

Abstract
Some studies have shown that gut microbiota along with its metabolites is closely associated with diabetic mellitus (DM). In this study we explored the relationship between gut microbiota and kidney injuries of early diabetic nephropathy (DN) and its underlying mechanisms. Male SD rats were intraperitoneally injected with streptozotocin to induce DM. DM rats were orally administered compound broad-spectrum antibiotics for 8 weeks. After the rats were sacrificed, their blood, urine, feces, and renal tissues were harvested for analyses. We found that compared with the control rats, DM rats had abnormal intestinal microflora, increased plasma acetate levels, increased proteinuria, thickened glomerular basement membrane, and podocyte foot process effacement in the kidneys. Furthermore, the protein levels of angiotensin II, angiotensin-converting enzyme, and angiotensin II type 1 receptor in the kidneys of DM rats were significantly increased. Administration of broad-spectrum antibiotics in DM rats not only completely killed most intestinal microflora, but also significantly lowered the plasma acetate levels, inhibited intrarenal RAS activation, and attenuated kidney damage. Finally, we showed that plasma acetate levels were positively correlated with intrarenal angiotensin II protein expression (r = 0.969, P < 0.001). In conclusion, excessive acetate produced by disturbed gut microbiota might be involved in the kidney injuries of early DN through activating intrarenal RAS.
Keywords: diabetic nephropathy, gut microbiota, plasma acetate, renin–angiotensin system, broad-spectrum antibiotics
Introduction
As a chronic microvascular complication of diabetes, diabetic nephropathy (DN) has become one of the major causes leading to the death of diabetic patients. According to the 8th IDF Diabetes Atlas, 425 million adults were diagnosed with diabetes worldwide in 2017, and this population has been expected to grow by 48% by 2045 [1]. The rapid growth of diabetes has led to a dramatically increasing prevalence of DN. In China, due to the rapid development of the social economy and lifestyle changes, the prevalence of diabetes is also on the rise. According to the latest survey data in China, the prevalence of diabetes in adults is 11.6%, and the total number of diabetic patients is as high as 114 million, nearly half of which also have DN [2]. However, due to the limited diagnosis of early DN, most of the newly diagnosed patients have already progressed to stage III or IV, which is basically irreversible. Therefore, it is of great importance to clarify the pathophysiological changes in the early stages of DN and formulate intervention strategies for clinical diagnosis and treatment.
The activation of the intrarenal renin–angiotensin system (RAS) has long been considered one of the initiators of DN. It has been reported that under diabetic conditions, the circulating RAS is normal or decreased, while local RAS is highly activated in the kidney. In addition, renal tissue is sensitive to angiotensin II (Ang II), leading to renal vasoconstriction, higher resistance of the glomerular efferent artery, and increased sodium and water reabsorption, all resulting in increased blood pressure and glomerular hypertension [3]. In addition, Ang II can also promote the phenotypic transformation of glomerular endothelial cells and podocytes, the deposition of extracellular matrix, and the secretion of inflammatory and profibrotic chemokines and factors, accelerating the progression of DN [4–7].
In recent years, the effect of gut microbiota on diabetes and its complications has aroused great interest. The gut microbiota of humans weighs ~1.5–2.0 kg, including ~100 trillion bacteria, the distribution density of which increases gradually from the proximal end to the distal intestine. The composition of gut microbiota in the host is associated with several factors, such as genetic and environmental influence and long-term dietary patterns, and this microbial community usually manifests as a state of equilibrium between different groups in the gut. While the host provides nutrients to the gut microbiota, the latter helps digest complex carbohydrates, producing immune molecules, short-chain fatty acids (SCFAs) and other metabolites that exert immune and metabolic functions [8, 9]. Studies have revealed notable differences in the gut microbiota between diabetic and healthy people [10]. In the intestines of healthy people, there are rich butyrate-producing bacteria, such as Escherichia coli, Clostridium, etc. In patients with type 2 diabetes (T2DM), butyrate-producing bacteria are significantly reduced compared with increasing opportunistic pathogens [11]. Karlsson et al. [12] have shown that postmenopausal women with T2DM in Europe have significant insulin resistance, partly because of a significant decrease in the abundance of Faecalibacterium prausnitzii and Roseburia, which are known as dwellers and butyrate producers of the human gut [13] and have been linked to improved insulin sensitivity and diabetes [14, 15]. Larsen et al. [16] found that the abundance of intestinal Firmicutes in adult male patients with T2DM decreased significantly, while the abundance of Bacteroides and Proteobacteria increased, and the ratio of Bacteroides/Firmicutes was correlated with patients’ glucose tolerance and blood-glucose (BG) levels. Compared with healthy controls, the abundance of Firmicutes increased in T1DM patients, while the abundance of Bacteroidetes decreased [17, 18]. In animal studies, treatment with a prebiotic (oligofructose) in high-fat-fed diabetic mice not only increased the bifidobacterial content of their guts but also improved their glucose tolerance and insulin resistance as well [19]. Therefore, the gut microbiota might be closely related to the occurrence and development of diabetes.
Recent studies have found that under the stimulation of injurious factors, the gut microbiota could produce excessive SCFAs such as acetate, mediate immune disorders and chronic inflammatory reactions of the host, and promote the occurrence of diseases such as diabetes, obesity, and inflammatory bowel disease [20–22]. The gut microbiota of high-fat-fed rats has been reported to promote insulin secretion and aggravate insulin resistance by synthesizing large amounts of acetate [23].
Studies have shown that SCFAs are involved in physiological pathways by binding to their receptors, G protein-coupled receptors (GPCRs) and olfactory receptors (Olfr). Reportedly, functional receptors of SCFAs include GPR43, GPR41, GPR109, Olfr78 [22, 24] and so on. Brown et al. [25] found that the smooth muscle cells of renal arteries express SCFA receptors, and among them, GPR41 and Olfr78 are relatively abundant. Intestinal-derived propionic acid can bind to the renal arteriolar Olfr78 and activate intrarenal RAS, further increasing the secretion of renin and angiotensin and thus regulating circulating and glomerular pressure.
Therefore, we speculated that in the development of diabetes, the gut microbiota was likely to produce excessive SCFAs, especially acetate, which could bind to renal-related signal receptors, thus activating intrarenal RAS and mediating the early pathophysiological processes of DN.
Materials and Methods
Experimental animals and measurement of general parameters
Eight-week-old healthy male Sprague-Dawley rats were kept under the following conditions: a constant 12-h photoperiod, temperature of 21–23 °C, and free access to food and water. After 2 weeks of adaptive feeding, these SD rats were randomly divided into three groups: the control group, diabetic mellitus (DM) group, and diabetic rats treated with antibiotics (DM + AB) (n = 10). The latter two groups were intraperitoneally injected with streptozotocin at a dose of 65.0 mg/kg (Sigma, USA), and BG levels were measured 3 days after the injection to confirm the establishment of diabetic models. All rats of the three groups were normally fed, while the DM + AB group were orally given compound antibiotic solution, consisting of 1 g/L ampicillin, 1 g/L neomycin, 0.5 g/L vancomycin, and 0.1 g/L amphotericin B. All rats were sacrificed after 8 weeks, with blood, urine, feces, and renal tissues harvested (kidney weights were measured at the time of sacrifice). The concentrations of BG were determined by a BG meter (Shanghai Johnson & Johnson Medical Devices Company). Serum creatinine and blood urea nitrogen (BUN) were measured by an automatic analyzer (Hitachi Tokyo, Japan). The rats were kept alone in metabolic cages to collect their 24-h urine samples, of which the level of 24-h urinary protein was quantitatively analyzed by the Lowry assay. The measurement of blood pressure in the three groups was performed at the Animal Core Facility of Nanjing Medical University.
The procedures for the animal experiments were approved by the Ethical Committee of Southeast University and followed the latest version of the Declaration of Helsinki.
Morphological analysis
After the kidneys were removed, the tissues were decapsulated and fixed in 4% paraformaldehyde and glutaraldehyde. After 48 h, fixed tissues were embedded in paraffin for observation of the pathological changes under light microscopy (Olympus, Japan) or embedded in 1% lanthanum nitrate for ultramicrostructural observation of the podocytes and glomerular basement membrane (GBM) under electron microscopy (JEM-1010, Japan). The tissues were cut into 2-μm-thick slices and then stained with periodic acid-Schiff (PAS) solution and wheat germ agglutinin (WGA) after removing the paraffin. Immunofluorescent staining of the tissue slices was performed using primary antibodies against Wilms’ tumor 1 (WT-1) and nephrin (Santa Cruz, USA) and examined by laser confocal microscopy (Leica, Germany).
Measurement of plasma acetate
The levels of plasma acetate were measured by gas chromatography. Briefly, 200 mmol/L 4-methylvaleric acid (Macklin, China) was used as an internal standard stock solution, while 1000 mM acetic acid (Sigma-Aldrich, USA) was used as the acetate standard stock solution. The diluent and extracting solvent were ethyl acetate. Different concentrations of acetate standards were prepared with 18 mmol/L internal standard. A total of 50 μL of plasma was spiked with 90 μL of 200 mM stock solution of internal standard and acidified with 10 μL of hydrochloric acid. After shaking for 30 s, the mixture was sonicated for 1 min and placed at 4 °C until layering. After centrifugation for 5 min at a speed of 12 500 r/min , the organic phase was filtrated using a 0.22-μm filter. A 1-μL injection of standards and filtrated samples was used for gas chromatography analysis with a DB-23 column (30 m × 0.250 mm, Agilent, USA) at a speed of 1 mL/min. The split ratio was 7:1. The carrier gas was nitrogen. The initial temperature was 90 °C, which increased to 150 °C (15 °C/min). It was then heated to 170 °C (5 °C/min), gradually increasing to 250 °C and waiting for 2 min. The standard curve was made according to the concentration and peak area of acetate standards to that of the internal standard. The plasma acetate concentration was then quantified.
16S ribosomal DNA (rDNA) sequencing analysis
The gene sequencing of gut microbiota was performed using 16S rDNA sequencing technology. Fresh fecal specimens were collected using sterile tweezers and tubes and stored at −80 °C. The total DNA of the fecal bacteria was extracted according to the manual of the QIAamp DNA Stool Mini kit (QIAGEN, Hilden, Germany). Briefly, feces were homogenized and lysed in ASL buffer. The mixture was centrifuged at 14 000 r/min for 1 min. An InhibitEX tablet was dissolved in the supernatant, followed by centrifugation. Proteinase K and ethanol were added to the supernatant. Lysates were then loaded onto the QIAamp spin column with a QIAamp membrane. The column was efficiently washed in two steps with repeated centrifugation. Lastly, purified DNA was eluted in low-salt buffer. PCR amplification of the variable region 4–5 of bacterial 16S rDNA was conducted through double eight cycles to yield detectable products. The amplicons were mixed and purified to construct the gene library after quantification by real-time PCR. Sequencing was performed on the Illumina MiSeq2 × 300 bp platform. Operational taxonomic unit picking (97% nucleotide sequence identity) was assigned using the ribosomal database project classifier. Chimeric sequences were removed using UCHIME. The alpha diversity analysis was performed with mothur (Version 1.33.3), while the beta diversity analysis was performed with R program (Version 3.2.3).
Measurement of circulating RAS
The levels of renin, Ang І, and Ang II in the plasma of the three groups were measured by a radioimmunoassay kit (Beijing North Institute of Biological Technology, China) in accordance with the instructions. The method for determining the level of plasma renin activity (PRA) is as previously described [26]: the optimal dose of trypsin (Invitrogen, USA) added was confirmed by constructing a dose-response curve, which was used for subsequent determination. Plasma samples were treated with trypsin and kept at 4 °C, pH 7.3, for 30 min for activation, after which trypsin inhibitor was added for 15 min at room temperature to terminate the activation. The levels of renin activation before and after the addition of trypsin were measured, and the PRA level of each sample was the total renin level by trypsin activation minus the renin level before trypsin addition.
Western blot analysis
Proteins were extracted from kidney tissues, and the protein concentration of each sample was measured. By adding lysate, loading buffer, and ddH2O, the protein concentration and volume of each sample were equal, and all samples were boiled at 100 °C for 10 min for storage. Gel electrophoresis was performed in a sodium dodecyl sulfate-polyacrylamide system, and gel transfer was performed using polyvinylidene fluoride membranes. The membranes were then immersed in blocking buffer for 1 h at room temperature and incubated overnight at 4 °C in primary antibodies against angiotensin-converting enzyme (ACE), angiotensinogen (AGT), Ang II, and Ang II type 1 receptor (AT1) (Santa Cruz, USA). After washing with 0.1% Tris buffered saline and tween, the membranes were incubated in horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Protein expression was observed using the ECL system (Bio-Rad, USA).
Statistical analysis
All data were processed using SPSS 19.0 (IBM, USA). Measurement data were expressed as the mean ± standard deviation (SD), t-test was used for statistical comparisons among the data of three groups, and the Spearman correlation test was used for correlation analysis. P values < 0.05 were considered statistically significant.
Results
General parameters of the three groups
Compared with the controls, the BG level of the DM group was significantly elevated, confirming that the STZ-induced diabetic model was established. Compared with the DM group, treatment with antibiotics caused an obviously reduced BG level in the DM + AB group (Fig. 1a), suggesting that gut microbiota might be closely related to the body’s high BG levels under diabetic conditions. The plasma insulin level in the three groups showed an opposite trend compared with that of BG (Fig. 1b).
Fig. 1.

General parameters of the three groups. a Blood-glucose levels. b Plasma insulin levels. *P < 0.01 compared with the control group, #P < 0.05 compared with the DM group.
Changes in gut microbiota in the three groups
The results of 16S rDNA gene sequencing showed that there were significant differences in the bacterial composition and abundance of gut microbiota between the control and DM groups. After the application of broad-spectrum antibiotics, most of the gut microbiota in the DM + AB group was killed (Fig. 2a). The subgroup sequencing analysis results of gut microbiota in each group have shown that the abundance of Blautia, Roseburia, and Paraprevotella in the colon of the DM group was significantly increased compared with that of the controls (Fig. 2b–d), while the abundance of Bacteroides was relatively decreased, suggesting that the composition and abundance of gut microbiota have both changed under diabetic status. The results of the DM + AB group further confirmed the bactericidal effect of the treatment with broad-spectrum antibiotics (Fig. 2e).
Fig. 2.
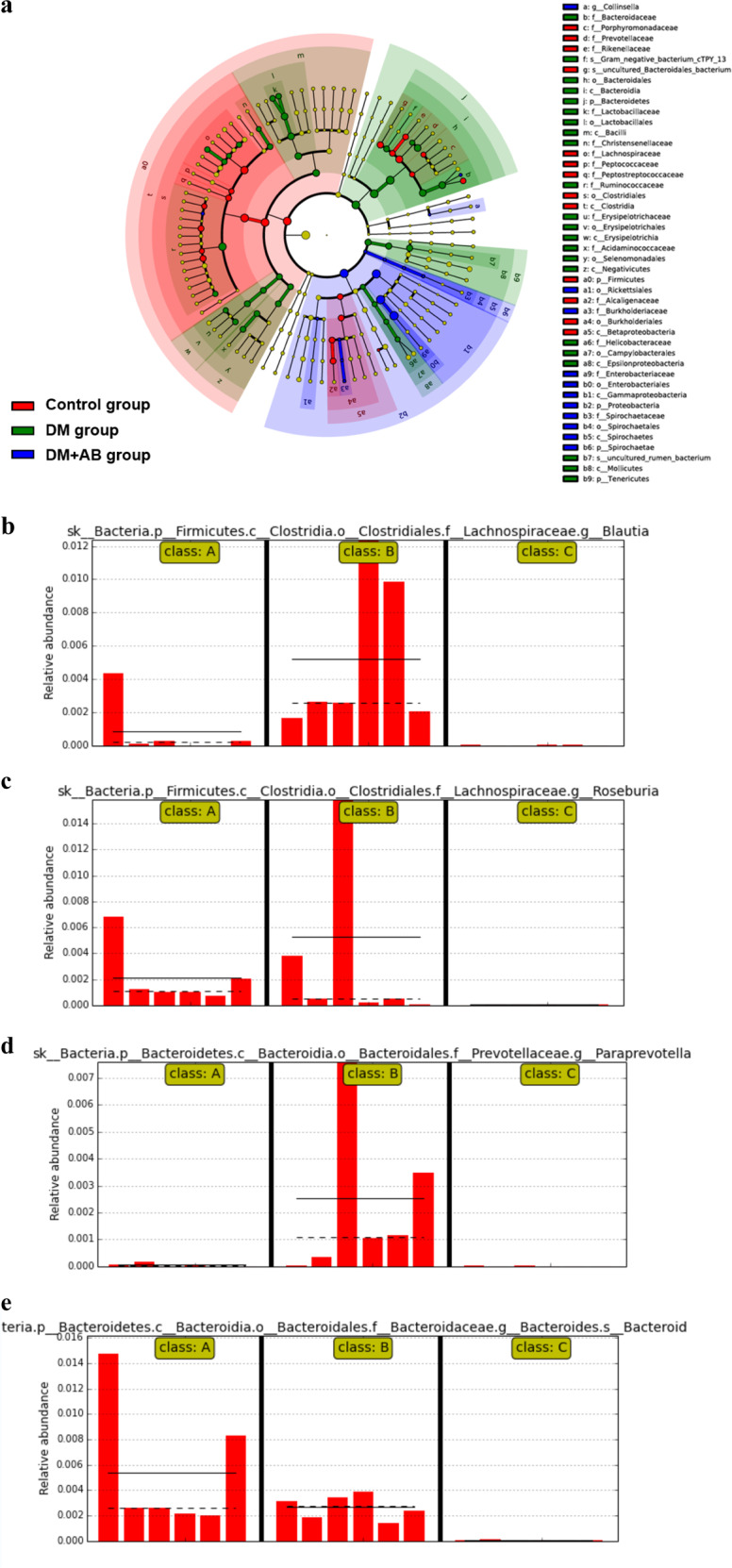
The changes in gut microbiota in the three groups. a Taxonomic cladogram derived from LEfSe analysis of 16S sequences. Red shaded areas indicate control-enriched taxa, green shaded areas indicate DM-enriched taxa, and blue shaded areas indicate DM + AB-enriched taxa. b–e Subgroup sequencing analysis of gut microbiota. Class A indicates the control group, Class B indicates the DM group, and Class C indicates the DM + AB group. The solid and dashed lines indicate the average and median relative abundance, respectively. b The relative abundance of Blautia in the gut microbiota of the three groups. c The relative abundance of Roseburia in the gut microbiota of the three groups. d The relative abundance of Paraprevotella in the gut microbiota of the three groups. e The relative abundance of Bacteroides in the gut microbiota of the three groups.
Treatment with antibiotics significantly reduced the level of plasma acetate
The results of gas chromatography analysis demonstrated significantly increasing plasma levels of acetate in the DM group, which might be due to the abnormalities of gut microbiota under diabetic conditions. Antibiotic intervention significantly lowered the plasma acetate level in the DM + AB group (Fig. 3).
Fig. 3.
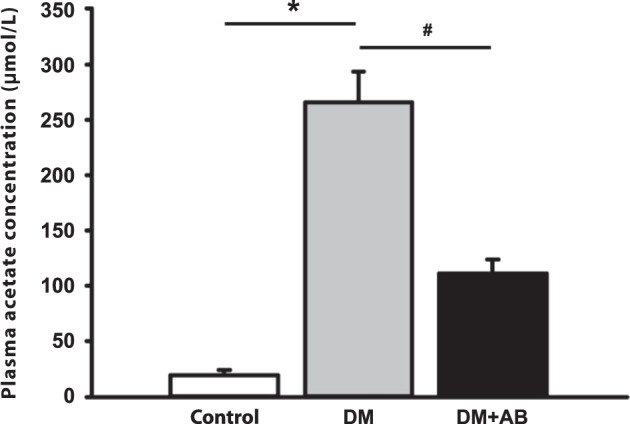
Treatment with antibiotics significantly reduced the level of plasma acetate. Plasma level of acetate. *P < 0.01 compared with the control group, #P < 0.05 compared with the DM group.
The effect of gut microbiota on renal injury of incipient DN
Compared with the controls, the ratio of kidney weight-to-body weight in both the DM group and the DM + AB group was significantly higher (Fig. 4a), indicative of renal hypertrophy under the state of diabetes. In terms of proteinuria, we observed a significantly increasing level of 24-h urine protein in the DM group compared with that of the control group. After the antibiotic intervention, the DM + AB group had a significant reduction in 24-h urine protein compared with that of the DM group (P < 0.05) (Fig. 4b). In addition, compared with the control group, the level of BUN increased in both the DM group and the DM + AB group (P < 0.01), but there was no significant difference in blood creatinine among the three groups (Fig. 4c, d), suggesting that this abnormality of gut microbiota has not yet developed to a sufficient degree to exert obvious effects on renal functions.
Fig. 4.

The effect of gut microbiota on renal indicators of incipient DN. a Kidney weight-to-body weight ratios. b 24-h urine protein. c Blood urea nitrogen (BUN) level. d Blood creatinine. *P < 0.01 compared with the control group, #P < 0.05 compared with the DM group.
We next observed renal pathological changes to further evaluate the degree of renal injury in the three groups. The results of PAS staining showed mild mesangial expansion among the kidneys in the DM group compared with that in the DM + AB group (Fig. 5a, b). Under electron microscopy, ultramicrostructural changes in the glomerular filtration membrane in each group could be observed. The GBM in the DM group was thickened, and there was fusion of fenestrated endothelium, along with partly merged and missing podocyte foot processes, which corresponds to the degree of renal injury in early DN. After the antibiotic intervention, the basement membrane thickening, endothelial fusion, and podocyte injury in the DM + AB group recovered to some extent (Fig. 5c). We performed WGA immunofluorescence staining to observe the changes in glomerular endothelium glycocalyx. Under laser confocal microscopy, we found that compared with the control group, the thickness of the glomerular endothelium was significantly reduced in the DM group. In the DM + AB group, the reduced glycocalyx was repaired after the antibiotic intervention (Fig. 5d). The immunofluorescence staining results also showed significantly decreased expression of glomerular podocyte-specific protein WT-1 and nephrin in the DM group compared with that in the control group, which was relatively recovered in the DM + AB group (Fig. 5e–g), suggesting that unbalanced gut microbiota might be a key factor resulting in injuries to the glomerular filtration membrane in early DN.
Fig. 5.

The effect of gut microbiota on renal injuries of incipient DN. a Pathological changes were assessed by PAS staining (original magnification, ×400). b The score of mesangial expansion was determined from histology sections. ***P < 0.01 compared with the control, #P < 0.05 compared with the DM group. c Changes in podocytes were evaluated by electron microscopy (original magnification, ×12 000). d The changes in glomerular endothelium glycocalyx were assessed by WGA staining (original magnification, ×400). e WT-1 and nephrin protein expression was evaluated by immunofluorescent staining (original magnification, ×400). The arrows indicate glomerular endothelium glycocalyx. f Quantitation of immunofluorescence staining for WT-1. *P < 0.05; compared with the control; ##P < 0.01 compared with DM. g Quantitation of immunofluorescence staining for nephrin. ***P < 0.001; compared with the control; #P < 0.05 compared with DM.
Intrarenal RAS is activated in early DN
The measurement of circulating RAS in the three groups showed no significant differences in PRA and the level of Ang І (Fig. 6a, b). However, compared with the DM group, the concentration of Ang II in the circulation was significantly reduced (Fig. 6c).
Fig. 6.

Intrarenal RAS is activated in early DN. Plasma renin activity level (a), Ang I level (b), and Ang II level (c). The protein expression levels of RAS were measured by Western blotting (d). The histograms represent the mean ± SD of the densitometric scans of the protein bands normalized to β-actin (e). Correlation analysis of plasma acetate levels and intrarenal Ang II expression levels (f) (r = 0.969, P < 0.001). *P < 0.01 compared with the control group, #P < 0.05 compared with the DM group.
The Western blot results to evaluate the degree of intrarenal RAS activation showed that compared with the control group, the protein expression of ACE, Ang II, and AT1R in the kidney of the DM group was significantly increased, and antibiotic treatment showed a suppressing effect on these three RAS-activating indicators (Fig. 6d, e). The expression of AGT showed an opposite trend, which might be a result of negative-feedback adjustment considering its role as a rate-limiting enzyme. This result suggests that the dysbiosis of the gut microbiota may be involved in the intrarenal RAS activation of early DN.
Correlation analysis between plasma acetate levels and intrarenal Ang II expression
To investigate the causal relationship between the dysbiosis of gut microbiota and intrarenal RAS activation, we further analyzed the correlation between the plasma acetate concentration and intrarenal Ang II protein expression as determined by Western blot (Fig. 6f). A positive correlation was observed (r = 0.969, P < 0.001).
Discussion
Most kidney diseases are characterized by an initial injury, followed by compensatory growth and associated functional alterations, usually manifested as renal hypertrophy [27]. Stages І and II of DN are mainly characterized by glomerular hypertension, hyperfiltration, and renal hypertrophy [28]. Renal hypertrophy is an important and significant marker of structural and functional changes in the kidney in early DN, mainly characterized by glomerular cell hypertrophy, thickening of the basement membrane, and an increase in the mesangial matrix. In this study, we confirmed that the experimental animals had developed corresponding renal injury under diabetic conditions by pathological observation.
The RAS can be divided into the circulatory and local RAS depending on its location of synthesis and action. In circulating RAS, Ang II is produced under the serial effects of renin secreted by the kidney, angiotensin from the liver, and ACE located in vascular endothelial cells. In certain organs or cells, the synthesis of RAS components is independent. Ang II can be produced in the intercellular space [29] under the effect of enzymes other than renin and ACE (such as chymase) and exerts many pathophysiological effects by activating AT1R [30]. The activation of RAS has always been considered an important factor in the development of DN, and local RAS seems to be more involved compared with the circulating RAS [31]. The kidney itself contains all the RAS components [32]. High glucose has been reported to promote the production of Ang II, which in turn leads to glomerular hyperfiltration and high permeability, as well as extracellular matrix deposition [33]. Clinical application of RAS inhibitors could significantly retard the development of proteinuria in DN patients and lower the incidence of ESRD. As a current first-line treatment for DN, the efficacy of RAS inhibitors in preventing DN is still very limited. The rising prevalence of DN has suggested that a deeper understanding of the molecular mechanisms underlying DNs is required to seek better treatment.
The relationship between gut microbiota and many metabolic diseases has been a hotpot for research in recent years. Under normal circumstances, Firmicutes and Bacteroidetes take up a large proportion of the whole microbiota in the gut, while the composition of other phyla varies individually due to several factors, such as genetics, diet, and antibiotic use. However, under pathological conditions, the species and abundance of the host gut microbiota would change significantly, mainly characterized by decreasing normally dominant bacteria and increasing pathogenic bacteria. It has also been shown in this study that compared with the control group, there was microecological dysbiosis of gut microbiota in the intestinal tract of diabetic rats. The application of antibiotic intervention is a common method used in studies of gut microbiota and its role in the development of diseases. In this study, we observed that the intervention of antibiotics affected many aspects of the DN animal model, from basic functions and pathological changes in the kidneys to the amount of products released from gut microbiota. The use of mixed antibiotics has ensured the killing effect of the bacteria in the intestinal tract of experimental animals, and in the meantime, it could also attenuate the renal injury of early DN, including reducing the amount of 24-h urine protein and mitigating the injury to the glomerular filtration membrane and the pathological changes in the tubules.
SCFAs are a major product of the fermentation of carbohydrates by gut microbiota, and there are three main types: acetate, propionate, and butyrate. The butyrate-producing microbiota is essential in maintaining the balance of the intestinal environment in humans. However, the number of butyrate-producing microbiota in diabetic patients is significantly reduced, while the number of other opportunistic pathogens shows an increasing trend. There is evidence that a butyrate-producing Clostridium genus could exert antidiabetic effects by increasing the production of butyrate and upregulating the expression of SCFA receptors in the gut [34]. As a main SCFA product of gut microbiota, acetate has demonstrated an intricate effect on the internal environment. It has been reported that acetate is almost undetectable in the blood of germ-free mice [23], while the content of acetate in animals with a high-fat diet is significantly increased, indicating that the level of acetate might be a potential indicator of the activity of gut microbiota. This study has shown that antibiotic intervention significantly reduced the relatively higher plasma level of acetate in diabetic rats, considering the alleviated renal lesions after antibiotic intervention. It can be speculated that overproduction of acetate might be adverse to the development of early DN. However, the causal relationship between gut microbiota dysbiosis and the development of DN remains to be elucidated, and more explorations may provide a new perspective and therapeutic target for the future diagnosis and treatment of DN.
RAS activation has been considered one of the important initiating factors in the early development of DN, yet the exact associations between gut microbiota and RAS activation remain to be elucidated. Pluznick et al. [24] found that signals from the gut microbiota, i.e., SCFAs, could be received by corresponding receptors expressed at renal small arterioles, further regulating the secretion of renin, which was involved in maintaining glomerular pressure. This process could be blocked by antibiotics or knockout of the SCFA receptor. Considering the characteristics of glomerular hypertension and hyperfiltration in the early stage of DN, the disordered gut microbiota is likely to generate excessive SCFAs, which bind to corresponding receptors in the kidney and regulate RAS, thus promoting the pathological changes in early DN. In this study, we have shown that in the early stage of DN, the expression of RAS in the kidney was significantly elevated, indicating RAS activation at this stage. After antibiotic intervention, the level of circulating Ang II was lowered, and the expression of RAS within the kidney was also significantly weakened, suggesting that there might be a causal relationship between the dysbiosis of gut microbiota and intrarenal RAS activation in early DN. Therapeutic intervention could be applied to change the composition of gut microbiota, focusing on the preservation of beneficial phyla, to create renoprotective prospects [35].
In summary, we have established a DN model to observe the changes in gut microbiota and its metabolite acetate in early DN and to further investigate the association between these changes and RAS activation in the kidney to elucidate the underlying mechanism of early renal injury in DN. Further exploration of the causal relationship and intricate mechanism of gut microbiota and RAS activation in the early development of DN are required to develop new prevention strategies for clinical early DN.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 81970629), the Jiangsu Province Social Development Project (BE2018744), the Project for Jiangsu Provincial Medical Talent (ZDRCA2016077), the Jiangsu Province Six Talent Peaks Project (2015-WSN-002), the Fundamental Research Funds for the Central Universities (KYCX18-0182, KYCX17-0169, KYZZ15-0061), the Jiangsu Province Ordinary University Graduate Research Innovation Project (SJZZ16-004), and the Joint Project of Southeast University and Pharmaceutical University of China (2242019K3DZ03).
Author contributions
CCL performed the research, analyzed the data, and wrote the paper; KLM designed and reviewed the paper; ZBH, RW, ZHH, JL, PPC, JXZ, XQL, BYY, and SJH assisted in the research; XZR and BCL analyzed and interpreted the data. All authors read and approved the final paper.
Competing interests
The authors declare no competing interests.
References
- 1.Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pr. 2018;138:271–81. doi: 10.1016/j.diabres.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 2.Xu Y, Wang L, He J, Bi Y, Li M, Wang T, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–59. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 3.Tamura J, Konno A, Hashimoto Y, Kon Y. Upregulation of renal renin–angiotensin system in mouse diabetic nephropathy. Jpn J Vet Res. 2005;53:13–26. [PubMed] [Google Scholar]
- 4.Kanasaki K, Taduri G, Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol. 2013;4:7. doi: 10.3389/fendo.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Ren Physiol. 2017;312:716–31. doi: 10.1152/ajprenal.00314.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miranda-Diaz AG, Pazarin-Villasenor L, Yanowsky-Escatell FG, Andrade-Sierra J. Oxidative stress in diabetic nephropathy with early chronic kidney disease. J Diabetes Res. 2016;2016:7047238. doi: 10.1155/2016/7047238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urushihara M, Kagami S. Role of the intrarenal renin–angiotensin system in the progression of renal disease. Pediatr Nephrol. 2017;32:1471–79.. doi: 10.1007/s00467-016-3449-7. [DOI] [PubMed] [Google Scholar]
- 8.Schroeder BO, Backhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079–89.. doi: 10.1038/nm.4185. [DOI] [PubMed] [Google Scholar]
- 9.Org E, Mehrabian M, Lusis AJ. Unraveling the environmental and genetic interactions in atherosclerosis: central role of the gut microbiota. Atherosclerosis. 2015;241:387–99. doi: 10.1016/j.atherosclerosis.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63:1513–21.. doi: 10.1136/gutjnl-2014-306928. [DOI] [PubMed] [Google Scholar]
- 11.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 12.Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 13.Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. 2010;12:304–14.. doi: 10.1111/j.1462-2920.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- 14.Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–57.. doi: 10.2337/db10-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 16.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5:e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11:46. doi: 10.1186/1741-7015-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82–91. doi: 10.1038/ismej.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50:2374–83.. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- 20.Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem Int. 2016;99:110–32. doi: 10.1016/j.neuint.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3:858–76.. doi: 10.3390/nu3100858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145:396–406. doi: 10.1053/j.gastro.2013.04.056. [DOI] [PubMed] [Google Scholar]
- 23.Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, et al. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. 2016;534:213–7. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pluznick J, Protzko R, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410–5. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown D, Sorscher EJ, Ausiello DA, Benos DJ. Immunocytochemical localization of Na+ channels in rat kidney medulla. Am J Physiol. 1989;256:366–9. doi: 10.1152/ajpcell.1989.256.2.C260. [DOI] [PubMed] [Google Scholar]
- 26.Kantorowicz L, Valego NK, Tang L, Figueroa JP, Chappell MC, Carey LC, et al. Plasma and renal renin concentrations in adult sheep after prenatal betamethasone exposure. Reprod Sci. 2008;15:831–8. doi: 10.1177/1933719108318599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hostetter TH. Progression of renal disease and renal hypertrophy. Annu Rev Physiol. 1995;57:263–78. doi: 10.1146/annurev.ph.57.030195.001403. [DOI] [PubMed] [Google Scholar]
- 28.Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes. 1983;32:64–78. doi: 10.2337/diab.32.2.S64. [DOI] [PubMed] [Google Scholar]
- 29.Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin–angiotensin system. Clin Sci. 2012;123:273–84. doi: 10.1042/CS20120089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–70. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 31.Wysocki J, Ye M, Khattab AM, Fogo A, Martin A, David NV, et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017;91:1336–46. doi: 10.1016/j.kint.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- 33.Carey RM, Siragy HM. The intrarenal renin–angiotensin system and diabetic nephropathy. Trends Endocrinol Metab. 2003;14:274–81.. doi: 10.1016/S1043-2760(03)00111-5. [DOI] [PubMed] [Google Scholar]
- 34.Jia L, Li D, Feng N, Shamoon M, Sun Z, Ding L, et al. Anti-diabetic effects of clostridium butyricum CGMCC0313.1 through promoting the growth of gut butyrate-producing bacteria in type 2 diabetic mice. Sci Rep. 2017;7:7046. doi: 10.1038/s41598-017-07335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120:1183–96. doi: 10.1161/CIRCRESAHA.117.309715. [DOI] [PMC free article] [PubMed] [Google Scholar]