

Abstract
CHS‐131 is a selective peroxisome proliferator‐activated receptor gamma modulator with antidiabetic effects and less fluid retention and weight gain compared to thiazolidinediones in phase II clinical trials. We investigated the effects of CHS‐131 on metabolic parameters and liver histopathology in a diet‐induced obese (DIO) and biopsy‐confirmed mouse model of nonalcoholic steatohepatitis (NASH). Male C57BL/6JRj mice were fed the amylin liver NASH diet (40% fat with trans‐fat, 20% fructose, and 2% cholesterol). After 36 weeks, only animals with biopsy‐confirmed steatosis and fibrosis were included and stratified into treatment groups (n = 12‐13) to receive for the next 12 weeks (1) low‐dose CHS‐131 (10 mg/kg), (2) high‐dose CHS‐131 (30 mg/kg), or (3) vehicle. Metabolic parameters, liver pathology, metabolomics/lipidomics, markers of liver function and liver, and subcutaneous and visceral adipose tissue gene expression profiles were assessed. CHS‐131 did not affect body weight, fat mass, lean mass, water mass, or food intake in DIO‐NASH mice with fibrosis. CHS‐131 improved fasting insulin levels and insulin sensitivity as assessed by the intraperitoneal insulin tolerance test. CHS‐131 improved total plasma cholesterol, triglycerides, alanine aminotransferase, and aspartate aminotransferase and increased plasma adiponectin levels. CHS‐131 (high dose) improved liver histology and markers of hepatic fibrosis. DIO‐NASH mice treated with CHS‐131 demonstrated a hepatic shift to diacylglycerols and triacylglycerols with a lower number of carbons, increased expression of genes stimulating fatty acid oxidation and browning, and decreased expression of genes promoting fatty acid synthesis, triglyceride synthesis, and inflammation in adipose tissue. Conclusion: CHS‐131 improves liver histology in a DIO and biopsy‐confirmed mouse model of NASH by altering the hepatic lipidome, reducing insulin resistance, and improving lipid metabolism and inflammation in adipose tissue.

Abbreviations
- α‐SMA
alpha‐smooth muscle actin
- Acox1
acyl‐coenzyme A oxidase 1
- ALT
alanine aminotransferase
- AMLN
amylin liver nonalcoholic steatohepatitis
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- BCAA
branched‐chain amino acids
- Ccl2
chemokine (C‐C motif) ligand 2
- Col1a1
collagen type I alpha
- DAG
diacylglycerol
- DIO
diet‐induced obese
- epiAT
epididymal adipose tissue
- Fabp4
fatty acid‐binding protein 4
- Fasn
fatty acid synthase
- Gal‐3
galectin‐3
- HP
hydroxyproline
- IHC
immunohistochemistry
- ipITT
intraperitoneal insulin tolerance test
- LSD
least significant difference
- Mlxipl
MLX interacting protein like
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- OGTT
oral glucose tolerance test
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PGC‐1a
peroxisome proliferator‐activated receptor gamma coactivator 1 alpha
- PPARγ
peroxisome proliferator‐activated receptor gamma
- scAT
subcutaneous adipose tissue
- sPLS‐DA
sparse partial least squares discriminant analysis
- T2D
type 2 diabetes
- TAG
triacylglycerol
- TC
total cholesterol
- TG
triglyceride
Nonalcoholic fatty liver disease (NAFLD) affects 20% to 30% of the general population and more than 50% of people with type 2 diabetes (T2D).( 1 , 2 ) NAFLD significantly increases liver and cardiovascular disease‐related mortality.( 1 , 2 ) NAFLD is characterized at an early stage by accumulation of free fatty acids and triglycerides (TGs) in the liver (nonalcoholic fatty liver [NAFL]).( 3 ) Patients with NAFL may often develop liver inflammation and hepatic injury (nonalcoholic steatohepatitis [NASH]). NASH, especially in people with T2D, can progress to liver fibrosis, cirrhosis, and liver failure.( 2 )
Treatment of NASH remains an unmet clinical need. Nonselective peroxisome proliferator‐activated receptor gamma (PPARᵧ) activators (i.e., thiazolidinediones) are antidiabetic drugs that have additionally demonstrated beneficial effects against NASH in randomized clinical trials.( 4 , 5 ) Their mechanism of function is not fully understood, but it seems that they achieve beneficial hepatic effects by improving insulin sensitivity, increasing the concentration of important hormonal mediators secreted by adipose tissue (i.e., adiponectin), reducing systemic and tissue‐specific low‐grade inflammation, ameliorating the circulating lipid profile (and consequently hepatic fat deposition), and finally inducing β‐oxidation (reviewed in Skat‐Rordam et al.( 6 )). Because direct PPARᵧ activation in the liver may promote steatosis, it is believed that most of the hepatoprotective effects of PPARᵧ activators are achieved indirectly through their actions on their main functional site, the adipose tissue.( 6 )
Based on their beneficial effects, the guidelines of the American Association for the Study of Liver Diseases recommend the use of pioglitazone to treat patients with biopsy‐proven NASH after discussing the risks and benefits of the treatment with the patient.( 7 ) However, the use of PPARᵧ activators in daily clinical practice has been limited due to their side effects (fluid retention, weight gain).( 8 , 9 ) CHS‐131 (previously known as INT131) is a selective PPARᵧ modulator that has demonstrated dose‐dependent antidiabetic effects with less fluid retention and weight gain compared to thiazolidinediones in phase II clinical trials.( 10 , 11 )
The aim of our study was to investigate the effects of CHS‐131 on metabolic parameters and liver pathology in a diet‐induced obese (DIO) and biopsy‐confirmed mouse model of NASH (DIO‐NASH mice) in order to assess whether CHS‐131 can be a novel therapeutic option with less side effects for the treatment of NAFLD.
Materials and Methods
Study Design
Five‐week‐old male C57BL/6JRj mice supplied by JanVier (France) were fed a high‐fat diet containing 40% fat with trans‐fat, 20% fructose, and 2% cholesterol (amylin liver NASH [AMLN] diet [D09100301]; Research Diets Inc., New Brunswick, NJ) for 36 weeks. A control group of mice of the same age were fed a regular chow diet. Three weeks before the start of the study, a prebiopsy was performed to confirm NASH according to criteria by Kleiner et al.( 12 ) and to include NASH‐affected mice only (steatosis score ≥2, fibrosis score ≥1). Animal welfare and housing as well as the procedure of liver biopsy and postsurgical follow‐up are described in the Supporting Information. One week before treatment initiation, mice were randomized in the following four treatment groups, each group consisting of at least 12 mice: (1) mice with a chow diet treated with vehicle; (2) mice with an AMLN diet treated with vehicle; (3) mice with an AMLN diet treated with low‐dose CHS‐131 (10 mg/kg); or (4) mice with an AMLN diet treated with high‐dose CHS‐131 (30 mg/kg), administrated per os (PO) using oral gavage. All treatments were performed once a day for a total of 12 weeks.
Tolerance Tests
An oral glucose tolerance test (OGTT) and intraperitoneal insulin tolerance test (ipITT) were performed in treatment weeks 7 and 10, respectively, following a 6‐hour fasting. For the ipITT, 0.5 U/kg rapid‐acting insulin (NovoRapid) was administrated; for the OGTT, 2 g/kg glucose PO was administered. Blood was collected 60 minutes before and up to 180 minutes after administration of glucose or insulin (see Supporting Information for details).
Biochemical Measurements
Plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), TGs, total cholesterol (TC), creatinine, and urea were measured using commercial kits (Roche Diagnostics) on the Cobas c 501 autoanalyzer according to the manufacturer’s instructions. Mouse insulin was measured in single determinations using the Meso Scale Diagnostics platform.
For liver hydroxyproline (HP; a protein marker of fibrosis) quantification, liver samples were homogenized in 6 M HCl and hydrolyzed to degrade collagen. Samples were centrifuged, and the HP content was measured in duplicate in the supernatant, using a colorimetric assay (QuickZyme Biosciences) according to the manufacturer’s instructions. All measurements were performed at the last week (week 12) of treatment. For liver TG and TC quantification, samples were homogenized and TG and TC were extracted in 5% Nonidet P40 by heating twice to 90°C. The samples were then centrifuged, and TG and TC content were measured in the supernatant, using commercial kits (Roche Diagnostics) on the Cobas c 501 autoanalyzer according to the manufacturer’s instructions.
Liver Pathology and Immunohistochemistry Staining
Details about liver pathology and immunohistochemistry (IHC) procedures are described in the Supporting Information. Briefly, liver biopsies were fixed in formalin, sectioned, and stained with hematoxylin and eosin (Dako, Glostrup, Denmark) or picrosirius red (Sigma‐Aldrich, Broendby, Denmark) and scored by a histopathology specialist blinded to treatment as recommended by Kleiner et al.( 12 ) Protein markers of fibrosis (i.e., collagen type I alpha [Col1a1], using antibody cat. #1310‐01 from SouthernBiotech, Birmingham, AL), fibrogenesis, and hepatic stellate cell activation (i.e., alpha‐smooth muscle actin [α‐SMA], using antibody cat. #Ab124964 from Abcam), and inflammation‐macrophage activation galectin‐3 (Gal‐3; using antibody cat. #125402 from BioLegend) were assessed by IHC. IHC staining was performed using standard procedures. The methods used for quantitative assessment of immunoreactivity and steatosis are described in the Supporting Information.
Echo Magnetic Resonance Imaging Body Composition
Mice body composition was assessed by an echo magnetic resonance imaging (MRI) 3‐1 body composition analyzer (EchoMRI, Houston, TX). Nonanesthetized mice were placed in a plastic tube inside the MRI scanner for approximately 80 seconds. Body composition as expressed by fat mass, fat‐free mass (lean mass), and water was recorded.
Gene Expression
RNA was isolated using trizol/chloroform extraction and RNeasy columns (RNeasy Plus Universal Mini Kit; Qiagen, Valencia, CA) and reverse transcribed to complementary DNA, using SSIV VILO Mastermix with ezDNase (Life Technologies, Grand Island, NY). Gene expression was assessed by quantitative polymerase chain reaction (qPCR), using Taqman custom array plates (Supporting Tables S1 and S2 for primers) (Life Technologies) using a QuantStudio 3 real‐time PCR system (Life Technologies).
Metabolomics‐Lipidomics
Samples were weighed and then soaked in 1:1 dichloromethane:methanol overnight at 4°C. The supernatants were subjected to a modified Bligh‐Dyer extraction using methanol/water/dichloromethane in the presence of deuterated internal standards. An ultrahigh performance liquid chromatography–tandem mass spectroscopy was performed and is described in detail in the Supporting Information. Raw data extraction, peak‐identification, and the quality control process were performed with the use of libraries based on authenticated standards (see Supporting Information for details).
Statistics
For single time‐point continuous data, one‐way analysis of variance (ANOVA) was performed followed by Fisher’s least significant difference (LSD) to compare treatments to the NASH+vehicle group. For multiple time‐point continuous data, two‐way ANOVA with factors time and treatment was performed. By using P < 0.05 for treatment, post‐hoc LSD comparing the different treatments with NASH+vehicle at each individual time point was performed. Data from categorical endpoints, such as histopathologic scoring values, were partitioned into a 2 × 2 contingency table consisting of mice that had lower versus same or higher scores and were analyzed with Fisher’s exact test. P values of the ANOVA are reported. One‐tailed P values in the post‐hoc LSD tests and in the Fisher‘s exact test are designated with stars for the metabolic and hepatic effects of CHS‐131 based on the a priori hypothesis that these effects will be one‐directional, i.e., beneficial and not detrimental.( 13 , 14 ) The analysis was performed with GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA).
Gene expression analysis was performed with ExpressionSuite Software v1.2 (Life Technologies). Relative expression as mean ± SEM compared to NASH mice treated with vehicle is reported. One‐way ANOVA with a post‐hoc two‐tailed LSD test for parametric values or Kruskal‐Wallis test followed by Dunn’s test for nonparametric tests was performed.
For metabolomics, values were normalized in terms of raw area counts; for lipidomics, values were normalized in terms of concentration (nmol/mg). For metabolomics, the values were also normalized by sample homogenate volume available. Both for metabolomics and lipidomics, each biochemical value was then rescaled to set the median equal to 1. Missing values were imputed with the minimum. Sparse partial least squares discriminant analysis (sPLS‐DA) was performed to visualize the data with the use of five components maximum, each containing 10 variables maximum. Pathway analysis was performed with metabolites identified with Human Metabolome Database identifications, with an enrichment analysis following a global t test and a topology analysis following the relative‐betweenness centrality. The Mus musculus pathway library of the Kyoto Encyclopedia of Genes and Genomes (October 2019) was used. Welch’s two‐sample t test was used to compare individual lipid or metabolite species or groups. MetaboAnalystR was used for the analysis.( 15 ) Two‐tailed P values are reported for gene expression and metabolomics analysis because the limited information available from previous studies could not inform the directionality of the outcomes.
Results
CHS‐131 Has No Substantive Effect on Body Weight, Body Composition, or Energy Intake but Did Significantly Improve Insulin Sensitivity
Mice fed an AMLN diet for 36 weeks (DIO mice) had a higher body weight due to increased energy intake compared to mice fed a chow diet (Fig. 1B,C). Body weight, energy intake, fat mass, lean mass, and water mass did not change significantly in response to either CHS‐131 dose in relation to the NASH‐vehicle‐treated group (Fig. 1B,D). DIO‐NASH mice had modestly elevated fasting blood glucose levels and profoundly increased fasting insulin levels compared to mice fed a chow diet (Fig. 1E,F). These mice had similar glucose levels during the OGTT and slightly elevated levels during the ipITT compared to chow‐fed mice. Thus, DIO‐NASH mice were mildly insulin resistant but not overtly diabetic. Treatment with CHS‐131 both in low and high doses improved insulin sensitivity, indicated by the lower fasting insulin levels and lower area under the curve in the ipITT (Fig. 1F) without affecting glucose levels during fasting or the OGTT (Fig. 1E).
FIG. 1.
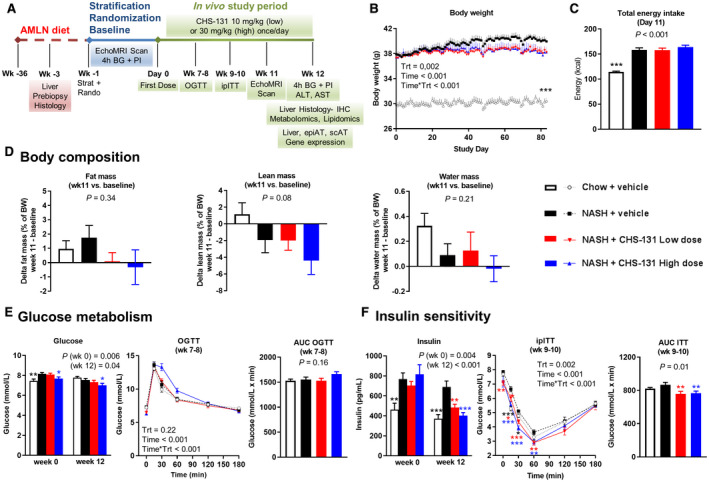
CHS‐131 improves insulin sensitivity without affecting body weight, body composition, energy intake, and glucose levels. (A) Schematic representation of the study design. (B) Body weight during the 12 weeks of treatment. Time is shown in minutes; Time*Trt indicates the interaction of parameters. (C) Total energy intake after 11 days of treatment. (D) Changes in body composition as percentage of body weight from baseline (start of treatment) up to week 11. (E) Glucose levels after 4 hours of fasting at start (week 0) and end (week 12) of treatment and during an OGTT at weeks 7‐8 (AUC of OGTT is also shown). (F) Insulin levels after 4 hours of fasting at start (week 0) and end (week 12) of treatment and during an ipITT at weeks 9‐10 (AUC of ipITT is also shown). One‐way ANOVA was performed for all single time‐point parameters (including body weight, which was analyzed only for the last study day) and two‐way ANOVA for parameters with multiple time points (OGTT, ipITT). For P < 0.05 (one‐way ANOVA) and for P < 0.05 for Trt (in two‐way ANOVA), one‐tailed *P < 0.05, **P < 0.01, ***P < 0.001, respectively, for post‐hoc LSD test for chow+vehicle (black stars) or NASH‐CHS‐131 low dose (red stars) or NASH‐CHS‐131 high dose (blue stars) compared to NASH+vehicle. Data show means ± SEMs. Abbreviations: AUC, area under the curve; BG, blood glucose; BW, body weight; PI, plasma insulin; min, minutes; Rando, randomized; Strat, stratification; Trt, treatment.
CHS‐131 Improves Plasma Levels of Liver Aminotransferases, TC, TG, and Adiponectin
DIO‐NASH mice had profoundly increased plasma levels of ALT, AST, and TC compared to mice fed a chow diet (Fig. 2A,B). Treatment with high‐dose CHS‐131 resulted in 37% lower levels of ALT, 29% of AST, and 20% of TC (Fig. 2A‐C). Plasma TGs were lower in DIO‐NASH mice compared to mice fed the chow diet, possibly due to increased uptake and accumulation of TGs in the liver. Treatment with CHS‐131 further decreased plasma TGs (Fig. 1B). Both low and high doses of CHS‐131 robustly increased plasma adiponectin levels (by 114% and 137% of mean levels, respectively) without affecting the elevated leptin levels in DIO‐NASH mice (Fig. 2C). Urea levels, as a marker of hydration, were not affected by CHS‐131 treatment (Fig. 2D).
FIG. 2.
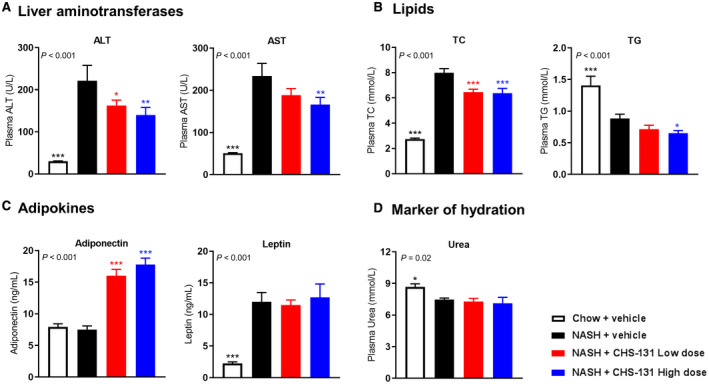
CHS‐131 reduces aminotransferases and TC and increases adiponectin levels. Plasma levels of (A) liver aminotransferases (ALT and AST), (B) lipids (TC and TG), (C) adipokines (adiponectin and leptin), and (D) urea (as marker of hydration) after 12 weeks of treatment. For P < 0.05 (one‐way ANOVA), one‐tailed *P < 0.05, **P < 0.01, ***P < 0.001 for post‐hoc LSD test for chow+vehicle (black stars) or NASH‐CHS‐131 low dose (red stars) or NASH‐CHS‐131 high dose (blue stars) compared to NASH+vehicle,. Data represent means ± SEMs.
CHS‐131 Improves the NAFLD Activity Score With Impacts on Lobular Inflammation and Hepatocellular Ballooning
A liver biopsy was performed after 33 weeks of the AMLN diet or normal chow, and mice were subsequently randomized after recovery at week 36 of the AMLN diet in the different treatment groups (Fig. 1A). After 33 weeks of the AMLN diet, DIO‐NASH mice had a high NAFLD activity score (NAS) (~5 points), whereas mice fed a chow diet had a very low score (11/12 mice with 0 points) (Supporting Fig. S1A). DIO‐NASH mice treated with high‐dose CHS‐131 had a significant reduction in the NAS (from baseline delta change in NAS, −0.77 points; seven mice with lower, five with the same, and one with higher NAS after treatment) compared to DIO‐NASH mice treated with vehicle (from baseline delta change in NAS, +0.15; two mice with lower, seven with the same, and four with higher NAS after treatment) (Fig. 3A,B; Supporting Fig. S1A). This finding was the result of combined improvement in lobular inflammation and hepatocellular ballooning (Fig. 3E,F; Supporting Fig. S1C‐E). Liver weight, lipid content, TG, TC, and steatosis score were not affected by treatment (Supporting Figs. S1B, S2, and S3D). Although fibrosis stage was not significantly altered (Fig. 3C; Supporting Fig. S1G,H), markers of liver fibrosis were reduced by 28% for HP, 24% for Col1a1, 21% for α‐SMA, and 18% for Gal‐3 (Fig. 4). Of note, the levels of Col1a1, α‐SMA, and Gal‐3 were strongly correlated (Supporting Table S3), indicating that mice treated with CHS‐131 that had the lowest Col1a1 also had the lowest α‐SMA and Gal‐3 levels.
FIG. 3.
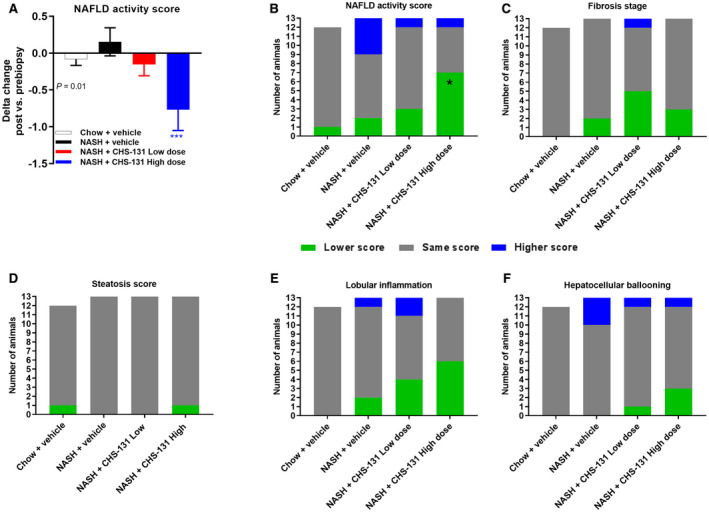
CHS‐131 reduces the NAFLD activity score mainly by improving lobular inflammation. (A) Comparison of histologic outcomes in posttreatment versus pretreatment biopsy. (B) Delta change of the NAS; number of animals with lower, same, or higher scores. NAS (C) fibrosis stage, (D) steatosis, (E) lobular inflammation, (F) hepatocellular ballooning. *** (blue stars) in (A) indicates P < 0.001 for post‐hoc LSD by one‐tailed ANOVA P = 0.01 and * (black star) in (B) indicates P < 0.05 for one‐tailed Fischer’s exact test compared to NASH+vehicle.
FIG. 4.
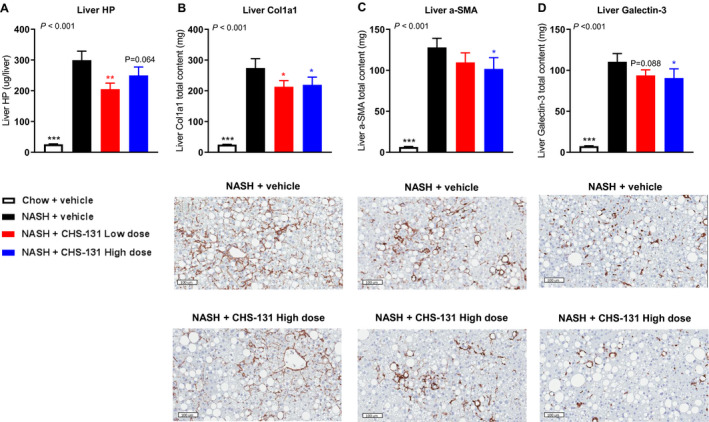
CHS‐131 does not affect the fibrosis stage but reduces markers of fibrosis, stellate cell activation, and inflammation. Liver content in (A) hydroxyproline, (B) Col1a1, (C) α‐SMA, and (D) Gal‐3. Images below (B‐D) represent IHC from NASH+vehicle and NASH+CHS‐131 high dose. For P < 0.05 (one‐tailed ANOVA), *P < 0.05, **P < 0.01, ***P < 0.001 for post‐hoc LSD test for chow+vehicle (black stars) or NASH‐CHS‐131 low dose (red stars) or NASH‐CHS‐131 high dose (blue stars) compared to NASH+vehicle. Data represent means ± SEMs.
CHS‐131 Normalizes the Changes in Lipid and Metabolite Composition in the Liver of DIO‐NASH Mice
A lipidomic analysis of liver tissues identified 987 different lipid species. sPLS‐DA demonstrated that with the use of two main components (Fig. 5A), each consisting of 10 lipid species, DIO‐NASH mice treated with CHS‐131 (blue dots at upper left) converge into separate clusters from DIO‐NASH mice treated with vehicle (green dots lower left) and mice fed a chow diet (red dots middle right), suggesting important differences in lipid profiles between the three groups. Component 1 (vertical separation) was responsible for the separation of chow mice from DIO‐NASH mice (both vehicle and CHS‐131 treated) and consisted of phosphatidylethanolamines (PEs) and phosphatidylcholines (PCs). Component 2 (horizontal separation) was responsible for the separation between vehicle and CHS‐131‐treated DIO‐NASH mice and consisted mainly of triacylglycerols (TAGs) and diacylglycerols (DAGs) and secondarily of PCs and PEs (Fig. 5A). Further analysis showed that despite the lack of change in total TGs and TCs (Supporting Fig. S2D), CHS‐131‐treated mice had lower concentrations in many TAGs (237 out of 518), DAGs (24 out of 54), and cholesterol ester (nine out of 26) species and higher concentrations in many PC esters, PE esters, and plasmalogens (Fig. 5B). Among the different TAG and DAG species, lower concentrations with CHS‐131 were observed in species with a large number of carbons in contrast to a trend for higher concentrations in species with smaller number of carbons (Fig. 5C), suggesting increased breakdown or reduced synthesis of the more complex lipids.
FIG. 5.
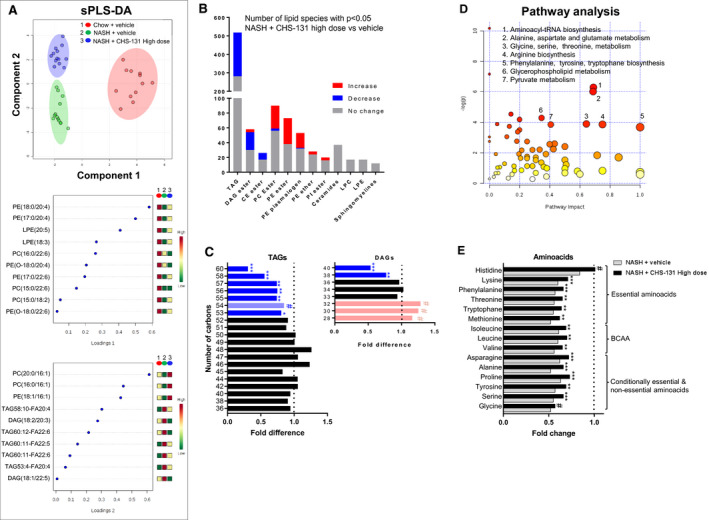
Effects of CHS‐131 treatment on hepatic lipidomic and metabolomic profile. (A) Scoreplot and loadings of the two main components in sPLS‐DA analysis. (B) Number of lipid species with significant increase, decrease, or no change in their hepatic concentrations after treatment with high‐dose CHS‐131 compared to vehicle. (C) Mean fold changes in hepatic concentrations of TAGs and DAGs according to the number of carbons after treatment with high‐dose CHS‐131 (columns) compared to vehicle (dotted line) in mice with NASH. (D) Score plot of pathway analysis indicating pathway impact (x axis) in relation to the P value (y axis) in metabolites in CHS‐131 versus vehicle‐treated mice with NASH. (E) Mean fold changes in amino acid concentrations in CHS‐131 (black columns) or vehicle‐treated (gray columns) NASH mice compared to non‐NASH chow‐fed mice (dotted line). # P = 0.05‐0.1, *P < 0.05, **P < 0.01, ***P < 0.001, two‐tailed Student t test or Mann‐Whitney test (for TAGs and DAGs) or Welch’s two‐sample t test (for amino acids) for the comparison with the NASH mice group treated with vehicle. Abbreviations: CE, cholesterol ester; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; PI, phosphatidylinositol; tRNA, transfer RNA.
Metabolomic analysis identified 592 known metabolites. Pathway analysis suggested that the most robust differences between the three groups were observed in amino acid synthesis and metabolism (Fig. 5D). DIO‐NASH mice treated with vehicle had ~50% lower concentration in 15 amino acids compared to mice fed a chow diet (essential, conditionally essential, and nonessential, including branched‐chain amino acids [BCAAs]). NASH mice treated with CHS‐131 still had lower levels in amino acids compared to chow mice but significantly increased levels when compared to NASH mice treated with vehicle (Fig. 5E).
CHS‐131 Alters Gene Expression Profiles Related to Mitochondrial Function and Inflammation in Adipose Tissue
We investigated whether CHS‐131 affected the expression of key genes involved in cellular metabolic procedures both in the liver and the adipose tissue. Mice treated with CHS‐131 demonstrated a profound increase in gene expression of key regulators of mitochondrial function. Specifically, the expression of uncoupling protein 1 (UCP1) and elongation of very long‐chain fatty acids‐like 3 (Elovl3), genes that are involved in browning and thermogenesis (Fig. 6A), were increased up to ~33‐fold in epididymal adipose tissue (epiAT) and up to ~80‐fold in subcutaneous adipose tissue (scAT) in mice treated with CHS‐131 (both with low and high doses) compared to mice treated with vehicle. Additionally in epiAT, a 2‐fold increase was observed in the gene expression of the major regulator of mitochondrial biogenesis, namely PPARγ coactivator 1 alpha (PGC‐1a), as well as in acyl‐coenzyme A (CoA) oxidase 1 (Acox1), which encodes the first enzyme of the fatty acid‐beta oxidation pathway that catalyzes the desaturation of acyl‐CoAs (Fig. 6B). The expression of genes (fatty acid synthase [Fasn], MLX interacting protein like [Mlxipl]) encoding proteins that stimulate the synthesis of long‐chain saturated fatty acids and TGs was reduced (both in epiAT and scAT for Fasn and mainly in scAT for Mlxipl) (Fig. 6D). These changes were observed in adipose tissue and together point toward increased browning and fatty acid oxidation and decreased long‐chain fatty acid and TG synthesis and agree with the lipid composition observed in the liver tissue of the CHS‐131‐treated mice. Of note, the expression of cell death‐inducing DNA fragmentation factor alpha‐like effector A (CIDEA) was also increased in epiAT and scAT, which suggests reduced lipolysis, and fatty acid‐binding protein 4 (Fabp4) was higher in the liver and tended to be higher in scAT (Fig. 6D; Supporting Fig. S3), which suggests increased lipid uptake in the CHS‐131‐treated mice. However, as described above, these changes did not result in higher body fat mass (Fig. 1D) or increased liver TGs and TCs (Supporting Fig. S2C,D). Regarding the expression of Ppars, no changes were observed in Pparg in epiAT, scAT (main expression sites), or in the liver (Fig. 6C; Supporting Fig. S3). The expression of Ppard was reduced both in epiAT and scAT, whereas the expression of Ppara was increased in epiAT but not in the liver (main expression site) (Fig. 6C; Supporting Fig. S3). Leptin receptor (Lepr) in epiAT as well as insulin receptor (Insr) and insulin receptor substrate 1 (Irs1) in scAT were reduced by ~50% (Fig. 6E). Finally, expression levels of the macrophage marker F4/80 and of the proinflammatory cytokine chemokine (C‐C motif) ligand 2 (Ccl2), which recruits monocytes, T cells, and dendritic cells, were reduced with a low dose of CHS‐131 in epiAT but not in scAT or in the liver (Fig. 6F; Supporting Fig. S3).
FIG. 6.
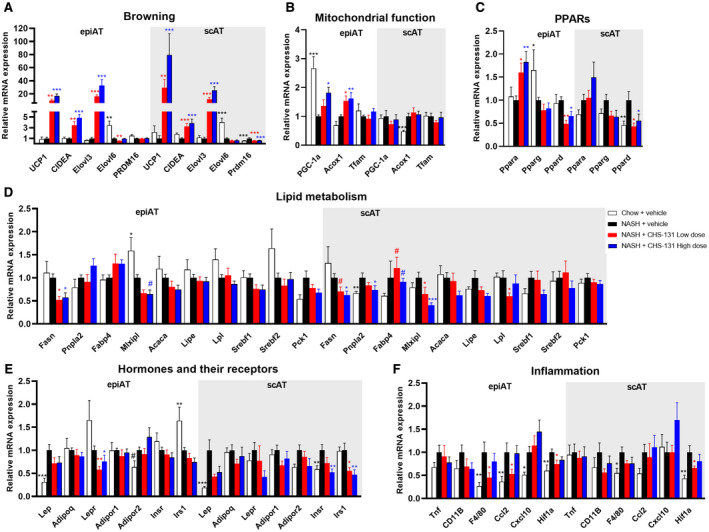
Effects of CHS‐131 treatment on the gene expression profile in epiAT and scAT. Relative mRNA expression levels of genes involved in (A) browning, (B) mitochondrial function, (C) PPARs, (D) lipid metabolism, (E) hormones and their receptors, (F) inflammation. For P < 0.05 with ANOVA or Kruskal‐Wallis test, # P = 0.05‐0.1, *P < 0.05, **P < 0.01, ***P < 0.001 for two‐tailed Fischer’s LSD or Dunn’s test for the comparison of each group (chow; NASH treated with CHS‐131 low dose or high dose) versus the NASH mice group treated with vehicle. Data represent means ± SEMs. Abbreviations: Acaca, acetyl‐coenzyme A carboxylase alpha; Adipoq, adiponectin, C1Q and collagen domain containing; Adipor, adiponectin receptor; CIDEA, cell death‐inducing DNA fragmentation factor alpha‐like effector; Cxcl10, C‐X‐C motif chemokine 10; Elovl, elongation of very long‐chain fatty acids‐like; Hif1a, hypoxia inducible factor 1 subunit alpha; Insr, insulin receptor; Irs1, insulin receptor substrate 1; Lep, leptin; Lepr, leptin receptor; Lipe, lipase, hormone sensitive; Lpl, lipoprotein lipase; mRNA, messenger RNA; Pck1, phosphoenolpyruvate carboxykinase 1; Pnpla2, patatin‐like phospholipase domain‐containing protein 2; PRDM, PR domain containing 16; Srebf, sterol regulatory element binding transcription factor; Tfam, transcription factor A, mitochondrial; Tnf, tumor necrosis factor; UCP1, uncoupling protein 1.
Discussion
We demonstrate herein that treatment with CHS‐131 improves liver histology by reducing lobular inflammation and decreasing hepatocyte ballooning in a diet‐induced and biopsy‐confirmed mouse model of NASH. Additionally, CHS‐131 ameliorates hepatic lipid composition by promoting a shift to TGs and DAGs with a lower number of carbons and increased hepatic amino acid levels. The beneficial effects of CHS‐131 on NASH are most probably achieved indirectly by changes in adipose tissue function, increased adiponectin levels, and systemic improvement of insulin sensitivity. Treatment with CHS‐131 did not lead to water retention and weight gain, which are common side effects observed in full PPARᵧ activators.
Previous studies have shown that CHS‐131 is distributed in higher concentrations in visceral adipose tissue and can normalize obesity‐related insulin‐signaling defects.( 13 , 14 ) In contrast to rosiglitazone, treatment of DIO mice for 14 days with CHS‐131 after a 16‐week high‐fat diet did not induce hepatic steatosis or hepatomegaly.( 14 ) In our study, we used an established mouse model of NASH( 16 , 17 ) in which liver inflammation and fibrosis occur in addition to liver steatosis, thus resembling the human phenotype of the disease. The most important finding of our study is the lower NAS score in mice treated with high‐dose CHS‐131 (equivalent to 10 mg rosiglitazone( 14 )) compared to vehicle. This lower score derives from an improvement of lobular inflammation and ballooning. Although steatosis in terms of lipid quantity did not change, intrahepatic lipid composition shifted to TGs and DAGs consisting of fatty acids with shorter chains (i.e., lower number of carbons). Shorter fatty acid chains have lower melting points, can be easily metabolized, and have been associated with reduced autophagy, improved liver status, and lower insulin resistance.( 18 , 19 ) This finding may be the result of changes in fatty acid synthesis, elongation, or oxidation with CHS‐131 treatment.( 20 ) In the liver, we observed an up‐regulation of Fabp4 and Fasn with CHS‐131 treatment, indicating increased fatty acid uptake and synthesis, and no changes in genes related to mitochondrial function and fatty acid oxidation. On the contrary, in adipose tissue, the expressions of Mlxipl, which encodes a transcription factor complex promoting TG synthesis, and Fasn were reduced by ~50%, whereas the expressions of PGC‐1a and Acox1, which stimulate fatty acid oxidation, were increased by ~80% and 60%, respectively.( 21 ) Thus, the changes of hepatic lipid composition (lipid quality) with CHS‐131 most probably resulted by the reduced fatty acid synthesis and increased fatty acid oxidation observed in adipose tissue and not in the liver.
An unexpected finding in our study is the decrease in amino acid concentrations in liver tissues with the AMLN diet; this decrease is partially restored with CHS‐131 treatment. In humans, especially the concentrations of circulating and hepatic BCAAs increase in progressive NAFLD( 22 ) and are high in obesity and T2D. In contrast, these metabolites decrease in liver cirrhosis, becoming exhausted by contributions from aminotransferase activity to ammonia detoxification and other pathways.( 23 ) Supplementation of BCAAs has been associated with improvement of hepatic steatosis, liver injury, glucose tolerance, and insulin sensitivity in several animal models.( 24 , 25 , 26 ) Albeit the partial restoration of amino acid levels by CHS‐131 treatment was not BCAA specific but was observed in all amino acids (including BCAAs), it still may contribute to the beneficial hepatic and metabolic effects of CHS‐131.
Despite the reduction in inflammatory foci with CHS‐131, we did not observe changes in the expression of genes encoding key regulators of inflammatory procedures in the liver. A 50% reduction of F4/80 and CCL2 was measured in epiAT with the lower but not the higher dose of CHS‐131. Previous studies involving shorter in vivo treatment or in vitro exposure to high‐dose CHS‐131 have demonstrated robust down‐regulation of multiple proinflammatory molecules in adipose tissue( 14 ) and in the brain( 27 ) but not in the liver.( 14 ) Thus, the reduction in hepatic inflammation is probably related to the down‐regulation of inflammatory procedures in adipose tissue. We also cannot rule out that changes in the expression of proinflammatory genes are more profound at the beginning of treatment (especially for the high CHS‐131 dose) or that posttranslational modifications contribute to the mitigation of hepatic and adipose tissue inflammation. This will explain both the histologic findings showing reduced lobular inflammation as well as the lower hepatic levels of Gal‐3, which is an established marker of macrophage activation and a potent mitogen of fibroblasts.( 28 )
Another important mediator of the metabolic and hepatic effects of PPARᵧ activators is adiponectin.( 29 , 30 ) Adiponectin stimulates fatty acid oxidation, decreases inflammation and fibrosis, and improves insulin sensitivity.( 31 , 32 , 33 ) Adiponectin levels progressively decrease from NAFL to NASH and have been used to develop noninvasive diagnostic tools for staging NAFLD.( 34 , 35 ) Treatment with CHS‐131 increased the circulating levels of adiponectin ~2.4‐fold, which is similar to the increase observed in humans treated with pioglitazone.( 30 )
In contrast to the beneficial effects on the NAS score, we observed more limited changes in fibrosis stages with CHS‐131. We did not observe a regression of fibrosis, which may demand a longer treatment period or may have been limited by the long duration of diet in our mouse model, but importantly fibrosis did not progress. Additionally, we observed lower levels in HP, Col1a1, α‐SMA, and Gal‐3, which are all sensitive markers of the short‐term effect of treatment on liver fibrosis. Specifically, α‐SMA and collagen type I increase with the regulation of quiescent hepatic stellate cell activation and their subsequent activation into myofibroblast‐like cells. The activated hepatic stellate cells are the main collagen‐producing cells in the liver.( 36 , 37 ) The lower α‐SMA levels with CHS‐131 treatment may suggest either lower activation or reduced proliferation and increased apoptosis of hepatic stellate cells, in line with findings described for full PPARγ agonists.( 38 ) In humans and according to several meta‐analyses, PPARγ activation seems to be more beneficial for the improvement of steatosis and necroinflammation than of fibrosis.( 39 , 40 , 41 ) However, as was recently shown, PPARγ activation may delay the progress of liver fibrosis in patients with T2D; these patients have more profound glucometabolic abnormalities, worse prognosis of NAFLD, and could probably benefit more from PPARγ activation( 42 ) compared to euglycemic and less insulin‐resistant individuals. Our mouse model was characterized by mild insulin resistance and no overt diabetes. CHS‐131 treatment improved insulin sensitivity and liver histology, showing that it can be effective even in milder conditions. However, it is plausible to expect that the benefit in NAFLD with CHS‐131 will be even more profound in overt diabetes and advanced insulin resistance.
An important limitation in the treatment with thiazolidinediones is the increase in body weight due to water retention, which is commonly observed and increases the risk of hospitalization for heart failure.( 8 , 9 ) Water retention can be even more profound in patients that are co‐administered insulin.( 43 ) In our study, CHS‐131 at low or high dose did not increase body weight, did not affect whole body fat and lean mass, and most importantly did not lead to increase water retention, in line with the findings observed with CHS‐131 in humans.( 10 ) Although we did not assess individually visceral, subcutaneous, or brown adipose tissue fat mass, we observe an up to 80‐fold stimulation of genes involved in adipose tissue browning and thermogenesis, which is much higher compared to the up‐regulation observed with short‐term CHS‐131 treatment (up to 10‐fold)( 14 ) or other PPARγ agonists.( 44 )
Several treatments against NAFLD are currently being evaluated in phase II and phase III clinical trials (reviewed in Polyzos et al.( 45 )), and among them are dual PPARα/δ (elafibranor), PPARα/γ (saroglitazar), or pan‐PPAR (lanifibranor) agonists. PPARα/δ agonists are safe but demonstrate rather modest, if any, improvement of NASH, insulin resistance, and dyslipidemia in humans.( 46 ) PPARα/γ or pan‐PPAR agonists may be effective but are associated with more severe side effects, mainly due to the full PPARγ agonism.( 46 ) Additionally, dual or triple agonism through one single medication offers less flexibility for dose adjustments, which makes it more challenging to achieve the right balance between benefit and side effects. Thus, if the beneficial effects of CHS‐131 are confirmed in humans with NAFLD, it may be an excellent therapeutic alternative for improving hepatic inflammation and NASH while preventing at the same time the development or treating and limiting the progression of diabetes. Additionally, it may be possible to combine CHS‐131 with medications that target the PPARα receptor because in our study the expression of PPARα is not affected in the liver and is increased in visceral adipose tissue by treatment with CHS‐131. Finally, it may be possible to combine CHS‐131 with medications that improve liver fibrosis but have a neutral or even negative metabolic effect by promoting dyslipidemia and increasing insulin resistance, such as farnesoid X receptor agonists (e.g., obeticholic acid).( 47 ) In these combinations, CHS‐131 may be able to improve metabolic status while further enhancing the beneficial effects of those medications in the liver.
In conclusion, we show that CHS‐131 can be an effective treatment in established NASH and is operational by improving metabolic status without increasing body weight gain or resulting in excessive water retention in mice. Future studies should evaluate the efficacy of CHS‐131 in humans with NAFLD who are metabolically at risk (those with obesity with insulin resistance or with T2D).
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Supported by Coherus Biosciences (grant number 2019‐01 to C.S.M.), the Deutsche Forschungsgemeinschaft (grant number 389891681 [PE2431/2‐1] to Ni.P.), and Supported by NIH (grant number K24DK081913 to C.S.M.).
Potential conflict of interest: Dr. Feigh and Dr. Veidal are employed by Gubra. Dr. Fleming and Dr. Rosen own stock in and are employed by Coherus. Dr. Mantzoros owns stock in, consults for, and received grants from Coherus. Dr. Veidal owns stock in and is employed by Gubra. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2019;69:2672‐2682. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta‐analysis. J Hepatol 2019;71:793‐801. [DOI] [PubMed] [Google Scholar]
- 3. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple‐hit pathogenesis of non‐alcoholic fatty liver disease (NAFLD). Metabolism 2016;65:1038‐1048. [DOI] [PubMed] [Google Scholar]
- 4. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al.; NASH CRN . Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C, et al. Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 2016;165:305‐315. [DOI] [PubMed] [Google Scholar]
- 6. Skat‐Rordam J, Hojland Ipsen D, Lykkesfeldt J, Tveden‐Nyborg P. A role of peroxisome proliferator‐activated receptor gamma in non‐alcoholic fatty liver disease. Basic Clin Pharmacol Toxicol 2019;124:528‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 8. Upadhyay J, Polyzos SA, Perakakis N, Thakkar B, Paschou SA, Katsiki N, et al. Pharmacotherapy of type 2 diabetes: an update. Metabolism 2018;78:13‐42. [DOI] [PubMed] [Google Scholar]
- 9. Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg Chem 2018;77:548‐567. [DOI] [PubMed] [Google Scholar]
- 10. DePaoli AM, Higgins LS, Henry RR, Mantzoros C, Dunn FL; INT131‐007 Study Group . Can a selective PPARgamma modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 2014;37:1918‐1923. [DOI] [PubMed] [Google Scholar]
- 11. Dunn FL, Higgins LS, Fredrickson J, DePaoli AM; INT131‐004 Study Group . Selective modulation of PPARgamma activity can lower plasma glucose without typical thiazolidinedione side‐effects in patients with type 2 diabetes. J Diabetes Complications 2011;25:151‐158. [DOI] [PubMed] [Google Scholar]
- 12. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 13. Lee DH, Huang H, Choi K, Mantzoros C, Kim YB. Selective PPARgamma modulator INT131 normalizes insulin signaling defects and improves bone mass in diet‐induced obese mice. Am J Physiol Endocrinol Metab 2012;302:E552‐E560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xie X, Chen W, Zhang N, Yuan M, Xu C, Zheng Z, et al. Selective tissue distribution mediates tissue‐dependent PPARgamma activation and insulin sensitization by INT131, a selective PPARgamma modulator. Front Pharmacol 2017;8:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chong J, Xia J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018;34:4313‐4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J, et al. Obese diet‐induced mouse models of nonalcoholic steatohepatitis‐tracking disease by liver biopsy. World J Hepatol 2016;8:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J Gastroenterol 2018;24:179‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang ME, Singh BK, Hsu MC, Huang C, Yen PM, Wu LS, et al. Increasing dietary medium‐chain fatty acid ratio mitigates high‐fat diet‐induced non‐alcoholic steatohepatitis by regulating autophagy. Sci Rep 2017;7:13999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yew Tan C, Virtue S, Murfitt S, Roberts LD, Phua YH, Dale M, et al. Adipose tissue fatty acid chain length and mono‐unsaturation increases with obesity and insulin resistance. Sci Rep 2015;5:18366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sassa T, Kihara A. Metabolism of very long‐chain fatty acids: genes and pathophysiology. Biomol Ther (Seoul) 2014;22:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagattin A, Hugendubler L, Mueller E. Transcriptional coactivator PGC‐1alpha promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci U S A 2010;107:20376‐20381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, et al. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 2015;47:603‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holecek M. Branched‐chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab (Lond) 2018;15:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hinault C, Mothe‐Satney I, Gautier N, Lawrence JC, Jr. , Van Obberghen E. Amino acids and leucine allow insulin activation of the PKB/mTOR pathway in normal adipocytes treated with wortmannin and in adipocytes from db/db mice. FASEB J 2004;18:1894‐1896. [DOI] [PubMed] [Google Scholar]
- 25. Honda T, Ishigami M, Luo F, Lingyun M, Ishizu Y, Kuzuya T, et al. Branched‐chain amino acids alleviate hepatic steatosis and liver injury in choline‐deficient high‐fat diet induced NASH mice. Metabolism 2017;69:177‐187. [DOI] [PubMed] [Google Scholar]
- 26. Macotela Y, Emanuelli B, Bang AM, Espinoza DO, Boucher J, Beebe K, et al. Dietary leucine—an environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS One 2011;6:e21187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Omeragic A, Saikali MF, Currier S, Volsky DJ, Cummins CL, Bendayan R. Selective peroxisome proliferator‐activated receptor‐gamma modulator, INT131 exhibits anti‐inflammatory effects in an EcoHIV mouse model. FASEB J 2020;34:1996‐2010. [DOI] [PubMed] [Google Scholar]
- 28. Sciacchitano S, Lavra L, Morgante A, Ulivieri A, Magi F, De Francesco GP, et al. Galectin‐3: one molecule for an alphabet of diseases, from A to Z. Int J Mol Sci 2018;19:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Mendonca M, Dos Santos BAC, de Sousa E, Rodrigues AC. Adiponectin is required for pioglitazone‐induced improvements in hepatic steatosis in mice fed a high‐fat diet. Mol Cell Endocrinol 2019;493:110480. [DOI] [PubMed] [Google Scholar]
- 30. Gastaldelli A, Harrison S, Belfort‐Aguiar R, Hardies J, Balas B, Schenker S, et al. Pioglitazone in the treatment of NASH: the role of adiponectin. Aliment Pharmacol Ther 2010;32:769‐775. [DOI] [PubMed] [Google Scholar]
- 31. Ishtiaq SM, Rashid H, Hussain Z, Arshad MI, Khan JA. Adiponectin and PPAR: a setup for intricate crosstalk between obesity and non‐alcoholic fatty liver disease. Rev Endocr Metab Disord 2019;20:253‐261. [DOI] [PubMed] [Google Scholar]
- 32. Liu X, Perakakis N, Gong H, Chamberland JP, Brinkoetter MT, Hamnvik OR, et al. Adiponectin administration prevents weight gain and glycemic profile changes in diet‐induced obese immune deficient Rag1‐/‐ mice lacking mature lymphocytes. Metabolism 2016;65:1720‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polyzos SA, Perakakis N, Mantzoros CS. Fatty liver in lipodystrophy: a review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019;96:66‐82. [DOI] [PubMed] [Google Scholar]
- 34. Boutari C, Perakakis N, Mantzoros CS. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol Metab (Seoul) 2018;33:33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perakakis N, Polyzos SA, Yazdani A, Sala‐Vila A, Kountouras J, Anastasilakis AD, et al. Non‐invasive diagnosis of non‐alcoholic steatohepatitis and fibrosis with the use of omics and supervised learning: a proof of concept study. Metabolism 2019;101:154005. [DOI] [PubMed] [Google Scholar]
- 36. Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, et al. Alpha‐SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig Liver Dis 2005;37:349‐356. [DOI] [PubMed] [Google Scholar]
- 37. Hou W, Syn WK. Role of metabolism in hepatic stellate cell activation and fibrogenesis. Front Cell Dev Biol 2018;6:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Panebianco C, Oben JA, Vinciguerra M, Pazienza V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: a putative synergy between retinoic acid and PPAR‐gamma signalings. Clin Exp Med 2017;17:269‐280. [DOI] [PubMed] [Google Scholar]
- 39. Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta‐analysis: pioglitazone improves liver histology and fibrosis in patients with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther 2012;35:66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Said A, Akhter A. Meta‐analysis of randomized controlled trials of pharmacologic agents in non‐alcoholic steatohepatitis. Ann Hepatol 2017;16:538‐547. [DOI] [PubMed] [Google Scholar]
- 41. Singh S, Khera R, Allen AM, Murad MH, Loomba R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: a systematic review and network meta‐analysis. Hepatology 2015;62:1417‐1432. [DOI] [PubMed] [Google Scholar]
- 42. Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila‐Pico C, Liu IC, et al. Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin Gastroenterol Hepatol 2018;16:558‐566.e2. [DOI] [PubMed] [Google Scholar]
- 43. Scheen AJ. Combined thiazolidinedione‐insulin therapy: should we be concerned about safety? Drug Saf 2004;27:841‐856. [DOI] [PubMed] [Google Scholar]
- 44. Sohn JH, Kim JI, Jeon YG, Park J, Kim JB. Effects of three thiazolidinediones on metabolic regulation and cold‐induced thermogenesis. Mol Cells 2018;41:900‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Polyzos SA, Kang ES, Boutari C, Rhee EJ, Mantzoros CS. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism 2020. Mar 6:154203 10.1016/j.metabol.2020.154203. [DOI] [PubMed] [Google Scholar]
- 46. Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab 2018;27:22‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dufour JF, Caussy C, Loomba R. Combination for therapy non‐alcoholic steatohepatitis: rationale, opportunities and challenges. Gut 2020. 10.1136/gutjnl-2019-319104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material