

Abstract
The expanded HTT CAG repeat causing Huntington’s disease (HD) exhibits somatic expansion proposed to drive the rate of disease onset by eliciting a pathological process that ultimately claims vulnerable cells. To gain insight into somatic expansion in humans, we performed comprehensive quantitative analyses of CAG expansion in ~50 central nervous system (CNS) and peripheral postmortem tissues from seven adult-onset and one juvenile-onset HD individual. We also assessed ATXN1 CAG repeat expansion in brain regions of an individual with a neurologically and pathologically distinct repeat expansion disorder, spinocerebellar ataxia type 1 (SCA1). Our findings reveal similar profiles of tissue instability in all HD individuals, which, notably, were also apparent in the SCA1 individual. CAG expansion was observed in all tissues, but to different degrees, with multiple cortical regions and neostriatum tending to have the greatest instability in the CNS, and liver in the periphery. These patterns indicate different propensities for CAG expansion contributed by disease locus-independent trans-factors and demonstrate that expansion per se is not sufficient to cause cell type or disease-specific pathology. Rather, pathology may reflect distinct toxic processes triggered by different repeat lengths across cell types and diseases. We also find that the HTT CAG length-dependent expansion propensity of an individual is reflected in all tissues and in cerebrospinal fluid. Our data indicate that peripheral cells may be a useful source to measure CAG expansion in biomarker assays for therapeutic efforts, prompting efforts to dissect underlying mechanisms of expansion that may differ between the brain and periphery.
Introduction
Huntington’s disease (HD) is a dominantly inherited, neurodegenerative disorder characterized by chorea, cognitive and psychiatric symptoms with onset typically in mid-life (1). A rarer percentage (1–15%) of cases presents as juvenile-onset disease with onset <21 years of age (2–5). The cause of the disease is the expansion over 35 repeats of a polymorphic CAG repeat tract within exon 1 of the HTT gene (6). The expanded HTT CAG repeat tract is highly unstable when transmitted to the next generation, accounting for the wide variation in expanded CAG repeat lengths among patients (7–12). Studies both in HD patient tissues and in HD mouse models have shown that the expanded HTT CAG repeat has a high propensity for further expansion in somatic cells, which occurs in a CAG length-, time- and tissue/cell-type-dependent manner (8,13–27).
Recent genome-wide association studies (GWAS) (28) have demonstrated that the length of the CAG repeat tract, rather than the length of the polyglutamine repeat, drives of the rate of onset of HD pathogenesis. This observation provided the framework for a two-step model of HD pathogenesis in which (1) the rate of phenotypic onset is determined by the rate of somatic CAG expansion, and (2) somatically expanded repeats trigger a toxic process(es) in vulnerable cells, resulting ultimately in clinical disease. One hypothesis, consistent with the genetic dominance in HD (29), and initially proposed based on computational modeling, is that disease onset occurs once the repeat has expanded beyond cell type-specific pathological thresholds in a critical proportion of cells (30), although potential threshold lengths and toxicity processes in different cell types are unknown. The role of somatic CAG expansion in driving the rate of pathogenesis is consistent with expansion being a CAG- and time-dependent process. This is strongly supported both by the association of longer somatic expansions in HD postmortem brain with earlier disease onset (21) and by the finding from GWAS that DNA mismatch repair (MMR) genes, known to control CAG expansion in mouse models (31–33), modify the rate of HD onset (28,34,35), with one (MSH3) also modifying the rate of progression (36,37).
Insight into the variation in HTT CAG expansion among tissues and ultimately between cell-types will allow a deeper understanding of the relationship of somatic expansion to HD pathogenesis, and a comparison with another CAG repeat expansion disease with distinct neuropathological and clinical features would help to elucidate components that may explain differences in cell-type and disease-specific vulnerabilities. HD mouse models have provided significant insight into the tissue specificity of HTT CAG expansion. Notably, the striatum and liver are highly prone to expansion, cerebellum exhibits relative stability, while overall, the brain tends to exhibit higher levels of CAG expansion than peripheral tissue (14–16,22–25). In patients, high levels of instability occur in the brain (17,21), and to the extent that somatic instability has been studied across tissues in HD patients, a consistent observation has been that the and that cerebellum is stable relative to other brain regions (8,13,17). Similar observations have been made in other CAG repeat expansion disorders (38–42). However, knowledge of tissue-specific patterns of HTT CAG instability in HD patients as well as other CAG repeat disorders is limited, as previous studies have focused on the analysis of a relatively small set of brain tissues and/or were performed using methodologies that lack the resolution to distinguish subtle differences in repeat length distributions. Here, to gain further insight into tissue-specific CAG expansion in HD pathogenesis, we have performed quantitative analyses of somatic expansion across a wide range of postmortem central nervous system (CNS) and peripheral tissues from seven individuals presenting with adult-onset HD and one juvenile-onset HD patient. We have also analyzed ATXN1 CAG repeat somatic expansion in postmortem brain tissues from an individual with a different inherited expansion disease spinocerebellar ataxia type 1 (SCA1), a disorder that features gait and limb ataxia, dysarthria and dysmetria and severe atrophy of the cerebellum and brainstem (43,44).
Results
Quantitative analyses of somatic HTT CAG instability in adult-onset HD patient postmortem tissues
To gain insight into levels of HTT CAG instability across a wide range of human tissues, we first analyzed instability in postmortem tissues from three individuals (HD1, HD2, HD3) with adult-onset HD who had undergone full-body autopsies (Table 1). Tissues were obtained from the New York Brain Bank (NYBB) (45) and were analyzed at the Massachusetts General Hospital (MGH, Boston, MA). The majority of the 26 CNS regions, post-mortem cerebrospinal fluid (CSF) and nine peripheral tissues were available for all three individuals, as well as testis or ovary (Supplementary Material, Table S1). For most of the samples, we extracted DNA and analyzed CAG instability from three separate tissue sub-pieces cut from the same block. We analyzed CAG instability by performing a quantitative assessment of CAG repeat length distributions from GeneMapper traces of HTT CAG repeat PCR products generated from ‘bulk’ genomic DNA, reflecting somatic length variation of the majority of alleles. This relatively high-throughput method allows quantification of subtle differences in repeat instability and correlates well with repeat length distributions obtained from analyses of single molecules (22). In each tissue, the degree of CAG expansion measured will reflect the net rate of CAG expansion in the cell types from which it is composed and which are captured in the bulk PCR assay, the length of time taken for the repeats to expand (i.e. age at death) and the degree of cell turnover and/or loss in each tissue.
Table 1.
HD individuals for analyses of CAG expansion in CNS and peripheral tissues
| Study | Individual | Gender | CAG repeat | CCG repeat | Pathological grade | Age at onset | Age of death | PMIa (h) |
|---|---|---|---|---|---|---|---|---|
| Boston | HD1 | M | 43/16 | 7/10 | 3 | 45 | 57 | 18:38 |
| Boston | HD2 | F | 44/17 | 7/7 | 4 | 42 | 66 | 5:30 |
| Boston | HD3 | M | 53/19 | 7/7 | 3 | 27 | 39 | 3:50 |
| Boston | HD4 | M | 140/20b | 7/7 | 3 | 4 | 6 | N/A |
| Bochum | HD5 | F | 44/17 | 7/7 | 2–3 | 43 | 55 | 110:00 |
| Bochum | HD6 | F | 46/17 | 7/7 | 3 | 40 | 50 | 21:00 |
| Bochum | HD7 | F | 50/18 | 7/7 | 3 | 25 | 40 | 20:00 |
| Bochum | HD8 | F | 53/19 | 7/10 | 3 | 36 | 45 | 8:00 |
aPostmortem interval (PMI) is given as hours:minutes. For Boston samples (New York Brain Bank) PMI is time to freezer; for Bochum samples PMI is time to autopsy.
bRepeat length in blood DNA, determined in this study. Repeat length is highly variable between tissues (see Fig. 7). The juvenile-onset patient is described in (62).
Pathological grade is according to Vonsattel et al. (1).
Examples of GeneMapper traces highlighting a range of instabilities are shown in Supplementary Material, Figure S1. The peaks to the left of the modal allele are predominantly the result of PCR slippage and do not differ substantially between tissues, while the expansion peaks to the right of the modal alleles differ between tissues and constitute somatic variation in CAG repeat length. We therefore quantified the expansion peaks (expansion index) for all the samples to obtain a tissue-wide profile of somatic expansion for each of the three individuals (Fig. 1A–C). On each graph, tissues are plotted in order of expansion index (mean value over the three sub-pieces of each tissue as appropriate) for the CNS and periphery separately. These profiles highlight a number of observations: (1) a wide variation in instability is notable within many of the brain regions. This is not the result of technical variation between PCR reactions (Supplementary Material, Fig. S2) and therefore reflects intra-tissue heterogeneity in sampling from a relatively large tissue block. (2) Overall, brain tissues show more instability than peripheral tissues. The exception to this is the liver, which shows relatively high instability; this is particularly striking for HD3 with 53 CAG repeats. A high expansion index is also seen in testis of individual HD3, capturing a long tail of highly expanded alleles (Supplementary Material, Fig. S1), which likely reflects the expansion-biased and CAG length-dependent instability that occurs in the male germline (12). (3) Broadly similar patterns of tissue-specific instability are observed in each patient, with multiple cortical regions and caudate/accumbens/putamen (neostriatum), tending to have relatively high instability, while midbrain (mesencephalon), hindbrain (metencephalon and myelencephalon), spinal cord and cerebellum tend to have lower instability. CAG instability can also be measured in genomic DNA extracted from postmortem CSF, with CSF exhibiting levels of expansion similar to those of the more stable tissues. To visualize better the tissues that exhibit the highest levels of somatic expansion and the extent to which these might be shared amongst the three individuals we ranked mean expansion indices for each individual (Fig. 1D). Within the top 10 most unstable tissues for each individual, five—prefrontal cortex (BA9), primary somatosensory cortex (BA3.1.2), temporal pole, primary visual cortex (BA17) and caudate/accumbens/putamen—were shared amongst all three individuals. Anterior cingulate/midcingulate cortex (BA24), primary motor cortex (BA4), globus pallidus/putamen, amygdala, head of hippocampal formation and hippocampal formation were shared between two of the three individuals, while caudal metencephalon, liver and testis were unique to HD3. Notably, the most unstable brain regions include those that are particularly vulnerable to the HD mutation, e.g. caudate/accumbens/putamen, BA4, BA9, BA3.1.2 and BA17 cortical regions, as well as the anterior BA24 and amygdala (1,46–48). We also find high levels of expansion in the temporal pole, and there is evidence for neuropathology of the temporal cortex as well as structural alteration in the temporal pole and temporal lobe (48–51).
Figure 1.
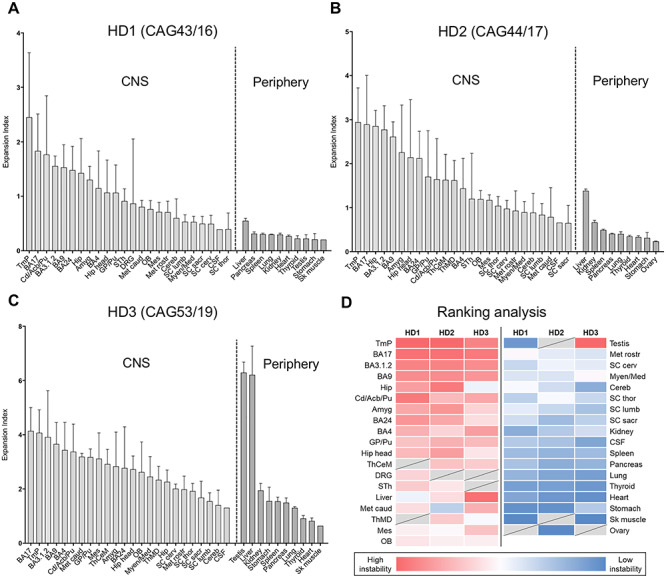
Quantitative analyses of somatic CAG expansion in adult-onset HD. (A–D) Expansion indices were quantified from GeneMapper peak height data of HTT CAG PCR products from individuals HD1, HD2 and HD3, analyzed in Boston, USA. Bars show mean expansion indices for each tissue, and error bars show standard deviation between tissue sub-pieces. For each individual, data are plotted in order of mean tissue expansion index, separately for the CNS and periphery. (D) Mean expansion indices were ranked for each individual and ranks displayed as heat map. Tissues are displayed in order of mean rank. Dark red = most expanded; white = median; dark blue = least expanded; gray = tissue not available. OB, olfactory bulb; BA9, prefrontal cortex; BA24, anterior cingulate/midcingulate cortex; BA4, primary motor cortex; BA3.1.2, primary somatosensory cortex; Tmp, temporal pole; BA17, primary visual cortex; Cd/Acb/Pu, caudate/accumbens/putamen; GP/Pu, globus pallidus/putamen; ThCeM, centromedial thalamic nucleus; ThMD, dorsomedial thalamic nucleus; STh, subthalamic nucleus; Amyg, amygdala; Hip head, hippocampal formation (head); Hip, hippocampal formation; Mes, mesencephalon; Met rostr, metencephalon rostral; Met caud, metencephalon caudal; Myen/Med, myelencephalon/medulla; Cereb, cerebellum; SC cerv, spinal cord cervical; SC thor, spinal cord thoracic; SC lumb, spinal cord lumbar; SC sacr, spinal cord sacral; DRG, dorsal root ganglion; CSF, cerebrospinal fluid; Sk muscle, psoas skeletal muscle. Numbers of samples for each tissue are shown in Supplementary Material, Table S1.
The majority of the brain regions from which we dissected the three tissue sub-pieces were heterogeneous. However, for the four spinal cord regions (cervical, thoracic, lumbar, sacral), we were able to readily distinguish white and gray matter separately and found significantly higher expansion indices in gray matter than white matter (2-way repeated measures ANOVA: F(2, 9) = 1.109, P = 0.0004; Fig. 2). Spinal cord white matter consists mostly of glial cells and myelinated axons, while the gray matter mainly contains neuronal somata, dendrites, synapses, glial cells and only a small number of nerve fibers. These data indicate that CAG expansion is greater in areas with abundant neurons and neuropil than in areas rich in myelinated fibers, consistent with previous observations of greater HTT CAG instability in neurons than in glia (19,20), and indicating that variation observed within other brain regions is likely due, in part, to varying proportions of gray and white matter.
Figure 2.
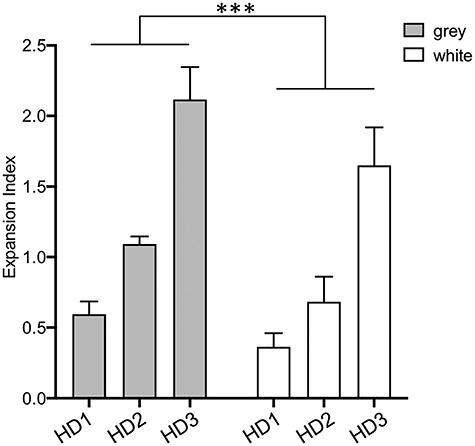
Comparison of spinal cord gray and white matter. Expansion indices were determined in spinal cord gray and white matter. Bars show the mean values across cervical, thoracic, lumbar and sacral regions of gray and white matter for each individual. Error bars represent standard deviation. Two-way ANOVA accounting for repeated measures across the four spinal cord regions from each individual: difference between gray and white matter P < 0.001 (***); difference between individuals P < 0.0001; difference between spinal cord subregion P = 0.42.
CAG expansion measures are robust across studies and capture CAG repeat-dependent biological variation
To provide an independent study, we also analyzed CAG repeat instability in postmortem tissues from an additional four adult-onset HD patients (HD5–8) (Table 1). Samples were collected and analyzed at the Ruhr University (Bochum, Germany). This sample set contained CNS and peripheral tissues that overlapped with a subset of those analyzed in Boston, with brain regions concentrated in the forebrain, as well as additional tissues not represented in the Boston study, e.g. retina, adrenal gland, jejunum, white adipose tissue and blood (Supplementary Material, Table S1). Either single or multiple pieces of tissues were sampled from the same region (Supplementary Material, Table S1). Expansion indices from these samples, determined as above, are shown in Figure 3, revealing broadly similar patterns of tissue instability to those in the Boston samples (Supplementary Material, Fig. S3). We note that for those regions in which more than one tissue sample was analyzed there was less intra-tissue variation in the Expansion index than in the Boston samples. This is likely due to sub-sampling from a smaller and more homogenous tissue block in the Bochum samples. In the brain, the primary visual cortex (BA17) shows high expansion indices in all samples investigated (Figs 1 and 3), whereas the retina presents with more moderate expansion levels (Fig. 3). This difference parallels neuroanatomical observations in HD of only mild changes in retinal thickness by optical coherence tomography (52–54) and the lack of histopathological changes or aggregates in postmortem retina (55). In contrast, there is considerable neuronal cell loss in BA17 and atrophy of associative visual cortices (56,57) suggesting that visual impairment in HD is more likely due to central visual perception. In the periphery, as for the Boston cohort (Fig. 1), liver exhibited the highest level of CAG expansion, while blood exhibited moderate expansion in relation to other peripheral tissues (Fig. 3). Mitochondrial dysfunction in hepatocytes of premanifest and manifest HD individuals has been documented using the C-methionine breath test (58,59). However, classical histopathological studies (60) showed no specific morphological alterations in HD postmortem liver tissue, and additional investigations of human HD liver are warranted to understand the hepatic involvement in HD. Among the peripheral organs, the kidney also shows high expansion indices in both cohorts (Figs 1 and 3). When kidney cortex and medulla were analyzed separately (Fig. 3C), the expansion index was much higher in the kidney medulla, indicating specific cell type(s) with different expansion propensities within these structures.
Figure 3.
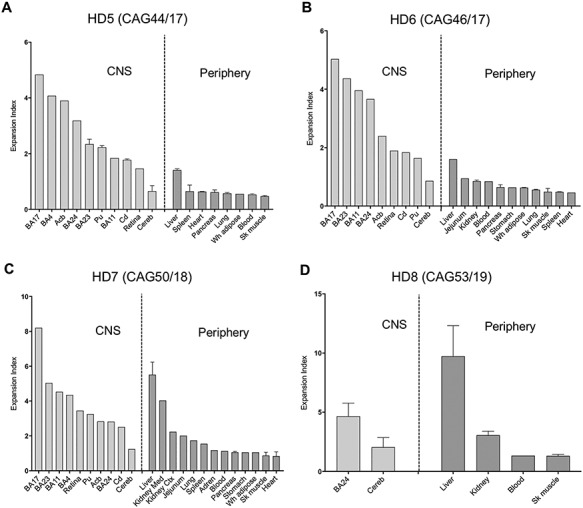
Quantitative somatic CAG expansion analyses of a replicate set of adult-onset HD tissues. (A-D) Expansion indices were quantified from GeneMapper peak height data of HTT CAG PCR products from individuals HD5, HD6, HD7 and HD8, analyzed in Bochum, Germany. Bars show mean expansion indices for each tissue, and error bars show standard deviation between tissue sub-pieces, other than HD5 blood where there are two technical PCR replicates of the same DNA sample. For each individual, data are plotted in order of mean tissue expansion index, separately for the brain and periphery. BA11, straight gyrus; BA24, anterior cingulate/midcingulate cortex; BA23, postcingulate cortex; BA4, primary motor cortex; BA3.1.2, primary somatosensory cortex; BA17, primary visual cortex; Cd, caudate; Acb, accumbens; Pu, putamen; Cereb, cerebellum; Kidney Med, kidney medulla; Kidney Ctx, kidney cortex; Sk muscle, sartorius or temporal skeletal muscle; Adren, adrenal gland; Wh adipose, white adipose tissue. Numbers of samples for each tissue are shown in Supplementary Material, Table S1.
The seven adult-onset individuals across the two studies spanned a range of repeat lengths from 43 to 53 CAGs. As CAG repeat length is the major driver of repeat expansion, we plotted expansion indices as a function of CAG repeat length for the subset of tissues represented in all seven individuals (Supplementary Material, Table S1, Fig. 4) to determine whether the predicted CAG length association could be detected. Remarkably, despite the two studies having been conducted independently and using different PCR assays for CAG repeat amplification (Materials and Methods), we were able to capture a strong positive association of expansion index with CAG length in many of the tissues across the seven individuals (Fig. 4). This was most apparent in many of the peripheral tissues, notably stomach, liver and pancreas, as well as in the cerebellum. BA4 and BA17 cortical regions showed the least robust CAG length-association across the sample continuum. It appears, therefore, that for tissues where there might be less cellular heterogeneity and/or less confounding by neuronal cell loss, that these measurements of CAG expansion across different laboratories and experimental conditions are sufficiently robust to capture biological variation with repeat length in a small number of samples.
Figure 4.
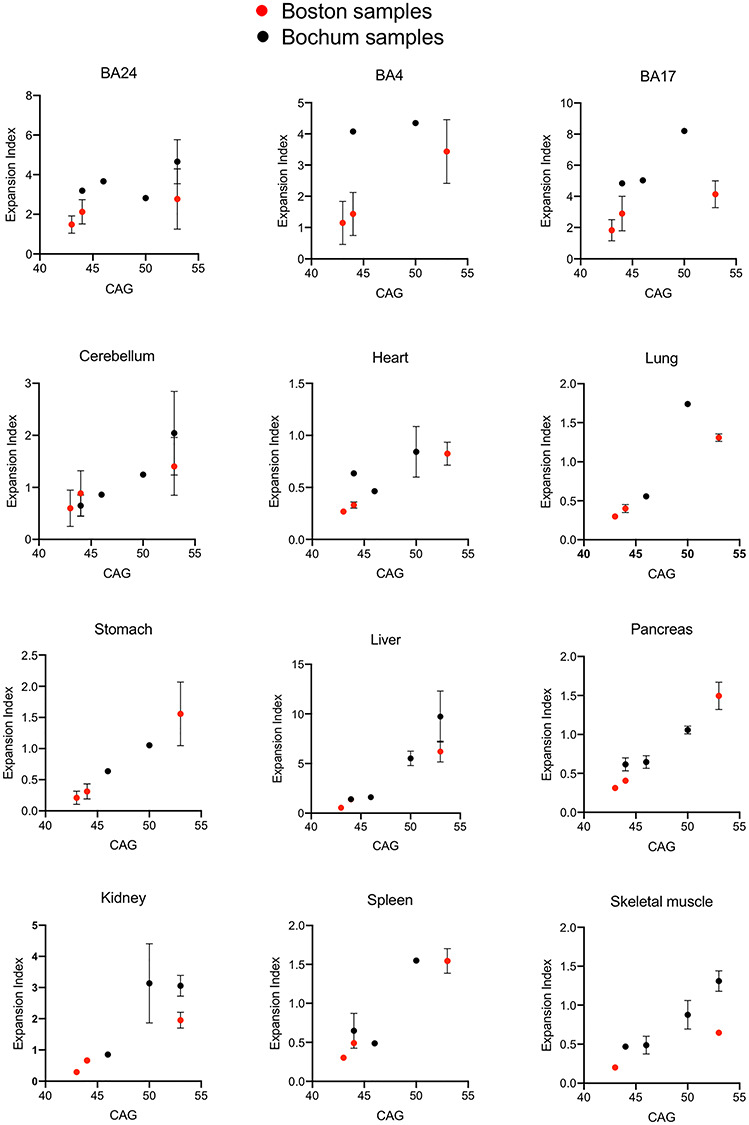
CAG length correlation across multiple tissues. Expansion indices were plotted as a function of CAG length for tissues shared between the Boston and Bochum studies in all seven adult-onset HD individuals (HD1–3, HD5–8). Points show the mean expansion index values and error bars show standard deviation.
Individual-specific CAG expansion is reflected across multiple brain and peripheral tissues
We also explored whether CAG instability in the periphery might reflect CAG instability in the brain. Figure 5 highlights how the expansion indices vary between the individuals HD1, HD2 and HD3 for a subset of high and low instability brain and peripheral tissues as well as CSF, including those that are most severely affected in the disease. Whereas the degree of expansion differs widely between tissues (note different y-axes on the graphs), the pattern of instability between the individuals is strikingly similar; in general, HD1 has the lowest instability, HD2 has an intermediate level of instability and HD3 has the highest instability. This is the typical pattern observed, however, there are exceptions, e.g. caudate/accumbens/putamen, and to a lesser extent, BA4, where the expansion index of HD2 is relatively low, and hippocampus where HD2’s expansion index is relatively high. Thus, it would appear that individual-specific expansion is largely captured across multiple tissues, regardless of the particular susceptibility towards expansion in that tissue. As instability is both a CAG length- and time-dependent process, it is likely that the long CAG repeat (53 CAGs) is a major contributor to the relatively high instability in HD3, while the slightly longer CAG repeat (44 versus 43 CAGs) and later age of death (66 versus 57 years) may contribute to the generally higher instability in HD2 relative to HD1. We speculate that the relatively low expansion index in caudate/accumbens/putamen and BA4 of HD2 may be due to advanced neuronal cell loss, as reflected by Vonsattel Grade 4 (Table 1). Similar comparisons in brain regions of individuals HD5–HD8 support both CAG length and cell loss in vulnerable tissues as contributors to postmortem somatic expansion levels (Supplementary Material, Fig. S4). Of note, in individuals HD5–HD7 the neostriatal sub-areas accumbens, caudate and putamen were analyzed separately (Fig. 3 and Supplementary Material, Fig. S4). The lower expansion index of the atrophic caudate in comparison with the morphologically less affected accumbens (1,61) and the relatively high expansion index in the accumbens of HD5 with Vonsattel grades 2–3 (Fig. 3 and Supplementary Material, Fig. S4), may be consistent with brain areas with severe neuronal loss and compensatory gliosis having lower levels of detectable expansions postmortem than better preserved areas.
Figure 5.
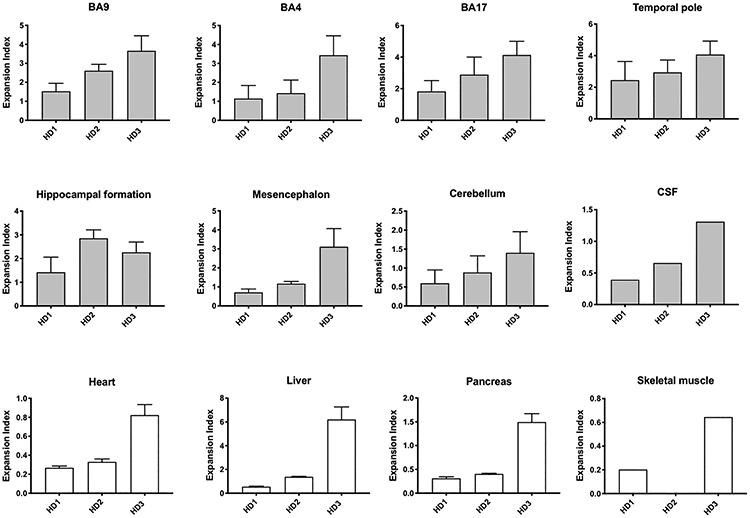
Inter-individual differences in somatic CAG expansion are largely reflected across multiple brain and peripheral tissues. Expansion indices were compared between individuals HD1, HD2 and HD3 in a number of brain and peripheral tissues, as well as post-mortem CSF. Bars show mean ± SD across the tissue sub-pieces. Skeletal muscle was not available from HD2.
We further assessed the relationship in CAG expansion between peripheral tissues that could potentially be obtained via biopsy in a living patient (skeletal muscle and liver) and brain regions (BA24, BA17, BA4 and cerebellum) that were available from individuals HD1–3 and HD5–8 (Supplementary Material, Table S1), finding significant correlations between brain region expansion and liver or skeletal muscle expansion (Fig. 6A). We also examined the relationship between blood and brain expansion in individuals HD5–8; while the sample number is very limited, it appears that the extent of an individual’s expansion in blood reflects the level of expansion in multiple brain regions (Fig. 6B). Relationships between expansion indices in all tissues relative to those in accessible peripheral tissues or in biofluids (blood, CSF) are shown in Supplementary Material, Figure S5. Finally, we compared expansion indices between cerebellum and BA9, exhibiting low and high instability respectively, from eight additional HD individuals with repeat lengths from 40 to 48 CAGs and found a highly significant correlation (R2 = 0.93, P < 0.0001) between the two measures (Fig. 6C). Together, these data support the idea that an individual’s propensity for somatic CAG expansion is reflected across multiple brain and peripheral tissues, including those that are accessible in patients, but indicate that other tissue specific factors, including neuronal cell loss may also contribute to differences in instability read-outs.
Figure 6.
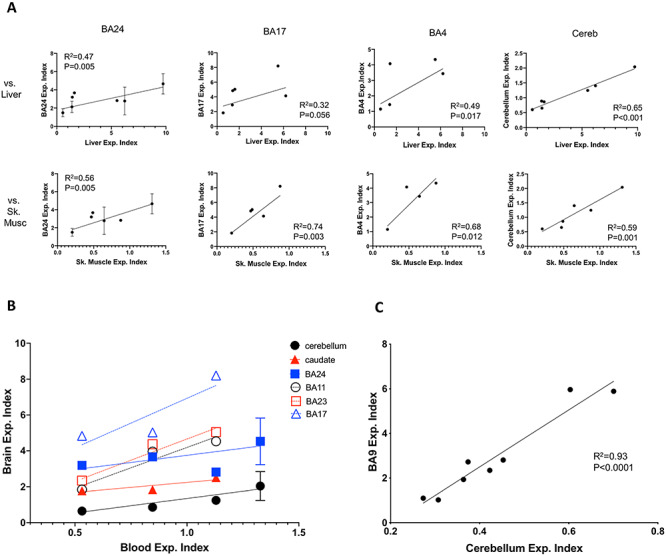
Correlations between expansion indices in different tissues. (A) Correlation between expansion indices in liver (top panels) or skeletal muscle (lower panels) and those in BA24, BA17, BA4 and cerebellum across individuals HD1–3 and HD5–8. N = 4–7 depending on the tissue comparison (see Supplementary Material, Table S1). (B) Comparison of Expansion indices in brain regions and in blood from individuals HD5–8. For replicate measurements, mean ± SD is plotted. Sample number is too low (N = 4 or N = 3 depending on the brain region) for meaningful statistical analyses, however, results of linear regressions of brain region versus blood expansion indices, meant to indicate trends are as follows: cerebellum R2 = 0.72, P = 0.0086; caudate R2 = 0.80, P = 0.11; BA24 R2 = 0.22, P = 0.35; BA11 R2 = 0.92, P = 0.18, BA23 R2 = 0.95, P = 0.026; BA17 R2 = 0.77, P = 0.32. Trend lines are shown on the graphs. (C) Correlation between expansion indices in cortex (BA9) and cerebellum from an additional eight individuals (CAG 40–48).
Extensive somatic instability in juvenile-onset HD
Juvenile-onset HD, caused by median CAG lengths of ~60–64 repeats, often has a different clinical presentation to adult-onset HD. This includes rigidity without chorea, seizures and cognitive decline as well as gait disturbance and ataxia that are indicative of cerebellar dysfunction (2–4). To gain insight into somatic CAG instability in juvenile HD, we profiled CAG instability in postmortem brain regions and peripheral tissues of a juvenile-onset patient (HD4) who presented with disease around 4 years of age and died at age 6 (62) (Table 1 and Supplementary Material, Table S1). Neuropathological examination of individual HD4 showed neuronal cell loss in the caudate, putamen and globus pallidus corresponding to Vonsattel grade 3 as well as prominent loss of Purkinje cells of the cerebellum with preservation of cerebellar granule cells (62). Genetic testing originally revealed 169 HTT CAG repeats (62). However, subsequent repeat sizing in blood DNA as part of the current study shows ~140 CAGs. GeneMapper profiles of brain and peripheral tissues from this individual are shown in Figure 7A, revealing extensive CAG repeat mosaicism. For each sample, the modal allele and the longest allele detected are plotted in Figure 7B. As the modal allele varies to such an extent between tissues, we cannot determine with certainty the size of the inherited CAG repeat length. We therefore did not quantify expansion indices in these samples, as anchoring this measurement on the modal allele length within each repeat length distribution will effectively ignore any contribution of somatic expansion that might have shifted the entire distribution of alleles. For the majority of the brain tissues, the bulk of the repeat length distribution (modal CAG allele) was ~140 CAGs. The striking exception is cerebellum, with a modal CAG repeat of ~110 CAGs. Modal allele length in the peripheral tissues was closer to ~130 CAGs, with distinct patterns of heterogeneity between the tissues, and in many cases a prominent bimodal distribution of alleles indicative of a subset of highly expandable cells, as in the liver where hepatocytes are the most unstable (23). Interestingly, the modal CAG length in blood (~140) was closer to those in the brain tissues than in the peripheral tissues. CAG lengths >200 were detected using this assay in BA9, BA24, BA3.1.2, temporal pole, BA17 and liver, consistent with findings that these are among the most unstable tissues in the adult-onset cases (Figs 1 and 3).
Figure 7.
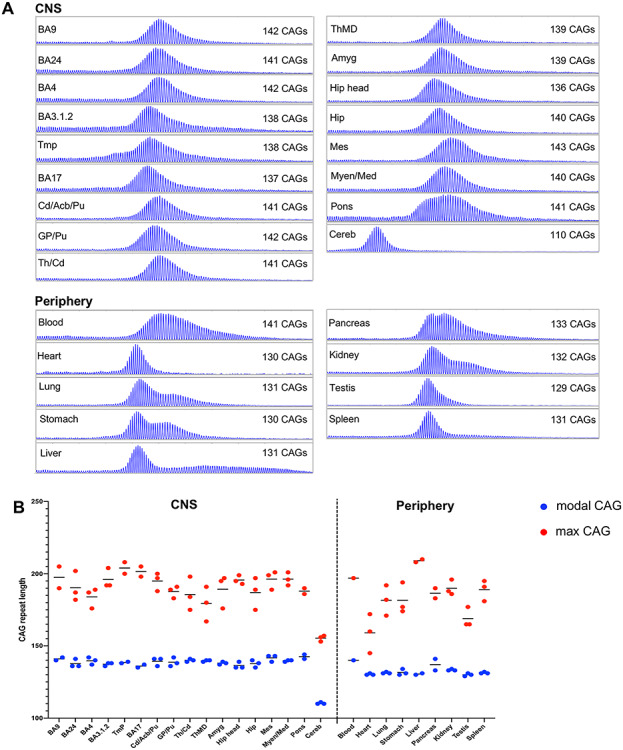
CAG expansion in a case of juvenile-onset HD. (A) GeneMapper profiles of expanded allele HTT PCR products from brain regions and peripheral tissues. The size of the modal allele for each trace is indicated. (B) Modal CAG repeat and maximum CAG repeat detectable (1% relative peak height threshold) are plotted for all tissue sub-piece replicates. Short horizontal lines show mean of tissue replicates. BA9, prefrontal cortex; BA24, anterior cingulate/midcingulate cortex; BA4, primary motor cortex; BA3.1.2, primary somatosensory cortex; Tmp, temporal pole; BA17, primary visual cortex; Cd/Acb/Pu, caudate/accumbens/putamen; GP/Pu, globus pallidus/putamen; Th/Cd, thalamus/caudate; ThMD, dorsomedial thalamic nucleus; Amyg, amygdala; Hip head, hippocampal formation (head); Hip, hippocampal formation; Mes, mesencephalon; Myen/Med, myelencephalon/medulla; Cereb, cerebellum.
Lower modal CAG lengths in cerebellum compared with other brain regions in juvenile-onset HD have previously been reported (8,63,64). However, this is to our knowledge the first indication that the cerebellum in juvenile-onset HD exhibits an even lower repeat length than peripheral tissues though this has been observed in other diseases (65,66). Note that we also observe a modal allele shift of one or two CAGs in some non-cerebellar brain regions of HD3 and HD5 with 53 CAGs and in the SCA1 individual (see Materials and Methods), and in ongoing analyses (unpublished), a repeat length-dependent divergence in cortical and cerebellar modal CAG lengths. It is therefore possible that the inherited repeat length in individual HD4 is closer to ~100 CAGs with the CAG length in postmortem cerebellum reflecting a low rate of somatic expansion relative to other tissues (Supplementary Material, Fig. S6). This is supported by the greater somatic expansion in several peripheral tissues, relative to cerebellum, in the adult-onset samples (Figs 1 and 3). Thus, there is nothing that qualitatively distinguishes the behavior of adult- and juvenile-onset repeat lengths, but rather that the rates of expansion of the longer juvenile-onset CAG repeats are sufficiently high to shift the entire repeat distribution. Alternatively, the low CAG length in postmortem cerebellum may be due to contraction from an inherited repeat length closer to ~130 CAGs during cerebellar development or aging (Supplementary Material, Fig. S6). Of note, a similar short modal CAG length in cerebellum, is not apparent in HD mouse models including the HttQ111 knock-in line harboring very similar repeat lengths to individual HD4 juvenile-onset patient, although the expansion propensities across tissues in mouse and human are similar (22,25). This may reflect insufficient time in the mouse for the bulk of the repeat lengths to diverge substantially, different repeat dynamics in mouse and human cerebellum or differences in cerebellar development.
Notably, testis of individual HD4 exhibited relative stability. Given our observation that testis exhibits levels of CAG expansion equivalent to those in liver for individual HD3 with 53 CAGs (Fig. 1), we might assume that at least a comparable level of expansion, presumably attributable to the germline, would also be detectable in testis of individuals with longer alleles. Thus, the apparent absence of extensive germline CAG expansion in this 6-year old individual suggests that substantial levels of male germline instability only occur post-puberty. A previous study indicated that instability in the male germline can occur both during and after meiosis as well as pre-meiotically (67). The lack of a strong influence of male age suggests that accrual of repeat length changes over successive spermatogonial cell divisions is not the major contributor to male germline expansions (12). Our data would appear to support meiotic or post-meiotic mechanisms underlying the bulk of male germline expansions, though we cannot exclude the possibility that 6 years is insufficient time for substantial expansions to accumulate in premeiotic spermatogonia.
Correlation of tissue-specific somatic expansion of ATXN1 and HTT CAG repeats
To gain insight into potential similarities or differences in the tissue specificity of somatic instability of a different disease-associated CAG repeat, we also measured somatic expansion of the CAG repeat tract in the ATXN1 gene that causes spinocerebellar ataxia type 1 (SCA1). We obtained a subset of the postmortem brain tissues that were analyzed in our HD samples—namely ‘cortex’ (of unspecified sub-region), BA9, BA24, BA17, caudate/accumbens/putamen, globus pallidus, thalamus, amygdala, hippocampal formation and cerebellum—from a SCA1 patient with a CAG repeat length of ~46 (see Materials and Methods) and quantified expansion indices from GeneMapper profiles of ATXN1 CAG repeats were quantified from GeneMapper traces as above (Fig. 8A and B). To compare better between the SCA1 and HD samples, we plotted ATXN1 CAG expansion indices against HTT CAG expansion indices, averaging values for the HD samples across the three tissue replicates for adult-onset individuals HD1, HD2 and HD3 (Fig. 8C). The two measures were highly correlated (R2 = 0.95; P < 0.0001), thereby providing strong support for disease locus independent trans-acting drivers of tissue-specific CAG repeat expansion.
Figure 8.
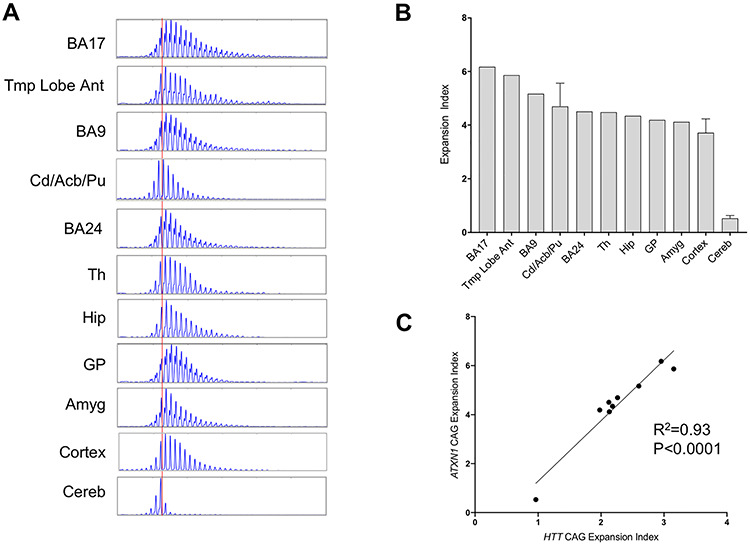
ATXN1 CAG expansion in brain tissues and correlation with HTT CAG expansion. (A) GeneMapper traces of ATXN1 CAG PCR products. The red vertical line shows the modal allele in cerebellum (CAG46). (B) Expansion indices calculated from GeneMapper peak height data of expanded allele ATXN1 CAG repeats. Three tissue pieces were analyzed for caudate/accumbens/putamen, cortex and cerebellum and bars show mean ± SD. BA9, prefrontal cortex; BA24, anterior cingulate/midcingulate cortex; Tmp Lobe Ant, anterior temporal lobe; BA17, primary visual cortex; Cd/Acb/Pu, caudate/accumbens/putamen; Th, thalamus; Hip: hippocampal formation; GP, globus pallidus; Amyg, amygdala; Cereb, cerebellum. (C) Correlation between mean ATXN1 and mean HTT CAG expansion indices (individuals HD1, HD2, HD3) for the shared tissues. Shared tissues used in the correlation are temporal pole (HD)/anterior temporal lobe (SCA1), BA17, caudate/accumbens/putamen, BA9, BA24, hippocampal formation, amygdala, globus pallidus/putamen (HD)/globus pallidus (SCA1) and cerebellum.
Discussion
Genetic data in HD patients support a two-step model for pathogenesis in which somatic CAG expansion drives the rate of phenotypic onset, triggering toxicity driver(s) that are responsible for the demise of vulnerable cell types (28). This model predicts that cell type- and disease-specific pathology in HD and in other CAG repeat expansion diseases will depend both on the rate of CAG expansion in a particular cell type and its vulnerability to the toxic entity/entities. Here, to gain insight into somatic CAG expansion in different tissues and their relationships to disease pathogenesis we have performed comprehensive quantitative analyses of CAG instability across a wide range of CNS regions and peripheral postmortem tissues of adult- and juvenile-onset HD individuals and in brain regions from an individual with SCA1. Our data support and expand upon previous studies (8,13,19–21,38–41), and demonstrate that HTT CAG repeat expansion occurs in all tissues and biofluids analyzed, though to different extents. These analyses did not permit detection of rare expansions that would require small pool PCR using sufficient numbers of molecules to detect low frequency events (19,21). Therefore, additional tissue-specific effects may be revealed by the detection of rare expansions. Next-generation sequencing approaches can also be used to capture somatic CAG length variation (68,69). Methods based upon sequencing of PCR amplicons (68) capture similar profiles of somatic expansion to fragment size-based analyses of PCR products, as performed in this study. Emerging PCR-free technologies can capture repeat length variation present in genomic DNA, without any confound of PCR bias (69,70) and can also capture rare events, though this will depend on the read depth attainable.
Significantly, we find similar tissue patterns of HTT CAG instability across the HD individuals, which strikingly, strongly correlate with ATXN1 CAG instability in the SCA1 individual. These data provide strong support for the presence of disease locus-independent trans factors that drive the rate(s) of CAG expansion in different cell types (22). Previous studies have uncovered potentially complex relationships between tissue instability and gene expression in mice, with no straightforward association with the expression of key DNA repair genes that drive CAG expansion (22,71). At the level of individual cell types, the trans factors driving CAG expansion are currently unknown.
The results of our study also reveal that tissue-specific patterns of CAG instability do not necessarily predict region- or disease-specific pathology, i.e. both HTT and ATXN1 instability are high in the cortex and in caudate/accumbens/putamen, regions that are the most vulnerable in HD, but less vulnerable in SCA1 where the most affected brain regions are the cerebellum and brainstem (44). Similarly, HTT CAG instability in liver is relatively high, yet although there is evidence for hepatic dysfunction in HD, liver does not exhibit obvious pathology (58–60). In support of a two-step model for disease pathogenesis, our data indicate that the vulnerability of a particular cell type depends on the nature of the toxic species and the repeat length(s) needed to elicit cellular toxicity. This provides an explanation for the high vulnerability of MSNs in HD compared with SCA1; thus, the HTT and ATXN1 CAG repeats expand at a similarly high rate in these neurons that are shown to have a high propensity for CAG instability (19,24), but HD toxicity is triggered at a lower repeat length(s) in MSNs than SCA1 toxicity. Similarly, this can explain the vulnerability of MSNs compared with hepatocytes in HD; although the CAG repeat expands at high rate in hepatocytes (23), these cells are much less sensitive to the toxic species than MSNs, with considerably longer repeat lengths likely needed to elicit hepatocyte degeneration. The nature of the toxic species in different cell types is not clear and may be different depending on the cell type. Potential drivers of cell dysfunction and death in HD include amino terminal fragments or full-length mutant Huntingtin, though roles for other entities including expanded RNA and RAN translation products have been proposed (72–74). In SCA1, the interaction of Ataxin1 with capicua drives Purkinje cell pathogenesis, however, different mechanisms of toxicity are likely in the brainstem (75,76).
While there is significant evidence that somatic CAG expansion in vulnerable neurons drives the rate of disease onset in HD, this is less obvious in SCA1 as little is understood of the degree of CAG expansion in cerebellar Purkinje cells, or in brainstem neurons that are highly vulnerable in SCA1 (44). As these neurons are relatively rare, they do not contribute significantly to an instability readout that is measured in bulk tissue. Notably, SNPs in two HD onset modifier genes (28) (FAN1, PMS2), with roles in CAG/CTG instability (77,78) (MacDonald/Loupe/Wheeler/Pinto unpublished data), were associated with age at onset in spinocerebellar ataxias, including SCA1 (79), indicating that somatic expansion may also drive the rate of onset of these other CAG repeat disorders. One prediction, therefore, is that cerebellar Purkinje neurons and vulnerable brainstem neurons are susceptible to CAG expansion. This was previously indicated for the ATN1 CAG repeat causing dentatorubral-pallidoluysian atrophy (DRPLA) in laser capture microdissection experiments that showed higher CAG expansion in cerebellar Purkinje cells than in cerebellar granule cells (40). Over the disease course, pathology in SCA1 extends beyond the cerebellum and brainstem, including involvement of the caudate, putamen and temporal lobe in much the same way that HD pathology extends beyond the caudate and putamen, also involving cerebellar Purkinje cells and brainstem nuclei (44,46,62,80,81), with disease manifestation extending to the periphery in HD (82,83). Therefore, somatic CAG expansion may drive the rate of various aspects of disease pathogenesis in many different cell types both in HD and in SCA1 over the courses of these diseases. Future analyses of CAG instability and disease phenotypes across a variety of different cell types and disease conditions will help to dissect: (1) the intrinsic cell type specific trans factors contributing to CAG expansion, as well as possible cis-acting modifiers (66,84), (2) the impact of somatic expansion on phenotypic expression and (3) potential cell type- and disease-specific repeat thresholds needed to elicit toxicity.
One goal of this study was to undertake a preliminary assessment of whether CAG instability in accessible peripheral tissues (e.g. skeletal muscle or liver) or in biofluids (e.g. blood or CSF) might provide a surrogate for CAG instability in brain tissues. This is important to understand for therapeutic trial biomarker read-outs of the efficacy of drugs targeting somatic expansion. Our data indicate that an individual’s propensity for somatic CAG expansion is reflected across multiple brain regions, peripheral tissues and biofluids regardless of their absolute level of instability, and that expansion in these peripheral tissues correlates with that in the brain. This observation is encouraging and suggests that despite exhibiting relatively low CAG instability, peripheral tissues/biofluids may be useful for readouts of an individual’s somatic expansion. Ideally, one would wish to use a neuronally derived read-out of somatic expansion to assess therapeutic efficacy of targeting instability in the brain. In this study, we were able to measure CAG expansion in three post-mortem CSF samples. The source of the DNA extracted from the CSF is currently unclear. As described (Materials and Methods), measures were taken to ensure that CSF was not contaminated with brain parenchyma or blood, however, the CSF was not spun, and it is possible that it could contain rare white blood cells if the harvest was not performed optimally. The level of expansion was low relative to the various brain regions examined, suggesting that the postmortem CSF is not enriched in DNAs from cell types containing highly expanded repeats that have succumbed to degeneration. Further studies of CAG instability in patient CSF, including in exosomes, are warranted.
In the periphery, liver may be the preferred accessible tissue over skeletal muscle as the greater level of instability in liver may provide a more sensitive readout. However, the patient burden of liver biopsy may make this an unrealistic option. Based on the current data, blood, previously shown to exhibit somatic CAG expansion (28,68), and with expansion levels between those of liver and muscle, would appear to be a reasonable option. However, the use of peripheral tissues/biofluids as a source for biomarker assays makes the assumption that the mechanisms underlying CAG instability are the same as in the brain. In the current study, although we find that peripheral and brain expansion are correlated, the principal contributor to CAG expansion that is discernible between individuals and that drives the correlations is constitutive CAG repeat length. Therefore, it is unknown whether other modifying factors that also contribute to individual-specific CAG expansion rates (28,68) act similarly across tissues/cell-types and whether therapeutics targeting such modifiers will produce similar effects in different tissues. Indeed, recent evidence suggests that there may be different effects of genetic modifiers of HTT CAG instability in blood and in the brain (68) and a brain-specific instability modifier has been described in a SCA1 knock-in mouse model (85). Further studies will therefore be needed to dissect the mechanisms of CAG instability that may differ between cell types.
Overall, our quantitative cross-tissue analyses of CAG expansion set the stage for future important avenues of investigation that are critical for understanding the underlying biology of CAG instability, its relationship to pathogenesis and for enabling clinical studies targeting the process of somatic CAG expansion.
Materials and Methods
Patient samples
Human postmortem tissue and DNA samples analyzed at the MGH were obtained and studied under an approved Institutional Review Board (IRB) protocol (2006P000090) from the New York Brain Bank (HD1–3), Ruhr-University Bochum (HD5–8), the University of Washington (HD4), the University of Minnesota (SCA1—consented at the University of Florida) and additional brain samples were obtained from the Harvard Brain Tissue Resource Center (Belmont, MA, USA) and the National Neurological Research Bank (Los Angeles, CA, USA) as part of historical collections obtained through the Huntington’s Disease Center Without Walls. Human HD tissue from individuals HD5 to HD8 was obtained by autopsy in the Institute of Anatomy, Ruhr-University Bochum (Germany). All donors had given written consent to examine the tissue for research purposes and the study was approved by the ethic committee of the Ruhr-University Bochum (Reg. No. 17-5939). All HD individuals had clinically manifest HD diagnosed by an experienced neurologist (CS). Cause of death for HD1 and HD2 is unknown, HD3 died from pneumonia with sepsis, HD4 died from aspiration pneumonia (62), HD5 committed suicide, HD6 and HD8 died from pneumonia and HD7 from renal cell carcinoma. Demographic and genetic data from patient samples HD1–8 are detailed in Table 1 and the tissues analyzed are shown in Supplementary Material, Table S1. Blood samples from HD5–8 were taken at the time of genetic testing (HD5 38 years, HD6 41 years, HD7 21 years, HD8 38 years).
Tissue preparation
Tissues analyzed at MGH were obtained as frozen blocks, or crushed frozen tissue, and Vonsattel grading (1) for HD1–3 was provided by the New York Brain Bank. To harvest the CSF, the whole, fresh brain was placed on a cutting board with the ventral aspect facing the prosector and with the frontal poles away from the prosector. A transverse cut was made through the distal end of the infundibulum and a distally truncated safety needle cap, used to prevent damage to the parenchyma and subsequent contamination of the CSF, attached to a clean 5 mL syringe was inserted into the infundibulum to draw the CSF into the syringe. To increase the yield of CSF, the occipital lobes were gently raised to drain the CSF toward the infundibulum. Processing of postmortem tissue at Ruhr-University Bochum was carried out as follows: after removing the brain stem with the cerebella, the two hemispheres were sagittally separated. One hemisphere was cut into coronal slices, small native (unfixed and not frozen) samples were dissected from defined brain areas, squashed and directly transferred to Tris buffer for subsequent DNA extraction. One cerebellum was dissected from the brain stem. Native samples from the cerebellar vermis, retina, peripheral organs and tissues were also directly transferred to Tris buffer. Most tissues from HD5-HD7 taken for DNA analyses were native. Additional samples were transferred to tubes, shock-frozen in 8% methylcylcohexan in 2-methylbutan (v/v) (−80°C) and stored at −80°C until used. The removed cerebellum was horizontally sectioned at the level of the cerebellar nuclei. The two cerebellar parts and the brain slices from which the samples were taken, were finally passive-frozen and stored at −80°C. Some frozen samples including all from HD8 were also taken for DNA analyses. To exclude differences, we compared several native and frozen tissue samples from the same area of the same patient and detected no obvious differences in CAG instability. The other brain hemisphere and the brain stem with the other cerebellum were fixed in 4% paraformaldehyde shaking gently for 4 weeks for later morphological reference. Fixed tissue was transferred to 0.5% paraformaldehyde and stored at 4°C until further used. The fixed hemisphere was cut into 1 cm thick coronal slices to rule out obvious neuropathologies and to assess Vonsattel grading.
Isolation of DNA
Genomic DNA was extracted using the Qiagen DNeasy Blood & Tissue kit according to the manufacturer’s protocol for HD5–8 and with the following modifications for other samples: samples were homogenized in 160 μL 1X Dulbecco’s phosphate buffered saline, volumes of proteinase K, Buffer AL and 100% ethanol were doubled and DNA was eluted in 125 μL of Qiagen Elution Buffer, preheated to 60°C, applied to the spin columns, and incubated at 56°C for 1 h before centrifugation. Isolation of DNA from 100 μL CSF was performed using the Qiagen Gentra Puregene kit, according to manufacturer’s instructions, but doubling the volume of all protocol reagents and including 1 μL of 20 mg/ml glycogen as a carrier in the DNA precipitation step.
Repeat length genotyping
Genotyping of the normal and expanded CAG repeats from samples from all HD individuals were determined at MGH using standardized assays for adult onset CAG repeats (86) or long CAG repeat lengths associated with juvenile-onset HD based on Milunsky et al. (87) and using primers HDXL-F (6-FAM-5′-CCT TCG AGT CCC TCA AGT CCT TC) and HDXL-R (5′-CGG CTG AGG CAG CAG CGG CTG T). Genotyping for the CCG repeat was performed using primers CCG1 (F) (6-FAM-5′-GCAGCAGCAGCAGCAACAGCCGCCACCGCC) and. CCG2 (R) (5′-CTTTCTTTGGTCGGTGCAGCGGCTCCTCAG) in a total reaction volume of 10 μL containing 2.5 mm MgCl2 (Applied Biosystems), 1× Buffer II (Applied Biosystems), 0.05 U Amplitaq Gold (Applied Biosystems), 0.5 mm each dATP, dCTP, dTTP, 0.25 mm dGTP and 0.5 mm 7-Deaza-2′-Deoxyguanosine 5′-Triphosphates (GE Healthcare), 1.0 ul of DMSO (Sigma) and 0.25 μM each primer with 60 ng of genomic DNA. Cycling conditions were 95°C 5 min, followed by 35 cycles of 95°C 1 min and 72°C 2 min. PCR products were run on an ABI 3730xL sequencer with GS500LIZ internal size standard and analyzed using GeneMapper v.5 software. All genotyping assays included samples with known, sequenced repeat lengths, for accurate sizing.
Analyses of somatic instability
For analyses of somatic instability in the Boston HD samples the HTT CAG repeat was PCR-amplified using the Qiagen Taq PCR Core kit with 0.8 μM CAG1 (F) (6-FAM-5′-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC), 0.8 μM Hu3 (R) (5′-GGCGGCTGAGGAAGCTGAGGA), 0.5 U Taq DNA Polymerase, 1× Qiagen PCR Buffer, 1× Q-Solution and 0.2 mm dNTP mix in a total reaction volume of 20 μL. Cycling conditions were 95°C 5 min, 30 cycles of 95°C 30 s, 65°C 30 s, 72°C 90 s, followed by 72°C for 10 min. PCR products were run on the Applied Biosystems 3730xl DNA Analyzer using GS500LIZ internal size standard and analyzed using GeneMapper v5. For analyses of somatic instability in the Bochum samples, the HTT CAG was PCR amplified using primers Hu4 (F) (6-FAM-5′-ATGGCGACCCTGGAAAAGCTGATGAA) and Hu5 (R) (5′-GGCGGTGGCGGCTGTTGCTGCTGCTGCTGC) as previously described (88,89). PCR products were resolved using the ABI 3500XL Genetic Analyzer (Applied Biosystems) using GeneMapper v4.1 software and GeneScan 500-ROX as internal size standard. For analyses of somatic instability in the SCA1 samples, the ATXN1 CAG repeat was amplified as previously described (90), but using the Qiagen Taq PCR Core kit with 0.8 μM REP1 (F) (6-FAM-5′ AACTGGAAATGTGGACGTAC), 0.8 μM REP2 (R) (5′-CAACATGGGCAGTCTGAG), 0.5 U Taq DNA Polymerase, 1 X Qiagen PCR Buffer, 1 X Q-Solution and 0.2 mm dNTP mix in a total reaction volume of 20 μL. PCR products were run on the Applied Biosystems 3730xl DNA Analyzer using GS500LIZ internal size standard and analyzed using GeneMapper v5. Repeat size was inferred based on the size of the PCR product in base-pairs. Expansion indices from GeneMapper peak height data were calculated as described (22), taking into account only expansion peaks to the right of the highest peak (modal allele) and using a 1% relative peak height threshold. Briefly, normalized peak heights were calculated by dividing each expansion peak height by the sum of the heights of the main allele plus all expansion peaks, the change in CAG length of each expansion peak from the main allele was determined, each normalized peak height was multiplied by the CAG change from the main allele, and these values were summed to generate an expansion index that represents the mean positive CAG repeat length change in the population of cells being analyzed. All samples analyzed from individual HD1, HD2, HD5, HD7 and HD8 had the same modal allele length (43, 44, 44, 46 and 50 respectively) For individual HD3, the modal allele was 53 CAGs for most tissues and 54 for a minority of tissues and for individual HD8 the modal allele length was 53 in all samples but one where it was 54 CAGs. For the SCA1 tissues, the modal allele length sized at 46 or 47 CAGs for the majority of samples and at 48 CAGs in three samples. These differences in modal allele length for the individuals with the longest repeat lengths very likely reflect somatic expansion, supported by all HD3 and HD8 peripheral tissues having the shorter modal repeat length (53) and the three tissue replicates of the more stable cerebellum in the SCA1 case sizing as 46 CAGs. A modal allele shift is also clearly apparent in the juvenile onset samples. Therefore, in order to account for somatic expansion that has shifted the modal repeat length, we have taken the lower/lowest repeat lengths in HD3, HD8 and in the SCA1 individual to represent most closely the inherited repeat lengths, and have calculated expansion indices based on these CAGs as the main allele. For HD4, we have not calculated expansion indices due to the high variation in modal allele length and uncertainty of the allele length that represents the inherited allele, and instead have plotted modal CAG and the maximum CAG detectable using a 1% relative peak height threshold to visually represent the variation across these tissue samples.
Statistical analyses
Statistical analyses were performed using GraphPad Prism v.8. Linear regression was used to assess the correlation between expansion index values in different tissues. To compare instability in spinal cord gray and white matter, we used 2-way ANOVA with region (gray versus white) and individual (HD1–3) as main effect variables, accounting for repeated measures across the four spinal cord regions.
Supplementary Material
Acknowledgements
Our gratitude goes to all donors and their families. The excellent technical assistance of Gudrun Rodepeter, Gabi Schlüter, Robert Nadgrabski and Claudia Schneider (Ruhr-University Bochum) is also gratefully acknowledged.
Conflict of Interest statement. V.C.W. is a scientific advisory board member of Triplet Therapeutics, a company developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease and Myotonic Dystrophy and of LoQus23 Therapeutics and has provided paid consulting services to Alnylam. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. T.D.B. receives licensing fees from Athena Diagnostics Inc. and is a consultant for Genentech.
Funding
This work was supported by the National Institutes of Health (NS049206 to V.C.W. and AG005136 to S.J., T.D.B., and C.D.K.); the CHDI Foundation; the Berman/Topper Family HD Career Development Fellowship from the Huntington’s Disease Society of America (R.M.P.); the Nancy and Buster Alvord Endowment (C.D.K.); The Department of Veterans Affairs Research Funds (T.D.B.). A.B., S.J., C.D.K. and T.D.B. are at the University of Washington HDSA Center of Excellence.
References
- 1. Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D. and Richardson E.P. (1985) Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol., 44, 559–577. [DOI] [PubMed] [Google Scholar]
- 2. Nance M.A. and Myers R.H. (2001) Juvenile onset Huntington's disease-clinical and research perspectives. Ment. Retard. Dev. Disabil. Res. Rev., 7, 153–157. [DOI] [PubMed] [Google Scholar]
- 3. Fusilli C., Migliore S., Mazza T., Consoli F., De Luca A., Barbagallo G., Ciammola A., Gatto E.M., Cesarini M., Etcheverry J.L. et al. (2018) Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol., 17, 986–993. [DOI] [PubMed] [Google Scholar]
- 4. Cronin T., Rosser A. and Massey T. (2019) Clinical presentation and features of juvenile-onset Huntington’s disease: a systematic review. J. Huntington’s Dis., 8, 171–179. [DOI] [PubMed] [Google Scholar]
- 5. Quarrell O., O'Donovan K.L., Bandmann O. and Strong M. (2012) The prevalence of juvenile Huntington's disease: a review of the literature and meta-analysis. PLoS Curr., 4, e4f8606b8742ef8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 7. Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M. and Abbott M. (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat. Genet., 4, 387–392. [DOI] [PubMed] [Google Scholar]
- 8. Telenius H., Kremer B., Goldberg Y.P., Theilmann J., Andrew S.E., Zeisler J., Adam S., Greenberg C., Ives E.J. and Clarke L.A. (1994) Somatic and gonadal mosaicism of the Huntington's disease gene CAG repeat in brain and sperm. [erratum appears in Nat Genet 1994 May;7(1):113]. Nat. Genet., 6, 409–414. [DOI] [PubMed] [Google Scholar]
- 9. MacDonald M.E., Barnes G., Srinidhi J., Duyao M.P., Ambrose C.M., Myers R.H., Gray J., Conneally P.M., Young A. and Penney J. (1993) Gametic but not somatic instability of CAG repeat length in Huntington's disease. J. Med. Genet., 30, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zuhlke C., Riess O., Bockel B., Lange H. and Thies U. (1993) Mitotic stability and meiotic variability of the (CAG)n repeat in the Huntington disease gene. Hum. Mol. Genet., 2, 2063–2067. [DOI] [PubMed] [Google Scholar]
- 11. Leeflang E.P., Zhang L., Tavare S., Hubert R., Srinidhi J., MacDonald M.E., Myers R.H., Young M., Wexler N.S. and Gusella J.F. (1995) Single sperm analysis of the trinucleotide repeats in the Huntington's disease gene: quantification of the mutation frequency spectrum. Hum. Mol. Genet., 4, 1519–1526. [DOI] [PubMed] [Google Scholar]
- 12. Wheeler V., Persichetti F., McNeil S., Mysore J., Mysore S., Macdonald M., Myers R., Gusella J., Wexler N. and Wexler T.U. (2007) Factors associated with HD CAG repeat instability in Huntington's disease. J. Med. Genet., 44, 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Rooij K.E., De Koning Gans P.A., Roos R.A., Van Ommen G.J. and Den Dunnen J.T. (1995) Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum. Genet., 95, 270–274. [DOI] [PubMed] [Google Scholar]
- 14. Mangiarini L., Sathasivam K., Mahal A., Mott R., Seller M. and Bates G.P. (1997) Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat. Genet., 15, 197–200. [DOI] [PubMed] [Google Scholar]
- 15. Wheeler V.C., Auerbach W., White J.K., Srinidhi J., Auerbach A., Ryan A., Duyao M.P., Vrbanac V., Weaver M., Gusella J.F. et al. (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet., 8, 115–122. [DOI] [PubMed] [Google Scholar]
- 16. Kennedy L. and Shelbourne P.F. (2000) Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum. Mol. Genet., 9, 2539–2544. [DOI] [PubMed] [Google Scholar]
- 17. Kennedy L., Evans E., Chen C., Craven L., Detloff P., Ennis M. and Shelbourne P. (2003) Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet., 12, 3359–3367. [DOI] [PubMed] [Google Scholar]
- 18. Veitch N.J., Ennis M., McAbney J.P., Shelbourne P.F. and Monckton D.G. (2007) Inherited CAG.CTG allele length is a major modifier of somatic mutation length variability in Huntington disease. DNA Repair (Amst), 6, 789–796. [DOI] [PubMed] [Google Scholar]
- 19. Shelbourne P.F., Keller-McGandy C., Bi W.L., Yoon S.R., Dubeau L., Veitch N.J., Vonsattel J.P., Wexler N.S., Arnheim N. and Augood S.J. (2007) Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet., 6, 1133–1142. [DOI] [PubMed] [Google Scholar]
- 20. Gonitel R., Moffitt H., Sathasivam K., Woodman B., Detloff P.J., Faull R.L. and Bates G.P. (2008) DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. USA., 105, 3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swami M., Hendricks A.E., Gillis T., Massood T., Mysore J., Myers R.H. and Wheeler V.C. (2009) Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet., 18, 3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee J.M., Zhang J., Su A.I., Walker J.R., Wiltshire T., Kang K., Dragileva E., Gillis T., Lopez E.T., Boily M.J. et al. (2010) A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst. Biol., 4, 29. doi: 10.1186/1752-0509-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J.M., Pinto R.M., Gillis T., St Claire J.C. and Wheeler V.C. (2011) Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One, 6, e23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kovalenko M., Dragileva E., St Claire J., Gillis T., Guide J.R., New J., Dong H., Kucherlapati R., Kucherlapati M.H., Ehrlich M.E. et al. (2012) Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington's disease knock-in mice. PLoS One, 7, e44273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larson E., Fyfe I., Morton A.J. and Monckton D.G. (2015) Age-, tissue- and length-dependent bidirectional somatic CAG*CTG repeat instability in an allelic series of R6/2 Huntington disease mice. Neurobiol. Dis., 76, 98–111. [DOI] [PubMed] [Google Scholar]
- 26. Geraerts F.C., Snell R.G., Faull R.L., Williams L., Jacobsen J.C. and Reid S.J. (2016) Comparison of Huntington's disease CAG repeat length stability in human motor cortex and cingulate Gyrus. J. Huntington's Dis., 5, 297–301. [DOI] [PubMed] [Google Scholar]
- 27. Ament S.A., Pearl J.R., Grindeland A., St Claire J., Earls J.C., Kovalenko M., Gillis T., Mysore J., Gusella J.F., Lee J.M. et al. (2017) High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington's disease CAG knock-in mice across multiple genetic backgrounds. Hum. Mol. Genet., 26, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Genetic Modifiers of Huntington's Disease (GeM_HD) Consortium (2019) CAG repeat not Polyglutamine length determines timing of Huntington's disease onset. Cell, 178, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J.M., Ramos E.M., Lee J.H., Gillis T., Mysore J.S., Hayden M.R., Warby S.C., Morrison P., Nance M., Ross C.A. et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaplan S., Itzkovitz S. and Shapiro E. (2007) A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput. Biol., 3, e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dragileva E., Hendricks A., Teed A., Gillis T., Lopez E.T., Friedberg E.C., Kucherlapati R., Edelmann W., Lunetta K.L., MacDonald M.E. et al. (2009) Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neurobiol. Dis., 33, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tome S., Manley K., Simard J.P., Clark G.W., Slean M.M., Swami M., Shelbourne P.F., Tillier E.R., Monckton D.G., Messer A. et al. (2013) MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington's disease mice. PLoS Genet., 9, e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pinto R.M., Dragileva E., Kirby A., Lloret A., Lopez E., St Claire J., Panigrahi G.B., Hou C., Holloway K., Gillis T. et al. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet., 9, e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Genetic Modifiers of Huntington's Disease (GeM_HD) Consortium (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell, 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J.M., Chao M.J., Harold D., Abu Elneel K., Gillis T., Holmans P., Jones L., Orth M., Myers R.H., Kwak S. et al. (2017) A modifier of Huntington's disease onset at the MLH1 locus. Hum. Mol. Genet., 26, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hensman Moss D.J., Pardinas A.F., Langbehn D., Lo K., Leavitt B.R., Roos R., Durr A., Mead S., TRACK-HD investigators, REGISTRY investigators et al. (2017) Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol., 16, 701–711. [DOI] [PubMed] [Google Scholar]
- 37. Flower M., Lomeikaite V., Ciosi M., Cumming S., Morales F., Lo K., Hensman Moss D., Jones L., Holmans P., the TRACK-HD investigators et al. (2019) MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain, 142, 1876–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chong S.S., McCall A.E., Cota J., Subramony S.H., Orr H.T., Hughes M.R. and Zoghbi H.Y. (1995) Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat. Genet., 10, 344–350. [DOI] [PubMed] [Google Scholar]
- 39. Zühlke C., Hellenbroich Y., Schaaff F., Gehlken U., Wessel K., Schubert T., Cervos-Navarro J., Pickartz H. and Schwinger E. (1997) CAG repeat analyses in frozen and formalin-fixed tissues following primer extension preamplification for evaluation of mitotic instability of expanded SCA1 alleles. Hum. Genet., 100, 339–344. [DOI] [PubMed] [Google Scholar]
- 40. Watanabe H., Tanaka F., Doyu M., Riku S., Yoshida M., Hashizume Y. and Sobue G. (2000) Differential somatic CAG repeat instability in variable brain cell lineage in dentatorubral pallidoluysian atrophy (DRPLA): a laser-captured microdissection (LCM)-based analysis. Hum. Genet., 107, 452–457. [DOI] [PubMed] [Google Scholar]
- 41. Cancel G., Gourfinkel-An I., Stevanin G., Didierjean O., Abbas N., Hirsch E., Agid Y. and Brice A. (1998) Somatic mosaicism of the CAG repeat expansion in spinocerebellar ataxia type 3/Machado-Joseph disease. Hum. Mut., 11, 23–27. [DOI] [PubMed] [Google Scholar]
- 42. Takano H., Onodera O., Takahashi H., Igarashi S., Yamada M., Oyake M., Ikeuchi T., Koide R., Tanaka H., Iwabuchi K. et al. (1996) Somatic mosaicism of expanded CAG repeats in brains of patients with dentatorubral-pallidoluysian atrophy: cellular population-dependent dynamics of mitotic instability. Am. J. Hum. Genet., 58, 1212–1222. [PMC free article] [PubMed] [Google Scholar]
- 43. Genis D., Matilla T., Volpini V., Rosell J., Davalos A., Ferrer I., Molins A. and Estivill X. (1995) Clinical, neuropathologic, and genetic studies of a large spinocerebellar ataxia type 1 (SCA1) kindred: (CAG)n expansion and early premonitory signs and symptoms. Neurology, 45, 24–30. [DOI] [PubMed] [Google Scholar]
- 44. Rub U., Schols L., Paulson H., Auburger G., Kermer P., Jen J.C., Seidel K., Korf H.W. and Deller T. (2013) Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol., 104, 38–66. [DOI] [PubMed] [Google Scholar]
- 45. Ramirez E.P.C., Keller C.E. and Vonsattel J.P. (2018) The New York Brain Bank of Columbia University: practical highlights of 35 years of experience. Handb. Clin. Neurol., 150, 105–118. [DOI] [PubMed] [Google Scholar]
- 46. Rüb U., Seidel K., Heinsen H., Vonsattel J.P., Dunnen W.F. and Korf H.W. (2016) Huntington's disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol., 26, 726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahveninen L.M., Stout J.C., Georgiou-Karistianis N., Lorenzetti V. and Glikmann-Johnston Y. (2018) Reduced amygdala volumes are related to motor and cognitive signs in Huntington's disease: the IMAGE-HD study. Neuroimage Clin., 18, 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nana A.L., Kim E.H., Thu D.C.V., Oorschot D.E., Tippett L.J., Hogg V.M., Synek B.J., Roxburg R., Waldvogel H.J. and Faull R.L.M. (2014) Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J. Huntington’s Dis., 3, 45–64. [DOI] [PubMed] [Google Scholar]
- 49. Martinez-Horta S., Sampedro F., Horta-Barba A., Perez-Perez J., Pagonabarraga J., Gomez-Anson B. and Kulisevsky J. (2020) Structural brain correlates of irritability and aggression in early manifest Huntington's disease. Brain Imaging Behav. Jan 2. doi: 10.1007/s11682-019-00237-x. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 50. Eddy C.M., Rickards H.E. and Hansen P.C. (2018) Through your eyes or mine? The neural correlates of mental state recognition in Huntington's disease. Hum. Brain Mapp., 39, 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rowley C.D., Tabrizi S.J., Scahill R.I., Leavitt B.R., Roos R.A.C., Durr A. and Bock N.A. (2018) Altered Intracortical T1-weighted/T2-weighted ratio signal in Huntington's disease. Front. Neurosci., 12, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kersten H.M., Danesh-Meyer H.V., Kilfoyle D.H. and Roxburgh R.H. (2015) Optical coherence tomography findings in Huntington's disease: a potential biomarker of disease progression. J. Neurol., 262, 2457–2465. [DOI] [PubMed] [Google Scholar]
- 53. Gatto E.M., Parisi V., Persi G., Fernandez Rey E., Cesarini M., Etcheverry J.L., Rivera P. and Squitieri F. (2018) Optical coherence tomography (OCT) study in Argentinean Huntington's disease patients. Int. J. Neurosci., 128, 1157–1162. [DOI] [PubMed] [Google Scholar]
- 54. Gulmez Sevim D., Unlu M., Gultekin M. and Karaca C. (2019) Retinal single-layer analysis with optical coherence tomography shows inner retinal layer thinning in Huntington's disease as a potential biomarker. Int. Ophthalmol., 39, 611–621. [DOI] [PubMed] [Google Scholar]
- 55. Petrasch-Parwez E., Saft C., Schlichting A., Andrich J., Napirei M., Arning L., Wieczorek S., Dermietzel R. and Epplen J.T. (2005) Is the retina affected in Huntington disease? Acta Neuropathol., 110, 523–525. [DOI] [PubMed] [Google Scholar]
- 56. Rüb U., Seidel K., Vonsattel J., Lange H.W., Eisenmenger W., Götz M., Del Turco D., Bouzrou M., Korf H.W. and Heinsen H. (2015) Huntington's disease (HD): neurodegeneration of Brodmann's primary visual area 17 (BA17). Brain Pathol., 25, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coppen E.M., Grond J.V., Hafkemeijer A., Barkey Wolf J.J.H. and Roos R.A.C. (2018) Structural and functional changes of the visual cortex in early Huntington's disease. Hum. Brain Mapp., 39, 4776–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stuwe S.H., Goetze O., Lukas C., Klotz P., Hoffmann R., Banasch M., Orth M., Schmidt W.E., Gold R. and Saft C. (2013) Hepatic mitochondrial dysfunction in manifest and premanifest Huntington disease. Neurology, 80, 743–746. [DOI] [PubMed] [Google Scholar]
- 59. Hoffmann R., Stüwe S.H., Oliver G., Matthias B., Peter K., Carsten L., Martin T., Christian B., Michael O. and Carsten S. (2014) Progressive hepatic mitochondrial dysfunction in premanifest Huntington's disease. Mov. Disord., 29, 831–834. [DOI] [PubMed] [Google Scholar]
- 60. Chiu E., Mackay I.R. and Bhathal P.B. (1975) Hepatic morphology in Huntington's chorea. J. Neurol. Neurosurg. Psychiatry, 38, 1000–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waldvogel H.J., Kim E.H., Tippett L.J., Vonsattel J.P. and Faull R.L.M. (2015) The neuropathology of Huntington's disease. Curr. Top. Behav. Neurosci., 22, 33–80. [DOI] [PubMed] [Google Scholar]
- 62. Latimer C.S., Flanagan M.E., Cimino P.J., Jayadev S., Davis M., Hoffer Z.S., Montine T.J., Gonzalez-Cuyar L.F., Bird T.D. and Keene C.D. (2017) Neuropathological comparison of adult onset and juvenile Huntington's disease with cerebellar atrophy: a report of a father and son. J. Huntington's Dis., 6, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aronin N., Chase K., Young C., Sapp E., Schwarz C., Matta N., Kornreich R., Landwehrmeyer B., Bird E. and Beal M.F. (1995) CAG expansion affects the expression of mutant Huntingtin in the Huntington's disease brain. Neuron, 15, 1193–1201. [DOI] [PubMed] [Google Scholar]
- 64. Kahlem P. and Djian P. (2000) The expanded CAG repeat associated with juvenile Huntington disease shows a common origin of most or all neurons and glia in human cerebrum. Neurosci. Lett., 286, 203–207. [DOI] [PubMed] [Google Scholar]
- 65. Ueno S., Kondoh K., Kotani Y., Komure O., Kuno S., Kawai J., Hazama F. and Sano A. (1995) Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (DRPLA). Hum. Mol. Genet., 4, 663–666. [DOI] [PubMed] [Google Scholar]
- 66. Lopez Castel A., Nakamori M., Tome S., Chitayat D., Gourdon G., Thornton C.A. and Pearson C.E. (2011) Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum. Mol. Genet., 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yoon S.R., Dubeau L., De Young M., Wexler N.S. and Arnheim N. (2003) Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc. Natl. Acad. Sci. USA., 100, 8834–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ciosi M., Maxwell A., Cumming S.A., Hensman Moss D.J., Alshammaria A.M., Flower M.D., Durr A., Leavitt B.R., Roos R.A.C., the TRACK-HD team et al. (2019) A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine, 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoijer H., Tsai Y.-C., Clark T.A., Kotturi P., Dahl N., Stattin E.-L., Bondeson M.L., Feuk L., Gyllensten U. and Ameur A. (2018) Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum. Mut., 39, 1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wieben E., Aleff R.A., Basu S., Sarangi V., Bowman B., McLaughlin I.J., Mills J.R., Butz M.L., Highsmith E.W., Ida C.M. et al. (2019) Amplification-free long-read sequencing of TCF4 expanded trinucleotide repeats in Fuchs Endothelial Corneal Dystrophy. PLoS One, 14, e0219446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mason A.G., Tome S., Simard J.P., Libby R.T., Bammler T.K., Beyer R.P., Morton A.J., Pearson C.E. and La Spada A.R. (2014) Expression levels of DNA replication and repair genes predict regional somatic repeat instability in the brain but are not altered by polyglutamine disease protein expression or age. Hum. Mol. Genet., 23, 1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jimenez-Sanchez M., Licitra F., Underwood B.R. and Rubinsztein D.C. (2017) Huntington's disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med., 7, a024240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Banez-Coronel M., Ayhan F., Tarabochia A.D., Zu T., Perez B.A., Tusi S.K., Pletnikova O., Borchelt D.R., Ross C.A., Margolis R.L. et al. (2015) RAN translation in Huntington disease. Neuron, 88, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fiszer A. and Krzyzosiak W.J. (2013) RNA toxicity in polyglutamine disorders: concepts, models, and progress of research. J. Mol. Med. (Berl.), 91, 683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rousseaux M.W.C., Tschumperlin T., Lu H.C., Lackey E.P., Bondar V.V., Wan Y.W., Tan Q., Adamski C.J., Friedrich J., Twaroski K. et al. (2018) ATXN1-CIC complex is the primary driver of cerebellar pathology in Spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron, 97, 1235–1243 e1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jafar-Nejad P., Ward C.S., Richman R., Orr H.T. and Zoghbi H.Y. (2011) Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc. Natl. Acad. Sci. USA., 108, 2142–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Goold R., Flower M., Moss D.H., Medway C., Wood-Kaczmar A., Andre R., Farshim P., Bates G.P., Holmans P., Jones L. et al. (2019) FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet., 28, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gomes-Pereira M., Fortune M.T., Ingram L., McAbney J.P. and Monckton D.G. (2004) Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet., 13, 1815–1825. [DOI] [PubMed] [Google Scholar]
- 79. Bettencourt C., Hensman-Moss D., Flower M., Wiethoff S., Brice A., Goizet C., Stevanin G., Koutsis G., Karadima G., Panas M. et al. (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol., 79, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Klaes A., Reckziegel E., Franca M.C. Jr., Rezende T.J.R., Vedolin L.M., Jardim L.B. and Saute J.A. (2016) MR imaging in Spinocerebellar ataxias: a systematic review. AJNR Am. J. Neuroradiol., 37, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Singh-Bains M.K., Mehrabi N.F., Sehji T., Austria M.D.R., Tan A.Y.S., Tippett L.J., Dragunow M., Waldvogel H.J. and Faull R.L.M. (2019) Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann. Neurol., 85, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Burg J.M., Bjorkqvist M. and Brundin P. (2009) Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol., 8, 765–774. [DOI] [PubMed] [Google Scholar]
- 83. Carroll J.B., Bates G.P., Steffan J., Saft C. and Tabrizi S.J. (2015) Treating the whole body in Huntington’s disease. Lancet Neurol., 14, 1135–1142. [DOI] [PubMed] [Google Scholar]
- 84. Nestor C.E. and Monckton D.G. (2011) Correlation of inter-locus polyglutamine toxicity with CAG*CTG triplet repeat expandability and flanking genomic DNA GC content. PLoS One, 6, e28260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hubert L. Jr., Lin Y., Dion V. and Wilson J.H. (2011) Xpa deficiency reduces CAG Trinucleotide repeat instability in neuronal tissues in a SCA1 mouse model. Hum. Mol. Genet., 20, 4822–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Warner J.P., Barron L.H. and Brock D.J. (1993) A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington's disease chromosomes. Mol. Cell Probes, 7, 235–239. [DOI] [PubMed] [Google Scholar]
- 87. Milunsky J.M., Maher T.A., Loose B.A., Darras B.T. and Ito M. (2003) XL PCR for the detection of large trinucleotide expansions in juvenile Huntington’s disease. Clin. Genet., 64, 70–73. [DOI] [PubMed] [Google Scholar]
- 88. Riess O., Thies U., Siedlaczck I., Potisek S., Graham R., Theilmann J., Grimm T., Epplen J.T. and Hayden M.R. (1994) Precise mapping of the brain alpha 2-adrenergic receptor gene within chromosome 4p16. Genomics, 19, 298–302. [DOI] [PubMed] [Google Scholar]
- 89. Rubinsztein D.C., Barton D.E., Davison B.C. and Ferguson-Smith M.A. (1993) Analysis of the huntingtin gene reveals a trinucleotide-length polymorphism in the region of the gene that contains two CCG-rich stretches and a correlation between decreased age of onset of Huntington's disease and CAG repeat number. Hum. Mol. Genet., 2, 1713–1715. [DOI] [PubMed] [Google Scholar]
- 90. Orr H.T., Chung M.Y., Banfi S., Kwiatkowski T.J. Jr., Servadio A., Beaudet A.L., McCall A.E., Duvick L.A., Ranum L.P. and Zoghbi H.Y. (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet., 4, 221–226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.