

Abstract
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or 2019 novel coronavirus (2019-nCoV), took tens of thousands of lives and caused tremendous economic losses. The main protease (Mpro) of SARS-CoV-2 is a potential target for treatment of COVID-19 due to its critical role in maturation of viral proteins and subsequent viral replication. Conceptually and technically, targeting therapy against Mpro is similar to target therapy to treat cancer. Previous studies show that GC376, a broad-spectrum dipeptidyl Mpro inhibitor, efficiently blocks the proliferation of many animal and human coronaviruses including SARS-CoV, Middle East respiratory syndrome coronavirus (MERS-CoV), porcine epidemic diarrhea virus (PEDV), and feline infectious peritonitis virus (FIPV). Due to the conservation of structure and catalytic mechanism of coronavirus main protease, repurposition of GC376 against SARS-CoV-2 may be an effective way for the treatment of COVID-19 in humans. To validate this conjecture, the binding affinity and IC50 value of Mpro with GC376 was determined by isothermal titration calorimetry (ITC) and fluorescence resonance energy transfer (FRET) assay, respectively. The results showed that GC376 binds to SARS-CoV-2 Mpro tightly (KD = 1.6 μM) and efficiently inhibit its proteolytic activity (IC50 = 0.89 μM). We also elucidate the high-resolution structure of dimeric SARS-CoV-2 Mpro in complex with GC376. The cocrystal structure showed that GC376 and the catalytic Cys145 of Mpro covalently linked through forming a hemithioacetal group and releasing a sulfonic acid group. Because GC376 is already known as a broad-spectrum antiviral medication and successfully used in animal, it will be a suitable candidate for anti-COVID-19 treatment.
Keywords: COVID-19, SARS-CoV-2, coronavirus (CoV), drug development, drug action, drug screening, protease inhibitor, structural biology
Introduction
Coronaviruses (CoVs) are severe pathogens that cause a wide range of diseases in human and animals. In December 2019, a novel viral pneumonia started in Wuhan, Hubei province of China. The main clinical symptoms included fever, breathing difficulties, and X-ray findings of infiltrative lung injury. The cause of this pneumonia was identified as a new coronavirus, officially called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The World Health Organization (WHO) announced the outbreak a Public Health Emergency of International Concern on 30 January and designated the coronavirus disease 2019 (COVID-19). On 11 March, WHO warned that the COVID-19 is a pandemic. Until 2 July 2020, COVID-19 is still an ongoing pandemic, which spread across the world with more than ten million confirmed cases and 500,000 people died in early July 2020. It is therefore an unmet medical need to develop effective treatments for the COVID-19 and several therapeutic targets have been identified including key protease such as main protease (Mpro) of the SARS-CoV-2 [1,2] and the host TMPRSS2 serine protease [3]. To this end, it is worthwhile to mention that the concept and approach to screen drugs to target proteases to treat COVID-19 is similar to those for development of target therapy for anti-cancer drugs. Indeed, TMPRSS2 serine protease that is critical for SARS-CoV-2 virus to enter host cells has been known to be activated in prostate cancer and is a therapeutic target to treat prostate cancer [4,5]. In addition, certain drugs from traditional herbal medicine that were used to treat cancer patients are commonly used to treat cornovirus-induced diseases [6].
Based on the results of the full-genome sequencing and phylogenic analysis, the RNA genome of SARS-CoV-2 shares about 82% identity with that of the severe acute respiratory syndrome (SARS) coronavirus [7], a betacoronavirus that belongs to lineage B of the subfamily Orthocoronavirinae within the family Coronaviridae. SARS-CoV-2 are enveloped and positive-sense single-stranded RNA viruses with the large viral RNA genome ranging from 27 to 32 kilobases in length [7]. The RNA genome is associated with the nucleocapsid (N) protein to form the viral capsid (helical for the genus coronavirus; tubular for the genus torovirus) for virion cryo-electron microscopy analysis [8]. The RNA genome is composed of gene 1 and genes 2-7. Gene 1, which is responsible for replication, has two open reading frames, ORF1a and ORF1b, and can be translated by cellular ribosomes to two polypeptides, termed pp1a and pp1b. The pp1a is then cleaved by viral proteinases to generate 11 nonstructural proteins. Some of the ribosomes translating ORF1a pause on a complex RNA structure (pseudoknot) in the overlap between ORF1a and ORF1b and then shift to ORF1b to generate a larger pp1b polyprotein, which is cleaved to 16 nonstructural proteins. Genes 2-7 encode structural proteins expressed by subgenomic mRNA [9-11]. The size of SARS-CoV-2 virion is 50-200 nm in diameter [12]. SARS-CoV-2 contains four structural proteins, the spike (S) protein (homotrimer), envelope (E) protein, membrane (M) protein, and N protein similar to other coronaviruses. The nucleocapsid protein holds the RNA genome, and the S, E, and M proteins make up the viral envelope [13]. Structural analysis showed that region of the receptor-binding domain of SARS-CoV-2 is similar to that of the SARS coronavirus [14]. Using reverse genetics methods, the spike protein of SARS-CoV-2 was shown to bind to the receptor angiotensin converting enzyme 2 (ACE2) with higher affinity than the original SARS virus strain [15].
SARS-CoV-2 maturation occurs via a highly complex cascade of proteolytic processing for viral gene expression and replication. The precursor polyprotein is proteolyzed by the Mpro (also known as 3CL protease or 3CLpro). Mpro belongs to the chymotrypsin-like protease family with recognition sequence at most sites of Leu-Gln↓ (Ser/Ala/Gly) (↓marks the cleavage site). Mpro is regarded as one of the best characterized drug targets among coronaviruses [16,17] as inhibition of Mpro blocks viral replication. The side effects of inhibitors against Mpro may be limited because no human proteases harbor similar cleavage site. Previously, Mpro inhibitor GC376, which exhibits broad-spectrum activity against human and animal coronaviruses, was evaluated for its efficacy in laboratory cats with feline infectious peritonitis (FIP). GC376 treatment led to full recovery of cats when treatment began at a stage of the disease that would be otherwise be fatal if left untreated [18]. We hypothesized that GC376 may be an effective therapeutic strategy against SARS-CoV-2.
To test our hypothesis, we first use differential scanning fluorimetry (DSF), isothermal titration calorimetry (ITC) and FRET-based enzyme kinetic assay to access the efficacy of GC376 against SARS-CoV-2 Mpro. Then, we solved the crystal structure of SARS-CoV-2 Mpro in complex with GC376, a dipeptidyl bisulfite adducts, and compared the substrate binding sites between Mpro apo-form and Mpro in complex with GC376. The structural characterization in this study provides a mechanistic insight into the inhibition mechanism of GC376 and support it as a promising drug candidate for the treatment of COVID-19. This work also provides a basis for structure-based drug design of broad-spectrum anti-coronaviral drugs.
Materials and methods
Cloning, expression and purification of SARS-CoV-2 main protease
The full-length gene encoding SARS-CoV-2 main protease (Mpro, Replicase polyprotein 1ab residues 3264-3569, UniProt accession: P0DTD1) with Escherichia coli codon usage was synthesized and subcloned into pSol SUMO vector using Expresso® Solubility and Expression Screening System (Lucigen). A pET16b plasmid encoding the fluorescent protein substrate of Mpro (CFP-TSAVLQSGFRKM-YFP) was synthesized and constructed for FRET based high-throughput screening assay. Each expression plasmid was transformed into E. coli BL21 (DE3) and then grown in Luria Broth medium at 37°C until OD600 reached between 0.6 and 0.8. Overexpression of Mpro or its fluorescent protein substrate was induced by the addition of 20% L-rhamnose or 0.5 mM IPTG and incubated for 18 hours at 20°C. The cell pellets were resuspended in sonication buffer [50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM tris (2-carboxyethyl) phosphine (TCEP), 1 mM phenylmethylsulfonyl fluoride (PMSF)] and lysed by sonication on ice. Following centrifugation at 28,000 g, 4°C for 30 min, the supernatant was loaded onto a HisTrap FF column (GE Healthcare), washed by sonication buffer containing 10 mM imidazole, and eluted with a 20-200 mM imidazole gradient in sonication buffer. TEV protease was used to remove the N-terminal SUMO fusion tag of Mpro. The Mpro and its substrate protein were further purified by size-exclusion chromatography.
Differential scanning fluorimetry (DSF)
DSF experiment was carried out on a CFX96 RT-PCR instrument (Bio-Rad) in a buffer comprising 25 mM Tris pH 8.0, 150 mM NaCl, 5X SYPRO Orange dye (Sigma-Aldrich), and 7.5 μM SARS-CoV-2 Mpro in the presence of GC376 or other potential protease inhibitors (TargetMol, Cat. No. L1100) at concentration of 120 μM. Fluorescence was monitored when temperature was gradually raised from 25 to 85°C in 0.3°C increments at 12-second intervals. Melt curve data were plotted using the Boltzmann model to obtain the temperature midpoint of unfolding of the protein using Prism 8.0 software (GraphPad).
FRET-based enzyme activity assay
Purified fluorescent protein substrate containing the cleavage site (indicated by the arrow,↓) of SARS-CoV-2 Mpro (CFP-TSAVLQ↓SGFRKM-YFP) was used for the fluorescence resonance energy transfer (FRET)-based enzyme activity assay. SARS-CoV-2 Mpro (0.5 μM) in assay buffer (20 mM Tris-HCl pH 7.8, 20 mM NaCl) was pre-incubated with different concentration of GC376 (0.1-10 μM) for 30 min at room temperature. The reaction was initiated by addition of 40 μM fluorescent protein substrate. The fluorescence signal of the CFP-TSAVLQ cleavage product was monitored at an emission wavelength of 474 nm with excitation at 434 nm using Synergy™ H1 hybrid multi-mode microplate reader (BioTek Instruments, Inc.). The first 15 min of the reaction was used to calculate initial velocity (V0) by linear regression. The IC50 was calculated by plotting the initial velocity against various concentrations of GC376 by use of a dose-response curve in Prism 8 software.
Isothermal titration calorimetry (ITC)
The binding of GC376 to SARS-CoV-2 Mpro was conducted on an ITC-200 instrument (MicroCal, Northampton, MA, USA) at 25°C. SARS-CoV-2 Mpro and GC376 were dissolved in assay buffer (20 mM Tris pH 8.0, 20 mM NaCl, 2% DMSO). Two-microliter aliquots of GC376 at a concentration of 1 mM in the syringe were injected into cells containing 60 μM SARS-CoV-2 Mpro at 3-min intervals. Data were fit to a one-site binding model using the commercial Origin 7.0 program to obtain ΔH, ΔS, and KD values.
Crystallization, data collection and structural determination of SARS-CoV-2 main protease with or without GC376
The crystallization of SARS-CoV-2 Mpro in ligand-free state and in complex with GC376 was performed by the sitting-drop vapor-diffusion method. For crystallization of SARS-CoV-2 Mpro in ligand-free state, the drops were set up by mixing 1 μl of SARS-CoV-2 Mpro at a concentration of 25 mg/ml with 1 μl of reservoir solution [0.1 M bicine pH 8.5, 20% (w/v) polyethylene glycol (PEG) 10000] and equilibrated against 300 μl of reservoir solution at 20°C. For crystallization of SARS-CoV-2 Mpro in complex with GC376, protein sample was incubated with 2 molar excess of GC376 at 4°C overnight, and then crystallized with mother liquid containing 0.1 M Bis-Tris propane pH 6.5, 0.02 M sodium/potassium phosphate, 20% (w/v) PEG 3350. All the crystals were soaked in reservoir solution containing an additional 25% glycerol for 15 s and were flash cooled in liquid nitrogen. The diffraction data for the crystals of SARS-CoV-2 Mpro in ligand-free state and in complex with GC376 were collected at the BL15A1 beamline at the National Synchrotron Radiation Research Center in Hsinchu, Taiwan, using a RAYONIX MX300HE CCD detector. The data were integrated, scaled, and merged using the HKL2000 software package [19]. The structure of the apo form of SARS-CoV-2 Mpro was solved by molecular replacement with the previously published crystal structure of SARS-CoV-2 Mpro (PDB entry 6LU7) [1] as search model using Phaser program. The structure was then refined by iterative manual model building in Coot [20], with refinement using the Phenix refinement module [21]. The structure of SARS-CoV-2 Mpro in complex with GC376 was then solved by molecular replacement using our apo form of SARS-CoV-2 Mpro. The data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
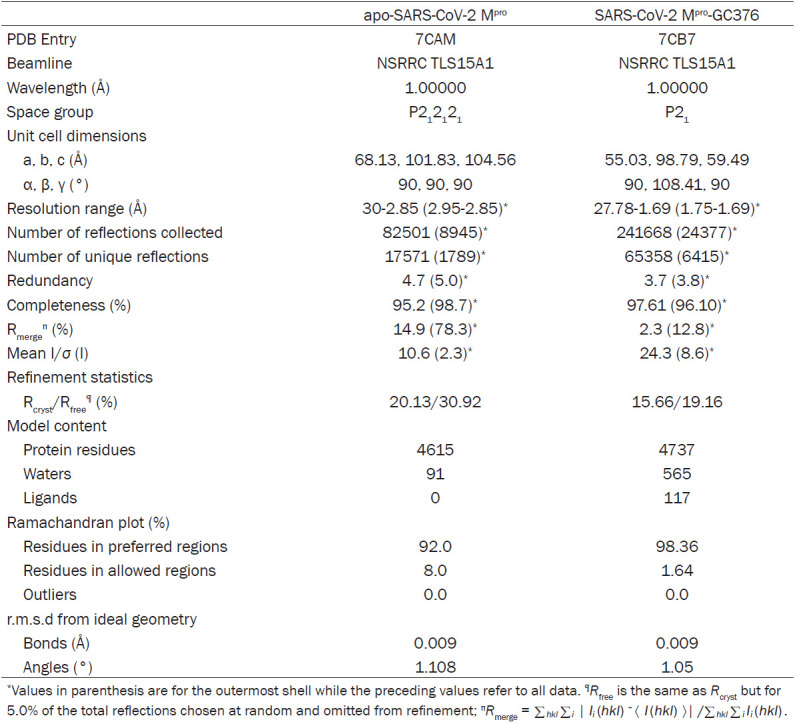
|
Results
GC376 is a highly potent drug against SARS-CoV-2 Mpro
To explore the use of potential protease inhibitors against SARS-CoV-2 Mpro, we evaluated the binding of drugs from a protease inhibitor library (TargetMol) and GC376 to SARS-CoV-2 Mpro by differential scanning fluorimetry (DSF). Most potential protease inhibitors, including lopinavir and ritonavir, caused little or no effect on the thermal stability of SARS-CoV-2 Mpro whereas GC376 caused a dramatic positive shift of 10.3°C beyond the control (Figure 1A). To validate the above results and test the dose-dependent effect of GC376, we repeated the experiment with different concentrations of GC376 (7.5-120 μM). Consistently, the melting temperature of SARS-CoV-2 Mpro increased with the increasing concentrations of GC376 (Figure 1B). The binding affinity of GC376 to SARS-CoV-2 Mpro was further determined by isothermal titration calorimetry (ITC). The titration curve suggests that one molecule of GC376 bound to one molecule of SARS-CoV-2 Mpro with a binding constant of about 1.6 μM (Figure 1C).
Figure 1.
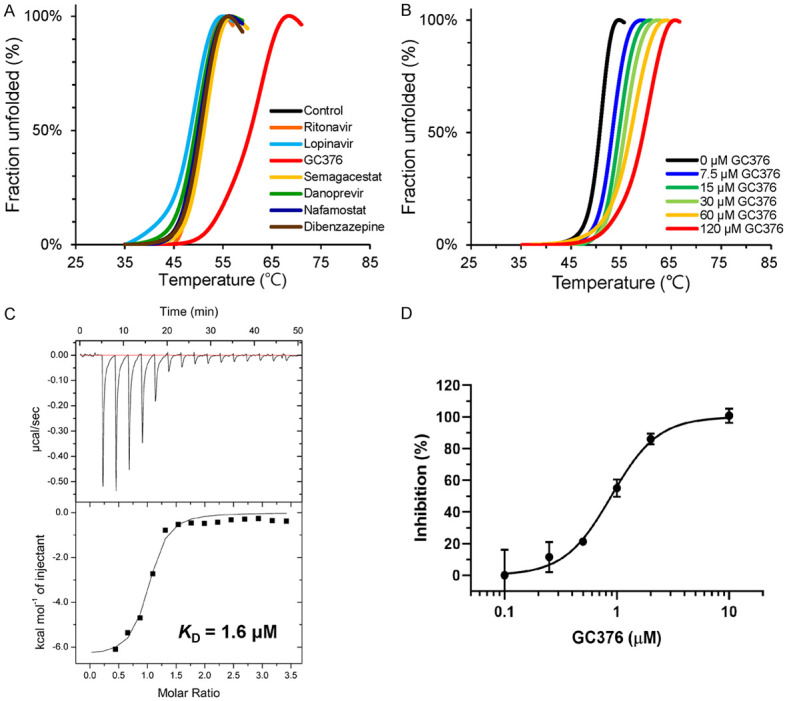
GC376 efficiently inhibit SARS-CoV-2 Mpro. A. Differential scanning fluorimetry of SARS-CoV-2 Mpro with various drugs. B. Differential scanning fluorimetry of SARS-CoV-2 Mpro with different concentrations of GC376. C. Binding affinity of GC376 to SARS-CoV2-Mpro determined by ITC. D. Dose-response curves of GC376 on inhibition of SARS-CoV-2 Mpro. Data are shown as mean ± S.D. from three independent replicates.
To assess the enzyme activity of SARS-CoV-2 Mpro and measure the inhibition efficiency of GC376, we established a fluorescence resonance energy transfer (FRET) assay using a fluorescent protein substrate as previously reported [22]. GC376 showed strong inhibition against SARS-CoV-2 Mpro with a half-maximal inhibitory concentration (IC50) of 0.89 μM (Figure 1D), which was similar to the IC50 values against Mpro observed in different coronaviruses, such as porcine epidemic diarrhea virus (PEDV; IC50 = 1.11 μM) [23], feline infectious peritonitis virus (FIPV; IC50 = 0.72 μM) [18], SARS-CoV Mpro (IC50 = 4.35 μM) [18], middle East respiratory syndrome coronavirus (MERS-CoV; IC50 = 1.56 μM) [18], and transmissible gastroenteritis coronavirus (TGEV; IC50 = 0.82 μM) [24].
Overall structure of SARS-CoV-2 Mpro
To structurally characterize SARS-CoV-2 Mpro, the protein was expressed recombinantly and purified to homogeneity (Figure S1A, S1B) to obtain protein as crystal for x-ray diffraction analysis. The crystal structure of SARS-CoV-2 Mpro in its ligand-free state was solved at a resolution of 2.85 Å (Table 1). As expected, the structure of SARS-CoV-2 Mpro is highly similar to that of SARS-CoV Mpro given that the Mpro of two viruses share 96.08% sequence identity. The structure shows that two Mpro molecules, named Molecule A and B per asymmetric unit, form a typical catalytically active and symmetrical homodimer (Figure 2A), which is consistent with the result of size-exclusion chromatography (Figure S1B). The three-dimensional structure of Molecule A and B are highly similar, with a root-mean-square deviation (RMSD) of 1.2 Å for all equivalent Cα atoms. Two molecules oriented perpendicular to one another (Figure 2A). Similar dimeric assembly was also observed in other Mpro from different coronaviruses [23,25,26].
Figure 2.
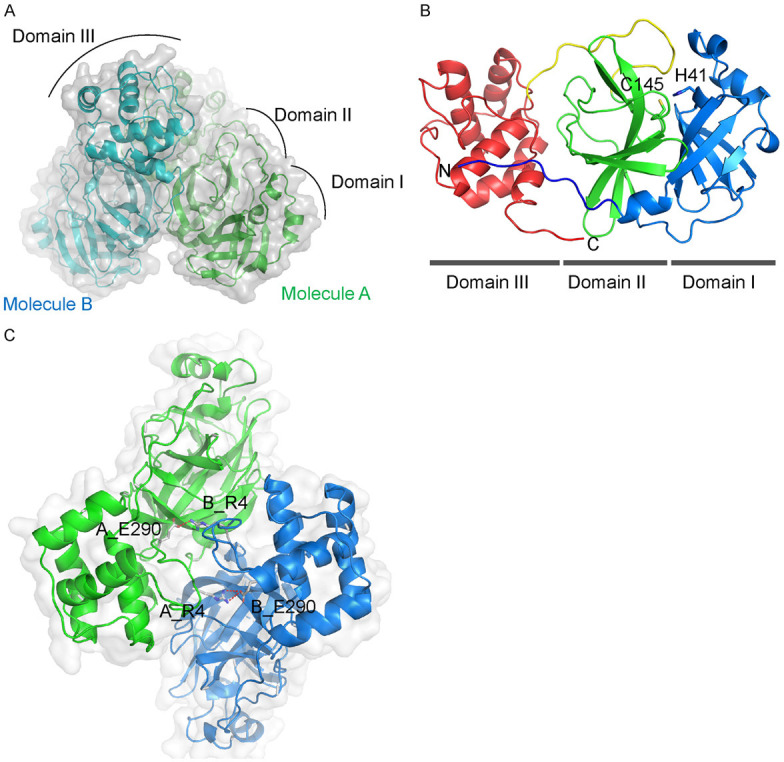
Crystal structure of SARS-CoV-2 Mpro in ligand-free form. A. Apo-SARS-CoV-2 Mpro forms a dimer (PDB code: 7CAM). B. Ribbon representation of one apo-SARS-CoV-2 protomer. Domain I (residues 10-100), II (residues 101-182), the connected loop (residues 183-199) and domain III (residues 200-304) are colored by blue, green, yellow and red, respectively. The residues involved in catalysis (Cys145, His41) are shown in sticks. C. The ionic interaction between Arg4 and Glu290 in the dimerization interface of SARS-CoV-2 Mpro. The side chains of the residues are shown in sticks and the ionic bonds in red.
Each SARS-CoV-2 Mpro molecule is composed of three domains (Figure 2B). Domain I (residues 10 to 100) and II (residues 101 to 182) have a chymotrypsin-like, two-β-barrel fold commonly found in the structure of Mpro of other human and animal coronaviruses, such as PEDV, MERS-CoV, and SARS-CoV [23,25,26]. Domain III (residues 200 to 304) of SARS-CoV-2 Mpro includes five α-helices that form a globular structure and is unique to CoV Mpro. Domains II and III are linked by a long loop (residues 183 to 199) (Figure 2B). Previous study of SARS-CoV Mpro suggested that dimerization is necessary for its catalytic activity [26]. The dimerization interface of SARS-CoV-2 Mpro is mainly maintained by an ionic interaction between Arg4 of one protomer and Glu290 of the other (Figure 2C) and allows the whole protein to form a buckle-like structure, which may help with stabilizing the protein. The substrate-binding pocket is located in the deep cleft between domains I and II and contains the catalytic dyad formed by His41 and Cys145 in the center (Figure 2B).
Crystal structure of SARS-CoV-2 Mpro in complex with inhibitor GC376
To decipher the inhibitory mechanism of GC376, we co-crystallized and solved the SARS-CoV-2 Mpro-GC376 complex at 1.69 Å resolution (Figure S1; Table 1). In the structure of SARS-CoV-2 Mpro in complex with GC376, the inhibitor is stabilized by a hydrogen bond network, including the side chain of H41, His163, Glu166, and Q189, the main chain of Phe140, Gly143, Cys145, and H164, and numerous hydrophobic interactions (Figures 3A, S2B). As previously reported, the backbone amide of Gly143 and Cys145 form an oxyanion hole to promote substrate binding [2]. Interestingly, GC376 is covalently linked to Cys145 as a hemiothioacetal at the catalytic center and can be fitted to the Fo-Fc electron-density map in two ways after bisulphite group leaving (Figure S2A). This phenomenon may be caused by the original bisulphite group leaving and the aldehyde group forming without the chiral center. It has been speculated that the facile transformation of aldehyde bisulfite (GC376) to aldehyde (GC373) occurred when the drug is administered to animal via intravenous and oral routes (Figure 3B) [24]. Cys145 can covalently link to the aldehyde group in two different directions and be stabilized by surrounding residues (Figure 3A). There are two ways to stabilize the aldehyde: 1) via the oxyanion hole formed by backbone amide of Cys145, Gly143 and partially Ser144 (Figures 3A, S2A) and 2) via the imidazole ring of His41 to stabilize the hemiothioacetal group (Figures 3A, S2A). Formation of both R and S enantiomers at the covalent binding site for GC376 has also been reported in the structural study of MERS-CoV Mpro in complex with GC376 [25]. In addition, structural alignment of apo-SARS-CoV-2 Mpro and SARS-CoV-2 Mpro with GC376 showed that the major structural difference is located in the loop region near the substrate-binding site indicated by the gold arrow in Figure 4A. The change in the loop region seemed to affect the dimerization interface. Another dominant structural shift was observed in domain III (Figure 4A). Moreover, the side chain of Arg4 of Molecule B from apo-SARS-CoV-2 Mpro was not easily assigned to the electron-density map compared with the SARS-CoV-2 Mpro-GC376 complex (Figure 4B).
Figure 3.
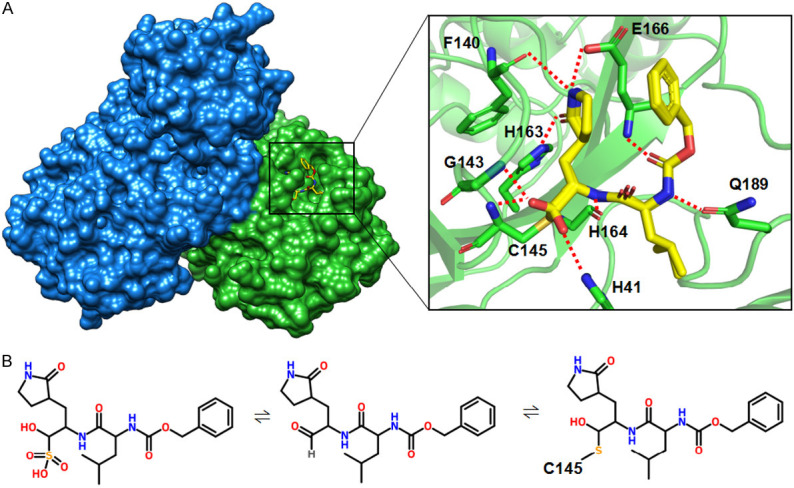
Crystal structure of SARS-CoV-2 Mpro in complex with GC376. A. Left, surface representation of dimeric SARS-CoV-2 Mpro in complex with GC376. Right, an enlarged view of the substrate-binding pocket. The residues involved in hydrogen boding with GC376 are shown in sticks. The H-bonds are shown in red. B. A proposed mechanism of the formation of the SARS-CoV-2 Mpro-GC376 covalent complex.
Figure 4.
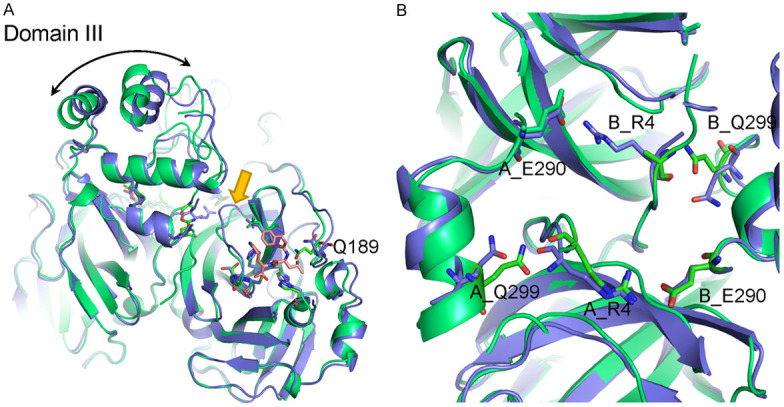
Structural comparison between apo-SARS-CoV-2 Mpro and SARS-CoV-2 Mpro with GC376. A. Structure alignment of SARS-CoV-2-Mpro with GC376 (green) and apo-protein (blue). B. Structural changes in the dimer interface of SARS-CoV-2 Mpro before and after binding with GC376.
Structural comparison of coronavirus Mpro-GC376 complexes across different species
To determine the structural differences, we superimposed SARS-CoV-2 Mpro-GC376 co-crystal structure in this study with published Mpro-GC376 complex from different coronaviruses. As shown in Figure 5A, SARS-CoV-2 Mpro adopts a similar fold common to the Mpro of MERS-CoV, PEDV and TGEV [23-25]. Domain I and II from the four CoV Mpro showed they overlapped well although a slight structural shift was observed in Domain III (Figure 5A). In the substrate-binding site, GC376 appears to assume a highly similar conformation in each coronavirus with one exception in which the benzoyl ring of GC376 from PEDV Mpro has different orientation (Figure 5B). The catalytic dyads are all conserved based on multiple sequence alignment and showed a general overlap (Figure 5C). The above observations are consistent with the comparable IC50 values of GC376 for the different CoV Mpro and suggested that the structure of Mpro in the coronaviruses examined is highly conserved and that GC376 may be a universal inhibitor to treat diseases caused by different coronaviruses.
Figure 5.
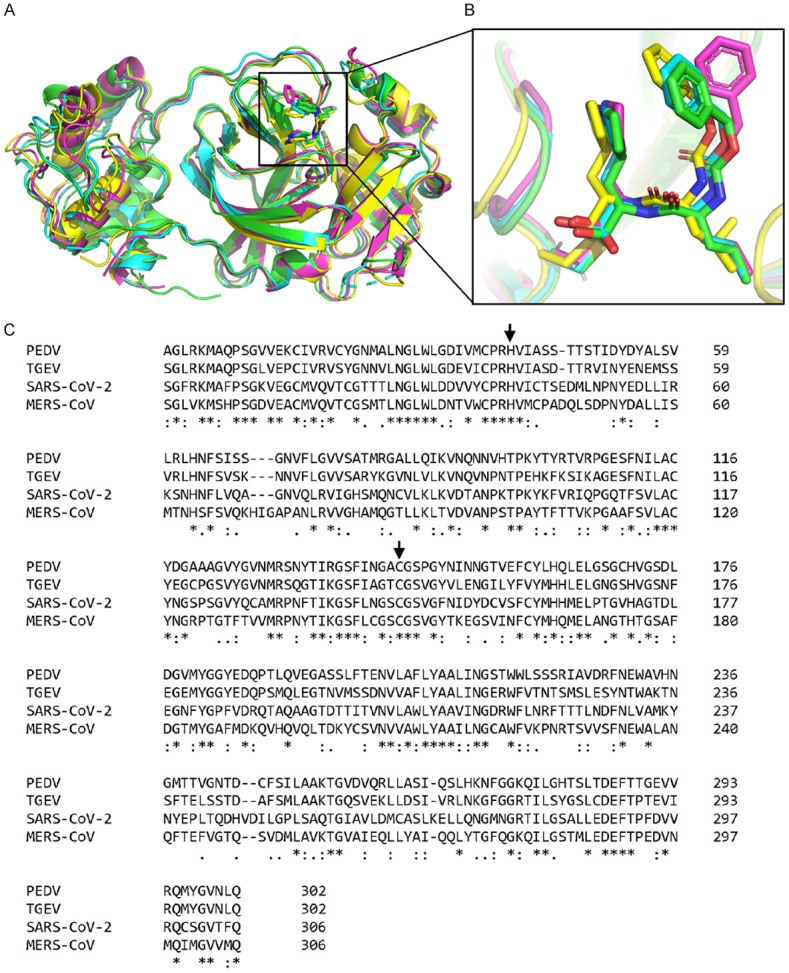
Structural comparison of CoV Mpros with GC376 across different species. A. Structural alignment of CoV Mpros with GC376 in different coronaviruses, including SARS-CoV-2 (green, PDB: 7CB7), MERS-CoV (cyan, PDB: 5WKJ), PEDV (magenta, PDB: 6L70) and TGEV (yellow, PDB: 4F49). B. An enlarged view of the substrate-binding pocket of different CoV Mpros with GC376. All GC376 are shown in sticks. C. Multiple sequence alignment of the Mpros of SARS-CoV-2, MERS-CoV, PEDV and TGEV. The catalytic dyad is indicated by an arrow.
Discussion
Since the SARS outbreak in 2002, a number of drugs have been developed to inhibit Mpro, a key anti-viral target responsible for maturation of functional viral proteins. In this study, we explore the potential of GC376, a broad-spectrum coronavirus inhibitor that has been used to treat FIP with high recovery rate, in inhibiting SARS-CoV-2 Mpro. The co-crystal structure of SARS-CoV-2 Mpro in complex with GC376 revealed that GC376 fits well in the substrate-binding pocket and is stabilized by numerous hydrophilic and hydrophobic interactions. In addition, GC376 can form a covalent bond with active site residue Cys145, revealing its ability to directly block the catalytic dyad and hence the proteolytic activity of Mpro. Thus, the structural information revealed in this study greatly supported GC376 as a promising drug candidate for the treatment of COVID-19.
Interestingly, a previous report suggested that Mpro of picornavirus-like supercluster, including picornaviruses, caliciviruses, and coronaviruses, all share several common characteristics, including a typical chymotrypsin-like fold and a cysteine residue as an active site nucleophile in the catalytic triad (or dyad), and that GC376 is highly effective against those viruses with IC50 values in the high nanomolar or low micromolar range in enzyme- and/or cell-based assays [24]. The structural comparison of Mpro-GC376 complexes across different species revealed in this study is also consistent with those observations and indicated that the inhibition of those viruses by GC376 occurs through a similar mechanism.
During the preparation of the current manuscript, a paper published in Cell Research by Wang’s group also indicated that GC376 is the most potent drug among the protease inhibitors they screened [27]. The antiviral activity of GC376 was tested against SARS-CoV-2 in the viral cytopathic effect (CPE) assay with EC50 value of 3.37 μM [27]. Together, the results from both studies suggest GC376 is worthy of further testing in clinical trials.
Moreover, a pre-investigational new drug request regarding the usage of GC376 for COVID-19 treatment in humans has been submitted to U.S. Food and Drug Administration by Anivive (Long Beach, CA, USA) based on the potency, safety, efficacy, and pharmacological properties of GC376 in four animal models [28]. Furthermore, studies from leading research universities involved in the evaluation of GC376 have shown that GC376 is stable in human plasma, highly efficient in clinical doses, and could prevent viruses from killing infected cells [29]. The findings from the current study also suggested that GC376 is a promising drug candidate for the treatment of COVID-19.
PDB deposition
The coordinates and structure factors of SARS-CoV-2 Mpro in ligand-free state and in complex with GC376 have been deposited in PDB with accession codes 7CAM and 7CB7, respectively.
Acknowledgements
We would like to thank the National Synchrotron Radiation Research Center (NSRRC, Taiwan) for assistance during the data collection. This research was supported by the Ministry of Science and Technology in Taiwan (108-2311-B-241-001) and Drug Development Center, China Medical University from the Ministry of Education in Taiwan.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 5.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2: ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006;66:3396–3400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 6.Huang ST, Lai HC, Lin YC, Huang WT, Hung HH, Ou SC, Lin HJ, Hung MC. Principles and treatment strategies for the use of Chinese herbal medicine in patients at different stages of coronavirus infection. Am J Cancer Res. 2020;10:2010–2031. [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sola I, Almazán F, Zúñiga S, Enjuanes L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol. 2015;2:265–288. doi: 10.1146/annurev-virology-100114-055218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 17.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y, Liu H, Galasiti Kankanamalage AC, Weerasekara S, Hua DH, Groutas WC, Chang KO, Pedersen NC. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016;12:e1005531. doi: 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chuck CP, Chong LT, Chen C, Chow HF, Wan DC, Wong KB. Profiling of substrate specificity of SARS-CoV 3CL. PLoS One. 2010;5:e13197. doi: 10.1371/journal.pone.0013197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye G, Wang X, Tong X, Shi Y, Fu ZF, Peng G. Structural basis for inhibiting porcine epidemic diarrhea virus replication with the 3C-like protease inhibitor GC376. Viruses. 2020;12:240. doi: 10.3390/v12020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y, Lovell S, Tiew KC, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, Chang KO. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86:11754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galasiti Kankanamalage AC, Kim Y, Damalanka VC, Rathnayake AD, Fehr AR, Mehzabeen N, Battaile KP, Lovell S, Lushington GH, Perlman S, Chang KO, Groutas WC. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur J Med Chem. 2018;150:334–346. doi: 10.1016/j.ejmech.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020:1–15. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anivive Lifesciences. Anivive repurposes veterinary drug GC376 for COVID-19 and submits Pre-IND to FDA [Google Scholar]
- 29.Anivive Lifesciences. Thanks to cats, one promising coronavirus treatment is already in development [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.