

Abstract
Patients with primary biliary cholangitis (PBC) with incomplete response to ursodeoxycholic acid are at risk of disease progression and need additional therapy. Obeticholic acid (OCA) was approved in Canada in May 2017, but its effectiveness in a real‐world setting has not been described. We sought to describe our experience with OCA in a Canadian cohort. OCA‐naive patients treated at two Canadian centers were included. Clinical and biochemical data were collected at OCA initiation and during follow‐up. Primary outcomes were changes in serum alkaline phosphatase (ALP), gamma‐glutamyl transferase (GGT), and total bilirubin (TB) over the duration of therapy. Secondary outcomes were changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), immunoglobulin M (IgM), platelets, and albumin; and achievement of the primary endpoint of the original phase 3 study that led to OCA approval (A Placebo‐Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis [POISE]), dose reductions, discontinuations, and tolerability. Repeated‐measures models were used to assess changes in biochemistry over time. Sixty‐four patients were included; 4 carried a diagnosis of overlap with autoimmune hepatitis. Mean age was 54.6 years, median ALP was 250 U/L, TB was 13 µmol/L, platelet count was 225 × 109/L, and 24% had liver stiffness measurements ≥16.9 kPa. There was a significant reduction in mean ALP of 55 U/L (P < 0.001), GGT of 138 U/L (P < 0.001), ALT of 11.9 U/L (P < 0.001), AST of 5.7 U/L (P < 0.05), and IgM of 0.70 g/L (P < 0.001) over 12 months; TB remained stable (P = 0.98). Forty‐four patients met POISE‐inclusion criteria, 39% (n = 17) of whom had 12‐month biochemical measurements. In this subset, 18% (n = 3/17) met the 12‐month POISE primary endpoint, but considering follow‐up to 19 months, 43% achieved this target (n = 9/21). Pruritus was the most commonly reported complaint. Conclusion: Use of OCA was associated with improvement in biochemical surrogates of outcome in PBC in a real‐world setting.
Patients with Primary Biliary Cholangitis with incomplete response to ursodeoxycholic acid are at risk of disease progression and need additional therapy. Obeticholic acid (OCA) was approved for use in Canada in May 2017, but its effectiveness in a real‐world setting has not been described. Herein we demonstrate that use of OCA was associated with improvement in biochemical surrogates of outcome in PBC in a real‐world setting.

Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CI
confidence interval
- GGT
gamma‐glutamyl transferase
- IgM
immunoglobulin M
- IQR
interquartile range
- LSM
liver stiffness measurement
- OCA
obeticholic acid
- PBC
primary biliary cholangitis
- POISE
A Placebo‐Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis
- TB
total bilirubin
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary biliary cholangitis (PBC) is an uncommon chronic cholestatic liver disease with an estimated prevalence of 318 per million people in Canada.( 1 ) The disease is characterized by progressive destruction of interlobular bile ducts and is clinically impactful with an increased risk of progression to advanced biliary fibrosis, end‐stage liver disease, and death. Until recently, ursodeoxycholic acid (UDCA) was the only approved therapy widely accepted to improve clinical outcomes in patients with PBC( 2 , 3 ); however, approximately 40% of patients have incomplete biochemical response to UDCA and remain at risk of disease progression.( 4 , 5 )
Alkaline phosphatase (ALP) and total bilirubin (TB) are validated biochemical surrogates of outcome and have been accepted as reasonably likely to predict outcome in patients with PBC. A large international cohort of patients with PBC demonstrated that ALP values greater than 1.5 times the upper limit of normal (ULN) were independently associated with a 2.0‐fold to 2.5‐fold increased risk of liver transplantation or death when compared to normal ALP values and that TB values greater than the ULN were associated with a 5.1‐fold to 10.7‐fold increased risk of liver transplantation or death when compared to normal serum TB values.( 6 )
Obeticholic acid (OCA) is a semisynthetic hydrophobic bile acid analogue that is a highly selective agonist for the farnesoid X receptor.( 7 ) The efficacy of OCA in patients with PBC was evaluated in a phase 3 randomized controlled trial, A Placebo‐Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis (POISE). The trial included patients with an incomplete response to UDCA, defined as ALP >1.67 × ULN or abnormal TB <2 × ULN, treated with 5 mg, 5 mg up‐titrated to 10 mg, or 10 mg OCA.( 8 ) Compared to 10% in the placebo group, 47% and 46% of patients in the 10mg and 5‐10mg treatment arms met the predefined primary endpoint of a reduction in ALP to less than 1.67 × ULN along with a 15% decline in ALP and normal bilirubin after 12 months of therapy (herein referred to as the POISE primary endpoint).( 8 ) In May 2017, Health Canada approved use of OCA in combination with UDCA for treatment of patients with PBC with an incomplete response to UDCA. Given the inherent differences in patient populations treated in clinical trials compared to routine clinical care,( 9 ) we sought to evaluate the effectiveness of OCA in patients with PBC in the first‐described real‐world cohort.
Participants and Methods
Study Design
A retrospective cohort study of patients with PBC with an incomplete response or intolerance to UDCA treated with OCA and followed at the Toronto Centre for Liver Disease or Centre Hospitalier de l’Université de Montréal was conducted. Electronic medical records were abstracted for clinical and biochemical parameters. Dates of diagnoses, treatment doses and duration, liver stiffness measurements (LSMs), and liver‐related complications were collected. Tolerability was reported as described in patient records.
Participants
Adult patients with an established clinical diagnosis of PBC based on accepted international guidelines( 2 ) and who were initiated on OCA based on the treating hepatologists’ discretion of individual risk were included. Patients were included between May 2017 and June 2019, beginning at the date of Health Canada’s approval of OCA and ending at the time of analysis. All patients were naive to prior treatment with OCA and were on stable weight‐based UDCA, as tolerated. Patients were censored at OCA discontinuation, although those who were dose reduced remained included. Dose escalation from 5 mg to 10 mg at 6 months was planned for all patients who did not reach the POISE primary endpoint at the 6‐month time point and were tolerant of therapy. Doses were adjusted to 5 mg weekly in patients with Child‐Pugh B/C cirrhosis, as per the label.
Outcomes
Primary outcomes were changes in serum ALP, TB, and gamma‐glutamyl transferase (GGT) over the duration of OCA therapy. Secondary outcomes were changes in alanine aminotransferase (ALT), aspartate aminotransferase (AST), immunoglobulin M (IgM), platelets, and albumin; achievement of the POISE primary endpoint; and treatment discontinuations, dose reductions, and tolerability.
Statistical Analysis
Univariate and multiple linear mixed‐effects regression models with random intercepts and random slopes were used to determine the effect of OCA therapy on serum biochemistry over time. Univariate linear mixed‐effects regression was performed with serum biochemistry as the outcome variable and duration of OCA treatment as the independent variable. Polynomial mixed‐effects regression was performed with serum biochemistry as the outcome variable and powers of time as the independent variables. Maximum likelihood estimation was performed using the restricted maximum likelihood approach to address potential bias due to missing measurements. Random intercepts and random slopes were used to allow a unique starting point and trajectory for each patient. Patients were stratified based on whether they met the inclusion criteria of the POISE study at OCA initiation and based on an LSM indicative of cirrhosis (i.e., ≥16.9 kPa). Linear mixed‐effects regression analysis was stratified by POISE inclusion criteria and an LSM indicative of cirrhosis to compare changes in serum biochemistry over time. ALP, ALT, AST, TB, GGT, and IgM were transformed by logarithm base 10 to satisfy assumptions of linearity. Statistical testing was performed at the 0.05 alpha level.
Ethics
This cohort is a subset of patients within the Canadian Network for Autoimmune Liver Disease. This study was approved by institutional research boards at the University Health Network and Centre Hospitalier de l’Université de Montréal, as per local regulation. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Results
Patient Cohort
In total, 108 patients were eligible for treatment with OCA based on the treating hepatologists’ discretion. Of these, 17% (n = 18) were excluded due to prior OCA exposure in clinical trials and a further 24% (n = 26) had not yet started therapy due to lack of access (n = 3), patient discretion (n = 5), or were waiting for insurance drug coverage (n = 18) (Fig. 1). Ultimately, 64 patients were initiated on OCA between August 2017 and June 2019, with a median of 13.1 months (range, 0.5‐22.5) on OCA therapy. At the time of analysis, 54 (84%) patients were treated for more than 3 months, 42 (66%) more than 6 months, 38 (59%) more than 9 months, 36 (56%) more than 12 months, and 24 (38%) more than 15 months. Forty‐five (70%) patients had biochemical measurements after 3 months, 38 (60%) after 6 months, 33 (52%) after 9 months, 29 (45%) after 12 months, and 16 (25%) after 15 months.
Fig. 1.
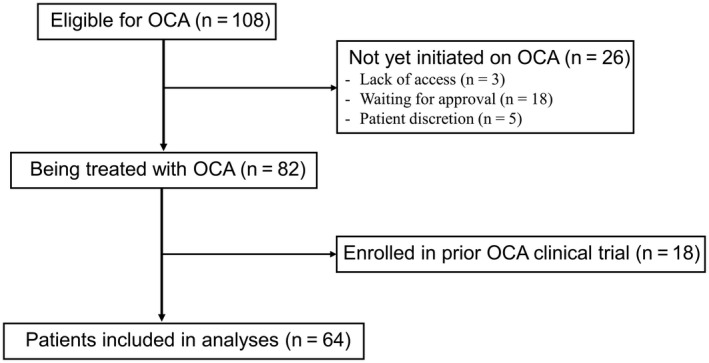
Patients eligible for inclusion in analyses.
Fifty‐seven (89%) patients from Toronto and 7 (11%) patients from Montreal were included. Mean age was 54.6 years (SD, 8.2), 95% (n = 61) were women, 89% (n = 57) were Caucasian, and 91% (n = 58) were positive for anti‐mitochondrial antibody. The majority (95%, n = 61) of patients were treated with UDCA, and 6.3% (n = 4) had an overlap with autoimmune hepatitis. The mean duration of disease was 11.0 years (SD, 6.3), and the mean duration of UDCA therapy was 9.7 years (SD, 6.3) (Table 1).
Table 1.
Demographic and Clinical Characteristics of a Real‐World OCA Cohort
| Characteristic at OCA Initiation | Real‐World Cohort (n = 64) |
|---|---|
| Mean age at OCA initiation, years (SD) | 54.6 (8.2) |
| Female sex, number (%) | 61 (95) |
| Caucasian ethnicity, number (%) | 57 (89) |
| Positive anti‐mitochondrial antibody, number (%) | 58 (91) |
| Autoimmune hepatitis overlap, number (%) | 4 (6.3) |
| Treated with UDCA, number (%) | 61 (95) |
| Mean duration of disease, months (SD) | 132 (75) |
| Mean duration of UDCA, months (SD) | 116 (75) |
| Median duration of OCA, months (IQR) | 13.1 (5.4‐18.5) |
| Median serum biochemistry*, † (IQR) | |
| ALP, U/L | 250 (203‐350) |
| ALT, U/L | 48 (37‐64) |
| AST, U/L | 46 (36‐63) |
| TB, μmol/L | 13 (10‐18) |
| Platelets, ×109/L | 225 (148‐255) |
| Albumin, g/L | 41 (39‐43) |
| IgM, g/L | 3.0 (2.3‐4.8) |
| GGT, U/L | 217 (154‐364) |
| Median GLOBE score (IQR) | 0.63 (0.06‐1.17) |
| Median predicted probability of liver transplant‐free survival | |
| 10 years, %/age‐matched % | 72.5/92.1 |
| Liver stiffness ‡ | |
| Median value, kPa (IQR) | 10.7 (8.0‐16.3) |
| ≥16.9 kPa, number/total number (%) | 14/59 (24) |
| Met POISE inclusion criteria, number (%)* | 44 (70) |
| Met biochemical response criteria, number (%) | |
| Paris‐II criteria | 7 (12) |
| Toronto criteria | 23 (37) |
One patient did not have any available biochemistry data at OCA initiation and was excluded from biochemical analyses and POISE stratification.
Serum biochemistry within 1 month of initiation was available for 50 (78%) patients. Most recent biochemistry from up to 6 months before initiation was used for the remaining patients. IgM data were not available for 13 (20%) patients and were taken from up to 1 year before initiation. GGT data were not available for 14 (22%) patients. AST data were not available for 5 (8%) patients.
Liver stiffness data within 1 year before OCA initiation were available for 54 (84%) patients, and 5 (7.8%) additional patients had measurements <16.9 kPa after initiation.
Clinical Characteristics at OCA Initiation
Biochemical measurements were considered at baseline if measured within 1 month of OCA initiation. If baseline data were unavailable, values from within 6 months before OCA initiation were used (Table 1). One patient did not have any available biochemistry data but was included in cohort descriptives. LSMs from the year before initiation were available for 84% (n = 54) of patients. An additional 7.8% (n = 5) had an LSM <16.9 kPa after initiation and were presumed to have an LSM <16.9 kPa at initiation. Median LSM was 10.7 kPa (interquartile range [IQR], 8.0‐16.3), with 24% (n = 14) of patients having an LSM indicative of cirrhosis (≥16.9 kPa). As per the GLOBE score (named for its use of data collected from across the globe), a validated risk assessment tool to estimate the future probability of liver transplant‐free survival,( 10 ) the median estimated liver transplant‐free survival in our cohort was 72.5% at 10 years compared to 92.1% in healthy controls.
Within the cohort, 70% (n = 44/63) of patients met the inclusion criteria of the POISE trial at OCA initiation and 30% (n = 19/63) of patients did not. The majority (91%, n = 40) of patients who met the POISE inclusion criteria had ALP values ≥1.67 × ULN, and 20% (n = 9) had an abnormal TB <2 × ULN. All patients who did not meet POISE inclusion criteria at OCA initiation (n = 19) had ALP <1.67 × ULN, 89% (n = 17/19) had normal TB values, and 11% (n = 2/19) had abnormal TB values greater than 2 × ULN (Supporting Table S1). All patients who did not meet POISE inclusion criteria were initiated on OCA based on clinical features of high‐risk disease, including presence of cirrhosis (79%, n = 15) either histologically proven (47%, n = 7) or with radiographic features consistent with cirrhosis (53%, n = 8), age <50 years at diagnosis (84%, n = 16), or both (63%, n = 12). Among POISE‐eligible patients, the median estimated probability of liver transplant‐free survival was 71% at 10 years as per the GLOBE score. Among patients who did not meet eligibility criteria for POISE, the median estimated probability of liver transplant‐free survival was 76% at 10 years (Supporting Table S1).
Compared to the POISE clinical trial cohort (5‐10 mg arm/10 mg arm), our patients were similar in age (mean ± SD, 54.6 ± 8.2 vs. 56.0 ± 11 years), sex (95% vs. 93%/86%), and proportion treated with UDCA (95% vs. 93%/92%), although patients in our cohort had a longer mean duration of disease (mean ± SD, 11.0 ± 6.3 years vs. 8.0 ± 6.0/9.0 ± 7.0 years). Regarding biochemical profiles at treatment initiation in our real‐world cohort, median ALP was 250 U/L (IQR, 203‐350), GGT was 217 U/L (IQR, 154‐364), ALT was 48 U/L (IQR, 37‐64), AST was 46 U/L (IQR, 36‐63), and TB was 13 μmol/L (IQR, 10‐18) (Table 1). In comparison, biochemical profiles at treatment initiation reported in the POISE clinical trial cohort (5‐10 mg arm/10 mg arm) were as follows: mean ± SD for ALP was 326 ± 116 U/L/316 ± 104 U/L, GGT was 253 ± 167 U/L/261 ± 207 U/L, ALT was 62 ± 39 U/L/56 ± 40 U/L, AST was 52 ± 25 U/L/51 ± 31 U/L, and TB was 10 ± 6 μmol/L/11 ± 7 μmol/L.( 8 ) Median LSM in our real‐world cohort was 10.7 kPa (IQR, 8.0‐16.3) compared to the reported mean ± SD for LSM in the POISE cohort of 10.7 ± 8.6 kPa/11.4 ± 8.2 kPa; the proportion of patients with an LSM ≥16.9 kPa was similar between cohorts (24% vs. 20%/19%).( 8 )
Changes in ALP, GGT, and TB
Mean ALP values declined significantly during OCA therapy. There was a reduction in mean ALP of 55 U/L (P < 0.01) from 274 U/L (95% confidence interval [CI], 242‐309) at initiation to 219 U/L (95% CI, 193‐248) at 12 months of therapy, equating to an ALP reduction of 0.41 × ULN (from 2.03 × ULN [95% CI, 1.79‐2.29] at initiation to 1.62 × ULN [95% CI, 1.43‐1.84] at 12 months), in keeping with a 20% reduction in mean ALP. Reductions in ALP were significant at 3, 6, 9, 12, and 15 months compared to initiation (P < 0.01 for all comparisons) (Fig. 2). ALP declined within the first 3 months (P < 0.01), stabilized between 3 and 6 months (P = 0.08), and further declined at each subsequent 3‐month interval up to 15 months (P < 0.01), achieving a value below 1.67 × ULN between 9 and 12 months. ALP continued to decline between 12 and 15 months and had not plateaued by the end of the analysis period (Table 2). Of patients with ≥12 months of follow‐up data (n = 29), 21% (n = 6) had normal ALP between 11 and 13 months and 28% (n = 8) had normal ALP at or before their last follow‐up when considering measurements up to 19 months.
Fig. 2.
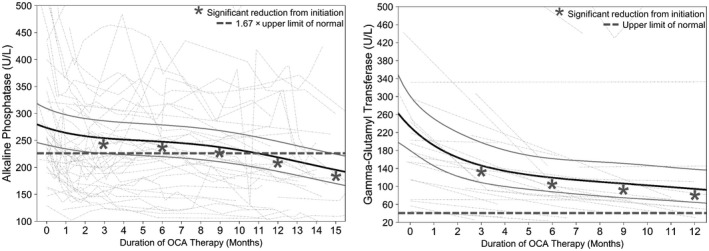
Estimated mean reductions in ALP and GGT from OCA initiation. The black solid line represents fixed effects, gray solid lines are 95% CI, and dotted lines are individual values of patients with at least two measurements. ALP and GGT at 3, 6, 9, and 12 months are reduced compared to initiation (P < 0.01 for all comparisons).
Table 2.
Estimated Mean Reductions in Serum Liver Tests Over the Duration of OCA Treatment
| Duration of OCA Therapy (Months) | ALP (U/L) (n = 63) | ALP Reduction From Initiation (U/L) | Significance of Reduction From Initiation | Significance of Reduction Compared to 3 Months Earlier |
|---|---|---|---|---|
| 0 | 274 | – | – | – |
| 3 | 254 | 20 | P < 0.01 | – |
| 6 | 248 | 26 | P < 0.01 | P = 0.08 |
| 9 | 237 | 37 | P < 0.01 | P < 0.01 |
| 12 | 219 | 55 | P < 0.01 | P < 0.01 |
| 15 | 195 | 79 | P < 0.01 | P < 0.01 |
| Duration of OCA Therapy (Months) | ALT (U/L) (n = 63) | ALT Reduction From Initiation (U/L) | Significance of Reduction From Initiation | Significance of Reduction Compared to 3 Months Earlier |
|---|---|---|---|---|
| 0 | 46.0 | – | – | – |
| 3 | 37.5 | 8.5 | P < 0.01 | – |
| 6 | 35.9 | 10.1 | P < 0.01 | P < 0.05 |
| 9 | 35.4 | 10.6 | P < 0.01 | P = 0.55 |
| 12 | 34.1 | 11.9 | P < 0.01 | P = 0.13 |
| 15 | 32.6 | 13.4 | P < 0.01 | P = 0.14 |
| Duration of OCA Therapy (Months) | AST (U/L) (n = 59) | AST Reduction From Initiation (U/L) | Significance of Reduction From Initiation | Significance of Reduction Compared to 3 Months Earlier |
|---|---|---|---|---|
| 0 | 46.2 | – | – | – |
| 3 | 43.7 | 2.5 | P < 0.05 | – |
| 6 | 42.6 | 3.6 | P < 0.05 | P = 0.17 |
| 9 | 41.6 | 4.6 | P < 0.05 | P = 0.20 |
| 12 | 40.5 | 5.7 | P < 0.05 | P = 0.17 |
| 15 | 39.8 | 6.4 | P < 0.05 | P = 0.53 |
| Duration of OCA Therapy (Months) | IgM (g/L) (n = 50) | IgM Reduction From Initiation (g/L) | Significance of Reduction From Initiation | Significance of Reduction Compared to 3 Months Earlier |
|---|---|---|---|---|
| 0 | 3.06 | – | – | – |
| 3 | 2.75 | 0.31 | P < 0.01 | – |
| 6 | 2.57 | 0.49 | P < 0.01 | P < 0.01 |
| 9 | 2.45 | 0.61 | P < 0.01 | P < 0.01 |
| 12 | 2.36 | 0.70 | P < 0.01 | P < 0.05 |
| 15* | – | – | – | – |
| Duration of OCA Therapy (Months) | GGT (U/L) (n = 49) | GGT Reduction From Initiation (U/L) | Significance of Reduction From Initiation | Significance of Reduction Compared to 3 Months Earlier |
|---|---|---|---|---|
| 0 | 233 | – | – | – |
| 3 | 147 | 86 | P < 0.01 | – |
| 6 | 119 | 114 | P < 0.01 | P < 0.05 |
| 9 | 106 | 127 | P < 0.01 | P = 0.10 |
| 12 | 95 | 138 | P < 0.01 | P = 0.12 |
| 15* | – | – | – | – |
Estimates obtained through mixed‐effects regression.
The 15‐month trends for IgM and GGT were not reported due to few measurements beyond 12 months.
Mean GGT values declined significantly during therapy. GGT measurements were available for 78% (n = 49) of patients, all within the Toronto cohort. There was a reduction in mean GGT values of 138 U/L (P < 0.01) from 233 U/L (95% CI, 180‐301) at initiation to 95 U/L (95% CI, 65‐138) at 12 months of therapy, equating to a GGT reduction of 3.4 × ULN and in keeping with a 59% reduction in mean GGT. Reductions in GGT were significant at 3, 6, 9, and 12 months when compared to initiation (P < 0.01 for all comparisons) (Fig. 2). GGT declined significantly by 86 U/L between baseline and 3 months (P < 0.01), continued to decline between 3 and 6 months (P < 0.05), and subsequently stabilized (Table 2).
Mean TB values remained stable at 13 μmol/L at all time points throughout 15 months of therapy (P = 0.98) (Fig. 3).
Fig. 3.
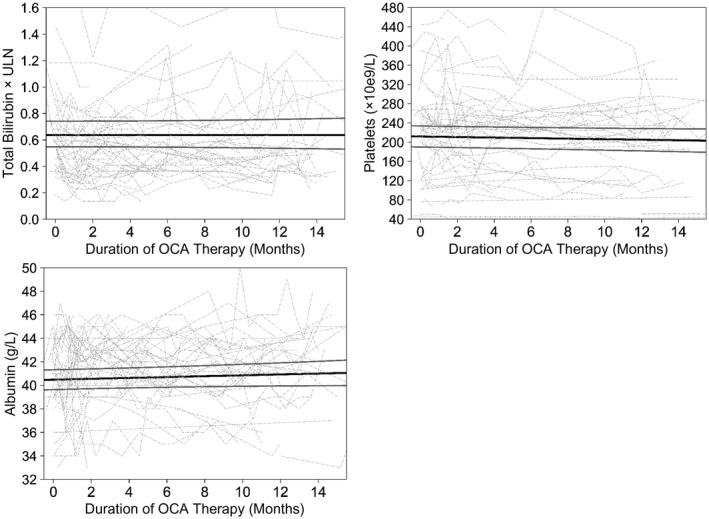
No change in mean TB, platelets, or albumin from OCA initiation. The black solid line represents fixed effects, gray solid lines are 95% CI, and dotted lines are individual values of patients with at least two measurements. TB, albumin, and platelets remained stable throughout OCA therapy (P = 0.98, P = 0.14, P = 0.12, respectively).
Changes in ALT, AST, IgM, Platelets, and Albumin
Mean ALT values declined significantly during therapy. There was a reduction in mean ALT values of 11.9 U/L (P < 0.01) from 46.0 U/L (95% CI, 40.5‐52.2) at initiation to 34.1 U/L (95% CI, 29.4‐39.7) at 12 months of therapy, in keeping with a 26% reduction. Reductions in ALT were significant at 3, 6, 9, 12, and 15 months when compared to initiation and normalized within 3 months (P < 0.01 for all comparisons) (Fig. 4). ALT declined in the first 6 months (P < 0.05) and subsequently stabilized, as may be expected due to its early normalization (Table 2).
Fig. 4.
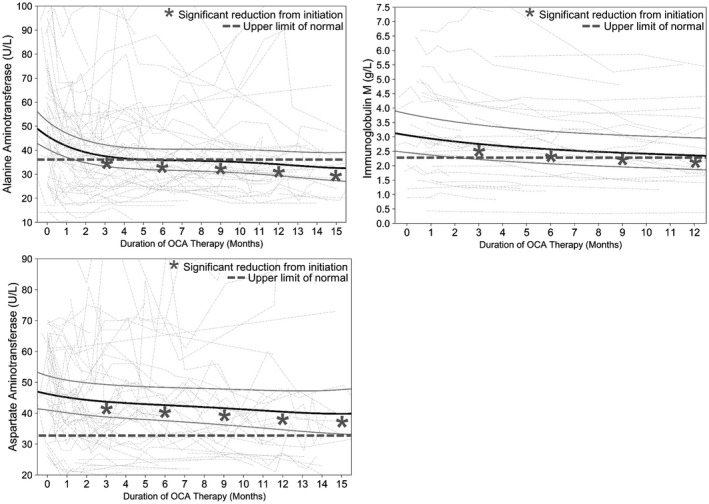
Estimated mean reductions in ALT, AST, and IgM from OCA initiation. The black solid line represents fixed effects, gray solid lines are 95% CI, and dotted lines are individual values of patients with at least two measurements. ALT, AST, and IgM at 3, 6, 9, and 12 months are reduced compared to initiation (P < 0.01, P ≤ 0.05, P < 0.01, respectively, for all comparisons).
Mean AST values declined significantly during therapy. There was a reduction in mean AST values from 46.2 U/L (95% CI, 41.0‐52.1) at initiation to 40.5 U/L (95% CI, 34.7‐47.3; P < 0.05) at 12 months of therapy, in keeping with a 12% reduction in mean AST. Reductions in AST were significant at 3, 6, 9, 12, and 15 months when compared to initiation (P < 0.05 for all comparisons) (Fig. 4). AST values declined by 2.5 U/L within the first 3 months (P < 0.05) and did not significantly decline between subsequent time points (Table 2).
Mean IgM values declined significantly during therapy. IgM measurements were available for 80% (n = 50) of patients. There was a reduction in mean IgM from 3.06 g/L (95% CI, 2.46‐3.81) at initiation to 2.36 g/L (95% CI, 1.87‐2.97; P < 0.01) at 12 months of therapy, in keeping with a 23% reduction in mean IgM. Reductions in IgM were significant at 3, 6, 9, and 12 months when compared to initiation (P < 0.01 for all comparisons) (Fig. 4) and significantly declined between each 3‐month follow‐up time point (Table 2).
Albumin and platelet values remained stable throughout 15 months of therapy (Fig. 3). Mean albumin values were 40 g/L at initiation and 41 g/L at 15 months (P = 0.14). Mean platelet count was 212 × 109/L at initiation and 204 × 109/L at 15 months (P = 0.12).
Outcomes Stratified by POISE Inclusion Criteria
Biochemical improvement was noted independent of meeting POISE inclusion criteria. Among patients deemed POISE eligible, a significant reduction in mean ALP of 70 U/L was observed, declining from 327 U/L (95% CI, 290‐369) to 257 U/L (95% CI, 226‐294) at 12 months (P < 0.01). A significant reduction in mean ALP of 34 U/L was also observed in the 19 patients who were not POISE eligible, declining from 180 U/L (95% CI, 150‐216) to 146 U/L (95% CI, 120‐178) at 12 months (P < 0.01) (Table 3).
Table 3.
Estimated Mean Reductions in Serum Liver Tests, Stratified by Clinical Trial (POISE) Inclusion Criteria
| ALP | POISE Eligible (n = 44) | Outside POISE Criteria (n = 19) | ||||||
|---|---|---|---|---|---|---|---|---|
| Duration of OCA (Months) | ALP (U/L) | Reduction (U/L) | P value | Duration of OCA (Months) | ALP (U/L) | Reduction (U/L) | P value | |
| 0 | 327 | – | – | 0 | 180 | – | – | |
| 12 | 257 | 70 | P < 0.01 | 12 | 146 | 34 | P < 0.01 | |
| ALT | Duration of OCA (Months) | ALT (U/L) | Reduction (U/L) | P value | Duration of OCA (Months) | ALT (U/L) | Reduction (U/L) | P value |
|---|---|---|---|---|---|---|---|---|
| 0 | 43.7 | – | – | 0 | 39.7 | – | – | |
| 12 | 32.7 | 11.0 | P < 0.01 | 12 | 34.4 | 5.3 | P = 0.21 |
| AST | Duration of OCA (Months) | AST (U/L) | Reduction (U/L) | P value | Duration of OCA (Months) | AST (U/L) | Reduction (U/L) | P value |
|---|---|---|---|---|---|---|---|---|
| 0 | 46.8 | – | – | 0 | 42.5 | – | – | |
| 12 | 40.8 | 6.0 | P = 0.09 | 12 | 39.9 | 2.6 | P = 0.46 |
| IgM | Duration of OCA (Months) | IgM (g/L) | Reduction (g/L) | P value | Duration of OCA (Months) | IgM (g/L) | Reduction (g/L) | P value |
|---|---|---|---|---|---|---|---|---|
| 0 | 3.20 | – | – | 0 | 2.49 | – | – | |
| 12 | 2.39 | 0.81 | P < 0.01 | 12 | 2.06 | 0.43 | P = 0.07 |
| GGT | Duration of OCA (Months) | GGT (U/L) | Reduction (U/L) | P value | Duration of OCA (Months) | GGT (U/L) | Reduction (U/L) | P value |
|---|---|---|---|---|---|---|---|---|
| 0 | 256 | – | – | 0 | 162 | – | – | |
| 12 | 106 | 150 | P < 0.01 | 12 | 63 | 99 | P < 0.01 |
Estimates obtained through linear mixed‐effects regression.
A significant reduction in mean GGT of 150 U/L was observed among POISE‐eligible patients, declining from 256 U/L (95% CI, 190‐345) to 106 U/L (95% CI, 66‐172) at 12 months (P < 0.01). A significant reduction in GGT of 99 U/L was also observed in patients who were not POISE eligible, declining from 162 U/L (95% CI, 105‐250) at initiation to 63 U/L (95% CI, 34‐119) at 12 months (P < 0.01).
A significant reduction in mean ALT of 11.0 U/L was observed among POISE‐eligible patients, declining from 43.7 U/L (95% CI, 37.6‐50.6) to 32.7 U/L (95% CI, 27.2‐39.2) at 12 months (P < 0.01). Patients who were not POISE eligible started with nearly normal ALT and did not experience a significant reduction in ALT (P = 0.21). Trends for AST and IgM in the POISE‐stratified cohort are shown in Table 3. There were no significant differences between the groups in changes of TB, platelets, or albumin over 12 months of therapy (P = 0.97, P = 0.19, P = 0.88, respectively).
Effectiveness of OCA in Advanced‐Stage PBC (LSM ≥16.9 kPa)
LSM from the year before initiation was available for 84% (n = 54) of patients, and an additional 7.8% (n = 5) of patients had LSM <16.9 kPa after initiation and were presumed to have LSM <16.9 kPa at initiation. Of patients with an LSM ≥16.9 kPa (n = 14), 85% (n = 12) were Child‐Pugh A, 7.1% (n = 1) were Child‐Pugh B, and 7.1% (n = 1) had insufficient data to calculate a Child‐Pugh score. All patients with LSM ≥16.9 kPa were initiated on appropriate doses, as recommended by the manufacturers’ label. Patients with Child‐Pugh A cirrhosis were prescribed 5 mg OCA daily or less at the discretion of the treating clinician. One patient with Child‐Pugh B cirrhosis was started at 5 mg weekly and did not tolerate up‐titration to 5 mg twice weekly.
Patients with LSM ≥16.9 kPa experienced a nominally significant reduction in mean ALP from 220 U/L (95% CI, 170‐285) at initiation to 211 U/L (95% CI, 157‐283) at 12 months (P < 0.05) compared to patients with an LSM <16.9 kPa (n = 45), who demonstrated a 67‐U/L reduction in mean ALP from 288 U/L (95% CI, 249‐332) to 221 U/L (95% CI, 191‐256) over 12 months (P < 0.01), although ALP at initiation was lower among those with advanced disease (Supporting Table S2).
Mean platelet count of patients with LSM ≥16.9 kPa declined from 131 × 109/L (95% CI, 104‐166) at initiation, which was subnormal, to 115 × 109/L (95% CI, 88‐151) at 12 months (P < 0.05), whereas platelet counts of patients with LSM <16.9 kPa remained stable (P = 0.11). Mean changes in TB were not statistically significant in either group (Supporting Table S2).
Achievement of POISE Primary Endpoint
In our cohort, 39% (n = 17) of patients who met the POISE inclusion criteria had at least 12 months of therapy and biochemical measurements between 11 and 13 months, allowing reasonable assessment of the POISE primary endpoint. Of these patients, 35% (n = 6) achieved an ALP value <1.67 × ULN, 65% (n = 11) had a normal bilirubin, 59% (n = 10) achieved at least a 15% reduction in ALP, and 18% (n = 3) achieved the composite POISE primary endpoint at 12 months of therapy (Fig. 5). Eighteen percent (n = 3) normalized ALP at 12 months. Of POISE‐eligible patients with biochemical measurements at or after 12 months, (n = 21), 43% (n = 9) achieved the primary endpoint at or before their last follow‐up when considering measurements up to 19 months after initiation (Fig. 5), with 19% (n = 4) of this group normalizing ALP at or before their last follow‐up.
Fig. 5.
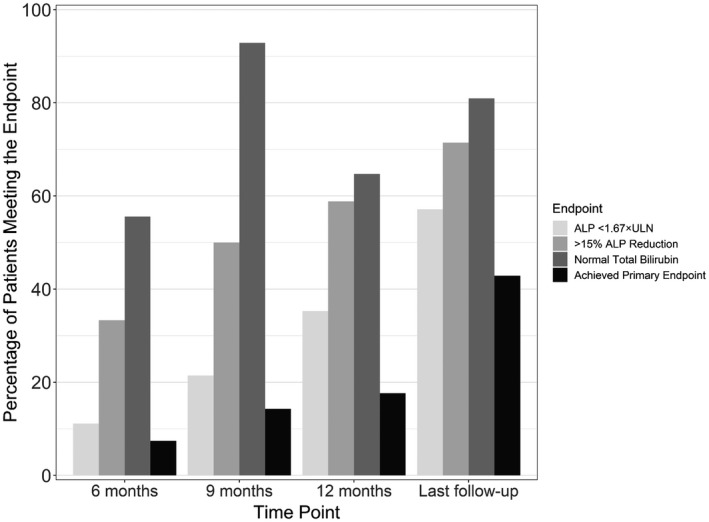
Achievement of clinical trial (POISE) primary endpoint during OCA therapy among patients who were POISE eligible. Time points are defined as ±1 month. Last follow‐up only includes patients with at least 12 months of therapy. Complete data allowing assessment of the endpoints were available for 61% (n = 27) of patients at 6 months, 32% (n = 14) at 9 months, 39% (n = 17) at 12 months, and 48% (n = 21) at or before last follow‐up.
All original analyses including those at 12, and up to 19 months were completed using observed values. Given the real‐world nature of the cohort, 4 POISE‐eligible patients with at least 12 months on therapy did not have observed measurements between 11 and 13 months but subsequently had measurements up to 19 months. To account for any potential bias at the 12‐month time point due to these missing measurements, achievement of the POISE primary endpoint was recalculated to include imputed values at 12 months for these patients with missing measurements. Imputed values were imputed based on individualized estimates from the mixed‐effects models. This analysis demonstrated similar results, with 38% (n = 8/21) achieving ALP <1.67 × ULN, 62% (n = 13/21) achieving normal TB, 62% (n = 13/21) achieving at least a 15% reduction in ALP, and 19% (n = 4/21) achieving the POISE primary endpoint.
Treatment Discontinuation, Dose Reduction, and Tolerability
Pruritus was the most commonly reported tolerability concern, occurring in 41% (n = 26) of patients, with 20.6% (n = 13) reporting new onset itch and 20.6% (n = 13) reporting worsening itch. Gastrointestinal upset was infrequent, with 2 patients reporting nausea (3.1%), 3 with constipation (4.7%), and 1 reporting bloating (1.6%). Two patients had suspected hepatotoxicity (3.1%), 1 patient reported polymenorrhea (1.6%), 2 patients reported headache (3.1%), 2 patients reported skin rash (3.1%), and 1 patient reported “sunburn feeling” (1.6%) (Supporting Table S3). Among those with established or emergent pruritus, an individualized approach to management was employed by the treating clinician, with a combination of dose reduction and/or reduced dosing interval generally followed by escalation of an antipruritic regimen (including binding resins, rifampicin, selective serotonin reuptake inhibitors, and/or gabapentin) or drug holiday.
In total, 17% (n = 11) discontinued therapy (compared to 9% in the POISE clinical trial), with 45% (n = 5) of those who stopped treatment doing so due to pruritus, similar to the 42% (n = 8/19) seen in the POISE trial.( 8 ) Of the other 55% (n = 6) who discontinued therapy, 2 stopped due to an incomplete response after 1 year of therapy, 2 due to skin rash, and 2 due to hepatotoxicity. Of those with suspected hepatotoxicity, the first was a 67‐year‐old woman diagnosed with PBC in 2004 with Child‐Pugh A cirrhosis and imaging evidence of intra‐abdominal midline varices without endoscopic evidence for esophagogastric varices. She was started on OCA 5 mg daily, which was up‐titrated to 10 mg daily after 6 months based on insufficient response. Five months after her dose increase, her ALT was noted to be 6.7 × ULN (baseline 1.1 × ULN and 1.2 × ULN at 6 months), ALP was 1.5 × ULN, and TB was 13 μmol/L in the absence of any other cause. OCA was discontinued, and 8 months later her liver tests returned to near baseline. The second patient was a 54‐year‐old woman diagnosed with PBC in 2007. By traditional scoring, she was Child‐Pugh B (score 7), although using Child‐Pugh modifications for cholestatic liver disease and associated higher bilirubin thresholds (bilirubin 4 mg/dL) she was Child‐Pugh A (score 6); thus, she was started on OCA 5 mg daily in July 2017. Four months later, she developed cholestatic jaundice with a TB of 147 μmol/L. OCA was promptly stopped, and a liver biopsy was performed that revealed severe cholestatic cirrhosis, severe ductopenia, severe cholestasis with ballooning and Mallory bodies, and interface hepatitis. The following year she developed complications, including ascites, hepatorenal syndrome, and encephalopathy; she received a liver transplant in October 2018.
Of patients who discontinued OCA, 73% (n = 8) were on 5 mg daily, 18% (n = 2) were on 5 mg weekly, and 9% (n = 1) were on 10 mg daily. Reduction from an initial dose of 5 mg daily to 5 mg weekly occurred in 4.7% (n = 3) of patients at a median of 3.7 months (range, 3.2‐6.1) of therapy: 1 due to worsening gastrointestinal symptoms, 1 due to increasing liver tests, and 1 due to nausea and headache (Supporting Table S3). Forty‐eight percent (n = 31) of patients had an increase from their initial dose at a median of 6.1 months (IQR, 5.6‐6.9) of therapy. An additional 25% (n = 16) of patients had at least 6 months of therapy but did not have their dose increased: 6 due to sufficient biochemical response, 2 due to tolerability concerns, 2 due to patient preference, 2 were pending a return clinic visit to consider up‐titration, 1 related to insurance coverage issues, and 3 for unknown reasons. Eighty‐one percent (n = 25/31) of patients with a dose increase were increased to 10 mg daily; 26% (n = 8) of these increases were subsequently reduced at a median of 12.0 months (IQR, 10.2‐12.6) of therapy: 5 due to pruritus, 1 due to increasing liver tests, 1 due to myalgia, and 1 for unknown reasons (Supporting Fig. S1).
Doses at the 12‐month time point for the 36 patients with at least 12 months of treatment were as follows: 17 (47%) on 10 mg daily, 15 (42%) on 5 mg daily, 2 (5.6%) on 5 mg and 10 mg daily alternating, 1 (2.8%) on 5 mg twice weekly, and 1 (2.8%) on 5 mg weekly.
Discussion
Herein, we report the first real‐world experience with OCA in a high‐risk cohort of patients with PBC. We demonstrate significant reductions in ALP, ALT, AST, GGT, and IgM within 3 months of treatment that were maintained throughout follow‐up. Of note, significant ALP reductions occurred regardless of whether patients met POISE inclusion criteria at initiation, and measures of liver function, including TB, albumin, and platelet values, remained stable throughout treatment. Biochemical trends in patients with overlap syndrome were consistent with the larger cohort. Nominal biochemical improvement was also observed in patients with cirrhotic‐stage disease.
Absolute reductions in ALP at 12 months were numerically less in our patients who were POISE eligible compared to the POISE clinical trial, although our cohort started with lower mean ALP values at treatment initiation. At 12 months of therapy, we observed a reduction in mean ALP of 70 U/L compared to 113‐130 U/L in the POISE trial, a reduction in mean ALT of 11.0 U/L compared to 21.3‐25.3 U/L, and a reduction in mean AST of 6.0 U/L compared to 13.0‐15.0 U/L.( 8 ) This may be a result of lower baseline biochemistries in our cohort or to the fact that 44% of our cohort had not yet reached 12 months on OCA. In addition, adherence was self‐reported by patients and is likely less robustly documented compared to a clinical trial setting.( 9 ) TB demonstrated statistically significant reductions over 12 months in the POISE trial, but this was not observed in our cohort despite having similar TB values at initiation.( 8 ) Finally, 18% of patients who were POISE eligible in our cohort met the POISE primary endpoint at 12 months, lower than the 46%‐47% observed in the clinical trial. However, 43% of patients who were POISE eligible achieved the POISE primary endpoint at or before the last follow‐up when considering measurements up to 19 months after initiation. It is possible that the discrepancy between 18% response at 12 months and 43% response by 19 months could be due to 4 patients missing measurements during the 11‐13‐month window. However, given that achievement of the POISE primary endpoint at 12 months was similar with and without imputation of missing 12‐month values, it is also possible that a slower response may be observed in the real world. Several factors could contribute to a potential slower response. These include suboptimal patient compliance in the real world, variability in follow‐up practice between physicians and across centers, a higher proportion of patients with cirrhosis than the POISE trial (24% vs. 20%),( 8 ) and biochemical differences in our cohort at initiation. There may also be unknown systematic differences between patients who were prescribed OCA at an earlier date and thus reached 12 months first. International collaborative efforts studying larger patient cohorts may shed light on these possibilities. This said, rates of achievement of the three individual components of the POISE primary endpoint (i.e., ALP <1.67 × ULN, normal TB, 15% reduction in ALP) at 12 months is encouraging.
A substantial minority (30%) of patients included in our cohort did not meet the POISE inclusion criteria at treatment initiation. All these patients were deemed at risk of disease progression on the basis of presence of cirrhosis or young age at diagnosis, both of which have been robustly demonstrated to be associated with poor outcomes in PBC.( 5 , 11 , 12 , 13 ). This highlights that risk in PBC is multifactorial and relates to biochemical response to UDCA in part but also to age and fibrosis stage.( 14 ) Additionally, further improvement in outcomes with subnormal bilirubin levels and ALP reductions below traditional thresholds has been suggested, emphasizing that improvement in biochemical parameters in this subset of patients is also clinically relevant. Inclusion of such patients in our analysis is a strength as it reflects a cohort of patients at risk who may be ineligible for clinical trials but may respond to and benefit from adjunctive therapy.
Data supporting use of OCA in patients with cirrhotic‐stage disease remain limited. Herein, we demonstrate that while nominal biochemical improvement is observed, it is not surprisingly less robust than in patients with a lower LSM. Further, we noted a reduction in platelet count in patients with an LSM ≥16.9 kPa, although bilirubin levels remained largely stable. This is in line with label recommendations suggesting dose reduction, namely in patients with Child‐Pugh B/C cirrhosis, to 5 mg weekly as the initial dose. Patients with platelet counts less than 150 × 109/L at initiation may have clinically relevant portal hypertension and thus may also benefit from a lower starting dose. A phase 4 study evaluating the safety of OCA in patients with PBC with advanced disease is currently recruiting,( 15 ) and results are eagerly awaited.
Worsening and emergent pruritus were each reported in 20.6% of patients, totaling 41.2% of our cohort and highlighting pruritus as a noteworthy tolerability issue in the real world, as it was in the clinical trial setting. In the POISE trial, similar rates of pruritus were reported with 56% in the 5‐10‐mg group and 68% in the 10‐mg group.( 8 ) However, pruritus‐related complaints in our cohort were described as in patient records as opposed to with a validated measurement tool, as in the POISE trial. Further, discontinuation rates due to pruritus were similar in real‐world and clinical trial settings. Suspected hepatotoxicity was noted in 2 patients, both of whom had cirrhosis at baseline. While liver injury was not reported in the POISE clinical trial, postmarketing reports of liver injury in patients treated with OCA, albeit generally at higher than recommended doses, led to a boxed warning by the Food and Drug Administration. Eaton et al.( 16 ) report on a small series of 8 patients (of whom 6 had PBC) who developed severe cholestatic injury with or without ascites on treatment with OCA. This was presumed to be drug injury that developed after a long latency period of 210 ± 104 days. Of note, nearly all patients who experienced potential drug injury had cirrhosis or features of portal hypertension at baseline; all were started at 5 mg daily and were on 5 mg daily, 10 mg daily, or 10 mg 3 times weekly at the time of development of jaundice. Here, we describe two patterns of potential drug injury in patients with cirrhosis and clinical evidence of portal hypertension despite otherwise compensated, albeit advanced, disease. This suggests that the Child‐Pugh score is reasonable, although imperfect in stratifying disease severity, and care must be taken to clinically assess individual disease severity and consider dose reduction in patients with advanced disease, potentially including those with evidence of portal hypertension. Dose reduction in patients with Child‐Pugh B/C cirrhosis should be ensured, as indicated by the drug label. Results from ongoing efforts aimed at assessing optimal dosing strategies in patients with advanced disease are eagerly awaited.
Peroxisome proliferator‐activated receptor agonists, including bezafibrate, remain an evidence‐based option for adjunctive therapy in patients with PBC with an incomplete response to UDCA, albeit off‐label to date. A phase 3 randomized trial of placebo with UDCA compared to bezafibrate with UDCA demonstrated normalization of liver tests in 31% of patients in the bezafibrate arm,( 17 ) and other agents in this class remain under investigation.( 18 ) Bezafibrate is not readily accessible throughout North America, and while patients treated with bezafibrate were not included in our analysis, a comparison between effectiveness of OCA and bezafibrate in the real world would be of clear value.
A primary limitation of this study is the limited follow‐up time that continues to accrue. This relates to the relatively recent approval of OCA in Canada as well as insurance‐related delays, resulting in 56% of patients completing 12 months of therapy. In addition, tolerability data were recorded in and collected from patient charts as opposed to with a validated measurement tool. Finally, we were unable to compare the demographics of patients initiated on OCA therapy to those of all patients potentially eligible for therapy because our inclusion criteria were limited to treated patients.
In this study, we demonstrated the real‐world effectiveness of OCA in improving important biochemical surrogates of outcome in patients with high‐risk PBC. We observed a reduction in biochemical markers of cholestasis independent of whether patients met POISE clinical trial inclusion criteria. Rates of pruritus were similar, although overall treatment discontinuation was higher and achievement of the POISE primary endpoint was potentially slower than observed in a clinical trial setting. These data highlight the utility of adjunctive therapy in an appropriately selected real‐world cohort.
Supporting information
Fig S1
Table S1‐3
Acknowledgment
We acknowledge the Canadian Network for Autoimmune Liver Disease for ethical approval and data collection.
Supported in part by unrestricted financial support from the Canadian Liver Foundation, Intercept Pharma Canada Inc., and the Toronto General and Western Hospital Foundation.
Funders had no influence on the study design, data collection, analysis and interpretation of data, or the decision to submit for publication.
Potential conflict of interest: Dr. Vincent received grants from Intercept, Gilead, CMED, GSK, and Novartis. Dr. Hirschfield consults for Intercept, Cymabay, Genfit, Roche, and GSK. Dr. Tsien consults for Lupin. Dr. Gulamhusein advises Intercept and is on the speakers’ bureau for Intercept and AbbVie. Dr. Mason is on the speakers’ bureau for Intercept and receives grants from Intercept and Merck. Dr. Hansen consults for and received grants from Intercept, Cymabay, Mirum, and Albireo and consults for ChemoMAB and Calliditas. Dr. Yoshida received grants from Gilead, Merck, Janssen, AbbVie, Intercept, and Genfit and is on the speakers’ bureau for Gilead Canada, Merck Canada, AbbVie Canada, Celgene Canada, and Intercept Canada. Dr. Janssen received grants from Intercept. Dr. Swain advises, is on the speakers’ bureau for, and received grants from Gilead and Intercept; he advises and received grants from Novartis and is on the speakers’ bureau for Abbott; he received grants from GSK, BMS, CymaBay, Genkyotex, and AstraZeneca. The other authors have nothing to report.
References
- 1. Yoshida EM, Mason A, Peltekian KM, Shah H, Thiele S, Borrelli R, et al. Epidemiology and liver transplantation burden of primary biliary cholangitis: a retrospective cohort study. CMAJ Open 2018;6:E664‐E670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the study of liver diseases. Hepatology 2019;69:394‐419. [DOI] [PubMed] [Google Scholar]
- 3. European Association for the Study of the Liver . EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017;67:145‐172. [DOI] [PubMed] [Google Scholar]
- 4. Parés A, Caballería L, Rodés J. Excellent long‐term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 2006;130:715‐720. [DOI] [PubMed] [Google Scholar]
- 5. Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, et al.; UK PBC Consortium . Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology 2013;144:560‐569.e7. [DOI] [PubMed] [Google Scholar]
- 6. Lammers WJ, van Buuren HR, Hirschfield GM, Janssen HL, Invernizzi P, Mason AL, et al.; Global PBC Study Group . Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow‐up study. Gastroenterology 2014;147:1338‐1349.e5. [DOI] [PubMed] [Google Scholar]
- 7. Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha‐ethyl‐chenodeoxycholic acid (6‐ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 2002;45:3569‐3572. [DOI] [PubMed] [Google Scholar]
- 8. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al.; POISE Study Group . A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 9. Kennedy‐Martin T, Curtis S, Faries D, Robinson S, Johnston J. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials 2015;16:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lammers WJ, Hirschfield GM, Corpechot C, Nevens F, Lindor KD, Janssen HL, et al.; Global PBC Study Group . Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology 2015;149:1804‐1812.e4 . [DOI] [PubMed] [Google Scholar]
- 11. Cheung AC, Lammers WJ, Murillo Perez CF, van Buuren HR, Gulamhusein A, Trivedi PJ, et al.; Global PBC Study Group . Effects of age and sex of response to ursodeoxycholic acid and transplant‐free survival in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol 2019;17:2076‐2084.e2. [DOI] [PubMed] [Google Scholar]
- 12. Roll J, Boyer JL, Barry D, Klatskin G. The prognostic importance of clinical and histologic features in asymptomatic and symptomatic primary biliary cirrhosis. N Engl J Med 1983;308:1‐7. [DOI] [PubMed] [Google Scholar]
- 13. Corpechot C, Carrat F, Poujol‐Robert A, Gaouar F, Wendum D, Chazouillères O, et al. Noninvasive elastography‐based assessment of liver fibrosis progression and prognosis in primary biliary cirrhosis. Hepatology 2012;56:198‐208. [DOI] [PubMed] [Google Scholar]
- 14. Murillo Perez CF, Hirschfield GM, Corpechot C, Annarosa Floreani, Mayo M, van der Meer A, et al.;GLOBAL PBC Study Group . Fibrosis stage is an independent predictor of outcome in primary biliary cholangitis despite biochemical treatment response. Aliment Pharmacol Ther 2019;50:1127‐1136. [DOI] [PubMed] [Google Scholar]
- 15. ClinicalTrials.gov . Study of OCA evaluating pharmacokinetics and safety in patients with PBC and hepatic impairment. https://clinicaltrials.gov/ct2/show/NCT03633227. Published August 16, 2018. Accessed August 24, 2019.
- 16. Eaton JE, Vuppalanchi R, Reddy R, Sathapathy S, Ali B, Kamath PS. Liver injury in patients with cholestatic liver disease treated with obeticholic acid. Hepatology 2019; 10.1002/hep.31017. [DOI] [PubMed] [Google Scholar]
- 17. Corpechot C, Chazouillères O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo‐controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med 2018;378:2171‐2181. [DOI] [PubMed] [Google Scholar]
- 18. ClinicalTrials.gov . Study to evaluate the efficacy and safety of elafibranor in patients with primary biliary cholangitis (PBC) and inadequate response to ursodeoxycholic acid. https://clinicaltrials.gov/ct2/show/NCT03124108. Published April 21, 2017. Accessed July 30, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1‐3