


Abstract
Divalent metal cations are essential to the structure and function of the ribosome. Previous characterizations of the ribosome performed under standard laboratory conditions have implicated Mg2+ as a primary mediator of ribosomal structure and function. Possible contributions of Fe2+ as a ribosomal cofactor have been largely overlooked, despite the ribosome's early evolution in a high Fe2+ environment, and the continued use of Fe2+ by obligate anaerobes inhabiting high Fe2+ niches. Here, we show that (i) Fe2+ cleaves RNA by in-line cleavage, a non-oxidative mechanism that has not previously been shown experimentally for this metal, (ii) the first-order in-line rate constant with respect to divalent cations is >200 times greater with Fe2+ than with Mg2+, (iii) functional ribosomes are associated with Fe2+ after purification from cells grown under low O2 and high Fe2+ and (iv) a small fraction of Fe2+ that is associated with the ribosome is not exchangeable with surrounding divalent cations, presumably because those ions are tightly coordinated by rRNA and deeply buried in the ribosome. In total, these results expand the ancient role of iron in biochemistry and highlight a possible new mechanism of iron toxicity.
INTRODUCTION
The translation system is responsible for the synthesis of all coded proteins and contains life's most conserved ribonucleic acids. The common core of the ribosome is universal to all life (1,2) and has been essentially invariant since the last universal common ancestor (3–5). Thus, ribosomes can be interrogated as molecular fossils (6–8). Because ribosomal structure and function are strongly dependent on divalent cations (M2+) (9), and because ribosomes originated long before the Great Oxidation Event (GOE), understanding ribosomal origins and evolution requires characterization of ribosomal interactions with M2+ ions under pre-GOE conditions (10–14).
In extant aerobic life, Mg2+ appears to be the dominant M2+ ion in the translation system. Hundreds of Mg2+ ions mediate ribosomal RNA (rRNA) folding and ribosomal assembly, in some instances binding to specific sites in the universal rRNA common core by direct coordination (9,15–17). Mg2+ ions facilitate association of the large ribosomal subunit (LSU) and small ribosomal subunit (SSU) (18), stabilize folded tRNA (19), maintain the reading frame during translation (20) and link ribosomal proteins (rProteins) to rRNA (21). Mg2+ also facilitates in-line cleavage (22–25), an RNA self-cleavage reaction (26–29).
Before the GOE, anoxia would have stabilized abundant Fe2+ in the biosphere and hydrosphere. Under pre-GOE conditions, Fe2+ would not have caused the damage to biomolecules that occurs today in the presence of O2, via Fenton chemistry (30). In Fenton chemistry, H2O2, a product of Fe2+ autoxidation by O2 (31), is reduced by Fe2+, generating hydroxyl radicals that can oxidatively damage nucleic acids (32–36). We recently reported that Fe2+ can fold RNA and mediate in vitro translation under ‘pre-GOE’ conditions: in the presence of abundant Fe2+ and in the absence of O2 (37). Based on these findings, we proposed that early ribosomal folding and catalysis used Fe2+ instead of, or in combination with Mg2+ and other M2+ ions. However, we found lower translation rates with anoxic Fe2+ than with Mg2+ (37). While lower translation rates with Fe2+ are consistent with ribosomal adaptation to Mg2+ over the last two billion years, this explanation alone appears insufficient because the common core of the ribosome, including the functional centers, has been conserved for over 3 billion years. One possible explanation for this finding is that non-oxidative damage of RNA mediated by Fe2+ is faster than with Mg2+.
Here, we demonstrate that Fe2+ can damage RNA by in-line cleavage, which is distinct from previously characterized oxidative processes. We discovered that this second, non-oxidative mechanism of Fe2+-mediated RNA damage by in-line cleavage can be more extensive in some conditions than oxidative damage. We show that anoxic Fe2+ is efficient in catalyzing in-line cleavage, cleaving rRNA far more rapidly and extensively than Mg2+. Given that the reaction is likely first-order with respect to the metal ion (38,39), the reaction rate constant (M−1 s−1) appears to be over 200 times greater for rRNA with Fe2+ than with Mg2+. The in-line mechanism of cleavage by Fe2+ was validated here by a variety of methods including reaction product characterization.
While metals such as Mg2+ cleave RNA, they are nonetheless essential for folding and function. In parallel with our experiments comparing Fe2+ and Mg2+ in-line cleavage, we investigated whether ribosomes from Escherichia coli grown in pre-GOE conditions associate functionally with Fe2+in vivo. We grew E. coli in anoxic conditions with ample Fe2+ in the growth media. We purified ribosomes from these bacteria and have probed their interactions with metals. We identified tightly bound M2+, which survive ribosomal purification. A small fraction (∼2%) of Fe2+ ions are not exchangeable with Mg2+ in solution and are detectable after purification involving repeated washes in high [Mg2+] buffers. We use these tightly bound ions as reporters for more general M2+ association in vivo. The data are consistent with a model in which certain M2+ ions are deeply buried and highly coordinated within the ribosome (16). Our results suggest that ribosomes grown in pre-GOE conditions contain ∼10 tightly bound Fe2+ ions compared to ∼1 Fe2+ ion in ribosomes from standard growth conditions. Ribosomes washed with Fe2+ contained significantly higher Fe2+ and showed more rRNA degradation than ribosomes washed with Mg2+. Our combined results show the capacity for Fe2+ to (i) associate with functional ribosomes in vivo and in vitro and (ii) mediate significant non-oxidative damage. Our results have significant implications for the evolution of rRNA and iron toxicity in disease.
MATERIALS AND METHODS
rRNA purification
A one-tenth volume of sodium acetate (3.0 M, pH 5.2) and an equal volume of 25:24:1 phenol:chloroform:isoamyl alcohol (Fisher BioReagents, pH 5.2) were added to purified ribosomes. The sample was vortexed and spun at 16,200 × g in a table-top centrifuge for 5 min. The top aqueous layer was transferred to a new tube and extracted twice with a 24:1 mixture of chloroform:isoamyl alcohol (Acros Organics) using the same procedure. rRNA was then precipitated by adding two volumes of 100% ethanol, followed by incubation at −20°C for 30 min. Precipitated rRNA was pelleted by centrifuging at 16,100 × g for 15 min. The pellet was washed with 70% ethanol and suspended in 0.1 mM sodium–EDTA (pH 8.0). rRNA concentrations were quantified by A260 (1A260 = 40 μg rRNA ml−1).
rRNA in-line cleavage reaction rates
Nuclease-free water (IDT) was used in all experiments involving purified or transcribed RNA. rRNA for in-line cleavage experiments was purified as above by phenol-chloroform extraction followed by ethanol precipitation of commercial E. coli ribosomes (New England Biolabs). All in-line cleavage reaction solutions were prepared and incubated in an anoxic Coy chamber (97% Ar, 3% H2 headspace). Fe2+ and Mg2+ solutions were prepared by dissolving a known mass of FeCl2·4H2O or MgCl2 salt in degassed water inside the Coy chamber. rRNA (0.5 μg μl−1) was suspended in degassed 20 mM N-(2-hydroxyethyl)piperazine-N’-2-ethanesulfonic acid (HEPES, pH 7.6), 30 mM KCl, 5% (v/v) glycerol [Invitrogen (UltraPure)], and either 25 mM of MgCl2 or 1 mM of FeCl2. Reactions were placed on a 37°C heat block and incubated for 4 days for MgCl2 and no M2+ and for 8 h for FeCl2. At each time point (0, 1.5, 3, 6, 12, 24, 48 and 96 h for the MgCl2 or no M2+ and 0, 7.5, 15, 30, 60, 120, 240 and 480 min for FeCl2) 4.5 μl aliquots were combined with 0.5 μl of 1 M sodium phosphate buffer pH 7.6 to precipitate the Fe2+ or Mg2+ from solution and stored at −80°C. Aliquots were defrosted on ice and combined with 2X Gel Loading Buffer II (Amicon) then loaded onto a 1% Tris/borate/EDTA agarose gel and run at 120 V for 1.25 h. The RNA in the gel was stained with GelStar™ (Lonza) and imaged with an Azure 6000 Imaging System (Azure Biosystems). Azurespot software was used as a pixel counter to create lane profiles. rRNA peaks were integrated by fitting to an Exponentially Modified Gaussian distribution using Igor Pro (v 7.08) (Supplementary Figure S1). Observed pseudo first-order rate constants (kobs) were found by taking the negative of the slope from the natural logarithm of the normalized peak area versus time plot. Reaction rate constants (k) were calculated by k = kobs/[M2+].
In-line cleavage banding patterns
a-rRNA (40), which is composed of the core of the LSU rRNA, was synthesized and purified as previously described. Lyophilized a-rRNA was resuspended in degassed nuclease-free water (IDT) inside the Coy chamber. Fe2+ and Mg2+ solutions were prepared by dissolving known amounts of FeSO4·7H2O or MgSO4 in degassed nuclease-free water inside the Coy chamber. To initiate the reaction, 1 mM (final concentration) of Mg2+ or Fe2+ was added to 0.02 μg μl−1 a-rRNA in 20 mM HEPES–Tris (pH 7.2) in a 37°C heat block. Samples were removed at 0, 0.25, 0.5 and 1 h for added Fe2+, and at 24 h for added Mg2+. Divalent cation chelation beads (Hampton Research) were added to quench the reactions. The chelation beads were removed using spin columns. RNA cleavage products were visualized using denaturing PAGE (6%, 8 M urea) run at 120 V for ∼1.3 h stained with SYBR Green II.
Fenton chemistry reactions
Purified rRNA from E. coli ribosomes (New England Biolabs) was obtained by phenol-chloroform extraction and ethanol precipitation as above. A stock solution of Fe/EDTA was prepared inside the Coy chamber by dissolving a known amount of FeCl2·4H2O salt in degassed water then mixing with EDTA in degassed water. The Fe/EDTA was removed from the Coy chamber for the Fenton reactions. rRNA was suspended to 0.5 μg μl−1 in 20 mM HEPES (pH 7.6), and 30 mM KCl, with 0% or 5% (v/v) glycerol and either 1 mM Fe/10 mM EDTA/10 mM ascorbate plus 0.3% (v/v) H2O2 or 10 mM EDTA as the reaction initiators wherein the initiators were separately dispensed onto the tube wall then vortexed with the other components. For the zero time points, reaction components were mixed in tubes containing the thiourea quenching agent at a final concentration of 100 mM. For non-zero time points, the reaction mixtures were prepared as bulk solutions and incubated at 37°C on a heat block, after which aliquots were removed at 0, 10 and 60 min and mixed with the thiourea quenching agent at a final concentration of 100 mM. The stopped solutions were immediately frozen and stored at −80°C. For analysis, samples were defrosted on ice, combined with 2X Gel Loading Buffer II, loaded onto a 1% Tris/borate/EDTA agarose gel and run at 120 V for 1.25 h.
Characterization of ApA cleavage products by HPLC
In-line cleavage reagents were prepared as described above in the Coy chamber. In duplicate reactions, 0.5 mM ApA RNA dinucleotide (5′ to 3′; TriLink BioTechnologies) was suspended in degassed 20 mM HEPES/NaOH (pH 7.6), 30 mM KCl, 5% (v/v) glycerol and combined with either water, 25 mM MgCl2 or 25 mM FeCl2 (final concentrations). Reactions were placed on a 37°C heat block with aliquots removed at 0, 0.25, 0.5, 1, 2, 4 and 8 days for no M2+ and Mg2+ samples and at 0, 0.25, 0.5, 1 and 2 days for Fe2+ samples. Aliquots were immediately quenched with 100 mM final concentration sodium phosphate pH 7.6, centrifuged at 2,000 × g for 1 min, and the ApA-containing supernatant was collected to avoid transfer of Fe or Mg phosphate precipitate to the High Performance Liquid Chromatography (HPLC) column. The samples were stored at −80°C prior to placement in the HPLC where they were held at 4°C. HPLC analyses were conducted on an Agilent 1260 Infinity HPLC with a DAD UV–Vis detector, with a path length of 1.0 cm. Products of the reactions were separated using a Kinetex XB-C18 column (150 × 2.1 mm, 2.6 μm particle size). The flow rate was 0.3 ml min−1 and the column temperature was held at 25°C. The mobile phase was 100% 0.1% formic acid (aq) for 5 min ramped to 55% acetonitrile over 25 min. The acetonitrile concentration was then ramped to 100% and was held as such for 10 min before returning to 0.1% formic acid (aq) for column equilibration for 15 min. We recorded the elution at 210, 220 and 260 nm wavelengths, with a 180–400 nm spectrum detected in 2 nm steps. To characterize reaction products, standards were spiked into product mixtures. The spiked standards were 0.5 mM ApA in 20 mM HEPES (pH 7.6), 30 mM KCl, 5% (v/v) glycerol with either water, 2.5 μM adenosine, 3′-adenosine monophosphate or 2′,3′-cyclic adenosine monophosphate.
Characterization of ApA cleavage products by LC–MS
ApA was prepared anoxically at 0.5 mM with 20 mM HEPES pH 7.6, 30 mM KCl, 5% (v/v) glycerol and 25 mM FeCl2, and then incubated at 37°C for 2 days. The sample was analyzed by liquid chromatography mass spectrometry using an Agilent 1290 HPLC; Agilent 1260 Autosampler and DAD UV–Vis detector; path length: 0.6 cm; Agilent 1260 quaternary pump and RID; column: Phenomenex Kinetex 2.6 mmxB-C18100Å, LC column 150 × 2.1 mm; column temp: 25°C; 10 μl injection with needle wash, 100 μl s−1 injection speed. The solvents were (A) 0.1% formic acid in LC–MS grade water and (B) LC–MS grade acetonitrile with a flow rates of 0.3 ml min−1; gradient: 5 min 100% A, 0% B; 20 min ramp to 45% A, 55% B; 10 min 0% A, 100% B; 1 min ramp 100% A, 0% B; 9 min 100% A, 0% B. Elutions were recorded at 210, 220 and 260 nm, with the entire spectrum (180–400 nm) detected in 2 nm steps. This system was coupled to an Agilent 6130 single quad MS Electrospray Ionization Mass Spectrometry system with scanning of ±65 to ±2,000 m/z and capillary voltage of 2.0 kV.
Cell culture and harvesting
Culturing media consisted of LB broth (10 g l−1 NaCl, 10 g l−1 tryptone, 5 g l−1 yeast extract) amended with 4 mM tricine, 50 mM sodium fumarate and 80 mM 3-(N-morpholino)propanesulfonic acid (MOPS; titrated with NaOH to pH 7.8). Fifty ml cultures containing all of these ingredients plus 0.25% (v/v) glycerol were inoculated from glycerol stocks of E. coli MRE600 cells and shaken overnight at 37°C with or without O2 and with either 1 mM FeCl2 or ambient Fe2+ [6–9 μM, measured by the ferrozine assay (41)]. Two ml of each overnight culture was used to inoculate 1-l cultures in the same conditions. These cultures were then orbitally shaken at 37°C to OD600 0.6–0.7. Aerobic cultures were grown in foil-covered Erlenmeyer flasks. Anaerobic fumarate-respiring cultures were inoculated into stoppered glass bottles containing medium that had been degassed with N2 for one hour to remove O2. Cells were then harvested by centrifugation at 4,415 × g for 10 min, washed in 20 ml buffer containing 10 mM Tris pH 7.4, 30 mM NaCl and 1 mM EDTA, and pelleted at 10,000 × g for 10 min. Cell pellets were stored at −80°C until ribosome purification.
Ribosome purification
The ribosome purification procedure was modified from Maguire et al. (42). All purification steps were performed in the Coy chamber unless otherwise noted. Buffers varied in their metal cation content. The typical wash buffer contained 100 mM NH4Cl, 0.5 mM EDTA, 3 mM β-mercaptoethanol, 20 mM Tris pH 7.5, 3 mM MgCl2 and 22 mM NaCl. For ‘Fe purification’ experiments, buffer was composed of 100 mM NH4Cl, 0.5 mM EDTA, 3 mM β-mercaptoethanol, 20 mM Tris pH 7.5, 1 mM FeCl2 and 28 mM NaCl. Sodium chloride concentrations were increased here to maintain the ionic strength of the buffer (131 mM). Elution buffers contained the same composition as the wash buffer except for NH4Cl (300 mM). Frozen cell pellets were resuspended in ribosome wash buffer and lysed in a BeadBug microtube compact homogenizer using 0.5 mm diameter zirconium beads (Benchmark Scientific). Cell lysate was transferred into centrifuge bottles inside the Coy chamber which were tightly sealed to prevent O2 contamination. Cell debris were removed by centrifuging outside of the Coy chamber at 30,000 × g for 30 min at 4°C. The soluble lysate was then transferred back into the chamber and loaded onto a column containing pre-equilibrated, cysteine-linked, SulfoLink™ Coupling Resin (Thermo Fisher Scientific). The resin was washed with 10 column volumes of wash buffer. Ribosomes were eluted into three 10 ml fractions with elution buffer. Eluted fractions were pooled inside the Coy chamber into ultracentrifuge bottles which were tightly sealed. Ribosomes were pelleted outside the chamber by centrifuging at 302,000 × g for 3 h at 4°C under vacuum in a Beckman Optima XPN-100 Ultracentrifuge using a Type 70 Ti rotor. Tubes containing ribosome pellets were brought back into the Coy chamber and suspended in buffer containing 20 mM HEPES (pH 7.6), 30 mM KCl and 7 mM β-mercaptoethanol, heat-sealed in mylar bags, and stored at −80°C. Ribosome concentrations were calculated with a NanoDrop spectrophotometer assuming 1A260 = 60 μg ribosome ml−1 (conversion factor provided by New England Biolabs). This conversion factor was used to estimate the molecular mass of bacterial ribosomes, from which molarity was calculated. Biological triplicates of each growth and purification method were taken for downstream analyses.
Ribosomal Fe content
Purified ribosomes were analyzed for iron content by total reflection X-ray fluorescence spectroscopy (TRXF) as described in Bray and Lenz et al. (37).
rProtein electrophoresis
For SDS-PAGE, purified ribosomes were normalized to 3.33 mg ml−1 in 2X SDS-PAGE loading buffer, heated at 95°C for 5 min, and then incubated on ice for 2 min. Samples were loaded onto a 12% SDS acrylamide gel with a 4% stacking gel and run at 180 V for 60 min.
In vitro translation
Translation reactions were based on the methods of Bray and Lenz et al. (37) with minor modifications. All 15 μl reactions contained 2.25 μl of purified ribosome samples normalized to 9 μg μl−1 (so that the final concentration of ribosomes in our reactions was 1.35 μg μl−1), 0.1 mM amino acid mix, 0.2 mM tRNAs, ∼0.2 μg μl−1 of dihydrofolate reductase mRNA, and 3 μl of factor mix (with RNA polymerase, and transcription/translation factors in 10 mM Mg2+) from the PURExpress® Δ Ribosome Kit (New England Biolabs). The reaction buffer was based on Shimizu et al. (43), with HEPES instead of phosphate buffer to avoid precipitation of metal phosphates. Buffer consisted of 20 mM HEPES (pH 7.3), 95 mM potassium glutamate, 5 mM NH4Cl, 0.5 mM CaCl2, 1 mM spermidine, 8 mM putrescine, 1 mM dithiothreitol (DTT), 2 mM adenosine triphosphate (ATP), 2 mM guanosine triphosphate (GTP), 1 mM uridine triphosphate (UTP), 1 mM cytidine triphosphate (CTP), 10 mM creatine phosphate (CP) and 53 μM 10-formyltetrahydrofolate. Divalent cation salts (MgCl2 or FeCl2) were added to 9 mM final concentration. The reaction buffer was lyophilized and stored at −80°C until resuspension in anoxic nuclease-free water immediately before experiments in the Coy chamber. Reaction mixtures were assembled in the chamber and run at 37°C in a heat block for 120 min. Reactions were quenched on ice to terminate translation (43) and stored on ice until they were assayed for the extent of protein synthesis. Protein synthesis was measured using a DHFR assay kit (Sigma-Aldrich), which measures the oxidation of NADPH (60 mM) to NADP+ by dihydrofolic acid (51 μM). Assays were performed by adding 5 μl of protein synthesis reaction to 995 μl of 1X assay buffer. The NADPH absorbance peak at 340 nm (Abs340) was measured in 15 s intervals over 2.5 min. The slope of the linear regression of Abs340 versus time was used to estimate protein activity (Abs340 min−1).
Protein characterization by LC–MS/MS
After ribosomal purification, samples were reduced with β-mercaptoethanol, and then alkylated with 14 mM iodoacetamide (HEPES, pH 7.6) for 30 minutes at room temperature in the dark. Alkylation was quenched with 5 mM dithiothreitol for 15 min at room temperature in the dark. Proteins were purified by the methanol/chloroform precipitation method and were then digested with trypsin in a buffer containing 5% acetonitrile, 1.6 M urea and 50 mM HEPES (pH 8.8) at 37°C with shaking overnight. The digestion was quenched with addition of trifluoroacetic acid to a final concentration of ∼0.2%. Peptides were purified by Stage-Tip (44) prior to LC–MS/MS analysis.
Peptides were dissolved in a solution containing 5% acetonitrile and 4% formic acid and loaded onto a C18-packed microcapillary column (Magic C18AQ, 3 μm, 200 Å, 75 μm × 16 cm, Michrom Bioresources) by a Dionex WPS-3000TPL RS autosampler (Thermostatted Pulled Loop Rapid Separation Nano/Capillary Autosampler). Peptides were separated by a Dionex UltiMate 3000 UHPLC system (Thermo Scientific) using a 112-minute gradient of 4–17% acetonitrile containing 0.125% formic acid. The LC was coupled to an LTQ Orbitrap Elite Hybrid Mass Spectrometer (Thermo Scientific) with Xcalibur software (version 3.0.63). MS analysis was performed with the data dependent Top15 method; for each cycle, a full MS scan with 60,000 resolution and 1 × 106 AGC (automatic gain control) target in the Orbitrap cell was followed by up to 15 MS/MS scans in the Orbitrap cell for the most intense ions. Selected ions were excluded from further sequencing for 90 s. Ions with single or unassigned charge were not sequenced. Maximum ion accumulation time was 1,000 ms for each full MS scan, and 50 ms for each MS/MS scan.
Raw MS files were analyzed by MaxQuant (version 1.6.2.3; 45). MS spectra were searched against the E. coli database from UniProt containing common contaminants using the integrated Andromeda search engine (46). Due to the unavailability of the proteome database for E. coli strain MRE-600, the database for strain K12 was used. It has been shown that the two strains have nearly identical ribosome associated proteins (47). All samples were searched separately and set as individual experiments. Default parameters in MaxQuant were used, except the maximum number of missed cleavages was set at 3. Label-free quantification was enabled with the LFQ minimum ratio count of 1. The match-between-runs option was enabled. The false discovery rates (FDR) were kept at 0.01 at both the peptide and protein levels.
The results were processed using Perseus software (48). In the final dataset, the reverse hits and contaminants were removed. The LFQ intensity of each protein from the proteinGroups table was extracted and reported. For the volcano plots showing differential regulation of proteins, the ratios used were from the LFQ intensities of samples from each of the three experiments. The cutoff for differential expression was set at 2-fold. P-values were calculated using a two-sided T-test on biological triplicate measurements with the threshold P-value of 0.05 for significant regulation. The raw files are available at http://www.peptideatlas.org/PASS/PASS01418 (username: PASS01418 and password: ZW2939nnw).
RESULTS
In-line cleavage of rRNA: Mg2+ and anoxic Fe2+
By manipulating reaction conditions, we could switch the mode of rRNA cleavage between Fenton and in-line mechanisms. In-line is the only possible mechanism of cleavage by Mg2+ due to its fixed oxidation state and inability to generate hydroxyl radicals. We confirm the expectation that Mg2+-mediated in-line cleavage reactions are not inhibited by anoxia or hydroxyl radical quenchers (Supplementary Figure S2).
We confirm here in a variety of experiments that RNA is degraded by in-line cleavage when incubated with Fe2+ under anoxic conditions (Figure 1A). Most of the experiments employed the rRNA of E. coli as substrate. A shorter RNA [a-RNA (40)] showed on a higher size resolution gel that RNA banding patterns and reaction products were nearly identical for Mg2+ and anoxic Fe2+ reactions (Figure 2), indicating that preferred sites of cleavage are the same for both metals. Common sites of cleavage are indications of common mechanisms of cleavage (23). Neither Mg2+ nor anoxic Fe2+ cleavage was inhibited by glycerol (5%), which is known to quench hydroxyl radical and to inhibit hydroxyl radical cleavage (49). By contrast, glycerol inhibited cleavage by Fe2+ under conditions that favor Fenton-type cleavage (Supplementary Figure S3). Glycerol did not inhibit Mg2+ in-line cleavage under any conditions (Supplementary Figure S2).
Figure 1.

In-line cleavage of rRNA in anoxia. In-line cleavage of purified rRNAs with (A) 1 mM Fe2+ (0–8 h) and (B) 25 mM Mg2+ (0–96 h). Reactions were conducted in an anoxic Coy chamber at 37°C in the presence of the hydroxyl radical quencher glycerol (5% v/v) and were analyzed by 1% agarose gel electrophoresis. Pseudo first-order rate plots were extracted from 23S and 16S rRNA band intensities for (C) 1 mM Fe2+ and (D) 25 mM Mg2+. The Mg2+ time axis is 10 times greater than the Fe2+ time axis. Error bars represent the SEM (standard error of the mean; n = 3).
Figure 2.
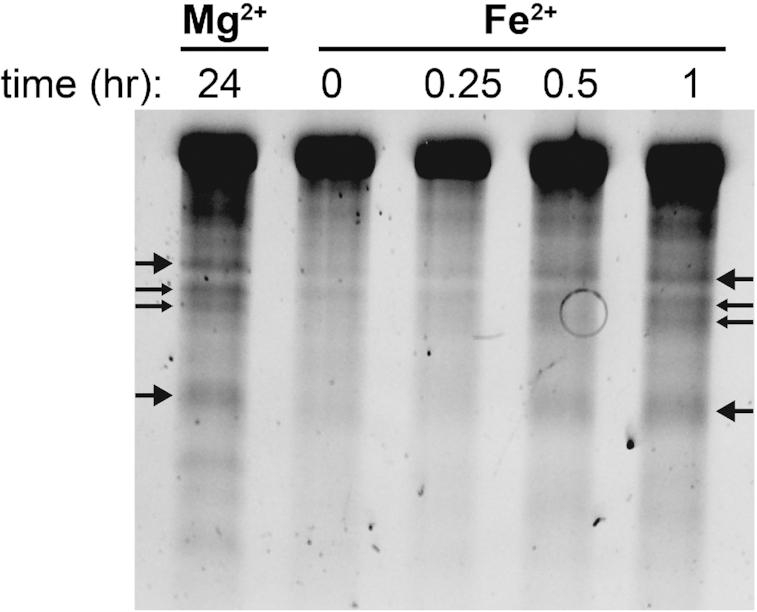
In-line cleavage banding patterns are the same for rRNA cleavage with Mg2+ and anoxic Fe2+. Several primary cleavage bands of a-rRNA (40) are indicated by arrows. This gel is 6% polyacrylamide, 8 M urea showing in-line cleavage mediated by 1 mM Mg2+ or 1 mM anoxic Fe2+ at 37°C for varying amounts of time. Reactions were run in 20 mM Tris–HEPES (pH 7.2).
In the absence of O2, cleavage rates are significantly greater for Fe2+ than for Mg2+. For 16S and 23S rRNAs, 1 mM Fe2+ caused significant in-line cleavage of rRNA after 30 min at 37°C. Both rRNAs were completely degraded after 2 hours in anoxic Fe2+ (Figure 1A). By contrast, when the M2+ ion was switched from 1 mM Fe2+ to 25 mM Mg2+, only a modest amount of in-line cleavage was observed after 6 h (Figure 1B). Fitting of the data to a pseudo first-order rate model (Figure 1C and D) reveals apparent rate constant for cleavage of the full-length 23S rRNA with Fe2+ is 67 × 10−5 s−1 and with Mg2+ is 5 × 10−5 s−1. The rate constants for cleavage of the 16S rRNA is 25 × 10−5 s−1 for Fe2+ and 3 × 10−5 s−1 for Mg2+ (Table 1). These apparent rate constants do not account for differences in metal concentration or in RNA length.
Table 1.
rRNA cleavage pseudo first-order rate constants
| Metal | rRNA | k obs (10−5 s−1) | SEM (10−5 s−1) |
|---|---|---|---|
| 1 mM Fe2+ | |||
| 23S | 67 | 12 | |
| 16S | 25 | 8.2 | |
| 25 mM Mg2+ | |||
| 23S | 5 | 1.1 | |
| 16S | 3 | 0.8 |
In sum, reactions with Mg2+ and anoxic Fe2+ showed a lack of inhibition by a hydroxyl radical quencher. By contrast, the quencher inhibited reactions with Fe2+ in the presence of O2. The observed rate constant for in-line cleavage for rRNA is ∼10 times greater for 1 mM Fe2+ than for 25 mM Mg2+. Under these conditions, in-line cleavage is expected to scale with metal concentration (38,39) so that that the reaction rate constant, k (M−1 s−1), is increased with Fe2+ by ∼300 times for the 23S and by ∼200 times for the 16S. We demonstrate that although Fe2+ interacts in the same way as Mg2+ with RNA, the cleavage potency of Fe2+ is greatly enhanced.
Characterization of in-line cleavage reaction products
To confirm that Fe2+ catalyzes non-oxidative in-line RNA cleavage, a series of cleavage reactions were performed on the dinucleotide ApA. The products of the reaction were characterized by HPLC via spiking with standards, and by LC-MS. A small RNA with only one possible cleavage site allowed us to identify specific cleavage products, which report on the mechanism of scission. In-line cleavage leads to a 2′,3′-cyclic phosphate upstream of the scission site and a downstream 5′-OH RNA fragment. Subsequently, the 2′,3′-cyclic phosphate can hydrolyze to either a 2′- or 3′- monophosphate. Conversely, oxidative cleavage of RNA by a hydroxyl radical that may be formed during iron oxidative processes abstracts a proton from a ribose sugar leading to a variety of products but not 2′,3′-cyclic phosphate, 2′-phosphate, or 3′-phosphate RNA fragments (50). Anoxic incubation of ApA with 25 mM Mg2+ or Fe2+ produces adenosine, 2′,3′-cyclic adenosine monophosphate (2′,3′-cAMP) and 3′-adenosine monophosphate (3′-AMP) (Figure 3A–D and Supplementary Figure S4). The repertoire of products is matched for the two metals, pointing to a common in-line cleavage mechanism.
Figure 3.
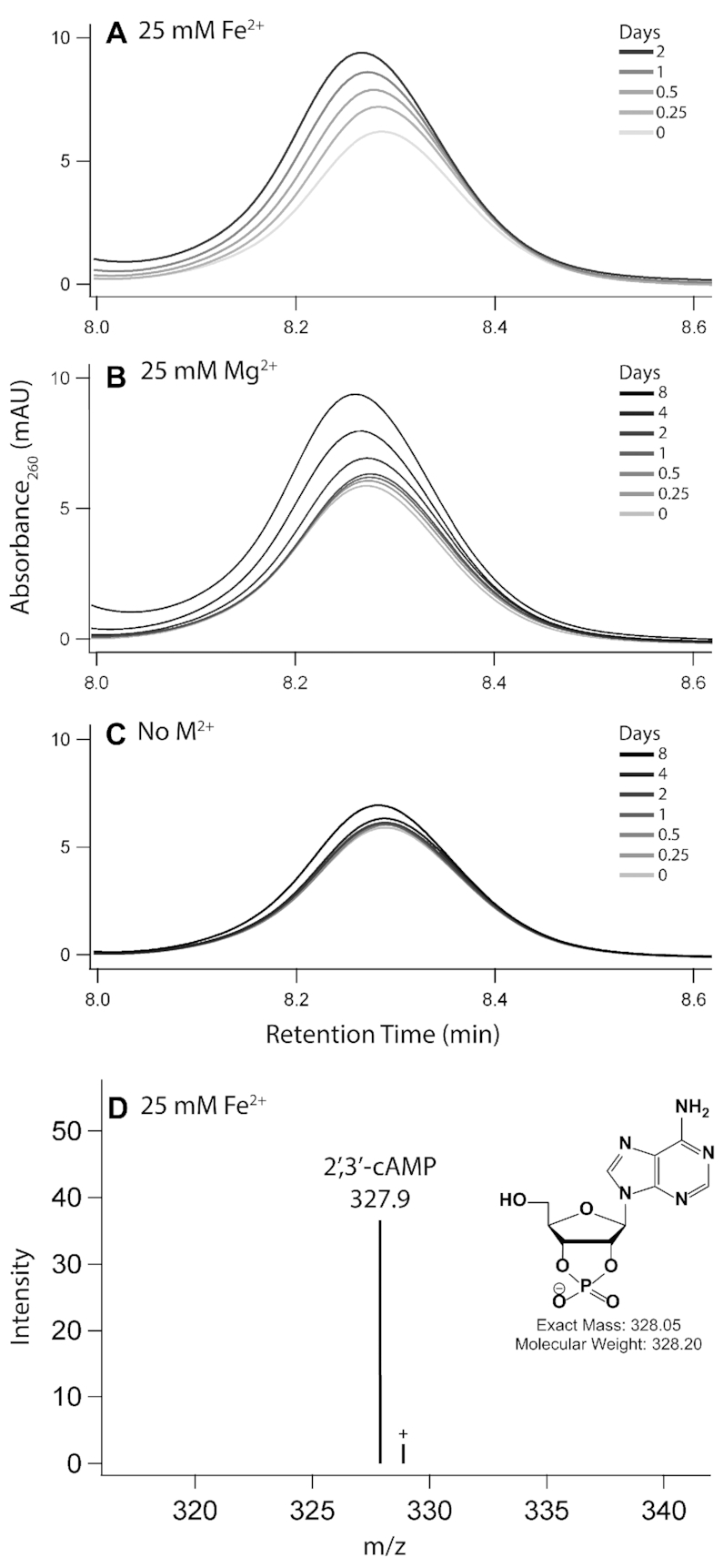
2′,3′-cAMP is formed upon incubation of ApA with Fe2+ or Mg2+. HPLC chromatograms show the accumulation of 2′,3′-cAMP, a direct product of in-line cleavage, upon incubation of ApA with either (A) 25 mM Fe2+, (B) 25 mM Mg2+ or (C) no metal as the negative control. Panel (D) shows identification of the 2′,3′-cyclic adenosine monophosphate by LC–MS of ApA incubated with 25 mM Fe2+ for 2 days. Labeled species correspond to [M−H]− ions. Reactions were incubated anoxically at 37°C in the presence of 5% (v/v) glycerol.
M2+ exchange during ribosomal purification
We hypothesized that O2 and Fe2+ content during bacterial growth could affect the iron content of ribosomes. However, the vast majority of ribosomal M2+ ions are exchangeable (51) and canonical ribosome purification procedures use high Mg2+ buffers (52) to maintain folding and stability. Therefore, spontaneous exchange of in vivo bound M2+ with those in the buffer occurs during purification, suggesting that the final Fe2+ content of purified ribosomes depends on the type of M2+ in the purification buffer. Indeed, ribosomes purified in solutions with 1 mM Fe2+ contained significantly higher Fe2+ than those purified in 3 mM Mg2+ regardless of growth condition (Figure 4). All ribosome samples purified in 1 mM Fe2+ contained similar Fe2+ (∼400–600 mol Fe mol−1 ribosome). These results show that the vast majority of ribosomal M2+ ions are exchangeable and that M2+ exchange takes place during purification.
Figure 4.
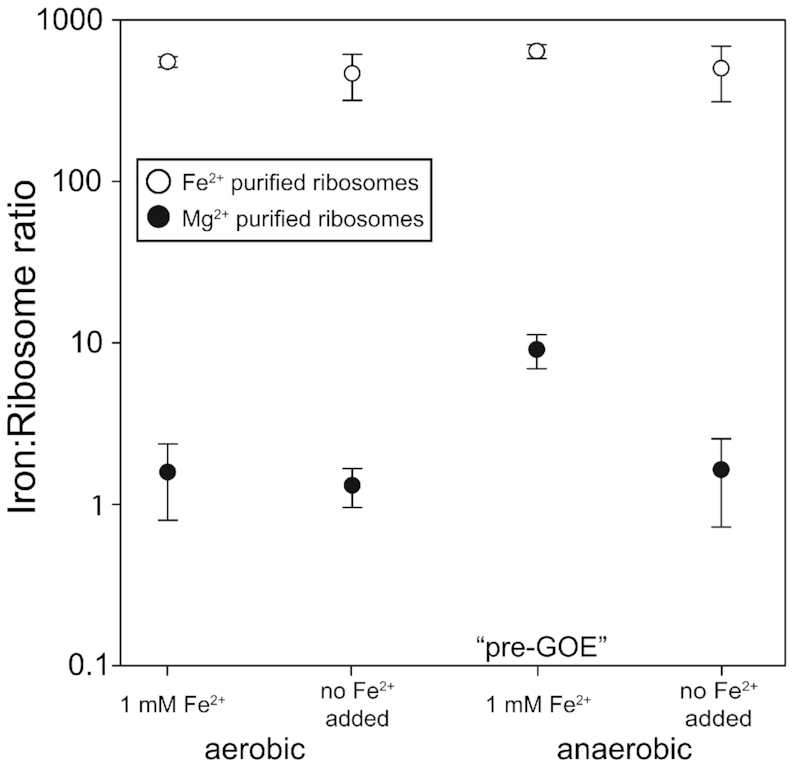
Iron content (mol Fe mol−1 ribosome) of purified ribosomes. E. coli were grown aerobically or anaerobically at 1 mM Fe2+ or ambient Fe2+ (6–9 μM, no Fe added), and purified in buffers containing either 3 mM Mg2+ (black circles) or 1 mM Fe2+ (white circles). Error bars represent the SEM (n = 3).
Tight ribosomal binding of a subset of M2+
A small subset of ribosomal M2+ ions are not exchangeable during purification. Ribosomes retain this subset of in vivo divalent cations after purification. We harvested E. coli in log phase from four growth conditions: oxic or anoxic with high Fe2+ in the medium (1 mM Fe2+), and oxic or anoxic without added Fe2+ in the growth medium (6–9 μM Fe2+). Ribosomes from E. coli grown in pre-GOE conditions (anoxic, high Fe2+) contained quantitatively reproducible elevated levels of Fe2+ after purification in solutions containing Mg2+. We detected around 9 mol Fe mol−1 ribosome from cells grown in pre-GOE conditions purified in solutions with high Mg2+ (Figure 4). The three other growth conditions yielded ribosomes containing near background levels of Fe2+ (<2 mol Fe mol−1 ribosome).
Quantitating translation
Ribosomes from all four growth conditions produced active protein in translation assays. Ribosomes were functional in vitro under standard conditions (with 10 mM Mg2+) and also in 8 mM Fe2+ + 2 mM Mg2+ under anoxia. Regardless of whether translation activity was assayed in the presence of 10 mM Mg2+ or 8 mM Fe2+ + 2 mM Mg2+, ribosomes synthesized aerobically in the absence of Fe2+ have higher activity than the ribosomes synthesized anaerobically in the absence of Fe2+ (P < 0.08; Supplementary Figure S5). The presence of 1 mM Fe2+ in the bacterial growth conditions did not affect ribosomal activity; the only differences are whether growth conditions are aerobic or anaerobic, and whether 10 mM Mg2+ or 8 mM Fe2+ + 2 mM Mg2+ are used in the assay. Translation was reduced in the presence of Fe2+ compared to Mg2+, consistent with our previous work (37). The translational activity of ribosomes harvested from anaerobic cells was slightly less than from those from aerobic cells. Ribosomes from all four growth conditions contained intact 23S, 16S and 5S rRNAs with purification in 3 mM Mg2+ (Figure 5A) resulting in a higher proportion of intact rRNA relative to purification in 1 mM Fe2+ (Figure 5B). Each purification also contained a full suite of rProteins as indicated by mass spectrometric analysis and by gel electrophoresis (Supplementary Figure S6). The protein composition of ribosomes from 1 mM Fe2+ growth conditions (Supplementary Figure S6B) was similar to that from Mg2+ growth conditions (Supplementary Figure S6A).
Figure 5.

Agarose gels (1%) showing rRNA from ribosomes purified in (A) 3 mM Mg2+ and (B) 1 mM Fe2+. The banding pattern suggests that rRNA is less degraded in ribosomes purified with 3 mM Mg2+ than in ribosomes purified with 1 mM Fe2+.
rProtein characterization
In addition to an oxidative mechanism, our results pointed to a non-oxidative cleavage mechanism of RNA with Fe2+. So, we next asked whether ribosomes might adopt different proteins to cope with high Fe2+ in both oxic and anoxic conditions. Ribosomes under all four growth conditions contained a full repertoire of rProteins, and were associated with additional proteins, as determined by mass spectrometry. These non-ribosomal proteins ranged in function from translation to central metabolism. Proteins from anaerobic pathways were generally more abundant in ribosomes from anaerobic cells while proteins from aerobic pathways were more abundant in ribosomes from aerobic cells (Supplementary Tables S1 and S2). Proteins for synthesis of enterobactin, an Fe3+-binding siderophore, were more abundant in ribosomes from aerobic cells and from those grown without the addition of Fe, while the bacterial non-heme ferritin subunit was more abundant in ribosomes from anaerobic cells regardless of the Fe2+ content in the media (Supplementary Table S2). Several proteins were differentially expressed in ribosomes grown in pre-GOE conditions relative to other growth conditions (Supplementary Figure S7). Notably, ribosomes grown anaerobically with high Fe2+ had five times the abundance of the protein YceD than ribosomes grown anaerobically without added Fe2+. Anaerobic high Fe2+ ribosomes had one third the abundance of the rProtein S12 methylthiotransferase protein RimO and rRNA LSU methyltransferase K/L protein RlmL than ribosomes from aerobically grown cells with 1 mM Fe2+.
DISCUSSION
Iron promotes rapid in-line cleavage of rRNA
Mg2+ is known to cleave the RNA phosphodiester backbone via an in-line mechanism (22,23). We have shown here that Fe2+, like Mg2+, can cleave RNA by a non-oxidative in-line mechanism. We used cleavage of 23S and 16S rRNA to determine the observed rate constants of both Mg2+- and Fe2+-mediated cleavage. The kobs, uncorrected for metal concentration, for in-line cleavage by Fe2+ is around 10 times greater than by Mg2+. Previous studies suggest that in-line cleavage is first-order with respect to metal concentration (38,39), allowing the calculation of a per molar metal reaction rate constant by k = kobs/[M2+]. Assuming this first-order relationship in our experiments, k with Fe2+ is ∼300 times greater for the 23S and ∼200 times greater for the 16S than with Mg2+. In Supplementary Table S3 we compare our results to literature k values of Mg2+ and Zn2+ cleavage taken under a range of conditions (38,39,53–57), normalizing for the number of cleavable phosphates in the RNA substrate. Changes in metal identity, RNA length, RNA folding, pH, and temperature result in differences in apparent rate constants. The values extend over four orders of magnitude. Rate enhancement by switching Mg2+ to another metal while other conditions are held constant is greater for Fe2+ than for Zn2+, highlighting the rapidity of cleavage by Fe2+.
Support for a non-oxidative in-line mechanism of cleavage of RNA by anoxic Fe2+ is provided by observations that the rate of the reaction is not attenuated by anoxia and that the sites of cleavage appear to be conserved for Mg2+ and anoxic Fe2+ (Figure 2). The absence of hydroxyl radical intermediates in the anoxic cleavage reaction is confirmed by the lack of inhibition by a hydroxyl radical quencher known to inhibit Fenton chemistry (32). Cleavage products of the RNA dinucleotide ApA include only those that are expected from an in-line mechanism and align with products formed with Mg2+. Among these is 2′,3′-cyclic phosphate, the hallmark of in-line attack of the bridging phosphate by the 2′-OH.
In-line cleavage is the dominant mechanism of Fe2+ cleavage when contributions from Fenton-mediated processes are minimized and is the only mechanism of Mg2+ cleavage. By contrast, in oxic environments, transient Fe2+ oxidation generates hydroxyl radicals (31) that cleave nucleic acids (30,32–35). Our results have significant implications for iron toxicity and human disease. The potency of Fe2+ in inducing rRNA cleavage may lead to decreased longevity of Fe2+-containing ribosomes. In fact, rRNA cleavage linked to Fe2+ oxidation, as in the human ribosome in Alzheimer's disease (58), or in yeast rRNA (59), could be in some measure attributable to Fe2+ in-line cleavage.
Fe2+ appears to be a potent all-around cofactor for nucleic acids. The combined results indicate that:
rRNA folds at a lower concentration of Fe2+ than Mg2+ (37),
at least a subset of ribozymes and DNAzymes are more active in Fe2+ than in Mg2+ (60,61),
the translation system is functional when Fe2+ is the dominant divalent cation (37),
at low concentrations of M2+, T7 RNA polymerase is more active with Fe2+ than with Mg2+ (62),
a broad variety of nucleic acid processing enzymes are active with Fe2+ instead of Mg2+ (62),
rates of in-line cleavage are significantly greater for Fe2+ than for Mg2+ (here), and
Fe2+ but not Mg2+ confers oxidoreductase functionality to some RNAs (17,63).
Why so fast?
Our previous DFT computations (62) help explain why Fe2+ is such a potent cofactor for RNA. Conformations and geometries of coordination complexes with water and/or phosphate are nearly identical for Fe2+ or Mg2+. However, differences between Mg2+ and Fe2+ are seen in the electronic structures of coordination complexes.
Firstly, because of low lying d orbitals, Fe2+ has greater electron withdrawing power than Mg2+ from first shell phosphate ligands. In coordination complexes with phosphate groups, the phosphorus atom is a better electrophile when M2+ = Fe2+ than when M2+ = Mg2+. This difference between Mg2+ and Fe2+ is apparent in both ribozyme reactions and in-line cleavage reactions.
Secondly, Fe(H2O)62+ is a stronger acid than Mg(H2O)62+; depletion of electrons is greater from water molecules that coordinate Fe2+ than from those that coordinate Mg2+. The lower pKa of Fe(H2O)62+ may promote protonation of the 5′-OH leaving group during cleavage. Metal hydrates with low pKas have been reported to induce RNA cleavage better than less acidic metal hydrates (22).
During in-line cleavage reactions, RNA coordinates M2+ or the M2+ hydrate (22,23). Indeed, studies of the in-line cleavage fragment patterns have previously been used to probe structural information on RNA molecules, such as metal-binding sites (26,27). We demonstrated with ApA that RNA secondary structure is not required for in-line cleavage. The same activities that drive in-line cleavage (e.g. 2′-OH activation and coordination of the leaving group) are thought to occur in metal-catalyzed ribozyme cleavage (64). Multiple ribozymes (60) and DNAzymes (61) have been observed to function with Fe2+ as a cofactor. Our results with Fe2+ in-line cleavage, and in-line cleavage in general, require no enzymatic activity.
The remarkably high cleavage activity of Fe2+ with RNA demonstrated here bears relevance to prebiotic chemistry and early biochemistry. Because these reactions are catalytic, they increase both forward and reverse reaction rates. RNA degradation through Fe2+ cleavage should be weighed against potential RNA polymerization and the benefits of increased function. The same dualism exists with Mg2+, but our work suggests higher stakes with Fe2+. At the extremes, without M2+, RNA cannot form complex folds and has few avenues for catalytic or functional activity while excessive M2+ degrades RNA. There theoretically exists some point of balance wherein M2+ is beneficially utilized with acceptable frequency of cleavage. Given the increased potency of cleavage with Fe2+ relative to Mg2+, this balancing point may be at a lower concentration of Fe2+ than Mg2+. Enhanced cofactor characteristics of Fe2+ may allow RNA to access more functions using less metal. On early Earth, heightened RNA cleavage in the presence of Fe2+ if balanced by a similar rate of RNA resupply would allow functional space to be explored in short time. RNAs would be selected that could cooperate with or tolerate a potent metal. Therefore, Fe2+ may have been a force for accelerated RNA evolution on early Earth.
Fe2+ associates with rRNA in vivo
Exchange of non-native metals for native metals is well-known during purification of proteins (51). We observe analogous phenomena with rRNA. Fe2+ can exchange with Mg2+ (and vice versa) during purification of ribosomes. Ribosomes purified in either Fe2+ or Mg2+ associate with 500–1000 M2+ ions that match the type of ion in the purification buffers. Our data support the tight association and lack of exchange of around 9 M2+ per ribosome. This subset of M2+ do not exchange during purification. The number of non-exchangeable M2+ closely matches the number of M2+ identified previously as a special class of deeply buried and highly coordinated M2+ in dinuclear microclusters (M2+-μc's) (16). Mg2+ ions in M2+-μc's are directly chelated by multiple phosphate oxygens of the rRNA backbone and are substantially dehydrated. M2+-μc's within the LSU provide a framework for the ribosome's peptidyl transferase center, the site of protein synthesis in the ribosome, suggesting an essential and ancient role for M2+-μc's in the ribosome. There are four dinuclear M2+-μc's in the LSU and one in the SSU, accounting for 10 M2+ (16). Displacement of these M2+ would require large-scale changes in ribosomal conformation. In sum, there are ten M2+ per ribosome that are expected to be refractory to exchange. We hypothesize that this subset M2+ are contained in M2+ -μc's, which can be occupied by either Mg2+ or Fe2+ (17), depending on growth conditions.
We also hypothesize that ribosomes harvested from aerobic cells have low Fe2+/Mg2+ ratios because of low intracellular Fe2+ availability and lability (65). This hypothesis is supported by our observation that the number of slow exchanging Fe2+ per ribosome from aerobic cells is near the baseline of our measurements. It appears that ribosomes harvested from pre-GOE conditions have high Fe2+/Mg2+ ratios because of high intracellular Fe2+ availability and lability, as indicated by the close match in the number of slowly exchanging Fe2+ per ribosome and the number of available M2+ sites in ribosomal M2+-μc's. In these experiments, we detect only the Fe2+ ions that do not exchange during purification.
Anoxic Fe2+ degrades rRNA within ribosomes
rRNA from all four growth conditions showed partial hydrolysis when ribosomes were purified in anoxic Fe2+. It appears that Fe2+ can mediate rRNA degradation by an in-line mechanism during ribosomal purification in anoxic Fe2+. Less rRNA cleavage was observed in ribosomes purified with Mg2+, which contain orders of magnitude lower Fe2+.
Summary
Here, we have shown for the first time that bacteria grown in pre-GOE conditions contain functional ribosomes with tightly bound Fe atoms. The ∼10 ribosomal Fe ions in ribosomes grown anoxically with high Fe2+ are likely deeply buried and specifically bound to rRNA. Depending on intracellular Fe lability, ribosomes may have higher Fe content in vivo given the high capacity for the ribosome to substitute ∼600 loosely bound Mg2+ ions for Fe2+. Furthermore, direct association of the rRNA with Fe atoms results in a fast rate of in-line cleavage. 1 mM Fe2+ gives a ∼10 times higher kobs than does 25 mM Mg2+ so that the assumed per molar metal rate constant is hundreds of times greater with Fe2+ than with Mg2+. This highlights a potential role of protection from in-line cleavage for rProteins and suggests that Fe2+ may drive rapid cycling of RNA between monomers and polymers. Our results support a model in which alternate M2+ ions, namely Fe2+, participated in the origin and early evolution of life: first in abiotic proto-biochemical systems, through potentially rapid rounds of formation and breakdown of RNA structures, and then within early cellular life up until the GOE (66). Our study also expands the role of Fe2+ in modern biochemistry by showing that extant life retains the ability to incorporate Fe into ribosomes. We surmise that extant organisms under certain environmental and cellular states may use Fe2+ as a ribosomal cofactor. In addition, obligate anaerobic organisms that have spent the entirety of their evolutionary history in permanently anoxic environments may still use abundant Fe2+ in their ribosomes in vivo.
DATA AVAILABILITY
The raw files are available at http://www.peptideatlas.org/PASS/PASS01418 (username: PASS01418 and password: ZW2939nnw).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Corinna Tuckey (New England BioLabs), Eric B. O’Neill, and Drs Anton Petrov, Roger M. Wartell, Thomas Tullius and Ada Yonath for helpful discussions.
Notes
Present address: Marcus S. Bray, Department of Biology, San Diego State University, San Diego, CA 92182, USA.
Contributor Information
Rebecca Guth-Metzler, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Marcus S Bray, NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA; School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Moran Frenkel-Pinter, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Suttipong Suttapitugsakul, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Claudia Montllor-Albalate, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Jessica C Bowman, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Ronghu Wu, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Amit R Reddi, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30332, USA.
C Denise Okafor, Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA.
Jennifer B Glass, NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA; Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30332, USA; School of Earth and Atmospheric Sciences, Georgia Institute of Technology, Atlanta, GA 30332, USA.
Loren Dean Williams, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA; NASA Center for the Origin of Life, Georgia Institute of Technology, Atlanta, GA 30332, USA; Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30332, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by National Aeronautics and Space Administration [NNX14AJ87G, NNX16AJ28G, NNX16AJ29G, 80NSSC18K1139] and National Science Foundation [MCB-1552791 to A.R.R.]. The TXRF instrument was supported by National Institutes of Health [ES025661 to A.R.R.]. Funding for open access charge: NASA [80NSSC18K1139].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bernier C.R., Petrov A.S., Kovacs N.A., Penev P.I., Williams L.D.. Translation: the universal structural core of life. Mol. Biol. Evol. 2018; 35:2065–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Melnikov S., Ben-Shem A., Garreau de Loubresse N., Jenner L., Yusupova G., Yusupov M.. One core, two shells: bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012; 19:560–567. [DOI] [PubMed] [Google Scholar]
- 3. Woese C.R. Translation: in retrospect and prospect. RNA. 2001; 7:1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noller H.F., Kop J., Wheaton V., Brosius J., Gutell R.R., Kopylov A.M., Dohme F., Herr W., Stahl D.A., Gupta R.. Secondary structure model for 23S ribosomal RNA. Nucleic Acids Res. 1981; 9:6167–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petrov A.S., Bernier C.R., Hsiao C., Norris A.M., Kovacs N.A., Waterbury C.C., Stepanov V.G., Harvey S.C., Fox G.E., Wartell R.M. et al.. Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bokov K., Steinberg S.V.. A hierarchical model for evolution of 23S ribosomal RNA. Nature. 2009; 457:977–980. [DOI] [PubMed] [Google Scholar]
- 7. Kovacs N.A., Petrov A.S., Lanier K.A., Williams L.D.. Frozen in time: the history of proteins. Mol. Biol. Evol. 2017; 34:1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Agmon I., Bashan A., Yonath A.. On ribosome conservation and evolution. Isr. J. Ecol. Evol. 2006; 52:359–374. [Google Scholar]
- 9. Klein D.J., Moore P.B., Steitz T.A.. The contribution of metal ions to the structural stability of the large ribosomal subunit. RNA. 2004; 10:1366–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anbar A.D. Elements and evolution. Science. 2008; 322:1481–1483. [DOI] [PubMed] [Google Scholar]
- 11. Hazen R.M., Ferry J.M.. Mineral evolution: mineralogy in the fourth dimension. Elements. 2010; 6:9–12. [Google Scholar]
- 12. Holland H.D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. London, Ser. B. 2006; 361:903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klein C. Some Precambrian banded iron-formations (BIFs) from around the world: their age, geologic setting, mineralogy, metamorphism, geochemistry, and origins. Am. Mineral. 2005; 90:1473–1499. [Google Scholar]
- 14. Holland H.D. The oceans; a possible source of iron in iron-formations. Econ. Geol. 1973; 68:1169–1172. [Google Scholar]
- 15. Bowman J.C., Lenz T.K., Hud N.V., Williams L.D.. Cations in charge: magnesium ions in RNA folding and catalysis. Curr. Opin. Struct. Biol. 2012; 22:262–272. [DOI] [PubMed] [Google Scholar]
- 16. Hsiao C., Williams L.D.. A recurrent magnesium-binding motif provides a framework for the ribosomal peptidyl transferase center. Nucleic Acids Res. 2009; 37:3134–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin S.Y., Wang Y.C., Hsiao C.. Prebiotic iron originates the peptidyl transfer origin. Mol. Biol. Evol. 2019; 36:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schuwirth B.S., Borovinskaya M.A., Hau C.W., Zhang W., Vila-Sanjurjo A., Holton J.M., Cate J.H.D.. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005; 310:827–834. [DOI] [PubMed] [Google Scholar]
- 19. Selmer M., Dunham C.M., Murphy F.V., Weixlbaumer A., Petry S., Kelley A.C., Weir J.R., Ramakrishnan V.. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006; 313:1935–1942. [DOI] [PubMed] [Google Scholar]
- 20. Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G.. A new understanding of the decoding principle on the ribosome. Nature. 2012; 484:256–259. [DOI] [PubMed] [Google Scholar]
- 21. Petrov A.S., Bernier C.R., Hsiao C., Okafor C.D., Tannenbaum E., Stern J., Gaucher E., Schneider D., Hud N.V., Harvey S.C. et al.. RNA-magnesium-protein interactions in large ribosomal subunit. J. Phys. Chem. B. 2012; 116:8113–8120. [DOI] [PubMed] [Google Scholar]
- 22. Forconi M., Herschlag D.. Metal ion-based RNA cleavage as a structural probe. Methods Enzymol. 2009; 468:91–106. [DOI] [PubMed] [Google Scholar]
- 23. Soukup G.A., Breaker R.R.. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999; 5:1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkler W., Nahvi A., Breaker R.R.. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002; 419:952–956. [DOI] [PubMed] [Google Scholar]
- 25. Winkler W.C., Nahvi A., Roth A., Collins J.A., Breaker R.R.. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004; 428:281. [DOI] [PubMed] [Google Scholar]
- 26. Dorner S., Barta A.. Probing ribosome structure by europium-induced RNA cleavage. Biol. Chem. 1999; 380:243–251. [DOI] [PubMed] [Google Scholar]
- 27. Winter D., Polacek N., Halama I., Streicher B., Barta A.. Lead-catalysed specific cleavage of ribosomal RNAs. Nucleic Acids Res. 1997; 25:1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pyle A.M. Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem. 2002; 7:679–690. [DOI] [PubMed] [Google Scholar]
- 29. Pan T., Uhlenbeck O.C.. In vitro selection of RNAs that undergo autolytic cleavage with lead (2+). Biochemistry. 1992; 31:3887–3895. [DOI] [PubMed] [Google Scholar]
- 30. Winterbourn C.C. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 1995; 82:969–974. [DOI] [PubMed] [Google Scholar]
- 31. Fischbacher A., von Sonntag C., Schmidt T.C.. Hydroxyl radical yields in the Fenton process under various pH, ligand concentrations and hydrogen peroxide/Fe(II) ratios. Chemosphere. 2017; 182:738–744. [DOI] [PubMed] [Google Scholar]
- 32. Dixon W.J., Hayes J.J., Levin J.R., Weidner M.F., Dombroski B.A., Tullius T.D.. Hydroxyl radical footprinting. Methods Enzymol. 1991; 208:380–413. [DOI] [PubMed] [Google Scholar]
- 33. Tullius T.D. Suslick K. Footprinting of nucleic acids by iron-based reagents. Comprehensive Supramolecular Chemistry. 1996; 5:Tarrytown, NY: Elsevier; 317–343. [Google Scholar]
- 34. Celander D.W., Cech T.R.. Visualizing the higher order folding of a catalytic RNA molecule. Science. 1991; 251:401–407. [DOI] [PubMed] [Google Scholar]
- 35. Li Z., Wu J., Deleo C.J.. RNA damage and surveillance under oxidative stress. IUBMB Life. 2006; 58:581–588. [DOI] [PubMed] [Google Scholar]
- 36. Shcherbik N., Pestov D.G.. The impact of oxidative stress on ribosomes: from injury to regulation. Cells. 2019; 8:1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bray M.S., Lenz T.K., Haynes J.W., Bowman J.C., Petrov A.S., Reddi A.R., Hud N.V., Williams L.D., Glass J.B.. Multiple prebiotic metals mediate translation. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:12164–12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Breaker R.R.. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′-Hydroxyl group. J. Am. Chem. Soc. 1999; 121:5364–5372. [Google Scholar]
- 39. Kuusela S., Lönnberg H.. Metal ions that promote the hydrolysis of nucleoside phosphoesters do not enhance intramolecular phosphate migration. J. Phys. Org. 1993; 6:347–356. [Google Scholar]
- 40. Hsiao C., Lenz T.K., Peters J.K., Fang P.-Y., Schneider D.M., Anderson E.J., Preeprem T., Bowman J.C., O’Neill E.B., Lie L.. Molecular paleontology: a biochemical model of the ancestral ribosome. Nucleic Acids Res. 2013; 41:3373–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riemer J., Hoepken H.H., Czerwinska H., Robinson S.R., Dringen R.. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004; 331:370–375. [DOI] [PubMed] [Google Scholar]
- 42. Maguire B.A., Wondrack L.M., Contillo L.G., Xu Z.. A novel chromatography system to isolate active ribosomes from pathogenic bacteria. RNA. 2008; 14:188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T.. Cell-free translation reconstituted with purified components. Nature Biotechnol. 2001; 19:751–755. [DOI] [PubMed] [Google Scholar]
- 44. Rappsilber J., Mann M., Ishihama Y.. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007; 2:1896–1906. [DOI] [PubMed] [Google Scholar]
- 45. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 46. Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M.. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome. Res. 2011; 10:1794–1805. [DOI] [PubMed] [Google Scholar]
- 47. Kurylo C.M., Alexander N., Dass R.A., Parks M.M., Altman R.A., Vincent C.T., Mason C.E., Blanchard S.C.. Genome sequence and analysis of Escherichia coli MRE600, a colicinogenic, nonmotile strain that lacks RNase I and the type I methyltransferase, EcoKI. Genome Biol. Evol. 2016; 8:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J.. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016; 13:731–740. [DOI] [PubMed] [Google Scholar]
- 49. Tullius T.D., Dombroski B.A., Churchill M.E., Kam L.. Hydroxyl radical footprinting: a high-resolution method for mapping protein-DNA contacts. Methods Enzymol. 1987; 155:537–558. [DOI] [PubMed] [Google Scholar]
- 50. Pogozelski W.K., Tullius T.D.. Oxidative strand scission of nucleic acids: routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998; 98:1089–1108. [DOI] [PubMed] [Google Scholar]
- 51. Handing K.B., Niedzialkowska E., Shabalin I.G., Kuhn M.L., Zheng H., Minor W.. Characterizing metal-binding sites in proteins with X-ray crystallography. Nat. Protoc. 2018; 13:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rivera M.C., Maguire B., Lake J.A.. Isolation of ribosomes and polysomes. Cold Spring Harb. Protoc. 2015; 2015:293–299. [DOI] [PubMed] [Google Scholar]
- 53. Kuusela S., Azhayev A., Guzaev A., Lönnberg H.. The effect of the 3′-terminal monophosphate group on the metal-ion-promoted hydrolysis of the phosphodiester bonds of short oligonucleotides. J. Chem. Soc. Perk. Trans 2. 1995; 6:1197–1202. [Google Scholar]
- 54. Kuusela S., Lönnberg H.. Zn2+-promoted hydrolysis of 3′,5′-dinucleoside monophosphates and polyribonucleotides. The effect of nearest neighbours on the cleavage of phosphodiester bonds. Nucleos. Nucleot. Nucl. 1996; 15:1669–1678. [Google Scholar]
- 55. Kuusela S., Lönnberg H.. Metal-ion-promoted hydrolysis of polyuridylic acid. J. Chem. Soc. Perk. Trans. 2. 1994; 11:2301–2306. [Google Scholar]
- 56. Ikenaga H., Inoue Y.. Metal (II) ion catalyzed transphosphorylation of four homodinucleotides and five pairs of dinucleotide sequence isomers. Biochemistry. 1974; 13:577–582. [DOI] [PubMed] [Google Scholar]
- 57. Breslow R., Huang D.-L.. Effects of metal ions, including Mg2+ and lanthanides, on the cleavage of ribonucleotides and RNA model compounds. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:4080–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Honda K., Smith M.A., Zhu X., Baus D., Merrick W.C., Tartakoff A.M., Hattier T., Harris P.L., Siedlak S.L., Fujioka H. et al.. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol. Chem. 2005; 280:20978–20986. [DOI] [PubMed] [Google Scholar]
- 59. Zinskie J.A., Ghosh A., Trainor B.M., Shedlovskiy D., Pestov D.G., Shcherbik N.. Iron-dependent cleavage of ribosomal RNA during oxidative stress in the yeast Saccharomyces cerevisiae. J Biol. Chem. 2018; 293:14237–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Athavale S.S., Petrov A.S., Hsiao C., Watkins D., Prickett C.D., Gossett J.J., Lie L., Bowman J.C., O’Neill E.B., Bernier C.R. et al.. RNA folding and catalysis mediated by iron (II). PLoS One. 2012; 7:e38024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moon W.J., Liu J.. Replacing Mg2+ by Fe2+ for RNA-cleaving DNAzymes. ChemBioChem. 2020; 21:401–407. [DOI] [PubMed] [Google Scholar]
- 62. Okafor C.D., Lanier K.A., Petrov A.S., Athavale S.S., Bowman J.C., Hud N.V., Williams L.D.. Iron mediates catalysis of nucleic acid processing enzymes: support for Fe(II) as a cofactor before the great oxidation event. Nucleic Acids Res. 2017; 45:3634–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hsiao C., Chou I.-C., Okafor C.D., Bowman J.C., O’Neill E.B., Athavale S.S., Petrov A.S., Hud N.V., Wartell R.M., Harvey S.C. et al.. RNA with iron (II) as a cofactor catalyses electron transfer. Nat. Chem. 2013; 5:525–528. [DOI] [PubMed] [Google Scholar]
- 64. Ward W.L., Plakos K., DeRose V.J.. Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem. Rev. 2014; 114:4318–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beauchene N.A., Mettert E.L., Moore L.J., Keleş S., Willey E.R., Kiley P.J.. O2 influences iron homeostasis. Proc. Natl. Acad. Sci. 2017; 114:12261–12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Okafor C.D., Bowman J.C., Hud N.V., Glass J.B., Williams L.D.. Folding and catalysis near life's origin: support for Fe2+ as a dominant divalent ion. Prebiotic Chemistry and Chemical Evolution of Nucleic Acids. 2018; Springer; 227–243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw files are available at http://www.peptideatlas.org/PASS/PASS01418 (username: PASS01418 and password: ZW2939nnw).