

Abstract
NMR spectroscopy is commonly used to infer site-specific acid dissociation constants (pKa) since the chemical shift is sensitive to the protonation state. Methods that probe atoms nearest to the functional groups involved in acid/base chemistry are the most sensitive for determining the protonation state. In this work, we describe a magic-angle-spinning (MAS) solid-state NMR approach to measure chemical shifts on the side chain of the anionic residues aspartate and glutamate. This method involves a combination of double quantum spectroscopy in the indirect dimension and REDOR dephasing to provide a sensitive and resolved view of these amino acid residues that are commonly involved in enzyme catalysis and membrane protein transport. To demonstrate the applicability of the approach, we carried out measurements using a microcrystalline soluble protein (ubiquitin) and a membrane protein embedded in lipid bilayers (EmrE). Overall, the resolution available from the double quantum dimension and confidence in identification of aspartate and glutamate residues from the REDOR filter make this method the most convenient for characterizing protonation states and deriving pKa values using MAS.
Keywords: magic-angle-spinning, solid-state NMR, acid dissociation constants, microcrystalline proteins, membrane proteins, EmrE
INTRODUCTION
Charged amino acids play key roles in biological functions, including protein folding and binding interactions1,2. In addition, acid/base chemistry at charged residues are involved in enzyme catalysis such as protein degradation and proton transport across cellular membranes3–7. Since the chemical shift is a sensitive reporter of the electrostatic environment, NMR spectroscopy has been a valuable experimental approach to probe site-specific protonation states and acid dissociation constants (pKa)8–18. The most effective NMR methods for inferring charged states rely on direct measurements of nuclei at the site of acid/base chemistry. For example, solution NMR methods have been developed for determining pKa values for the anionic residues aspartate and glutamate by recording 13C chemical shifts of the side chain carboxyl carbon in the indirect dimension (i.e., HCABGCO pulse sequence19). This experiment was employed to determine asymmetric protonation sites at a conserved aspartate residue in the HIV protease dimer when bound to an asymmetric inhibitor19. Although this experiment has been used with success for small and medium sized soluble proteins, application of the method to macromolecular systems is limited due to the requirement of multiple magnetization transfers that reduce sensitivity. Furthermore, this solution NMR experiment is not applicable for solid-like samples such as microcrystalline proteins or membrane proteins in liposomes, which are ideally studied using solid-state NMR spectroscopy.
In this article, we present a magic-angle-spinning (MAS) solid-state NMR method to directly observe the carboxyl groups of aspartate and glutamate using double quantum spectroscopy20. A new pulse sequence is presented by combining double quantum spectroscopy with REDOR dephasing to sensitively distinguish aspartate and glutamate side chains from those of asparagine and glutamine. As proof of concept, we applied the method to microcrystalline ubiquitin and a mutant of the multidrug efflux pump EmrE reconstituted into lipid bilayers. This method offers improved sensitivity and resolution relative to available methods for performing pH titration experiments for deriving pKa values directly at the side chain carboxyl carbon.
MATERIALS AND METHODS
Preparation of Ubiquitin and Crystallization
Uniformly labeled 13C/15N ubiquitin protein was expressed and purified as previously published21–23. In brief, ubiquitin was expressed in BL21(DE3) E. coli bacteria in the presence of uniformly labeled 13C6 glucose and 15N ammonium chloride in minimal media (M9). Purification involved resuspending bacterial cells in 25 mM Tris buffer, pH 7.5 and 0.04 mg/mL lysozyme. Following sonication for 30 mins, the lysate was centrifuged for 20 min at a speed of 48,000 x g at 4 °C. The supernatant was dialyzed against 50 mM sodium acetate buffer (pH 4.1) overnight at 4 °C and heated at 70 °C for 10 mins. Precipitated protein was removed by centrifugation for 40 min at a speed of 70,000 x g at 4 °C. The supernatant was concentrated and purified using size exclusion chromatography with a Superdex 75 column (GE Healthcare). For preparation of the solid-state NMR samples, 25 mg/ml ubiquitin (10 mg total) in 20 mM sodium citrate at pH 4.3 was crystallized by dropwise addition of 2-methyl-2,4-pentanediol (MPD) to a final concentration of 60%. The sample was incubated overnight at 4 °C. The microcrystalline samples were packed into a 3.2 mm MAS rotor with sample spacers to prevent dehydration.
Preparation of EmrE Samples
Protein expression and purification of the E14Q mutant of EmrE (EmrEE14Q) has been described previously3,24–26. In brief, uniformly labeled 13C/15N EmrEE14Q protein was obtained by expression of BL21(DE3) E. coli bacteria in the presence of uniformly labeled 13C6 glucose and 15N ammonium chloride in minimal media (M9). EmrEE14Q was expressed as a fusion construct with maltose binding protein and purified using amylose affinity chromatography and size exclusion chromatography in n-dodecyl-β-D-maltopyranoside (DDM, Anatrace). Purified EmrEE14Q was reconstituted in 1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine (O-14:0-PC) (Avanti Polar Lipids) by removing DDM detergent using Bio-Beads SM-2 resin (Bio-Rad). Proteoliposomes were pelleted by ultra-centrifugation for 12 hours at 436,000 x g using a TLA-100 rotor (Beckman-Coulter) and packed into a 3.2 mm MAS rotor using sample spacers to prevent dehydration. Proteoliposomes were in 150 mM sodium phosphate and 20 mM sodium chloride and were buffer exchanged to give a range of pH values from 1.7 to 11.0.
Solid State NMR Spectroscopy
All NMR experiments were carried out using an Agilent DD2 NMR spectrometer operating at a 1H frequency of 600 MHz (14.1 T) using a 3.2 mm triple resonance MAS probe manufactured by Black Fox, LLC. The sample temperature was set to 5 °C and −5 °C for ubiquitin and EmrEE14Q, respectively. The MAS rate was 8333 ± 5 Hz. The 1H-13C cross polarization time was set to 0.5 msec (ubiquitin) or 1.45 msec (EmrEE14Q) using a tangent ramp27 on 1H with field strengths of ω/2π = ~66.7 kHz (1H, middle of pulse) and ω/2π = 50 kHz (13C). For REDOR dephasing28, 15N inversion was achieved by composite 90°-180°-90° pulses29,30,31 using a 5 μsec 90° pulse. For double quantum experiments, an SPC-5 pulse train of 0.48 msec was utilized for each of the conversion and reconversion steps. During the SPC-5 element20, continuous wave 1H decoupling was applied at a strength of ω/2π = 100 kHz. All other 1H decoupling periods utilized SPINAL-6432 at ω/2π = 100 kHz. The indirect dimension spectral width was 8.333 kHz for the double quantum indirect dimension and 100 kHz for the direct 13C dimension. The 13C offset was 99.81 ppm and the 15N offset was 120.03 ppm. Note that the relevant aspartate and glutamate peaks in the double quantum dimension were aliased and subsequently corrected using the circular shift function in NMRPipe33. Spectral referencing in the 13C dimension was carried out by setting the CH2 resonance of adamantane to 40.48 ppm34.
RESULTS AND DISCUSSION
Statistical Distribution of Anionic Amino Acid Side Chains
Single quantum 2D spectroscopy such as 13C/13C correlations can be used to distinguish aspartate and glutamate side chain carboxyl groups in the β1 immunoglobulin binding domain of protein G35. We wondered whether an alternative approach to single quantum correlations, through the use of double quantum spectroscopy, would provide improved resolution to probe functionally important anionic residues. The use of double quantum spectroscopy typically involves evolution of double quantum coherences in indirect dimensions, which are correlated with single quantum coherences in the direct dimension (i.e., DQSQ experiment). Since the double quantum shifts appear at the additive chemical shift frequencies of the two nuclei in the double quantum coherence, we envisioned that the indirect dimension of the DQSQ experiment would nicely resolve side chain carboxyl groups of aspartate and glutamate residues from backbone carbonyl sites that often share overlapping 13C resonances. To test this theory, we analyzed statistical distributions of chemical shifts36,37 and plotted these in the form of a DQSQ spectrum (Figure 1). This plot shows that the DQSQ experiment provides frequency isolation for the side chain amino acids of aspartate, glutamate, asparagine, and glutamine relative to the backbone carbonyl sites. Namely, the indirect double quantum dimension evolves under the additive chemical shifts of for aspartate and asparagine and for glutamate and glutamine, which separates these resonances from the backbone sites corresponding to (Figure 1). More significantly, the evolution of the additive chemical shifts in the indirect dimension increases the effective resolution between protonated and deprotonated side chain chemical shifts for aspartate and glutamate. Specifically, the average chemical shift differences between charged and uncharged sites in the two dimensions of the DQSQ experiment are 6.2 and 3.2 ppm for aspartate and 7.6 and 4.1 ppm for glutamate. This can be compared to chemical shift differences of 3.0 and 3.2 ppm for aspartate and 3.5 and 4.1 ppm for glutamate in a single quantum based 2D experiment. From this analysis, DQSQ spectroscopy is anticipated to improve the spectral dispersion of aspartate and glutamate side chains to provide insight into protonation states of proteins during a pH titration to determine pKa values.
Figure 1.
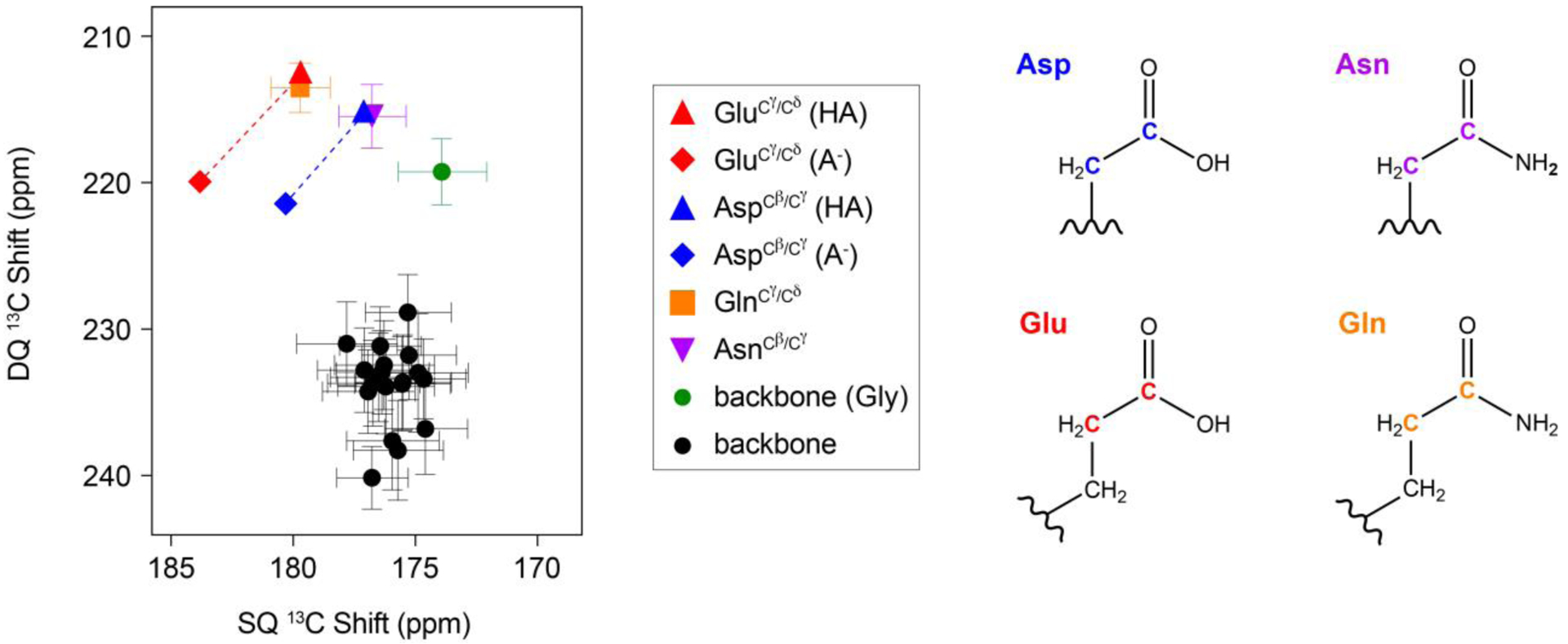
Projection of chemical shifts onto a DQSQ spectrum using the chemical shift statistics from the BMRB database37 and Platzer et al.36. Errors reflect the standard deviation from the chemical shift assignments of these residues. Chemical shifts of aspartate and glutamate side chains were taken from Platzer et al.36 because both protonated and deprotonated chemical shifts were reported. The structures of the side chains of aspartate, asparagine, glutamate, and glutamine are color coded with the DQSQ plot.
Combination of REDOR Filter with DQSQ Spectroscopy to Distinguish Aspartate and Glutamate from Asparagine and Glutamine
While the 2D DQSQ experiment is expected to provide enhanced resolution for observing side chain carboxyl sites between protonated and deprotonated states, the statistical distribution of chemical shifts in Figure 1 does not inherently distinguish aspartate and glutamate from asparagine and glutamine side chains for all protonation states. However, these residue types differ in bonding at the terminal carbon: –NH2 in asparagine and glutamine or –OH in aspartic acid and glutamic acid (Figure 1). Thus, the application of 15N REDOR pulses can be used to distinguish asparagine and glutamine from aspartic acid and glutamic acid through a loss in signal intensity. A suitable pulse sequence that combines DQSQ and REDOR is shown in Figure 2. This sequence, referred to as DQSQ-REDOR, utilizes SPC-520 for double quantum conversion and reconversion back to the single quantum coherence for detection followed by REDOR dephasing28 on the 15N channel. The only requisite for the DQSQ-REDOR experiments is a uniformly 13C/15N labeled protein, which is the same sample as those used to obtain resonance assignments. Therefore, this experiment has the potential for enhancing resolution for aspartate and glutamate residues while selectively reducing overlapping resonances with the goal of studying biologically important mechanisms. Note that filtering approaches have been previously used to reduce spectral complexity both at backbone38,39 and side chain sites9. It should also be noted that the REDOR period was also placed before the DQSQ element; however, this produced poorer dephasing relative to DQSQ-REDOR.
Figure 2.
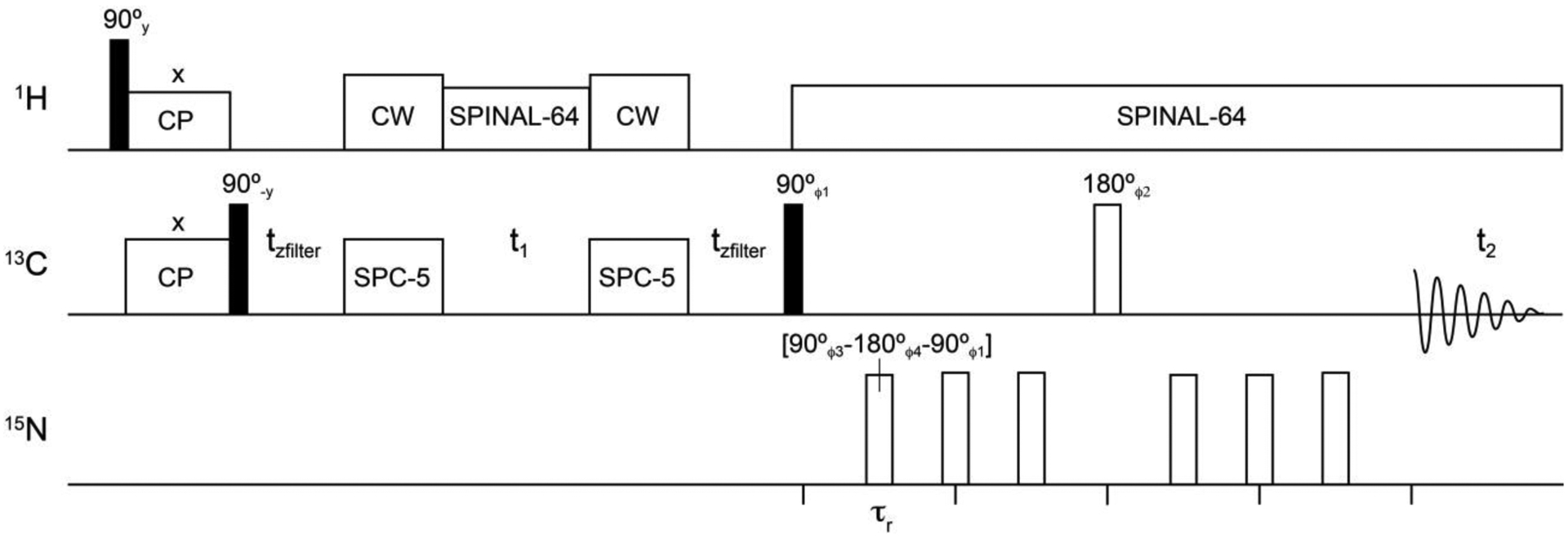
DQSQ-REDOR pulse sequence. All 15N REDOR dephasing pulses were applied as composition pulses (90°−180°−90°) with the indicated phases. A 4-step phase cycle was implemented as follows: ϕ1 = [x, y, -x, -y], ϕ2 = [y, -x, -y, x], ϕ3 = [y, y, -y, -y], ϕ4 = [x, x, -x, -x], and ϕreceiver = [x, -y, -x, y]. τr is the rotor period (120 μsec) and tzfilter is the z-filter time (120 μsec).
Application of DQSQ-REDOR to Microcrystalline Ubiquitin
To test the effectiveness of DQSQ and DQSQ-REDOR to resolve side chain anionic residues, we applied these experiments to uniformly 13C/15N microcrystalline ubiquitin as a model protein where the primary sequence contained several asparagine, aspartate, glutamine, and glutamate residues (Figure 3A). The DQSQ spectrum of ubiquitin displayed several isolated peaks corresponding to these residue side chains that were predicted from the statistical distribution of chemical shifts (Figure 3B). Namely, residues Glu18, Asn25, Gln31, Asp32, Asp39, Gln41, Asp58, and Gln62 in ubiquitin were identified from previously reported assignments21,40. Note that five other glutamate residues in the primary sequence were not observed at 5 °C and with the employed DQ mixing time, which is consistent with a prior study40 and likely stems from the disordered nature of these side chains. For the residues that were observed, the DQSQ spectrum showed dispersed side chains of asparagine, aspartate, glutamine, and glutamate and the glycine backbone from other backbone carbonyls within ubiquitin. Based on our experimental result and the statistical distribution of chemical shifts in Figure 1, we anticipate the DQSQ experiment will be a valuable method in mechanistic studies for probing side chains of essential asparagine, aspartate, glutamine, and glutamate residues.
Figure 3.
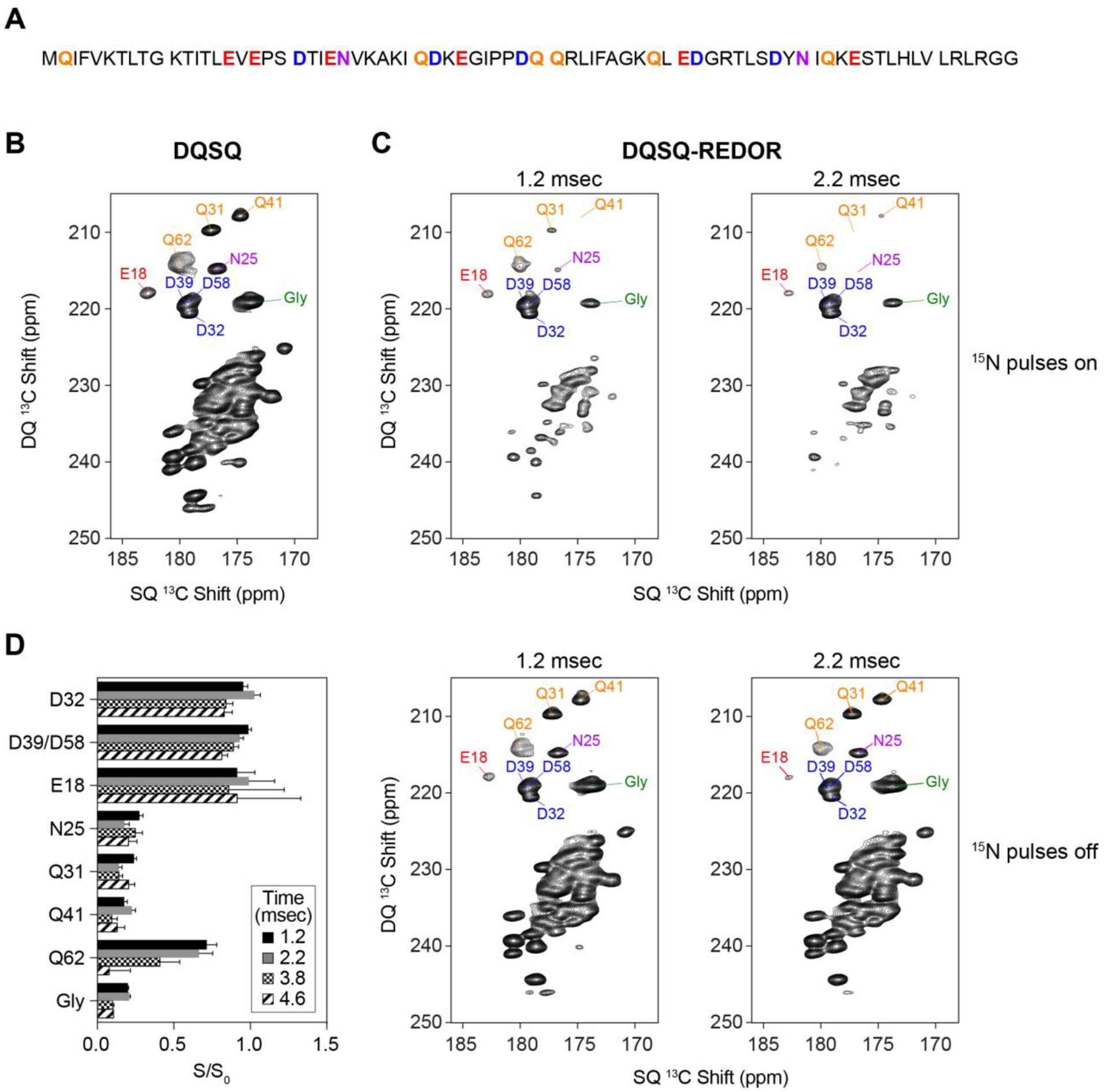
DQSQ and DQSQ-REDOR applied to microcrystalline ubiquitin. (A) Primary sequence of ubiquitin with aspartate, asparagine, glutamate, and glutamine are color coded with residue types as in Figure 1. (B) DQSQ and (C) DQSQ-REDOR with dephasing periods of 1.2 msec and 2.2 msec. Top spectra in panel C were collected with application of 15N dephasing pulses, while the bottom spectra were collected with the same time period without 15N REDOR pulses. All peak labels are color coded with residue types as in Figure 1. (D) Intensity ratios at different dephasing times of select side chains (Asn, Asp, Gln, Glu) and backbone peaks (Gly) in the presence of 15N pulses (S) divided by those in the absence of 15N pulses (S0).
Next, we explored whether the DQSQ-REDOR pulse sequence (Figure 2) would be a suitable technique to distinguish side chain carboxyls within aspartate and glutamate from side chain carbonyls within asparagine and glutamine residues. Since one-bond distances for Cγ−Nδ2 (asparagine) and Cδ−Nε2 (glutamine) are 1.3 Å, a REDOR dephasing period of ~1.2 msec would lead to efficient dephasing for an ordered site. Using this dephasing time, we collected two DQSQ-REDOR datasets corresponding to experiments with and without 15N REDOR 180° pulses (Figure 3C, top and bottom). These spectra showed significant reduction of backbone signal intensity when REDOR pulses were applied (S) compared to when REDOR pulses were turned off (S0). Quantification of these spectra showed that most asparagine and glutamine side chains displayed intensity ratios (S/S0) less than ~0.25 (Figure 3D), which validated the method. However, the S/S0 values were not effectively dephased for all non-carboxyl sites at a dephasing period of 1.2 msec (e.g., Gln62), which could stem from mobility of the side chain leading to a reduced effective dipolar coupling or imperfect REDOR dephasing. To identify mobile sites, we also employed longer dephasing time periods of 2.2, 3.8 and 4.6 msec. These data showed that progressively longer REDOR dephasing times reduced S/S0 for Gln62 (Figure 3C, 3D), while maintaining S/S0 values for aspartate and glutamate side chains greater than ~0.8. The observation of Gln62 as a mobile side chain is potentially required for its function in polyubiquination41. We also observed that several side chain asparagine and glutamine residues (e.g., Asn25, Gln31, Gln41) maintained S/S0 values less than ~0.25, which means these residues were efficiently dephased at the shortest and longest dephasing times. Thus, for asparagine and glutamine sites that are well ordered, the DQSQ-REDOR method can be used to distinguish anionic residues, which increases the signal to noise relative to experiments with longer dephasing times. Notably, a previous method that combined REDOR dephasing and DARR spectroscopy to observe side chain aspartate and glutamate residues required a dephasing period of 3.8 msec to completely dephase asparagine and glutamine side chains9. Thus, shorter REDOR dephasing times available with DQSQ-REDOR will achieve higher signal to noise ratios for confidently identifying aspartate and glutamate residues. However, we emphasize that highly mobile residues that are not detected in cross-polarization based techniques also cannot be studied using the DQSQ-REDOR method.
Lastly, we compared our DQSQ-REDOR method with the 2D 13C/13C DARR experiment42 for ubiquitin (Figure 4). The DARR spectrum was obtained by using a 20 msec mixing time while the DQSQ-REDOR spectrum was carried out with 1.2 msec of dephasing (same spectrum as in Figure 3C). As expected, the sensitivity of DARR was higher than that of the DQSQ-REDOR method since DARR does not employ double quantum or REDOR filters. The two resolvable signals in the DARR spectrum showed 1.4-fold and 2.9-fold better sensitivity for Asp32 and Glu18, respectively, for the same total time to collect each dataset. However, as seen in Figure 4, the DARR spectrum was quite congested and most of the aspartate residues were not clearly resolvable. Therefore, while a DARR spectrum provided for better sensitivity, the major advantage of DQSQ-REDOR is the reduced spectral overlap that enabled identification of aspartate and glutamate residues in a straightforward manner.
Figure 4.
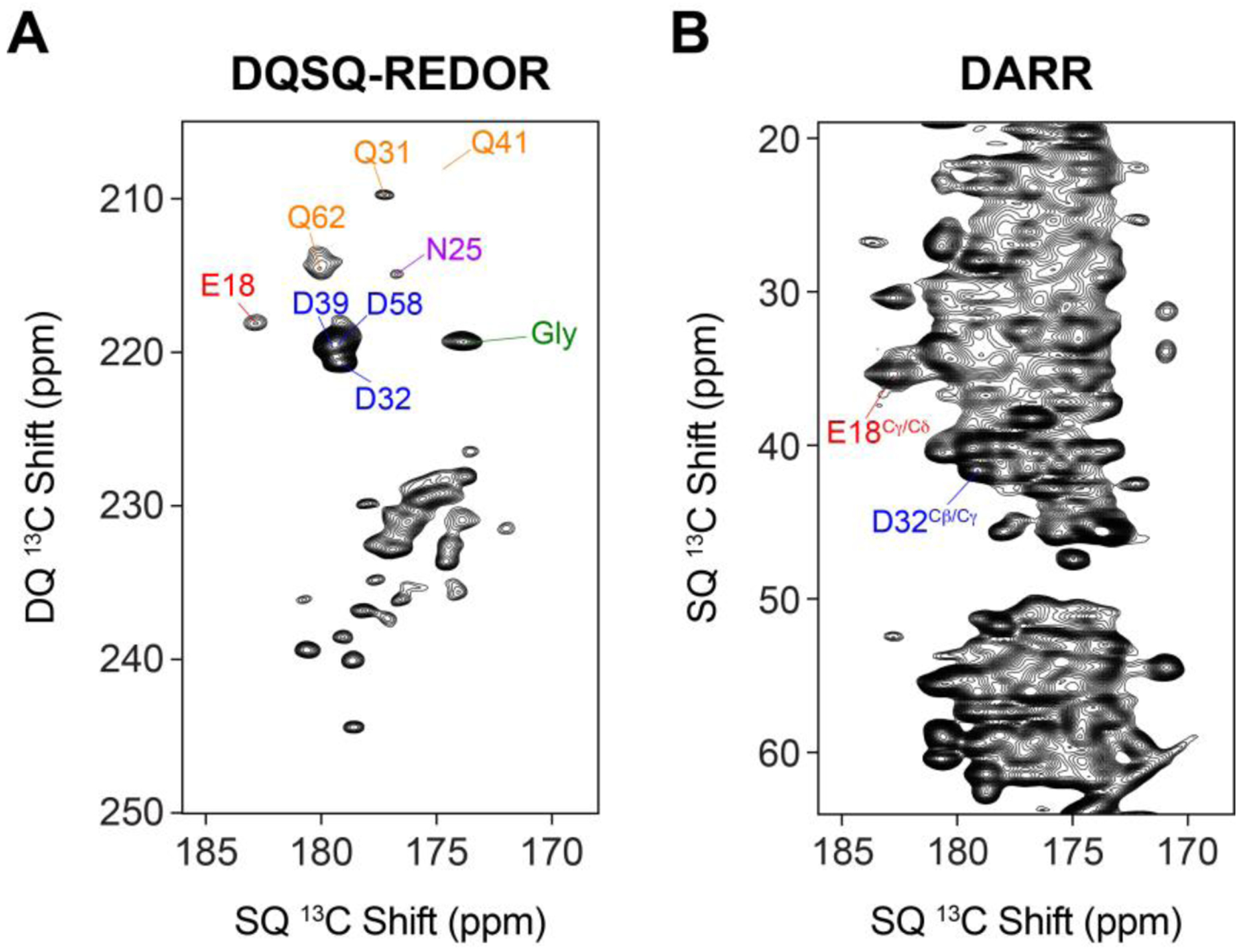
Comparison of (A) DQSQ-REDOR with 1.2 msec 15N dephasing and (B) DARR 13C/13C correlation with 20 msec mixing time on a microcrystalline sample of uniformly 13C, 15N ubiquitin. Note that the total time to collect the spectra were the same.
Application of DQSQ Method to a Membrane Protein in Lipid Bilayers
pH titrations are commonly employed to derive pKa values to discern roles of carboxyl residues in biological function. To demonstrate the value of DQSQ-REDOR for determining a pKa value using MAS solid-state NMR, we applied these experiments to the E14Q mutant of the membrane protein EmrE (EmrEE14Q) in lipid bilayers. This mutant was selected to serve as a simpler model system than wild-type EmrE due to the role of pH dependent changes in structure and dynamics stemming from acid/base chemistry at Glu143,4,25. Similar to the ubiquitin sample, EmrEE14Q contains asparagine and glutamine (Asn2, Gln14, Gln81, Asn102) and aspartate and glutamate residues (Glu25 and Asp84) that serve to test the robustness of the method for distinguishing these residue types within a membrane protein (Figure 5A).
Figure 5.
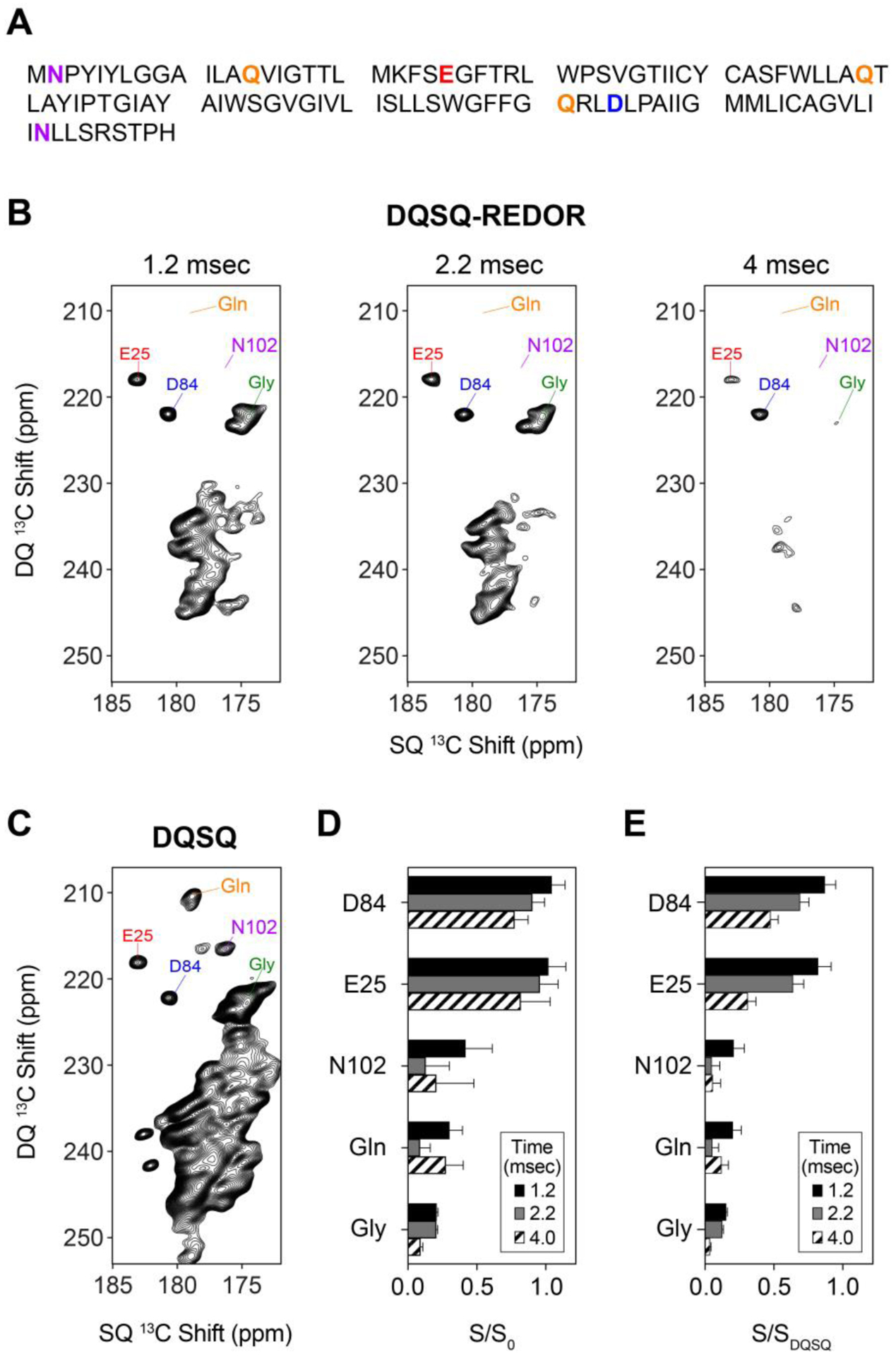
(A) Primary sequence of EmrEE14Q with aspartate, asparagine, glutamate, and glutamine are color coded with residue types as in Figure 1. (B) DQSQ-REDOR and (C) DQSQ spectra of uniformly labeled 13C/15N EmrEE14Q in O-14:0-PC liposomes at a pH value of 5.0. The REDOR dephasing times in panel B are indicated on top of each spectrum. (D) Intensity ratios (S/S0) of select side chains (Asn, Asp, Gln, Glu) and backbone peaks (Gly) in the presence of 15N pulses (S) divided by those in the absence of 15N pulses (S0). (E) Intensity ratios (S/SDQSQ) of select side chains (Asn, Asp, Gln, Glu) and backbone peaks (Gly) in the presence of 15N REDOR pulses (S) divided by those in the DQSQ spectrum (SDQSQ).
Uniformly 13C/15N EmrEE14Q was reconstituted in ether-14:0-PC (O-14:0-PC) to ensure lipid stability during the pH titration24,43. Proteoliposomes were initially prepared at a pH value of 5.0. DQSQ-REDOR experiments were collected using dephasing times of 1.2, 2.2 and 4.0 msec in the presence an absence of 15N REDOR pulses. At a dephasing time of 1.2 msec with REDOR pulses on, several peaks including the side chains of glutamine and the backbone of glycine residues were found to be significantly attenuated in intensity (Figure 5B). In fact, many of the asparagine and glutamine signals gave very low signal intensities relative to a reference DQSQ spectrum (Figure 5C). The relative intensities remained low in intensity with longer dephasing times of 2.2 and 4.0 msec, which is observed in the quantification of intensity ratios (S/S0) (Figure 5D). To the contrary, S/S0 values for Glu25 and Asp84 remained greater than ~0.75 at all mixing times (Figure 5D). These results indicate effective dephasing at a short time period of 1.2 msec and demonstrates the effectiveness of the method for distinguishing side chain carboxyl sites of aspartate and glutamate from side chain carbonyl sites of asparagine and glutamine. Lastly, note that detection of aspartate and glutamate signals using DQSQ-REDOR with a 1.2 msec dephasing time retained excellent sensitivity relative to DQSQ, as evidenced by the intensity ratios of ~0.8 for Glu25 and Asp84 when comparing signal intensities of DQSQ-REDOR and the DQSQ experiment (S/SDQSQ) (Figure 5E).
We next performed a pH titration on EmrEE14Q as proof of principle of the method to detect pH dependent chemical shift changes. For these measurements, we used DQSQ-REDOR at a dephasing time of 2.2 msec for each pH value within the range 1.7 to 11.0 (Figure 6). However, we note that it is possible to perform the titration experiments using only DQSQ once an aspartate or glutamate side chain of interest has been identified and assigned using DQSQ-REDOR and triple resonance experiments, respectively. The pH titration showed that Glu25 experienced chemical shift perturbations in the fast exchange regime such that the peak moved by ~4.5 ppm in the direct dimension and ~8.5 ppm in the indirect dimension. No other peaks in the spectrum had similarly large chemical shift perturbations over this pH range. The observed pH dependent chemical shifts of Glu25 were in agreement with previously reported chemical shifts of model tripeptides containing a glutamate residue where the deprotonated chemical shifts are downfield (Cδ = 183.8 ppm; Cγ = 36.1) relative to the protonated side chain (Cδ = 179.7 ppm; Cγ = 32.7)36. Using the chemical shift changes in both the single quantum and double quantum dimensions, we fitted an apparent pKa of Glu25 to be 4.4 ± 0.3 using a modified Henderson-Hasselbalch equation3. Note that we did not observe changes in chemical shifts for Asp84 over the pH range of 3.0 to 11.0, implying this residue has a pKa value below ~3.0 and would be deprotonated under all physiologically relevant pH conditions.
Figure 6.
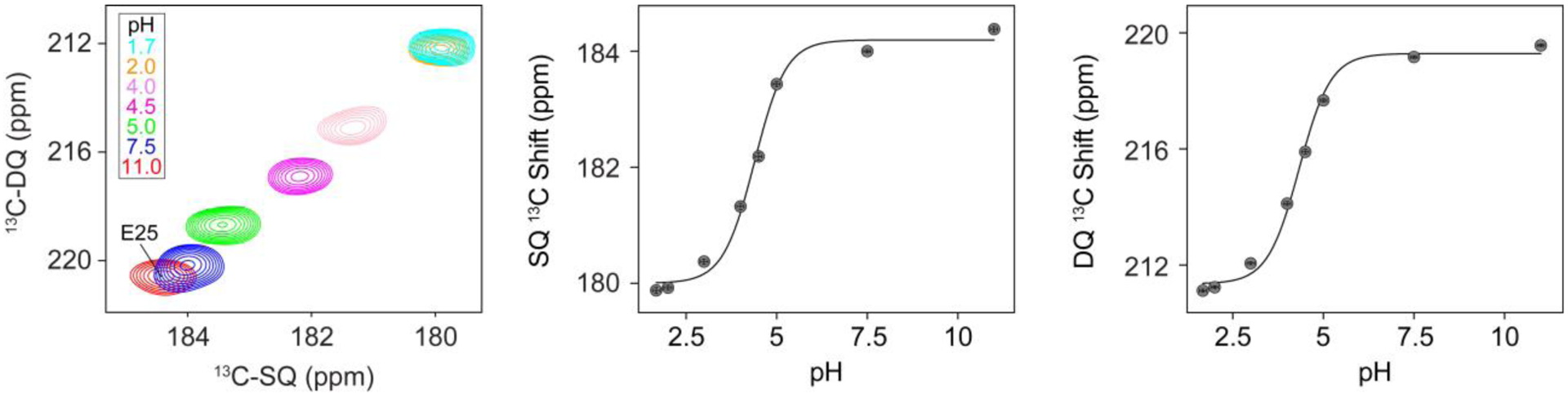
Spectra and subsequent pKa fitting of pH-dependent chemical shift changes for Glu25 within EmrEE14Q. Left: DQSQ spectra with 2.2 msec REDOR dephasing for Glu25 over the indicated pH values. Center and right: experimental data points of single quantum chemical shifts in the direct dimension (middle) and double quantum chemical shifts in the indirect dimension (right). The continuous line is the best global fit using a modified Henderson-Hasselbalch equation3 that yielded an apparent pKa value of 4.4 ± 0.3 for Glu25.
In summary, DQSQ offers excellent separation of the side chains of asparagine, aspartate, glutamine, and glutamate residues relative to backbone amide sites. Inclusion of a REDOR period into this sequence enabled aspartate and glutamate side chains to be easily distinguished from asparagine and glutamine side chain residues that often overlap within the DQSQ spectrum. The DQSQ-REDOR offers excellent sensitivity since it can be applied with a short REDOR dephasing time of ~1.2 msec that is sufficient for identifying anionic side chains through the comparison of signal intensities in the presence and absence of REDOR dephasing pulses. Thus, DQSQ-REDOR is anticipated to be a valuable method for characterizing catalytically important anionic residues with MAS solid-state NMR spectroscopy.
CONCLUSION
We proposed and validated a DQSQ-based pulse sequence for distinguishing asparagine, aspartate, glutamine, and glutamate side chains in proteins. Inclusion of a REDOR dephasing period into this sequence (i.e., DQSQ-REDOR) was effective at distinguishing aspartate and glutamate side chains from asparagine and glutamine. The DQSQ-REDOR method has an intrinsic resolution advantage due to the double quantum chemical shift indirect dimension that better resolves protonated side chains of aspartate and glutamate from their deprotonated states. Since 13C chemical shifts of carboxyl sites are sensitive to the protonation state, this method can be employed in microcrystalline proteins and membrane proteins to infer charged states of anionic side chains as well as in pH titrations to quantify pKa values.
ACKNOWLEDGEMENT
The NMR methodology was supported by NSF grant MCB 1902449 (to N.J.T.). Application to EmrE was funded by NIH grant R01 AI108889 (to N.J.T.). A.S.H. was supported from a Dean’s Dissertation Fellowship from New York University.
REFERENCES:
- 1.Tollinger M, Crowhurst KA, Kay LE & Forman-Kay JD Site-specific contributions to the pH dependence of protein stability. Proc Natl Acad Sci U S A 100, 4545–50 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wells JA, Powers DB, Bott RR, Graycar TP & Estell DA Designing substrate specificity by protein engineering of electrostatic interactions. Proc Natl Acad Sci U S A 84, 1219–23 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gayen A, Leninger M & Traaseth NJ Protonation of a glutamate residue modulates the dynamics of the drug transporter EmrE. Nat Chem Biol 12, 141–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrison EA, Robinson AE, Liu Y & Henzler-Wildman KA Asymmetric protonation of EmrE. J Gen Physiol 146, 445–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masureel M et al. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat Chem Biol 10, 149–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fluman N, Ryan CM, Whitelegge JP & Bibi E Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol Cell 47, 777–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fluman N & Bibi E Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim Biophys Acta 1794, 738–47 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Jehle S et al. Spectral editing: selection of methyl groups in multidimensional solid-state magic-angle spinning NMR. J Biomol NMR 36, 169–77 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Schmidt-Rohr K, Fritzsching KJ, Liao SY & Hong M Spectral editing of two-dimensional magic-angle-spinning solid-state NMR spectra for protein resonance assignment and structure determination. J Biomol NMR 54, 343–53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reggie L, Lopez JJ, Collinson I, Glaubitz C & Lorch M Dynamic nuclear polarization-enhanced solid-state NMR of a 13C-labeled signal peptide bound to lipid-reconstituted Sec translocon. J Am Chem Soc 133, 19084–6 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Nielsen JE Chapter 5 - Analyzing Protein NMR pH-Titration Curves in Annual Reports in Computational Chemistry, Vol. 4 (eds. Wheeler RA& Spellmeyer DC) 89–106 (Elsevier, 2008). [Google Scholar]
- 12.Oregioni A, Stieglitz B, Kelly G, Rittinger K & Frenkiel T Determination of the pKa of the N-terminal amino group of ubiquitin by NMR. Sci Rep 7, 43748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pielak RM & Chou JJ Influenza M2 proton channels. Biochimica et Biophysica Acta (BBA) - Biomembranes 1808, 522–529 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J et al. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci U S A 103, 6865–70 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu F, Schmidt-Rohr K & Hong M NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J Am Chem Soc 134, 3703–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartik K, Redfield C & Dobson CM Measurement of the individual pKa values of acidic residues of hen and turkey lysozymes by two-dimensional 1H NMR. Biophys J 66, 1180–4 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIntosh LP et al. The pKa of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C-NMR study of bacillus circulans xylanase. Biochemistry 35, 9958–66 (1996). [DOI] [PubMed] [Google Scholar]
- 18.Oda Y et al. Individual ionization constants of all the carboxyl groups in ribonuclease HI from Escherichia coli determined by NMR. Biochemistry 33, 5275–84 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Wang YX et al. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry 35, 9945–50 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Hohwy M, Rienstra CM, Jaroniec CP & Griffin RG Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. The Journal of Chemical Physics 110, 7983–7992 (1999). [Google Scholar]
- 21.Zech SG, Wand AJ & McDermott AE Protein structure determination by high-resolution solid-state NMR spectroscopy: application to microcrystalline ubiquitin. J Am Chem Soc 127, 8618–26 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Banigan JR & Traaseth NJ Utilizing afterglow magnetization from cross-polarization magic-angle-spinning solid-state NMR spectroscopy to obtain simultaneous heteronuclear multidimensional spectra. J Phys Chem B 116, 7138–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazar GA, Desjarlais JR & Handel TM De novo design of the hydrophobic core of ubiquitin. Protein Sci 6, 1167–78 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banigan JR, Leninger M, Her AS & Traaseth NJ Assessing Interactions Between a Polytopic Membrane Protein and Lipid Bilayers Using Differential Scanning Calorimetry and Solid-State NMR. J Phys Chem B 122, 2314–2322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho MK, Gayen A, Banigan JR, Leninger M & Traaseth NJ Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J Am Chem Soc 136, 8072–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gayen A, Banigan JR & Traaseth NJ Ligand-induced conformational changes of the multidrug resistance transporter EmrE probed by oriented solid-state NMR spectroscopy. Angew Chem Int Ed Engl 52, 10321–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldus MG, D. G.; Hediger, S.; Meier, B. H. Efficient 15N- 13C Polarization Transfer by Adiabatic-Passage Hartmann-Hahn Cross Polarization. Journal of Magnetic Resonance 118, 140–144 (1996). [Google Scholar]
- 28.Gullion T & Schaefer J Rotational-echo double-resonance NMR. 1989. J Magn Reson 213, 413–7 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Levitt MH Short perspective on “NMR population inversion using a composite pulse” by Levitt MH and Freeman R [J. Magn. Reson. 33 (1979) 473–476]. J Magn Reson 213, 274–5 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Levitt MH & Freeman R NMR population inversion using a composite pulse. Journal of Magnetic Resonance (1969) 33, 473–476 (1979). [DOI] [PubMed] [Google Scholar]
- 31.Sinha N, Schmidt-Rohr K & Hong M Compensation for pulse imperfections in rotational-echo double-resonance NMR by composite pulses and EXORCYCLE. J Magn Reson 168, 358–65 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Fung BM, Khitrin AK & Ermolaev K An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson 142, 97–101 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–93 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Morcombe CR & Zilm KW Chemical shift referencing in MAS solid state NMR. J Magn Reson 162, 479–86 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Schmidt HL, Shah GJ, Sperling LJ & Rienstra CM NMR Determination of Protein pK(a) Values in the Solid State. J Phys Chem Lett 1, 1623–1628 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Platzer G, Okon M & McIntosh LP pH-dependent random coil (1)H, (13)C, and (15)N chemical shifts of the ionizable amino acids: a guide for protein pK a measurements. J Biomol NMR 60, 109–29 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Ulrich EL et al. BioMagResBank. Nucleic Acids Res 36, D402–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banigan JR, Gayen A & Traaseth NJ Combination of (1)(5)N reverse labeling and afterglow spectroscopy for assigning membrane protein spectra by magic-angle-spinning solid-state NMR: application to the multidrug resistance protein EmrE. J Biomol NMR 55, 391–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Traaseth NJ & Veglia G Frequency-selective heteronuclear dephasing and selective carbonyl labeling to deconvolute crowded spectra of membrane proteins by magic angle spinning NMR. J Magn Reson 211, 18–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Igumenova TI et al. Assignments of carbon NMR resonances for microcrystalline ubiquitin. J Am Chem Soc 126, 6720–7 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Suryadinata R et al. Molecular and structural insight into lysine selection on substrate and ubiquitin lysine 48 by the ubiquitin-conjugating enzyme Cdc34. Cell Cycle 12, 1732–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takegoshi K, Nakamura S & Terao T 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chemical Physics Letters 344, 631–637 (2001). [Google Scholar]
- 43.Leninger M, Sae Her A & Traaseth NJ Inducing conformational preference of the membrane protein transporter EmrE through conservative mutations. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]