

Abstract
Poly (ADP-ribose) polymerase (PARP) has recently emerged as a central mediator in cancer resistance against numerous anticancer agents to include chemotherapeutic agents such as microtubule targeting agents and DNA damaging agents. Here, we describe AMXI-5001, a novel, highly potent dual PARP1/2 and microtubule polymerization inhibitor with favorable metabolic stability, oral bioavailability, and pharmacokinetic properties. The potency and selectivity of AMXI-5001 were determined by biochemical assays. Anticancer activity either as a single-agent or in combination with other antitumor agents was evaluated in vitro. In vivo antitumor activity as a single-agent was assessed in a triple-negative breast cancer (TNBC) model. AMXI-5001 demonstrates comparable IC50 inhibition against PARP and microtubule polymerization as clinical PARP inhibitors (Olaparib, Rucaparib, Niraparib, and Talazoparib) and the potent polymerization inhibitor (Vinblastine), respectively. In vitro, AMXI-5001 exhibited selective antitumor cytotoxicity across a wide variety of human cancer cells with much lower IC50s than existing clinical PARP1/2 inhibitors. AMXI-5001 is highly active in both BRCA mutated and wild type cancers. AMXI-5001 is orally bioavailable. AMXI-5001 elicited a remarkable In vivo preclinical anti-tumor activity in a BRCA mutated TNBC model. Oral administration of AMXI-5001 induced complete regression of established tumors, including exceedingly large tumors. AMXI-5001 resulted in superior anti-tumor effects compared to either single agent (PARP or microtubule) inhibitor or combination with both agents. AMXI-5001 will enter clinical trial testing soon and represents a promising, novel first in class dual PARP1/2 and microtubule polymerization inhibitor that delivers continuous and synchronous one-two punch cancer therapy with one molecule.
Keywords: PARP inhibitor, microtubule inhibitor, BRCA, homologous recombination, cancer therapy, synthetic lethality, breast cancer, malignancy, AMXI-5001
Introduction
According to American Cancer Society Cancer Facts and Figures from 2019, cancer is the second leading cause of death in the United States [1]. Despite the introduction of many novel therapeutics including immunotherapy, biologic, and targeted therapies, most patients with metastatic cancer benefit from any given therapy for a limited time only. This limited benefit is primarily due to intolerable side effects and resistance (primary or acquired) to standard therapies used in treating advanced cancers. Causes underlying the resistance include tumor mediated factors, interference from the tumor or immune environment, and epigenetic factors. Thus, advanced cancers continue to be serious, life-threatening, and incurable. Developing more effective and tolerable therapies to treat most cancers remains an unmet medical need.
Synthetic lethality is an emerging strategy in cancer treatment that seeks to exploit the oncogenic features of tumor cells rather than overcome them. Synthetic lethality occurs when essential and overlapping molecular pathways that control cellular functions are simultaneously inhibited [2-4]. Exploiting synthetic lethality may result in greater effects than inhibiting each pathway alone.
PARP and microtubules are attractive targets for many anti-cancer drugs. Microtubules play critical roles in cell structure, function, and processes including transport, migration and mitosis. Microtubule targeting agents (MTAs) inhibit the function of cellular microtubules by promoting polymerization and depolymerization. PARP1 and PARP2 proteins are involved with repairing single-strand DNA breaks that lead to toxic double-stranded breaks if not repaired prior to replication [2,5-7]. Drugs that inhibit the PARP proteins cause multiple double-stranded breaks that lead to cell death.
A synergistic role of PARP and microtubule targeting agents is emerging in cancer therapy. Simultaneous targeting of PARP and microtubule polymerization may result in an amplified and prolonged DNA damage, an enhanced sensitization of cancers, and a broad anti-tumor efficacy with reduced risk for both cancer drug resistance and dose limiting peripheral neuropathy associated with microtubule targeting agents [8-13].
Emerging evidence suggests that microtubule targeting agents may exert a potentiating effect on PARP inhibitor-induced DNA damage [13]. Recent studies demonstrate that MTAs interfere with the trafficking of proteins critical for the double-strand DNA damage repair to include ATM, ATR, DNA-PK, Rad50, Mre11, p95/NBS1, p53, 53BP1, and p63 [13]. By impairing the trafficking of these critical double-stranded DNA damage repair proteins, MTAs may synergize with DNA damaging agents and the PARP inhibitors, which inhibit the alternative DNA repair pathway (base excision or single-strand repair), thereby resulting in prolonged DNA damage and enhanced cytotoxicity.
PARP inhibitors may enhance the anti-tumor activity of MTAs. In keeping, following microtubule inhibition by MTAs, PARP1 is auto-ribosylated and thus stimulated. Subsequently, the stimulated PARP interacts with and stabilizes important mitotic checkpoint proteins such as E3 ubiquitin ligase (CHFR), and thus leads to inhibition of cell cycle progression to M phase in replicating cancer cells [10,12]. Consequently, cancer cell avoidance of mitotic catastrophe induced by MTAs implicates an important role of PARP activity in cancer cell resistance to these drugs. There are currently at least seven PARP inhibitors at various stages of clinical development [2,14]. In addition to blocking PARP catalytic action, certain PARP inhibitors induce trapping of PARP proteins on DNA, thus generating toxic DNA-adducts [2]. These toxic DNA-adducts interfere with replication, causing cell death preferentially in cancer cells, which grow faster than non-cancerous cells.
Recent studies show that sustained DNA damage occurs with the combination of MTAs with either DNA damaging agents or radiation [15,16]. This finding provides a likely explanation for why combinations involving a MTA and a DNA damaging agent have emerged “empirically” as active combinations in the majority of cancers. Moreover, The DNA damage repair inhibitors, such as PARP inhibitors, may sensitize cancers to MTAs as significantly increased apoptosis has been shown in cancer cells resistant to MTAs with combination treatment of a PARP inhibitor and a MTA.
These combined results suggest that MTAs may synergize with PARP inhibition in both DNA-double-strand break repair proficient and defective cancers, owing to their exacerbation of the DNA damage phenotype [13]. Given this supportive evidence, PARP inhibitors have been proposed to kill cancer cells resistant to MTAs in combinatorial regimens with MTAs [10,12].
MTA-induced peripheral neuropathy is a major dose limiting toxicity for which no therapy is approved. A role for PARP inhibition in sensory neurotransmission has been established [8]. Recent reports demonstrate that PARP inhibition protects against microtubule targeting agent-induced painful neuropathy in rats [9], and thus supports that PARP inhibition may represent a novel therapeutic approach for this dose limiting toxicity associated with microtubule targeting agents.
Collectively, these findings suggest that simultaneous inhibition of PARP and microtubules may confer a broad and potent anti-tumor efficacy with reduced risk for MTA-induced dose limiting toxicity and cancer drug resistance. Accordingly, synchronous inhibition of PARP and microtubules may result in a synthetically lethal anti-cancer therapeutic strategy by ensuring a synchronous ‘one-two punch’ cancer therapy.
Here, we report the characteristics of a novel dual PARP1/2 and Microtubule Polymerization inhibitor, AMXI-5001. AMXI-5001 exhibits many of the biochemical and cytotoxic profiles found with either or both the clinically approved PARP1/2 inhibitors such as Olaparib, Rucaparib, and Niraparib, and Talazoparib, and the microtubule inhibitors polymerization inhibitors such as Vinblastine, Vincristine and Colchicine. However, AMXI-5001 is able to achieve antitumor cell responses and elicit DNA repair biomarkers at much lower concentrations than all current clinical PARP1/2 inhibitors, an effect consistent with its dual target inhibition and enhanced biochemical potency. Moreover, the favorable metabolic stability, oral bioavailability, and pharmacokinetic properties of AMXI-5001 suggest that it is a useful addition to existing targeted agents in oncology.
Results
AMXI-5001 potently and selectively inhibits PARP1 and PARP2
AMXI-5001 was engineered using a medicinal chemistry approach, to generate novel bifunctional drug-like small molecules that synchronously inhibit the catalytic activity of PARP1 and microtubule polymerization (Figure 1A) (manuscript in preparation).
Figure 1.

AMXI-5001 is a potent PARP1/2 inhibitor. (A) Structure of AMXI-5001 HCl. (B-D) The inhibitory action of the test compounds towards PARP1 (B, C) and PARP2 (D) was determined using a commercially available microplate assay (Universal Colorimetric PARP Assay from Trevigen, Inc) for PARP1 and (BPS BIOSCIENCE INC) for PARP2. To determine the IC50 value for the inhibitors, the compounds were tested using increasing concentrations of the test compounds and the average absorbance of each inhibitor concentration was plotted against the log of the concentration of inhibitor using the GraphPad Prism 6 program. Subsequently, the IC50s for the PARP1 or PARP2 inhibition were determined after non-linear fit using GraphPad Prism. The inhibitory effects of both the free base form (AMXI-5001-FB) and the HCl salt form (AMXI-5001-HCl) of AMXI-5001 were evaluated. DMSO was used as a negative control. Clinically approved PARP inhibitors Olaparib, Talazoparib, Niraparib, or Rucaparib were used as a positive control for PARP1 inhibition. Paclitaxel was also used as a negative control. (E) AMXI-5001 reduced PAR levels in situ in MDA-MB-436 human breast cancer cells: Cells were grown for 24 hr in media with or without varying concentrations of AMXI-5001, Olaparib, or Talazoparib. Cell lysates were prepared and the PAR levels were assessed by HT PARP In Vivo Pharmacodynamic Assay. The normalized ratio of pg PAR/ug total protein for cells treated with increasing doses of test compounds (AMXI-5001, Olaparib, or Talazoparib) over normalized ratio for DMSO Control-treated cells, were plotted against the compound concentrations using non-linear fit using GraphPad Prism. (F) AMXI-5001 profoundly inhibited the PAR levels in situ in MDA-MB-436 human breast cancer cells. Cells were grown for 4 hr in media with or without varying concentrations of AMXI-5001, Olaparib, or Talazoparib. Cell lysates were analyzed by western blot with an anti-PAR or anti-actin antibodies. AMXI-5001 inhibited cellular PAR expression in a comparable manner as the clinically approved PARPi. DMSO-treated (DMSO CTL) or untreated cells (Untreated CTL) cells were used as negative controls. (G) AMXI-5001 treatment induced a strong and dose dependent chromatin binding of both PARP1, and to a lesser extent PARP2: MDA-MB-436 cell lines were co-treated for 3 hr with 0.01% MMS and vehicle controls or increasing doses of AMXI-5001. Subsequently, cell lysates were prepared and fractionated into nuclear-soluble and chromatin-bound fractions then analyzed by western blot with an anti-PARP1 or anti-PARP2. Anti-H3 and anti TOP1 antibodies were used as control loading for chromatin- bound or soluble nuclear fractions, respectively.
The inhibitory effect of the AMXI-5001 compound towards PARP1 was determined using a commercially available microplate assay kit (Universal Colorimetric PARP Assay from Trevigen, Inc., Gaithersburg, MD). Clinically approved PAPR inhibitors (Olaparib, Talazoparib, Niraparib, or Rucaparib) were used as a positive control for PARP inhibition. Since AMXI-5001 structure also comprises a microtubule targeting moiety, Paclitaxel was used as a negative control for PARP inhibition. As shown in Figure 1A and 1B, AMXI-5001 inhibited PARP1 enzymatic activity with a very low IC50 of ~5 nmol/L. In a side-by-side comparison, we found that AMXI-5001 has comparable IC50 inhibition profile against PARP1 to other clinically approved PARP inhibitors to include Olaparib, Talazoparib, Rucaparib, and Niraparib (Figure 1B, 1C and Table S1). In contrast, Paclitaxel did not affect PARP1 enzymatic activity (Figure 1C). AMXI-5001 was originally synthesized as a free base form. Subsequently and in an attempt to improve AMXI-5001 solubility, its hydrochloride salt form was identified and selected. As shown in Figure 1B, both the free base and HCL salt forms of the compound have equivalent effect against PARP1 enzymatic activity.
Most PARP1 inhibitors are known to also inhibit the homologous enzyme PARP2 due to the sequence similarity of PARP1 and -2 catalytic domains [17]. We found that AMXI-5001 inhibited PARP2 to a similar extent than the clinically approved PARP inhibitors Olaparib and Talazoparib, with IC50 of 0.05, 0.03, and 0.09 nmol/L, respectively (Figure 1D; Table S2).
PARP1 and 2 are nuclear enzymes that synthesize PAR chains on target proteins as a form of posttranslational modification. To assess the ability of AMXI-5001 to inhibit intracellular PARP activity, we exposed MDA-MB-436 breast cancer cells to the alkylating agent methyl methanesulfonate (MMS) to induce PAR synthesis and examined the ability of AMXI-5001 to inhibit PAR formation both by the validated HT PARP in vivo Pharmacodynamic ELISA Assay (Trevigen, Inc., Gaithersburg, MD), and by western blotting analysis. The clinically approved PARP inhibitors Olaparib and Talazoparib were used as positive controls for cellular PAR synthesis inhibition.
Using the HT PARP In Vivo Pharmacodynamic Assay, we found that AMXI-5001 inhibited intracellular PAR formation with an IC50 of 7 nmol/L (Figure 1E; Table S3). AMXI-5001 was comparable to Olaparib and modestly less potent than Talazoparib, which inhibited the cellular PAR formation with IC50s of 8, and 3 nmol/L, respectively (Figure 1E and Table S3). The effect of AMXI-5001 on the cellular PAR levels, and thus PARP activity, was also assessed by standard western blotting procedures (as described in the experimental section). As shown in Figure 1F, AMXI-5001 profoundly inhibited cellular PAR levels in an equivalent manner as the clinically approved PARPi (Olaparib and Talazoparib) (Figure 1F). In contrast treatment with MTA (Paclitaxel and Vinblastine) had no effect on the cellular PAR levels (Not shown).
In agreement with our HT PARP in vivo pharmacodynamic assay results, quantification of the PAR protein bands densities also showed that AMXI-5001 inhibited intracellular PAR formation with a very low IC50 of 10 nmol/L (Figure S1 and Table S4). In the western blot analysis, Talazoparib, inhibited the cellular PAR formation with IC50s 3 nmol/L (Figure S1 and Table S4).
The main mechanisms by which PARP inhibitors exert their cytotoxicity are accumulation of unrepaired single-strand breaks (SSBs) resulting from catalytic PARP inhibition, and PARP trapping at the replication fork. Most PARPis trap PARP1- and PARP2-DNA complexes at DNA damage sites that arise spontaneously and/or are produced by DNA damaging agents [18]. Different PARPis may vary in their specificity for PARP enzymes and PARP trapping activity.
To examine whether AMXI-5001 induces trapping of PARP-DNA complexes at DNA damage sites, we fractionated cell lysates into nuclear-soluble and chromatin-bound fractions. Cell lysates were prepared from human cancer cells (MDA-MB-436 or Ovcar-8) treated with vehicle controls or increasing doses of AMXI-5001 or PARPis. Clinically approved PAPRis, (Olaparib, Talazoparib, Niraparib, and Rucaparib) were used as positive controls for PARP-DNA complexes trapping. To increase base damage, we co-treated the cultured cells with low dose of the classic alkylating agent MMS (0.01%). Under drug-free conditions, most PARP1 was in the nuclear-soluble fraction (Figures 1F and S2, S3, S4 and S5). AMXI-5001 increased PARP1- and PARP2-chromatin binding (Figures 1F and S2, S3, S4 and S5). Notably, increasing the concentration of AMXI-5001 greatly induced PARP1 accumulation in the chromatin-bound fraction both in the breast cancer cells (MDA-MB-436) (Figure 1G) and in the ovarian cancer cells (Ovcar-8) (Figure S2). Similar results were also obtained in other cancer cells of various origins (Not shown). Under these conditions, PAR levels were also reduced by AMXI-5001 with similar potency as the clinical PARPis (Olaparib and Talazoparib) (Figures S3A and S3B). Moreover, In human cancer cells, AMXI-5001 induced chromatin binding of both PARP1 and PARP2 in the presence of the DNA damaging agent MMS (0.01%) (Figures 1G and S3 and S4). In a side-by-side comparison, we found that AMXI-5001 induces a dose dependent cellular PARP1 and PARP-2 trapping to DNA to a similar extent than Talazoparib in cells treated with MMS (Figures S4 and S5). However, AMXI-5001 produced substantially more potent PARP1 trapping than all the other clinical PARPis (Olaparib, Niraparib and Rucaparib) (Figures S4 and S5). At concentrations greater than 0.01 mM, both AMXI-5001 and Talazoparib resulted in a strong PARP1-DNA complex trapping with an approximatively 10 fold greater potency than Olaparib, albeit, AMXI-5001 was slightly more effective than Talazoparib (Figures S4, S5 and S6). In contrast, Niraparib and Rucaparib were only slightly more or less effective, respectively, in PARP1-DNA complex trapping when compared to Olaparib (Figures S5 and S6A). In keeping with our results in the PARP catalytic activity inhibition studies, both the free base and HCl salt forms of AMXI-5001 exhibited equipotent cellular PARP1-DNA complex trapping (Figure S7).
AMXI-5001 binds to the catalytic domain of human PARP1: Co-crystal structure of AMXI-5001 in complex with human PARP1
PARP1 is a polypeptide whose activation requires binding as a homodimeric protein to a nicked DNA. This polypeptide possesses a highly conserved organization consisting of three main domains: a N-terminal DNA-binding domain which acts a DNA nick sensor, a central portion designated as the automodification domain (AMD) and which contains regions for dimerization and for modulating interactions with DNA and with proteins, and a C-terminal region, representing the most conserved part of the enzyme, and capable of catalyzing poly ADP-ribose (PAR) synthesis and of binding to target proteins [19]. In turn, this active site, also known as cat PARP, can be divided into an acceptor (adenosine) site and a donor (NIC) site. The acceptor site is occupied by the ADP moiety of PAR and the donor by NAD+. In the donor site, three subsites are described: a NIC-ribose (NI) binding site, a phosphate (PH) binding site and an adenine-ribose (AD) site [19].
To shed some light on the molecular mechanism of action of AMXI-5001, we performed co-crystal structure studies of AMXI-5001 in complex with Human PARP1 protein using Charles River’s structural biology platform support Charles River’s structural biology platform. Briefly, Screening for crystallization conditions was carried out with AMXI-5001 compound to obtain crystals suitable for X-ray data collection. Subsequently X-ray data was collected and structure solved and refined to 2.5 Å. AMXI-5001 was clearly bound in the active site with the interactions of the Human PARP1 active site residues with the ligand highlighted in Figure S8. Of particular note were the interactions of Asp 770 and Arg 878 with the amide tail and the stacking interactions of Tyr907 and Tyr896 with the aromatic moieties of the ligand. It should be noted that The Structure Solution had 2 molecules in the asymmetric unit. The solution clearly showed one copy of ligand for each molecule of PARP1 in the asymmetric unit. Moreover, the two molecules in the asymmetric unit have been independently fitted and refined resulting in slight conformational differences (Not shown).
AMXI-5001 is a week tankyrase inhibitor
The PARP protein superfamily, particularly PARP1 and PARP2, has wide-ranging roles in cellular processes such as DNA repair, opening of the chromatin and transcription of various genes. Recent reports highlighting the role of another family member, tankyrase 1 (TNKS1; also known as PARP5A and ARTD5), in the control of oncogenic WNT signaling have fueled interest in the development of additional inhibitors to target this enzyme class [20,21]. Tankyrase function is also implicated in other processes such as the regulation of telomere length, lung fibrogenesis and myelination, suggesting that tankyrase inhibitors could have broad clinical utility.
The effect of AMXI-5001 towards TNKS1 was determined using a commercially available Tankyrase 1 colorimetric activity assay (BPS BIOSCIENCE INC). XAV939 a small molecule inhibitor of TNKS1, and Olaparib were used as controls for TNK1 inhibition. AMXI-5001 exhibited only a weak inhibition of TNKS1 activity when compared to XAV939 (Figure S9 and Table S5). Olaparib also demonstrated a weak TNKS1 inhibitory activity compared to XAV939, albeit stronger than AMXI-5001. AMXI-5001 inhibited TNKS1 activity with an IC50 value >800-fold lower than its corresponding IC50 towards either PARP1 or PARP2 enzymes.
AMXI-5001 is a potent tubulin polymerization inhibitor
Microtubules have pivotal roles in fundamental cellular processes, such as mitosis, cell division, intracellular transport and cell migration [22,23]. They are highly dynamic filaments assembled from αβ-tubulin heterodimers [24-27]. MTAs interact with tubulin and alter one or more of the three characteristic phases of polymerization, namely nucleation, growth and steady state equilibrium.
To evaluate the direct effect of AMXI-5001 on tubulin polymerization, we used a standard cell free fluorescence-based tubulin polymerization assay, which generates a polymerization curve representing the three phases of microtubule formation. Vinblastine was used as a positive control for tubulin polymerization inhibition, and Paclitaxel was used as a positive control for tubulin polymerization enhancement.
As shown in Figure 2A, AMXI-5001 inhibited tubulin polymerization in a dose-dependent manner, thereby indicating that AMXI-5001 inhibited the polymerization of tubulin in a direct manner. Both AMXI-5001 and Vinblastine reduced the nucleation phase, and decreased the Vmax and polymer in comparable fashion (Figure 2). The Vmax value was decreased by 4.9, 4.2, and 2.1 fold in the presence of AMXI-5001 at 10 μM, 5 μM and 0.5 μM, respectively, as compared to DMSO control (CTL) (Table S6). The Vmax value was decreased by 5.3 fold in the presence of Vinblastine. Conversely, the addition of paclitaxel at 5 μM final concentration eliminated the nucleation phase, enhanced the Vmax by 4 fold and increased the maximum fluorescence of the reaction.
Figure 2.
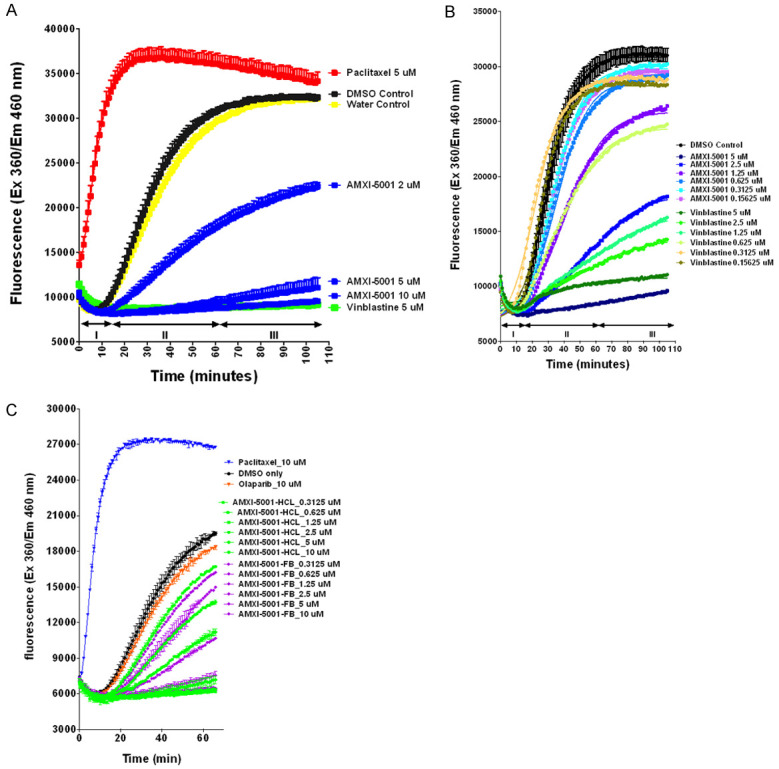
AMXI-5001 is a potent microtubule polymerization inhibitor. Tubulin polymerization assay using the fluorescence-based tubulin polymerization assay (BK011P). A. Tubulin was incubated alone (Control), with Paclitaxel or Vinblastine. Each condition was tested in duplicate. Polymerization was measured by excitation at 360 nm and emission at 460 nm. The three Phases of tubulin polymerization are marked for the control (DMSO and Water) polymerization curve; I: nucleation, II: growth, III: steady state equilibrium. Data were plotted using the GraphPad Prism 6 program. B. AMXI-5001 and Vinblastine reduced the nucleation phase, and decreased the Vmax in dose dependent manner. Tubulin was incubated alone (DMSO Control), or in the presence of inhibitors (AMXI-5001 or Vinblastine), at increasing doses ranging from 0.16 μM to 5 μM. Each condition was tested in duplicate. C. Equipotent microtubule polymerization inhibition between free base (FB) and HCl- salt forms of AMXI-5001. Tubulin was incubated alone (DMSO Control), or in the presence of inhibitors either free base form or HCl salt form of AMXI-5001 at increasing doses ranging from 0.3125 μM to 10 μM. Each condition was tested in duplicate.
To measure the compound’s IC50, we conducted the standard BK011P Tubulin polymerization assay in presence of increasing doses of either AMXI-5001 or Vinblastine (Figure 2B). The Vmax were determined as described in the experimental section. The Vmax of the DMSO control was set to 100% polymerization. The percentages of compounds Vmax over DMSO control Vmax were plotted against the compound concentrations, and IC50s were determined (Figure S10).
The Vmax value was decreased in dose dependent manner in presence of either AMXI-5001 or Vinblastine (Figure 2B). AMXI-5001 inhibited tubulin polymerization with an IC50 comparable to Vinblastine (Table S7).
As shown in Figures 2C, S11 and Table S8, both the salt and free base forms of AMXI-5001 strongly inhibited tubulin polymerization with an equipotent inhibitory activity and an IC50 of 0.92 μM and 0.86 μM, respectively. Paclitaxel induced a marked (~2 fold) increase in the polymerized (P) tubulin fraction as compared to DMSO control (Figure 2C). In contrast, Olaparib had no effect on tubulin polymerization (Figure 2C).
Since AMXI-5001 has an inhibitory effect on microtubule dynamics in vitro, in cell free assay, we sought to determine its influence on the dynamics of cellular microtubules in dividing A549 cells as described in the experimental section. Paclitaxel was used as a positive control for tubulin polymerization enhancement; and Vinblastine was used as a positive control for tubulin polymerization inhibition. Medium alone (untreated) or medium with 0.1% DMSO were used as negative controls. AMXI-5001 depolymerized microtubules in a dose-dependent manner and in comparable manner as Vinblastine (Figures 3 and S12). When treated with 0.05 μM AMXI-5001, the polymerization of microtubules was moderately inhibited (Figures 3 and S12). However, when cells were treated with 0.5 μM AMXI-5001, the microtubules polymerization was significantly inhibited; Moreover, when cells were treated with 5 μM AMXI-5001 (Figures 3 and S12), almost all microtubules were depolymerized compared with the control group. Analysis of Paclitaxel-treated cells showed the formation of thickened microtubule bundles that encircled the nucleus (Figures 3 and S12), consistent with a tubulin polymerization enhancement and increased microtubule polymers mass. Given that, AMXI-5001 is a dual PARP and microtubule inhibitor, we investigated cellular microtubule dynamics in cells treated with clinical PARP inhibitors Olaparib and Talazoparib (Figures 3 and S12), and showed that PARP inhibition does not affect the status of cellular microtubule polymerization compared to controls.
Figure 3.
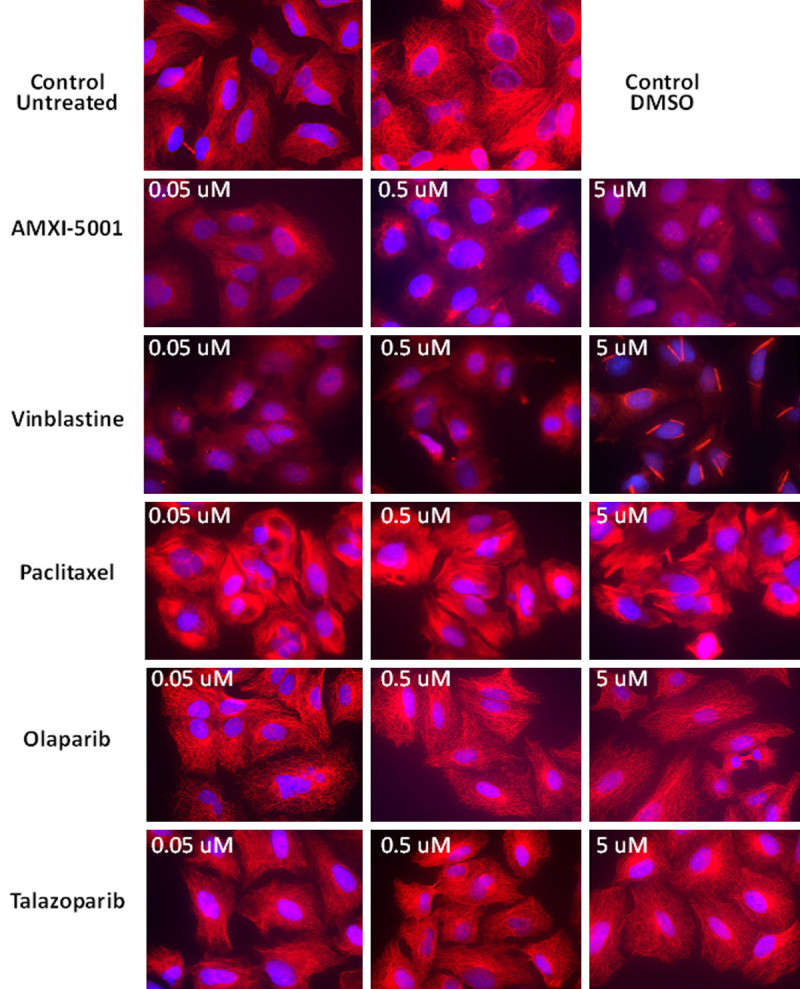
Comparaison of the anti-microtubule effect for AMXI-5001 versus standard microtubule targeting agents and PARP inhibitors. A549 cells were treated with either AMXI-5001, Vinblastine, Paclitaxel, Olaparib or Talazoparib at 0.05 μM, 0.5 μM, or 5 μM for 24 h. 0.1% DMSO or PARP inhibitors (Olaparib and Talazoparib) were used as a negative control. Vinblastine was used as a positive control for tubulin polymerization inhibition. Paclitaxel was used as a positive control for tubulin polymerization enhancement. Samples were then prepared as in the “Experimental” section, and the status of microtubules was observed using a Zeiss AxioImager 2 microscope; 63x objective, DAPI and RFP channels. Microtubule filaments are stained in red, and cell nuclei are stained in blue. AMXI-5001 inhibited the polymerization of microtubules in comparable manner as Vinblastine. PARP inhibitors does not affect the status of cellular microtubule polymerization when compared to controls.
In vitro, the equilibrium between the dimeric and polymeric forms of tubulin can be altered by different effecters and microtubule targeting agents (MTA) which alter the stability of tubulin dimers or the polymerization process. Three unique binding sites for MTAs are known and are responsible for the interactions and pharmacologic effect of paclitaxel, vinblastine and colchicine [28-30]. Paclitaxel, which preferably binds to polymeric tubulin as opposed to its dimeric form. Paclitaxel, a classic type of microtubule-stabilizing agent, binds at the inner surface of the β -subunit known as the Taxane -binding domain. Vinblastine and colchicine are microtubule destabilizing agents, which prefer to bind to dimeric tubulin. Vinblastine binds to the β-tubulin subunit at a distinct region known as the Vinca alkaloid -binding domain, while colchicine binds to the β-subunit at the interface with α-monomer of the same tubulin molecule.
The Competitive MS binding assay was conducted to characterize the binding site of AMXI-5001 on tubulin. Tubulin was incubated with colchicine, vinblastine and paclitaxel. Both colchicine- and vinblastine-tubulin binding studies were examined in the absence of GTP without preincubation. Paclitaxel-tubulin binding studies were performed after pre-incubation of tubulin with GTP for 1 h. Nocodazole, vincristine and docetaxel are well-known competitors for colchicine, vinblastine and paclitaxel, respectively, binding to tubulin. As shown in Figure S13A-C, the MS competitive binding assay demonstrated that Nocodazole, vincristine and docetaxel effectively competed for the colchicine-, vinblastine- and paclitaxel-tubulin binding sites, respectively, indicating that competitive displacement of the known ligand can be quantified and identified using this assay. The MS competitive binding method was further employed to determine the binding site of AMXI-5001 (Figure S13A-C) on tubulin. AMXI-5001 is a novel dual PARP and anti-tubulin agent that potently inhibits tubulin polymerization as demonstrated above. AMXI-5001 microtubule targeting moiety is a benzimidazole known to interact with Colchicine binding site on tubulin. Varying concentrations of AMXI-5001were used to compete with colchicine-, vinblastine- and paclitaxel-tubulin binding. Figure S13A-C show that AMXI-5001 competed specifically with colchicine-tubulin binding, but not to the Vinca alkaloid or paclitaxel sites, suggesting that AMXI-5001 binds to the colchicine-binding site on tubulin. AMXI-5001 competitive binding was less potent than that of Nocodazole (Figure S13A). Vinca alkaloid. These results indicate that AMXI-5001 specifically binds only to the colchicine-binding site, but not to Vinca alkaloid - or Taxane sites.
The effect of the AMXI-5001 treatment on the tubulin polymerization in situ were evaluated using the cell-based intracellular tubulin polymerization assay. Both AMXI-5001 and Vinblastine treatments resulted in a significant decrease in polymerized (P) tubulin, and thus microtubule assembly, compared with the negative control DMSO-treated cells (Figure S14). Conversely, Paclitaxel induced a marked (~2 fold) increase in the polymerized (P) tubulin fraction as compared to DMSO control. The density of the (P) tubulin protein band was decreased in dose dependent manner in presence of either AMXI-5001 or Vinblastine (Figures S14, S15). Both AMXI-5001 and Vinblastine strongly inhibited in situ tubulin polymerization with an IC50 of 0.67 mM and 0.17 mM, respectively (Figure S14 and Table S9).
Furthermore, treatment with AMXI-5001, or with Paclitaxel resulted in a significant decrease in the cellular levels of total tubulin protein expression, as compared to DMSO treatment (Figure S16). The density of the total tubulin protein band was decreased in dose dependent manner in presence of AMXI-5001 or Paclitaxel (Figure S16). In contrast vinblastine and the benzimidazoles (Nocodazole, Flubendazole, or Mebendazole) induced only a very modest inhibition of the cellular total tubulin protein expression. AMXI-5001 and Paclitaxel strongly inhibited in situ tubulin polymerization with an IC50 of 0.26 mM and 0.02 mM, respectively (Figure S17 and Table S10). IC50s for Vinblastine or Colchicine site- bonding benzimidazoles (Nocodazole, Flubendazole, and Mebendazole) could not be determined. These data confirm that AMXI-5001 engages the microtubule target inside the cells leading to microtubule destabilization. These findings also suggest that the levels of total alpha/beta-tubulin could be a potential simple and reliable pharmacodynamic marker of AMXI-5001-induced microtubule polymerization inhibition. in keeping, a previous study has reported a decrease in total a alpha/beta-tubulin levels in PBMCs from patients treated with ixabepilone (a microtubule polymerization stabilizer) [31].
AMXI-5001 has no significant off target effect
The potential for AMXI-5001 to inhibit off target kinases was assessed in a panel of 156 recombinant human kinase activity and binding assays including cytoplasmic and receptor tyrosine kinases, serine/threonine kinases and lipid kinases. The kinase profiling assays were performed using Life Technologies’ SelectScreen® Profiling Service, (Thermo Fisher Scientific, Madison, WI), with a broad coverage of the human kinome. AMXI-5001 was tested at 8 µM in triplicate against each kinase and the mean % inhibition values was determined. At the concentration of 8 mM, AMXI-5001 did not have any significant inhibitory effects on most of the kinases tested (Data not shown).
Furthermore, AMXI-5001 was also profiled in radioligand binding assays as part of in vitro compound selection to detect potential off-target activity. AMXI-5001 was evaluated at a concentration of 10 µM against 38 targets in a binding screening assay that included primary molecular targets, transmembrane and soluble receptors, ion channels, and monoamine transporters. No receptors or enzymes were inhibited ≥50% (Data not shown).
AMXI-5001 targets tumor cells with or without defects in homologous recombination
The objective of this study was to evaluate the cell growth inhibitory activity of AMXI-5001 as well as clinical PARPis (Olaparib, Tlazoparib, Niraparib and Rucaparib), and MTAs (Paclitaxel and Vinblastine) in a panel of 110 cancer cell lines using the Cell Titer Glo assay. The panel included a wide variety of cancer cell lines of various origins, with proficient DNA damage response genes (BRCA-1 or BRCA-2) or defective for BRCA-1 or BRCA-2 expression or expressing mutant forms of the two genes.
Following a 3 days treatment, AMXI-5001 resulted in a robust cytotoxicity in a wide variety of human cancer cell lines (110 lines) of various origins, with very low IC50s and a potency that is far superior than all clinical PARPis. AMXI-5001 potency was 20, to >10000-fold more than that of the clinical PARPis. Importantly, AMXI-5001 was highly active in both BRCA1/2-deficient cancer cells (Homologous recombination (HR) deficient) and BRCA1/2 wild type cancer cells (HR proficient) (Table S11). Cell lines with BRCA1/2 mutations or low expression of homologous recombination repair (HRR) genes/proteins were more sensitive (IC50 ranging from 18 nM to 26 nM) (Table S11) to cell growth inhibitory activity by AMXI-5001, as compared to HR proficient cancer lines (IC50 ranging from 4 nM to >5000 nM) (Table S11). In contrast, clinic PARP inhibitors were mostly inactive in HR proficient cancers and only modestly active in HR deficient cancers (Table S11).
On account of the different doubling time between the various tested cancer cell lines, we have also assessed AMXI-5001 anti-growth potency in a 6 days treatment cell viability assay. 6 days drug exposure, resulted in a dramatic increase in the apparent drug potency as reflected by a decreased IC50 in the majority of the tested lines, as compared 3 days drug exposure (Table S12).
Moreover, AMXI-5001 inhibited colony formation in a various cell lines, including ovarian, non-small cell lung, and prostate. AMXI-5001 inhibited colony formation with stronger potency (5 to 200-fold greater) than all the clinical PARPis or standard chemotherapy carboplatin, as determined by a decreased IC50, in all the tested lines (Figures S18, S19 and S20 and Tables S13, S14 and S15).
One of the hallmarks of aggressive tumor cells is an ability to migrate and to invade surrounding tissue. The ability of AMXI-5001 to inhibit cell migration which may be an important process in tumor metastasis, was tested using in vitro scratch assay that assess cell migration by recovery of the scratch wound in A549 lung cancer cells. AMXI-5001 effectively inhibited scratch wound recovery in A549 cells as compared to control-treated cells (Figures S21, S22). Importantly, AMXI-5001 demonstrated greater cell migration inhibition than both the clinical PARPis, Olaparaib and Talazoparib. AMXI-5001 showed a comparable inhibitory effect to MTAs, against scratch wound recovery (Figures S21, S22). However, Clinical MTAs inhibitory effect was largely due to a significant cell death induction by these agents (Figures S21, S22).
The effect of AMXI-5001 on cell-cycle progression in dividing MDA-MB-436, OVCAR8 and A549 cells, was evaluated following 24 Hrs treatment with medium alone (untreated CTL) or medium containing either 0.1% DMSO (DMSO CTL), or increasing concentrations of AMXI-5001. Clinical MTAs (Paclitaxel and Vinblastine) were used as positive controls for cell cycle arrest. Cell cycle analyses revealed that similar to Vinblastine, AMXI-5001 induced a concentration dependent cell cycle arrest in G2/M phase in all three tested cancer cells (Figures S23, S24 and S25). 5 mM Paclitaxel also induced G2/M cell cycle arrest in all three tested cancer cells (Figures S24 and S25). Furthermore, dose-response analysis of AMXI-5001 effect on the cell cycle, showed that low (equal or lower than 2 mM) concentrations induced cell cycle arrest chiefly at G2/M phase, while increasingly higher (greater than 2 mM) concentrations caused arrest at late, mid or early S phase in addition to G2M phase arrest (Figures S24 and S25). In contrast, drug- induced S-phase arrest was not observed with either the low and high Vinblastine concentrations, or with a high concentration (5 mM) of Paclitaxel (Figures S24 and S25). Cell cycle arrest in S-phase with high doses of AMXI-5001, implies that the treated cells are unable to duplicate their DNA due to a significant DNA damage induced by AMXI-5001 potent and simultaneous PARP and microtubule polymerization inhibition in situ.
AMXI-5001 effect on cellular checkpoint, regulatory, and signaling proteins
To better understand the mechanisms by which S and G2/M arrest are regulated in response to AMXI-5001, western blot analyses were performed on cell lysates to assess the status of various checkpoint-related proteins. Cells were incubated with AMXI-5001 at various concentrations that causes cell cycle arrest after a 24 h incubation. In MDA-MB-436 cells and A549 cells, AMXI-5001 treatment resulted in modulation of its direct targets as shown by decreased levels of PAR and total tubulin (Figure S26A). In addition, AMXI-5001 treatment increased phosphorylation of cell cycle checkpoint kinases CHK1 and CHK2 at ser296Chk1 and thr68Chk2 sites, respectively, indicating activation of these kinases (Figure S26A, S26B). Clinical PARPi (Olaparib and Talazoparib) also activated both CHK1 and CHK2, whereas clinical MTAs (Vinblastine and Paclitaxel) had no effect on either of these kinases (Figure S26B). CHK1 is implicated in several checkpoints of the cell cycle acting as a key player in the signal transduction pathway activated in response to either or both DNA damage and mitotic spindle damage. CHK2 is primarily involved in cell cycle checkpoints in response to DNA double-strand breaks (DSBs). Furthermore, AMXI-5001 induced modulation of the phosphorylation levels of serine 10 in histone H3 (pH3) in a dose dependent manner (Figure S26A, S26C, S26D). There was no significant overall change in total H3 protein expression (Figure S26A). At low doses (less than 1 mM), AMXI-5001 resulted in an increase in the pH3 level, suggesting a cell cycle arrest in prometaphase (G2/M phase) (Figure S26A, S26D). However, treatment with higher AMXI-5001 concentrations (>1 mM) triggers loss of pH3 signal (Figure S26A, S26D). Clinical PARPis (Olaparib and Talazoparib) induced inhibition of cellular PAR but had no significant effect on pH3 level (Figure S26D). Cell treatment with clinical MTAs (Vinblastine and Paclitaxel) resulted in decreased tubulin levels and increased pH3 levels (Figure S26C), suggesting that AMXI-5001-induced pH3 modulation is principally due to its anti-microtubule activity. AMXI-5001-induced pH3 loss at higher concentration, could be attributed to either or both enhanced rates of apoptosis and significant increase in DNA damage and cell cycle arrest at S phase, as demonstrated by increased expression of p21, a growth arrest and a proapoptotic protein (Figure S26E). In keeping, AMXI-5001 also induced an enhanced expression of rH2AX, a marker for double strand DNA damage (Figure S26F).
Previous studies demonstrated a role for PARP activity in cancer cell resistance to clinical MTAs [12]. These studies showed that microtubule inhibition by MTAs results in stimulation of cellular PARP1 activity. Subsequently, the activated PARP stabilizes important mitotic checkpoint proteins such as E3 ubiquitin ligase (CHFR), and thus lead to inhibition of cell cycle progression to M phase in the replicating cancer cells [12]. Consequently, cancer cells avoid mitotic catastrophe induced by MTAs. Our results showed that treatment with AMX-5001, a dual PARP and microtubule polymerization inhibitor, results in decreased expression of CHFR protein in cancer cells (Figure S26F). These results are exciting as they indicate that AMXI-5001dual mechanism of action may override the potential for cancer cells resistance to microtubule damage through its synchronous inhibition of both PARP and microtubule polymerization in the same cells. In agreement with these findings, AMXI-5001 also resulted in activation of Cyclin A-associated kinases, CDK1 and CDK2, as indicated by decreased phosphorylation of CDK1 at Tyr15 site (an inhibitory site), and increased phosphorylation of CDK2 at Tyr160 site (an activating site) (Figure S26A, S26B). CDK1 and CDK2, participate in regulating cellular progression into mitosis. CDK2 drives the progression of cells into the S- and M-phases of the cell cycle. Cdk1 is a central regulator that drives cells through G2 phase and mitosis. Activation of CDK1/2 with AMXI-5001, suggests that the AMXI-5001-treated cancer cells may progress to the irreversible M phase before the AMXI-5001-induced damage of mitotic spindle and/or DNA is resolved, and thus resulting in an enhanced cancer cell death. In agreement with this, previous investigations reported that cancer cells sensitivity to MTAs requires CDK1/2 kinase activity [32,33].
Treatment with chemotherapies including MTAs, DNA-damaging agents, and PARPis are known to result in neoantigen expression in various human cancers. The chemotherapy-induced neoantigen expression may increase cancer cells immunogenicity and thus prime cancers to respond to immunotherapy. We sought to assess the effect of AMXI-5001 treatment on some predicted immunomodulatory neoantigen expression in various cancer cell lines including. Our results showed that AMXI-5001 treatment induced a concentration-dependent increase protein expression of either or both the natural killer (NK) cells activating antigens DR4 and CD155, and the T regulatory cells immunomodulatory antigen PDL-L1 (Figure S27A-C). In contrast, neither clinical PARPis (Olaparib ad Talazoparib) or Paclitaxel had any significant effect on the protein expression of these antigens as assessed in MDA-MB-436 breast cancer cells (Figure S27C). However, Vinblastine, a microtubule polymerization inhibitor, resulted in increased expression of both DR4 and PDL1 in a similar manner as AMXI-5001 (Figure S27C). These results suggest that the increased expression of death receptors and PD-L1 is chiefly associated with its inhibitory effect on the microtubule polymerization.
Our aforementioned findings are interesting as they suggest potential synergy of AMXI-5001 in combination with the immunotherapy to include the immune checkpoint PD1/PDL-1 inhibitors. In addition, AMXI-5001-induced expression of NK activating antigens (DR4/5 and CD155) is exciting as it indicates a potential for AMXI-5001 to illicit a NK driven anti-tumor response. Furthermore, death receptors including DR4/5 can selective trigger apoptosis in cancer cells through interaction with TNF-related apoptosis-inducing ligand (TRAIL) on NK cells. In keeping, AMXI-5001 demonstrated synergistic cytotoxicity, in vitro, when combined with TRAIL in various cancer cells (Figure S28; Table S16). In contrast, there was no significant synergy in combination of TRAIL with other clinical PARP inhibitors (Olaparib and Talazoparib).
Moreover, increased expression of CD155 (necl-5) on cancer cells, may induced NK and T cell-mediated anti-tumor immunity through its interaction with CD226 on these immune cells. Previous studies have shown that tumor cells with higher CD155 expression are more susceptible to CD226-induced killing [34,35]. In addition, CD155-CD226 interaction can mediate the inhibition of CD155-positive tumor metastasis by NK [36]. Cell surface expression for both DR4/5 and PD-L1 antigens is required for binding to their respective ligands/receptors on the target cells and thus exerting their full effect. Flow cytometry analysis demonstrated a marked increase in cell surface expression of both Death receptors DR4 and DR5, as well as PD-L1, in A549 cells following either 24 Hr or 48 Hr treatment with AMXI-5001 (Figures S29 and S30). The abovementioned increase in cell surface expression for these antigens was demonstrated by the conspicuous shift to the right (increase) in fluorescence intensity for cells treated with AMXI-5001 (1 mM or 5 mM) compared to vehicle treated controls (Figures S29 and S30).
Synergistic anticancer activity of AMXI-5001 with approved anticancer therapies in vitro
To evaluate the potential synergistic effect of AMXI-5001 when combined with some selected clinically approved anticancer therapies to include DNA damaging chemotherapies (Gemcitabine, Cisplatin or Etoposide) or Topoisomerase inhibitor (Topotecan) or PI3K inhibitor (Idelalisib). Our results showed that a 1/10 ratio combination of AMXI-5001 with the aforementioned anticancer therapies resulted in more potent anti-proliferative effect in MDA-MB-436 cell line than either single agents alone (Figure S31 and Table S17). Moreover, AMXI-5001 demonstrated stronger synergy when combined with Gemcitabine or Topotecan (Figure S31 and Table S17).
Metabolism and PK properties of AMXI-5001
Currently, there are no approved orally bioavailable microtubule targeting agent. One of the objectives of our dual PARP and microtubule inhibitor discovery program was to develop an orally bioavailable dual PARP and microtubule inhibitor and to improve metabolic stability, PK properties and oral bioavailability over existing PARP1/2 inhibitors. In vitro metabolism studies of AMXI-5001 in hepatocytes from rats, dogs, cynomolgus monkeys, and humans demonstrated that AMXI-5001 had excellent liver stability; The half-life of AMXI-5001 (2 μM) during incubation with hepatocytes (500,000 cells/mL) from Sprague-Dawley rats, beagle dogs, cynomolgus monkeys, or humans was estimated to be 217 min, 812 min, 185 min, and 417 min, respectively. After incubation 1 μM concentration for 120 min at 37°C, the percentage of AMXI-5001 remaining was 63.6%, 85.4%, 64.1%, and 78.5% for rat, dog, monkey, and human hepatocytes, respectively (Not shown). These data suggest AMXI-5001 will have human clearance approximately similar to that of the animal species.
AMXI-5001 has been given orally to rats and dogs at various dose levels (manuscript in preparation). AMXI-5001 was absorbed and bioavailable in all species tested. In rats and dogs, exposure increased with the increase in dose level, supporting the use of these species in the toxicity studies of AMXI-5001. A variety of formulations were tested, eventually leading to selection of 10% TPGS (D-α-tocopherol polyethylene glycol-1000-succinate; Vitamin E) in 0.01 N HCl, pH 2.1-2.3, because of its suitable toxicity profile and ability to deliver adequate AMXI-5001 systemic exposure. AMXI-5001 demonstrated an absolute bioavailability in rats and dogs of 31% and 64%, respectively with this formulation, and PK properties that would predict a human half-life that is sufficient to support a regimen of twice daily administration (manuscript in preparation).
In vitro studies assessing the potential for inhibition of human cytochrome P450 enzymes (CYP450s) showed that AMXI-5001 did not inhibit any of the major human hepatic CYP450 enzymes CYP1A2, CYP2B6, CYP2D6, and CYP3A4/5. There was weak (and not time-dependent) inhibition of CYP2C9, and CYP2C19 (Data not shown).
Overall, AMXI-5001 demonstrated excellent metabolic stability, oral bioavailability and PK properties.
Anti-tumor effect of AMXI-5001 oral administration in xenograft tumor models
AMXI-5001 pharmacokinetic assessment
To support the in vivo primary pharmacodynamic studies in xenograft models, pharmacokinetic parameters of AMXI-5001 were determined in female BALB/C mice. AMXI-5001 free base or hydrochloride salt forms were formulated either as N-methyl-pyrrolidine/carboxymethylcellulose (NMP/CMC) suspension or in 10% D-α-tocopherol polyethylene glycol-1000-succinate; Vitamin E (TPGS) suspension, respectively and administered orally. AMXI-5001 was absorbed and bioavailable in this strain of mice, enabling its testing in murine xenograft models with oral dosing (Table S18).
Initial 5-day study in MDA-MB-436 xenografts
AMXI-5001 (HCl salt form) was formulated in 10% TPGS and administered orally to female athymic nude mice bearing established MDA-MB-436 xenograft tumors, an aggressive basal breast carcinoma cell line cell line that harbors BRCA1 deletion and is BRCA1 deficient. When tumors reached average volume of 350 mm3, AMXI-5001 suspension was administered orally at doses of 12.5, 25, or 50 mg/kg/dose BID for 5 days. Five days of twice per day (BID) oral administration of AMXI-5001 caused a rapid and marked tumor growth inhibition at doses of 25 and 50 mg/kg/dose BID, and clear tumor regression at 50 mg/kg/dose BID compared to the vehicle group (Figure S32A).
Moreover, plasma and tumor tissue bioanalyses revealed a dose dependent increase in AMXI-5001 plasma concentration and a corresponding dose dependent increase in AMXI-5001 tumor concentration (Figure S32B). However, there was no significant changes in AMXI-5001 concentration in either plasma or tumor tissues between tissues harvested after 4th and 10th repeat oral dose (Figure S32B). These results suggest that there is no significant increase in the accumulation of AMXI-5001 in either blood or tumor tissue with repeat treatment.
Western Blot analyses of tumor lysates showed that AMXI-5001 modulates its targets in vivo in a dose dependent manner (Figure S33). Particularly, AMXI-5001 inhibition of its targets was more evident at 25 and 50 mg/kg BID doses as indicated by a noticeable decrease in polymeric adenosine diphosphate (ADP) ribose (PAR) expression, a surrogate marker for PARP activity, and in total tubulin expression, a surrogate marked for microtubule polymerization inhibition (Figure S33). This provides support for the hypothesis that the anti-tumor efficacy of AMXI-5001 potentially results from inhibition of both PARP and microtubule polymerization. In general, there was a good correlation between increases in plasma and tumor drug concentrations and increased inhibition of either or both of these AMXI-5001 targets at the doses tested.
Confirmatory 31-day study in MDA-MB-436 xenografts
The efficacy of AMXI-5001 seen in the 5-day study was confirmed in this subsequent study of a longer dosing duration. MDA-MB-436 xenografts tumors were established by inoculation of female athymic nude mice with 3.5 × 106 MDA-MB-436 cells subcutaneously in the third mammary fat. When the tumors reached approximately 100 mm3, mice were randomized to five treatment groups (8 animals per group): 1) Vehicle control, oral; 2) 10 mg/kg PO BID AMXI-5001 on 5-day ON/2-day OFF cycles, oral; 3) 50 mg/kg PO BID AMXI-5001 on 5-day ON/2-day OFF cycles, oral; 4) 50 mg/kg BID Olaparib on 5-day ON/2-day OFF cycles, oral; 5) 1 mg/kg vinblastine once weekly (Q1W), intraperitoneally. All animals were treated over a 31-day dosing period. Tumor size for each animal was measured twice a week (Figure 4A).
Figure 4.

A. Effect of AMXI-5001 on MDA-MB-436 Xenograft Growth and Body Weight in Female Athymic Nude Mice (31-Day Dosing). A. MDA-MB-436 xenografts tumors were established by inoculation of female athymic nude mice with 3.5 × 106 MDA-MB-436 cells subcutaneously in the third mammary fat. When the tumors reached approximately ~100 mm3, mice were randomized to five treatment groups (8 animals per group): 1) Vehicle control administered BID orally on 5 day ON and 2 day OFF cycles, or AMXI-5001 administered at 2) 10 and 3) 50 mg/kg orally BID on 5 day ON and 2 day OFF cycles, or 4) Olaparib at 50 mg/k BID on 5 day ON and 2 day OFF cycles, or 5) vinblastine at IP 1 mg/kg once a week. All animals were treated over a 31 day dosing period. Tumor size for each animal was measured twice a week. B. Body weights were recorded twice a week for animals in each group. C. Histologic therapy response. Histological analysis using H&E staining at low (10 ×) and high (20 ×) magnifications of representative paraffin embedded MDA-MB-436 xenograft tumor sections form mice treated with: vehicle control, AMXI-5001 at 10 mg/kg or 50 mg/kg BID, Olaparib at 50 mg/kg BID 5 day on 2 day off cycles, vinblastine at 1 mg/kg once a week. All treatments were administered over 31 days dosing period.
AMXI-5001 exhibited significant, dose-dependent tumor growth inhibition. In addition to tumor growth inhibition, AMXI-5001 also exhibited clear tumor regression in this model. For the 50-mg/kg AMXI-5001 group, all tumors experienced a complete regression. By Day 31 of the dosing schedule, tumors were either too small to be accurately measured or non-palpable. Comparison to Olaparib revealed that the anti-tumor growth effect observed with AMXI-5001 at a dose of either 10 or 50 mg/kg/dose BID was superior to Olaparib at a dose of 50 mg/kg/dose BID. In contrast to AMXI-5001, Olaparib did not exhibit any tumor regression activity in this model and vinblastine given at a dose of 1 mg/kg Q1W showed no significant effect on tumor growth in this model (Figure 4A).
Animals treated with AMXI-5001 doses of 10 and 50 mg/kg/dose BID on 5-day ON/2-day OFF cycles for 31 days, did not display any physical symptoms of compound-related toxicity, nor did Olaparib and vinblastine treated animals. There was no significant effect on the body weight during the entire course of treatment in any group (Figure 4B).
In order to assess treatment related histological changes, paraffin sections of harvested tumors taken at the end of treatment were stained with hematoxylin-eosin (H&E). H&E staining revealed dramatic hemorrhagic tumor necrosis in tumors treated with AMXI-5001 at 50 mg/kg dose (Figure 4C). The harvested tumors from the mice treated with AMXI-5001 at 50 mg/kg were nonpalpable by the end of the treatment. Tumor sections from this group showed a few remaining xenograft cancer cells dispersed through extensive fibrosis, which were surrounded by skin, skeletal muscle and fat from the host animal. At higher magnification a photograph of these tumors showed a significant treatment effect with evidence of necrosis and hemorrhage, with hemosiderin filled macrophage accumulation, cholesterol granuloma, chronic inflammation and fibrosis (Figure 4C). The tumors from mice treated with AMXI-5001 at 10 mg/kg also showed reduced density of xenograft cells with areas of necrosis, inflammation, and fibrosis. Tumor sections from vehicle-treated, Olaparib-treated or vinblastine-treated mice, contained mostly vital tumor cells without any fibrosis (Figure 4C).
At the end of the study, tumors were resected and processed for analysis of microtubule filament formation. Sections from paraffin embedded tumors were stained with an antibody specific to alpha/beta-tubulin, and visualized by fluorescent microscopy. Cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). Repeat oral dose of AMXI-5001 at either 10 mg/kg or 50 mg/kg BID resulted in a striking dose dependent inhibition of microtubule filament formation in MDA-MB-436 cells derived tumors (Figure S34). Vinblastine treatment also induced a marked inhibition of microtubule filament in MDA-MB-436 tumors. Conversely, repeat oral treatment with Olaparib at 50 mg/kg BID dose showed no effect on tumor cells microtubule filaments formation.
Pharmacodynamic parameters for AMXI-5001 treatment, were also determined using lysate prepared with xenograft tumor specimens from each group in this study. For PARP inhibition, PAR levels were evaluated, and for microtubule destabilization, total alpha/beta tubulin expression levels were assessed by western blot analyses with their specific corresponding antibodies. AMXI-5001 treatment at either 10 or 50 mg/kg inhibited dramatically, and in a dose dependent manner, PAR expression in MDA-MB-436 derived tumors (Figure S35). AMXI-5001 treatment also resulted in a significant and dose dependent decrease in the expression levels of total alpha/beta-tubulin in MDA-MB-436 derived tumors (Figure S35). Olaparib treatment resulted in a significant inhibition of PAR expression but had no effect on tubulin expression in MDA-MB-436 derived tumors. In contrast, vinblastine treatment showed no effect on levels of PAR or total tubulin expression in MDA-MB-436 derived tumors (Figure S35). Consistent with PARP inhibition effect, both AMXI-5001 treatment and Olaparib treatment resulted in a conspicuous inhibition of the tumor protein expression of the cell cycle checkpoint protein (CHFR), as compared to vehicle control treatment (Figure S35).
Collectively, the aforementioned results, indicate that AMXI-5001 modulates in a dose dependent manner, both its intended targets in vivo in tumors. These results also suggest that antitumor activity of AMXI-5001 might be attributed to its dual mechanism of action through its synchronous inhibition of both PARP and microtubule polymerization in the tumor cells.
Complete regression of established large tumors (600-1300 mm3) in MDA-MB-436 xenografts with single agent AMXI-5001 treatment
Based on the anti-tumor efficacy in the above study in MDA-MB-436 xenografts when tumors were staged at 100-150 mm3 before initiation of dosing, the anti-tumor effect of AMXI-5001 was tested in larger, more well-established tumors (Figure 5A).
Figure 5.
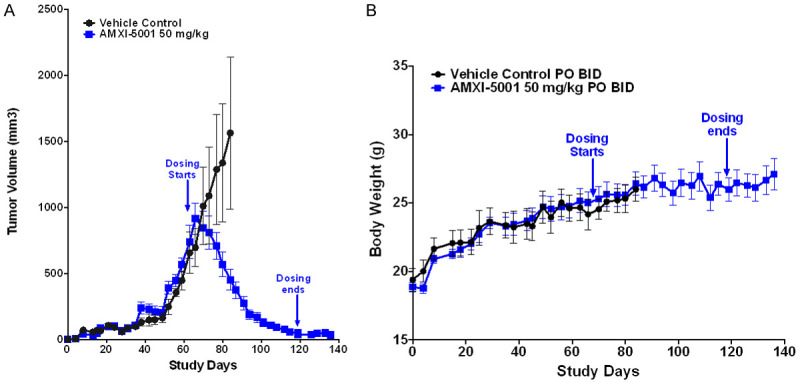
Effect of AMXI-5001 on Large MDA-MB-436 Xenograft Growth and Body Weight in Female Athymic Nude Mice. A. AMXI-5001 causes uniformly growth inhibition and complete regression without recurrence of large MDA-MB-436 mammary fat xenograft tumors in all treated mice. Five mice bearing large tumors (Tumor size ranging from ~554 to ~1318 mm3) in each group. B. Body weights were recorded twice a week for animals in each group. No significant effect on the body weight during the entire course of treatment with AMXI-5001 at 50 mg/kg as compared to vehicle control treated group was observed.
At the end of the 31-day dosing period of the above described study, 5 out of 8 mice from the control group with tumor sizes of 554-1318 mm3 received AMXI-5001 at a dose of 50 mg/kg/dose BID on 5-day ON/2-day OFF cycles, 2 mice from the vinblastine group, and 1 mouse from the control group (tumor size 458-953 mm3) received vehicle control on the same schedule. Remarkably, all large tumors treated with AMXI-5001 exhibited gradual and approximatively complete tumor regression starting in the first week of treatment initiation (Figure 5A). Notably, following AMXI-5001 treatment, there was no sign of tumor recurrence 2 weeks after mice stopped treatment (Figure 5A). Conversely, in the new vehicle treated group, tumors continued to grow rapidly and the mice were terminated, less than three weeks after vehicle treatment was initiated, because of excessive tumor growth and the consequent poor animal health. There was no significant effect on the body weight during the entire course of treatment with AMXI-5001 at 50 mg/kg/dose BID as compared to vehicle control treated group (Figure 5B).
Superior antitumor effect of AMXI-5001 compared to combination therapy of single-agent PARP and microtubule inhibitors in MDA-MB-436 xenograft model
The objective of this study was to compare the in vivo anti-tumor efficacy of AMXI-5001 treatment to either single agent Olaparib (a clinical PARP inhibitor), or single agent paclitaxel (a potent clinical microtubule polymerization inhibitor), and to combination therapy of Olaparib and paclitaxel in MDA-MB-436 Xenograft model. Olaparib and paclitaxel were administered at clinically relevant doses and schedules.
MDA-MB-436 xenografts tumors were established by inoculation of female athymic nude mice with 3.5 × 106 MDA-MB-436 cells subcutaneously. Mice were randomized when the tumors reached approximately an average of 220 mm3 prior to dosing (Figure 6).
Figure 6.
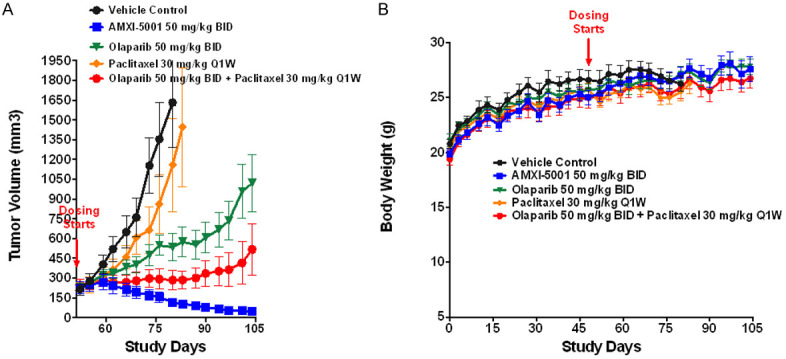
Effect of AMXI-5001, Olaparib, Paclitaxel, and Olaparib/Paclitaxel on MDA-MB-436 Xenograft Growth and Body Weight in Female Athymic Nude Mice (56-Day Dosing). MDA-MB-436 xenografts tumors were established by inoculation of female athymic nude mice with 3.5 × 106 MDA-MB-436 cells subcutaneously in the third mammary fat. When the tumor size average reached approximately ~200 mm3, mice were randomized to five treatment groups (8 animals/group): vehicle control, administered BID orally on 5 day ON and 2 day OFF cycles, AMXI-5001 (50 mg/kg) orally BID on 5 day ON and 2 day OFF cycles, Olaparib (50 mg/kg) BID on 5 day ON and 2 day OFF cycles, paclitaxel IV (30 mg/kg) once a week, or a combination therapy of Olaparib (50 mg/kg PO BID 5 day ON 2 day OFF) with paclitaxel (30 mg/kg) IV once per week. A. Tumor size for each animal was measured twice a week. B. Body weights were recorded twice a week for animals in each group.
AMXI-5001 induced complete or near-complete tumor regression in all treated animals, and this effect was superior to either single agent Olaparib or paclitaxel, or combination treatment with both agents (Figure 6A). In this tumor model, single agent paclitaxel treatment displayed weak anti-tumor activity, whereas treatment with single agent Olaparib induced a clear but modest inhibition of the tumor growth. Combination therapy of Olaparib and paclitaxel resulted in a greater tumor growth inhibition than either of these single agents alone. However, the combination therapy of Olaparib and paclitaxel failed to induce tumor regression and the tumors continued to grow, albeit, at slower rate than the vehicle treated or single agent (Olaparib or paclitaxel) treated tumors. AMXI-5001 was much more effective in this model than the combination of single agent Olaparib with single agent paclitaxel.
Animals treated with AMXI-5001, single agent Olaparib, or combination treatment of Olaparib with paclitaxel did not show any notable treatment-related toxicity (Figure 6B). However, 3 out of 8 mice in the single-agent paclitaxel group showed lethargy, lack of response to external stimuli, loss of consciousness, and labored respiration within a few minutes after first intravenous (IV) administration of paclitaxel (30 mg/kg). The 3 affected animals in this group were euthanized. Nevertheless, all remaining 5 animals in the single agent paclitaxel treated group did not show any notable treatment-related toxicity for the duration of treatment. There were no significant treatment-related effects on body weight during the entire course of treatment with any of the treatment modalities as compared to the vehicle control treatment.
Discussion
Standard treatment available to most patients with advanced cancers is inadequate with most approved therapies for advanced cancers having response rates ≤ 20-30% and modest survival benefits. The ineffectiveness of standard therapies for advanced cancers has a number of causes, including that cancers are quite heterogeneous, especially at the molecular level. There are ongoing advances in our understanding of the genetic and molecular drivers of cancer as well as our ability to molecularly characterize cancers [37-39].
Over the past 50 years, chemotherapy, to include microtubule targeting agents (MTAs), and DNA damaging agents (DDAs), has been and remains the standard of care in cancer treatment. Despite all the recent advances in targeted cancer therapy, these chemotherapeutic agents are still the most potent agents to kill cancer cells owing to their ability to target critical mechanisms for cancer cells proliferation, DNA repair, mitosis, migration, and cytoskeleton. Unfortunately, the dose limiting toxicity along with drug resistance, remain the most significant barriers to delivering curative doses for cancer with these agents.
Preclinical studies have demonstrated that PARP inhibitors act synergistically with other chemotherapy agents, including platinum compounds and microtubule targeting agents. However, due to limited dosing schedule options for chemotherapy agents, combination of single agent PARP inhibitors with chemotherapy do not allow for a synchronous and continuous effect of these mutually reinforcing treatments. Accordingly, synchronous delivery of both PARP inhibition and chemotherapy may result in a synthetically lethal anti-cancer therapeutic strategy, and is therefore a desirable profile for a small molecule dual-inhibitor. In addition, synchronous delivery of both PARP inhibition and chemotherapy with one drug, could theoretically accentuate the efficacy of these mutually reinforcing therapies at lower doses, thereby decreasing off-target side effects and, consequently, increasing the therapeutic indices of either therapy.
Here we describe a novel and first in class small-molecule dual PARP and microtubule polymerization inhibitor AMXI-5001. AMXI-5001 was designed to inhibit PARP and the microtubule polymerization synchronously and thus sensitize cancer cells to spindle damage resulting in suppression of mitotic checkpoints and cell death. Microtubule inhibition may also sensitize cancer cells to PARP inhibition by impairing the trafficking of critical DNA repair proteins, and resulting in prolonged DNA damage and enhanced cell death.
In vitro, AMXI-5001 inhibits with high potency both PARP and tubulin polymerization. AMXI-5001 shows comparable IC50 inhibition profiles against PARP1/2 and microtubule polymerization as clinical PARP inhibitors (Olaparib, Rucaparib, Niraparib, and Talazoparib) and the potent polymerization inhibitor (vinblastine), respectively. AMXI-5001 resulted in a robust cytotoxicity across a wide variety of human cancer cell lines (110 lines) of various origins, with very low IC50s (low nanomolar concentrations) and a potency that is far superior than all currently approved PARP inhibitors (Olaparib, Rucaparib, Niraparib, and Talazoparib). Importantly, AMXI-5001 was highly active in both BRCA mutated and thus homologous recombination deficient and BRCA wild type and HR proficient cancer cells. In contrast, the clinic PARPis which were mostly inactive in HR proficient cancers and modestly active in HR deficient cancers when used at micromolar concentrations. AMXI-5001 potency was 20, to >10000-fold more than that of the clinical PARPis. Cell lines with BRCA1/2 mutations or low expression of homologous recombination repair (HRR) genes/proteins were more sensitive (IC50 ranging from 18 nM to 26 nM) (Figure S7) to cell growth inhibitory activity by AMXI-5001, as compared to HR proficient cancer lines (IC50 ranging from 4 nM to >5000 nM).
Furthermore, AMXI-5001 treatment results in PARP1 and PARP2 trapping to chromatin in human cancer cells lines. AMXI-5001 is a very potent PARP-DNA complexes trapper, far superior (~10 fold greater) than most of the clinically approved PARP inhibitors (Olaparib, Rucaparib, Niraparib, and veliparib) and comparable in potency to Talazoparib (the most potent clinical PARP inhibitor). The PARP-DNA complexes trapping process may interfere with DNA replication, causing cell death preferentially in cancer cells, which grow faster than non-cancerous cells. Preclinical studies have demonstrated that PARPi trapping ability contributes significantly to their monotherapy activity in vitro and potentiate their synergistic activity with chemotherapy including DNA damaging agents in vivo [40].
Also, AMXI-5001 demonstrated strong anti-tubulin polymerization activity, with a comparable potency and IC50 to Vinblastine, an FDA approved and a clinically-used potent microtubule destabilizer compound. Noteworthy, through its simultaneous PARP and microtubule polymerization inhibition, AMXI 5001 may mitigate the peripheral neuropathy commonly associated with MTAs. MTAs-induced peripheral neuropathy is a major dose limiting toxicity for which no therapy is approved. Recent reports demonstrated that PARP inhibition protects against microtubule targeting agent-induced painful neuropathy in rats [9], and thus support that PARP inhibition may represent a novel therapeutic approach for the dose limiting toxicity associated with MTAs. Additionally, unlike the standard MTAs used in cancer treatment, AMXI-5001 is a non-taxane and non-vinca alkaloid microtubule inhibitor. AMXI-5001 targets the colchicine-binding motif on tubulin, and thus inhibits its polymerization into microtubules. Microtubule inhibitors that target the colchicine binding site are emerging as promising anticancer with reduced risk for dose limiting neuropathy. Therefore, AMXI-5001 has a potential to lowering the risk of neuropathy commonly associated with the microtubule inhibition.
AMXI-5001 demonstrated a remarkable In vivo preclinical anti-tumor activity in BRCA mutated triple negative breast cancer (TNBC) model, a cancer with currently no effective therapy. Oral administration of single agent AMXI-5001 induced complete regression of established tumors, including exceedingly large tumors (Figures 4, 5 and 6). Most importantly, none of the AMXI-5001 treated mice had tumor re-growth until the end of the study, two weeks after AMXI-5001 dosing stopped. Furthermore, AMXI-5001 resulted in superior anti-tumor effects when compared to either single agent (PARP inhibitor (Olaparib) or microtubule targeting agent (Paclitaxel or Vinblastine)) or combination treatment with both agents which were given at clinically relevant doses (Figures 4A, 6A). Importantly, the AMXI-5001 anti-tumor effect was achieved with tolerable toxicity, evidence of PARP and microtubule inhibition in vivo in tumors and favorable PK properties that allow twice-a-day oral dosing in human patients.
AMXI-5001 is the first in class small-molecule inhibitor reported to date that offers a continuous and synchronous PARP and microtubule polymerization inhibitions, and thus results in synthetic lethality, particularly in cancer cells vulnerable to DNA damage. The discovery and characterization of AMXI-5001 as an orally bioavailable dual PARP and microtubule polymerization inhibitor, provides a welcome addition to the oncology field and we believe the pharmacological properties of AMXI-5001 warrant further investigation, and its advancement into clinical studies in cancer patients.
Materials and methods
Drugs and cell lines
Synthesis of AMXI-5001 is described elsewhere (patent). Olaparib, rucaparib, and veliparib, Paclitaxel, Vincristine, Colchicine, Nocodazole, Mebendazole, docetaxel and Carboplatin were obtained from Selleck Chemicals, LLC. Olaparib was obtained from LC laboratories, LLC Talazoparib (BMN-673) and Rucaparib were obtained from MedChemExpress, Inc. Cell Titer Glo reagent was purchased from Promega. Human TRAIL antibodies were obtained from R&D systems. The colorimetric Universal PARP Assay, HT PARP In Vivo Pharmacodynamic Assay II, and Anti-PAR antibody from BPS Trevigen Inc. Tubulin polymerization assay (BK011P) and Anti-alpha/beta tubulin antibody were obtained from Cytoskeleton, Inc. PD-L1, DR4 antibodies and all other antibodies used in this study were purchased from Cell signaling Technology, Inc.
All cell lines were obtained from American Type Culture Collection and maintained as exponentially growing monolayers according to the supplier’s instructions.
PARP enzyme assays
The inhibitory action of the test compounds towards PARP1 was determined using a commercially available microplate assay kit (Universal Colorimetric PARP Assay from Trevigen, Inc., Gaithersburg, MD) and in accordance with the instructions provided by the manufacturer. Briefly, stock solutions of the various test compounds were made in dimethyl sulfoxide (DMSO). For the assay, each strip well was filled with 10 µL of the inhibitor solution, 20 μL of diluted PARP1 enzyme (providing 0.5 Unit/well), and 25 µL of PARP Cocktail (consisting of biotinylated NAD, activated DNA in Tris-Cl pH 8.0, and EDTA). The strip wells were incubated at room temperature for 60 min, and then washed 4 times with phosphate buffered saline (PBS: Na2HPO4, NaH2PO4, and NaCl) and 0.1% Triton X-100, and 2 times with PBS. Then, 50 µL of diluted Strep-HRP (blocking solution) was added to each well, and the strips were further incubated at room temperature for 60 min. After washing the wells 2 times each with PBS and with 0.1% Triton X-100, and 2 times with PBS they were mixed with 50 µL of TACS-Sapphire™ colorimetric substrate, and allowed to stand in the dark for 15 min. After stopping the reaction by adding 50 µL of 0.2 N hydrochloric acid (HCl) to each well, the absorbance was measured at 450 nm. Parallel experiments were conducted by substituting the test solution with an equivalent volume of DMSO to verify the effect of the vehicle on the enzyme activity. All the assays were conducted in at least two separate occasions, each time in duplicates Subsequently, the IC50s for the PARP1 inhibition were determined after non-linear fit using GraphPad Prism. The results of these studies are reported in μM. DMSO was used as a negative control. Clinically approved PAPR inhibitors (olaparib, talazoparib, niraparib, or rucaparib) were used as a positive control for PARP inhibition. In some instances, paclitaxel was also used as a negative control.
PARP2 inhibition assay
The inhibitory action of the test compounds towards PARP2 was determined using a commercially available microplate assay kit (BPS BIOSCIENCE INC., San Diego, CA) and in accordance with the instructions provided by the manufacturer. Subsequently, the IC50s for the PARP2 inhibition were determined after non-linear fit using GraphPad Prism.
HT PARP in vivo pharmacodynamic assay
The HT PARP In Vivo Pharmacodynamic Assay (Trevigen, Inc. #4520-096-K) is a high-throughput, chemiluminescent ELISA designed to quantify poly (ADP-ribose) (PAR) in cellular extracts. The assay employs a two-site sandwich technique in which two different anti-PAR antibodies are used to capture and detect the target analyte. This assay is useful for measuring PAR in extracts from peripheral blood mononuclear cells (PBMC), cultured cells, and tissues. Additionally, this assay can be used to monitor the efficacy of PARPi or anti-cancer drugs on cellular PAR formation and cancer cell cytotoxicity. The inhibitory effect of AMXI-5001 on cellular PAR formation was quantified with this HT PARP In Vivo Pharmacodynamic Assay in accordance with the instructions provided by the manufacturer. Each ELISA plate contains serial dilutions of purified PAR standard used to plot the PAR standard curve. The net mean RLU (Relative Light Units) values of the PAR standards were calculated by subtracting the background (without PAR) from the RLU values, then plotted as a function of the corresponding PAR values (pg/ml). The PAR standard curve was plotted using the GraphPad Prism 6 program (GraphPad Software Inc., San Diego, CA). Typically, the linear dynamic range for the PAR standard curve is from 10 to 1000 pg/ml. The net RLU values for each cell lysate sample was calculated by subtracting the background from the RLU values. Subsequently, the PAR levels in each sample was determined using the PAR standard curve. The PAR level of DMSO-treated control was set to 100% PAR level. The percentages of test compounds-treated samples over DMSO-treated control PAR levels were plotted against the compound concentrations. Subsequently, the IC50s for the cellular PAR formation inhibitors were determined after non-linear fit using GraphPad Prism.
Cellular PAR western blot assay
The effect of AMXI-5001 on the cellular PAR levels was also assessed by standard western blotting procedures. Briefly, cancer cells were cultured overnight in 6 wells plates, followed by incubation with vehicle control or test compound for 24 h. The cells were then washed and cell lysates were prepared for western blotting as described below. Protein concentration in each cell lysate was quantified using BCA method. Immunoblotting was subsequently performed using standard procedures. A total of 10 μg of protein was resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with Anti-PAR (Trevigen, MD) and secondary antibodies (Jaskson ImmunoResearch Inc). Clinically approved PARPi (olaparib, talazoparib, or rucaparib) were used as a positive control for PAR inhibition and γ H2AX induction. β-actin (Cell Signaling Technology, MA) was used as the loading control.
Cellular PARP-DNA trapping assays
To assess the PARP trapping ability of AMXI-5001, standard cellular trapping assays were performed as previously described [18]. Briefly, cancer cells were cultured overnight in 6 wells plates, followed by cotreatment with alkylating agent methyl methanesulfonate (MMS) and vehicle control, or cotreatment with alkylating agents and varying concentrations of the test PARPi for 1 or 3 hr. Cells were then washed and collected by trypsinization. Subsequently chromatin fractions were prepared using Thermo Scientific subcellular protein fractionation kits (#PI78840) per manufacturer protocol. Samples were normalized for protein concentration and analyzed by immunoblotting by anti-PARP1, anti-TOP1, and anti-H3. Clinically approved PARPi (olaparib, talazoparib, or rucaparib) were used as a positive control for PARP-DNA trapping. To quantify the PARPi-induced PARP1-DNA trapping in human cancer cells, densitometry was performed on immunoblots. DNA-bound PARP1 levels were normalized to total cellular PARP1 levels (chromatin-bound PARP1+ Unbound PARP1). Each experiment was conducted at least three independent times. Representative results are depicted in the result section below.
Crystal structure of AMXI-5001 in complex with human PARP1
The crystal structure of AMXI-5001 compound in complex with Human PARP1 protein was determined using Charles River’s structural biology platform support Charles River’s structural biology platform (Report ATLAS-AMXI-5001-PRE-CLIN PHARM-PARP1 structure AMXI-5001). Briefly, PARP1 (residues 611-1011) was cloned into pET24 and expressed in E.coli (BL21DE3), then PARP1 protein was purified. Subsequently, crystallization studies were conducted. The formed co-crystal of the PARP protein complexed with AMXI-5001 was collected and analyzed examined by Synchrotron X-ray diffraction analysis. X-ray data was collected and processed using AUTOPROC to a resolution of 2.43 Å and the structure solved by molecular replacement using PHASER using the PDB structure 5WRZ as a search model. The model was then rebuilt in COOT and refined using REFMAC and anisotropic B factors. The ligand dictionary was prepared using ACEDRG and modelled into the protein active site and further rounds of TLS and isotropic B factor refinement and placement of solvent molecules carried out.
Tankyrase 1 activity assay
The effect of AMXI-5001 towards TNKS1 was determined using a commercially available Tankyrase 1 colorimetric activity assay (#80573, BPS BIOSCIENCE INC) in accordance with the instructions provided by the manufacturer.
In vitro tubulin polymerization assay
An in vitro fluorescence-based tubulin polymerization assay kit (#BK011, Cytoskeleton, Inc) was used according to the manufacturer’s protocol for monitoring the time-dependent polymerization of tubulin to microtubules. The reaction mixture, containing porcine brain tubulin, fluorescent reporter and GTP in presence or absence of test compounds, was prepared. Tubulin polymerization was followed by monitoring fluorescence enhancement due to the incorporation of a fluorescent reporter into microtubules as polymerization proceeded. Fluorescence emission (excitation at 360 nm and elimination at 460 nm wavelength) was measured for one hour and 45 minutes at one min intervals in a Biotek Synergy Plate Reader. Under the conditions described above, approximately 45% of the tubulin is polymerized, leaving flexibility for detecting enhancers and inhibitors of polymerization. DMSO was used as a negative control. Paclitaxel was used as a positive control for tubulin polymerization enhancement. Vinblastine was used as a positive control for tubulin polymerization inhibition. The incorporation of the fluorescent reporter into microtubules as polymerization proceeded was measured. The fluorescence absorbance, at 360 nm excitation and 460 nm elimination wavelengths, was plotted against the reaction time using the GraphPad Prism 6 program (GraphPad Software Inc., San Diego, CA). The Vmax were determined after non-linear fit using GraphPad Prism. The Vmax of DMSO control was set to 100% polymerization The Vmax of the DMSO control was set to 100% polymerization. The percentages of compounds Vmax over DMSO control Vmax were plotted against the compound concentrations. Subsequently, The IC50s for the tubulin polymerization inhibitors were determined after non-linear fit using GraphPad Prism.
Cellular microtubule polymerization Immunocytochemistry assay
The effects of drugs on cellular microtubules were observed using an immunocytochemistry assay with the anti-alpha/beta tubulin: (ATN02: sheep polyclonal, Cytoskeleton) as previously described [41].
Competitive MS binding assay
To determine the binding site of AMXI-5001 on tubulin, competitive MS binding assay was conducted as previously described [42] but with slightly different conditions for Colchicine, vinblastine and paclitaxel binding. This assay was used to assess the tubulin binding of Colchicine, Vinblastine and Paclitaxel and to identify which of these three binding sites that AMXI-5001 binds. The method involves a very simple step of separating the unbound ligand from macromolecules using ultrafiltration. The unbound ligand in the flow through fraction can be accurately determined using highly sensitive and specific HPLC-MS/MS method. Briefly, Nocodazole Colchicine, and Vinblastine were incubated with tubulin in the incubation buffer at 37°C for 1 h. Paclitaxel was incubated with performed tubulin in the incubation buffer at 37°C for 1 hr. Performed tubulin was prepared by incubating tubulin with GTP in the incubation buffer at 37°C for 1 hr. Varying concentrations of Nocodazole, vincristine and Docetaxel were used to compete with Colchicine-, Vinblastine- and Paclitaxel-tubulin binding, respectively. After incubation, reaction samples were centrifuged in an Ultracel-30 microconcentrators. The flow through (unbound ligands) was collected and analyzed by HPLC MS/MS, as described below. Varying concentrations of AMXI-5001 were examined to individually compete with colchicine-, vinblastine- and paclitaxel-tubulin binding. The ability of the competitor or AMXI-5001 to inhibit the binding of ligands was expressed as a percentage of unbound ligand control in the absence of any competitor.
Tubulin protein expression assay
The effect of AMXI-5001 on the levels of total tubulin was evaluated by western blot analysis as described above. Briefly, MDA-MB-436 breast cancer cells were seeded into 6-well plates and cultured until attached (~5 hours), then changed to serum-free medium and cultured overnight. Cells were treated in 1% FBS medium for 24 hrs with either control DMSO (0.1%) or varying concentrations of test drugs. Treated cells were then lyzed in M-PER buffer in wells, scrapped cells off wells. and rocked at 4°C for 10 minutes. The cell lysates were centrifuged at 14 kg, 4°C, 10 minutes and supernatants were harvested. The total protein concentration was assessed by BCA quantification method. 6 ug of each of these lysates was added to 4x sample buffer, in 3:1 mixture ratio, and boiled at 95-100° for 10 minutes, cooled down and centrifuged for 10 seconds. Subsequently, the sample were loaded on SDS-PAGE gels, and western blots were probed with antibodies specific for total alpha/beta-tubulin or GAPDH as described above. The densities of the detected tubulin and actin protein bands were quantified with ImageJ software (NIH). For each sample, the tubulin band density was normalized to the GAPDH band density. Normalized band density of DMSO control was set to 100% density. The percentages of compounds band density over DMSO control band density were plotted against the compound concentrations. Subsequently, The IC50s for the tubulin polymerization inhibition were determined after non-linear fit using GraphPad Prism. DMSO was used as a negative control. Paclitaxel was used as a positive control for tubulin polymerization enhancement. Vinblastine was used as a positive control for tubulin polymerization inhibition. Benzimidazoles (Nocodazole, Flubendazole, and Mebendazole) were used as controls for Colchicine-site binding tubulin polymerization inhibitors.
In vitro kinase inhibition assays
The potential for AMXI-5001 to inhibit off target kinases was assessed in a panel of 156 recombinant human kinase activity and binding assays including cytoplasmic and receptor tyrosine kinases, serine/threonine kinases and lipid kinases. The kinase profiling assays were performed using Life Technologies’ SelectScreen® Profiling Service, (Thermo Fisher Scientific, Madison, WI), with a broad coverage of the human kinome. The kinase activity assays measure peptide phosphorylation (Z’-LYTE®) or ADP production (Adapta®) while the kinase binding assays monitor displacement of ATP site-binding probes (LanthaScreen®). The ATP concentrations used in the activity assays were within 2-fold of the experimentally determined apparent Michaelis constant (K m app) value for each kinase while the competitive binding tracer concentrations used in the binding assays were within 3-fold of the experimentally determined dissociation constant (K d) values. AMXI-5001 was tested at 8 µM in triplicate against each kinase and the mean % inhibition values are reported. For selected kinases, 10-point inhibitor titrations were carried out using the same kinase assays as used in the single point tests in order to determine the inhibitor concentration providing 50% inhibition (IC50). Details regarding the kinase proteins used and the assay protocols are on file and also available online (thermofisher.com).
Cell titer Glo assay
Antiproliferative activity of AMXI-5001 was evaluated in a panel of 110 cancer cell lines of various origins, either proficient or defective for BRCA-1 or BRCA-2 expression or expressing mutant forms of the two genes. Proliferation was assessed after cells were exposed for 3 days or 6 days with AMXI-5001 doses ranging from 8 nM to 5 μM. Briefly, 5000 or 1000 cells (In 3 days or 6 days exposure, respectively) were cultured in a 96-well plate and incubated at 37°C and 5% CO2 prior to adding serial dilutions of AMXI-5001. Cells were then incubated for 3-6 days at 37°C and 5% CO2. Cell viability was assessed by Cell Titer Glo assay. The number of living cells was determined by reading the plate on a GloMax Luminametor. Cell growth was expressed as percentage growth with respect to vehicle (DMSO control) treated cells. The concentration required to inhibit cell growth by 50% (IC50) was determined after non-linear fit using GraphPad Prism. DMSO treated cells were used as a vehicle control. MTAs (Paclitaxel and Vinblastine) as well as clinical PARPis (Olaparib, Talazoparib, Niraparib, and Rucaparib) were used as comparative controls.
Clonogenic assay
The effect of AMXI-5001 on cell viability was also evaluated using colony formation assay in cells lines of ovarian, non-small cell lung cancer, and prostate origin. In brief, cell lines were incubated in 0.1% (v/v) DMSO (vehicle control) or 0.008, 0.04, 0.2, 1, 5 mM AMXI-5001 at 37°C until > 50 colonies have formed (6-28 days). Seeding cell density was determined from preliminary experiments and is defined as seeding density producing linear cell growth after cell incubation in 0.1% (v/v) DMSO (vehicle control). Cell viability was determined using the colony formation assay by counting Crystal Violet-stained colonies using ImageJ software. IC50 values were defined as the AMXI-5001 concentrations producing 50% cell viability when compared to vehicle control cells (100% cell viability). Clinical PARPis (Olaparib, Talazoparib, Niraparib, and Rucaparib) were used as comparative controls.
Wound healing assay/scratch assay
The ability of AMXI-5001 to inhibit cell migration was tested using in vitro scratch assay that assess cell migration by recovery of the scratch wound in A549 lung cancer cells grew into monolayer. Briefly, A549 lung cancer cells were seeded onto 12-well plates. When the cell confluence reached about 80% and above, scratch wounds were made by scraping the cell layer across each culture plate using the tip of 200 μl pipette. After wounding, the debris was removed by washing the cells with PBS. Wounded cultures were incubated in culture medium alone (Untreated control), or medium containing 0.1% (v/v) DMSO (DMSO control) or 0.04, 0.2, 1 mM AMXI-5001 at 37°C for 24 hr. Subsequently, 3 fields (40 Å~) were randomly picked from each scratch wound and visualized by microscopy to assess cell migration ability. Clinical PARPis (Olaparib and Talazoparib) and clinical MTAs (Paclitaxel and Vinblastine) were used as positive controls for cell migration inhibition.
Cell cycle analysis
The effect of AMXI-5001 on cell-cycle progression in dividing MDA-MB-436, OVCAR8 and A549 cells, was evaluated. Briefly, the cells were grown in T75 flasks and treated with 0.1% DMSO (vehicle control), Vinblastine, or AMXI-5001 at varying concentrations for 24 Hr. After incubation, the cells were trypsinized, washed with PBS and fixed. After fixation, the cells were centrifuged, washed with once with PBS and stained with propidium iodide. Finally, the distributions of treated cells at different phases of cell cycle (G1, S, G2/M) were analyzed using flow cytometry. The percentage of cells in each mitotic phase pre- and post-treatment with AMXI-5001 for 24 Hr was determined. MTAs (Paclitaxel and Vinblastine) were used as positive controls for cell cycle arrest.
Western blot analysis of cellular checkpoint and signaling proteins
To better understand the mechanisms by which AMXI-5001 regulates the cell cycle arrest in cancer cells, standard western analysis was performed on cell lysates to assess the status of various checkpoint-related proteins and cell signaling proteins (Figure 3). Briefly, cancer cells were cultured overnight in 6 wells plates in serum free medium, followed by incubation with vehicle control or test compound for 24 h. The cells were then washed and cell lysates were prepared for western blotting as described below. Protein concentration in each cell lysate was quantified using BCA method. Immunoblotting was subsequently performed using standard procedures. a total of 10 μg of protein was resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with checkpoint proteins corresponding antibodies, and secondary antibodies. Clinically approved PARPi (Olaparib, Talazoparib) or MTAs (Paclitaxel, Vinblastine) were used as controls. β-actin and GAPDH (Cell Signaling Technology) were used as the loading control.
Flow cytometry analysis-cell surface marker staining
A549 cells were cultured overnight in 100 mm culture dishes, followed by incubation with vehicle control or AMXI-5001 at 1 mM or 5 mM for either 24 hr or 48 hr then harvested by trypsinization, as described below in method section. For PDL1 and death receptors (DR4 and DR5) cell surface staining, harvested cells were washed with PBS, and stained with respective antibodies in cell staining buffer and processed for flow cytometry analysis. (BD FACS Calibur). BD CellQuest Pro software (Franklin Lakes, NJ) was used for flow cytometric data analysis.
Pharmacokinetic evaluations of AMXI-5001 in mice
The objective of these studies was to determine pharmacokinetic and bioavailability profile following a single dose oral administration of either free base form or HCl salt forms of AMXI-500 in BALB/c mice. The In-Life procedures for these studies were conducted using Explora Bioloabs contract research services (San Diego, CA). The in vitro bioanalyses of the plasma samples were performed using Integrated Analytical Solutions contract research services (Berkeley, CA). Briefly, AMXI-5001 free base and HCl salt form were formulated either as NMP/CMC suspension or in 10% TPGS suspension, respectively and administered orally to female BALB/c mice. The formulations protocols used for the PK studies are described below. The bioavailability for AMXI-5001 free base form was assessed following a single oral dose administration at either 50 or 100 mg/kg per mouse. The bioavailability for AMXI-5001 HCL form was assessed following a single oral dose administration at 50 mg/kg per mouse. Blood samples were collected at pre-dose, 0.5, 1, 2, 4, 8 and 24 hours after AMXI-5001 single dose oral administration. For each mouse, one-two time points were assigned. Each group had 3 blood samples per time point. The first blood collection was survival bleed and the second blood collection was terminal. Blood samples were collected into tubes containing K2EDTA anticoagulant and stored on wet ice until centrifuged and processed for plasma. Plasmas were stored at -80°C until analysis. The peak concentration (Cmax), the time to maximum concentration (Tmax), the half-life, and the AUC were determined from composite mean plasma concentration-time data. All doses and plasma concentrations of AMXI-5001 were presented as free base.
Plasma samples were analyzed by high performance liquid chromatography (HPLC) in conjunction with a triple quadrupole mass spectrometer that used electrospray ionization in tandem with positive ionization (MS/MS). The lower limit of quantitation (LLOQ) was 5 ng/ml. The quantitative range of the assay was 5 to 10000 ng/ml.
The pharmacokinetic modeling was performed using PK Solver software (Version 2.0) and the following PK parameters were reported: Cmax: Maximum (peak) plasma concentration DN-Cmax: Dose-normalized Cmax; Tmax: Time at Cmax; AUC: Area under the concentration-time curve; DN-AUC: Dose-normalized AUC; T1/2: Terminal half-life; and VF: Apparent volume of distribution. The PK parameters were determined based on a Non-Compartmental PK model Analysis of Plasma Data after Extravascular Input.
Pharmacodynamic evaluations of AMXI-5001 in mice bearing MDA-MB-436 tumors
The primary objective of this study was to determine the relationship between AMXI-5001 dose, plasma concentration, tumor concentration and inhibition of PARP and microtubule polymerization In vivo in MDA-MB-436 Xenograft Tumors. The In-Life procedures for this study were conducted using Explora Bioloabs contract research services (San Diego, CA). The in vitro bioanalyses of the plasma samples were performed using Integrated Analytical Solutions contract research services (Berkeley, CA). Briefly, AMXI-5001 HCL salt form was formulated in 10% TPGS suspension, as described below and administered orally to female athymic nude mice bearing established MDA-MB-436 Xenograft Tumors (When tumors reach average volume of 500 mm3). AMXI-5001 suspension was administered orally BID at 6.25, or 12.5, or 25 mg/kg per mouse for 5 days. At 3 hr post the 4th and 10th dose, n=3 mice per group, or each AMXI-5001-dose and the vehicle control group, were euthanized and blood collected in EDTA tubes for plasma processing. At necropsy the tumor tissues from all groups, as well as stomach, small intestine, large intestine, caecum, kidneys, and liver from the vehicle control group and the group treated with AMXI-5001 at 50 mg/kg BID, were individually collected, placed into cryotubes and flash frozen in liquid nitrogen. The frozen plasma and tissue samples were kept at -80°C until bioanalysis for AMXI-5001 concentration. Plasma samples were analyzed by high performance liquid chromatography (HPLLC) in conjunction with a triple quadrupole mass spectrometer that used electrospray ionization in tandem with positive ionization (MS/MS). The lower limit of quantitation (LLOQ) was 5 ng/ml. The quantitative range of the assay was 5 to 10000 ng/ml.
In vivo xenograft models
In the present studies, the MDA-MB-436 (triple negative human breast carcinoma with BRCA1 mutation) was utilized to explore the efficacy and potency of AMXI-5001 with regard to tumor growth inhibition and regression in vivo. The In-Life procedures for these studies were conducted using the Explora Biolabs contract research services (San Diego, CA). The standard experimental design for these studies involved twice daily (BID) daily oral administration (PO) of AMXI-5001, following a 5-day ON and 2-day OFF cycles, beginning when the established solid tumors were staged (~100 -150 mm3 for most xenografts, ~700 mm3 and up to 1500 mm3 for xenograft regression studies). Throughout the dosing period of 10-60 days, tumor size and body weight were measured twice weekly.
At the end of the study, tumors were resected and processed for analysis of microtubule filament formation. Sections from paraffin embedded tumors were stained with an antibody specific to for alpha/beta-tubulin, and visualized by fluorescent microscopy.
In order to assess treatment related histological changes, paraffin sections of harvested tumors taken at the end of treatment were also stained with hematoxylin-eosin (H&E) and analyzed by a certified pathologist.
Pharmacodynamic parameters for AMXI-5001 treatment, were also determined using lysate prepared with xenograft tumor specimens from each group in this study. For poly {adenosine diphosphate (ADP)}-ribose polymerase (PARP) inhibition (PARPi), Poly (ADP-ribose) (PAR) levels were evaluated, and for microtubule destabilization, total alpha/beta tubulin expression levels were assessed by Western blot analyses with their specific corresponding antibodies.
Disclosure of conflict of interest
AMXI-5001 is currently developed by AtlasMedx, Inc. Lemjabbar-Alaoui Hassan, Csaba Peto, and David Jablons may benefit financially from the development of AMXI-5001 through patents held jointly with University of California, San Francisco and AtlasMedx, Inc.
Supporting Information
References
- 1.Society AAC: American Cancer Society. Cancer Facts & Figures 2020. Atlanta, GA: American Cancer Society; 2020. volumes. [Google Scholar]
- 2.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science (New York, NY) 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613–623. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 4.Canaani D. Application of the concept synthetic lethality toward anticancer therapy: a promise fulfilled? Cancer Lett. 2014;352:59–65. doi: 10.1016/j.canlet.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 5.Fouquin A, Guirouilh-Barbat J, Lopez B, Hall J, Amor-Gueret M, Pennaneach V. PARP2 controls double-strand break repair pathway choice by limiting 53BP1 accumulation at DNA damage sites and promoting end-resection. Nucleic Acids Res. 2017;45:12325–12339. doi: 10.1093/nar/gkx881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610–621. doi: 10.1038/nrm.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ronson GE, Piberger AL, Higgs MR, Olsen AL, Stewart GS, McHugh PJ, Petermann E, Lakin ND. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat Commun. 2018;9:746. doi: 10.1038/s41467-018-03159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balko RA, Hendrickson AW, Grudem ME, Klampe CM, Jatoi A. Can Poly (ADP-ribose) polymerase inhibitors palliate paclitaxel-induced peripheral neuropathy in patients with cancer? Am J Hosp Palliat Care. 2019;36:72–75. doi: 10.1177/1049909118786958. [DOI] [PubMed] [Google Scholar]
- 9.Brederson JD, Joshi SK, Browman KE, Mikusa J, Zhong C, Gauvin D, Liu X, Shi Y, Penning TD, Shoemaker AR, Giranda VL. PARP inhibitors attenuate chemotherapy-induced painful neuropathy. J Peripher Nerv Syst. 2012;17:324–330. doi: 10.1111/j.1529-8027.2012.00413.x. [DOI] [PubMed] [Google Scholar]
- 10.Brodie SA, Li G, Harvey D, Khuri FR, Vertino PM, Brandes JC. Small molecule inhibition of the CHFR-PARP1 interaction as novel approach to overcome intrinsic taxane resistance in cancer. Oncotarget. 2015;6:30773–30786. doi: 10.18632/oncotarget.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL, Carmichael J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15:R88. doi: 10.1186/bcr3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kashima L, Idogawa M, Mita H, Shitashige M, Yamada T, Ogi K, Suzuki H, Toyota M, Ariga H, Sasaki Y, Tokino T. CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J Biol Chem. 2012;287:12975–84. doi: 10.1074/jbc.M111.321828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poruchynsky MS, Komlodi-Pasztor E, Trostel S, Wilkerson J, Regairaz M, Pommier Y, Zhang X, Kumar Maity T, Robey R, Burotto M, Sackett D, Guha U, Fojo AT. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A. 2015;112:1571–1576. doi: 10.1073/pnas.1416418112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 15.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solier S, Pommier Y. The nuclear gamma-H2AX apoptotic ring: implications for cancers and autoimmune diseases. Cell Mol Life Sci. 2014;71:2289–2297. doi: 10.1007/s00018-013-1555-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliver AW, Ame JC, Roe SM, Good V, de Murcia G, Pearl LH. Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res. 2004;32:456–464. doi: 10.1093/nar/gkh215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Soto M, Perez JM. Poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors in cancer chemotherapy. Recent Pat Anticancer Drug Discov. 2006;1:39–53. doi: 10.2174/157489206775246430. [DOI] [PubMed] [Google Scholar]
- 20.Kim MK. Novel insight into the function of tankyrase. Oncol Lett. 2018;16:6895–6902. doi: 10.3892/ol.2018.9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riffell JL, Lord CJ, Ashworth A. Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov. 2012;11:923–936. doi: 10.1038/nrd3868. [DOI] [PubMed] [Google Scholar]
- 22.Etienne-Manneville S. Microtubules in cell migration. Annu Rev Cell Dev Biol. 2013;29:471–499. doi: 10.1146/annurev-cellbio-101011-155711. [DOI] [PubMed] [Google Scholar]
- 23.Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem. 2012;287:43359–43369. doi: 10.1074/jbc.M112.423905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16:711–726. doi: 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- 25.Bowne-Anderson H, Zanic M, Kauer M, Howard J. Microtubule dynamic instability: a new model with coupled GTP hydrolysis and multistep catastrophe. Bioessays. 2013;35:452–461. doi: 10.1002/bies.201200131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 27.Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 28.Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435:519–522. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang SH, Hung YE, Hung L, Robey RW, Sackett DL, Linehan WM, Bates SE, Fojo T, Poruchynsky MS. Evidence for microtubule target engagement in tumors of patients receiving ixabepilone. Clin Cancer Res. 2007;13:7480–7486. doi: 10.1158/1078-0432.CCR-06-2883. [DOI] [PubMed] [Google Scholar]
- 32.Bae T, Weon KY, Lee JW, Eum KH, Kim S, Choi JW. Restoration of paclitaxel resistance by CDK1 intervention in drug-resistant ovarian cancer. Carcinogenesis. 2015;36:1561–1571. doi: 10.1093/carcin/bgv140. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama S, Torikoshi Y, Takahashi T, Yoshida T, Sudo T, Matsushima T, Kawasaki Y, Katayama A, Gohda K, Hortobagyi GN, Noguchi S, Sakai T, Ishihara H, Ueno NT. Prediction of paclitaxel sensitivity by CDK1 and CDK2 activity in human breast cancer cells. Breast Cancer Res. 2009;11:R12. doi: 10.1186/bcr2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, Bottino C, Moretta A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004;64:9180–9184. doi: 10.1158/0008-5472.CAN-04-2682. [DOI] [PubMed] [Google Scholar]
- 35.Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, Falco M, Lanino E, Pierri I, Zambello R, Bacigalupo A, Mingari MC, Moretta A, Moretta L. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112) Blood. 2005;105:2066–2073. doi: 10.1182/blood-2004-09-3548. [DOI] [PubMed] [Google Scholar]
- 36.Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol. 2010;184:902–911. doi: 10.4049/jimmunol.0903225. [DOI] [PubMed] [Google Scholar]
- 37.Markman M. The promise and perils of ‘targeted therapy’ of advanced ovarian cancer. Oncology. 2008;74:1–6. doi: 10.1159/000138349. [DOI] [PubMed] [Google Scholar]
- 38.Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. 2018;15:353–365. doi: 10.1038/s41571-018-0002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168:584–599. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, Abraham VC, Algire MA, Shi Y, Olson AM, Johnson EF, Wilsbacher JL, Maag D. PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res. 2019;17:409–419. doi: 10.1158/1541-7786.MCR-18-0138. [DOI] [PubMed] [Google Scholar]
- 41.Jenks AD, Vyse S, Wong JP, Kostaras E, Keller D, Burgoyne T, Shoemark A, Tsalikis A, de la Roche M, Michaelis M, Cinatl J Jr, Huang PH, Tanos BE. Primary cilia mediate diverse kinase inhibitor resistance mechanisms in cancer. Cell Rep. 2018;23:3042–3055. doi: 10.1016/j.celrep.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li CM, Lu Y, Ahn S, Narayanan R, Miller DD, Dalton JT. Competitive mass spectrometry binding assay for characterization of three binding sites of tubulin. J Mass Spectrom. 2010;45:1160–1166. doi: 10.1002/jms.1804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.