


Abstract
Trypanosoma brucei is a parasitic protozoan that undergoes a complex life cycle involving insect and mammalian hosts that present dramatically different nutritional environments. Mitochondrial metabolism and gene expression are highly regulated to accommodate these environmental changes, including regulation of mRNAs that require extensive uridine insertion/deletion (U-indel) editing for their maturation. Here, we use high throughput sequencing and a method for promoting life cycle changes in vitro to assess the mechanisms and timing of developmentally regulated edited mRNA expression. We show that edited CYb mRNA is downregulated in mammalian bloodstream forms (BSF) at the level of editing initiation and/or edited mRNA stability. In contrast, edited COIII mRNAs are depleted in BSF by inhibition of editing progression. We identify cell line-specific differences in the mechanisms abrogating COIII mRNA editing, including the possible utilization of terminator gRNAs that preclude the 3′ to 5′ progression of editing. By examining the developmental timing of altered mitochondrial mRNA levels, we also reveal transcript-specific developmental checkpoints in epimastigote (EMF), metacyclic (MCF), and BSF. These studies represent the first analysis of the mechanisms governing edited mRNA levels during T. brucei development and the first to interrogate U-indel editing in EMF and MCF life cycle stages.
INTRODUCTION
Trypanosoma brucei is a flagellated, parasitic protozoan and the causative agent of human African trypanosomiasis and one of the parasitic trypanosomes that cause nagana in cattle and other domesticated livestock in 36 sub-Saharan African countries, where over 70 million people are at risk of infection (1,2). The parasite has a digenetic life cycle in which it transitions through several developmental stages, each with distinct cell morphologies and transcriptomic, proteomic, and metabolic profiles (3,4). T. brucei is transmitted to its mammalian hosts by the bite of infected tsetse flies. Infective metacyclic form (MCF) trypomastigotes are injected into the mammalian bloodstream and subcutaneous tissues when the tsetse fly takes a blood meal. The shift in temperature and nutritional environment initiates differentiation from MCF to proliferative, slender bloodstream form (BSF) trypomastigotes. As the slender BSF parasites divide and parasitemia increases, a subpopulation of parasites responds to increasing concentrations of stumpy induction factor by differentiating into the quiescent, stumpy BSF (5–8). The stumpy BSF parasites are unable to revert back to proliferating slender BSF and are pre-adapted for survival in the tsetse fly insect vector (9–13). In the tsetse fly midgut, stumpy BSF must rapidly transition to the proliferative procyclic form (PCF) to survive the new nutritional environment and insect immune response (11,14–18). PCF trypomastigotes begin to migrate towards the salivary glands. During this journey, the PCF differentiates into the epimastigote form (EMF). EMF parasites attach to the epithelium of the salivary gland and proliferate to colonize the salivary glands. Asymmetrical cell division of the epithelium-bound EMF generates a daughter cell that matures into infective, quiescent MCF that are pre-adapted for survival in the mammalian host (19).
For the parasites to survive in distinct hosts, they modulate their metabolism to exploit the various nutrients available in the mammalian tissues or the tsetse fly. A major component of this metabolic shift is the developmental regulation of mitochondrial function. While T. brucei resides in the tsetse fly midgut as the PCF, it must survive in a niche that is deprived of glucose due to the rapid catabolism of glucose by the tsetse fly (11). Instead, the parasite relies heavily on proline metabolism to fuel oxidative phosphorylation in the mitochondrion (11). As a consequence, the electron transport chain (ETC) of PCF mitochondria is robust and resembles the classical ETC found in many other model eukaryotic organisms (20,21). However, once T. brucei enters into the mammalian bloodstream, it is able to utilize the high abundance of glucose and, once differentiated to the slender BSF, the parasite relies primarily on aerobic glycolysis to meet its ATP demand (21–24). This is coupled with the downregulation of many mitochondrial functions in BSF, including the lack of cytochromes (25). Nevertheless, the mitochondrial FoF1-ATP synthase and its mitochondrially-encoded subunit, ATPase subunit 6 (A6), are still essential in BSF parasites, where the FoF1-ATP synthase runs in reverse and hydrolyzes ATP to generate mitochondrial membrane potential (26).
Similar to other eukaryotes, the T. brucei mitochondrial genome encodes subunits of several respiratory complexes of the ETC. However, unlike many other eukaryotes, 12 out of 18 mitochondrially encoded mRNAs in T. brucei are non-translatable immediately following their transcription because they do not encode functional open reading frames (ORFs) (27–31). To achieve translatable ORFs in these select mitochondrial mRNAs, uridine residues must be post-transcriptionally added to or removed from the mRNA in a complex process called uridine insertion/deletion (U-indel) mRNA editing. For several mRNAs that are edited throughout their lengths, termed pan-edited mRNAs, modification is so extensive that abundant uridine insertions nearly double the size of the mRNA. The precise number and position of uridine insertions/deletions is directed by small guide RNAs (gRNAs) encoded in the mitochondrial genome (32,33). These gRNAs are utilized in a sequential order by the RNA editing machinery to modify the mitochondrial mRNA in a 3′-to-5′ direction by annealing with the target mRNA by Watson–Crick and G–U base pairing (32,33). Catalysis of U-indel editing is executed by three RNA editing catalytic complexes (RECCs): two that catalyze uridine insertion and one that catalyzes uridine deletion (34–36). Also essential to the editing process are the RNA helicase 2 complex (REH2C) and variants of the non-catalytic RNA editing substrate-binding complex (RESC), which organize and facilitate the dynamic network of mRNA–gRNA, mRNA–protein, gRNA–protein and protein–protein interactions that characterize the editing process (27–30,37–49). U-indel editing appears to be a relatively inefficient process, as the bulk of the steady state mitochondrial mRNA pool is undergoing editing, and the resulting partially edited mRNAs typically contain regions of mis-edited sequence, called junctions, at the leading edge of the canonically edited region (42).
Because a subset of mitochondrially encoded gene products translated from edited mRNAs is essential in both PCF and BSF, the process of U-indel editing is also essential in both life cycle stages (50). Indeed, due to the dramatic metabolic changes that T. brucei undergoes during its life cycle, U-indel mRNA editing and the parasite life cycle are intimately linked. It has been long documented that several edited mRNAs exhibit remarkable changes in abundance between PCF and BSF T. brucei, although some discrepancies exist between older data and a more recent study with regard to specific mRNAs (51–58). Despite this long standing knowledge, the mechanisms by which the parasite regulates the accumulation of edited mitochondrial mRNAs during development remain unknown. The mitochondrial mRNAs and gRNAs are transcribed in both PCF and BSF, suggesting that at least one aspect of editing regulation occurs through regulated gRNA utilization, potentially involving interaction of specific gRNAs and/or mRNAs with the editing machinery (59–62). Protein components of the mRNA editing machinery are also similarly expressed in various life cycle stages (63). Subtle differences in the functions of some RECC proteins between PCF an BSF have been described; however, their contribution to the developmental regulation of editing is not known (35,64,65).
In this study, we revisit the mechanisms underlying the developmental regulation of edited mRNA abundance in T. brucei, employing recent advances in our knowledge and technology. Combining high throughput sequencing (HTS) with an in-depth, custom bioinformatics tool, we provide the first comparison of edited mRNA sequences in PCF and BSF at the single nucleotide level. Examination of these partially edited mRNA sequences in the context of recently reported comprehensive gRNA libraries allows us to assess patterns of gRNA utilization in the two life cycle stages (61,62). Our data demonstrate that there are different mechanisms at play governing the downregulation of edited CYb and COIII mRNAs in BSF, and cell line specific differences in the mechanisms abrogating COIII mRNA editing in BSF, including the possible utilization of terminator gRNAs that preclude the 3′ to 5′ progression of editing. We further examine the developmental timing of changing mitochondrial mRNA levels in detail, using overexpression of RBP6 in PCF to drive cells through metacyclogenesis in vitro (66). These studies represent the first analysis of U-indel editing in EMF and MCF T. brucei, and they reveal several transcript-specific developmental checkpoints.
MATERIALS AND METHODS
Generation of transgenic cell lines and cell culture conditions
The PCF T. brucei 29–13 cell line, which transgenically expresses T7 RNA polymerase and the tetracycline repressor, was grown in vitro at 27°C in SDM-80 medium containing hemin (7.5 μg/ml) and 10% fetal bovine serum (FBS). The cells were grown in the presence of 15 μg/ml G418/neomycin and 25 μg/ml hygromycin B. The pLew100v5 vector for RBP6 expression was linearized with NotI and transfected into 29–13 cells as described previously (67), and cells were positively selected with 2.5 μg/ml phleomycin. RBP6OE cells were grown in SDM-80 medium containing no glucose, further supplemented with 50 mM N-acetylglucosamine to block uptake of residual glucose molecules from 10% FBS. The induction of ectopically expressed RBP6 protein was induced by the addition of 10 μg/ml of tetracycline into the culture medium (66). Cell densities were measured using the Z2 Cell Counter (Beckman Coulter Inc.). Throughout the analyses, cells were maintained in the exponential mid-log growth phase (between 2 × 106 and 1 × 107 cells/ml). BSF T. brucei 427, MiTat 90–13 (a kind gift from F. Bringaud's lab), single-marker (SM-1) (cultivated for the last 15 years in Read lab), and SM-2 (cultivated for the last 10 years in Zikova lab) cell lines were grown in vitro at 37°C with 5% CO2 in HMI-11 medium supplemented with 10% FBS. Throughout the analyses, BSF cells were maintained in culture below 1 × 106 cells/ml. For the in vitro differentiation of monomorphic BSF cells to PCF, the protocol was adapted from the published reports (14,68,69) with the following modifications. On day zero, BSF cells were transferred to no-glucose SDM80 medium supplemented with 15% FBS, and the differentiation was triggered by adding 3 mM citrate, 3 mM cis-aconitate, 10 mM glycerol, 0.2 mM 2-mercaptoethanol, 28.2 mg/l bathocuproine, 182 mg/ml cysteine and by changing the temperature to 27°C (5). The cell density was maintained between 4 × 106 and 6 × 106 cells/ml. After 2 days, the glucose-containing SDM79 medium was added to the differentiated cells to support cell growth.
Western blot analysis and immunofluorescence
Whole cell lysates were separated by SDS-PAGE and transferred to PVDF membrane (Thermo Fisher Scientific, Waltham, MA, USA). Protein blots were probed with monoclonal anti-mitochondrial hsp70 (1:2000) (70) and anti-AOX (1:100; gifted by Dr M. Chaudhuri) primary antibodies, as well as polyclonal anti-GPEET (1:1000; gifted by Dr I. Roditi), anti-trCOIV (1:1000) (71) and anti-RBP6 (1:5000; gifted by Dr C. Tschudi) primary antibodies. Protein signals were generated by the incubation with HRP-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies (1:5000; Bio Rad) and visualized using the Pierce ECL system and ChemiDoc instrument (Bio Rad). Immunofluorescence for procyclin (GPEET) and BARP expression or dextran uptake was performed as previously described (71).
RNA isolation and quantitative RT-PCR analysis
Total RNA was isolated from 1 × 108 cells lysed in 1 ml of TRIzol reagent (Ambion), phenol-chloroform extracted, and ethanol-precipitated. The resulting RNA pellet was incubated with 4 U of rDNase I (Ambion) at 37°C for 1 h to remove DNA contamination. The purity and integrity of the RNA were confirmed using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) and RNA agarose gel electrophoresis. DNase-treated RNA was converted to cDNA using random hexamer primers and iScript reverse transcriptase (Bio-Rad). Primers for the selected targets for qRT-PCR analysis are listed in Supplementary Table S1 (40,58,72–74). Real-time PCR amplification was performed on a CFX Real Time System thermocycler (BioRad) and the data were analyzed on the BioRad CFX Manager software. For all qRT-PCR comparative analyses in this study, we measured three biological replicates with three technical replicates for each sample. Values were normalized to the average of three controls: TERT, PFR2 and actin mRNA (58,73).
High throughput sequencing and bioinformatics analyses
RNA from PCF 29–13 and BSF 427, 90–13, SM-1 and SM-2 cell lines was isolated, DNase-treated, and converted to gene-specific cDNA using primers specific for the editing domain of the minimally-edited cytochrome b (CYb), the 3′ region of the editing domain for cytochrome oxidase subunit III (COIII), and the 3′ region of the editing domain for ATPase subunit 6 (A6) mRNAs. The primer sequences used for the generation of these cDNAs are listed in the supplemental data (Supplementary Table S1) (74). The cDNAs were PCR-amplified within the linear range to ensure that subpopulations of short sequences were not disproportionately amplified in our samples (42). The PCR products were column-purified and eluted with 10 mM Tris pH 8.0 and subjected to Illumina MiSeq sequencing as described (74). For PCF 29–13 samples we sequenced five biological replicates, and for BSF samples (427, 90–13, SM-1 and SM-2) we sequenced three biological replicates for each cell line. The number of standard reads (reads that contain no non-T matches) and non-standard reads (reads containing non-T matches) for each sample are listed in the supplemental data (Supplementary Table S2). For each sample, all standard reads were normalized to 100 000 reads. This ensures that each sample can be compared to one another based on their normalized read counts. The normalized reads were then analyzed using the Trypanosome RNA Editing Alignment Tool (TREAT) developed in our laboratory to determine the relative proportion of pre-edited, partially edited, and fully edited reads in each sample based on user-defined templates (42,43). Through a combination of Sanger and Illumina sequencing, we identified alternative sequences at the extreme 3′ ends of both A6 and COIII mRNAs, as indicated by pink sequences in Figures 4A, 5A, 6A and 7A. We identified gRNAs that could direct these alternative sequences (purple in Figures 4A, 5A, 6A, 7A) by searching the PCF and BSF gRNA databases from the Koslowsky lab using their published algorithm (61,62,75). Exacerbated pause sites (EPSs) were determined as previously described (43). The sequences of gRNAs that could potentially guide the editing of junction sequences at specific COIII mRNA editing stop sites (ES32 in BSF 427 and ES37 in BSF SM-1) were predicted as previously described (61). The sequencing data used in this study have been deposited in the Sequence Read Archive under accession number PRJNA597932.
Figure 4.

Editing profiles of COIII and A6 mRNA intermediates organized by editing stop site. Editing peak profiles for PCF 29–13 (upper left), BSF 427 (upper right), BSF SM-1 (lower left), and BSF SM-2 (lower right) for (A) COIII mRNA intermediates and (B) A6 mRNA intermediates. The Y axes represent the number of normalized read counts for mRNAs at each editing stop site (note some differences in scales). Red diamonds indicate the final major peak in COIII mRNA or the most prominent peak in A6 mRNA. The yellow diamonds in (B) indicate peaks at ES26 generated by alternative A6 mRNA editing in BSF 427 and SM-2 at ES27, ES29 and ES33.
Figure 5.

Statistical and bioinformatic analyses of COIII mRNA editing intermediates at the final prominent EPS in PCF and BSF 427 cells. (A) Schematic illustrating the positions of exacerbated pause sites (EPSs) in BSF COIII mRNA in relation to the positions of gRNA families. gRNA anchor regions are illustrated as a box while the coding information region is illustrated with a line. The hatchmarked boxes at gRNA 5′ and 3′ ends represent variation in gRNA lengths within a gRNA family. The BSF 427 cells have an alternative editing consensus sequence that differs from the canonical sequence (shown in pink). The alternative guide RNA (1-Alt) that directs the editing of this alternative sequence is shown as a purple gRNA. Every 10 ESs are indicated above the sequence. Underscores at ES10 and ES40 are inserted to clarify the position of the ES. The yellow shaded regions indicate where the reverse COIII HTS primer anneals. The red diamond represents the final prominent EPS at ES32 in this cell line. (B) Sequence alignments of the 10 most abundant junction sequences at ES32 in COIII mRNA of PCF and BSF 427 cells. The canonical edited sequence is shown red and white letters with gray or black shading. Junction sequences are shown in black letters with yellow shading. Non-canonical consensus sequences are shown in blue letters with gray shading. To the right of each sequence is the average normalized read counts (Count) for the sequence and the junction length (JL). (C) Schematic of terminator guide RNAs found in PCF (gRNAs A–E) annealed to COIII mRNA with the non-productive terminator consensus junction sequence at ES32. The ratio of the counts for the canonical gRNAs that direct the editing past ES32 and the terminator gRNAs are shown as determined by previously published gRNA reads (61,62).
Figure 6.

Statistical and bioinformatic analyses of COIII mRNA editing intermediates at the final prominent EPS in PCF and BSF SM-1 cells. (A) Schematic illustrating the positions of exacerbated pause sites (EPSs) in BSF COIII mRNA in relation to the position of gRNAs. Symbols as in Figure 5. The red diamond represents the final prominent EPS at ES37 in this cell line. (B) Sequence alignments of the 10 most abundant junction sequences at ES37 in COIII mRNA of PCF and BSF SM-1 cells. Symbols and shading as in Figure 5. (C) Schematic of terminator guide RNAs found in PCF (gRNAs A–C) and BSF (gRNAs D–E) annealed to COIII mRNA with the non-productive consensus junction sequence at ES37. The ratio for the counts for the canonical gRNAs that direct the editing past ES37 and the terminator gRNAs are shown as determined by previously published gRNA reads (61,62).
Figure 7.

Statistical and bioinformatics analysis of COIII mRNA intermediates at the final prominent EPS in PCF and BSF SM-2 cells. (A) Schematic illustrating the positions of exacerbated pause sites (EPSs) in BSF COIII mRNA in relation to the position of guide RNAs (gRNAs). Symbols as in Figure 5. The red diamond represents the final prominent EPS at ES53. (B) Sequence alignments of the 10 most abundant junction sequences at ES53 in COIII mRNA of PCF and BSF SM-2 cells. Symbols and shading as in Figure 5.
RESULTS
Four edited mRNAs are consistently and dramatically regulated during the T. brucei life cycle
In this study, we aimed to characterize the developmental regulation of edited mitochondrial mRNAs at the single nucleotide level to provide insight into their mechanisms of regulation and to determine whether they are subject to common or distinct mechanisms. Because a recent study using qRT-PCR found that the relative levels of specific edited mRNAs can be strain-specific (58), we began by analyzing mRNA levels in multiple BSF cell lines compared to those in PCF. We isolated RNA from BSF Lister 427 strain (the parental line for all other cell lines used here), the MiTat 90–13 cell line, and two SM cell lines grown in independent laboratories for over 10 years (termed SM-1 and SM-2). qRT-PCR was performed using three biological replicates for each cell line and values were normalized to the average of three control mRNAs: TERT, PFR2 and actin (73). We measured pre-edited and edited versions of eight mitochondrial mRNAs, including both pan-edited and moderately edited mRNAs, and those reported to be either developmentally or constitutively edited during the T. brucei life cycle (58,76). We also measured total mRNA levels for RNAs that undergo moderate editing (CYb, COII and MURF2) using primers outside the edited region. For pan-edited mRNAs (COIII, A6, RPS12, ND7–3′, and ND8), we measured ‘total’ mRNAs levels using primers corresponding to the 5′ never-edited domain and pre-edited sequence at the extreme 5′ end of the molecule as previously described (40), since the majority of a given pan-edited mRNA population typically remains pre-edited in this region due to the inefficiency of the editing process.
Our results show consistent, dramatic decreases (in most cases >95%) in the abundances of edited CYb, COII and COIII mRNAs across all four BSF samples compared to PCF (Figure 1). Edited CYb mRNA was undetectable in all but the parental 427 strain, and even in this strain edited CYb mRNA was greatly reduced compared to PCF (0.056 ± 0.012). Edited COII mRNA was decreased ∼100-fold in all BSF samples. Edited COIII mRNA was undetectable in both SM samples, and was present at very low levels in the 427 and 90–13 cell lines (0.010 ± 0.003 and 0.010 ± 0.001 of PCF levels, respectively). In no case did we observe a concomitant increase in pre-edited mRNA. In stark contrast to CYb, COII and COIII mRNAs, edited ND8 mRNA was substantially increased in all BSF cell lines to varying degrees ranging from 19- to 1468-fold. Furthermore, determination of relative levels of total CYb, COII, COIII and ND8 mRNAs demonstrates that, despite some fluctuations, modulation of the abundances of the corresponding edited mRNAs is not simply due to a similar decrease (CYb, COII and COIII) or increase (ND8) in total mRNA levels. Finally, when examining the 3′ domain of ND7, we observed significant cell line-specific differences in edited mRNA levels, including a >10-fold increase in SM-1 and a >10-fold decrease in 427 compared to PCF. Even the two SM samples that had been cultured separately for over a decade exhibited a 10-fold difference in the degree to which edited ND7–3′ mRNA is increased in BSF. Thus, the separate culture conditions experienced by the four cell lines in Figure 1 over the past decade has permitted divergence in editing patterns where this is tolerated. These data further suggest that those mRNAs whose developmental patterns are consistent between cell lines (CYb, COII, COIII and ND8) remain so because these patterns are physiologically constrained.
Figure 1.

Quantitative RT-PCR analysis of PCF and BSF mRNAs. Relative mRNA levels for total, pre-edited (Pre) and edited (Edit) mitochondrial mRNAs in four BSF cell lines (427, 90–13, SM-1 and SM-2) relative to those in the 29–13 PCF cell line. Values were normalized to the average of three controls (TERT, PFR2 and actin). Data shown were generated from three biological replicates of each sample with three technical replicates per biological replicate. The gray shaded area represents the range from one log2 increase (2.00) and one log2 decrease (0.50). Error bars indicate standard error of the mean.
Having shown changes in the patterns of editing in BSF compared to PCF that are conserved among cell lines, we next asked whether these effects are reversible, consistent with their being physiologically relevant effects. All of the BSF cell lines used here are monomorphic and thus incapable of traversing the normal pathway through stumpy forms to PCF. Nevertheless, they can be triggered to differentiate directly to PCF cells by the addition of citrate/cis-aconitate to the media and a shift in temperature to 27°C (5). To test reversibility of the observed editing effects, we used this method to differentiate BSF 427 and SM-2 cells and demonstrated hallmark signs of differentiation to PCF, such as the expression of the cytochrome oxidase subunit IV (trCOIV), downregulation of the alternative oxidase (AOX) and expression of GPEET procyclin (Figure 2A). We then harvested RNA from the cells 2, 4 or 6 days post-citrate/cis-aconitate/27°C and compared total, pre-edited and edited mRNA levels to those in PCF 29–13 by qRT-PCR. Of the mRNAs that were consistently downregulated in BSF, edited COII mRNA was fully restored to PCF levels by day 2 (Figure 2B) and edited COIII mRNA was partially restored by day 6 (Figure 2C) in both cell lines. In contrast, edited CYb mRNA levels remained unchanged throughout the time course (Figure 2D). The modest decrease in edited A6 mRNA in BF was gradually restored (Figure 2E), while the increased edited ND8 mRNA levels observed in BSF gradually decreased throughout the timecourse towards PCF levels in both cell lines, despite the increase in total ND8 mRNA levels (Figure 2F). Together, these data show that monomorphic BSF cell lines have at least some capacity to release the physiological constraints on mRNA editing operating in this stage upon a differentiation signal. Interestingly, the differences in the timing and extent of COII, COIII and CYb mRNA increases indicates that distinct transcript-specific mechanisms, and potentially distinct signals, are in effect during this transition.
Figure 2.

Reversibility of BSF-stage edited mRNA levels. (A) Western blot analysis of undifferentiated BSF 427 cells, BSF 427 cells treated with 3 mM citrate/3 mM cis-aconitate (CCA) for 2, 4 or 6 days, and PCF cells. Western blots of SM-2 cells showed similar trends (not shown). (B–F) Quantitative RT-PCR analysis of relative mRNA levels for total, pre-edited (Pre), and edited (Edit) (B) COII, (C) COIII, (D) CYb, (E) A6 and (F) ND8 mRNAs in two undifferentiated BSF cell lines (427 and SM-2) and BSF 427 and SM-2 cells treated with CCA for 2, 4 or 6 days relative to those in the PCF 29–13 cell line. Undifferentiated BSF cells compared to PCF are used as a reference starting point for each cell line. Values were normalized to the average of three controls (TERT, PFR2 and actin). Data shown were generated from two biological replicates of each sample with four technical replicates per biological replicate. The gray shaded area represents the range from one log2 increase (2.00) and one log2 decrease (0.50). Error bars indicate standard error of the mean.
CYb and COIII mRNA populations exhibit different patterns of change in BSF
While edited CYb and COIII mRNAs are very dramatically decreased or undetectable in the four BSF cell lines tested, total mRNA populations are unchanged or decreased to a lesser degree (Figure 1), indicating that the remaining CYb and COIII mRNAs in BSF are either pre-edited or partially edited. A predominantly pre-edited mRNA population in BSF would indicate a defect in editing initiation and/or increased decay of edited mRNA in this life cycle stage. Alternatively, the accumulation of partially edited mRNAs would reflect a defect in the 3′ to 5′ progression of editing, and identification of distinct partially edited intermediates may highlight specific defects in editing progression. To provide insight into the mechanisms by which edited CYb and COIII mRNA abundances are decreased in BSF, we employed Illumina MiSeq HTS coupled with bioinformatic analysis using the Trypanosome RNA Editing Alignment Tool (TREAT) developed in our laboratory (42). We designed primers that specifically amplify the edited domains of CYb and COIII mRNA in order to amplify the majority of mRNA intermediates (Figure 3A). As a control, we similarly analyzed the edited domain of A6 mRNA, as this edited mRNA was only moderated decreased in BSF (Figure 1), and editing of A6 mRNA is essential in both PCF and BSF life cycle stages (26,50,77). The edited domain of CYb mRNA is small enough to sequence in its entirety, so the primers were designed to hybridize to the 5′ and 3′ never-edited regions (Figure 3A). The edited domains of pan-edited COIII and A6 mRNAs are too large to sequence in their entirety using MiSeq, so we amplified the 3′ edited regions of these two mRNAs using forward primers designed to anneal the pre-edited sequence near the middle of the transcript and reverse primers anneal to the 3′ never-edited regions (Figure 3a). We expect a large proportion of intermediates to be amplified by these primer pairs as a result of the 3′ to 5′ directionality of mRNA editing. Due to this primer placement, ‘fully’ edited COIII or A6 mRNA in HTS/TREAT refers to mRNA intermediates that are canonically edited up to the pre-edited forward primer. Because qRT-PCR revealed decreases in many total mRNA populations in BSF compared to PCF (Figure 1), and our previous HTS/TREAT studies were performed on PCF (40–43,74), we first validated HTS/TREAT analysis for BSF mRNA populations. To this end, we performed a down-sampling experiment on PCF A6 mRNA reads to determine the read count threshold for data integrity (78). We found that the data profiles were extremely consistent as low as 10,000 randomized standard reads (Supplementary Figure S1). The standard read count for each BSF replicate for each mRNA was well above this threshold, with the lowest standard read count being above 100,000 and most being several times this (Supplementary Table S1); thus, we have confidence in the detailed analyses of the BSF samples.
Figure 3.
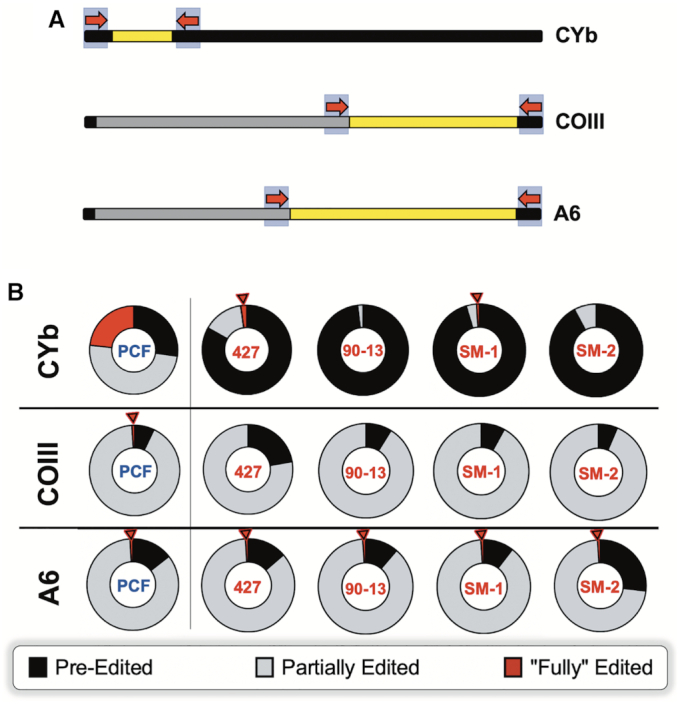
Proportions of pre-edited, partially edited, and fully edited CYb, COIII, and A6 mRNA in procyclic and bloodstream cells. (A) Schematic for the placement of CYb, COIII and A6 high throughput sequencing (HTS) primer pairs. The primers are represented by red arrows. The never-edited regions of the mRNAs are indicated in black; the pre-edited regions are indicated in gray; the edited regions of the mRNAs are indicated in yellow. (B) Pie charts illustrating the breakdown for the percentage of CYb, COIII and A6 mRNAs by editing status in procyclic (PCF) 29–13 and bloodstream cell lines 427, 90–13, SM-1 and SM-2 as determined by HTS/TREAT analysis. Red arrows denote samples containing fully edited mRNA reads. Note that ‘fully’ edited reads for A6 and COIII refer to those reads that are canonically fully edited up to the end of the forward primer.
To interrogate the mechanisms regulating edited RNA populations during the life cycle, we first asked what proportion of the CYb, COIII and A6 mRNA populations is pre-edited, partially edited, and fully edited in PCF and BSF. Figure 3B shows that 23.2% of the PCF CYb mRNA population is fully edited, consistent with our recently published data (74). In contrast, BSF CYb mRNA is overwhelmingly pre-edited, ranging from 83.1% in cell line 427 to 92–98% in the remaining BSF cell lines. Thus, either the initiation of CYb mRNA editing is significantly impaired in BSF and/or the stability of CYb mRNAs that have entered the editing pathway is compromised in BSF. When we analyzed COIII mRNAs, a different picture emerged. Here, we observed little or no increase in pre-edited mRNA, and the majority of the COIII mRNA population is partially edited in both PCF and BSF (Figure 3B). Additionally, while some fully edited COIII reads were detected in PCF, none were found in any BSF cell line (Figure 3B). Absence of fully edited mRNA together with a large population of partially edited mRNAs indicates that the initiation of COIII mRNA editing is not impaired in BSF, but rather there is a defect in editing progression. Finally, we analyzed the proportions of differentially edited A6 mRNAs, knowing that edited A6 mRNA is required in both PCF and BSF. Consistent with this notion, we observed that the proportions of pre-edited, partially edited, and fully edited A6 mRNAs are comparable in PCF and BSF (Figure 3B). From the above data, we conclude that the abundance of edited CYb and COIII mRNAs in BSF are regulated by distinct mechanisms, consistent with their differing abilities to be released from constraint upon differentiation (Figure 2). Whereas edited CYb mRNA abundance is downregulated in BSF by inhibition of editing initiation and/or decreased stability of partially edited intermediates, COIII mRNA is regulated by the inhibition of editing progression.
The progression of COIII mRNA editing is efficiently inhibited in the 3′ region of the mRNA
To compare COIII mRNA editing progression in PCF and BSF and to determine where along the COIII mRNA sequence the editing progression defects are localized, we utilized TREAT to analyze the COIII mRNA reads from PCF and BSF cell lines. TREAT defines any space between two non-T nucleotides as an editing site (ES) (42). The ESs are numbered in order from 3′ to 5′ to coincide with the directionality of mRNA editing. An editing stop site is the 5′-most ES of continuous and uninterrupted canonical editing; thus, every ES that is 3′ of the editing stop site is also canonically edited (42). The region 5′ of the editing stop site can match the pre-edited sequence, a junction sequence in the process of active editing, or an alternative edited sequence that does not match the published canonical edited sequence (31).
To analyze editing progression, we normalized COIII and A6 mRNA standard reads to 100,000. Using TREAT, we then sorted the reads according to their editing stop sites, and plotted the relative frequencies of editing stop sites to create an editing peak profile for COIII and A6 in PCF and three BSF cell lines (427, SM-1 and SM-2) (Figure 4) (43). In this readout, editing proceeds from right to left (3′ to 5′) with increasing ES number, and peaks represent ESs at which canonical editing frequently pauses. The length of the entire read, within the boundaries of the upstream pre-edited and downstream never-edited primers (Figure 3A), is shown. The peak profile of COIII mRNA editing in PCF shows that even though there are some intrinsic barriers to COIII mRNA editing, as indicated by particularly tall peaks where editing frequently pauses, the editing of COIII mRNA proceeds 5′ into the transcript (Figure 4A, top left). In BSF, we observed a different pattern in which all editing stop sites were clustered in the 3′ portion of the COIII reads, indicative of a near complete halt of editing progression beyond these sites (Figure 4A). In contrast, although A6 mRNA populations exhibit some larger pauses in editing, canonical editing progresses throughout the length of the A6 read in both PCF and BSF (Figure 4B). We note the differences in scale between PCF and BSF A6 normalized reads in Figure 4B due to large peaks at ES26 in some BSF samples. The apparent editing stop site at ES26 in BSF 427 and SM-2 is the result of an alternative sequence at ES27, ES29 and ES33, which is designated by TREAT as an editing stop site at ES26 (Figure 4B, yellow diamond). Although canonical edited A6 sequence continues for the remainder of the reads 5′ of ES33 in BSF 427 and SM-2, designation of ES26 as an editing stop site diminishes the apparent relative abundance of more 5′ editing stop sites in the BSF 427 and SM-2 cell lines (Figure 4). Overall, the comparisons of editing progression between PCF and BSF presented in Figure 4 demonstrate that BSF cells specifically and completely inhibit the progression of COIII mRNA editing early in the process, thereby depleting the fully edited COIII mRNA pool. A6 mRNA is not subject to this regulation, presumably due to the essentiality of the A6 protein product in the BSF life cycle stage.
Different termination mechanisms exist for COIII mRNA in distinct BSF cell lines
The data demonstrate that canonical COIII mRNA editing appears to abruptly terminate at specific ESs in the BSF cell lines (Figure 4A). We next wanted to address the mechanism(s) of editing attenuation by determining the editing actions at these termination sites. To this end, we compared the frequency of mRNAs with a given editing stop site in BSF to that in PCF to identify exacerbated pause sites (EPSs) in the BSF cell lines (43). In this study, we define an EPS as an editing stop site with a normalized mRNA read count that is statistically more abundant in BSF than in PCF, signifying ESs where canonical COIII mRNA editing pauses more frequently in BSF than it does in PCF (Supplementary Table S3). We plotted the BSF EPSs on a schematic depicting edited COIII mRNA aligned with its published cognate gRNAs (62), or with a gRNA that directs an alternative 3′ edited sequence that we identified here by Sanger sequencing and Illumina MiSeq in all cell lines used in this study (gRNA 1-Alt). This allows us to visualize where COIII EPSs are localized for the 427 (Figure 5A), SM-1 (Figure 6A) and SM-2 (Figure 7A) cell lines relative to cognate gRNAs. Editing stop sites are denoted by diamonds, with ‘prominent’ EPSs (defined here as EPSs that have a BSF-to-PCF normalized read count ratio greater than or equal to 1.50 and have a BSF normalized read count greater than or equal to 1000) highlighted in black. We were particularly interested in identifying the actions at the final (5′ most) major EPS at which canonical editing ends in each BSF cell line, and these 5′-most prominent EPSs are designated as red diamonds for each cell line (Figures 5A, 6A, 7A).
When analyzing COIII mRNA intermediates for BSF 427, we identified seven prominent EPSs within the region of the first three gRNAs, with the final prominent EPS at ES32 (Figure 5A). Strikingly, ES32 marks the end of the region directed by gRNA-2. Therefore, we wanted to examine the sequences 5′ of this EPS to determine whether editing halts altogether, indicating that gRNA-2 cannot be removed or gRNA-3 is not utilized, or whether a common junction sequence can be identified, suggesting alternative gRNA utilization. We used TREAT to identify the ten most abundant junction sequences of mRNA intermediates at ES32 in BSF 427 and compared them to the mRNA intermediates at ES32 in PCF. We observed a strong consensus in the junction sequences in BSF 427, while this consensus sequence rarely appeared in PCF (Figure 5B, blue u's). The abundance of mRNA intermediates with this consensus sequence and the number of uridines added over several ESs, strongly suggests that a non-canonical gRNA is utilized to generate the consensus sequence in BSF. Our data suggest that such a gRNA acts as a terminator, being fully utilized in BSF to generate an edited sequence to which the third gRNA is unable to anchor to continue editing. Interestingly, we also noted that the PCF junction sequences, although shorter, appeared to match the beginning of the consensus sequence observed in BSF (Figure 5B). Therefore, it is possible that the same non-canonical gRNA can begin to direct editing in PCF, albeit infrequently (Figure 5B, count), but even in these cases the gRNA is not fully utilized.
To identify a potential non-canonical terminator gRNA that could direct the BSF 427 alternative sequence at ES32, we searched previously reported PCF and BSF gRNA transcriptomic databases (61,62,75). We readily identified a family of such gRNAs in the three reported PCF databases (Figure 5C). Remarkably, when comparing the reported abundance of the predicted terminator gRNA to that of the canonical gRNA for this region in the combined PCF databases, we found that the terminator gRNA population was 5-fold more abundant than the canonical gRNAs that encode for the same region in PCF, suggesting that a mechanism for preferential utilization of the canonical gRNA exists in PCF (Figure 5c). While the canonical gRNAs directing editing in this region were detected in the BSF gRNA database, we could not identify a viable gRNA that could adequately anchor to the canonical sequence up to ES32 and encode a significant portion of the terminator sequence in this life cycle stage. It is possible that the terminator gRNA simply was not detected in the previous BSF gRNA sequencing studies. Alternatively, we cannot rule out that the BSF EATRO 164 strain sequenced in the previous studies utilizes a different COIII junction sequence at ES32 or terminates COIII editing at a different editing site. It is also possible that the same terminator gRNA is used in the EATRO 164 strain but is rapidly consumed in the editing process (46). Together, the data in Figure 5 demonstrate that BSF 427 cells can efficiently terminate canonical COIII mRNA editing at ES32, likely by preferentially utilizing an alternative terminator gRNA more efficiently than the canonical gRNA in this region. This same terminator gRNA is present in PCF, but it appears to be inefficiently utilized, as primarily the first two or three editing sites of the non-canonical consensus sequence appear in PCF intermediates.
Editing peak profile analysis revealed distinct COIII mRNA editing pausing profiles in different BSF cell lines (Figure 4), suggesting the existence of different pausing mechanisms. To identify the mechanisms by which COIII editing progression is terminated in SM-1 and SM-2 cell lines, we performed similar EPS and junction sequence analyses on COIII mRNAs these lines. In SM-1, we identified eight prominent EPSs, with the final prominent EPS at ES37 (Figure 6A and B). We used TREAT to identify the ten most abundant junction sequences of mRNA intermediates at ES37 in BSF SM-1 and compared them to the mRNA intermediates at ES37 in PCF. In PCF, vanishingly few mRNAs contain editing stop site 37, and many of those that do possess small junctions typical of standard editing (Figure 6b). Longer PCF junctions at ES37 were clearly intermediates in the canonical editing pathway that were mis-edited at ES38. In contrast, we identified a clear consensus sequence in BSF at ES37 that was not present in the PCF junction sequences (Figure 6B), suggesting that, similar to the 427 cells, SM-1 cells also utilize a terminator gRNA. However, unlike the ES32 terminator gRNA in 427 cells (Figure 5), the ES37 terminator gRNA in SM-1 cells does not appear to be utilized at all in PCF. When searching for terminator gRNAs that could anchor to the canonical sequence up to ES37 and direct the consensus edited sequence observed in the SM-1 junctions, we identified several such gRNAs in both PCF and BSF that possess a predicted anchor sequence in a region analogous to that of the canonical gRNA-3 (Figure 6C). When comparing the ratio of canonical-to-terminator gRNA populations in PCF and BSF (61,62), we found that in PCF the terminator and canonical gRNA populations were present in similar amounts while in BSF that the canonical gRNA was far more abundant than the terminator gRNA despite the lack of canonical gRNA utilization. These data suggest that, in SM-1 cells, the final prominent EPS arises due to a terminator gRNA that is selectively utilized in BSF but that is hardly utilized at all in PCF.
Similar analyses of SM-2 cells identified seven prominent EPSs within the region of the first four gRNAs (Figure 7A). The final prominent EPS for SM-2 cells was identified at ES53. Notably, this ES is at the end of the region directed by the fourth gRNA (gRNA-4; Figure 7A). Analysis of the junction sequences at ES53 in PCF and BSF SM-2 shows that the most abundant sequence 5′ of ES53 is pre-edited in both life cycle stages; however, mRNAs with this sequence are 6-fold more abundant in BSF than in PCF (Figure 7B, counts, JL of 0). Sequences with junctions are also far less abundant in PCF and the junctions are generally much shorter than those in BSF (Figure 7B). Bioinformatic analysis of COIII mRNA sequences in the BSF 90–13 cell line revealed a similar pattern to that observed in SM-2 (data not shown). Based on the abundance of sequences lacking junctions 5′ of the final prominent editing stop site at ES53, we conclude that COIII mRNA editing is frequently terminated in BSF SM-2 and 90–13 cells through either inhibition of gRNA-4 removal or inhibition of gRNA-5 annealing. This process appears to occur relatively infrequently in PCF. Taken together, our single nucleotide level analysis of COIII mRNA editing progression reveals multiple distinct mechanisms whereby BSF cells pause the 3′ to 5′ progression of editing near the 3′ end of the transcript.
Editing actions at a major pause site in A6 mRNA are comparable in PCF and BSF
Having shown that the editing actions at final prominent EPSs in COIII mRNAs differ substantially between BSF and PCF (Figures 5–7), we wanted to confirm that this pattern is not an intrinsic property of BSF editing at major pause sites. Thus, we examined the junction sequences of A6 mRNA intermediates. Unlike COIII mRNA, where the EPSs are clustered in the 3′ end of the transcript, the editing peak profile of A6 mRNA shows that canonical editing proceeds past major peaks and extends throughout the length of the mRNA region investigated in both PCF and BSF (Figure 4B). TREAT analysis of A6 mRNAs from cell lines 427 and SM-2 revealed an alternative editing pattern at ES27, ES29, and ES33 in a substantial proportion of the population, 5′ of which editing returns to the canonical sequence (Figure 4B, yellow diamonds). Because of this complication, we began by analyzing the editing patterns in BSF SM-1 compared to PCF. Pause sites in A6 mRNA editing that are exacerbated in BSF SM-1 compared to those in PCF are distributed throughout the sequence, as shown in Figure 8A, and the most prominent EPS occurs at ES58 (Figures 4B and 8A; Supplementary Table S4). We used TREAT to determine the ten most abundant junction sequences at ES58 in PCF and BSF SM-1, revealing very similar profiles in the two life cycle stages (Figure 8B). Indeed, the top three most abundant ES58 junctions are identical in PCF and BSF SM-1, and these account for 97% of PCF sequences and 99% of BSF SM-1 sequences at this editing stop site. After accounting for alternative editing between ES27 and ES33, the prominent EPS in BSF 427 and SM-2 lines also occurs at ES58 (Figure 4B, red diamonds), and TREAT analysis of junctions arising at ES58 in BSF 427 and SM-2 lines showed similar patterns to those in SM-1 (Supplementary Figure S2). Thus, the major pausing in A6 mRNA editing at ES58 does not induce distinctive editing patterns between PCF and BSF and, while prominent EPSs in COIII mRNA can vary between BSF cell lines, this flexibility appears absent in A6 mRNA. Importantly, the major pause site at ES58 in A6 mRNA, although exacerbated in BSF, does not cause the BSF cells to terminate editing of this mRNA.
Figure 8.

Statistical and bioinformatics analysis of A6 mRNA editing intermediates at ES58 in PCF and BSF SM-1 cells. (A) Schematic illustrating the positions of exacerbated pause sites (EPSs) in BSF A6 mRNA in relation to the position of guide RNAs (gRNAs). Symbols as in Figure 5. The red diamond represents the most prominent EPS in SM-1 cells at ES58. (B) Sequence alignments of the 10 most abundant junction sequences at ES58 in A6 mRNA of PCF and BSF SM-1 cells. Symbols and shading as in Figure 5.
Edited mRNAs are subject to transcript-specific control points during the T. brucei life cycle
Having shown that the mechanisms governing the decreased abundance of edited CYb and COIII mRNAs in BSF are different, we next asked if the timing of their regulation during the T. brucei life cycle also differs. In addition, we wanted to determine if the relative abundances of edited mRNAs progressively decrease (COII, COIII, CYb) or increase (ND8 and potentially ND7–3′) as PCF parasites progress through the life cycle towards BSF, or if distinct control points exist. To do this, we utilized a tetracycline-regulatable system that allowed us to inducibly overexpress RBP6 in PCF to trigger metacyclogenesis (66). Cells were induced with doxycycline to overexpress RBP6 for 2–8 days (Figure 9A). In agreement with the published study (71), the differentiation of PCF cells progressed efficiently through the procyclin-positive EMF cells (day 2) and BARP-positive EMF (day 4) to MCF cells (days 6 and 8) (Figure 9B). The individual life cycle stages were assessed by the position of the nucleus and kinetoplast since EMF displays the kinetoplast in an anterior position relative to the nucleus. Mature EMF cells were further identified by immunostaining for the specific surface marker BARP, while MCF cells were identified by the ability to uptake fluorescent dextran, a hallmark for MCF due to the increased endocytosis (Figure 9B). We isolated RNA from uninduced PCF cells, from RBP6-induced cells for 2 days when EMF parasites dominate the culture, and from day 8 when the culture was a mixed population of PCF and MCF parasites (40% MCF/40% PCF) but have the highest resemblance to the pure MCFs at the RNA and protein levels (4,71).
Figure 9.

Quantitative RT-PCR analysis of induced RBP6OE cells compared to uninduced PCF. (A) Western blot analysis of RBP6 levels at 0–8 days post-induction. Mitochondrial (mt) Hsp70 is a loading control. (B) Immunofluorescence of RBP6OE cells on day 2 post-induction (using anti-procyclin antibody), day 4 post-induction (using anti-BARP antibody) and day 6 post-induction (using internalized fluorescently-labeled dextran). DNA was visualized by DAPI staining. (C, D) Relative mRNA levels for total, pre-edited, and edited mitochondrial mRNAs between cells that were induced to ectopically express RBP6 for (C) 2 days to generate an EMF-dominant culture or (D) 8 days to generate a mixed culture of MCF and PCF cells compared to the uninduced PCF cells. Values were normalized to the average of three controls (TERT, PFR2, and actin). Data shown were generated from three biological replicates of each sample with three technical replicates per biological replicate. The gray shaded areas represent the range from one log2 increase (2.00) and one log2 decrease (0.50). Error bars indicate standard error of the mean.
We performed qRT-PCR analysis to compare the relative levels of total, pre-edited, and edited mRNAs after 2 or 8 days post-induction. In cultures harboring primarily EMF, the levels of edited ND8 and ND7–3′ mRNAs increased 5- to 8-fold compared to those in PCF, while the levels of other RNAs measured exhibited little or no change (Figure 9C). In cultures containing approximately a 1:1 ratio of MCF-to-PCF, the abundances of many mitochondrial mRNAs, including total mRNA populations, were substantially increased compared to levels in PCF cultures (Figure 9D). Surprisingly, changes in the day 8 population included an almost 6-fold increase in the abundance edited COIII mRNA (Figure 9C), despite the substantial downregulation of this edited mRNA in BSF, indicating that the stimulus for decreasing editing of COIII mRNA occurs in BSF. In contrast to COIII mRNA, relative levels of total and edited CYb mRNAs were modestly decreased following 8 days of RBP6 expression. Thus, while both edited CYb and edited COIII mRNAs undergo a dramatic decrease upon the transition to BSF, these mRNAs are not governed by the same regulatory mechanisms during the PCF-MCF-BSF transition.
DISCUSSION
Significant transcript-specific differences in the abundances of specific edited mRNAs between T. brucei PCF and BSF life cycle stages have been known for decades (76); however, the mechanisms by which these changes are effected have remained completely obscure. Several scientific advances of the past decade permit us to now return to this intriguing question with a new arsenal of approaches. These advances include the HTS revolution that provided comprehensive gRNA libraries and several bioinformatic approaches to analyze mitochondrial mRNA populations (31,42,61,62,79–81), thereby allowing us to gain single nucleotide level insight into changes in editing progression during the life cycle. A second critical advance is the ability to trigger metacyclogenesis in PCF in vitro by overexpression of a single RNA-binding protein (66), which allowed us to perform the first analysis of mRNA editing throughout the T. brucei life cycle, including EMF and MCF stages. Here, we demonstrate that the developmental downregulation of edited CYb mRNA abundance in BSF begins during metacyclogenesis. This downregulation is significantly exacerbated in BSF, by a reduction in editing initiation and/or edited mRNA stability. In contrast, the downregulation of edited COIII mRNA abundance begins in BSF, when elevated edited COIII mRNA levels fall through efficient impairment of editing progression near the 3′ end of the transcript. A more global analysis of mitochondrial RNA levels throughout the PCF-EMF-MCF transition revealed transcript-specific control points in both EMF and MCF stages.
Developmental regulation of mRNA editing and its connection to metabolism
As T. brucei progresses through the life cycle from PCF to BSF, nutrient availability changes and the parasite adapts to extreme environmental changes. Furthermore, the parasite must also be somewhat pre-emptive about its metabolic adaptations as some of these environmental switches are quite shocking (11). Since mRNA editing is essential for the production of translatable mRNAs that encode proteins involved in mitochondrial bioenergetics, mRNA editing is intimately linked to metabolism. Our data show the abundance of several edited mitochondrial mRNAs fluctuates throughout the life cycle (Figures 1 and 9). PCF parasites must replicate in the amino acid-rich, glucose-depleted tsetse fly midgut, and consequently, they rely on oxidative phosphorylation for the bulk of ATP production. This is reflected in the presence of several edited mitochondrial mRNAs in PCF that encode proteins connected to an active ETC (CYb, complex III; COII and COIII, complex IV). Conversely, as slender BSF parasites have access to abundant glucose, they utilize primarily glycolysis and deprioritize the ETC, thereby permitting the dramatic decrease in edited mRNAs encoding ETC components in this stage in all cell lines tested here. In cultures comprised of 1:1 MCF-to-PCF, both edited COIII and edited CYb mRNAs are present, although edited CYb is beginning to decrease. Persistence of these edited mRNAs suggests that oxidative phosphorylation is still active in MCF parasites, and its drastic downregulation is not yet permissible due to the lack of glucose in the tsetse fly salivary glands where they reside in vivo. In fact, active oxidative phosphorylation in MCF was recently confirmed in a thorough study on the mitochondrial metabolic dynamics in cells differentiated in response to RBP6 overexpression (71). Persistence of complex III and IV components, and presumably increased abundance and/or activity of complex I, in MCF may also be a compensatory mechanism to counteract another pre-adaptation for BSF that occurs during metacyclogenesis. Namely, at this stage, the trypanosome alternative oxidase (AOX) is upregulated (4), but its activity is not coupled to proton translocation and ATP production by oxidative phosphorylation, so it competes with complexes III/IV for reducing power (71).
We observed a very different pattern of developmental regulation of edited mRNAs that encode subunits for NADH-ubiquinone oxidoreductase (complex I) (Figures 1 and 9). The abundances of edited mRNAs encoding ND7 and ND8 were upregulated early on the pathway from PCF to BSF, exhibiting 5- to 8-fold increases in EMF stage parasites. Edited ND8 mRNA continued increasing in MCF and was dramatically upregulated in all BSF cell lines tested. In contrast, edited ND7 mRNA decreased slightly in MCF and was inconsistent between BSF cell lines, even decreasing in one BSF cell line (427) compared to PCF. The necessity and function of complex I in T. brucei is still unclear. Previous studies have reported that although complex I in PCF parasites has NADH-ubiquinone oxidoreductase activity and participates in oxidative phosphorylation, its activity is non-essential (82). Despite the upregulation of edited mRNAs that encode complex I subunits and the reported assembly of the complex in BSF, complex I does not appear to have NADH-ubiquinone oxidoreductase activity or to be essential in BSF (23). It was later found that another type II NADH dehydrogenase (NDH2) is necessary for acetate production, and complex I assists NDH2 in maintaining NADH/NAD+ redox balance to maintain an environment conducive for acetate production, but even this assistance function appears non-essential (24). Therefore, the physiological rationale for the consistent and dramatic upregulation of edited ND8 mRNA in EMF, MCF and BSF parasites remains enigmatic. Overall, the pre-adaptation of T. brucei EMF and MCF to BSF complex I editing patterns, as well as the persistence of edited mRNAs encoding cytochrome components in EMF and MCF appears to be conserved in trypanosomatids, as it is reminiscent of observations made decades ago in T. congolense (55).
Regulation of edited CYb mRNA levels
Our data clearly demonstrate that the decreased abundances of edited CYb and COIII mRNAs in BSF occur through distinct mechanisms. In BSF, the predominantly pre-edited CYb mRNA population could arise through inhibition of editing initiation and/or rapid destabilization of CYb mRNAs that enter the editing pathway in this life cycle stage. Several lines of evidence suggest that a combination of these effects is at play. Destabilization of some portion of the partially and fully edited CYb population apparently takes place in BSF, as reflected in the decreased total CYb mRNA levels in BSF compared to PCF (Figure 1). However, the level to which the total CYb mRNA population is decreased in BSF is not large enough to account for decay of the ∼75% of CYb mRNAs that are partially or totally edited in PCF in at least two cell lines (Figure 3B). These data predict additional regulation at the step of editing initiation. An effect on editing initiation could involve the catalytic component of the editing machinery, RECC, as complementation studies demonstrated that mutants of several RECC proteins have enhanced effects in BSF compared to PCF (35,64,65). However, the basis for these phenotypes is not known and they are not transcript-specific. Other factors likely to impact edited CYb mRNA levels in BSF are KRBP16 and KMRP1/2, both of which are strictly required to maintain edited CYb mRNA levels in PCF T. brucei with considerable specificity (74,83,84). A recent study from our laboratory suggests that KRBP16 promotes the initiation of CYb mRNA editing, while KMRP1/2 stabilizes partially and fully edited CYb mRNAs in PCF T. brucei (74). Although these factors are present in both PCF and BSF stages, their functions may be regulated by posttranslational modifications. For example, KRBP16 is methylated on three arginine residues in PCF, and this modification is required for its interaction with CYb mRNA (85,86). The methylation status of KRBP16 in other life cycle stages is not known. KMRP1/2 undergoes multiple phosphorylation events, and phosphorylation of serine-198 on KMRP1 is over 10-fold increased in PCF compared to BSF (87). This modification may impact KMRP1/2 protein–protein or protein–RNA interactions, particularly those involving CYb mRNA. Future studies will address the roles of KRBP16 and KMRP1/2 and their posttranslational modification in the regulation of CYb mRNA editing throughout the life cycle.
Regulation of edited COIII mRNA
We demonstrate here that developmental regulation of COIII mRNA editing is regulated at the level of 3′ to 5′ editing progression. We identified distinct mechanisms by which editing progression is halted in three different BSF cell lines, although all three cell lines similarly effect these mechanisms near the 3′ end of the COIII transcript. In BSF 427 and SM-1 cell lines, control of editing progression entails preferential gRNA utilization (Figures 5 and 6). These cells utilize alternative gRNAs to derail canonical editing by introducing edited RNA sequences to which the subsequent canonical gRNAs are unlikely to anchor. To our surprise, terminator gRNA populations appear equally as or substantially more abundant than the corresponding canonical gRNAs in PCF (61,62), indicating the presence of active mechanisms governing utilization of specific gRNAs in a life cycle stage-specific manner. One protein in the RESC component of the editing holoenzyme, RESC14 (formerly MRB7260; new nomenclature summarized in reference 49), also forms distinct complexes with the gRNA-binding proteins, RESC1/2 (formerly GAP1/2), and has been implicated in selective gRNA utilization in PCF (40). It will be important to examine whether RESC14 depletion in PCF impacts the COIII termination sites identified here or the association of terminator gRNAs with the editing machinery. Interestingly, a recent genome-wide study identified a large population of non-canonical gRNAs of unknown function encoded in the T. brucei mitochondrial genome (88). Expression of most of these small RNAs, which were present at 40% of the levels of canonical gRNAs, was confirmed by transcriptome analysis. Our data suggest that one function of these ‘orphan’ gRNAs may be to act as editing terminators under specific conditions and at specific points in the life cycle. While our data are consistent with the terminator gRNA model, they do not exclude the possibility of another model. We cannot rule out that it is not the use of the terminator gRNA that is promoted in BSF, but rather the utilization of the canonical COIII gRNA is specifically prohibited in BSF. The halt of canonical editing at this point would allow the utilization of alternative gRNAs that happen to be present in the population and that can anchor to the now free COIII mRNA anchor sequence. This non-productive gRNA utilization would serve to further derail canonical editing. Additional experimental study is needed to discern which of the two scenarios are at play.
We identified a second mechanism for regulating editing progression in the BSF SM-2 line. Here, canonical COIII editing is completely halted at the end of the region directed by gRNA-4, and the overwhelming abundance of mRNAs lacking any editing events past this point indicates an arrest in editing overall. Thus, there appears to be a barrier involving either inefficient removal of the fully utilized gRNA-4 or the recruitment, availability, or annealing of gRNA-5. This process serves as an intrinsic barrier to the progression of editing in both PCF and BSF SM-2 but is greatly exacerbated in BSF SM-2 cells. COIII gRNA-5 is reportedly abundant in BSF, indicating that altered gRNA-5 levels do not govern its differential utilization during the life cycle (62). Indeed, gRNA transcriptome studies in PCF and BSF T. brucei revealed no apparent trend relating gRNA abundance and developmental editing patterns (61,62), again consistent with regulated utilization of gRNAs. Notably, in vitro differentiation of BSF 427 and SM-2 cell lines, which utilize different mechanisms for the inhibition of COIII mRNA editing progression, to PCF showed that both cell lines are able to substantially restore editing of COIII mRNA.
Using A6 mRNA as a control for an edited mRNA that maintains high levels throughout the life cycle, we found that the editing pattern of this mRNA is relatively strictly maintained. Whereas COIII mRNA editing exhibited substantial flexibility in its patterns of editing progression between life cycle stages and between BSF cell lines, A6 mRNA editing patterns were more rigid in all cell lines examined. The conservation of specific pausing patterns and junction sequences at distinct pause sites in A6 mRNA likely reflects the essentiality of the A6 protein product in both PCF and BSF. Understanding the mechanisms by which these patterns are maintained in the face of enormous gRNA diversity may provide insight into how these mechanisms are subverted in developmentally regulated mRNAs such as COIII.
Perspective
Altogether, this study offers the first insight into the mechanistic regulation of mRNA editing during the T. brucei life cycle, and suggests a dynamic orchestration of RNA-protein interactions in this process. Several lines of study demonstrate the concurrent action of multiple distinct, reversible, and transcript-specific processes. The precise interplay of signals and mechanisms that determine the preferential utilization of the gRNAs during regulation of COIII mRNA editing, and likely that of other mRNAs, is still unknown. It is possible that life cycle stage-specific protein or RNA modifications, expression and affinity of different RNA-binding proteins, and/or thermodynamic differences in RNA–RNA, protein–protein or RNA–protein interactions between BSF and PCF play significant roles. Further investigations of editing progression and macromolecular interactions in pleomorphic strains during the slender BSF-stumpy BSF-PCF transition and during the RBP6-stimulated PCF-to-MCF transition will be highly informative in our understanding of the dynamic and reversible nature of mRNA editing regulation during T. brucei development.
DATA AVAILABILITY
BioProject: PRJNA597932 https://dataview.ncbi.nlm.nih.gov/object/PRJNA597932?reviewer=skkf1ukebuoluctnjqliudbrop.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Donna Koslowsky and Laura Kirby for providing their program and databases for identification of alternative and terminator gRNAs and Rachel Simpson for designing primers for HTS/TREAT analyses. We are grateful to the University at Buffalo Center for Computational Research, especially Andrew Bruno, and the University at Buffalo Genomics and Bioinformatics Core for their support.
Contributor Information
Joseph T Smith Jr., Department of Microbiology and Immunology, University at Buffalo – Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY 14203, USA.
Eva Doleželová, Institute of Parasitology, Biology Centre Czech Academy of Science, České Budejovice, Czech Republic.
Brianna Tylec, Department of Microbiology and Immunology, University at Buffalo – Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY 14203, USA.
Jonathan E Bard, Genomics and Bioinformatics Core, University at Buffalo, Buffalo, NY 14203, USA.
Runpu Chen, Department of Computer Science and Engineering, University at Buffalo, Buffalo, NY 14260, USA.
Yijun Sun, Department of Microbiology and Immunology, University at Buffalo – Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY 14203, USA.
Alena Zíková, Institute of Parasitology, Biology Centre Czech Academy of Science, České Budejovice, Czech Republic.
Laurie K Read, Department of Microbiology and Immunology, University at Buffalo – Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY 14203, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R01 GM129041 to L.K.R.]; Czech Science Foundation [17-22248S]; ERD fund [CZ.02.1.01/0.0/0.0/16_019/0000759 to A.Z.]. Funding for open access charge: National Institute of General Medical Sciences [R01GM129041].
Conflict of interest statement. None declared.
REFERENCES
- 1. Simarro P.P., Cecchi G., Franco J.R., Paone M., Diarra A., Ruiz-Postigo J.A., Fèvre E.M., Mattioli R.C., Jannin J.G.. Estimating and mapping the population at risk of sleeping sickness. PLoS Negl. Trop. Dis. 2012; 6:e1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franco J.R., Simarro P.P., Diarra A., Jannin J.G.. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014; 6:257–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matthews K.R. The developmental cell biology of Trypanosoma brucei. J. Cell. Sci. 2005; 118:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christiano R., Kolev N.G., Shi H., Ullu E., Walther T.C., Tschudi C.. The proteome and transcriptome of the infectious metacyclic form of Trypanosoma brucei define quiescent cells primed for mammalian invasion. Mol. Microbiol. 2017; 106:74–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vassella E., Reuner B., Yutzy B., Boshart M.. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell. Sci. 1997; 110:2661–2671. [DOI] [PubMed] [Google Scholar]
- 6. Savill N.J., Seed J.R.. Mathematical and statistical analysis of the Trypanosoma brucei slender to stumpy transition. Parasitology. 2004; 128:53–67. [DOI] [PubMed] [Google Scholar]
- 7. McDonald L., Cayla M., Ivens A., Mony B.M., Macgregor P., Silvester E., McWilliam K., Matthews K.R.. Non-linear hierarchy of the quorum sensing signalling pathway in bloodstream form African trypanosomes. PLoS Pathog. 2018; 14:e1007145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sollelis L., Marti M.. A major step towards defining the elusive stumpy inducing factor in Trypanosoma brucei. Trends Parasitol. 2019; 35:6–8. [DOI] [PubMed] [Google Scholar]
- 9. van Grinsven K.W.A., Van Den Abbeele J., Van den Bossche P., van Hellemond J.J., Tielens A.G.M.. Adaptations in the glucose metabolism of procyclic Trypanosoma brucei isolates from tsetse flies and during differentiation of bloodstream forms. Eukaryot. Cell. 2009; 8:1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rico E., Rojas F., Mony B.M., Szoor B., Macgregor P., Matthews K.R.. Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei. Front. Cell Infect. Microbiol. 2013; 3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mantilla B.S., Marchese L., Casas-Sánchez A., Dyer N.A., Ejeh N., Biran M., Bringaud F., Lehane M.J., Acosta-Serrano A., Silber A.M.. Proline metabolism is essential for Trypanosoma brucei brucei survival in the tsetse vector. PLoS Pathog. 2017; 13:e1006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wargnies M., Bertiaux E., Cahoreau E., Ziebart N., Crouzols A., Morand P., Biran M., Allmann S., Hubert J., Villafraz O. et al.. Gluconeogenesis is essential for trypanosome development in the tsetse fly vector. PLoS Pathog. 2018; 14:e1007502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiu Y., Milanes J.E., Jones J.A., Noorai R.E., Shankar V., Morris J.C.. Glucose signaling is important for nutrient adaptation during differentiation of pleomorphic African trypanosomes. mSphere. 2018; 3:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ziegelbauer K., Quinten M., Schwarz H., Pearson T.W., Overath P.. Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro. Eur. J. Biochem. 1990; 192:373–378. [DOI] [PubMed] [Google Scholar]
- 15. Ziegelbauer K., Stahl B., Karas M., Stierhof Y.D., Overath P.. Proteolytic release of cell surface proteins during differentiation of Trypanosoma brucei. Biochemistry. 1993; 32:3737–3742. [DOI] [PubMed] [Google Scholar]
- 16. Rolin S., Paindavoine P., Hanocq-Quertier J., Hanocq F., Claes Y., Le Ray D., Overath P., Pays E.. Transient adenylate cyclase activation accompanies differentiation of Trypanosoma brucei from bloodstream to procyclic forms. Mol. Biochem. Parasitol. 1993; 61:115–125. [DOI] [PubMed] [Google Scholar]
- 17. Matthews K.R., Gull K.. Evidence for an interplay between cell cycle progression and the initiation of differentiation between life cycle forms of African trypanosomes. J. Cell Biol. 1994; 125:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nolan D.P., Rolin S., Rodriguez J.R., Van Den Abbeele J., Pays E.. Slender and stumpy bloodstream forms of Trypanosoma brucei display a differential response to extracellular acidic and proteolytic stress. Eur. J. Biochem. 2000; 267:18–27. [DOI] [PubMed] [Google Scholar]
- 19. Rotureau B., Subota I., Buisson J., Bastin P.. A new asymmetric division contributes to the continuous production of infective trypanosomes in the tsetse fly. Development. 2012; 139:1842–1850. [DOI] [PubMed] [Google Scholar]
- 20. Acestor N., Zíková A., Dalley R.A., Anupama A., Panigrahi A.K., Stuart K.D.. Trypanosoma brucei mitochondrial respiratome: composition and organization in procyclic form. Mol. Cell Proteomics. 2011; 10:M110.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zíková A., Verner Z., Nenarokova A., Michels P.A.M., Lukes J.. A paradigm shift: The mitoproteomes of procyclic and bloodstream Trypanosoma brucei are comparably complex. PLoS Pathog. 2017; 13:e1006679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bienen E.J., Maturi R.K., Pollakis G., Clarkson A.B.. Non-cytochrome mediated mitochondrial ATP production in bloodstream form Trypanosoma brucei brucei. Eur. J. Biochem. 1993; 216:75–80. [DOI] [PubMed] [Google Scholar]
- 23. Surve S., Heestand M., Panicucci B., Schnaufer A., Parsons M.. Enigmatic presence of mitochondrial complex I in Trypanosoma brucei bloodstream forms. Eukaryot. Cell. 2012; 11:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Surve S.V., Jensen B.C., Heestand M., Mazet M., Smith T.K., Bringaud F., Parsons M., Schnaufer A.. NADH dehydrogenase of Trypanosoma brucei is important for efficient acetate production in bloodstream forms. Mol. Biochem. Parasitol. 2017; 211:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vickerman K. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 1985; 41:105–114. [DOI] [PubMed] [Google Scholar]
- 26. Schnaufer A., Clark-Walker G.D., Steinberg A.G., Stuart K.. The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function. EMBO J. 2005; 24:4029–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hashimi H., Zimmer S.L., Ammerman M.L., Read L.K., Lukes J.. Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol. 2013; 29:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aphasizheva I., Aphasizhev R.. U-insertion/deletion mRNA-Editing holoenzyme: definition in sight. Trends Parasitol. 2016; 32:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Read L.K., Lukes J., Hashimi H.. Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley Interdiscip. Rev. RNA. 2016; 7:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cruz-Reyes J., Mooers B.H.M., Doharey P.K., Meehan J., Gulati S.. Dynamic RNA holo-editosomes with subcomplex variants: Insights into the control of trypanosome editing. Wiley Interdiscip Rev RNA. 2018; 9:e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimmer S.L., Simpson R.M., Read L.K.. High throughput sequencing revolution reveals conserved fundamentals of U-indel editing. Wiley Interdiscip. Rev. RNA. 2018; 9:e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blum B., Bakalara N., Simpson L.. A model for RNA editing in kinetoplastid mitochondria: ‘Guide’ RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990; 60:189–198. [DOI] [PubMed] [Google Scholar]
- 33. Seiwert S.D., Stuart K.. RNA editing: transfer of genetic information from gRNA to precursor mRNA in vitro. Science. 1994; 266:114–117. [DOI] [PubMed] [Google Scholar]
- 34. Carnes J., Soares C.Z., Wickham C., Stuart K.. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J. Biol. Chem. 2011; 286:19320–19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDermott S.M., Guo X., Carnes J., Stuart K.. Differential editosome protein function between life cycle stages of Trypanosoma brucei. J. Biol. Chem. 2015; 290:24914–24931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McDermott S.M., Luo J., Carnes J., Ranish J.A., Stuart K.. The architecture of Trypanosoma brucei editosomes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6476–E6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ammerman M.L., Hashimi H., Novotná L., Cicová Z., McEvoy S.M., Lukes J., Read L.K.. MRB3010 is a core component of the MRB1 complex that facilitates an early step of the kinetoplastid RNA editing process. RNA. 2011; 17:865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ammerman M.L., Downey K.M., Hashimi H., Fisk J.C., Tomasello D.L., Faktorová D., Kafková L., King T., Lukes J., Read L.K.. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res. 2012; 40:5637–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ammerman M.L., Tomasello D.L., Faktorová D., Kafková L., Hashimi H., Lukes J., Read L.K.. A core MRB1 complex component is indispensable for RNA editing in insect and human infective stages of Trypanosoma brucei. PLoS One. 2013; 8:e78015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McAdams N.M., Simpson R.M., Chen R., Sun Y., Read L.K.. MRB7260 is essential for productive protein-RNA interactions within the RNA editing substrate binding complex during trypanosome RNA editing. RNA. 2018; 24:540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McAdams N.M., Harrison G.L., Tylec B.L., Ammerman M.L., Chen R., Sun Y., Read L.K.. MRB10130 is a RESC assembly factor that promotes kinetoplastid RNA editing initiation and progression. RNA. 2019; 25:1177–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simpson R.M., Bruno A.E., Bard J.E., Buck M.J., Read L.K.. High-throughput sequencing of partially edited trypanosome mRNAs reveals barriers to editing progression and evidence for alternative editing. RNA. 2016; 22:677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simpson R.M., Bruno A.E., Chen R., Lott K., Tylec B.L., Bard J.E., Sun Y., Buck M.J., Read L.K.. Trypanosome RNA editing mediator complex proteins have distinct functions in gRNA utilization. Nucleic Acids Res. 2017; 45:7965–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weng J., Aphasizheva I., Etheridge R.D., Huang L., Wang X., Falick A.M., Aphasizhev R.. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol. Cell. 2008; 32:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Madina B.R., Kumar V., Mooers B.H.M., Cruz-Reyes J.. Native variants of the MRB1 complex exhibit specialized functions in kinetoplastid RNA editing. PLoS One. 2015; 10:e0123441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aphasizheva I., Zhang L., Wang X., Kaake R.M., Huang L., Monti S., Aphasizhev R.. RNA binding and core complexes constitute the U-insertion/deletion editosome. Mol. Cell. Biol. 2014; 34:4329–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Acestor N., Panigrahi A.K., Carnes J., Zíková A., Stuart K.D.. The MRB1 complex functions in kinetoplastid RNA processing. RNA. 2009; 15:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang Z., Faktorová D., Křížová A., Kafková L., Read L.K., Lukes J., Hashimi H.. Integrity of the core mitochondrial RNA-binding complex 1 is vital for trypanosome RNA editing. RNA. 2015; 21:2088–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aphasizheva I., Alfonzo J., Carnes J., Cestari I., Cruz-Reyes J., Göringer H.U., Hajduk S., Lukes J., Madison-Antenucci S., Maslov D.A. et al.. Lexis and grammar of mitochondrial RNA processing in trypanosomes. Trends Parasitol. 2020; 36:337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schnaufer A., Panigrahi A.K., Panicucci B., Igo R.P., Wirtz E., Salavati R., Stuart K.. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science. 2001; 291:2159–2162. [DOI] [PubMed] [Google Scholar]
- 51. Feagin J.E., Jasmer D.P., Stuart K.. Developmentally regulated addition of nucleotides within apocytochrome b transcripts in Trypanosoma brucei. Cell. 1987; 49:337–345. [DOI] [PubMed] [Google Scholar]
- 52. Feagin J.E., Stuart K.. Developmental aspects of uridine addition within mitochondrial transcripts of Trypanosoma brucei. Mol. Cell. Biol. 1988; 8:1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Souza A.E., Myler P.J., Stuart K.. Maxicircle CR1 transcripts of Trypanosoma brucei are edited and developmentally regulated and encode a putative iron-sulfur protein homologous to an NADH dehydrogenase subunit. Mol. Cell. Biol. 1992; 12:2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Souza A.E., Shu H.H., Read L.K., Myler P.J., Stuart K.D.. Extensive editing of CR2 maxicircle transcripts of Trypanosoma brucei predicts a protein with homology to a subunit of NADH dehydrogenase. Mol. Cell. Biol. 1993; 13:6832–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Read L.K., Stankey K.A., Fish W.R., Muthiani A.M., Stuart K.. Developmental regulation of RNA editing and polyadenylation in four life cycle stages of Trypanosoma congolense. Mol. Biochem. Parasitol. 1994; 68:297–306. [DOI] [PubMed] [Google Scholar]
- 56. Corell R.A., Myler P., Stuart K.. Trypanosoma brucei mitochondrial CR4 gene encodes an extensively edited mRNA with completely edited sequence only in bloodstream forms. Mol. Biochem. Parasitol. 1994; 64:65–74. [DOI] [PubMed] [Google Scholar]
- 57. Priest J.W., Hajduk S.L.. Developmental regulation of Trypanosoma brucei cytochrome c reductase during bloodstream to procyclic differentiation. Mol. Biochem. Parasitol. 1994; 65:291–304. [DOI] [PubMed] [Google Scholar]
- 58. Gazestani V.H., Hampton M., Shaw A.K., Salavati R., Zimmer S.L.. Tail characteristics of Trypanosoma brucei mitochondrial transcripts are developmentally altered in a transcript-specific manner. Int. J. Parasitol. 2018; 48:179–189. [DOI] [PubMed] [Google Scholar]
- 59. Koslowsky D.J., Riley G.R., Feagin J.E., Stuart K.. Guide RNAs for transcripts with developmentally regulated RNA editing are present in both life cycle stages of Trypanosoma brucei. Mol. Cell. Biol. 1992; 12:2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Riley G.R., Myler P.J., Stuart K.. Quantitation of RNA editing substrates, products and potential intermediates: Implications for developmental regulation. Nucleic Acids Res. 1995; 23:708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koslowsky D., Sun Y., Hindenach J., Theisen T., Lucas J.. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res. 2014; 42:1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kirby L.E., Sun Y., Judah D., Nowak S., Koslowsky D.. Analysis of the Trypanosoma brucei EATRO 164 bloodstream guide RNA transcriptome. PLoS Negl. Trop. Dis. 2016; 10:e0004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Urbaniak M.D., Guther M.L.S., Ferguson M.A.J.. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One. 2012; 7:e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McDermott S.M., Stuart K.. The essential functions of KREPB4 are developmentally distinct and required for endonuclease association with editosomes. RNA. 2017; 23:1672–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McDermott S.M., Carnes J., Stuart K.. Editosome RNase III domain interactions are essential for editing and differ between life cycle stages in Trypanosoma brucei. RNA. 2019; 25:1150–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kolev N.G., Ramey-Butler K., Cross G.A.M., Ullu E., Tschudi C.. Developmental progression to infectivity in Trypanosoma brucei triggered by an RNA-binding protein. Science. 2012; 338:1352–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wirtz E., Leal S., Ochatt C., Cross G.A.. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999; 99:89–101. [DOI] [PubMed] [Google Scholar]
- 68. Brun R., Schönenberger M.. Stimulating effect of citrate and cis-aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z. Parasitenkd. 1981; 66:17–24. [DOI] [PubMed] [Google Scholar]
- 69. Overath P., Czichos J., Haas C.. The effect of citrate/cis-aconitate on oxidative metabolism during transformation of Trypanosoma brucei. Eur. J. Biochem. 1986; 160:175–182. [DOI] [PubMed] [Google Scholar]
- 70. Panigrahi A.K., Zíková A., Dalley R.A., Acestor N., Ogata Y., Anupama A., Myler P.J., Stuart K.D.. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol. Cell Proteomics. 2008; 7:534–545. [DOI] [PubMed] [Google Scholar]
- 71. Doleželová E., Kunzová M., Dejung M., Levin M., Panicucci B., Regnault C., Janzen C.J., Barrett M.P., Butter F., Zíková A.. Cell-based and multi-omics profiling reveals dynamic metabolic repurposing of mitochondria to drive developmental progression of Trypanosoma brucei. PLoS Biol. 2020; 18:e3000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Carnes J., Trotter J.R., Ernst N.L., Steinberg A., Stuart K.. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:16614–16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brenndörfer M., Boshart M.. Selection of reference genes for mRNA quantification in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010; 172:52–55. [DOI] [PubMed] [Google Scholar]
- 74. Tylec B.L., Simpson R.M., Kirby L.E., Chen R., Sun Y., Koslowsky D.J., Read L.K.. Intrinsic and regulated properties of minimally edited trypanosome mRNAs. Nucleic Acids Res. 2019; 47:3640–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kirby L.E., Koslowsky D.. Mitochondrial dual-coding genes in Trypanosoma brucei. PLoS Negl. Trop. Dis. 2017; 11:e0005989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schnaufer A., Domingo G.J., Stuart K.. Natural and induced dyskinetoplastic trypanosomatids: how to live without mitochondrial DNA. Int. J. Parasitol. 2002; 32:1071–1084. [DOI] [PubMed] [Google Scholar]
- 77. Zíková A., Schnaufer A., Dalley R.A., Panigrahi A.K., Stuart K.D.. The FoF1-ATP synthase complex contains novel subunits and is essential for procyclic Trypanosoma brucei. PLoS Pathog. 2009; 5:e1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vitter J. Random sampling with a reservoir. ACM Trans. Math. Softw. TOMS. 1985; 11:37–57. [Google Scholar]
- 79. Carnes J., McDermott S., Anupama A., Oliver B.G., Sather D.N., Stuart K.. In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases. Nucleic Acids Res. 2017; 45:4667–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gerasimov E.S., Gasparyan A.A., Kaurov I., Tichý B., Logacheva M.D., Kolesnikov A.A., Lukes J., Yurchenko V., Zimmer S.L., Flegontov P.. Trypanosomatid mitochondrial RNA editing: Dramatically complex transcript repertoires revealed with a dedicated mapping tool. Nucleic Acids Res. 2018; 46:765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kirby L.E., Koslowsky D.. Cell-line specific RNA editing patterns in Trypanosoma brucei suggest a unique mechanism to generate protein variation in a system intolerant to genetic mutations. Nucleic Acids Res. 2019; 49:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Verner Z., Cermáková P., Skodová I., Kriegová E., Horváth A., Lukes J.. Complex I (NADH:ubiquinone oxidoreductase) is active in but non-essential for procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2011; 175:196–200. [DOI] [PubMed] [Google Scholar]
- 83. Vondrusková E., van den Burg J., Zíková A., Ernst N.L., Stuart K., Benne R., Lukes J.. RNA interference analyses suggest a transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J. Biol. Chem. 2005; 280:2429–2438. [DOI] [PubMed] [Google Scholar]
- 84. Fisk J.C., Presnyak V., Ammerman M.L., Read L.K.. Distinct and overlapping functions of MRP1/2 and RBP16 in mitochondrial RNA metabolism. Mol. Cell. Biol. 2009; 29:5214–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Goulah C.C., Pelletier M., Read L.K.. Arginine methylation regulates mitochondrial gene expression in Trypanosoma brucei through multiple effector proteins. RNA. 2006; 12:1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goulah C.C., Read L.K.. Differential effects of arginine methylation on RBP16 mRNA binding, guide RNA (gRNA) binding, and gRNA-containing ribonucleoprotein complex (gRNP) formation. J. Biol. Chem. 2007; 282:7181–7190. [DOI] [PubMed] [Google Scholar]
- 87. Urbaniak M.D., Martin D.M.A., Ferguson M.A.J.. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 2013; 12:2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cooper S., Wadsworth E.S., Ochsenreiter T., Ivens A., Savill N.J., Schnaufer A.. Assembly and annotation of the mitochondrial minicircle genome of a differentiation-competent strain of Trypanosoma brucei. Nucleic Acids Res. 2019; 47:11304–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BioProject: PRJNA597932 https://dataview.ncbi.nlm.nih.gov/object/PRJNA597932?reviewer=skkf1ukebuoluctnjqliudbrop.