant without the dnaA mutation. At 30°C, the WT and all of the mutant combinations grow similarly. At 42°C, the WT and dnaA(46)ΔrnhA double mutant grew as expected. The ΔrnhAΔuup double mutant, unencumbered with a temperature sensitive DnaA protein, also grew. The dnaA(46) single mutant does not grow, again as expected. The Δuup dnaA(46) control double mutant did not grow. Most important, when Δuup was added to dnaA(46)ΔrnhA, the growth observed in the dnaA(46)ΔrnhA double mutant was entirely eliminated. As growth in this mutant is dependent on RecG function, the result provides another possible connection between Uup and RecG.
Figure 10.
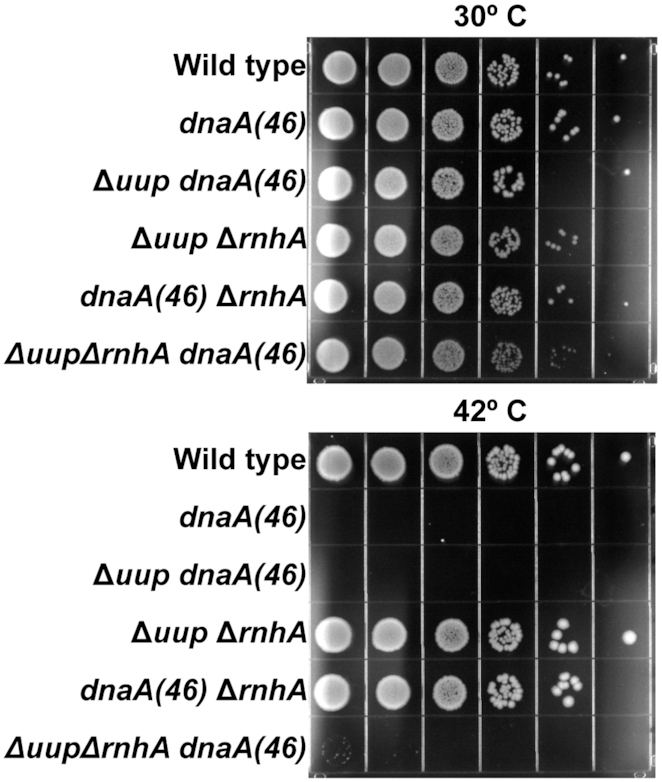
Spot plates grown at permissive (30°C) or restrictive (42°C) temperature. The dnaA(46) allele cannot grow at 42°C unless accompanied by rnhA deletion shown in the first and fourth rows of both plates. Adding a uup deletion to dnaA(46)ΔrnhA strain restores temperature sensitive growth as shown on the last row of both plates.
Mutations in priA and PrecA suppress ΔradDΔrecG defect by mitigating ΔrecG effects but still exhibit high SOS induction
The suppression of recG sensitivity to damaging agents by a uup deletion made us question if all accumulated suppressors are directed at alleviating the consequences of deleting recG. The double mutants of ΔrecG priA S278A, ΔrecG priA A520P, and ΔrecG PrecAwere made and treated with cipro or NFZ (Figure 11). All three suppressors were able to rescue survival of the recG mutant. Figure 2 shows that despite the presence of a suppressor as a result of extended growth prior to the experiment (see discussion of Figure 2), a ΔradDΔrecG strain still shows high SOS induction. We wanted to determine if SOS induction again occurred when a defined suppressor was present, by incorporating both priA and PrecA suppressor mutations into a ΔradD strain before deleting recG. We found that both priA A520P and the PrecA mutation (in the ΔradDΔrecG background) exhibited increased levels of SOS in the absence of stress. The priA S278A mutant, however, does not (Supplemental Figure S1). The results suggest that the main effect of the suppressors is to abrogate the deleterious effects of the recG deletion. The results also indicate that the priA suppressor mutations are not all equivalent in their effects on PriA activity.
Figure 11.

Sensitivity assays exploring ΔrecG cells with either uup, priA S278A, priA A520P or PrecA mutation added. Compound, dose and dilution are labeled at the top of each plate. LB, cipro and NFZ are Luria Broth, ciprofloxacin and nitrofurazone, respectively.
DISCUSSION
This work leads to two major conclusions with several subsidiary observations. One major result is that loss of both RadD and RecG function generates a severe growth defect in E. coli under normal growth conditions in rich media but otherwise in the absence of stress. At least some cells survive to generate suppressor mutations. This indicates that replisome challenges requiring either RecG or RadD intervention are a feature of virtually every replication cycle. The second conclusion is that RadD and Uup are both important functions in the repair processes involving replication fork stalling and the processing of postreplication gaps. RadD is essential to growth in the absence of RecG. Uup appears to function in a pathway or pathways that also feature RecG, likely acting upstream of RecG in at least some key situations.
Subsidiary observations include the following: (i) RadD and RecG function in distinct pathways that both contribute to maintenance of genomic integrity during replication. The work highlights the importance of RadD in at least one of those pathways. (ii) Proteins that create structures or situations requiring the action of RadD or RecG include (but are probably not limited to) RecA, RecO, RecF, Uup and PriA. (iii) Suppression of the ΔrecGΔradD phenotype relieves the barrier to growth. However, the cells remain very sensitive to DNA damaging agents. The RadD and RecG proteins play an important role in DNA repair that cannot be completely bypassed by alternative pathways. (iv) There appears to be some set of lesions or replication barriers for which pathways involving RecG or RadD are the primary paths to repair. At least for these events, RadD and RecG are among the first responders. Translesion (TLS) DNA synthesis repair pathways are still intact, but they are unable to overcome the damage that persists in a ΔradDΔrecG strain.
The identification of suppressors allows us to outline likely paths for DNA intermediate processing (Figure 12A). The initial DNA substrate generated during replication or as a result of replisome stalling is processed by the RecA recombinase via the RecFOR pathway. With all proteins present, the RecG or RadD-dependent pathways facilitate productive repair and resolution. In the absence of both proteins, persistent DNA intermediates will become targets for deleterious processing due to the perturbation of normal repair flow. The types of branched DNA intermediates likely to be targets for these resolution pathways, or at least some of them, are shown in Figure 12B.
Figure 12.

(A) Functional scheme showing how each gene fits into the various stages of intermediate processing. In the absence of RecG or RadD, a buildup of intermediates leads to toxic processing that is dependent on Uup and PriA. (B) Schematic demonstrating repair activities facilitated by RecG or RadD in a postreplication gap and stalled replication fork. Either protein may be capable of branch migrating Holliday Junctions formed in postreplication gaps or to revert a regressed fork into a substrate suitable for replication restart.
The general view of two repair pathways, one with RecG (sometimes in partnership with Uup) and the other with RadD, is based not only on the growth defect and suppression patterns, but also on the DNA damage sensitivity patterns and observed effects on SDR. Many of the deleterious effects of a recG deletion depend upon the continued presence of Uup. The DNA damage sensitivity to Cipro, NFZ, HU and MMC exhibited in a ΔrecG strain is greatly ameliorated if uup is also deleted. In addition, the SDR that supports oriC-independent growth in a strain lacking RnaseH is suppressed if Uup is missing. While not constituting final proof, all of these observations lead to an obvious hypothesis: that Uup functions upstream of RecG.
Even if our hypothesis that Uup functions upstream of RecG is correct in some contexts, Uup is not required in all situations in which RecG contributes. We cite four examples of data indicating that Uup is not needed for RecG function in all contexts: (a) In no case are the effects of a uup deletion as phenotypically deleterious as a recG deletion. (b) A lack of Uup eliminates growth in a dnaA(46)ΔrnhA double mutant at nonpermissive temperatures. A ΔrnhAΔrecG double mutant cannot be constructed, with no viability at any temperature with or without a dnaA(46) mutation. (c) Whereas a ΔrecGΔradD strain cannot grow, a ΔuupΔradD strain grows well in the absence of stress (33). (d) Eliminating Uup does not affect the sensitivity of a recG deletion to UV irradiation or Trimethoprim although it does suppress the ΔrecG sensitivity to a number of other agents. Overall, the results suggest an association of Uup with RecG that is limited to particular situations or substrates.
The very strong growth defect in a ΔradDΔrecG strain, coupled to the reliable generation of numerous suppressor mutations, provides a powerful experimental entre into the workings of the underlying repair pathways. The subsidiary observations come largely from the identity of the suppressor mutations. Spontaneous suppressors identified to date compromise the function of PriA (many) or arise in the recA promoter so as to lower RecA expression (one). It is unlikely that we have saturated the possibilities for suppression. The concentration of suppressors in the priA gene may simply reflect a multitude of mutational paths to suitable functional priA suppressors. Many single nucleotide polymorphisms in the priA gene appear to alter PriA function in a suitable manner. Facile success in priA can have the consequence of obscuring other avenues to suppression. Alternatives might require a more unlikely mutational change or complete inactivation of a gene or genes. By exploring a few logical possibilities, we have found additional suppressors that affect RecA loading onto SSB-coated ssDNA (elimination of RecO or RecF) or eliminate Uup function. These suppressors do not immediately suggest a common mechanistic origin. Reduction in RecA-mediated fork reversal at a stalled replisome, or RecA-mediated strand exchange in a postreplication gap, may reduce the numbers of branched intermediates requiring intervention by the RecG or RadD helicase functions. Rescue of the ΔradDΔrecG strain's viability by eliminating the RecA-loading functions RecO or RecF supports this idea. In the absence of RecG or RadD function to restore reversed forks or resolve RecA intermediates in post replication gaps, PriA may engage in toxic activity (28). PriA has figured prominently in the suppression of recG phenotypes in earlier studies (15,20,21,31,62,64).
The uup suppression is more difficult to explain mechanistically but may arise from the putative functional relationship between Uup and RecG. Based entirely on its structural relationship to UvrA and its documented binding to Holliday junctions in vitro, we have hypothesized that Uup is a DNA scanner that binds to Holliday junctions. Thus bound, Uup may recruit other repair functions to deal with the bound DNA species, with RecG now a prime candidate for recruitment. Based on the positive effects of a uup deletion on the DNA damage sensitivity of a strain missing RecG function, a plausible (but doubtless not unique) scenario can be put forward as a working hypothesis. Uup scans DNA for Holliday junctions and binds to them. RecG is recruited, and then migrates the branch to either restore a fork structure or resolve an intermediate in postreplication gap repair. If RecG is missing, Uup may bind to the Holliday junction in such a way as to block or constrain other potential paths of resolution. If Uup is also missing, the deleterious effect of RecG loss is ameliorated as other paths take over. RadD represents an important component of the major alternative path.
The work further defines the function of the enigmatic helicase RadD. Like RecG, RadD appears to be involved in many repair processes. Deletion of both helicases results in a nearly inviable strain unless accompanied by suppressor mutations. The requirement for both proteins can be explained by a few different mechanisms. (i) RadD has complementary activity to RecG. RadD can bind fork structures, suppress crossover products, and has an interaction with the replisome hub protein SSB (33,35). These observations, while seemingly disparate, become logical when combined with the severe growth defect of the ΔradDΔrecG strain. RadD can supplement for lost RecG function at an abandoned fork or resolve D-loops formed in gaps by RecA to prevent SDR initiation. (ii) RadD can be viewed as a first responder to replisome roadblocks. This idea establishes RadD as a ‘housekeeping’ helicase localized to the replisome or gaps through its SSB interaction.
The dependence of cells on either RadD or RecG present exciting new avenues of study. RadD can possibly provide insights on how specific lesions dictate repair pathways. Uup may be a modulator of RecG. Further investigation of these ideas is currently underway.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Christian Rudolph for the generous donation of the pJJ100 plasmid.
Contributor Information
Zachary J Romero, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706, USA.
Stefanie H Chen, Biotechnology Program, North Carolina State University, Raleigh, NC 27695, USA.
Thomas Armstrong, Molecular Horizons Institute and School of Chemistry, University of Wollongong, Wollongong, Australia; Illawarra Health and Medical Research Institute, Wollongong, Australia.
Elizabeth A Wood, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706, USA.
Antoine van Oijen, Molecular Horizons Institute and School of Chemistry, University of Wollongong, Wollongong, Australia; Illawarra Health and Medical Research Institute, Wollongong, Australia.
Andrew Robinson, Molecular Horizons Institute and School of Chemistry, University of Wollongong, Wollongong, Australia; Illawarra Health and Medical Research Institute, Wollongong, Australia.
Michael M Cox, Department of Biochemistry, University of Wisconsin-Madison, Madison, WI 53706, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [RM1 GM130450 to M.M.C., A.v.O.]; Australian Research Council (Laureate Research Fellowship to A.v.O.); National Health and Medical Research Council [APP1165135 to A.R., A.v.O.]. Funding for open access charge: National Institutes of Health [RM1 GM130450].
Conflict of interest statement. None declared.
REFERENCES
- 1. Courcelle J., Belle J.J., Courcelle C.T.. When replication travels on damaged templates: bumps and blocks in the road. Res. Microbiol. 2004; 155:231–237. [DOI] [PubMed] [Google Scholar]
- 2. Lovett S.T. Filling the gaps in replication restart pathways. Mol. Cell. 2005; 17:751–752. [DOI] [PubMed] [Google Scholar]
- 3. Michel B., Sandler S.J.. Replication restart in bacteria. J. Bacteriol. 2017; 199:e00102-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kath J.E., Jergic S., Heltzel J.M., Jacob D.T., Dixon N.E., Sutton M.D., Walker G.C., Loparo J.J.. Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc. Natl Acad. Sci. U.S.A. 2014; 111:7647–7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kath J.E., Chang S., Scotland M.K., Wilbertz J.H., Jergic S., Dixon N.E., Sutton M.D., Loparo J.J.. Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res. 2016; 44:1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nelson S.W., Benkovic S.J.. Response of the bacteriophage T4 replisome to non-coding lesions and regression of a stalled replication fork. J. Mol. Biol. 2010; 401:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McInerney P., O’Donnell M.. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 2004; 279:21543–21551. [DOI] [PubMed] [Google Scholar]
- 8. Mezzina M., Menck C.F., Courtin P., Sarasin A.. Replication of simian virus 40 DNA after UV irradiation: evidence of growing fork blockage and single-stranded gaps in daughter strands. J. Virol. 1988; 62:4249–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henrikus S.S., Wood E.A., McDonald J.P., Cox M.M., Woodgate R., Goodman M.F., van Oijen A.M., Robinson A.. DNA polymerase IV primarily operates outside of DNA replication forks in Escherichia coli. PLoS Genet. 2018; 14:e1007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson A., McDonald J.P., Caldas V.E.A., Patel M., Wood E.A., Punter C.M., Ghodke H., Cox M.M., Woodgate R., Goodman M.F. et al.. Regulation of mutagenic DNA polymerase V activation in space and time. PLoS Genet. 2015; 11:e1005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M.. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012; 19:417–423. [DOI] [PubMed] [Google Scholar]
- 12. Yeeles J.T., Poli J., Marians K.J., Pasero P.. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013; 5:a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robu M.E., Inman R.B., Cox M.M.. RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl Acad. Sci. USA. 2001; 98:8211–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bradley A.S., Baharoglu Z., Niewiarowski A., Michel B., Tsaneva I.R.. Formation of a stable RuvA protein double tetramer is required for efficient branch migration in vitro and for replication fork reversal in vivo. J. Biol. Chem. 2011; 286:22372–22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaktaji R.P., Lloyd R.G.. PriA supports two distinct pathways for replication restart in UV-irradiated Escherichia coli cells. Mol. Microbiol. 2003; 47:1091–1100. [DOI] [PubMed] [Google Scholar]
- 16. Baharoglu Z., Petranovic M., Flores M.J., Michel B.. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 2006; 25:596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weaver G.M., Mettrick K.A., Corocher T.A., Graham A., Grainge I.. Replication fork collapse at a protein-DNA roadblock leads to fork reversal, promoted by the RecQ helicase. Mol. Microbiol. 2019; 111:455–472. [DOI] [PubMed] [Google Scholar]
- 18. Poole L.A., Cortez D.. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017; 52:696–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gregg A.V., McGlynn P., Jaktaji R.P., Lloyd R.G.. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell. 2002; 9:241–251. [DOI] [PubMed] [Google Scholar]
- 20. Lloyd R.G., Rudolph C.J.. 25 years on and no end in sight: a perspective on the role of RecG protein. Curr. Genet. 2016; 62:827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGlynn P., Lloyd R.G.. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl Acad. Sci. U.S.A. 2001; 98:8227–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singleton M.R., Scaife S., Wigley D.B.. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001; 107:79–89. [DOI] [PubMed] [Google Scholar]
- 23. Warren G.M., Stein R.A., McHaourab H.S., Eichman B.F.. Movement of the RecG motor domain upon DNA binding is required for efficient fork reversal. Int. J. Mol. Sci. 2018; 19:3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whitby M.C., Ryder L., Lloyd R.G.. Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell. 1993; 75:341–350. [DOI] [PubMed] [Google Scholar]
- 25. Whitby M.C., Lloyd R.G.. Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J. Biol. Chem. 1998; 273:19729–19739. [DOI] [PubMed] [Google Scholar]
- 26. Marians K.J. PriA-directed replication fork restart in Escherichia coli. Trends Biochem. Sci. 2000; 25:185–189. [DOI] [PubMed] [Google Scholar]
- 27. Sandler S.J., Marians K.J.. Role of PriA in replication fork reactivation in Escherichia coli. J. Bacteriol. 2000; 182:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Windgassen T.A., Wessel S.R., Bhattacharyya B., Keck J.L.. Mechanisms of bacterial DNA replication restart. Nucleic Acids Res. 2018; 46:504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Windgassen T.A., Leroux M., Sandler S.J., Keck J.L.. Function of a strand-separation pin element in the PriA DNA replication restart helicase. J. Biol. Chem. 2019; 294:2801–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Windgassen T.A., Leroux M., Satyshur K.A., Sandler S.J., Keck J.L.. Structure-specific DNA replication-fork recognition directs helicase and replication restart activities of the PriA helicase. Proc. Natl Acad. Sci. U.S.A. 2018; 115:E9075–E9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Al-Deib A.A., Mahdi A.A., Lloyd R.G.. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J. Bacteriol. 1996; 178:6782–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F.U., Kerner M.J., Frishman D.. Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics. 2008; 9:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romero Z.J., Armstrong T.J., Henrikus S.S., Chen S.H., Glass D.J., Ferrazzoli A.E., Wood E.A., Chitteni-Pattu S., van Oijen A.M., Lovett S.T. et al.. Frequent template switching in postreplication gaps: suppression of deleterious consequences by the Escherichia coli Uup and RadD proteins. Nucleic Acids Res. 2020; 48:212–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen S.H., Byrne R.T., Wood E.A., Cox M.M.. Escherichia coli radD (yejH) gene: a novel function involved in radiation resistance and double-strand break repair. Mol. Microbiol. 2015; 95:754–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen S.H., Byrne-Nash R.T., Cox M.M.. Escherichia coli RadD protein functionally interacts with the single-stranded DNA-binding protein. J. Biol. Chem. 2016; 291:20779–20786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Negro V., Krin E., Aguilar Pierle S., Chaze T., Giai Gianetto Q., Kennedy S.P., Matondo M., Mazel D., Baharoglu Z.. RadD contributes to R-loop avoidance in sub-MIC tobramycin. MBio. 2019; 10:e01173-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buss J.A., Kimura Y., Bianco P.R.. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 2008; 36:7029–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shereda R.D., Bernstein D.A., Keck J.L.. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 2007; 282:19247–19258. [DOI] [PubMed] [Google Scholar]
- 39. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bernhardt T.G., de Boer P.A.. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol. Microbiol. 2004; 52:1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahdi A.A., Buckman C., Harris L., Lloyd R.G.. Rep and PriA helicase activities prevent RecA from provoking unnecessary recombination during replication fork repair. Genes Dev. 2006; 20:2135–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sliusarenko O., Heinritz J., Emonet T., Jacobs-Wagner C.. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 2011; 80:612–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weisemann J.M., Weinstock G.M.. Direct selection of mutations reducing transcription or translation of the recA gene of Escherichia coli with a recA-lacZ protein fusion. J. Bacteriol. 1985; 163:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weisemann J.M., Weinstock G.M.. The promoter of the recA gene of Escherichia coli. Biochimie. 1991; 73:457–470. [DOI] [PubMed] [Google Scholar]
- 45. Cox M.M. The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 2003; 57:551–577. [DOI] [PubMed] [Google Scholar]
- 46. Lusetti S.L., Cox M.M.. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 2002; 71:71–100. [DOI] [PubMed] [Google Scholar]
- 47. Lenhart J.S., Brandes E.R., Schroeder J.W., Sorenson R.J., Showalter H.D., Simmons L.A.. RecO and RecR are necessary for RecA loading in response to DNA damage and replication fork stress. J. Bacteriol. 2014; 196:2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morimatsu K., Wu Y., Kowalczykowski S.C.. RecFOR proteins target RecA protein to a DNA gap with either DNA or RNA at the 5′ terminus: implication for repair of stalled replication forks. J. Biol. Chem. 2012; 287:35621–35630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Webb B.L., Cox M.M., Inman R.B.. Recombinational DNA repair: the RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell. 1997; 91:347–356. [DOI] [PubMed] [Google Scholar]
- 50. Wyatt H.D., West S.C.. Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 2014; 6:a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dickman M.J., Ingleston S.M., Sedelnikova S.E., Rafferty J.B., Lloyd R.G., Grasby J.A., Hornby D.P.. The RuvABC resolvasome. Eur. J. Biochem. 2002; 269:5492–5501. [DOI] [PubMed] [Google Scholar]
- 52. Muller B., West S.C.. Processing of Holliday junctions by the Escherichia coli RuvA, RuvB, RuvC and RecG proteins. Experientia. 1994; 50:216–222. [DOI] [PubMed] [Google Scholar]
- 53. Michel B., Grompone G., Florès M.J., Bidnenko V.. Multiple pathways process stalled replication forks. Proc. Natl Acad. Sci. U.S.A. 2004; 101:12783–12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seigneur M., Bidnenko V., Ehrlich S.D., Michel B.. RuvAB acts at arrested replication forks. Cell. 1998; 95:419–430. [DOI] [PubMed] [Google Scholar]
- 55. Wentzell L.M., Maxwell A.. The complex of DNA gyrase and quinolone drugs on DNA forms a barrier to the T7 DNA polymerase replication complex. J. Mol. Biol. 2000; 304:779–791. [DOI] [PubMed] [Google Scholar]
- 56. Olive P.L. Nitrofurazone-induced DNA damage to tissues of mice. Chem. Biol. Interact. 1978; 20:323–331. [DOI] [PubMed] [Google Scholar]
- 57. Ona K.R., Courcelle C.T., Courcelle J.. Nucleotide excision repair is a predominant mechanism for processing nitrofurazone-induced DNA damage in Escherichia coli. J. Bacteriol. 2009; 191:4959–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sogo J.M., Lopes M., Foiani M.. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002; 297:599–602. [DOI] [PubMed] [Google Scholar]
- 59. Giroux X., Su W.L., Bredeche M.F., Matic I.. Maladaptive DNA repair is the ultimate contributor to the death of Trimethoprim-Treated cells under aerobic and anaerobic conditions. Proc. Natl Acad. Sci. U.S.A. 2017; 114:11512–11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997; 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Torrey T.A., Kogoma T.. Genetic analysis of constitutive stable DNA replication in rnh mutants of Escherichia coli K12. Mol. Gen. Genet. 1987; 208:420–427. [DOI] [PubMed] [Google Scholar]
- 62. Midgley-Smith S.L., Dimude J.U., Taylor T., Forrester N.M., Upton A.L., Lloyd R.G., Rudolph C.J.. Chromosomal over-replication in Escherichia coli recG cells is triggered by replication fork fusion and amplified if replichore symmetry is disturbed. Nucleic Acids Res. 2018; 46:7701–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hong X., Cadwell G.W., Kogoma T.. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J. 1995; 14:2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Michel B., Sinha A.K., Leach D.R.F.. Replication fork breakage and restart in Escherichia coli. Microbiol. Mol. Biol. Rev. 2018; 82:e00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guyer M.S., Reed R.R., Steitz J.A., Low K.B.. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb. Symp. Quant. Biol. 1981; 45:135–140. [DOI] [PubMed] [Google Scholar]
- 66. Blattner F.R., Plunkett G.R., Bloch C.A., Perna N.T., Burland V., Riley M., Collado V.J., Glasner J.D., Rode C.K., Mayhew G.F. et al.. The complete genome sequence of Escherichia coli K-12. Science. 1997; 277:1453–1474. [DOI] [PubMed] [Google Scholar]
- 67. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hirota Y., Mordoh J., Jacob F.. On the process of cellular division in Escherichia coli. 3. Thermosensitive mutants of Escherichia coli altered in the process of DNA initiation. J. Mol. Biol. 1970; 53:369–387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.