

Abstract
Cocaine is one of the most abused illicit drugs worldwide. It is well known that the dopamine (DA) transporter is its major target; but cocaine also acts on other targets including nicotinic acetylcholine receptors (nAChRs). In this study, we investigated the effects of cocaine on a special subtype of neuronal nAChR, α3β4-nAChR expressed in native SH-SY5Y cells. α3β4-nAChR-mediated currents were recorded using whole-cell recordings. Drugs were applied using a computer-controlled U-tube drug perfusion system. We showed that bath application of nicotine induced inward currents in a concentration-dependent manner with an EC50 value of 20 µM. Pre-treatment with cocaine concentration-dependently inhibited nicotine-induced current with an IC50 of 1.5 μM. Kinetic analysis showed that cocaine accelerated α3β4-nAChR desensitization, which caused a reduction of the amplitude of nicotine-induced currents. Co-application of nicotine and cocaine (1.5 μM) depressed the maximum response on the nicotine concentration-response curve without changing the EC50 value, suggesting a non-competitive mechanism. The cocaine-induced inhibition of nicotine response exhibited both voltage- and use-dependence, suggesting an open-channel blocking mechanism. Furthermore, intracellular application of GDP-βS (via recording electrode) did not affect cocaine-induced inhibition, suggesting that cocaine did not alter receptor internalization. Moreover, intracellular application of cocaine (30 µM) failed to alter the nicotine response. Finally, cocaine (1.5 μM) was unable to inhibit the nicotine-induced inward current in heterologous expressed α6/α3β2β3-nAChRs and α4β2-nAChRs expressed in human SH-EP1 cells. Collectively, our results suggest that cocaine is a potent blocker for native α3β4-nAChRs expressed in SH-SY5Y cells.
Keywords: cocaine, nicotinic acetylcholine receptor, patch-clamp, open-channel block, SH-SY5Y cells
Introduction
Cocaine addiction is a serious public health problem worldwide that affects normal brain function, leading to alterations in memory as well as behavioral and neuronal physiology. Although many approaches have been pursued to address cocaine addiction, cocaine remains one of the most abused illicit drugs worldwide, and there are currently no approved medications to treat cocaine addiction.
It is well known that cocaine increases intrasynaptic dopamine via its inhibition of dopamine transporters, producing a rewarding effect [1, 2]. However, cocaine also acts on other brain targets involved in reward, such as neuronal nicotinic acetylcholine receptors (nAChRs) [3–5]. Epidemiological studies suggest that smoking increases the intake of cocaine and that cocaine use, in turn, increases cigarette consumption [6]. However, little is known about the mechanisms that would support such interactions. In laboratory animals, stimulation of nAChRs by nicotine increases cocaine-induced locomotor sensitization [7], while blockade with a nonselective nAChR antagonist such as mecamylamine reduces cocaine reinforcement in rats [8]. Additionally, pretreatment with nicotine reduces cocaine-conditioned place preference (CPP) established in rats, while mecamylamine slightly attenuates cocaine-CPP in rats [9–11]. The roles of the α4β2 nAChRs in the modulation of cocaine self-administration are still controversial. It has been reported that α4β2 nicotinic receptor desensitizing compounds can decrease the self-administration of cocaine and methamphetamine in rats [12], while in the monkey cocaine self-administration model, varenicline, a partial agonist of α4β2 nAChRs and a full agonist of an α7 nAChRs, did not attenuate the reinforcing effects of cocaine [13].
Although the interactions between cocaine and nAChRs have been established in Xenopus oocytes expressing system [3, 4], the specific pharmacological effects of cocaine on native nAChR subtypes are still unclear. Behavioral experiments showed that local injection of a selective α4β2 nAChR antagonist (dihydro-β-erythroidine, DHβE) into the ventral tegmental area (VTA) prevents cocaine-induced locomotor activity [14], while the α7-selective antagonist methyllycaconitine (MLA) did not alter cocaine-induced behavioral sensitization [15]. Recently, nAChRs containing the α6 subunit (α6* nAChRs), but not those containing the α4 subunit, were shown to play a role in the induction of cocaine-conditioned reward [16], and we have recently reported that cocaine blocks human α6* nAChRs expressed in human SH-EP1 cells [17]. The α3β4 nAChR may also be an important target that mediates the effects of cocaine. Although most widely expressed in the peripheral nervous system, α3β4 nAChRs are also highly expressed in some brain regions such as the medial habenula, interpeduncular nucleus, and fasciculus retroflexus and moderately expressed in the VTA, dorsolateral tegmentum and basolateral amygdala [18–21]. Several studies have shown that α3β4 nAChRs play a role in drug-induced reward and reinforcement [22–26]. A recent study demonstrated that inhibition of α3β4 nAChRs by high-affinity α3β4-nAChR ligands attenuates cocaine-induced CPP and behavioral sensitization [27], suggesting that α3β4 nAChRs may participate in the rewarding effect of cocaine.
The role of α3β4 nAChRs in drug addiction behaviors is intriguing. However, little is known about the mechanisms that support the interactions between cocaine and α3β4 nAChRs. In the present study, we used patch-clamp whole-cell recording to evaluate the acute effects of cocaine on native α3β4 nAChRs expressed in SH-SY5Y cells, and we also explored the mechanisms underlying these pharmacological effects.
Materials and methods
Cell culture
Human SH-SY5Y neuroblastoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin and 2 mM glutamine. Previous studies demonstrated that native α3β4 nAChRs were stably expressed in SH-SY5Y cells. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and then plated in 35-mm dishes for patch-clamp recording.
Expression of human neuronal α4β2 nAChRs in SH-EP1 human epithelial cells
Heterologous expression of human α4β2 nAChRs has been previously described [28, 29]. Briefly, human nAChR α4 and β2 subunits, subcloned into pcDNA3.1-zeocin or hygromycin vectors, respectively, were introduced into native nAChR-null SH-EP1 cells [30] to create the SH-EP1-hα4β2 cell line. Cells were maintained as low-passage-number cultures in a medium augmented with 0.5 mg/mL zeocin and 0.4 mg/mL hygromycin and were passaged once per week by splitting cultures 1/10 just as they reached confluence to maintain the cells in a state of proliferative growth.
Expression of human neuronal α6/α3β2β3 nAChRs in human SH-EP1 cells
The human epithelial cell line expressing α6/α3 chimera β2β3 nAChRs (α6N/α3Cβ2β3 nAChRs) was developed by Dr. Whiteaker’s laboratory [31], and its pharmacological response properties have been profiled recently [32]. α6/α3 denotes a chimeric subunit composed of the extracellular, ligand-binding domain of the human α6 subunit fused to the first transmembrane domain and followed by the sequence of the human α3 nAChR subunit; this approach reproducibly increases expression compared to that of native α6 subunits while retaining α6-like pharmacology. Consensus-sequence β2 and β3 human nAChR subunit clones were also used. Wild-type SH-EP1 cells were transfected with nAChR subunit clones using the cationic polymer Effectene (Qiagen, Valencia, CA, USA). These cells were maintained in DMEM (Invitrogen) with 10% horse serum (Invitrogen), 5% fetal bovine serum (HyClone, GE Healthcare Life Sciences, Pittsburgh, PA, USA), 1 mM sodium pyruvate, and 4 mM L-glutamine, supplemented with 0.25 mg/mL zeocin, 0.13 mg/mL hygromycin B, and G418 at 0.6 mg/mL, ensuring positive selection of transfectants. Low-passage-number (1−40 from our frozen stocks) cultures were used to ensure phenotypic stability. Cells were typically passaged once per week by splitting just-confluent cultures 1:20−1:40 to maintain cells in proliferative growth.
Patch-clamp whole-cell current recordings and data acquisition
Standard whole-cell current recordings were performed as previously described, using an application system that enables fast application and removal of drugs [33, 34]. Briefly, SH-SY5Y cells and two types of transfected SH-EP1 cells were prepared in 35-mm culture dishes without polylysine coating, plated on the bottoms of the dishes, and later placed on the stage of an inverted microscope (Axiovert 200; Zeiss, Germany). Cells were perfused with standard external solution (2 mL/min) and bubbled with 95% O2/5% CO2. Glass microelectrodes with 3–5 MΩ resistance between the pipette and extracellular solution were used to form tight seals (>1 GΩ) on the surface of the cells, and standard whole-cell current recording was initiated by applying suitable suction and then waiting 5 to 10 min for the exchange of the pipette solution and the cytosol. Subsequently, recorded cells were lifted up gently from the bottoms of the culture dishes, which improved both solution exchange and accurate evaluation of the differences in the kinetics of agonist-induced whole-cell currents. Before capacitance and resistance compensation, access resistance (Ra) was measured; experiments with Ra values of less than 20 MΩ were accepted. Pipette and whole-current capacitance were both minimized, and series resistance was compensated routinely to 80%. The recorded cells were voltage clamped at a holding potential of −60 mV in SH-SY5Y cells and −40 mV in two types of transfected SH-EP1 cells. Inward currents induced by nicotine were measured (200B amplifier; Molecular Devices, Sunnyvale, CA, USA). Current signals were typically filtered at 2 kHz, acquired at 5 kHz, and displayed and digitized online (Digidata 1440 A series A/D board; Molecular Devices, Sunnyvale, CA, USA). Data acquisition and analyses were performed using pClamp10.0 (Molecular Devices), and results were plotted using Prism 5.0 software (GraphPad Software, Inc., CA, USA). All experiments were performed at room temperature (22 ± 1 °C).
Solutions and drug application
The standard external solution for SH-SY5Y and SH-EP1 cells contained (in mM) 120 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 25 D-glucose, and 10 HEPES and had a pH of 7.4, adjusted with Tris base. The pipette solution for SH-SY5Y cells contained (in mM) 140 K-gluconate, 10 HEPES, 0.2 EGTA, 5 KCl, 2 MgCl2, 4 Na2ATP, 0.3 Na3GTP, and Na2-phosphocreatine and had a pH of 7.3. For traditional whole-cell recording in SH-EP1 cells, the pipette solution contained (in mM) 110 dibasic Tris phosphate, 28 Tris base, 11 EGTA, 2 MgCl2, 0.5 CaCl2, and 4 Na-ATP and had a pH of 7.3. In the experiments, nicotine was used as the test agonist and induced whole-cell current responses. Nicotine was quickly applied to recorded cells using a computer-controlled U-tube drug perfusion system and could fully surround the recorded cells within 30 ms, as in our previous reports [33–35]. The interval between drug applications (2 min) was optimized specifically to ensure the stability of nAChR responsiveness (e.g., no functional rundown).
Homology modeling and ligand docking
The amino acid sequences of the human α3 (NP_000734) and β4 (NP_000741) nAChR subunits and the crystal structure of the human α4β2 nAChR (PDBID: 5kxi; Chain A: α4; Chain B: β2; Chain C: β2; Chain D: α4; Chain E: β2) were downloaded from the NCBI website. Using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/ clustalo/), the α3 sequence was aligned with the α4 sequence (Chain A) from the crystal structure, and the β4 sequence was aligned with the β2 sequence (Chain B) from the crystal structure. The unaligned regions of the α3 and β4 subunits (mainly the signal peptide and the M3-M4 intracellular loop) were removed. The homology models of all five chains were built with ICM Pro 1.7 (MolSoft, San Diego, CA, USA; Chain A: α3; Chain B: β4; Chain C: β4; Chain D: α3; Chain E: β4). Five model chains were combined to form a pentameric structure. The receptor was further converted to an ICM object, and energy was minimized using MolMechanics function in ICM Pro. The cocaine molecule was built from the crystal structure, with a PDBID of 2pgz. The docking was performed using ICM Pro with a docking space manually adjusted to cover the transmembrane domain of the receptor. The top-scored pose was used for presentation using Discovery Studio 4.0 Visualizer (BIOVIA, San Diego, CA, USA).
Data analysis and statistics
To analyze the effects of cocaine on the nAChR whole-cell current responses, we measured the peaks of inward currents. Data are presented as the means ± standard errors. Statistical analysis was performed using paired t-tests when evaluating data obtained from a single cell or Student’s t-test (unpaired values) or one-way ANOVA followed by Student-Newman-Keuls test when comparing data obtained from different cells; all these tests were conducted using GraphPad 5.0 software. P values less than 0.05 were considered significant. Curve fitting for agonist and antagonist concentration-response data were performed by GraphPad 5.0 software (GraphPad Software, Inc., CA, USA) using the logistic equation to provide fits for maximal and minimal responses, EC50 or IC50 values, and Hill coefficients.
Results
Nicotine produces a concentration-response curve in native α3β4 nAChRs in SH-SY5Y cells, and cocaine inhibits α3β4 nAChR-mediated currents in a concentration-dependent manner
Patch-clamp whole-cell current recording in voltage-clamp mode was performed, and bath application of nicotine induced an inward current at a holding potential (VH) of −60 mV. In order to obtain the concentration-response relationship of nicotine-induced currents, whole-cell currents induced by different concentrations of nicotine were measured from the same recorded cell (Fig. 1a). From the 6 cells tested, normalized (to the response to 1000 μM nicotine) whole-cell current peak amplitudes in response to nicotine (Fig. 1b), when plotted as a function of agonist concentration, were sigmoid and well fitted by the logistic equation for a single-site model. Fits to the data yielded an EC50 value and a Hill coefficient of 20 µM and 1.02 for nicotine, demonstrating that nicotine acts as an agonist of α3β4 nAChRs in SH-SY5Y cells. Then, we determined whether cocaine affected α3β4 nAChR function in SH-SY5Y cells using patch-clamp whole-cell recording combined with fast drug application by a U-tube apparatus. At a VH of −60 mV, bath application of 20 µM nicotine (nicotine EC50 concentration for α3β4 nAChRs) induced an inward current, and the application of 20 µM nicotine after 30 s pretreatment with different concentrations (0.1–30 µM) of cocaine reduced α3β4 nAChR-mediated currents (Fig. 1c). According to the fitting results from the concentration-response curves (Fig. 1d), the IC50 value and Hill coefficient for cocaine-mediated suppression of whole-cell peak currents were 1.56 µM and 1.04, respectively.
Fig. 1.

Effects of cocaine on the nicotine-induced currents in native α3β4 nAChRs expressed in SH-SY5Y cells. a Under voltage-clamp conditions, α3β4-nAChR-mediated whole-cell currents recorded at a VH of −60 mV were induced by different concentrations of nicotine. All typical traces in this and the following figures were collected as single traces (not averaged trace). b Nicotine concentration-response curves plotted for normalized peak current induced by 1000 µM nicotine, indicated by *. c Under voltage-clamp conditions, α3β4 nAChR-mediated whole-cell currents recorded at a VH of −60 mV were induced by 20 µM nicotine alone (indicated by open horizontal bars above the traces) and 20 µM nicotine plus 30 s pretreatment with 1, 3 or 30 µM cocaine (indicated by open horizontal bars above the traces). d Concentration-dependent inhibition of peak nicotinic responses by cocaine at the indicated concentrations. Each point represents the average response from eight cells. Data in (b) and (d) are presented as the mean ± SEM
Effects of cocaine on α3β4 nAChR-mediated currents induced by different nicotine concentrations
In order determine the mechanisms (competitive or noncompetitive) of how cocaine inhibits nicotine-induced responses, the nicotine concentration-response relationship curves in the presence and absence of 1.5 µM cocaine were compared. As shown in Fig. 2a, nicotine-induced inward currents increased with increasing nicotine concentrations, forming a sigmoid-shaped curve (open circle symbols, Fig. 2c). In the presence of 1.5 µM cocaine, nicotine-induced currents were reduced compared to those for nicotine exposure alone (solid circle symbols, Fig. 2b). Unpaired t-test analysis for each nicotine concentration pair with 1.5 µM cocaine showed highly significant differences between nicotine alone and nicotine plus cocaine at all tested nicotine concentrations, including 10 (inhibitory rate = 40.8% ± 12.6%), 100 (inhibitory rate = 40.8% ± 7.7%) and 1000 µM (inhibitory rate = 35.8% ± 4.2%, Fig. 2c, black circle symbols). The inhibitory rates of these different concentrations of cocaine against nicotine-induced currents were not significantly different (one-way ANOVA, F(2, 15) = 0.09, P = 0.91). For normalized nicotine concentration-response curves with and without cocaine, the EC50 value was not significantly different between the nicotine group and the nicotine plus cocaine group (Fig. 2d). In the presence of nicotine alone or nicotine plus 1.5 µM cocaine, the EC50 values of the groups were 21.55 ± 4.39 µM and 22.79 ± 6.03 µM, respectively, which were not significantly different (unpaired t-test, t(16) = 0.165, P = 0.87). Collectively, the data show that cocaine reduces the maximal response of the nicotine concentration-response curve (Fig. 2c) but does not change the nicotine EC50 value (Fig. 2d), suggesting noncompetitive inhibition.
Fig. 2.

Effects of cocaine on nicotine-induced currents mediated by α3β4 nAChRs in SH-SY5Y cells. Typical traces of responses to the indicated concentrations of nicotine in the absence of cocaine (a, indicated by horizontal black bar) or with 30 s cocaine pretreatment (b, indicated by open horizontal bar). The traces from (a) and (b) were recorded from the same cell. c Statistical comparison of the inhibitory rate of cocaine against nicotine-induced current. All symbols were normalized to the peak amplitude of 1000 µM nicotine-induced current, to form the concentration-response curves for responses to the indicated concentrations of nicotine either alone (open symbols) or coapplied with 1.5 µM cocaine (filled symbols). The data are represented as the mean ± SEM. d Bar graph shows that cocaine equally inhibited different concentrations of nicotine-induced inward currents (**P < 0.01)
Kinetic analysis of cocaine-induced inhibition in α3β4 nAChR-mediated currents
In further tests, we examined kinetic changes in α3β4 nAChR-mediated whole-cell current after acute exposure to 1.5 µM cocaine. As shown in Fig. 3, cocaine did not alter the rising slope of the 20 µM nicotine-induced whole-cell current (Fig. 3a, b) but increased the decay rate (Fig. 3a, right traces). Statistical analysis demonstrated that after 1.5 µM cocaine exposure, α3β4 nAChR channel activation did not alter the rising slope of the current (from −0.60 ± 0.10 to −0.58 ± 0.12 pA/ms, paired t-test, P > 0.05, n = 8, Fig. 3b), but channel inactivation was accelerated, as measured by the current decay constant (tau values from 628.4 ± 135.3 to 161.6 ± 22.1 ms, paired t-test, P < 0.05, n = 8, Fig. 3c). These results suggest that acute cocaine application inhibits nicotine-induced currents by accelerating channel inactivation.
Fig. 3.

Kinetic analysis of cocaine-induced inhibition of α3β4 nAChR-mediated currents. a Typical traces of nicotine-induced whole-cell currents before (i, black) and after (ii, red) cocaine exposure. ai + ii shows a comparison of current rising and decay kinetics after superimposition of ai and aii with similar peak amplitudes. b Single exponential fitting of the whole-cell current rising time during nicotine exposure before (black) and after (red) cocaine exposure and showed no significant difference after cocaine exposure. c Cocaine significantly accelerated the whole-cell current decay constant. *P < 0.05 (baseline vs. cocaine exposure)
Use dependence of cocaine suppression of α3β4 nAChR-mediated currents
It has been reported that cocaine inhibits nAChRs through an open-channel block mechanism. Under patch-clamp recording conditions, one feature of open-channel block is use dependence. Thus, we examined whether cocaine-induced inhibition in α3β4 nAChR-mediated current is use-dependent. To exclude the phenomenon of nAChR desensitization caused by nicotine itself, we applied 5 doses of nicotine alone for 1 s each at 30 s intervals, and we found that the second nicotine response was reduced (82.7% ± 5.2%) compared to the first one; however, the difference was not statistically significant (paired t-test, t(7) = 1.92, P = 0.09), and subsequent nicotine responses were stabilized at this level without further rundown (Fig. 4a top trace and 4d red curve). Under the same conditions, repetitive applications of nicotine (20 µM) in the continuous presence of 1.5 µM cocaine led to gradual decreases in responses induced by nicotine (Fig. 4b). In a variant of the same protocol, 1.5 µM cocaine was applied only for the 1st and 5th traces of nicotine application, whereas the standard extracellular solution took the place of the cocaine solution during the second, third and fourth traces. With this protocol, the recorded cell was exposed only two times (first and fifth) of nicotine (Fig. 4c), assuming nAChRs were less frequently used compared to five doses of nicotine. Fig. 4d summarizes the nicotine current amplitude changes of the control, 5-exposure and 2-exposure protocols. Comparison of the amplitude ratios of the fifth and first current traces between these two protocols demonstrated that in the presence of 1.5 µM cocaine, the ratio was 61.2% ± 8.8% after repeated challenges to 5 nicotine exposures, while the ratio was 96.7% ± 13.7% after two nicotine exposures (unpaired t-test, t(15) = 6.42, P < 0.05, Fig. 4e). These results indicate that cocaine-mediated suppression of α3β4-nAChR function was use-dependent.
Fig. 4.

Cocaine use-dependently inhibits nicotine response. SH-SY5Y α3β4 nAChRs cell responses when repeatedly exposed to 20 µM nicotine with 1 s exposure at 30 s intervals, indicated by black down arrows (a); when 20 µM nicotine was applied in the presence of 1.5 µM cocaine, indicated by an open horizontal bar (b); or when nicotine and 1.5 µM cocaine were applied for 1 s only at the first and fifth traces, where the gray down arrows indicate application of standard extracellular solution (c). d Line graph comparing the effects of cocaine during five applications of nicotine alone (red line), five applications of nicotine plus cocaine (black line), and two applications of nicotine plus cocaine (blue line). e Bar graph (n = 8) comparing the ratio of the fifth trace to the first trace during repetitive exposure to nicotine alone, nicotine plus five applications cocaine or nicotine plus two applications of cocaine (the first and fifth exposures were nicotine plus cocaine). *P < 0.05 for the difference in effects assessed under the two different nicotine challenge protocols
Voltage dependence of cocaine inhibition in α3β4 nAChR-mediated currents
Another feature of open-channel block is voltage dependence. Thus, we examined whether cocaine showed different inhibitory rates at two different holding potentials. The effects of changes in the holding potential (VH) on cocaine inhibition were tested in the α3β4 nAChRs. As shown in Fig. 5a, the application of 1.5 µM cocaine at a standard VH of −60 mV resulted in approximately 50% inhibition of the peak inward current induced by nicotine. The application of the same concentration of cocaine at a VH of −20 mV decreased the observed inhibition to approximately 5% of the control value (Fig. 5b). Statistical analysis demonstrated that after 1.5 µM cocaine exposure at a VH of −60 mV, the responses to nicotine were 55.7% ± 10.9% of the responses induced by nicotine alone (paired t-test, t(7) = 2.81, P = 0.02, n = 8). When the VH was raised to −20 mV, the responses to nicotine after 1.5 µM cocaine exposure were 92.5% ± 21.1% of the responses induced by nicotine alone (paired t-test, t(7) = 0.35, P = 0.74, n = 8). The responses to nicotine at VH of −20 mV were 35.9% ± 8.5% (n = 8) of the responses induced by nicotine alone at VH of −60 mV (Fig. 5c). These results demonstrated the voltage dependence of the inhibition of α3β4 nAChRs by cocaine.
Fig. 5.
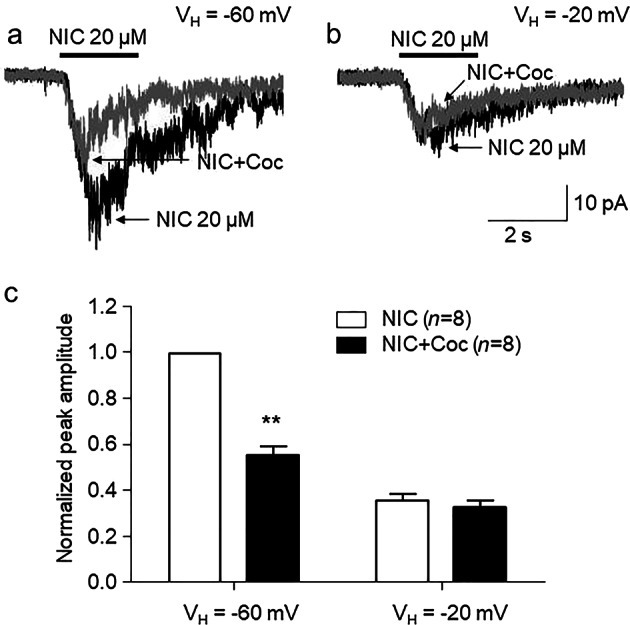
Voltage dependence of cocaine inhibition in α3β4 nAChR-mediated current a. Typical traces show the whole-cell current responses of SH-SY5Y α3β4 cells to 20 µM nicotine alone (black trace) or coapplied with 1.5 µM cocaine (gray trace) at the indicated VH. b Bar graph comparing the effects of cocaine during exposure to nicotine at the indicated VH (n = 8), ***P < 0.001 (baseline vs. cocaine exposure at a VH of −60 mV)
Cocaine inhibition of the α3β4 nAChR-mediated current did not occur via an intracellular mechanism
In order to test the hypothesis that cocaine inhibition of α3β4 nAChR-mediated current is mediated via intracellular mechanisms, cocaine (30 µM) was added to the pipette solution and applied to cells intracellularly throughout whole-cell recordings. Under these conditions, bath application of 20 µM nicotine induced stable responses. After conversion to whole-cell recording mode followed by a waiting period of more than 20 min, which allowed full loading of cocaine into the cell, there were no declines in responses to nicotine. However, these responses decreased when nicotine and cocaine (1.5 µM) were coapplied extracellularly to the same recorded cell. In these same recorded cells, bath application of cocaine (30 µM) completely blocked nicotine-induced inward currents (Fig. 6a). Statistical analysis also demonstrated that after intracellular application of 30 µM cocaine, bath application of 1.5 µM cocaine also inhibited nicotine-induced responses by 43.52% ± 10.48% (paired t-test, t(6) = 3.49, P = 0.01; Fig. 6b). These results suggest that intracellular mechanisms are not involved in cocaine modulation of α3β4 nAChR function.
Fig. 6.
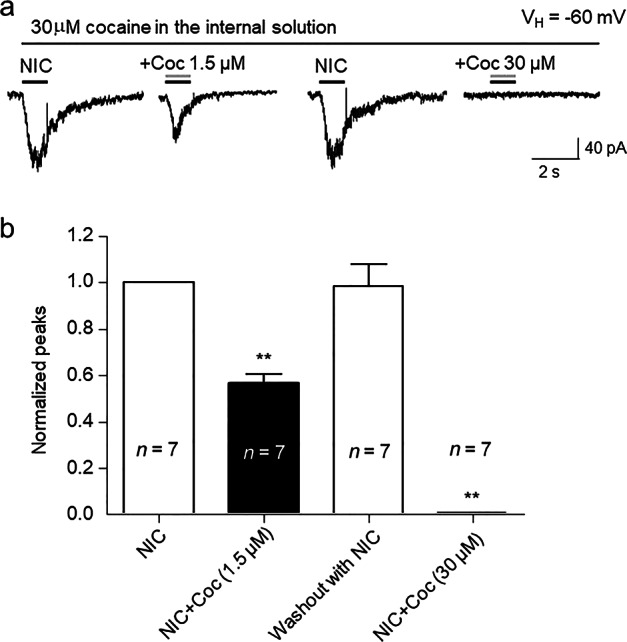
Intracellular application of cocaine did not reduce α3β4-nAChR function. a After the establishment of whole-cell patch-clamp recording, 30 µM cocaine was perfused into the recorded cell over 20 min, while whole-cell inward currents mediated by α3β4 nAChRs to 20 µM nicotine were recorded in the absence or the presence of cocaine coapplied with nicotine extracellularly through the perfusion system. b Bar graph summarizing the effects of cocaine supplied under the conditions specified in a on peak current responses (normalized to the response to 20 µM nicotine alone), **P < 0.01 (baseline vs. cocaine 1.5 µM exposure and nicotine washout vs. cocaine 30 µM exposure)
Effects of receptor internalization on cocaine-induced inhibition of α3β4-nAChR function
Since endocytosis or exocytosis of several transmembrane receptors occurs on the order of seconds to minutes through processes modulated by agonists and antagonists, we evaluated the effects of cocaine in SH-SY5Y cells preloaded with 600 µM GDP-βS (added to the pipette solution) for 20 min following conversion to whole-cell recording configuration, which has been reported to prevent AMPA receptor internalization [36]. Preloading cells with GDP-βS for 20 min had no effect on nicotine-induced currents or on cocaine-induced suppression of α3β4 nAChR function (Fig. 7). Statistical analysis showed that acute cocaine (1.5 µM) application still significantly inhibited nicotine-induced currents after intracellular application of 600 µM GDP-βS (one-way ANOVA followed by the Student-Newman-Keuls test, F(2, 26) = 4.79, P = 0.016). These results support the hypothesis that the modulatory effect of cocaine on α3β4 nAChRs does not occur via receptor internalization.
Fig. 7.
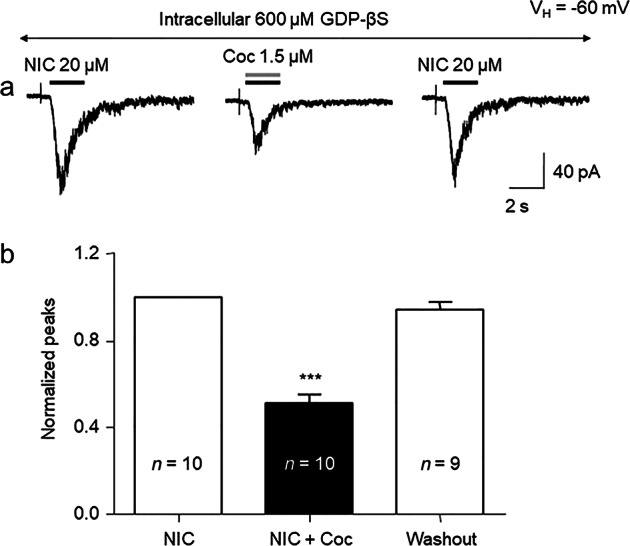
Intracellular application of GDP-βS does not affect cocaine-mediated inhibition of α3β4-nAChR function. a Typical whole-cell inward current responses to 20 µM nicotine alone (horizontal black bar) or under the condition of nicotine plus 1.5 µM cocaine coapplication (horizontal open bar) were recorded after 600 µM GDP-βS was loaded into the recorded cell during the 20 min after conversion to the whole-cell recording mode. b Bar graph summarizing the effects of 1.5 µM cocaine on responses to 20 µM nicotine (peak currents normalized to those induced by 20 µM nicotine alone), ***P < 0.001 (baseline vs. cocaine exposure)
Effects of cocaine on α4β2-containing and α6-containing nAChRs
The effects of 1.5 µM cocaine on the function of native α3β4 nAChRs in SH-SY5Y cells and on α4β2 or α6/α3β2β3 nAChRs heterologously expressed in SH-EP1 cells were evaluated. The EC50 concentrations of nicotine for α6/α3β2β3 (1 µM) [32], α4β2 (3 µM) [37], and α3β4 (20 µM, see Fig. 1) were used. As shown in Fig. 8a, at a VH of −40 mV (approximately the resting potential of SH-EP1 cells), 1.5 µM cocaine was unable to inhibit nicotine-induced inward currents mediated by α6/α3β2β3 (Fig. 8ai) or α4β2 (Fig. 8aii) nAChRs in SH-EP1 cells. However, the same concentration of cocaine blocked approximately 50% of nicotine-induced inward current mediated by α3β4 nAChRs in SH-SY5Y cells (Fig. 8aiii) at a VH of −60 mV. Statistical analysis also demonstrated that 1.5 µM cocaine significantly inhibited nicotine-induced inward currents mediated by α3β4 nAChRs in SH-SY5Y cells (paired t-test, t(7) = 3.16, P = 0.02) but had no effect on α6 (paired t-test, t(19) = 0.12, P = 0.91) and α4β2 nAChRs (paired t-test, t(7) = 0.42, P = 0.68) in SH-EP1 cells (Fig. 8b).
Fig. 8.
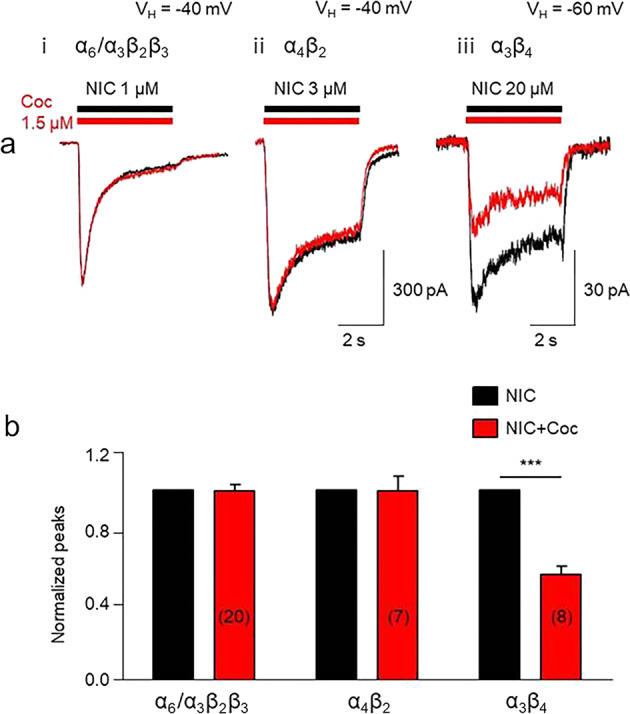
Effects of cocaine on different nAChR subtypes. a Typical whole-cell inward current responses of α6/α3β2β3 nAChRs (i) to 1 µM nicotine (EC50 concentration), α4β2 nAChRs (ii) to 3 µM nicotine (EC50 concentration), or α3β4 nAChRs (iii) to 20 µM nicotine (EC50 concentration) applied alone (horizontal black bar) or coapplied with 1.5 µM cocaine (horizontal red bar). b The bar graph indicates that 1.5 µM cocaine selectively inhibited α3β4 nAChR-mediated currents (normalized peak current responses to the challenge agonist alone), ***P < 0.001 (baseline vs. cocaine exposure for the α3β4 nAChRs
The cocaine molecule docks with the channel domain of the homology model of the human α3β4 nAChR with subunit stoichiometry of (α3)2(β4)3
To explore the possible binding site of cocaine on α3β4 nAChRs, we performed homology modeling of human α3β4 nAChR with the subunit stoichiometry of (α3)2(β4)3 using the crystal structure of human α4β2 as the template and ligand docking to the transmembrane channel domain docked to the channel domain of the α3β4 homology model. As shown in Fig. 9, the highest-scored position of the docked cocaine is located in the center of the channel pore, below the channel gate but above the channel selectivity filter, and completely occludes the ion-conducting pore. The location of the docked cocaine in the pore domain is very similar to the location of the GABAA receptor channel blocker picrotoxin in the pore [38]. Thus, our docking result is consistent with a channel-blocking effect of cocaine on α3β4 nAChRs.
Fig. 9.
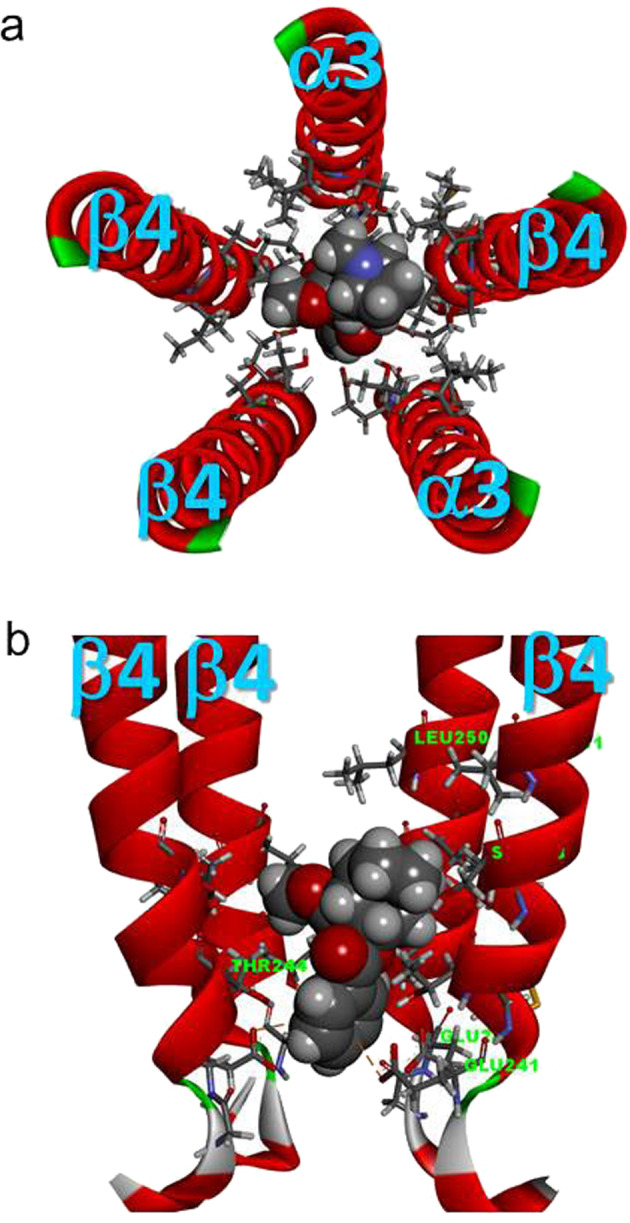
Cocaine molecule docked to the channel domain of the homology model of the human α3β4 nAChR with the subunit stoichiometry of (α3)2(β4)3. a Top view (from the extracellular side) of the channel-lining domain (with only M2 domains for clarity). The docked cocaine is located in the center of the pore and has a potential hydrogen bond with α3Ser247 (Chain A). b Side view of the channel-lining domain, with one α3 subunit (Chain D) removed from the front for clarity. The docked cocaine was located immediately above the selectivity filter (β4Glu241) up to the level at the 6′ position (α3Ser247, β4Ser248) of the second transmembrane domain (M2) with two potential π-anion interactions with two β4Glu241 residues of Chains C and D. The docked cocaine molecule was below the putative channel gate at the 9′ position (α3Leu250, β4Leu251)
Discussion
In the present study, we found that cocaine potently blocks α3β4-nAChR function. Using natively expressed α3β4 nAChRs in SH-SY5Y cells, we demonstrated that bath application of cocaine inhibits α3β4 nAChR-mediated whole-cell currents in a concentration-dependent manner with an IC50 value of 1.5 µM. Kinetic analysis showed that cocaine accelerated α3β4 nAChR desensitization, which caused a reduction in the amplitude of nicotine-induced currents. Cocaine inhibited α3β4 nAChRs in a noncompetitive manner. Cocaine inhibition of α3β4 nAChRs was also use-dependent and voltage-dependent. In addition, cocaine-induced inhibition is mediated neither via intracellular sites nor through changes in receptor internalization. Finally, potent cocaine inhibition occurs specifically in α3β4 nAChRs but not in α4β2- or α7 nAChRs. These studies provide experimental evidence that cocaine directly inhibits native α3β4 nAChR function.
Cocaine inhibits native α3β4-nAChR function in SH-S5Y5 cells
Previous research found that cocaine inhibits heterologously expressed nAChR function in Xenopus oocytes. For example, Damaj et al. reported that cocaine inhibits heterologously expressed nAChR-mediated currents in a concentration-dependent manner with an IC50 range of 4.4–6.9 µM for α4β2 nAChRs and 22–42.3 µM for α3β2 nAChRs [3]. They also found that the cocaine-induced inhibition of nAChRs functions through a noncompetitive mechanism. Francis et al. systematically compared cocaine-induced inhibition in different subtypes of nAChRs expressed in oocytes and found that α4β4 nAChRs exhibited the highest sensitivity, with an IC50 of 2 µM, while α3β2 showed the lowest sensitivity, with an IC50 of 60 µM. They also found that cocaine inhibits α3β4 nAChRs with an IC50 of 6 µM [4]. By using transfected α3β4 nAChRs in HEK 293 cells, Krivoshein et al. compared the possible mechanisms of cocaine- and MK801-induced inhibition of α3β4 nAChRs and found that both tested compounds exhibited noncompetitive inhibition under closed and open-channel conditions [39]. In addition to these expressed nAChRs, Acevedo-Rodriguez et al. found that cocaine also inhibits native (β2-containing) nAChRs in midbrain dopaminergic neurons with an IC50 of 19.8 µM [5]. Collectively, these previous data suggest that cocaine serves as a low-potency antagonist of different subtypes of nAChRs. Thus far, however, whether cocaine modulates native α3β4-nAChR function is unknown. The present study provides experimental evidence that cocaine blocks native α3β4 nAChR function in a concentration-dependent manner with an IC50 value of 1.5 µM. This concentration is even lower than the IC50 for cocaine-induced inhibition in dopamine transporters, indicating that native α3β4 nAChR is highly sensitive to cocaine.
Mechanisms of cocaine-induced inhibition of native α3β4 nAChRs
Krivoshein et al. reported the mechanisms of cocaine-induced inhibition in transfected α3β4 nAChRs in HEK 293 cells [39]. We believe that the present study exhibits some differences from Dr. Krivoshein’s report. First, to explore the possible mechanisms of cocaine-induced inhibition in native α3β4 nAChRs, we compared nicotine concentration-response curves with or without 1.5 µM cocaine. In our nicotine concentration-response curve, 1.5 µM cocaine reduced the maximal nicotine concentration-induced current without changing the nicotine EC50 value, suggesting noncompetitive inhibition. This finding is consistent with the cocaine-induced inhibition observed in other subtypes of nAChRs [4]. Second, we demonstrated that cocaine-induced inhibition was dependent on use and voltage. This suggests that its suppression of nicotine-induced currents involves an agonist-induced transition of nAChRs to conformations having a higher affinity for the antagonist and allowing it free access to the binding site(s) [40]. It is reasonable to conclude that this transition is to an open-channel state. Third, through kinetic analysis, we found that cocaine accelerated α3β4 nAChR desensitization during nicotine exposure, again suggesting that it blocked open channels. Finally, we applied either cocaine or GDP-βS intracellularly through the recording electrode and found that these manipulations did not affect α3β4 nAChR function. Together, these findings suggest that cocaine has access to α3β4 nAChRs in the resting state but principally acts as a noncompetitive antagonist, with a presumed binding site within the channel pore.
To visualize the potential mechanism whereby cocaine blocks the channel, we performed applied homology modeling and simulated ligand docking to the transmembrane domain of α3β4 nAChR with the stoichiometry and arrangement of α3β4β4α3β4 (Fig. 9a, extracellular view counterclockwise). The resulting top-scored docking pose of cocaine was located in the center of the channel pore. The docked cocaine was located immediately above the selectivity filter (β4Glu241/α3Glu240) at the −1′ position, contacting residues at the 2′ (β4Thr244/α3Thr243), 5′ (β4Ile247/α3Ile246), and 6′ (β4Thr248/α3Thr247) positions (Fig. 9b). This site is consistent with the location of a typical channel blocker in the pore, below the channel gate and immediately above the narrowest part of an open-channel selectivity filter.
The potential clinical significance of cocaine’s suppression of α3β4-nAChR function
Cocaine inhibits natural nAChR-mediated currents in VTA DA neurons and consequently increases the ratio of phasic to tonic DA release, thus potentially enhancing its reinforcing effects [5]. On the other hand, blocking nAChRs may mediate cocaine craving and addiction [41, 42]. Although α3β4 nAChRs are most highly expressed in the medial habenula, interpeduncular nucleus and fasciculus retroflexus, they are expressed at low levels in the mesolimbic reward circuitry [43] and are involved in the rewarding effects of cocaine and possibly other psychostimulants [22–24, 44]. This idea is supported by recent studies showing that α3β4-selective nAChR ligands AT-1001 and AT-1012, which do not induce a place preference or aversion in mice per se, significantly and dose-dependently suppress the acquisition of CPP induced by a low dose of cocaine [27].
Cocaine addiction is a serious social, medical, and economic problem worldwide. Countless attempts have been made to find compounds that will displace abused drugs, such as cocaine, from their sites of action in the nervous system. Thus far, no satisfactory drugs exist for the treatment of cocaine reward and dependence. Therefore, the finding of cocaine-directed inhibition of native α3β4 nAChRs is highly significant for understanding the targets and mechanisms of cocaine and potential therapeutics for its abuse. Additionally, the high selectivity of cocaine for α3β4 nAChRs might contribute to the cardiovascular effects of cocaine. It is well known that the α3β4 subunit combination is prevalent in the nAChRs of autonomic ganglia neurons. nAChRs with the α3β4 combination are also called “ganglia-type nAChRs”. The inhibition of α3β4 nAChRs by a low dose of cocaine may result in the modulation of cholinergic pathways in the neurons of autonomic ganglia, thereby inducing the possible adverse cardiovascular effects of cocaine. Furthermore, recent studies showed that α3β4 nAChRs are predominantly expressed in human chromaffin cells, where they play a role in exocytosis [45]. This may be one of the mechanisms involved in the cardiovascular effects of cocaine. Therefore, the finding of cocaine-directed inhibition of native α3β4 nAChRs is highly significant for understanding the targets and mechanisms of cocaine and potential therapeutics for its abuse.
In conclusion, using patch-clamp whole-cell recordings in cultured SH-SY5Y cells, we demonstrate that cocaine potently inhibits α3β4 nAChR-mediated whole-cell currents with an IC50 value of 1.5 µM. Cocaine-induced inhibition of the α3β4 nAChR-mediated current occurs in a noncompetitive manner through an open-channel block mechanism. This inhibition is not mediated by the alteration of α3β4 nAChR internalization. In addition, the same concentration (1.5 µM) of cocaine fails to alter α4β2 and α6* nAChR function. Based on these experimental results, we conclude that cocaine is a potent blocker of native α3β4 nAChRs in SH-SY5Y cells.
Acknowledgements
This work was supported by grants from Barrow Neuroscience Foundation and NIDA (DA11064). Authors thank Kellen Vu for his assistance with editing the English for this manuscript.
Author contributions
ZGM, NJ, YBH, XKM, MG, YCC, performed experiments, collected and analyzed data. JBE: prepared cell cultures. RJL, PW, JN: designed experiments revised manuscript. JW: designed experiments, data analysis, made all figures, wrote and revised manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Jones SR, Garris PA, Wightman RM. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J Pharmacol Exp Ther. 1995;274:396–403. [PubMed] [Google Scholar]
- 2.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 3.Damaj MI, Slemmer JE, Carroll FI, Martin BR. Pharmacological characterization of nicotine’s interaction with cocaine and cocaine analogs. J Pharmacol Exp Ther. 1999;289:1229–36. [PubMed] [Google Scholar]
- 4.Francis MM, Vazquez RW, Papke RL, Oswald RE. Subtype-selective inhibition of neuronal nicotinic acetylcholine receptors by cocaine is determined by the alpha4 and beta4 subunits. Mol Pharmacol. 2000;58:109–19. doi: 10.1124/mol.58.1.109. [DOI] [PubMed] [Google Scholar]
- 5.Acevedo-Rodriguez A, Zhang L, Zhou F, Gong S, Gu H, De Biasi M, et al. Cocaine inhibition of nicotinic acetylcholine receptors influences dopamine release. Front Synaptic Neurosci. 2014;6:19. doi: 10.3389/fnsyn.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgins ST, Budney AJ, Hughes JR, Bickel WK, Lynn M, Mortensen A. Influence of cocaine use on cigarette smoking. JAMA. 1994;272:1724. doi: 10.1001/jama.1994.03520220018016. [DOI] [PubMed] [Google Scholar]
- 7.Schoffelmeer AN, De Vries TJ, Wardeh G, van de Ven HW, Vanderschuren LJ. Psychostimulant-induced behavioral sensitization depends on nicotinic receptor activation. J Neurosci. 2002;22:3269–76. doi: 10.1523/JNEUROSCI.22-08-03269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blokhina EA, Kashkin VA, Zvartau EE, Danysz W, Bespalov AY. Effects of nicotinic and NMDA receptor channel blockers on intravenous cocaine and nicotine self-administration in mice. Eur Neuropsychopharmacol. 2005;15:219–25. doi: 10.1016/j.euroneuro.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Levine A, Huang Y, Drisaldi B, Griffin EA, Jr., Pollak DD, Xu S, et al. Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine. Sci Transl Med. 2011;3:107ra09. doi: 10.1126/scitranslmed.3003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sershen H, Hashim A, Lajtha A. Differences between nicotine and cocaine-induced conditioned place preferences. Brain Res Bull. 2010;81:120–4. doi: 10.1016/j.brainresbull.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 11.Zachariou V, Caldarone BJ, Weathers-Lowin A, George TP, Elsworth JD, Roth RH, et al. Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharmacology. 2001;24:576–89. doi: 10.1016/S0893-133X(00)00224-4. [DOI] [PubMed] [Google Scholar]
- 12.Levin ED, Rezvani AH, Wells C, Slade S, Yenugonda VM, Liu Y, et al. alpha4beta2 Nicotinic receptor desensitizing compounds can decrease self-administration of cocaine and methamphetamine in rats. Eur J Pharmacol. 2019;845:1–7. [DOI] [PMC free article] [PubMed]
- 13.Gould RW, Czoty PW, Nader SH, Nader MA. Effects of varenicline on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J Pharmacol Exp Ther. 2011;339:678–86. doi: 10.1124/jpet.111.185538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Champtiaux N, Kalivas PW, Bardo MT. Contribution of dihydro-beta-erythroidine sensitive nicotinic acetylcholine receptors in the ventral tegmental area to cocaine-induced behavioral sensitization in rats. Behav Brain Res. 2006;168:120–6. doi: 10.1016/j.bbr.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Zanetti L, de Kerchove D’Exaerde A, Zanardi A, Changeux JP, Picciotto MR, Zoli M. Inhibition of both alpha7* and beta2* nicotinic acetylcholine receptors is necessary to prevent development of sensitization to cocaine-elicited increases in extracellular dopamine levels in the ventral striatum. Psychopharmacology. 2006;187:181–8. doi: 10.1007/s00213-006-0419-y. [DOI] [PubMed] [Google Scholar]
- 16.Sanjakdar SS, Maldoon PP, Marks MJ, Brunzell DH, Maskos U, McIntosh JM, et al. Differential roles of alpha6beta2* and alpha4beta2* neuronal nicotinic receptors in nicotine- and cocaine-conditioned reward in mice. Neuropsychopharmacology. 2015;40:350–60. doi: 10.1038/npp.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen D, Gao F, Ma X, Eaton JB, Huang Y, Gao M, et al. Cocaine directly inhibits alpha6-containing nicotinic acetylcholine receptors in human SH-EP1 cells and mouse VTA DA neurons. Front Pharmacol. 2019;10:72. doi: 10.3389/fphar.2019.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–63. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry DC, Xiao Y, Nguyen HN, Musachio JL, Davila-Garcia MI, Kellar KJ. Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82:468–81. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- 20.Quick MW, Ceballos RM, Kasten M, McIntosh JM, Lester RA. Alpha3beta4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology. 1999;38:769–83. doi: 10.1016/S0028-3908(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 21.Zhu PJ, Stewart RR, McIntosh JM, Weight FF. Activation of nicotinic acetylcholine receptors increases the frequency of spontaneous GABAergic IPSCs in rat basolateral amygdala neurons. J Neurophysiol. 2005;94:3081–91. doi: 10.1152/jn.00974.2004. [DOI] [PubMed] [Google Scholar]
- 22.Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, et al. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13:368–73. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4:e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saccone NL, Wang JC, Breslau N, Johnson EO, Hatsukami D, Saccone SF, et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res. 2009;69:6848–56. doi: 10.1158/0008-5472.CAN-09-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlaepfer IR, Hoft NR, Collins AC, Corley RP, Hewitt JK, Hopfer CJ, et al. The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol Psychiatry. 2008;63:1039–46. doi: 10.1016/j.biopsych.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khroyan TV, Yasuda D, Toll L, Polgar WE, Zaveri NT. High affinity alpha3beta4 nicotinic acetylcholine receptor ligands AT-1001 and AT-1012 attenuate cocaine-induced conditioned place preference and behavioral sensitization in mice. Biochem Pharmacol. 2015;97:531–41. doi: 10.1016/j.bcp.2015.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, et al. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–94. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004;279:37842–51. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- 30.Lukas RJ, Norman SA, Lucero L. Characterization of nicotinic acetylcholine receptors expressed by cells of the SH-SY5Y human neuroblastoma clonal line. Mol Cell Neurosci. 1993;4:1–12. doi: 10.1006/mcne.1993.1001. [DOI] [PubMed] [Google Scholar]
- 31.Breining SR, Melvin M, Bhatti BS, Byrd GD, Kiser MN, Hepler CD, et al. Structure-activity studies of 7-heteroaryl-3-azabicyclo[3.3.1]non-6-enes: a novel class of highly potent nicotinic receptor ligands. J Med Chem. 2012;55:9929–45. doi: 10.1021/jm3011299. [DOI] [PubMed] [Google Scholar]
- 32.Chen DJ, Gao FF, Ma XK, Shi GG, Huang YB, Su QX, et al. Pharmacological and functional comparisons of alpha6/alpha3beta2beta3-nAChRs and alpha4beta2-nAChRs heterologously expressed in the human epithelial SH-EP1 cell line. Acta Pharmacol Sin. 2018;39:1571–81. doi: 10.1038/aps.2017.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, George AA, Schroeder KM, Xu L, Marxer-Miller S, Lucero L, et al. Electrophysiological, pharmacological, and molecular evidence for alpha7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J Pharmacol Exp Ther. 2004;311:80–91. doi: 10.1124/jpet.104.070417. [DOI] [PubMed] [Google Scholar]
- 34.Zhao L, Kuo YP, George AA, Peng JH, Purandare MS, Schroeder KM, et al. Functional properties of homomeric, human alpha 7-nicotinic acetylcholine receptors heterologously expressed in the SH-EP1 human epithelial cell line. J Pharmacol Exp Ther. 2003;305:1132–41. doi: 10.1124/jpet.103.048777. [DOI] [PubMed] [Google Scholar]
- 35.Yang K, Hu J, Lucero L, Liu Q, Zheng C, Zhen X, et al. Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol. 2009;587(Pt 2):345–61. doi: 10.1113/jphysiol.2008.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–58. doi: 10.1016/S0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, et al. Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4-containing nicotinic receptors. J Physiol. 2006;576(Pt 1):103–18. doi: 10.1113/jphysiol.2006.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Durkin KA, Casida JE. Structural model for gamma-aminobutyric acid receptor noncompetitive antagonist binding: widely diverse structures fit the same site. Proc Natl Acad Sci U S A. 2006;103:5185–90. doi: 10.1073/pnas.0600370103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krivoshein AV, Hess GP. Mechanism-based approach to the successful prevention of cocaine inhibition of the neuronal (alpha 3 beta 4) nicotinic acetylcholine receptor. Biochemistry. 2004;43:481–9. doi: 10.1021/bi034838l. [DOI] [PubMed] [Google Scholar]
- 40.Zhao X, Salgado VL, Yeh JZ, Narahashi T. Kinetic and pharmacological characterization of desensitizing and non-desensitizing glutamate-gated chloride channels in cockroach neurons. Neurotoxicology. 2004;25:967–80. doi: 10.1016/j.neuro.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Crunelle CL, Miller ML, Booij J, van den Brink W. The nicotinic acetylcholine receptor partial agonist varenicline and the treatment of drug dependence: a review. Eur Neuropsychopharmacol. 2010;20:69–79. doi: 10.1016/j.euroneuro.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Williams MJ, Adinoff B. The role of acetylcholine in cocaine addiction. Neuropsychopharmacology. 2008;33:1779–97. doi: 10.1038/sj.npp.1301585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, et al. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–11. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 44.Costello MR, Reynaga DD, Mojica CY, Zaveri NT, Belluzzi JD, Leslie FM. Comparison of the reinforcing properties of nicotine and cigarette smoke extract in rats. Neuropsychopharmacology. 2014;39:1843–51. doi: 10.1038/npp.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albillos A, McIntosh JM. Human nicotinic receptors in chromaffin cells: characterization and pharmacology. Pflug Arch. 2018;470:21–27. doi: 10.1007/s00424-017-2073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]