

SUMMARY
Despite the development of second-generation antiandrogens, acquired resistance to hormone therapy remains a major challenge in treating advanced prostate cancer. We find that cancer-associated fibroblasts (CAFs) can promote antiandrogen resistance in mouse models and in prostate organoid cultures. We identify neuregulin 1 (NRG1) in CAF supernatant, which promotes resistance in tumor cells through activation of HER3. Pharmacological blockade of the NRG1/HER3 axis using clinical-grade blocking antibodies re-sensitizes tumors to hormone deprivation in vitro and in vivo. Furthermore, patients with castration-resistant prostate cancer with increased tumor NRG1 activity have an inferior response to second-generation antiandrogen therapy. This work reveals a paracrine mechanism of antiandrogen resistance in prostate cancer amenable to clinical testing using available targeted therapies.
In Brief
Zhang et al. find that cancer-associated fibroblasts promote antiandrogen resistance in prostate cancer by secreting NRG1 to activate HER3 signaling in prostate cancer cells. Blockade of the NRG1/HER3 axis can re-sensitize prostate cancer models to antiandrogen therapy.
Graphical Abstract
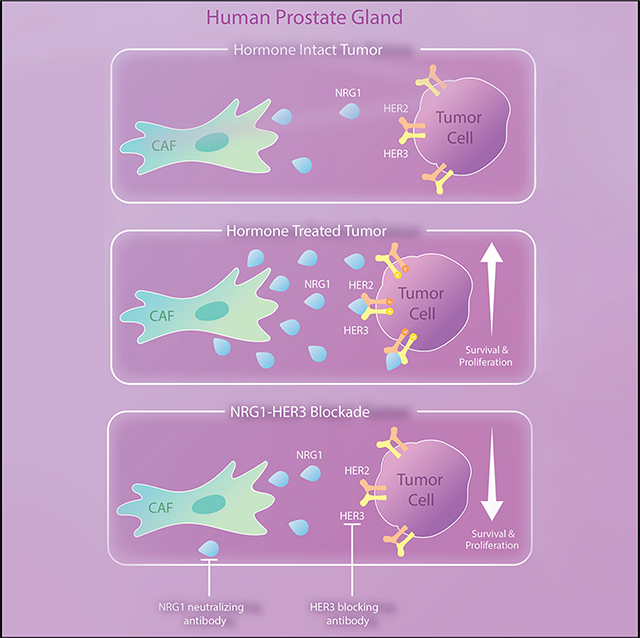
INTRODUCTION
Androgen receptor (AR) is a lineage survival factor for luminal cancer cells in prostate tumor that plays an essential role in cancer progression and drug resistance (Watson et al., 2015). Although second-generation antiandrogens have significantly improved patient survival, patients rarely achieve complete response even with combinations of the most potent AR signaling inhibitors (Montgomery et al., 2017; Taplin et al., 2014). Understanding survival mechanisms in persisting tumor cells is critical to achieve complete response. A state of drug tolerance or persistence has been characterized in lung adenocarcinoma and melanoma, where transcriptional, epigenetic, or metabolic changes induced by treatment render previously susceptible tumors insensitive to the therapy (Smith et al., 2016; Zhang et al., 2019).
In prostate cancer, efforts to understand resistance to AR-targeted therapy have focused mainly on cell-intrinsic mechanisms (Antonarakis et al., 2014; Arora et al., 2013; Balbas et al., 2013; Ku et al., 2017; Mu et al., 2017; Shah et al., 2017), but there is increasing evidence implicating the microenvironment (stroma and inflammatory cells) as a driver of drug resistance in various cancers (Klemm and Joyce, 2015; Zhang et al., 2013). Examples include melanoma, glioma, breast, lung, lymphoma, and prostate cancer, where secretion of various growth factors (hapatocyte growth factor [HGF], WNTs) and cytokines (interleukin-6 [IL-6], IL-8) have been implicated in resistance to kinase inhibitors or to chemotherapy (Crawford et al., 2009; Gilbert and Hemann, 2010; Kodack et al., 2017; Straussman et al., 2012; Su et al., 2018; Sun et al., 2012). In prostate cancer, tumor-infiltrating B lymphocytes and myeloid-derived immune suppressor cells (MDSCs) promote castration-resistant prostate cancer (CRPC) through production of inflammatory cytokines, such as IL-23 (Ammirante et al., 2010; Calcinotto et al., 2018). Fibroblast growth factors (FGFs) can also play a role in CRPC, through autocrine or paracrine production (Bluemn et al., 2017). The ability of microenvironment cells to promote drug resistance is likely linked to the role of these cells in development and tumor initiation. For example, PTEN loss, TGFBRII loss, or nuclear factor κB activation in stroma can elicit early neoplastic changes in mammary, prostate and pancreatic epithelium (Bhowmick et al., 2004; Erez et al., 2010; Franco et al., 2011; Kiskowski et al., 2011; Trimboli et al., 2009). Indeed, AR expression in prostate stroma plays a crucial role in morphogenesis and maturation of a normal prostate gland (Cunha, 1994; Cunha and Chung, 1981).
Further investigation of cancer-associated stromal cells has converged on the concept of reactive stroma, now documented in multiple cancers, including pancreas, prostate, breast, and colorectal tumors (Calon et al., 2015). In breast and colorectal cancer, increased reactive stroma is associated with poorer clinical outcome (Finak et al., 2008; Isella et al., 2015). Here, we investigate the role of reactive stroma in prostate cancer and the implications for response to AR-targeted therapy.
RESULTS
Reactive Stroma in Localized Prostate Cancer Is Associated with Higher Tumor Grade and PI3K-AKT Pathway Activation
Using a prostate-specific reactive stroma gene signature (Dakhova et al., 2009), we first examined the prostate cancer The Cancer Genome Atlas (TCGA) dataset (Cancer Genome Atlas Research Network, 2015) for associations with cancer and with tumor grade. The reactive stroma score was significantly higher in tumor samples compared with normal prostate gland (p = 2 × 10−6) (Figure S1A) and also in higher-grade tumors (p = 2×10−4) based on Gleason score (Figure S1B). This association was confirmed using a second signature enriched for genes expressed in high-grade tumor-associated stroma (Tyekucheva et al., 2017) (p = 3 × 10−6) (Figure S1C). We further validated the association of reactive stroma with primary prostate cancer using immunohistochemical staining for alpha smooth muscle actin (α-SMA) and vimentin in an independent cohort from MSKCC (Figures S1D–S1H). Notably, phosphatidylinositol 3-kinase (PI3K)-AKT-mammalian target of rapamycin and receptor tyrosine kinase (RTK) signaling were among the top pathways enriched in high-stroma score patients based on gene set enrichment analysis (GSEA) of the TCGA dataset (Figure S1I).
Cancer-Associated Fibroblasts Promote Castration Resistance and Antiandrogen Resistance in an Androgen-Dependent PCa Model
To follow up on these clinical associations, we modeled tumor-stroma interaction using the patient-derived xenograft model CWR22Pc, which is initially castration sensitive but can progress to castration resistance (Dagvadorj et al., 2008). We found that a cell line derived from this model has a population of murine fibroblasts. To obtain a pure epithelial population, we used flow cytometry with species-specific antibodies against surface antigens to individually purify the human cancer (epithelial) cells from the mouse fibroblasts, which we named CWR22Pc-EP, hereafter called 22Pc-EP (Mu et al., 2017) and CWR22Pc-cancer-associated fibroblast (22Pc-CAF), respectively (Figures 1A and S1J). Interestingly, when 22Pc-EP cells were isolated from 22Pc-CAF, acquisition of bicalutamide (Bic) resistance was substantially delayed compared with the mixed epithelial/fibroblast parental CWR22Pc population (152 versus 48 days). Notably, enzalutamide (Enz) resistance was never observed in 22Pc-EP cells, which remained growth arrested at day 229, whereas the mixed epithelial/fibroblast parental CWR22Pc cells acquired resistance to Enz at day 88 (Figures 1B and S1K). Based on this observation, we hypothesized that CAFs may contribute to the acquisition of an antiandrogen-resistant state.
Figure 1. Cancer-Associated Fibroblasts Promote Antiandrogen Resistance in an Androgen-Dependent PCa Model.

(A) Schematic diagram depicting the origin and characteristics of the CWR22Pc model (Dagvadorj et al., 2008). CWR22Pc contains both murine cancer-associated fibroblasts (22Pc-CAF) and human cancer cells (22Pc-EP), as described previously (Mu et al., 2017).
(B) Bar graph showing time to development of resistance to Bic (10 μM) or Enz (1 μM) in CWR22Pc and 22Pc-EP.
(C) Top: cartoon showing 22Pc-EPeGFP + 22Pc-CAFtdTomato co-culture or 22Pc-EPeGFP mono-culture. Bottom: fluorescent images of co-culture assay showing number of 22Pc-EPeGFP cells (green) and 22Pc-CAFtdTomato cells (red) in the presence of Bic (10 μM), Enz (1 μM), or vehicle (Veh, DMSO) on day 30 (n = 3).
(D–F) Quantification of eGFP fluorescence signal intensity from (C) using relative fluorescence units (RFU) in three experimental conditions: Veh (D), Bic (E), or Enz (F) (**p < 0.01, *p < 0.05; n.s., not significant. Student’s t test).
(G) Growth of CWR22Pc or 22Pc-EP tumor xenografts in castrated mice (n = 5 mice per group, **p < 0.01, multiple t test).
Data are represented as mean ± SD (D–F), or mean ± SEM (G). See also Figure S1.
We next developed a co-culture assay to model CAF/cancer cell interactions in vitro and in vivo. We labeled 22Pc-EP with eGFP and 22Pc-CAF with tdTomato using viral transduction (Figure S1L). 22Pc-EPeGFP cells were cultured either with or without 22Pc-CAFtdTomato cells in the presence of vehicle, Bic, or Enz, and eGFP fluorescence intensity was measured to quantify the relative 22Pc-EP cell number (Figure 1C). We observed significantly more 22Pc-EPeGFP cells in the co-culture condition following antiandrogen treatment but no difference with vehicle treatment (Figures 1D–1F). Thus, the CAFs confer a pro-growth/survival signal to 22Pc-EP cells only in the setting of AR blockade. We then evaluated whether 22Pc-CAF also promotes castration-resistance in vivo. To this end, we first grafted parental CWR22Pc (with CAFs) or 22Pc-EP (without CAFs) into castrated male mice to mimic the clinical scenario of chronic androgen-deprivation therapy (ADT) and found that parental CWR22Pc tumors grew significantly faster than 22Pc-EP (Figure 1G). To further test the contribution of CAFs to castration resistance, 22Pc-EPeGFP cells were grafted into castrated mice with or without 22Pc-CAFtdTomato cells. Like the CAF-containing parental CWR22Pc, 22Pc-EP tumors grew significantly faster when co-injected with CAFs (Figure S1M). Analysis of tumors from the co-injection group by immunofluorescence (of eGFP or tdTomato) revealed infiltration of 22Pc-CAF in the tumor mass, suggestive of interactions between CAFs and adjacent tumor cells in vivo (Figure S1N). Taken together, these data establish that 22Pc-CAF can promote antiandrogen and castration resistance of 22Pc-EP.
CAF-Secreted Factors Promote Antiandrogen Resistance
We posited that the growth-promoting effects of CAFs on cancer cell growth during antiandrogen treatment could be occurring through two possible mechanisms: fibroblast-epithelial cell-to-cell contact or secretion of CAF-derived soluble factors. To test the latter hypothesis, we collected conditioned media from either 22Pc-CAF (22Pc-CAFCM) or 22Pc-EP (22Pc-EPCM) and tested their ability to stimulate the growth of 22Pc-EP cells treated with androgen deprivation (modeled by the use of 5% charcoal-dextran stripped FBS, hereafter CSS) or antiandrogens (Bic or Enz). 22Pc-CAFCM promoted resistance of 22Pc-EP to CSS and antiandrogens, while 22Pc-EPCM or serum-free media (negative control) did not (Figures 2A and 2B), indicating that one or more soluble factor(s) secreted by CAFs was responsible for growth. Furthermore, concentrating the 22Pc-CAFCM by 2- to 4-fold increased the resistance-promoting activity, suggesting a dose-dependent effect (Figure S2A). Exposure of 22Pc-CAFCM to either heat (95°C) or proteinase K abolished the ability of 22Pc-CAFCM to rescue the growth phenotypes (Figures 2C, 2D, and S2B), suggesting that the soluble factors responsible for promotion of resistance were proteins. 22Pc-CAFCM also promoted resistance to CSS and Enz in two additional androgen-dependent human prostate models, the VCaP cell line and the patient-derived cancer organoid MSK-PCa2 (Figures S2C–S2F).
Figure 2. CAF-Secreted Factors Promote Antiandrogen Resistance.

(A) Growth of 22Pc-EP cells in CSS media supplemented with either conditioned media from 22Pc-EP (hereafter, 22Pc-EPCM) or from 22Pc-CAF (hereafter, 22Pc-CAFCM) assessed by CellTiter-Glo on day 4 using relative luminescence unit (RLU). Media Ctrl, serum-free media.
(B) Growth of 22Pc-EP cells in FBS media supplemented with either 22Pc-EPCM or 22Pc-CAFCM treated with Veh (DMSO), Bic (10 μM), or Enz (0.1 μM). CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
(C) Growth of 22Pc-EP cells in CSS media supplemented with either control or heat-inactivated 22Pc-EPCM or 22Pc-CAFCM. CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(D) Growth of 22Pc-EP cells in FBS media supplemented with heat-inactivated 22Pc-EPCM or 22Pc-CAFCM treated with Veh (DMSO), Bic (10 μM), or Enz (0.1 μM). CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
(E) Growth curve of 22Pc-EP cells in CSS media supplemented with 22Pc-EPCM or 22Pc-CAFCM. CellTiter-Glo reading on days 1, 4, 7, and 10.
(F) Growth curve of 22Pc-EP cells in FBS media supplemented with 22Pc-EPCM or 22Pc-CAFCM treated with Enz (0.1 μM). CellTiter-Glo reading on day 1, 7, and 10.
(G) qRT-PCR analysis of AR target genes in 22Pc-EP cells in CSS media treated with Veh (DMSO), DHT (1 nM), or 22Pc-CAFCM for 24 h (normalized to ACTB).
(H) qRT-PCR analysis of AR target genes in 22Pc-EP cells in FBS media treated with Veh (DMSO), Enz (0.1 μM), or Enz +22Pc-CAFCM for 24 h (normalized to ACTB).
Assays were performed with three biological replicates. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05; n.s., not significant. (A–D) One-way ANOVA, (E–F) multiple t test with false discovery rate of 1%, (G–H) Student’s t test. Data are represented as mean ± SD. See also Figure S2.
AR signaling is critical for prostate oncogenesis, and AR pathway reactivation in advanced PCa is one of the primary mechanisms of acquired resistance to AR-targeted therapies (Watson et al., 2015). Therefore, we next asked if secreted protein factors from 22Pc-CAF reactivate AR signaling in 22Pc-EP even in the presence of Enz. We observed sustained growth of 22Pc-EP cells in CSS or in Enz in the presence of 22Pc-CAFCM (Figures 2E and 2F). Notably, several canonical AR target genes remained suppressed despite abundant AR mRNA expression (Figures 2G, 2H, and S2G). Collectively, these results suggest that secreted protein factors from 22Pc-CAF promote antiandrogen resistance in 22Pc-EP through a mechanism that does not immediately result in AR reactivation.
Biochemical Fractionation of CAF-Secreted Resistance Activity Implicates Neuregulin 1
To identify the key protein(s) present in 22Pc-CAFCM responsible for antiandrogen resistance we undertook a biochemical fractionation approach and analyzed the resolved fractions in two parallel assays: (1) the 22Pc-EP growth assay as a readout of antiandrogen resistance activity and (2) activation of human RTKs in 22Pc-EP cells. The rationale for the RTK assay was based on previous work from our group and others demonstrating that RTK activation enables pro-tumorigenic AR bypass signaling in PCa (Carver et al., 2011; Gao et al., 2016; Mellinghoff et al., 2004). Furthermore, the GSEA of reactive prostate stroma revealed growth factor binding and RTK activity as highly enriched pathways (Figures S1I, S2H, and S2I).
To carry out the purification, serum-free 22Pc-CAFCM was collected, concentrated, and applied to a Q-Superose anion exchange column, from which we eluted two protein peaks by using 30% and 100% high-salt buffer B (termed Q30 and Q100, respectively; see the STAR Methods for further details) (Figure 3A). Resistance-promoting activity resided in fraction Q30, but not Q100 (Figure 3B). Further resolving of Q30 by gradient elution on a Q-Superose column yielded fractions Q6–Q8 which promote 22Pc-EP growth in CSS (Figure 3C). In parallel, results from an RTK array showed that 22Pc-CAFCM strongly activated HER2 and HER3 in 22Pc-EP (Figure S3A). This result, as well as FGF receptor (FGFR) activation, was confirmed by western blot (Figures 3D and S3B). (We tested FGFR based on recent data implicating FGF in CRPC [Bluemn et al., 2017].) Analysis of Q30 and its subfractions, as well as Q100, for RTK activity revealed that HER3 (and downstream AKT) phosphorylation activity was present in Q30 but not Q100, and specifically in the Q6–Q10 subfractions of Q30. The fact that the resistance-promoting activity elutes in precisely the same fractions as the HER3 phosphorylation activity (Figure 3E) suggests that two are functionally linked.
Figure 3. Biochemical Fractionation of CAF-Secreted Resistance Activity Implicates NRG1.

(A) Schematic diagram showing fraction purification and resistance activity in 22Pc-CAFCM.
(B) Growth of 22Pc-EP cells in CSS media supplemented with purified fractions from input (22Pc-CAFCM), Q30, or Q100. CellTiter-Glo reading on day 4. Ctrl, PBS.
(C) Growth of 22Pc-EP cells in CSS media supplemented with sub-purified fractions (Q3–Q17) from Q30. CellTiter-Glo reading on day 4. Input, Q30; Ctrl, PBS.
(D) Western blot analysis of HER3 and FGFR activation in 22Pc-EP after stimulation with FBS media, 22Pc-CAFCM, Q30 or Q100. Ctrl, PBS. β-Actin serves as loading control.
(E) Western blot analysis of HER3-AKT activation in 22Pc-EP after stimulation with different Q fractions and analysis of secreted NRG1 in corresponding fractions. Ctrl, PBS; Input, Q30; FT, flow through. Total AKT serves as loading control.
(F) Western blot analysis of HER3-AKT activation in 22Pc-EP after stimulation with 22Pc-CAFCM in the presence of a commercial HER3-blocking antibody (10 or 30 μg/mL) or immunoglobulin G (IgG) (30 μg/mL). Total AKT serves as loading control.
(G) Growth of 22Pc-EP cells in CSS media supplemented with 22Pc-CAFCM treated with a commercial HER3-blocking antibody (10 or 30 μg/mL) or IgG (30 μg/mL). CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(H) Western blot analysis of NRG1 from 22Pc-EPCM or 22Pc-CAFCM (concentrated 1×, 10×, 20×, or 50×).
(I) qRT-PCR analysis of Nrg1 expression in CWR22Pc (n = 8) or 22Pc-EP (n = 10) tumor xenografts using mouse-specific primers.
(J) Representative images showing RNA in situ hybridization (ISH) analysis of mouse Nrg1 expression in CWR22Pc or 22Pc-EP tumor xenografts. T, tumor; S, stroma; mouse-specific Nrg1 probe, brown dots.
Assays were performed with three biological replicates. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05; n.s., not significant. (B and C) One-way ANOVA compared with Ctrl group, (G and I) Student’s t test. Data are represented as mean ± SD. See also Figure S3.
To determine whether HER3 activation contributes to resistance, we treated 22Pc-EP cells with an HER3-blocking antibody or with small-molecule inhibitors to HER2 (lapatinib and neratinib) or FGFR (AZD4547, BGJ398, PD173074). The HER3-blocking antibody and the HER2 kinase inhibitors blocked the resistance-promoting activity of 22Pc-CAFCM, whereas FGFR inhibitors did not, despite pharmacological blockade of FGFR kinase activity (Figures 3F, 3G, and S3C–S3E). Since NRG1 (neuregulin 1) is the principal ligand for HER3 (Mei and Nave, 2014), we next asked if NRG1 could be detected in active Q subfractions. Indeed, we observed NRG1 protein exclusively in the active Q subfractions (Q6–Q10) from 22Pc-CAFCM (Figure 3E) but not in 22Pc-EPCM (Figures 3H and S3F). To determine if other neuregulin family members are also expressed, we measured mRNA levels of Nrg1–4 by qRT-PCR and found that Nrg1 is the dominantly expressed neuregulin in 22Pc-CAF (Figure S3G).
Taken together, these data suggest that NRG1 acts in a paracrine manner to activate HER3 in tumor cells in vivo. Consistent with this model, Nrg1 mRNA levels (detected using mouse-specific Nrg1 primers) are significantly increased in parental CWR22Pc tumor xenografts (which contain 22Pc-CAF) compared with 22Pc-EP tumors (Figure 3I). To determine the spatial relationship between Nrg1-expressing mouse cells and ERBB3-expressing tumor cells, we performed RNA in situ hybridization (RNA-ISH). Using a murine-specific RNA-ISH probe, Nrg1 mRNA was detected in the stromal compartment in both models (CWR22Pc and 22Pc-EP), but with a stronger signal in parental CWR22Pc xenografts (Figure 3J, brown dots). No signal was detected using a human NRG1 RNA-ISH probe, as expected (Figure S3H). To visualize the spatial expression pattern of Nrg1 relative to ERBB3, we applied multi-color RNA-fluorescence in situ hybridization (FISH) staining and found that murine Nrg1 (green) co-localized with the stromal marker Vim (white), but not with human ERBB3 (red) staining (Figure S3I), providing further support for the model that NRG1 expressed by tumor infiltrating CAFs acts on cancer cells through a paracrine mechanism.
NRG1-HER3 Signaling Confers Antiandrogen Resistance
Having established that HER3 activation is required for the resistance-promoting activity of CAFs, we performed additional experiments to determine if NRG1 is the responsible factor. NRG1 is known to have >30 isoforms, all of which share the consensus epidermal growth factor (EGF)-like domain (Mei and Nave, 2014; Mei and Xiong, 2008). To determine if loss of NRG1 in 22Pc-CAF impairs their ability to promote resistance, we targeted the EGF-like domain of Nrg1 using CRISPR/Cas9 to disrupt all isoforms (Figure 4A). Conditioned media from sgNrg1 22Pc-CAF had significantly reduced capacity to activate HER3/AKT phosphorylation and to promote resistance to CSS or Enz in 22Pc-EP compared with conditioned media from 22Pc-CAF expressing a non-targeting sgRNA (Figures 4B–4D). As an independent confirmation of its essential role in conditioned media, we immunoprecipitated NRG1 from the 22Pc-CAFCM and found that NRG1-depleted conditioned media lost the ability to activate HER3-AKT and to promote resistance to CSS (Figures S4A and S4B). A clinical-grade NRG1α/β neutralizing antibody YW538.24.71 (Genentech) also blocked the ability of 22Pc-CAFCM to activate HER3/AKT as well as to promote resistance of 22Pc-EP to Enz or CSS (Figures 4E–4G), as did the clinical-grade HER3-blocking antibody AMG888 (Figures 4H–4J). To determine if NRG1 itself was sufficient to promote antiandrogen resistance in 22Pc-EP or if other ErbB RTK ligands, such as EGF, could substitute for NRG1, we treated 22Pc-EP with increasing concentrations of each. Recombinant NRG1 activated HER3/AKT and promoted resistance to CSS or Enz in 22Pc-EP. EGF also induced AKT phosphorylation (without inducing HER3 phosphorylation) but was not sufficient to confer resistance (Figures 4K–4M). Recombinant NRG1 also promoted resistance to CSS or Enz in three additional human prostate models (22Rv1, LAPC4, and VCaP) (Figures S4C–S4F) and in organoids derived from three genetically engineered mouse models (Trp53-KO, Rb1-KO and PtenΔ/Δ-Rosa26-ERG) (Figures S4G–S4L).
Figure 4. NRG1 Promotes Antiandrogen Resistance in Androgen-Dependent PCa Models.

(A) Western blot analysis of NRG1 expression in 22Pc-CAF in which Nrg1 was deleted using CRISPR/Cas9. sgNT, non-targeting guide control; sgNrg1 (no. 1–4), four independent guides targeting Nrg1. β-Actin serves as loading control.
(B) Western blot analysis of HER3-AKT activation in 22Pc-EP after stimulation with either sgNT- or sgNrg1-22Pc-CAFCM. Total AKT serves as loading control.
(C) Growth of 22Pc-EP cells in CSS media supplemented with either sgNT- or sgNrg1-22Pc-CAFCM. CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(D) Growth of 22Pc-EP cells in FBS media supplemented with either sgNT- or sgNrg1-22Pc-CAFCM treated with Enz (0.1 μM) or Veh (DMSO). The Enz group was normalized to the Veh group. CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
(E) Western blot analysis of HER3-AKT activation in 22Pc-EP cells after stimulation with 22Pc-CAFCM in the presence of either NRG1 neutralizing antibody YW538.24.71 (1, 10, or 100 μg/mL) or IgG (100 μg/mL). Total AKT serves as loading control.
(F) Growth of 22Pc-EP cells in CSS media supplemented with 22Pc-CAFCM treated with YW538.24.71 (1, 10, or 20 μg/mL) or IgG (20 μg/mL). CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(G) Growth of 22Pc-EP cells in Enz (0.1 μM) or Veh (DMSO) containing FBS media supplemented with 22Pc-CAFCM treated with YW538.24.71 (1, 10, or 20 μg/mL) or IgG (20 μg/mL). The Enz group was normalized to the Veh group. CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
(H) Western blot analysis of HER3-AKT activation in 22Pc-EP after stimulation with 22Pc-CAFCM in the presence of an HER3-blocking antibody AMG888 (1 or 10 μg/mL) or IgG (10 μg/mL). Total AKT serves as loading control.
(I) Growth of 22Pc-EP cells in CSS media supplemented with 22Pc-CAFCM treated with AMG888 (1 or 10 μg/mL) or IgG (10 μg/mL). CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(J) Growth of 22Pc-EP cells in Enz (0.1 μM) or Veh (DMSO) containing FBS media supplemented with 22Pc-CAFCM treated with AMG888 (1 or 10 μg/mL) or IgG (10 μg/mL). The Enz group was normalized to the Veh group. CellTiter-Glo reading on day 7. Media Ctrl: serum-free media.
(K) Western blot analysis of HER3-AKT activation in 22Pc-EP after stimulation with either recombinant NRG1 or EGF (10 or 50 ng/mL). Total AKT serves as loading control.
(L) Growth of 22Pc-EP in CSS media supplemented with either recombinant NRG1 or EGF (10 or 50 ng/mL). CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(M) Growth of 22Pc-EP cells in Enz (0.1 μM) or Veh (DMSO) containing FBS media treated with recombinant NRG1 or EGF (10 or 50 ng/mL). The Enz group was normalized to the Veh group. CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
Assays were performed with three biological replicates. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05; n.s., not significant, Student’s t test. Data are represented as mean ± SD. See also Figure S4.
Having established paracrine NRG1-HER3 signaling as a primary driver of in vitro resistance in these models, we next explored the physiological relevance of this signaling using in vivo mouse xenograft models. We first documented increased levels of NRG1 (stroma) and phospho-HER3 (tumor cells) in lysates from CWR22Pc xenografts grown in castrated mice compared with intact mice (Figures 5A and 5B). We then treated established, castration-resistant CWR22Pc xenografts with a blocking antibody to HER3 (AMG888) or a HER2 kinase inhibitor (neratinib) and observed potent growth inhibition, as well as tumor regressions with combination therapy (Figures 5C and 5D). The NRG1-neutralizing antibody (YW538.24.71) also had potent antitumor activity in this assay, given alone or in combination with neratinib (Figures 5E–5G). We extended these findings to a castration-sensitive model of CWR22Pc, showing that NRG1 blockade significantly enhanced the antitumor effect of castration, either alone or in combination with neratinib (Figures 5H and 5I). Corresponding analysis of tumor lysates confirmed reduction of pHER2/pHER3 in mice treated with NRG1 or HER kinase inhibitors (Figures S5A–S5C). Thus, the dependence of prostate tumor cells on stromal-derived NRG1 translates to in vivo models and can potentially be exploited for therapeutic benefit using clinical-grade inhibitors of the NRG1-HER3 signaling axis.
Figure 5. NRG1-HER3 Signaling Confers Antiandrogen Resistance In Vivo.

(A) qRT-PCR analysis of Nrg1 expression in hormone-intact (n = 7) or castration-resistant (n = 8) CWR22Pc tumors using mouse-specific primers.
(B) Western blot analysis of NRG1 expression and HER3-AKT activation in hormone-intact or castration-resistant CWR22Pc tumors. Total AKT serves as loading control. Bands were quantified using ImageJ and normalized to mean of β-actin intensity in each group. Green, hormone intact; red, castrated.
(C) Growth of castration-resistant CWR22Pc tumor xenografts in castrated mice, treated with AMG888 (20 mg/kg), neratinib (20 mg/kg), or vehicle. Treatment started when the average tumor size reached 150 mm3 (n = 5 mice per group).
(D) Waterfall plot showing growth of individual tumors from (C).
(E) Growth of castration-resistant CWR22Pc tumor xenografts in castrated mice, treated with YW538.24.71 (25 mg/kg), neratinib (20 mg/kg), or vehicle. Treatment started when the average tumor size reached 150 mm3 (n = 5 mice per group).
(F) Waterfall plot showing growth of individual tumors from (E).
(G) Boxplot showing tumor size at week 6 in single-agent neratinib or YW538.24.71 versus combination treatment groups from (E). Each dot represents individual tumors, upper/lower/median values are labeled.
(H) Growth of castration-sensitive CWR22Pc tumor xenografts in intact mice, treated with castration plus either YW538.24.71 (25 mg/kg), neratinib (20 mg/kg), or vehicle. Treatment started when the average tumor size reached 200 mm3 (n = 5 mice per group).
(I) Boxplot showing tumor size at week 6 in single-agent neratinib or YW538.24.71 versus combination treatment groups in (H). Labeling is the same as in (G). ****p < 0.0001, ***p < 0.001, *p < 0.05; n.s., not significant. (A, G, and I) Student’s t test, data are represented as mean ± SD, (C, E, and H) multiple t test, data are represented as mean ± SEM. See also Figure S5.
NRG1 Activates a Subset of AR Target Genes
Our earlier analysis of five canonical AR target genes suggested that NRG1 preserves tumor cell viability without restoring AR target gene expression (Figures 2G and 2H). To address this question more comprehensively, we performed whole-transcriptome analysis of 22Pc-EP cells treated with recombinant NRG1, Enz, or both (Figures 6A and 6B) and generated an AR signature, defined as Enz-suppressed genes (p < 0.05, log2 fold change>2) (Figure S6A; Table S1). GSEA using this 22Pc-EP-derived AR signature, as well as two independent AR signatures, showed that AR transcriptional activity is not enriched by NRG1 treatment (Figures 6C, S6B, and S6C), as suggested by our earlier analysis of a limited number of canonical AR target genes. However, comparison of NRG1- versus AR-regulated transcriptomes revealed 1,917 co-regulated genes (p < 0.05), which subdivide into 4 major clusters by unsupervised clustering (Figures 6D–6I; Tables S2 and S3). Cluster 1 is of particular interest because these genes are suppressed by Enz but restored by NRG1 (Figure 6J) and therefore may play a functional role in maintaining tumor cell viability. Of note, this cluster is enriched for genes involved in amino acid and folate metabolism based on GO term pathway analysis (log2 fold change>0.5, 103 out of 308) (Figure S6D).
Figure 6. NRG1 Activates a Subset of AR Target Genes.

(A) MA plot showing differentially expressed genes (n = 4,978, adjusted p value < 0.05) in 22Pc-EP cells treated with Enz (0.5 μM, 48 h) or Veh (DMSO). Top 15 up- or downregulated genes were labeled. Canonical AR targets FKBP5 and NKX3–1 were also labeled.
(B) MA plot showing differentially expressed genes (n = 5,690, adjusted p value < 0.05) in 22Pc-EP cells treated with recombinant NRG1 (10 ng/mL, 48 h) or Veh (PBS). Top 15 up- or downregulated genes were labeled.
(C) GSEA of AR signature between Veh (DMSO)- versus NRG1-treated group in 22Pc-EP cells.
(D) Venn diagram showing the number of overlapping genes that are co-regulated by both AR and NRG1 (n = 1,971, adjusted p value < 0.05).
(E) Heatmap showing unsupervised clustering of expression of 1,971 genes across 4 conditions (Veh, Enz, NRG1, or Enz + NRG1). Four distinct clusters were identified (clusters 1–4), each representing AR and NRG1 co-regulated genes in the same or opposite directions.
(F–I) Venn diagram showing the number of AR and NRG1 co-regulated genes (adjusted p value < 0.05 for both conditions) in each direction (clusters 1–4). Cluster 1 (F), cluster 2 (G), cluster 3 (H), and cluster 4 (I).
(J) Dot plot showing fold change values (log2) of genes in each of clusters 1–4. In clusters 1 and 3, genes with log2 fold change >1 were labeled. In clusters 2 and 4, genes with log2 fold change >1.5 were labeled.
Androgen Deprivation Therapy Induces NRG1 Expression in Stroma of Prostate Cancer Patients
To gain insight into the potential clinical relevance of these findings, we examined NRG1 expression in a cohort of 43 patients with localized prostate cancer who underwent radical prostatectomy surgery, 23 of whom received neoadjuvant ADT (Tables S4 and S5). For this analysis we developed an immunohistochemical (IHC) assay to detect NRG1 expression in formalin-fixed tissue, including tissue microarrays, as described in the STAR Methods (Figure 7A). Using this assay we detected NRG1 staining in 5 of 23 patients (22%) who received ADT before prostatectomy and in 0 of 20 patients who were hormonally intact at the time of surgery (p = 0.0265) (Figure 7B; Tables S6 and S7). NRG1 staining was observed in stromal cells in four of the five positive cases. The fifth case had NRG1-positive tumor cells, and one case had evidence of NRG1 positivity in both tumor and stroma. We suspect the failure to detect NRG1 expression by IHC in the stroma of hormonally intact patients, despite clear evidence of NRG1 expression at the RNA level by RNA-FISH and RNA-PCR, is due to reduced sensitivity of the IHC assay (Figure S7A). Direct comparisons of both assays across a larger cohort are warranted.
Figure 7. ADT Induces NRG1 Expression in the Stroma of Prostate Cancer Patients.

(A) Representative images showing H&E (left, middle) and immunohistochemistry (right) analysis of stromal NRG1 staining in radical prostatectomy specimens from high-grade primary PCa patients. Asterisk denotes areas of intraductal carcinoma (no. 1) or invasive cancer (no. 2).
(B) Top: pie chart showing percentage of NRG1 positivity in hormone-intact or neoadjuvant ADT-treated groups, and table showing number of patients with NRG1-positive biopsies in hormone-intact or neoadjuvant ADT-treated group.
(C) Western blot analysis of NRG1 and CAF markers (PDFGRα, FAP, vimentin, and α-SMA) in five independent patient-derived primary PCa CAFs (pCAFs). pCAF no. 1 is from a neoadjuvant ADT-treated patient and pCAFs no. 2–5 are from hormone-intact patients. HSP90 serves as loading control. Bands were quantified using ImageJ and normalized to mean of HSP90 intensity in each group.
(D) Western blot analysis of HER3-AKT activation in 22Pc-EP cells stimulated with conditioned media from patient-derived primary CAFs (pCAFCM). Total AKT serves as loading control. Bands were quantified using ImageJ and normalized to mean of total AKT intensity in each group.
(E) Growth of 22Pc-EP cells in CSS media supplemented with pCAFCM. CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(F) Growth of 22Pc-EP cells in FBS media supplemented with pCAFCM and treated with Enz (0.1 μM). The Enz group was normalized to the Veh group. CellTiter-Glo reading on day 7. Media Ctrl, serum-free media.
(G) Growth of 22Pc-EP cells in CSS media supplemented with pCAFCM treated with YW538.24.71 (10 μg/mL) or AMG888 (10 μg/mL). CellTiter-Glo reading on day 4. Media Ctrl, serum-free media.
(H) Growth of VCaP or VCaP + pCAF no. 1 co-injected tumor xenografts in castrated mice treated with YW538.24.71 (25 mg/kg), neratinib (20 mg/kg), or vehicle. Treatment started when tumors reached 200 mm3 (n = 5 mice per group).
(I) qRT-PCR analysis of NRG1 mRNA expression in pCAF no. 1 treated with CSS, Enz (10 μM), or Veh (DMSO) on day 7. NRG1 expression is normalized to ACTB.
(J) Western blot analysis of NRG1 protein in pCAFs no. 2 and 3 treated with CSS, Enz (10 μM), or Veh (DMSO) on day 7. Cyclophilin B serves as loading control. Assays were performed with three biological replicates. ****p < 0.0001, *p < 0.05; n.s., not significant. (E–G) One-way ANOVA compared with Ctrl/Veh/IgG group, (H) one-way ANOVA compared with VCaP alone + Vehicle group, (I) Student’s t test compared with Ctrl group, (E–G and I) data are represented as mean ± SD, (H) data are represented as mean ± SEM. Ctrl, serum-free media. See also Figure S7 and Tables S4–S7.
To further address the question of stromal-derived NRG1 expression in clinical samples, we generated primary CAFs from five PCa patients with high-risk, localized disease who underwent radical prostatectomy surgery, as described in the STAR Methods. As expected, these patient-derived CAFs express PDGFRα, FAP (canonical CAF markers), vimentin, and α-SMA (stromal lineage). Notably, all five patient-derived CAF cultures (pCAFs) expressed NRG1 protein (Figure 7C). Furthermore, conditioned media from each pCAF culture activated HER3/AKT phosphorylation and promoted resistance to CSS or Enz in 22Pc-EP cells, which was efficiently blocked using NRG1-neutralizing antibody (YW538.24.71) or HER3-blocking antibody (AMG888) (Figures 7D–7G). To determine if pCAFs can promote in vivo tumor growth, we co-injected human pCAF isolate no. 1 (selected based on high NRG1 expression) with human VCaP prostate cancer cells in the xenograft assay. Co-injection of pCAF no. 1 cells significantly enhanced the growth of VCaP tumors in castrated mice, and this acceleration in growth was completely reversed by treatment with NRG1-blocking antibody (Figure 7H).
To examine if upregulation of NRG1 is induced by hormone therapy, we treated freshly isolated primary CAFs from CWR22Pc tumors or pCAFs with CSS or Enz. CSS and Enz both induced NRG1 mRNA and protein expression after 7 days, with the highest induction seen with the combination of CSS plus Enz (Figures 7I, 7J, S7B, and S7C), a finding confirmed in pCAF isolates from 15 of 18 additional patients (Figure S7D). To investigate the mechanism underlying NRG1 upregulation following AR inhibition, we performed time course experiments and observed consistent changes in NRG1 levels 7 days after androgen withdrawal but not after 24 h (Figure S7E). NRG1 levels returned to baseline after replenishing androgen in the culture media but also after 7 days (Figures S7F and S7G). This delayed response to AR pathway perturbation suggests an indirect mechanism of NRG1 regulation, which is further supported by our failure to detect AR peaks in the NRG1 promoter or enhancer in datasets from prostate cancer-derived CAFs, whereas strong AR peaks are present in the FKBP5 promoter region (Figure S7H). Thus, NRG1 is expressed in tumor-associated stromal cells of primary prostate cancers at increased levels after ADT treatment, and these levels are sufficient to promote resistance to ADT in vitro and in vivo.
NRG1 Activity Is Associated with Unfavorable Treatment Outcome in CRPC Patients
To determine whether NRG1 influences response to antiandrogen therapy, we took advantage of a recently published cohort of genomically annotated CRPC patients with associated treatment-response data to second-generation antiandrogens (Abida et al., 2019). Tissue samples from these patients are not available for in situ measures of NRG1 expression; therefore, we used a previously reported NRG1 activity gene signature derived by ex vivo exposure of breast cancer cells to NRG1 (Nagashima et al., 2007). First we validated that NRG1 mRNA expression (by RNA sequencing [RNA-seq]) is positively correlated with the NRG1 activity score in two prostate cancer cohorts (TCGA and SU2C, representing localized and metastatic disease, respectively) (Figures S8A and S8B). The NRG1 activity score also correlated with a CAF signature score, consistent with stroma as the likely source of NRG1 (Figures S8C and S8D). Using the NRG1 signature score, we then asked whether increased NRG1 activity in patients is associated with clinical response to second-generation antiandrogen therapy in a cohort of 56 CRPC patients previously treated with enzalutamide or abiraterone on whom tumor RNA-seq data were available within 30 days before treatment (2 out of 56 were excluded due to NRG1 signature <0). Pearson correlation analysis showed that NRG1 signature score is negatively correlated with time on treatment (p = 0.005) (Figure 8A). To further dissect this correlation, we plotted the NRG1 activity score of each patient, which revealed a Gaussian-like distribution, then subdivided the cohort into upper and lower halves using the median (Figure 8B). Patients in the upper half had a significantly shorter time to progression on either enzalutamide or abiraterone compared with the patients in the lower half (p = 0.034) (Figure 8C). This result is further supported by Cox hazards ratio analysis showing significantly reduced hazards related to a low NRG1 signature score (Figure 8D). This negative correlation was also seen using a second NRG1 activity signature derived by ex vivo treatment of 22Pc-EP prostate cancer cells (p = 0.036) (Figure 8E and Table S8). Finally, GSEA of the NRG1 activity-high versus -low patients revealed enrichment of signatures for RTK and PI3K signaling and reduced hormone receptor signaling (Figures S8E–S8G).
Figure 8. NRG1 Activity Is Associated with Unfavorable Treatment Outcome in CRPC Patients.

(A) Pearson correlation analysis of NRG1 signature score versus time on treatment for first line androgen receptor signaling inhibitors (ARSI) of a 54 mCRPC patient cohort (p = 0.005).
(B) Histogram showing frequency distribution of NRG1 signature score in the same patient cohort. Dotted line denotes median cutoff.
(C) Probability of treatment duration of the high and low (median separation) NRG1 signature (Nagashima et al., 2007) groups among 54 patients. p value (0.034) was calculated using log rank test.
(D) Cox hazard ratio analysis of the NRG1 signature score high and low groups of 54 patients. p value (0.019) was calculated using log rank test.
(E) Probability of treatment duration of the high and low (median separation) prostate-specific NRG1 signature (22Pc-EP) groups among 54 patients. p value (0.036) was calculated using log rank test.
DISCUSSION
Studies of resistance to antiandrogen therapy in prostate cancer have primarily focused on cell-autonomous mechanisms that collectively point to the central role of sustained AR signaling, even in late-stage CRPC. These include well-documented mechanisms, such as AR gene amplification/mutation and AR splice variants (Watson et al., 2015), as well as tandem duplication events involving the AR enhancer (Quigley et al., 2018; Takeda et al., 2018; Viswanathan et al., 2018). Collectively these mechanisms may explain up to ~80% of CRPC cases, but there is growing evidence for microenvironmental sources as additional contributors to antiandrogen resistance. Immune cells, specifically myeloid-derived suppressor cells, are one such source and can drive CRPC progression through production of the cytokine IL-23 (Calcinotto et al., 2018). Stromal-derived growth factors have also been implicated, specifically HGF and FGFs (Humphrey et al., 1995; Hwang et al., 2011; Kwabi-Addo et al., 2004). Here, we document a critical role of NRG1, also stromal derived, together with its receptor HER3, and we provide a clear strategy toward targeted intervention using clinical-grade blocking antibodies.
A unique feature of our work is the discovery of NRG1 through an unbiased biochemical fractionation approach in which we screened for the antiandrogen resistance factor produced by murine CAFs that persist during in vitro culture of the CWR22Pc prostate cancer cell line. Specifically, we show that CAF-derived NRG1 is required for CWR22Pc tumor cells to develop resistance to enzalutamide or to ADT. Mechanistic studies using multiple cell lines (VCaP, LAPC4, and 22Rv1), mouse and human cancer organoids (P53-KO, RB-KO, PTENΔ/Δ -Rosa26-ERG, and MSKPCa2) and in vivo xenograft models (CWR22Pc and VCaP) establish the importance of NRG1-HER3 kinase signaling as a critical driver of antiandrogen resistance. Furthermore, we observed significant antitumor activity, including tumor regressions, using clinical-grade neutralizing antibodies against NRG1 and HER3, as well as HER2-specific kinase inhibitors. In primary prostate cancer clinical samples, we show that NRG1 is synthesized by adjacent stromal cells (by RNA-FISH), with evidence (by IHC) that these levels are higher in patients who received neoadjuvant ADT. This finding is consistent with activation of HER3 observed in a subset of patients who underwent prostatectomy after neoadjuvant ADT (Gao et al., 2016). We also observed increased NRG1 mRNA expression in both mouse and patient-derived primary CAFs when they are given antiandrogen treatments in culture, suggesting that AR signaling negatively regulates NRG1 expression through mechanisms that need further investigation. Importantly, in a cohort of genomically profiled CRPC patients with associated treatment response data, we found that patients with higher NRG1 activity develop resistance earlier than those with lower NRG1 activity. The collective evidence suggests that NRG1 expression in prostate stromal cells (which are AR positive) is upregulated in patients receiving neoadjuvant ADT and, through activation of HER3 signaling in tumor cells, may contribute to their persistence. We also have evidence, through single-cell analysis of normal prostate tissue, that stromal-derived NRG1 can function as a survival factor for luminal cells independent of AR activation (Karthaus et al., 2020). Of note, AR has been implicated in CAF biology in another mesenchymal tissue, dermal fibroblasts, where its loss results in CAF activation (Clocchiatti et al., 2018).
The fact that NRG1 protects tumor cells from androgen withdrawal without full restoration of downstream AR signaling is intriguing in light of our earlier work documenting links between RTKs and AR activation (Carver et al., 2011). In that context, impaired AR pathway activation is seen in tumor cells with PTEN loss due to increased PI3K signaling, which results in reduced HER2/HER3 expression through downstream transcriptional effects. The effects of NRG1 activation reported here occur in wild-type PTEN models where baseline PI3K activity is low but potently activated by NRG1 through HER2/HER3. We postulate that this hyperactivated PI3K signal contributes to reduced AR activity. However, we also identify a set of genes co-regulated by NRG1 and AR (and not previously recognized as AR targets) implicated in amino acid and folate metabolism that warrant further functional investigation.
In addition to the role of paracrine NRG1 production described here in prostate cancer, there is growing evidence that autocrine NRG1 expression plays a role in other tumor types. NRG1 is specifically implicated as a driver in squamous cancers (e.g., esophageal or squamous lung cancer) through its role as a direct target gene of the basal epithelial lineage-defining transcription factor TP63 (Hegde et al., 2019). More commonly, secretion of NRG1 has been observed in various cancers, including ovarian, non-small cell lung, and melanoma, as well as brain metastasis (Capparelli et al., 2018; Hegde et al., 2013; Kodack et al., 2017; Sheng et al., 2010; Wilson et al., 2011). Among the most compelling are translocations that fuse the NRG1 genomic locus to a transcriptionally active gene partner, as seen in invasive mucinous adenocarcinomas of the lung and other tumor types. Furthermore, dramatic responses have been reported in such patients after receiving afatinib or anti-HER3-blocking antibody therapy (Dhanasekaran et al., 2014; Drilon et al., 2018; Heining et al., 2018; Jones et al., 2019; Jonna et al., 2019).
As to whether the clinical success of anti-HER2/3 therapy in tumors with NRG1 might translate to prostate cancer, it is worth noting that previous clinical trials of HER2 inhibitors in prostate cancer were disappointing (Agus et al., 2007; de Bono et al., 2007; Lara et al., 2004; Morris et al., 2002; Sridhar et al., 2010; Whang et al., 2013; Ziada et al., 2004). However, these studies lacked current insights into which patient population is most likely to benefit and were not specifically designed to test the hypothesis raised here. Furthermore, the HER2 therapies tested in these trials are not optimal for blocking NRG1-mediated activation of HER3/4, as is now clear from more recent studies (Drilon et al., 2018; Wilson et al., 2011). The insights emerging from our work suggest a different translational strategy. Wild-type PTEN status could be a patient selection biomarker, based on the mutual exclusivity of increased NRG1 and PTEN mutant tumors (data not shown), which is interesting in light of a similar mutual exclusivity between NRG1 translocations and KRAS mutation in pancreas cancer (Heining et al., 2018; Jones et al., 2019). In the neoadjuvant setting, one can envision combination therapy with ADT plus anti-HER3 antibody in patients with increased NRG1 and/or phospho-HER3 levels after a short trial of ADT alone. Finally, it is worth noting that clinical trials of HER3-targeted antibody drug conjugates, such as U3–1402, have shown clinical activity (Janne et al., 2019).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Charles L. Sawyers (sawyersc@mskcc.org).
Materials Availability
All cell lines, plasmids and other reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
RNA-seq data has been deposited in the Sequence Read Archive (SRA) with the accession numbers GSE147976 and also listed in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-NRG1 | Cell Signaling Technology | Cat# 2573; RRID: AB_1031011 |
| Rabbit monoclonal anti-EGFR | Cell Signaling Technology | Cat# 4267; RRID: AB_2246311 |
| Rabbit monoclonal anti-HER3 | Cell Signaling Technology | Cat# 4754; RRID: AB_10691324 |
| Rabbit monoclonal anti-HER2 | Cell Signaling Technology | Cat# 2165; RRID: AB_10692490 |
| Rabbit monoclonal anti-AKT | Cell Signaling Technology | Cat# 4691; RRID: AB_915783 |
| Rabbit monoclonal anti-β-actin | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| Rabbit monoclonal anti-CyclophilinB | Cell Signaling Technology | Cat# 43603; RRID: AB_2799247 |
| Rabbit monoclonal anti-Vimentin | Cell Signaling Technology | Cat# 5471; RRID: AB_10692897 |
| Rabbit monoclonal anti-PDGFRα | Cell Signaling Technology | Cat# 3164; RRID: AB_2162351 |
| Rabbit monoclonal anti-phospho-EGFR Tyr1068 | Cell Signaling Technology | Cat# 3777; RRID: AB_2096270 |
| Rabbit monoclonal anti-phospho-HER2 Tyr1221/1222 | Cell Signaling Technology | Cat# 2243; RRID: AB_490899 |
| Rabbit monoclonal anti-phospho-HER3 Tyr1289 | Cell Signaling Technology | Cat# 4791; RRID: AB_2099709 |
| Rabbit Phospho-FGF Receptor (Tyr653/654) antibody | Cell Signaling Technology | Cat# 3471; RRID: AB_331072 |
| Rabbit monoclonal anti-phospho-AKT S473 | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Rabbit monoclonal anti-phospho-AKT T308 | Cell Signaling Technology | Cat# 4056; RRID: AB_331163 |
| Rabbit Phospho-FRS2-α (Tyr436) Antibody | Cell Signaling Technology | Cat# 3861; RRID: AB_2231950 |
| Rabbit monoclonal anti-HSP90 | Cell Signaling Technology | Cat# 4877; RRID: AB_2233307 |
| Anti-alpha smooth muscle Actin antibody [E184] | Abcam | Cat# ab32575; RRID: AB_722538 |
| Anti-Fibroblast activation protein, alpha antibody | Abcam | Cat# ab28244; RRID: AB_732312 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | Cat# 114608; RRID: AB_313599 |
| Mouse Anti-Human CD326 (EpCAM) Monoclonal Antibody, FITC Conjugated Clone HEA-125, | Miltenyi Biotec | Cat# 130-080-301; RRID: AB_244192 |
| Biological Samples | ||
| Prostate cancer Neoadjuvant ADT samples | Dana Farber Cancer Institute | Gao et al., 2016 |
| Radical prostatectomy samples | MSKCC Tissue Bank IRB #06-107 and #17-472 | https://www.mskcc.org/research/ski/core-facilities/pathology |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GlutaMax Supplement | Gibco | Cat# 35050061 |
| 1M HEPES Solution | Gibco | Cat# 15630080 |
| 100mM Sodium Pyruvate | Gibco | Cat# 11360-070 |
| Penicillin-streptomycin | Sigma Aldrich | Cat# P0781-100ML |
| Puromycin | Gibco | Cat# A1113803 |
| Blasticidin | Gibco | Cat# A1113903 |
| Human Neuregulin-1 (hNRG-1) | Cell Signaling Technology | Cat# 5218 |
| Human recombinant EGF | StemCell Technology | Cat# 78006.1 |
| Human recombinant FGF1 | StemCell Technology | Cat# 78187 |
| Human recombinant FGF2 | StemCell Technology | Cat# 78003.1 |
| Trizol | Ambion | Cat# 15596018 |
| 20x NuPAGE MES SDS Buffer | Novex | Cat# NP0002 |
| 1x Bolt Transfer Buffer | Novex | Cat# BT00061 |
| Neratinib | LC Laboratories | Cat# n-6404, NRT-104 |
| Enzalutamide | Selleckchem | Cat# S1250 |
| Bicalutamide | Selleckchem | Cat# S1190 |
| AMG888 | Amgen/Daiichi Sankyro | N/A |
| YW538.24.71 | Genentech | Cat# OR-216518 |
| 100% Methanol | Thermo Fisher | Cat# A412-20 |
| Fetal Bovine Serum, charcoal stripped | Omega Scientific | Cat# FB-11 lot #761007 |
| Fetal Bovine Serum | Omega Scientific | Cat# FB-11 lot#101943 |
| TrypLE Express | Gibco | Cat# 12605-010 |
| Critical Commercial Assays | ||
| 2X PowerUpTM SYBRTM Green Master Mix | Thermo Fisher | Cat# 11766500 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| MycoAlertTM PLUS Mycoplasma Detection kit | Lonza | Cat# LT07-710 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | Cat# 11668500 |
| Qiagen MinElute PCR purification kit | Qiagen | Cat# 28004 |
| Cell Titer glo assay | Promega | Cat# G9243 |
| Deposited Data | ||
| RNA-seq | GEO | GSE147976 |
| Experimental Models: Cell Lines / Organoids | ||
| CWR22Pc | Dagvadorj et al., 2008 | N/A |
| CWR22Pc-EP (22Pc-EP) | This paper | N/A |
| CWR22Pc-CAF (22Pc-CAF) | This paper | N/A |
| VCaP | ATCC | Cat# CRL-2876, RRID: CVCL_2235 |
| 22Rv1 | ATCC | ATCC Cat# CRL-2505, RRID:CVCL_1045 |
| LAPC4 | Klein et al., 1997 | N/A |
| Patient derived CAFs | MSKCC | IRB protocol: 12-001, 12-245, 90-040, 15-331 |
| Experimental Models: Organisms/Strains | ||
| C.B-Igh-1b/IcrTac-Prkdcscid mouse | Taconic | Cat# CB17SC-M |
| Oligonucleotides | ||
| sgNT (non-targeting guide): Forward-5’- CACCGGGCCAGTGTGGCCGTTACGC Reverse-5’- AAACGCGTAACGGCCACACTGGCCC |
This paper | N/A |
| sgNrg1-Guide 1 Forward-5’- CACCGAGTATCTTGAGGGGTTTGAC Reverse-5’- AAACGTCAAACCCCTCAAGATACTC |
This paper | N/A |
| sgNrg1-Guide 2 Forward-5’- CACCGAGGCGAGTGCTTCATGGTGA Reverse-5’- AAACTCACCATGAAGCACTCGCCTC |
This paper | N/A |
| sgNrg1-Guide 3 Forward-5’- CACCGCCACATCTACATCCACGACT Reverse-5’- AAACAGTCGTGGATGTAGATGTGGC |
This paper | N/A |
| sgNrg1-Guide 4 Forward-5’- CACCGGAGATGGCTGGTCCCAGTCG, Reverse-5’- AAACCGACTGGGACCAGCCATCTCC |
This paper | N/A |
| qRTPCR primer for human NRG1 | Qiagen | Cat# PPH01151F |
| qRTPCR primer for human FKBP5 | Qiagen | Cat# PPH02277A |
| qRTPCR primer for human NKX3-1 | Qiagen | Cat# PPH02267C |
| qRTPCR primer for human PMEPA1 | Qiagen | Cat# PPH01013B |
| qRTPCR primer for human SLC45A3 | Qiagen | Cat# PPH15695A |
| qRTPCR primer for human TRPM8 | Qiagen | Cat# PPH17934F |
| qRTPCR primer for mouse Nrg1 | Qiagen | Cat# PPM57587C |
| qRTPCR primer for mouse Nrg2 | Qiagen | Cat# PPM04520G |
| qRTPCR primer for mouse Nrg3 | Qiagen | Cat# PPM03007A |
| qRTPCR primer for mouse Nrg4 | Qiagen | Cat# PPM04712B |
| qRTPCR primer for mouse specific Actb | Qiagen | Cat# PPM02945B |
| qRTPCR primer for human specific Actb Forward-5’- CACCAACTGGGACGACAT Reverse-5’- ACAGCCTGGATAGCAACG |
This paper. | N/A |
| Recombinant DNA | ||
| SGEP-Renilla | Fellmann C et al. 2013 | N/A |
| QCXIP-tdTomato | Clontech | Cat# 9136-1 |
| p-tdTomato | Clontech | Cat# 632531 |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| Software and Algorithms | ||
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| R Studio | N/A | https://www.rstudio.com/ |
| Tumor measuring system Peira TM900 | Peira bvba, Belguim | |
| HISAT (v 2.0.1) | Kim et al., 2016 | http://ccb.jhu.edu/software/hisat2/index.shtml |
| Sambamba (v0.6.6) | Tarasov et al., 2015 | http://lomereiter.github.io/sambamba/ |
| Featurecount (v1.4.6) | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| DEseq2 (v1.6.3) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| PANTHER | Mi et al., 2019 | http://www.pantherdb.org |
| Samtools (v1.3) | Li et al., 2009 | http://samtools.sourceforge.net |
| Hclust | Müllner, 2013 | http://danifold.net/fastcluster.html |
| Pheatmap | R Core Team, 2016 | https://cran.r-project.org/web/packages/pheatmap/index.html |
| Other | ||
| HiTrap Q HP anion exchange chromatography column | GE Healthcare | Cat# 17115401 |
EXPERIMENTAL MODEL AND SUBJECT DETAIL
Generation of 22Pc-EP and 22Pc-CAF Models
The CWR22Pc prostate cancer cell line was kindly provided by Marja Nevalainen (Dagvadorj et al., 2008). We found that this cell line contained a subpopulation of cells with fibroblast-like morphology that were human EpCAM-negative and confirmed to be of mouse origin. In order to purify tumor cells and mouse fibroblasts, we plated CWR22Pc at 400–800 cells per well (6-well) in 50% conditioned media. Numerous multi-clonal, cancer epithelial islands visually free of fibroblasts were isolated by cloning cylinders and then pooled to derive the pure epithelial subline, CWR22Pc-EP, in short 22Pc-EP. Human EpCAM-negative cancer-associated mouse fibroblasts were obtained by performing mouse specific H-2Kb and H-2Db MHC class I sorting (Biolegend #114608) and the FACs purified cancer-associated fibroblasts were termed as CWR22Pc-CAF, in short 22Pc-CAF. Purified 22Pc-EP and 22Pc-CAF cells were transduced with eGFP (SGEP-Renilla) (Fellmann et al., 2013) or tdTomato (QCXIP-tdTomato, Clontech #9136–1). tdTomato was derived from vector p-tdTomato (Clontech #632531) and cloned into the AgeI and EcoRI sites of QCXIP retroviral vector. Both were and selected with 1 μg/mL puromycin (Gibco #A1113803) for 5 days.
Other Cell Lines and Organoids Models
LNCaP cells were purchased from ATCC (#CRL-1740™). VCaP cells were purchased from ATCC (#CRL-2876™). 22Rv1 cells were purchased from ATCC (#CRL-2505™). LAPC4 cells were generated in the Sawyers laboratory (Klein et al., 1997). MSK-PCa2 human prostate cancer organoid was generated by Gao et al. at MSKCC (Gao et al., 2014). Trp53Δ/Δ and Rb1Δ/Δ mouse organoid was generated from GEMM mice by introducing a lentiviral-expressing Cre recombinase (Ku et al., 2017). PtenΔ/Δ-Rosa26-ERG organoid was generated from GEMM mice (Chen et al., 2013). All organoids were maintained according to established organoid culture protocol (Karthaus et al., 2014). All cell lines and organoids were periodically tested negative for mycoplasma (Lonza #LT07–318).
Xenograft Experiment
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Memorial Sloan Kettering Cancer Center. For CWR22Pc, 22Pc-EP and VCaP xenograft experiments, 2 × 106 cells were mixed into a 50% Matrigel suspension (Corning #356237) and injected subcutaneously (100 μl/injection) into flanks of castrated male C.B-17 scid mice at age 6–8 weeks (Taconic). For co-injection experiments, 5 × 105 22Pc-EP-eGFP and 5 × 105 22Pc-CAF-tdTomato cells were mixed and grafted into the mice of the same genetic background. For all xenograft experiments, 5 mice per group were grafted at both flanks (10 tumors per group). Tumor measurement began when tumors became palpable and was performed weekly using the tumor measuring system Peira TM900 (Peira bvba, Belguim). For drug treatment experiments (Figure 5), 20 mg/kg neratinib (0.5% methyl cellulose + 0.4% Tween80) was given by oral gavage 5 times a week. 20 mg/kg AMG888 (PBS) was given by intraperitoneal injection twice a week. 25 mg/mL YW538.24.71 (PBS) was given by intraperitoneal injection once a week.
Clinical Specimen
Informed consent was obtained from all subjects before any patient-related studies. All patient derived tissue was collected in compliance with rules and regulations of Memorial Sloan Kettering Cancer Center (IRB: 12–001, 12–245, and 90–040). Patient information was de-identified prior to any analysis.
METHOD DETAILS
Isolation of Primary CAFs
Isolation of cancer-associated fibroblasts (CAFs) from mouse tumors or patient samples was performed with previously established protocol with modifications (Seluanov et al., 2010; Sharon et al., 2013). For patient-derived CAFs, human tissue acquisition and usage was conducted under approved IRB protocol numbers: 12–001, 12–245, and 90–040. Specifically, tumors are minced and dissociated in RPMI-1640 with FBS (10%), PenStrep (1%), L-glutamine (1%), sodium pyruvate (1%) and HEPES pH=7.6 (1%) plus 0.5% collagenase Type I, 305U/mg (Worthington #LS004197) for 1 h at 37°C in a thermo-shaker. The digested tumor-cell mixture was filtered through a 100 μm filter (Corning #352360) and then spun down at 400g for 1.5 min. Depending on the pellet size, the pellet containing mixture of cells were plated on either a 6-well plate or a 10-cm dish that allows fibroblasts to attach and grow. After 2–3 passages, a limited dilution protocol was performed and cells were plated in a high dilution in a 96-well plate, single clones were expanded later. Validation of fibroblast identity was performed at the protein level by flow cytometry analysis for fibroblast surface markers.
Cell Culture
Cell lines used in this study were maintained in a 37°C and 5% CO2 incubator. CWR22Pc, 22Pc-EP, 22Pc-CAF and patient-derived primary CAF cells were cultured in RPMI-1640 with FBS (10%), PenStrep (1%), L-glutamine (1%), sodium pyruvate (1%) and HEPES pH=7.6 (1%). VCaP cells were cultured in DME-HG with FBS (10%), PenStrep (1%), L-glutamine (1%), sodium pyruvate (1%) and HEPES pH=7.6 (1%). 22Pc-EP cells were cultured on collagen I-coated plates. (Fisher Scientific #356450). All serums used in cell culture came from Omega Scientific (FBS, #FB-11, #lot:101943; CSS, #FB-11, #lot: 761007).
Quantitative Co-Culture Assays
Day 1: 22Pc-EPeGFP (2500 cells/well) or 22Pc-EPeGFP (2500 cells/well) plus 22Pc-CAFtdTomato (150 cells/well) were plated into black walled, collagen I collated 96-well plate (Corning™ #356700) to reduce fluorescent background. Day 2: Bicalutamide (10 μM) or Enz (1 μM) or vehicle (DMSO) was added into each well in triplicates. Fresh media and drug were replaced every 3 days, and images were taken every 7 days using a ZEISS ZEN Widefield microscope. Individual images were stitched using an automated program from MetaMorph. Fluorescent intensity was quantified using MetaMorph software (MetaMorph Inc). Assays were repeated with at least two independent biological replicates.
Conditioned Media Collection
Day 0: 4×106 CAFs or cancer cells were plated in 10cm dish. Day 1: cells were washed twice with PBS and replaced with serum free media. Day 3: the first batch of conditioned media was collected and replaced with serum free media. Collected conditioned media was filtered with a 0.45 μm filter (Millex, #SLHA033SS) to remove cell debris and then stored at 4°C. Day 5: the second batch of conditioned media was collected and filtered. Media from the first and second collections were combined and then concentrated to a 10x (for assays) or 50x stock (for purification) using Vivaspin™ protein concentrator spin columns (Sartorius #VS15T02, #VS6002). Concentrated conditioned media could be stored at 4°C for 2 weeks, or up to 6 months at - 80°C without significant activity loss.
Conditioned Media Assays
Antiandrogen Assay
Day 0: 22Pc-EP (3000 cells/well) or VCaP (5000 cells/well) were plated in 96-well plates. Day 1: conditioned media (10x) was mixed with 10%FBS-containing media at a 1:1 ratio. Antiandrogens (Enz 0.1 μM or Bic 10 μM) or DMSO was added into the culture (1:1000 dilution). Day 4: media and drugs in each well was replaced. Day 7: cell viability/number was measured by CellTiter-Glo luminescent cell viability assay (Promega #G9243). All assays were repeated in at least two independent biological replicates.
Androgen Deprivation Assay
Day 0: 22Pc-EP (3000 cells/well), VCaP (5000 cells/well), LAPC4 (5000 cells/well) or MSK-PCa2 (3000 single-cell organoids/well) cells were plated in 96-well plates. Day 1: conditioned media (10x) was first diluted with serum free media into a working solution (2.5x) and then mixed with 10% CSS media (charcoal-dextran stripped FBS, hereafter CSS) at a 1:1 ratio. The final experimental media contains 5% CSS and 1.25x conditioned media. For MSK-PCa2, CSS media was replaced with DHT- and EGF- deficient human prostate organoid media. Day 4: cell viability/number was measured by CellTiter-Glo luminescent cell viability assay (Promega #G9243).
Antiandrogen (Enzalutamide or Bicalutamide) Assay
Day 0: 22Pc-EP (3000 cells/well), VCaP (5000 cells/well), LAPC4 (5000 cells/well) or MSK-PCa2 (3000 single-cell organoids/well) cells were plated in a 96-well plate in triplicates. Day1: conditioned media (10x) was first diluted with serum free media into a working solution (2.5x) and then mixed with 10% FBS media at a 1:1 ratio. The final experimental media contains 5% FBS and 1.25x conditioned media (For MSK-PCa2, FBS media was replaced with DHT- and EGF- deficient human prostate organoid media). The mixture was added into the 96-well plate (100μL/mL). Antiandrogen (Enz or Bic) or Veh (DMSO) was also added to the plate. Day 4: media and drug was replaced. For growth curve analysis (Figures 2E and 2F), cell viability/number was measured by CellTiter-Glo luminescent cell viability assay (Promega #G9243). Day 7: for viability assay or growth curve analysis, cell viability/number was measured by CellTiter-Glo luminescent cell viability assay (Promega #G9243). Enz dosage: 22Pc-EP (0.1 μM), VCaP and MSK-PCa2 (1 μM), LAPC4 and 22Rv1 (10 μM). Bic dosage: 22Pc-EP (10 μM).
RTK Signaling
Day 0: 22Pc-EP (106 cells/well) cells were plated in a 6-well plate. Day 1: cells were serum starved for 1hr with serum free RPMI-1640 media and stimulated with conditioned media for 10 minutes in a 37°C and 5% CO2 incubator. 10%FBS and serum-free RPMI-1640 media were used as control. Cells were then washed with cold PBS on ice and lysates were collected for western blot. Following experimental procedure can be found in western blot method section.
Growth Factor Assay
The procedure was the same as Conditioned Media Assay but growth factors were added directly into the cell culture with corresponding culture media. Growth factors used were listed: NRG1 (Cell Signaling Technology #5218) and EGF (Stemcell Technology #78006.1).
Cell Growth Assay
CellTiter-Glo luminescent cell viability assay (Promega #G9243) was carried out in a 96-well plate format per manufacturer’s instructions. Luminescent signal representing relative number of cells was recorded as RLU (relative light units) according to manufacturer’s instructions. CellTiter-Glo 2.0 reagent was aliquoted into working solutions, stored at - 80°C and thawed to room temperature at each assay time point. Equal volume of reagent was added into each well of 96-well plates using a multi-channel pipette. Plates were incubated in room temperature on an orbital shaker for 10 minutes to stabilize the reaction. Luminescence was read by GloMax 96 Microplate Luminometer. Cells were seeded in 100 μl/well of media in triplicate per condition on day 0 and media was replaced every 3 days. The baseline level of luminescence that was measured on day 1 was subtracted from each corresponding plate at other time points to determine the relative cell growth (increase in luminescence signal). All growth assays were repeated in at least two independent biological replicates.
3D Organoid Growth Assay
Human and mouse organoids were trypsinized into single cell solution and counted. MSK-PCa2 (5000 cells), Trp53-KO (2000 cells), Rb1-KO (2000 cells) or PTENΔ/Δ -Rosa26-ERG (2000 cells) single organoids were seeded in 4×15ul Matrigel domes (Corning #356231) in a 48-well plate with 300 ul organoid culture media and media was replenished every 3 days. After 6 days, media was withdrawn and 100 μl cell recovery solution (Corning #354253) was added. The organoid plate was then incubated at 4°C on a rotator for 60 minutes. Equal volume (100μl) of CellTiter-Glo reagents (Promega #G7571) was added into the organoid suspension, mixed and incubated in room temperature on an orbital shaker for 15 minutes to stabilize the reaction. A total 200μl reaction volume was transferred to a 96-well plate for CellTiter-Glo assay.
Tissue Microarray
For prostate tissue microarray staining, archival formalin-fixed paraffin-embedded (FFPE) material was used under an IRB-approved protocol (15–331). For hormone naive primary prostate adenocarcinoma tissues (20 patients in total), tumor tissue was obtained from radical prostatectomy specimens. The Gleason scores ranged from 7 (3+4) to 9 (4+5). For neoadjuvant ADT treated primary prostate adenocarcinoma tissues (23 patients in total), tumor tissue was obtained from radical prostatectomy specimens after neoadjuvant ADT treatment. The Gleason scores ranged from 7 (3+4) to 10 (5+5). Please refer detailed information in Table S1. Each case was represented at least in duplicate (two cores per case) on the TMA. Most cases were represented in triplicate (three cores per case).
NRG1 Immunohistochemistry Analysis in Clinical Specimens
NRG1 antibody (1:200, Cell Signaling Technology #2573) was used in human tissue immunohistochemistry. Human NRG1 immunohistochemistry was performed by Department of Pathology at MSKCC using the anti-NRG1 antibody (Cell Signaling Technology #2573) at a 1:200 dilution, on a bond III automated immunestainer (Leica Microsystems, IL, USA). Formalin-fixed paraffin-embedded (FFPE) tissue sections were de-paraffinized and endogenous peroxidase was inactivated. Antigen retrieval was performed using the Bond Epitope Retrieval Solution 1 (ER1) at 99–100°C for 60 minutes (Leica Microsystems). Sections were then incubated sequentially with the primary antibody overnight, post-primary for 15 minutes and polymer for 25 minutes, followed by a 10-minute colorimetric development with diaminobenzidine (DAB) (Bond Polymer Refine Detection; Leica Microsystems). FFPE material from CHL-1 human melanoma cell line xenografts with known levels of NRG1 were used as positive controls. FFPE materials from a SKBR3 human breast cancer cell line that do not express NRG1 were used as negative controls. IHC staining result was evaluated by a pathologist with experience in genitourinary pathology (A.G.). NRG1 expression was considered positive when there was cytoplasmic membranous or granular staining in the tumor or stromal cells. NRG1 staining intensity was scored following a three-tiered system (negative= 0, weak=1, and strong=2). The immunohistochemistry detection of anti-human α-SMA and vimentin antibody was performed at the Molecular Cytology Core Facility at Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems). Tissue microarrays were purchased from US Biomax (#PR243d and #PR481). The tissue sections were blocked first for 30 min in MOM Blocking reagent (Vector Labs; #: MKB-2213) in PBS. A mouse α-SMA antibody (Sigma #A5228) was used in a 1 μg/mL concentration and incubated for 3 hours and followed by 30 minutes incubation with biotinylated anti-mouse secondary (M.O.M. Kit, Vector Labs, #BMK-2202), in 1:200 dilution. The Blocker D, Streptavidin- HRP and DAB detection kit (Ventana Medical Systems) were used according to the manufacturer’s instructions. For vimentin; the tissue sections were blocked first for 30 min in mouse IgG Blocking Reagent (Vector Labs; #MKB-2213) in PBS. The primary antibody incubation (mouse vimentin anti-human antibody (Vector Lab #VP-V684, concentration 0.1 μg/mL) was done for 3.5 hours and was followed by a 52-minute incubation of biotinylated mouse Secondary (M.O.M. Kit, Vector Labs, #BMK-2202), in 1:200 dilution. The Blocker D, Streptavidin-HRP and DAB detection kit (Ventana Medical Systems) were used according to the manufacturer’s instructions.
CRISPR-Cas9 Gene Targeting
Ten pairs of guides against the EGF-like domain of mouse NRG1 (exon2 or exon3) were designed using the guide design tool found here: (https://zlab.bio/guide-design-resources/) with the input of mouse Nrg1 EGF-like domain sequence:
AAGTGTGCGGAGAAGGAGAAAACTTTCTGTGTGAATGGAGGCGAGTGCTTCATGGTGAAGGACCTGTCAAACCCCTCAAGATACTTGTGCAAGTGCCCAAATGAGTTTACTGGTGATCGTTGCCAAAACTAC.
After CRISPR-deletion efficiency validation, we chose 4 pairs for functional experiments. Guide sequences were cloned into the Lentiviral CRISPR/Cas9 vectors that were previously described (Mu et al., 2017; Zhang et al., 2020). All CRISPR-guide sequences and vector information were listed in the Key Resources Table.
Retroviral Transduction
Lentiviral or retroviral transduction of cells for gRNA was performed as previously described (Mu et al., 2017). Cells were selected with 1 μg/mL puromycin for 5 days or with 10 μg/mL blasticidin for 5 days. To generate Nrg1-KO 22Pc-CAF, transduced cells were first selected with blasticidin for Cas9-expression and then sorted by flow cytometry to enrich for the mRFP-positive population carrying the gRNA against mouse Nrg1.
Western Blot
Cell lysates were collected using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific # PI78501), in the presence of 1% protease inhibitor cocktail set (Calbiochem/EMD #539134) and 1% phosphatase inhibitor cocktail set (Millipore #524636). For tumor tissues, T-PER Tissue Protein Extraction Reagent (Thermo Fisher Scientific #78501) was used. Protein concentration was measured with Pierce™ BCA Protein Assay Kit (ThermoFisher #23225) following manufacturer’s instructions. Three volumes of proteins were mixed with 4x NuPAGE LDS Sample Buffer (Thermo Fisher Scientific #NP0008) and boiled at 95°C for 5 minutes. Boiled protein samples were stored at −80°C until usage. Proteins were run on pre-cast gels (Invitrogen). Full-range Rainbow protein marker was used (Fisher Scientific #RPN800E). Gels were run using 1x MOPS running buffer (Teknova #M1088) at 150 volts. Gels were transferred using home-made 1x transfer buffer. Nitrocullulose membrane paper (Immobilon #IPVH00010) was used for transfer and was activated in 100% methanol (Fisher Scientific #A412–20). Transfer was performed at 4°C for 1.5 hour at 90 volts and the membrane was then blocked with5%non-fat milk for 1 hour prior to addition of primary antibody. Membrane was washed with 1X TBST (Teknova #T9511). Antibodies used were listed in detail in Key Resources Table.
Therapeutic Antibody
YW538.24.71 was acquired from Genentech through an MTA request (ID #OR-216518). AMG888 (LoRusso et al., 2013) was kindly provided by Dr. Sarat Chandarlapaty at Memorial Sloan Kettering Cancer Center.
Column Purification of Conditioned Media
50x conditioned media from 22Pc-CAF was collected as described above. Total 5ml (50x) conditioned media was first diluted into 20ml with buffer A (20mM Hepes, pH 7.5, 15mM NaCl) as Input and injected into HiTrap Q HP anion exchange chromatography column (GE Healthcare #17115401). 500μl of Input was saved for future analysis. During sample loading, 5ml of flow through (FT) was collected and saved for further analysis. After samples were loaded on the column, the first elution was performed with5%of buffer B (20mM Hepes, pH 7.5, 1M NaCl) for 5ml to remove weakly bound proteins. A second elution was performed with 5ml of 30% Buffer B. 500 μl of Q30 was saved for future analysis. Concurrently, sample collection was initiated with 1ml per fraction. Then the third elution was performed with 5ml of 100% buffer B (Q100), with 1ml/fraction. 500 μl of Q100 was saved for future analysis. A total of 4.5 ml from fractions Q30 was pooled, dialyzed against Buffer A, and re-injected into a second HiTrap Q HP anion exchange column. A gradient elution from 10% to 45% buffer B was applied and 18 fractions were collected with 1 ml/fraction. Then the final elution was applied with 100% buffer B and 4 fractions were collected with 1ml/fraction. All fractions were used immediately for assays, or stored short-term at 4°C and used within 1 week. For protein analysis, 4x loading buffer was added into each fraction and samples were boiled at 95°C for 5 minutes.
Gene Expression Analysis by qRT-PCR
Total RNA from live cells or frozen tissue was extracted using PureLink™ RNA Mini Kit (Thermo Fisher Scientific #12183025) following manufacturer’s instructions. RNA was diluted into 200 ng/μL with DEPC-treated water (Thermo Fisher Scientific #AM9916). Reverse transcription was performed using the high capacity cDNA Reverse Transcriptase Kit (Fisher Scientific, #4368813), following manufacturer’s instructions. Real-time quantitative qRT-PCR analysis was performed using QuantiFast SYBR Green PCR Kit (QIAGEN #204057) and with QuantStudio 6 Flex Real-Time PCR System. Gene expression was normalized to ACTB or GAPDH. RT2 qRT-PCR Primer Assays and QuantiTect Primer Assays from QIAGEN were used to perform all gene expression analysis. Individual primers are listed in the Key Resources Table. To distinguish mouse or human gene expression from xenograft tissue, we designed human specific qRT-PCR primers for ACTB: Forward-5’-CACCAACTGGGACGACAT, Reverse-5’-ACAGCCTGGATAGCAACG and used mouse specific qRT-PCR primers for Actb (QIAGEN #PPM02945B-200). See all qRT-PCR primer sequences in Key Resources Table. All gene expression assays were repeated in at least two independent biological replicates.
Sample and Library Preparation for RNA-seq
1×106 22Pc-EP cells was plated in 6-well plate, growing in regular RPMI-1640 containing 10% FBS. Enzalutamide (500nM) or recombinant NRG1 peptide (10 ng/mL) was added the next day. After 48 hours, cells were trypsinized and collected by spinning at 500 g for 1.5 min at 4°C. Cells were then washed once with cold 1X PBS and spinned down at 500 g for 1.5 min at 4°C. After discarding supernatant, cells were lysed using 50 mL cold lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-360) and spinned down immediately at 500 g for 10 min, 4°C. RNA was extracted using PureLink™ RNA Mini Kit (Thermo Fisher Scientific #12183025) following manufacturer’s instructions. RNA was diluted into 200 ng/μL with DEPC-treated water (Thermo Fisher Scientific #AM9916). RNA-Seq libraries were prepared using the Illumina TruSeq stranded mRNA kit, with 10 cycles of PCR amplification, starting from 500 ng of total RNA, at the Genome Technology Center (GTC) at New York University Langone Medical Center. Barcoded RNA-Seq were run as paired-end read 50 nucleotides in length on the Illumina NovaSeq 6000 and Poly-A selection was performed.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods
GraphPad Prism 8.0 was used for statistical calculations. For all comparisons between two groups of independent datasets, multiple t tests were performed, p value and standard error of the mean (SEM) were reported. For all comparisons among more than two groups (>2), one-way or two-way ANOVA were performed, p values and SEM were reported; and p values were adjusted by multiple testing corrections (Bonferroni) when applicable. Results from all in vitro assays are representatives of at least two independent biological repeats. In all figures, not significant (n.s.), p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) and p < 0.0001 (****). The usage of all statistical approaches was examined by our bioinformatics collaborators. All bioinformatic analysis and comparisons are described in detail below.
Analysis of RNA-seq Data
Raw Data Processing and Quantification
For each read, the first 6 and the last nucleotides were trimmed to the point where the Phred score of an examined base fell below 20 using in-house scripts. If, after trimming, the read was shorter than 45 bp, the whole read was discarded. Trimmed reads were mapped to the human reference genome (hg38) with HISAT2 v2.1.0 indicating that reads correspond to the reverse complement of the transcripts. Alignments with quality score below 20 were excluded from further analysis. Gene counts were produced with StringTie v1.3.3b and the Python script “prepDE.py” provided in the package. StringTie was limited to reads matching the reference annotation GENCODE v27. After obtaining the matrix of read counts, differential expression analysis was conducted, and normalized counts were produced using DESeq2. P values were adjusted for multiple testing using the Benjamini-Hochberg procedure. In all following downstream analysis, p value was adjusted by multiple hypothesis testing (represented as adjusted p value) and FDR was calculated. Differential expressed gene list was generated based on adjusted p value<0.05 and FDR<0.1 (Figures 6A and 6B).
AR Signature
A list of Enz suppressed genes in 22PC-EP was used to construct an AR signature gene list (DMSO versus Enz condition, log2 fold change>2 or <−2, adjusted p value < 0.05). Top 24 ranking genes was selected. GP2 (ranking=25th, log2 fold change=1.996, adjusted p value=3.71×10−5) and FKBP5 (ranking=26th, log2 fold change=1.972, adjusted p value=2.31×10−51) were also included in this AR signature gene list. The final AR signature consists of 26 genes (Figure S6A). AR output score was calculated by the quantification of the composite expression of this 26-gene signature in each condition following method in TCGA (Cancer Genome Atlas Research Network, 2015) (Figure 6C). AR score with two other AR signature gene lists was calculated similarly (Arora et al., 2013; Hieronymus et al., 2006) (Figures S6B and S6C).
GO Term Enrichment Analysis
Gene Ontology Enrichment Pathway analysis was performed using PANTHER to determine molecular and biological functional categories that were enriched in both AR and NRG1 co-activated genes. The input gene lists were generated from the overlapping of differentially up-regulated genes in two comparisons based on adjusted p value<0.05 (FBS+Enz compared to FBS+DMSO, FBS+-NRG1 compared to FBS+DMSO) which consists of 303 genes in total. 303 genes were further filtered by log2 Fold Change>0.5. Cutoff values of FDR<0.05 was used to select top enriched pathways.
Gene Set Enrichment Analysis (GSEA)
GSEA statistical analysis was carried out with publicly available software from the Broad Institute (http://www.broadinstitute.org/gsea/index.jsp). Weighted GSEA enrichment statistic and Signal2Noise metric for ranking genes were used.
Stroma Gene Signature and Pathway Enrichment Analysis in Patients
Analysis in primary prostate cancer patient cohort was performed in cBioportal.org and Python environment using the The Cancer Genome Altas (TCGA) database (Cancer Genome Atlas Research Network, 2015; Cerami et al., 2012; Gao et al., 2013). Reactive prostate stroma signature gene list was obtained from Dakhova et al., and was used to calculate reactive stromal signature score (Figures S1A, S1B, S2H, and S2I) (Dakhova et al., 2009). High-grade prostate tumor associated stroma gene list was obtained from Tyekucheva et al., and was used to calculate high-grade tumor stroma signature score (Figures S1C, S8C, and S8D) (Tyekucheva et al., 2017). All tumor-stroma signature score was calculated using the same method (Tyekucheva et al., 2017). Specifically, we used the ssGSEA algorithm to assign an enrichment score of genes in each gene list above for each sample. We compared the ssGSEA score of high-grade tumor stroma signature and reactive stroma signature in patients stratified by histology (tumor vs normal) and Gleason score (low (<7) vs high (>=7)). Higher ssGSEA scores correspond to more joint upregulation of genes in each signature. Gene signatures were subsetted to the genes measured in the TCGA dataset. Differential gene expression analysis was first performed between patients with high reactive stroma scores and low reactive stroma scores (p value<0.05). GO-term pathway enrichment analysis was then performed with the list of differentially expressed genes (p value< 0.05, FDR<0.25). Gene set enrichment analysis was performed with GSEA version 4.0.0 from the Broad Institute at MIT. Tumor purity was calculated using ABSOLUTE method (Carter et al., 2012).
Time on Drug Treatment Analysis with NRG1 Signature
Processed 444 SU2C metastatic prostate cancer patient cohort (Abida et al., 2019). RNA-seq data and enzalutamide/abiraterone treatment data were downloaded from cBioPortal (http://www.cbioportal.org/). 128 patients of this cohort with metastatic CRPC have baseline biopsy and matched clinical data. 75 patients of this 128 sub-cohort have gene expression data captured by poly-A RNA-seq. 56 patients of this 75 sub-cohort have records of time on treatment with either enzalutamide or abiraterone. NRG1 signature gene list was obtained from Nagashima et al., and calculated using the Single Sample Gene Set Enrichment Analysis (ssGSEA) method (Figures 8 and S8) (Nagashima et al., 2007). Histogram of NRG1 signature score distribution was generated with the Seaborn package in Python. The probability of treatment duration figure was generated using the Kaplan Meier method and Log-Rank test implemented in the Lifelines package in Python. Pearson correlation analysis was conducted between time on first-ARSI (androgen receptor signaling inhibitor) and NRG1 signature score (p = 0.005). Cox Hazard Ratio analysis was performed by R Studio (Version 1.1.453).
Supplementary Material
Highlights.
CAF-derived NRG1 confers antiandrogen resistance in prostate cancer
Pharmacological blockade of the NRG1-HER3 axis induces tumor regression
NRG1 is upregulated in prostate stroma in patients after androgen deprivation therapy
High NRG1 activity and reduced sensitivity to second-generation antiandrogen therapy
Significance.
Research into how prostate cancers develop resistance to antiandrogen therapy has primarily focused on tumor intrinsic mechanisms involving the androgen receptor. Here, we show that non-malignant stromal cells can promote antiandrogen resistance in a paracrine fashion through secretion of the HER3 ligand NRG1. In patients with localized prostate cancer, stromal expression of NRG1 is increased after treatment with androgen deprivation therapy before surgical resection, potentially protecting tumor cells from castration-induced cell death. In patients with advanced disease, NRG1-mediated activation of HER3 signaling is associated with a shorter duration of response to second-generation antiandrogen therapy. Our results provide rationale for targeting a tumor survival factor produced by the microenvironment in combination with tumor-focused antiandrogen therapy to improve treatment outcomes in prostate cancer.
ACKNOWLEDGMENTS
We thank cBioportal and the TCGA research network for providing us access to genomic and transcriptomic data. We thank Elizabeth Adams Whicher for critical input in revising the manuscript. We thank Rohit Bose, Phil Iaquinta, J. Joshua Smith, and all members of the Sawyers laboratory for comments, Memorial Sloan Kettering Cancer Center core facilities, Ning Fan, Mesruh Turkekul, Afsar Barlas, Dmitry Yarilin, Sho Fujisawa, and Sharon Huang from the MSKCC Molecular Cytology Core Facility and Juan Qiu and Qing Chang from the MSKCC Antitumor Assessment Core Facility. We thank Ann Bialik who performed NRG1 IHC staining in clinical specimens. We thank Sarat Chandalarpaty and Neal Rosen who provided us the AMG888 (U3-1287) antibody. We thank Han Yuan and Xiaoyi Li who provided expertise and suggestions for bioinformatics analysis, and Samir Zaidi who helped perform alignment and gene quantification for 22Pc-EP RNA-seq experiment. This work is partially supported by National Cancer Institute (R01CA155169-04 and R01CA19387-01 to C.L.S., R01 CA166413 and R01 CA204232 to X.J., R00CA218885-04 to P.M., P30CA008748 and U54OD020355 to E.d.S.), Howard Hughes Medical Institute (DT0712 to C.L.S.), NIH/NCI/MSKCC Spore in Prostate Cancer (P50 CA092629-14 to C.L.S.), NCI/MSKCC Support Grant/Core Grant (P30CA008748-49 and P3CA008748-49-S2 to C.L.S.) and the Starr Cancer Consortium (I9-A9- 071 to C.L.S.), Department of Defense (PC170900 to P.M., W81XWH-18-1-0379 to S.P.B and J.W.R.), Cancer Prevention Research Institute (CPRIT RR170050 to P.M., RP150596 to S.B.), the Vallee Foundation, the Prostate Cancer Foundation (17YOUN12 to P.M., 17YOUN10 to W.R.K. and 18YOUNG24 to J.W.R.), Dutch Cancer Foundation (to W.R.K.), Welch Foundation (I-2005-20190330 to P.M.), the WorldQuant Foundation, The Pershing Square Sohn Cancer Research Alliance, the NIH (1R01MH117406), UTSW Deborah and W.A. Tex Moncrief, Jr., Scholar (to P.M.), UTSW Harold C. Simmons Cancer Center Pilot Award, USA (to P.M.), The molecular cytology core at MSKCC (P30 CA008748). Z.Z. is supported by the NCI Predoctoral to Postdoctoral Fellow F99/K00 Transition Award (F99CA223063).
Footnotes
DECLARATION OF INTERESTS
C.L.S. and J.W. are co-inventors of enzalutamide and apalutamide and may be entitled to royalties. C.L.S. serves on the Board of Directors of Novartis and is a co-founder of ORIC Pharmaceuticals. He is a science advisor to Agios, Beigene, Blueprint, Column Group, Foghorn, Housey Pharma, Nextech, KSQ, Petra, and PMV. W.A. reports consulting for Clovis Oncology, Janssen, MORE Health, and ORIC Pharmaceuticals, and received honoraria from CARET and travel accommodations from GlaxoSmith Kline, Clovis Oncology, and ORIC Pharmaceuticals. D.E.R. reports having consulting or advisory role (uncompensated) from Genentech/Roche, Janssen Oncology, and TRACON Pharma, and received research funding from: AstraZeneca (Inst); Celgene (Inst); Ferring (Inst); Genentech/Roche (Inst); Janssen Oncology (Inst); Medivation/Astellas/Pfizer (Inst); Millennium (Inst); Novartis (Inst); Taiho Pharmaceutical (Inst); Takeda (Inst); TRACON Pharma (Inst). W.R.K. is a coinventor on patent WO2012168930A2 filed by Koninklijke Nederlandse Akademie Van Wetenschappen that covers organoid technology.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2020.06.005.
REFERENCES
- Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al. (2019). Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. U S A 116, 11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agus DB, Sweeney CJ, Morris MJ, Mendelson DS, McNeel DG, Ahmann FR, Wang J, Derynck MK, Ng K, Lyons B, et al. (2007). Efficacy and safety of single-agent pertuzumab (rhuMAb 2C4), a human epidermal growth factor receptor dimerization inhibitor, in castration-resistant prostate cancer after progression from taxane-based therapy. J. Clin. Oncol 25, 675–681. [DOI] [PubMed] [Google Scholar]
- Ammirante M, Luo JL, Grivennikov S, Nedospasov S, and Karin M (2010). B-Cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 464, 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. (2014). AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. (2013). Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155, 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, and Sawyers CL (2013). Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2, e00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, and Moses HL (2004). TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303, 848–851. [DOI] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 32, 474–489.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, De Bernardis G, Losa M, Mirenda M, Pasquini E, et al. (2018). IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 559, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DV, Byrom D, et al. (2015). Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet 47, 320–329. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2015). The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli C, Purwin TJ, Heilman SA, Chervoneva I, McCue PA, Berger AC, Davies MA, Gershenwald JE, Krepler C, and Aplin AE (2018). ErbB3 targeting enhances the effects of MEK inhibitor in wild-type BRAF/NRAS melanoma. Cancer Res. 78, 5680–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. (2011). Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, et al. (2013). ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med 19, 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clocchiatti A, Ghosh S, Procopio MG, Mazzeo L, Bordignon P, Ostano P, Goruppi S, Bottoni G, Katarkar A, Levesque M, et al. (2018). Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation. J. Clin. Invest 128, 5531–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, Kaminker J, and Ferrara N (2009). PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 15, 21–34. [DOI] [PubMed] [Google Scholar]
- Cunha GR (1994). Role of mesenchymal-epithelial interactions in normal and abnormal development of the mammary gland and prostate. Cancer 74, 1030–1044. [DOI] [PubMed] [Google Scholar]
- Cunha GR, and Chung LW (1981). Stromal-epithelial interactions. I. Induction of prostatic phenotype in urothelium of testicular feminized (Tfm/y) mice. J. Steroid Biochem 14, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Dagvadorj A, Tan SH, Liao Z, Cavalli LR, Haddad BR, and Nevalainen MT (2008). Androgen-regulated and highly tumorigenic human prostate cancer cell line established from a transplantable primary CWR22 tumor. Clin. Cancer Res 14, 6062–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, and Ittmann M (2009). Global gene expression analysis of reactive stroma in prostate cancer. Clin. Cancer Res 15, 3979–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Bellmunt J, Attard G, Droz JP, Miller K, Flechon A, Sternberg C, Parker C, Zugmaier G, Hersberger-Gimenez V, et al. (2007). Open-label phase II study evaluating the efficacy and safety of two doses of pertuzumab in castrate chemotherapy-naive patients with hormone-refractory prostate cancer. J. Clin. Oncol 25, 257–262. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran SM, Balbin OA, Chen G, Nadal E, Kalyana-Sundaram S, Pan J, Veeneman B, Cao X, Malik R, Vats P, et al. (2014). Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat. Commun 5, 5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Somwar R, Mangatt BP, Edgren H, Desmeules P, Ruusulehto A, Smith RS, Delasos L, Vojnic M, Plodkowski AJ, et al. (2018). Response to ERBB3-directed targeted therapy in NRG1-rearranged cancers. Cancer Discov. 8, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, and Hanahan D (2010). Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 17, 135–147. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. (2008). Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med 14, 518–527. [DOI] [PubMed] [Google Scholar]
- Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS 2nd, Revelo MP, Bhowmick NA, and Hayward SW (2011). Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 71, 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Ye H, Gerrin S, Wang H, Sharma A, Chen S, Patnaik A, Sowalsky AG, Voznesensky O, Han W, et al. (2016). ErbB2 signaling increases androgen receptor expression in abiraterone-resistant prostate cancer. Clin. Cancer Res 22, 3672–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, and Hemann MT (2010). DNA damage-mediated induction of a chemoresistant niche. Cell 143, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde GV, de la Cruz C, Giltnane JM, Crocker L, Venkatanarayan A, Schaefer G, Dunlap D, Hoeck JD, Piskol R, Gnad F, et al. (2019). NRG1 is a critical regulator of differentiation in TP63-driven squamous cell carcinoma. eLife 8, 10.7554/eLife.46551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde GV, de la Cruz CC, Chiu C, Alag N, Schaefer G, Crocker L, Ross S, Goldenberg D, Merchant M, Tien J, et al. (2013). Blocking NRG1 and other ligand-mediated Her4 signaling enhances the magnitude and duration of the chemotherapeutic response of non-small cell lung cancer. Sci. Transl. Med 5, 171ra118. [DOI] [PubMed] [Google Scholar]
- Heining C, Horak P, Uhrig S, Codo PL, Klink B, Hutter B, Frohlich M, Bonekamp D, Richter D, Steiger K, et al. (2018). NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 8, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al. (2006). Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330. [DOI] [PubMed] [Google Scholar]
- Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT, and Day ML (1995). Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am. J. Pathol 147, 386–396. [PMC free article] [PubMed] [Google Scholar]
- Hwang CI, Choi J, Zhou Z, Flesken-Nikitin A, Tarakhovsky A, and Nikitin AY (2011). MET-dependent cancer invasion may be preprogrammed by early alterations of p53-regulated feedforward loop and triggered by stromal cell-derived HGF. Cell Cycle 10, 3834–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, et al. (2015). Stromal contribution to the colorectal cancer transcriptome. Nat. Genet 47, 312–319. [DOI] [PubMed] [Google Scholar]
- Janne PA, Yu HA, Johnson ML, Steuer CE, Vigliotti M, Iacobucci C, Chen S, Yu C, and Sellami DB (2019). Safety and preliminary antitumor activity of U3–1402: a HER3-targeted antibody drug conjugate in EGFR TKIresistant, EGFRm NSCLC. J. Clin. Oncol 37, 9010. [Google Scholar]
- Jones MR, Williamson LM, Topham JT, Lee MKC, Goytain A, Ho J, Denroche RE, Jang G, Pleasance E, Shen Y, et al. (2019). NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin. Cancer Res 25, 4674–4681. [DOI] [PubMed] [Google Scholar]
- Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, Ma PC, Nieva JJ, Spira AI, Vanderwalde AM, et al. (2019). Detection of NRG1 gene fusions in solid tumors. Clin. Cancer Res 25, 4966–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Hofree M, Choi D, Linton EL, Turkekul M, Bejnood A, Carver B, Gopalan A, Abida W, Laudone V, et al. (2020). Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 368, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, et al. (2014). Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskowski MA, Jackson RS 2nd, Banerjee J, Li X, Kang M, Iturregui JM, Franco OE, Hayward SW, and Bhowmick NA (2011). Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 71, 3459–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, Lamb DJ, Marcelli M, Belldegrun A, Witte ON, and Sawyers CL (1997). Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat. Med 3, 402–408. [DOI] [PubMed] [Google Scholar]
- Klemm F, and Joyce JA (2015). Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 25, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodack DP, Askoxylakis V, Ferraro GB, Sheng Q, Badeaux M, Goel S, Qi X, Shankaraiah R, Cao ZA, Ramjiawan RR, et al. (2017). The brain microenvironment mediates resistance in luminal breast cancer to PI3K inhibition through HER3 activation. Sci. Transl. Med 9, eaal468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwabi-Addo B, Ozen M, and Ittmann M (2004). The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 11, 709–724. [DOI] [PubMed] [Google Scholar]
- Lara PN Jr., Chee KG, Longmate J, Ruel C, Meyers FJ, Gray CR, Edwards RG, Gumerlock PH, Twardowski P, Doroshow JH, and Gandara DR (2004). Trastuzumab plus docetaxel in HER-2/neu-positive prostate carcinoma: final results from the California Cancer Consortium Screening and Phase II Trial. Cancer 100, 2125–2131. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- LoRusso P, Janne PA, Oliveira M, Rizvi N, Malburg L, Keedy V, Yee L, Copigneaux C, Hettmann T, Wu CY, et al. (2013). Phase I study of U3–1287, a fully human anti-HER3 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res 19, 3078–3087. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, and Nave KA (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, and Xiong WC (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci 9, 437–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, and Sawyers CL (2004). HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 6, 517–527. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery B, Tretiakova MS, Joshua AM, Gleave ME, Fleshner N, Bubley GJ, Mostaghel EA, Chi KN, Lin DW, Sanda M, et al. (2017). Neoadjuvant enzalutamide prior to prostatectomy. Clin. Cancer Res 23, 2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, Reuter VE, Kelly WK, Slovin SF, Kenneson K, Verbel D, Osman I, and Scher HI (2002). HER-2 profiling and targeting in prostate carcinoma. Cancer 94, 980–986. [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müllner D (2013). fastcluster: fast hierarchical, agglomerative clustering routines for R and Python. J Stat Softw 53, 1–18. [Google Scholar]
- Nagashima T, Shimodaira H, Ide K, Nakakuki T, Tani Y, Takahashi K, Yumoto N, and Hatakeyama M (2007). Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J. Biol. Chem 282, 4045–4056. [DOI] [PubMed] [Google Scholar]
- Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018). Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174, 758–769.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Vaidya A, and Gorbunova V (2010). Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp 2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Wang P, Wongvipat J, Karthaus WR, Abida W, Armenia J, Rockowitz S, Drier Y, Bernstein BE, Long HW, et al. (2017). Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. eLife 6, 10.7554/eLife.27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon Y, Alon L, Glanz S, Servais C, and Erez N (2013). Isolation of normal and cancer-associated fibroblasts from fresh tissues by fluorescence activated cell sorting (FACS). J. Vis. Exp e4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, Moustafa Z, Thomas RK, Greulich H, Schinzel A, et al. (2010). An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 17, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque MP, et al. (2016). Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 29, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar SS, Hotte SJ, Chin JL, Hudes GR, Gregg R, Trachtenberg J, Wang L, Tran-Thanh D, Pham NA, Tsao MS, et al. (2010). A multicenter phase II clinical trial of lapatinib (GW572016) in hormonally untreated advanced prostate cancer. Am. J. Clin. Oncol 33, 609–613. [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. (2012). Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, et al. (2018). CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856 e816. [DOI] [PubMed] [Google Scholar]
- Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L, and Nelson PS (2012). Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med 18, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Spisak S, Seo JH, Bell C, O’Connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szallasi Z, et al. (2018). A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 174, 422–432 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Montgomery B, Logothetis CJ, Bubley GJ, Richie JP, Dalkin BL, Sanda MG, Davis JW, Loda M, True LD, et al. (2014). Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. J. Clin. Oncol 32, 3705–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al. (2009). Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyekucheva S, Bowden M, Bango C, Giunchi F, Huang Y, Zhou C, Bondi A, Lis R, Van Hemelrijck M, Andren O, et al. (2017). Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun 8, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J, et al. (2018). Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 174, 433–447 e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, and Sawyers CL (2015). Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, Wallen EM, Crane JM, Moore DT, Grigson G, et al. (2013). A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol. Oncol 31, 82–86. [DOI] [PubMed] [Google Scholar]
- Wilson TR, Lee DY, Berry L, Shames DS, and Settleman J (2011). Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 20, 158–172. [DOI] [PubMed] [Google Scholar]
- Zhang WC, Wells JM, Chow KH, Huang H, Yuan M, Saxena T, Melnick MA, Politi K, Asara JM, Costa DB, et al. (2019). miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab 1, 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, and Massague J (2013). Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154, 1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E, Chen CC, Lee YS, Zhang Y, Wang C, et al. (2020). Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell 37, 584–598.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziada A, Barqawi A, Glode LM, Varella-Garcia M, Crighton F, Majeski S, Rosenblum M, Kane M, Chen L, and Crawford ED (2004). The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. Prostate 60, 332–337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data has been deposited in the Sequence Read Archive (SRA) with the accession numbers GSE147976 and also listed in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-NRG1 | Cell Signaling Technology | Cat# 2573; RRID: AB_1031011 |
| Rabbit monoclonal anti-EGFR | Cell Signaling Technology | Cat# 4267; RRID: AB_2246311 |
| Rabbit monoclonal anti-HER3 | Cell Signaling Technology | Cat# 4754; RRID: AB_10691324 |
| Rabbit monoclonal anti-HER2 | Cell Signaling Technology | Cat# 2165; RRID: AB_10692490 |
| Rabbit monoclonal anti-AKT | Cell Signaling Technology | Cat# 4691; RRID: AB_915783 |
| Rabbit monoclonal anti-β-actin | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| Rabbit monoclonal anti-CyclophilinB | Cell Signaling Technology | Cat# 43603; RRID: AB_2799247 |
| Rabbit monoclonal anti-Vimentin | Cell Signaling Technology | Cat# 5471; RRID: AB_10692897 |
| Rabbit monoclonal anti-PDGFRα | Cell Signaling Technology | Cat# 3164; RRID: AB_2162351 |
| Rabbit monoclonal anti-phospho-EGFR Tyr1068 | Cell Signaling Technology | Cat# 3777; RRID: AB_2096270 |
| Rabbit monoclonal anti-phospho-HER2 Tyr1221/1222 | Cell Signaling Technology | Cat# 2243; RRID: AB_490899 |
| Rabbit monoclonal anti-phospho-HER3 Tyr1289 | Cell Signaling Technology | Cat# 4791; RRID: AB_2099709 |
| Rabbit Phospho-FGF Receptor (Tyr653/654) antibody | Cell Signaling Technology | Cat# 3471; RRID: AB_331072 |
| Rabbit monoclonal anti-phospho-AKT S473 | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Rabbit monoclonal anti-phospho-AKT T308 | Cell Signaling Technology | Cat# 4056; RRID: AB_331163 |
| Rabbit Phospho-FRS2-α (Tyr436) Antibody | Cell Signaling Technology | Cat# 3861; RRID: AB_2231950 |
| Rabbit monoclonal anti-HSP90 | Cell Signaling Technology | Cat# 4877; RRID: AB_2233307 |
| Anti-alpha smooth muscle Actin antibody [E184] | Abcam | Cat# ab32575; RRID: AB_722538 |
| Anti-Fibroblast activation protein, alpha antibody | Abcam | Cat# ab28244; RRID: AB_732312 |
| PE anti-mouse H-2Kb/H-2Db Antibody | Biolegend | Cat# 114608; RRID: AB_313599 |
| Mouse Anti-Human CD326 (EpCAM) Monoclonal Antibody, FITC Conjugated Clone HEA-125, | Miltenyi Biotec | Cat# 130-080-301; RRID: AB_244192 |
| Biological Samples | ||
| Prostate cancer Neoadjuvant ADT samples | Dana Farber Cancer Institute | Gao et al., 2016 |
| Radical prostatectomy samples | MSKCC Tissue Bank IRB #06-107 and #17-472 | https://www.mskcc.org/research/ski/core-facilities/pathology |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GlutaMax Supplement | Gibco | Cat# 35050061 |
| 1M HEPES Solution | Gibco | Cat# 15630080 |
| 100mM Sodium Pyruvate | Gibco | Cat# 11360-070 |
| Penicillin-streptomycin | Sigma Aldrich | Cat# P0781-100ML |
| Puromycin | Gibco | Cat# A1113803 |
| Blasticidin | Gibco | Cat# A1113903 |
| Human Neuregulin-1 (hNRG-1) | Cell Signaling Technology | Cat# 5218 |
| Human recombinant EGF | StemCell Technology | Cat# 78006.1 |
| Human recombinant FGF1 | StemCell Technology | Cat# 78187 |
| Human recombinant FGF2 | StemCell Technology | Cat# 78003.1 |
| Trizol | Ambion | Cat# 15596018 |
| 20x NuPAGE MES SDS Buffer | Novex | Cat# NP0002 |
| 1x Bolt Transfer Buffer | Novex | Cat# BT00061 |
| Neratinib | LC Laboratories | Cat# n-6404, NRT-104 |
| Enzalutamide | Selleckchem | Cat# S1250 |
| Bicalutamide | Selleckchem | Cat# S1190 |
| AMG888 | Amgen/Daiichi Sankyro | N/A |
| YW538.24.71 | Genentech | Cat# OR-216518 |
| 100% Methanol | Thermo Fisher | Cat# A412-20 |
| Fetal Bovine Serum, charcoal stripped | Omega Scientific | Cat# FB-11 lot #761007 |
| Fetal Bovine Serum | Omega Scientific | Cat# FB-11 lot#101943 |
| TrypLE Express | Gibco | Cat# 12605-010 |
| Critical Commercial Assays | ||
| 2X PowerUpTM SYBRTM Green Master Mix | Thermo Fisher | Cat# 11766500 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| MycoAlertTM PLUS Mycoplasma Detection kit | Lonza | Cat# LT07-710 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | Cat# 11668500 |
| Qiagen MinElute PCR purification kit | Qiagen | Cat# 28004 |
| Cell Titer glo assay | Promega | Cat# G9243 |
| Deposited Data | ||
| RNA-seq | GEO | GSE147976 |
| Experimental Models: Cell Lines / Organoids | ||
| CWR22Pc | Dagvadorj et al., 2008 | N/A |
| CWR22Pc-EP (22Pc-EP) | This paper | N/A |
| CWR22Pc-CAF (22Pc-CAF) | This paper | N/A |
| VCaP | ATCC | Cat# CRL-2876, RRID: CVCL_2235 |
| 22Rv1 | ATCC | ATCC Cat# CRL-2505, RRID:CVCL_1045 |
| LAPC4 | Klein et al., 1997 | N/A |
| Patient derived CAFs | MSKCC | IRB protocol: 12-001, 12-245, 90-040, 15-331 |
| Experimental Models: Organisms/Strains | ||
| C.B-Igh-1b/IcrTac-Prkdcscid mouse | Taconic | Cat# CB17SC-M |
| Oligonucleotides | ||
| sgNT (non-targeting guide): Forward-5’- CACCGGGCCAGTGTGGCCGTTACGC Reverse-5’- AAACGCGTAACGGCCACACTGGCCC |
This paper | N/A |
| sgNrg1-Guide 1 Forward-5’- CACCGAGTATCTTGAGGGGTTTGAC Reverse-5’- AAACGTCAAACCCCTCAAGATACTC |
This paper | N/A |
| sgNrg1-Guide 2 Forward-5’- CACCGAGGCGAGTGCTTCATGGTGA Reverse-5’- AAACTCACCATGAAGCACTCGCCTC |
This paper | N/A |
| sgNrg1-Guide 3 Forward-5’- CACCGCCACATCTACATCCACGACT Reverse-5’- AAACAGTCGTGGATGTAGATGTGGC |
This paper | N/A |
| sgNrg1-Guide 4 Forward-5’- CACCGGAGATGGCTGGTCCCAGTCG, Reverse-5’- AAACCGACTGGGACCAGCCATCTCC |
This paper | N/A |
| qRTPCR primer for human NRG1 | Qiagen | Cat# PPH01151F |
| qRTPCR primer for human FKBP5 | Qiagen | Cat# PPH02277A |
| qRTPCR primer for human NKX3-1 | Qiagen | Cat# PPH02267C |
| qRTPCR primer for human PMEPA1 | Qiagen | Cat# PPH01013B |
| qRTPCR primer for human SLC45A3 | Qiagen | Cat# PPH15695A |
| qRTPCR primer for human TRPM8 | Qiagen | Cat# PPH17934F |
| qRTPCR primer for mouse Nrg1 | Qiagen | Cat# PPM57587C |
| qRTPCR primer for mouse Nrg2 | Qiagen | Cat# PPM04520G |
| qRTPCR primer for mouse Nrg3 | Qiagen | Cat# PPM03007A |
| qRTPCR primer for mouse Nrg4 | Qiagen | Cat# PPM04712B |
| qRTPCR primer for mouse specific Actb | Qiagen | Cat# PPM02945B |
| qRTPCR primer for human specific Actb Forward-5’- CACCAACTGGGACGACAT Reverse-5’- ACAGCCTGGATAGCAACG |
This paper. | N/A |
| Recombinant DNA | ||
| SGEP-Renilla | Fellmann C et al. 2013 | N/A |
| QCXIP-tdTomato | Clontech | Cat# 9136-1 |
| p-tdTomato | Clontech | Cat# 632531 |
| lentiCRISPR v2 | Addgene | Cat# 52961 |
| Software and Algorithms | ||
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| R Studio | N/A | https://www.rstudio.com/ |
| Tumor measuring system Peira TM900 | Peira bvba, Belguim | |
| HISAT (v 2.0.1) | Kim et al., 2016 | http://ccb.jhu.edu/software/hisat2/index.shtml |
| Sambamba (v0.6.6) | Tarasov et al., 2015 | http://lomereiter.github.io/sambamba/ |
| Featurecount (v1.4.6) | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| DEseq2 (v1.6.3) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| PANTHER | Mi et al., 2019 | http://www.pantherdb.org |
| Samtools (v1.3) | Li et al., 2009 | http://samtools.sourceforge.net |
| Hclust | Müllner, 2013 | http://danifold.net/fastcluster.html |
| Pheatmap | R Core Team, 2016 | https://cran.r-project.org/web/packages/pheatmap/index.html |
| Other | ||
| HiTrap Q HP anion exchange chromatography column | GE Healthcare | Cat# 17115401 |