

Abstract
Parkinson’s disease (PD) is a multifactorial disorder characterized by progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the presence of Lewy bodies (LBs) consisting of misfolded α-synuclein protein. The etiology of PD is still not clear but systemic inflammation is proved to trigger and exacerbate DA neurons degeneration. Toll-like receptor 4 (TLR4) is a pattern-recognition receptor (PRR) and plays a major role in promoting the host immune. TLR4-mediated signal pathways induce the release of many inflammatory cytokines. It is reasonable to hypothesize that TLR4 is the mediator in microglia contributing to the damage of DA neurons in the SNpc. In this study, we evaluated the role of TLR4 in the chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)/probenecid mouse model. Both TLR4-deficient and wild-type (WT) mice were injected with probenecid (250 mg/kg, i.p.) followed by injection of MPTP (25 mg/kg, s.c.) every 4 days for 10 times. From D43 to D47, the behavioral performance in pole test and wire hang test was assessed. Then the mice were euthanized, and SN and striatum were dissected out for biochemical tests. We showed that compared with MPTP-treated WT mice, TLR4 deficiency significantly attenuated MPTP-induced motor deficits and TH-protein expression reduction in SNpc and striatum, suppressed MPTP-induced α-synuclein abnormality and neuroinflammation mediated through oxidative stress, glial activation, NF-κB and the NLRP3 inflammasome signaling pathways. These findings highlight the neuroprotective effect of TLR4-pathways in the chronic MPTP-induced PD mouse model.
Keywords: Parkinson’s disease, Toll-like receptor 4, MPTP/probenecid mouse model, α-synuclein, oxidative stress, glial activation, NF-κB, neuroinflammation
Introduction
Parkinson’s disease (PD) is an age-related neurodegenerative disease that is typified by resting tremor, slowness of movement, postural instability, and muscle rigidity [1]. The most typical pathological characteristics of PD are the progressive death of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the emergence of Lewy bodies (LBs) composed of aggregated and misfolded presynaptic protein α-synuclein [2]. The causes of sporadic PD remain controversial, but many data indicate that significant inflammatory responses, including activated microglia and increased cytokine expression, have been reported in the SN of PD patient brains. Thus, neuroinflammation is regarded as a key contributor to the pathogenesis of PD [3].
α-Synuclein protein was initially shown to be involved in PD pathogenesis. Point mutations and overexpression of the α-synuclein gene SNCA have been linked with familial PD [4]. In the healthy brain, α-synuclein is available in its native conformation of a soluble monomer, which exerts its physiological function in neuron presynaptic termini [5]. Upset of this balance may mediate the transformation of α-synuclein into aggregated or disease-related forms. Although the potential mechanisms that induce the aggregation of α-synuclein are still uncertain, α-synuclein aggregation is well accepted to play a central role in the neuronal death and neuroinflammation of PD [6]. Existing studies suggest that aggregated α-synuclein triggers abnormal microglia activation and enhances prion-like cell-to-cell spread of misfolded α-synuclein [7].
Toll-like receptors (TLRs) are a family of pattern-recognition receptors (PRRs) that have a close relationship with host immune responses. TLRs can be activated by pathogen-associated molecular patterns (PAMPs) but also endogenous damage-associated molecular patterns (DAMPs) [8]. Microglia are the major phagocytes and serve immune-like functions in the brain. TLR4 is highly expressed on the microglia membrane and is highly involved in neuroinflammation during central nervous system (CNS) injury [9]. α-Synuclein is able to activate microglia as a DAMP, and TLR4 is involved in the inflammatory response induced by α-synuclein, inciting the production of pro-inflammatory cytokines [10]. Additionally, a recent study indicated that TLR4 is upregulated in the MPTP-treated mouse model [11–13]. Therefore, TLR4 is likely the mediator in microglia promoting the release of inflammatory molecules, thereby injuring DA neurons in the SNpc.
Interleukin (IL)-1β exerts a key role in the regulation of immune and inflammatory responses, and the production of active IL-1β is modulated by inflammasomes [14]. The inflammasome is a multiprotein complex that senses intracellular infectious pathogens as well as various host danger signals [15]. The NLRP3 inflammasome is the most extensively studied inflammasome in the CNS. The assembly of the inflammasome leads to the cleavage of procaspase-1 to mature caspase-1, and active caspase-1 shears pro-IL-1β into the bioactive form IL-1β [16]. Recently, α-synuclein fibrils have been demonstrated to activate the NLRP3 inflammasome, resulting in the production of IL-1β [17].
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neurotoxin that induces PD-like features, including unique behavior, dopamine neuron degeneration, upregulation of α-synuclein levels, and microglia and astrocyte activation in the SNpc and striatum [18, 19]. MPTP is capable of penetrating the blood–brain barrier (BBB) and is catalyzed into the toxic form MPP+ by monoamine oxidase B (MAO-B) in glial cells. MPP+ can be transferred into DA neurons, thereby inhibiting mitochondrial complex I and inducing neuronal degeneration [20]. The chronic MPTP/probenecid model is an improvement over the acute and subacute models and has an advantage in its ability to allow for the exploration of PD disease progression and mechanisms [21]. Based on these discoveries, we aimed to investigate the role of TLR4 in the chronic mouse MPTP model to determine whether TLR4-deficient mice are less vulnerable to MPTP toxicity than wild-type mice.
Materials and methods
Reagents and antibodies
MPTP hydrochloride and probenecid were obtained from Sigma-Aldrich (St Louis, MO, USA). The primary antibodies against NF-κB/p65, α-synuclein (C-20), tyrosine hydroxylase (TH), caspase-1, and ASC were purchased from Santa Cruz Biotechnology (CA, USA). p-NF-κB/p65 was purchased from Cell Signaling Technology (Beverly, MA, USA). The anti-aggregated α-synuclein (5G4) antibody was ordered from EMD Millipore (Darmstadt, Germany). The nitrated α-synuclein (Tyr125/Tyr133) antibody was obtained from Thermo (Waltham, MA, USA). The phosphorylated α-synuclein (Ser129), NLRP3, 8-OHdG and IL-1β antibodies were purchased from Abcam (Cambridge, MA, USA). The anti-GFAP antibody was purchased from Dako (Glostrup, Denmark), and the Iba-1 antibody was purchased from Wako (Osaka, Japan). The monoclonal antibody against β-actin was obtained from Sigma-Aldrich (St. Louis, MO, USA). All horseradish peroxidase-conjugated secondary antibodies were purchased from KPL (Gaithersburg, MD, USA). All antibodies are shown in Table 1.
Table 1.
The information of the antibodies used in the present work
| Antibody | Source | Dilutions | Company |
|---|---|---|---|
| NF-κB/p65 | Mouse | 0.388889 | Santa Cruz Biotechnology |
| α-Synuclein (C-20) | Rabbit | 0.388889 | Santa Cruz Biotechnology |
| TH | Mouse | 0.180556 | Santa Cruz Biotechnology |
| Caspase-1 | Mouse | 0.388889 | Santa Cruz Biotechnology |
| ASC | Rabbit | 0.388889 | Santa Cruz Biotechnology |
| p-NF-κB/p65 | Rabbit | 0.736111 | Cell Signaling Technology |
| α-Synuclein (5G4) | Mouse | 0.736111 | EMD Millipore |
| n-α-Synuclein | Mouse | 0.736111 | Thermo |
| p-α-Synuclein | Rabbit | 0.736111 | Abcam |
| NLRP3 | Rabbit | 0.736111 | Abcam |
| IL-1β | Rabbit | 0.736111 | Abcam |
| 8-OHdG | Mouse | 0.111111 | Abcam |
| β-Actin | Mouse | 0.736111 | Sigma |
| GFAP | Rabbit | 0.180556 | Dako |
| Iba-1 | Rabbit | 0.180556 | Wako |
Animals
Male TLR4-deficient mice (TLR4-/-, C57BL/10ScNJNju, aged 8 weeks) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Male wild-type C57BL/6 mice (8 weeks, Vital River Laboratory Animal Technology Co. Ltd, Beijing, China) were used in the experiments. All animals were fed under a 12-h light/dark cycle at room temperature 22 ± 1 °C. The animals had free access to standard diet and water. All experiments were carried out in compliance with the guidelines established by the National Institutes of Health for the care and use of laboratory animals and were approved by the Animal Care Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences.
MPTP treatment
After 1 week of acclimatization, TLR4-/- mice and wild-type (WT) mice were randomly divided into control and model groups, respectively. Then, the mice were pretrained in the pole test and wire hang test. The chronic mouse model was prepared as follows (Fig. 1). The mice in the model group were treated with an intraperitoneal injection of probenecid (250 mg/kg dissolved in 0.1 M NaOH-Tris-HCl, pH = 7.4). After 1 h, the mice were subcutaneously injected with MPTP-HCl (25 mg/kg, dissolved in 0.9% saline). Control mice were injected with equal volumes of saline. After injection, heat pads and radiators were used to keep the mice warm. MPTP/p injections were performed every 4 days on a 40-day schedule. From Day 43 to Day 47, all groups were assessed for behavioral performance by the pole test and wire hang test.
Fig. 1.
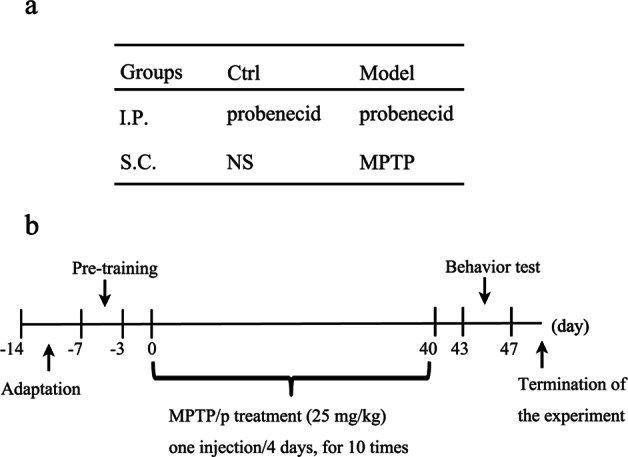
Schematic of the experimental procedure. a Treatment of each group. b Schedule for this study
Behavior testing
Pole test
The pole test was performed to evaluate motor function as described in our previous work [13]. This apparatus includes a 50-cm-long wooden pole wrapped in gauze and a 0.5-cm diameter ball on the top. The pole was placed in the plastic cage, which was filled with padding to protect the mice. The mice were trained on the pole apparatus to ensure that all mice could climb down as soon as they were put on the ball. The time spent climbing down from the pole was recorded.
Wire hang test
The wire hang test is used to assess the muscular strength of mice. The method was modified from a previous study [22]. A single animal was positioned on a wire lid of a standard mouse cage, and then the lid was inverted. The vertical distance between the wire lid and the bottom of the cage was 37 cm. The time that the mouse stood with at least two limbs on the lid was tested. With 180 s per trial, each mouse was tested three times, and the longest hang time was recorded.
Tissue preparation
Animals were anesthetized with chloral hydrate (400 mg/kg, i.p.). For biochemical tests, dissected tissues of the SN and striatum were rapidly isolated and stored at −80 °C. For the histologic analysis, mice were transcardially perfused with 0.1 M phosphate-buffered saline followed by 4% paraformaldehyde. The collected brain tissues were preserved in 4% paraformaldehyde and embedded in paraffin 24 h later. Paraffin sections (4 μm thick) of the midbrain and striatum were cut for immunofluorescence and immunohistochemical procedures.
Western blotting assay
Lysis buffer was prepared with a protease inhibitor cocktail and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO, USA), and the striatal tissues were homogenized with tenfold (w/v) cold lysis buffer in an ice bath for 30 min. Then, the protein extracts were centrifuged for 30 min at 12 000 × g, and the supernatant was collected. Protein concentrations were measured using a BCA kit (Applygen Technologies Inc., Beijing, China). The protein supernatant was denatured in loading buffer and boiled in water for 10 min, and the protein samples were stored at −20 °C. Then, 30 μg of protein from each group was separated by 15% SDS-PAGE gels and electrophoresis and subsequently transferred onto a PVDF membrane (Millipore, Boston, MA, USA). BSA (3%; Sigma-Aldrich, St. Louis, MO, USA) was used to block the membranes for 2 h at room temperature. The membranes were then incubated with primary antibodies overnight at 4 °C. Details of the antibodies used in the present work are shown in Table 1. After the membranes were washed three times with tris-buffered saline containing 0.1% Tween-20 (TBST), they were incubated with the horseradish peroxidase-conjugated secondary antibody (1:5000, KPL, Gaithersburg, MD, USA) for 1.5 h at room temperature. The protein levels were detected by the enhanced chemiluminescence plus detection system (ImageQuant LAS4000 mini, GE, Fairfield, CT, USA) and were quantified with image analysis software (Quantity One, Toyobo, Japan).
Immunohistochemistry and immunofluorescence
Paraffin slices were placed successively into dimethylbenzene for 10 min and different ethanol concentrations for 5 min each. After deparaffinization and rehydration, slices were subjected to citrate buffer (0.1 M, pH 6.0) at 95 °C for 10 min for antigen retrieval. After the tissue was washed three times with phosphate-buffered saline containing 0.2% Tween-20 (PBST) for 10 min, the sections were treated with 0.5% Triton X-100 for 10 min and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. For immunohistochemistry, slides were incubated in 3% hydrogen peroxide (H2O2) for 10 min to remove endogenous peroxidase activity before blocking nonspecific antigens. After blocking, the sections were incubated with anti-TH (1:250), anti-Iba-1 (1:200), or anti-GFAP (1:200) antibodies overnight at 4 °C. For immunofluorescence, the slides were then incubated with secondary antibodies (AlexaFluor 488-conjugated goat anti-mouse antibody and AlexaFluor 546-conjugated goat anti-rabbit antibody, 1:500 dilution, Invitrogen, Carlsbad, CA, USA) and Hoechst 33342 (1:1000 dilution, Life Technologies, Grand Island, NY, USA) for 1 h. All images were acquired using a confocal microscope (Leica TCS SP2, Solms, Germany). For immunohistochemistry, samples were incubated with the horseradish peroxidase-conjugated secondary antibody (1:200, KPL, Gaithersburg, MD, USA) for 1 h, and then samples were detected with 3,3'-diaminobenzidine (DAB) for 2–3 min. Finally, the sections were cover-slipped with neutral balsam and observed with an Olympus BA51 photomicroscope (Tokyo, Japan). Image Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA) was used for cell counting.
ROS assessment
Dihydroethidium (DHE) staining was used to evaluate reactive oxygen species (ROS) generation according to the manufacturer’s protocol. After dewaxing and dehydration, brain slices were incubated with DHE (100 μM, Sigma-Aldrich, St. Louis, MO, USA) for 90 min in the dark, followed by three washes with phosphate-buffered saline (PBS). The slides were mounted with neutral balsam and then visualized by a confocal laser scanning microscope (Leica TCS SP2, Solms, Germany).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad, San Diego, CA, USA). Results are expressed as the mean ± SD and were statistically analyzed by two-way ANOVA followed by Bonferroni’s multiple comparisons test. The ‘n’’ indicates the number of animals or tissue sections used from different animals. Statistical differences were considered significant at P < 0.05.
Results
TLR4 deficiency attenuated the motor impairment induced by MPTP
To determine the effects of TLR4 on motor function, pole tests and wire hang tests were performed. The behavioral evaluation with the pole test showed that MPTP-injected WT mice spent a longer time to complete the test than the mice not given MPTP (P < 0.05). However, the MPTP-treated TLR4-deficient mice performed better than the MPTP-treated WT mice (P < 0.05; Fig. 2a). The wire hang test was used to measure muscular strength. WT mice treated with MPTP had a significant reduction in hanging times compared to mice in the control group (P < 0.01). However, no significant differences were found between the control mice and the MPTP-treated TLR4-deficient mice. Compared with MPTP-treated WT mice, the MPTP-treated TLR4-deficient mice exhibited an obvious improvement in wire hang performance (P < 0.01; Fig. 2b).
Fig. 2.

TLR4 deficiency attenuated the motor impairment induced by MPTP. a Behavioral changes were assessed using a pole test. b Behavioral changes were tested using the wire hang test. The data are expressed as the mean ± SD (n > 6). *P < 0.05, **P < 0.01 vs. control group of WT mice; #P < 0.05, ##P < 0.01 vs. MPTP-treated WT mice
TLR4 deficiency ameliorated the reduction in TH-protein expression and TH-positive dopamine neurons damaged by MPTP
TH is an enzyme involved in DA synthesis; therefore TH+ cells represent dopamine neurons. The results of immunohistochemical analysis in the SNpc showed that the number of TH-positive cells was markedly decreased in both WT (P < 0.01) and TLR4-deficient mice (P < 0.01) after MPTP exposure. However, TLR4-deficient mice had an increased number of surviving TH-positive cells in comparison to WT mice (P < 0.01; Fig. 3a, c). Immunohistochemical staining results in the striatum indicated that MPTP treatment resulted in a loss of dopaminergic terminals in WT (P < 0.01) and TLR4-deficient mice (P < 0.05). The density of TH-positive fibers exhibited a significant change in both genotype groups after MPTP treatment (P < 0.05; Fig. 3b, d). Furthermore, TLR4 deficiency ameliorated the loss of TH-positive cells, which was also confirmed by Western blotting results (Fig. 3e).
Fig. 3.

TLR4 deficiency reversed the reduction in the TH protein level and number of TH-positive dopamine neurons induced by MPTP. a and c Immunohistochemical staining and quantitative analysis of TH in the SNpc (scale bar = 500 μm, 200 μm). b and d Immunohistochemical staining and quantitative analysis of TH in the striatum (scale bar = 1000 μm, 200 μm). e Western blotting results for TH protein levels in the striatum. The data are expressed as the mean ± SD (n = 3). **P < 0.01 vs. control group of WT mice; &P < 0.05, &&P < 0.01 vs. control group of TLR4-deficient mice; #P < 0.05, ##P < 0.01 vs. MPTP-treated WT mice
TLR4 deficiency suppressed the activation of astrocytes and microglia induced by MPTP
We examined MPTP-induced astrocyte and microglia activation in the SNpc using immunofluorescence analysis. As shown in Fig. 4, MPTP exposure significantly increased the number of both astrocytes and microglia in WT (P < 0.01) and TLR4-deficient mice (P < 0.05). In addition, TLR4-deficient mice exhibited obviously fewer activated astrocytes and microglia than the MPTP-treated WT mice (P < 0.01).
Fig. 4.
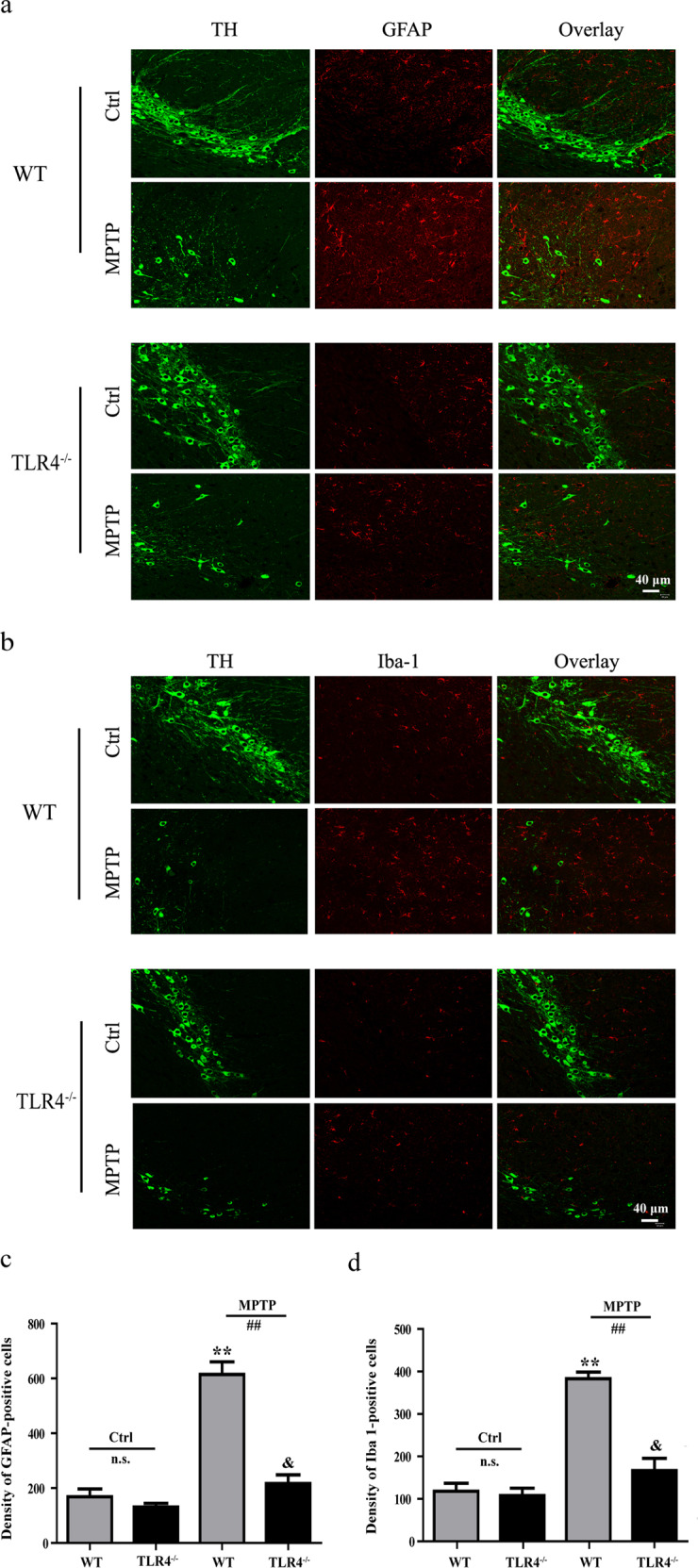
TLR4 deficiency suppressed the activation of astrocytes and microglia induced by MPTP. a and b Immunofluorescence staining of GFAP- and Iba-1-immunoreactive cells in the SNpc (scale bar = 40 μm). c and d Quantitative analysis of GFAP- and Iba-1-immunoreactive cells. The data are expressed as the mean ± SD (n = 3). **P < 0.01 vs. control group of WT mice; &P < 0.05 vs. control group of TLR4-deficient mice; ##P < 0.01 vs. MPTP-treated WT mice
TLR4 deficiency exerted a protective effect on MPTP-induced α-synuclein abnormalities
We detected that disease-associated α-synuclein was upregulated in WT mice after MPTP treatment with the anti-aggregated α-synuclein (5G4) antibody (P < 0.01; Fig. 5a). The 5G4 antibody has a high reactivity for disease-associated α-synuclein and does not recognize the monomeric form of α-synuclein [23–25]. Post translational modifications of α-synuclein have been shown to participate in the disease course of PD. The levels of nitrated-α-synuclein and the phosphorylation of α-synuclein at Ser129 were determined across groups. Similar to the levels of aggregated α-synuclein, the levels of nitrated-α-synuclein (P < 0.01) and phosphorylated-α-synuclein (P < 0.05) were notably higher in MPTP-treated WT mice than in untreated WT mice (Fig. 5b, c). However, there was no evidence of an increase in disease-related α-synuclein expression or phosphorylation or oxidation of α-synuclein in MPTP-treated TLR4-deficient mice.
Fig. 5.

TLR4 deficiency exerted a protective effect on MPTP-induced α-synuclein abnormalities. a–c The protein levels of aggregated α-synuclein (5G4), nitrated α-synuclein, and phosphorylated Ser129 α-synuclein were analyzed by Western blotting. β-actin protein levels served as loading controls. The data are expressed as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 vs. control group of WT mice; #P < 0.05, ##P < 0.01 vs. MPTP-treated WT mice
TLR4 deficiency ameliorated the activation of MPTP-induced NF-κB and the NLRP3 inflammasome signaling pathways
The NLRP3 inflammasome requires two signaling steps to be activated: priming (signal 1) stimulates the TLR4/NF-κB pathway, inducing the transcription of pro-IL-1β. Activation signal 2 triggers NLRP3, ASC and caspase-1 protein complex formation. Then, active caspase-1 generates the mature cytokine IL-1β (Fig. 6a). Hence, we tested the protein levels of NLRP3 inflammasome components by Western blotting analysis. Our results showed that the MPTP-treated WT mice exhibited upregulation of the protein contents of NLRP3 (P < 0.01; Fig. 6b, c), ASC (P < 0.01; Fig. 6b, d), caspase-1 (p20) (P < 0.01; Fig. 6b, e), IL-1β (P < 0.05; Fig. 6b, f) and p-NF-κB/p65 (P < 0.05; Fig. 6b, g) compared to untreated WT mice. Moreover, TLR4 deficiency obviously suppressed the activation of NF-κB and the NLRP3 inflammasome signaling pathways induced by MPTP.
Fig. 6.

TLR4 deficiency ameliorated the activation of MPTP-induced NF-κB and the NLRP3 inflammasome signaling pathways. a Priming and subsequent activation of NLRP3 inflammasome. b–g The protein levels of NLRP3, ASC, caspase-1 (pro- and p20), IL-1β, and p-NF-κB/65 were analyzed by Western blotting. β-Actin protein levels served as loading controls. The data are expressed as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 vs. control group of WT mice; #P<0.05, ##P < 0.01 vs. MPTP-treated WT mice
TLR4 deficiency alleviated the ROS impairment and oxidative stress induced by MPTP
We further studied the production of ROS in the SNpc induced by MPTP using immunofluorescence analysis. As shown in Fig. 7a, MPTP treatment induced the overproduction of ROS in WT mice (P < 0.01), while TLR4 deficiency ameliorated the MPTP-induced ROS impairment (P < 0.01). Inflammation is almost synonymous with oxidative stress. The model groups showed severe oxidative damage with augmented 8-hydroxyguanosine staining following MPTP treatment (P < 0.01), and TLR4 deficiency markedly attenuated the effects of MPTP on oxidative stress (P < 0.01; Fig. 7b).
Fig. 7.

TLR4 deficiency alleviated the MPTP-induced ROS impairment and oxidative stress. a The production of ROS in the SNpc was detected by immunofluorescence (scale bar = 50 μm). b Immunohistochemical results for 8-hydroxyguanosine (scale bar = 200 μm). The data are expressed as the mean ± SD (n = 3). **P < 0.01 vs. control group of WT mice; &P < 0.05 vs. control group of TLR4-deficient mice; ##P < 0.01 vs. MPTP-treated WT mice
Discussion
The present study highlights the neuroprotective effect of the TLR4 pathway in the chronic MPTP-induced PD mouse model (Fig. 8). Our data indicated that TLR4-deficient mice were partially protected against MPTP toxicity by attenuating motor deficits and reducing α-synuclein dysfunction and neuroinflammation, including activation of glia, NF-κB, and the NLRP3 inflammasome signaling pathways.
Fig. 8.
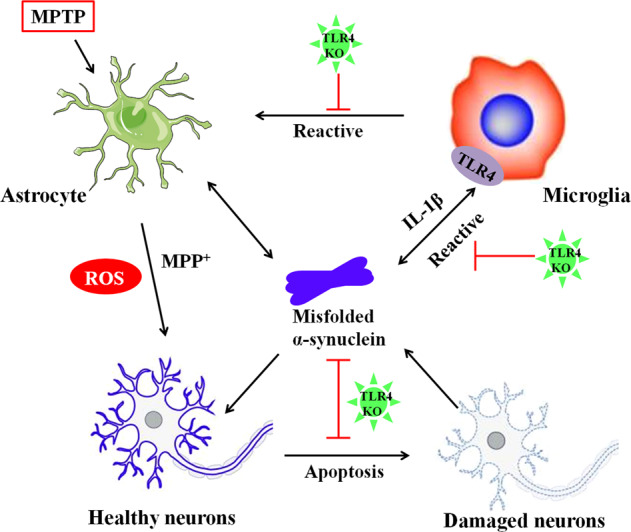
A schema for the neuroprotective effect of TLR4 pathways in the chronic MPTP-induced PD mouse model. TLR4-deficient mice can protect against MPTP toxicity by ameliorating dopamine neuron loss and reducing α-synuclein abnormalities and neuroinflammation
TLR4 is a pattern-recognition receptor that recognizes various DAMPs and subsequently evokes immune responses [26]. Recent studies have observed that TLR4 is overexpressed in the SN of MPTP-treated mice [11, 27]. Moreover, TLR4 is highly expressed in microglia, which are more abundant in the SNpc than in other brain regions [28]. MPTP can cross the BBB and be metabolized into the toxic form MPP+ in astrocytes, thereby resulting in neuron death. Extracellular α-synuclein released from damaged neurons is capable of acting as a DAMP, and α-synuclein-induced microglia activation is linked to TLR4-related signal pathways [29]. Neuroinflammation characterized by abnormal microglial activation and inflammatory cytokine release can further enhance the loss of neurons. Both inhibition of the activation and knockout of the TLR4 gene have been shown to reduce the inflammatory response, indicating the involvement of the TLR4-mediated inflammatory response in the pathophysiology of PD [30]. In support of these findings, in this study, TLR4-deficient mice exhibited an increased number of surviving TH-positive neurons and a notably lower number of activated glial cells following MPTP treatment compared to WT mice.
α-Synuclein is an unfolded, soluble protein in the brain and is mainly expressed in neurons where it regulates synaptic plasticity and dopamine release in physiological conditions [31]. The presence of LBs is the pathological hallmark of PD, and α-synuclein is a main component of LBs [32]. Although the identified mechanisms remain obscure, monomeric α-synuclein can transform into aggregates. Aggregated α-synuclein exacerbates neuron death by inhibiting tubulin polymerization and activating the inflammatory response [33]. Neuroinflammation might also trigger α-synuclein misfolding, thereby promoting the prion-like spread of α-synuclein via upregulating its release and uptake [7]. Additionally, modifications of α-synuclein, especially nitrative α-synuclein, play a critical role in modulating α-synuclein inclusion formation and stabilization [34]. Moreover, α-synuclein phosphorylated at Ser129 closely participates in PD pathogenesis. Approximately 90% of the total α-synuclein in LBs is phosphorylated at Ser129; however, <4% is phosphorylated in the healthy brain [35]. In conclusion, α-synuclein in different states may be a promising target for therapeutic strategies against PD neurodegeneration. In our study, we observed that treatment with MPTP was accompanied by aggregated, phosphorylated, and nitrated α-synuclein. Compared with MPTP-treated WT mice, MPTP-treated TLR4-deficient mice exhibited an amelioration of α-synuclein abnormalities.
Nigrostriatal dopaminergic neurons are vulnerable to oxidative damage and inflammatory attacks. Neuroinflammation is considered a crucial contributor to the pathogenesis of PD [36]. Abnormal activated glial cells secrete pro-inflammatory cytokines such as Tumor necrosis factor-α (TNF-α) and IL-1β, which are expressed abundantly in the serum of PD patients [37]. IL-1β is a master pro-inflammatory cytokine that is involved throughout the pathological process of PD [38]. The production and maturation of IL-1β are modulated by the activation of inflammasomes. All inflammasomes are generally composed of three major components, including a PRR, apoptosis-associated speck-like adapter protein containing a CARD (ASC) and the downstream effector enzyme caspase-1 [15]. As of now, the NLRP3 inflammasome is the most extensively investigated inflammasome in the CNS. Recent research has revealed an essential role of the NLRP3 inflammasome during the pathogenesis of PD. si-NLRP3 suppresses the activation of microglia and restores the loss of DA neurons in the SNpc after MPTP injection [39]. The triggering of the NLRP3 inflammasome involves a two-step process. Signal 1 of the NLRP3 inflammasome is induced by priming the TLR4/NF-κB pathway, thereby enhancing the transcription of pro-IL-1β and NLRP3. Then, signal 2 assembles the inflammasome complex and transforms pro-IL-1β to mature IL-1β [40]. Excessive production of ROS may aggravate inflammatory responses by activating pro-inflammatory signaling pathways. In the present study, MPTP treatment increased the expression of NLRP3, ASC, caspase-1, IL-1β, p-NF-κB/p65 and ROS in WT mice. Consistent with our assumption, TLR4 deficiency displayed comparable effects on the regulation of all of these molecular changes.
Conclusion
Taken together, the data in the present study demonstrated that the MPTP-induced mouse model of PD displayed dopaminergic neuron deficiency, α-synuclein abnormality, and neuroinflammation. Furthermore, there were significant differences between WT mice and TLR4-deficient mice. Our findings indicated that TLR4-deficient mice were able to protect against MPTP toxicity, demonstrated by an attenuation of motor deficits and a reduction in α-synuclein dysfunction and neuroinflammation. According to our data, the contribution of TLR4-mediated pathways to PD may open up new avenues for therapeutic potential. Further studies are still needed to explore whether pharmacological modulation of the TLR4 pathway is a prospective strategy for PD therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81573640 and 81773925), the Beijing Natural Science Foundation (7161011), and the Drug Innovation Major Project 2018ZX09711-001-001 and 2018ZX09711-001-001-003.
Author contributions
YHY and NHC designed research; QHS performed research; FFL, SW and XLZ contributed new analytical tools and reagents; YC analyzed data; QHS wrote the paper. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Yu-he Yuan, Email: yuanyuhe@imm.ac.cn.
Nai-hong Chen, Email: chennh@imm.ac.cn.
References
- 1.Erro R, Stamelou M. The motor syndrome of Parkinson’s disease. Int Rev Neurobiol. 2017;132:25–32. doi: 10.1016/bs.irn.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Zeng XS, Geng WS, Jia JJ, Chen L, Zhang PP. Cellular and molecular basis of neurodegeneration in Parkinson disease. Front Aging Neurosci. 2018;10:109. doi: 10.3389/fnagi.2018.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivekanantham S, Shah S, Dewji R, Dewji A, Khatri C, Ologunde R. Neuroinflammation in Parkinson’s disease: role in neurodegeneration and tissue repair. Int J Neurosci. 2015;125:717–25. doi: 10.3109/00207454.2014.982795. [DOI] [PubMed] [Google Scholar]
- 4.Kay DM, Factor SA, Samii A, Higgins DS, Griffith A, Roberts JW, et al. Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147b:1222–30. doi: 10.1002/ajmg.b.30758. [DOI] [PubMed] [Google Scholar]
- 5.Grozdanov V, Danzer KM. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 2018;373:175–82. doi: 10.1007/s00441-017-2775-9. [DOI] [PubMed] [Google Scholar]
- 6.Bridi JC, Hirth F. Mechanisms of alpha-Synuclein induced synaptopathy in Parkinson’s disease. Front Neurosci. 2018;12:80. doi: 10.3389/fnins.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lema Tome CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease-is there a link? Mol Neurobiol. 2013;47:561–74. doi: 10.1007/s12035-012-8267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beraud D, Maguire-Zeiss KA. Misfolded alpha-synuclein and toll-like receptors: therapeutic targets for Parkinson’s disease. Park Relat Disord. 2012;18(Suppl 1):S17–20. doi: 10.1016/S1353-8020(11)70008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aloisi F. Immune function of microglia. Glia. 2001;36:165–79. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 10.Drouin-Ouellet J., St-Amour I., Saint-Pierre M., Lamontagne-Proulx J., Kriz J., Barker R. A., Cicchetti F. Toll-Like Receptor Expression in the Blood and Brain of Patients and a Mouse Model of Parkinson's Disease. International Journal of Neuropsychopharmacology. 2014;18(6):pyu103–pyu103. doi: 10.1093/ijnp/pyu103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M, et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep. 2013;3:1393. doi: 10.1038/srep01393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Ying, Zhang Qiu-shuang, Shao Qian-hang, Wang Shuo, Yuan Yu-he, Chen Nai-hong, Wang Hong-bo. NLRP3 inflammasome pathway is involved in olfactory bulb pathological alteration induced by MPTP. Acta Pharmacologica Sinica. 2019;40(8):991–998. doi: 10.1038/s41401-018-0209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang QS, Heng Y, Chen Y, Luo P, Wen L, Zhang Z, et al. A novel bibenzyl compound (20C) protects mice from 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine/Probenecid toxicity by regulating the alpha-synuclein-related inflammatory response. J Pharmacol Exp Ther. 2017;363:284–92. doi: 10.1124/jpet.117.244020. [DOI] [PubMed] [Google Scholar]
- 14.Shaftel SS, Griffin WS, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao QH, Zhang XL, Yang PF, Yuan YH, Chen NH. Amyloidogenic proteins associated with neurodegenerative diseases activate the NLRP3 inflammasome. Int Immunopharmacol. 2017;49:155–60. doi: 10.1016/j.intimp.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 16.Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer HD. Caspase-1: the inflammasome and beyond. Innate Immun. 2014;20:115–25. doi: 10.1177/1753425913484374. [DOI] [PubMed] [Google Scholar]
- 17.Gustot A, Gallea JI, Sarroukh R, Celej MS, Ruysschaert JM, Raussens V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem J. 2015;471:323–33. doi: 10.1042/BJ20150617. [DOI] [PubMed] [Google Scholar]
- 18.Tristao FS, Amar M, Latrous I, Del-Bel EA, Prediger RD, Raisman-Vozari R. Evaluation of nigrostriatal neurodegeneration and neuroinflammation following repeated intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration in mice, an experimental model of Parkinson’s disease. Neurotox Res. 2014;25:24–32. doi: 10.1007/s12640-013-9401-8. [DOI] [PubMed] [Google Scholar]
- 19.Wu KC, Liou HH, Lee CY, Lin CJ. Down-regulation of natural resistance-associated macrophage protein-1 (Nramp1) is associated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)/1-methyl-4-phenylpyridinium (MPP( + ))-induced alpha-synuclein accumulation and neurotoxicity. Neuropathol Appl Neurobiol. 2019;45:157–73. doi: 10.1111/nan.12493. [DOI] [PubMed] [Google Scholar]
- 20.Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc. 2007;2:141–51. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- 21.Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/S0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 22.Olivan S, Calvo AC, Rando A, Munoz MJ, Zaragoza P, Osta R. Comparative study of behavioural tests in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp Anim. 2015;64:147–53. doi: 10.1538/expanim.14-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovacs GG, Wagner U, Dumont B, Pikkarainen M, Osman AA, Streichenberger N, et al. An antibody with high reactivity for disease-associated alpha-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012;124:37–50. doi: 10.1007/s00401-012-0964-x. [DOI] [PubMed] [Google Scholar]
- 24.Unterberger U, Lachmann I, Voigtlander T, Pirker W, Berghoff AS, Flach K, et al. Detection of disease-associated alpha-synuclein in the cerebrospinal fluid: a feasibility study. Clin Neuropathol. 2014;33:329–34. doi: 10.5414/NP300796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maetzler W, Pilotto A, Apel A, Deuschle C, Kuebart G, Heinzel S, et al. In vivo markers of Parkinson’s disease and dementia with Lewy bodies: current value of the 5G4 alpha-synuclein antibody. Acta Neuropathol. 2014;128:893–5. doi: 10.1007/s00401-014-1364-1. [DOI] [PubMed] [Google Scholar]
- 26.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Panaro MA, Lofrumento DD, Saponaro C, De Nuccio F, Cianciulli A, Mitolo V, et al. Expression of TLR4 and CD14 in the central nervous system (CNS) in a MPTP mouse model of Parkinson’s-like disease. Immunopharmacol Immunotoxicol. 2008;30:729–40. doi: 10.1080/08923970802278557. [DOI] [PubMed] [Google Scholar]
- 28.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–9. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao QH, Yan WF, Zhang Z, Ma KL, Peng SY, Cao YL, et al. Nurr1: A vital participant in the TLR4-NF-kappaB signal pathway stimulated by alpha-synuclein in BV-2 cells. Neuropharmacology. 2019;144:388–99. doi: 10.1016/j.neuropharm.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H, Wang Y, Liang X, Xing X, Xu X, Zhou C. Toll-like receptor 4 knockdown attenuates brain damage and neuroinflammation after traumatic brain injury via inhibiting neuronal autophagy and astrocyte activation. Cell Mol Neurobiol. 2018;38:1009–19. doi: 10.1007/s10571-017-0570-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J, et al. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun. 2016;479:881–6. doi: 10.1016/j.bbrc.2016.09.109. [DOI] [PubMed] [Google Scholar]
- 32.Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease-a systematic review. Acta Neurol Scand. 2014;130:59–72. doi: 10.1111/ane.12247. [DOI] [PubMed] [Google Scholar]
- 33.Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–9. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Norris EH, Giasson BI, Ischiropoulos H, Lee VM. Effects of oxidative and nitrative challenges on alpha-synuclein fibrillogenesis involve distinct mechanisms of protein modifications. J Biol Chem. 2003;278:27230–40. doi: 10.1074/jbc.M212436200. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Deng Y, Qing H. The phosphorylation of alpha-synuclein: development and implication for the mechanism and therapy of the Parkinson’s disease. J Neurochem. 2015;135:4–18. doi: 10.1111/jnc.13234. [DOI] [PubMed] [Google Scholar]
- 36.Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience. 2015;302:89–102. doi: 10.1016/j.neuroscience.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Stojkovska I, Wagner BM, Morrison BE. Parkinson’s disease and enhanced inflammatory response. Exp Biol Med (Maywood) 2015;240:1387–95. doi: 10.1177/1535370215576313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dursun E, Gezen-Ak D, Hanagasi H, Bilgic B, Lohmann E, Ertan S, et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J Neuroimmunol. 2015;283:50–7. doi: 10.1016/j.jneuroim.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Qiao C, Zhang Q, Jiang Q, Zhang T, Chen M, Fan Y, et al. Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson’s disease. J Neuroinflamm. 2018;15:193. doi: 10.1186/s12974-018-1236-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu J, Tao XY, Teng P, Zhang Y, Guo CL, Hu L, et al. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J Neuroinflamm. 2017;14:228. doi: 10.1186/s12974-017-0997-0. [DOI] [PMC free article] [PubMed] [Google Scholar]