

Abstract
Inflammation is a significant component of Alzheimer’s disease pathology. While neuroprotective microglia are important for containment/clearance of Amyloid plaques and maintaining neuronal survival, Alzheimer inflammatory microglia may play a detrimental role by eliciting tau pathogenesis and accelerating neurotoxicity. Regulatory T cells have been shown to suppress microglia-mediated inflammation. However, the role of regulatory T cells in ameliorating the proinflammatory immune response in Alzheimer’s disease requires further investigation. Forty-six patients with Alzheimer disease, 42 with mild cognitive impairment and 41 healthy controls were studied. The phenotypes of peripheral regulatory T cells were assessed with multicolour flow cytometry. Regulatory T cells were co-cultured with responder T cells and proliferation was determined by 3H-thymidine incorporation. In separate experiments, regulatory T cells were added to induced pluripotent stem cell-derived pro-inflammatory macrophages and changes in interleukin-6/tumour necrosis-alpha transcripts and protein levels were measured. Freshly isolated regulatory T cells were expanded ex vivo in the presence of CD3/CD28 expander beads, interleukin-2 and rapamycin to promote their suppressive function. We found that the suppressive function of regulatory T cells on responder T-cell proliferation was compromised at the Alzheimer disease stage, compared with mild cognitive impairment and healthy controls. CD25 mean fluorescence intensity in regulatory T-cell population was also reduced in Alzheimer dementia patients. Regulatory T cells did not suppress pro-inflammatory macrophages at baseline. Following ex vivo expansion, regulatory T-cell suppression of responder T-cell proliferation and pro-inflammatory macrophage activation increased in both patients and controls. Expanded regulatory T cells exerted their immunoregulatory function on pro-inflammatory macrophages through a contact-mediated mechanism. In conclusion, regulatory T-cell immunophenotype and function are compromised in Alzheimer’s disease. Following ex vivo expansion, the immunomodulatory function of regulatory T cells is enhanced even at advanced stages of Alzheimer’s disease. Restoration of regulatory T-cell function could be explored as a means to modulate the inflammatory status of Alzheimer’s disease.
Keywords: regulatory T cells, Alzheimer’s disease, inflammation, immune system, dementia
The suppressive function of regulatory T cells is compromised throughout the course of Alzheimer. Following ex vivo expansion, the immunoregulatory capacity of regulatory T cells was substantially enhanced through a cell contact-mediated mechanism. Restoration of regulatory T-cell dysfunction offers a unique therapeutic option to modulate neuroinflammation in Alzheimer’s disease.
Graphical Abstract
Graphical Abstract.

Introduction
Recent findings, including the discovery of risk genes involved in inflammation signalling, indicate that inflammation is critical for the onset and progression of Alzheimer’s disease (Marioni et al., 2018; Jansen et al., 2019). Mounting evidence, including the presence of vascular and parenchymal T cells in post-mortem brains, suggests the potential involvement of T cells in the development of neuroinflammation in Alzheimer’s disease (Giubilei et al., 2003; Zhang et al., 2003; Ferretti et al., 2016; Merlini et al., 2018). Regulatory T lymphocytes (Tregs) are the subset of T cells that are critically involved in suppressing inflammation (Henkel et al., 2013; Okeke et al., 2014; Xie et al., 2015). Tregs are not only important for maintaining immune balance in the periphery but also contribute to self-tolerance and immune privilege in the central nervous system (He and Balling, 2013; Liesz et al., 2013; Huang et al., 2020). This cell population has been recently found to be dysfunctional in several neurodegenerative disorders (Saunders et al., 2012; Beers et al., 2017).
Innate immune cell population has now been identified as key players in Alzheimer’s disease pathology. Microglia are the initial responders to neuronal release of amyloid-β. While early activation of microglia is important for containing and phagocyting amyloid plaques and for sustaining neuronal survival, chronic stimulation in combination with genetic susceptibility skews these cells towards a defective subtype (Hickman et al., 2008; Daria et al., 2017; Kang et al., 2018; Nordengen et al., 2019). This subtype is characterized by a unique expression profile consisting of up-regulation of certain inflammatory genes and down-regulation of homeostatic gene expressions (Venegas et al., 2017; Del-Aguila et al., 2019; Mathys et al., 2019; Gray et al., 2020). This ongoing inflammation may facilitate phosphorylation and truncation of tau (Lee et al., 2010; Sy et al., 2011; Maphis et al., 2015). Subsequent release of truncated, phosphorylated tau may also enhance immune cell activation and further promote the release of inflammatory mediators resulting in a self-propagating cascade of neuronal injury, glial activation, synaptic dysfunction and cell death (Khandelwal et al., 2012; Nilson et al., 2016; Rajendran and Paolicelli, 2018). Tregs have been shown to steer the differentiation of macrophages and microglia towards a neuroprotective state (Reynolds et al., 2009; Li et al., 2010; Henkel et al., 2013). In a murine model of Alzheimer’s disease, early depletion of peripheral Tregs accelerated the onset of cognitive deficits (Dansokho et al., 2016). Conversely, amplification of these cells showed neuroprotective effects and restored cognitive functions (Baek et al., 2016; Dansokho et al., 2016). As the role of Tregs in Alzheimer disease patients is poorly understood, we evaluated the peripheral blood population and function of Tregs during the disease course. We found a failure in suppressive activity of Tregs at the clinical Alzheimer dementia stage, which could shift the immune system response towards a pro-inflammatory state, both in the periphery and in the brain. Further, we evaluated the potential of ex vivo expansion of Tregs to restore homeostasis.
Materials and methods
Patient recruitment and Alzheimer-defined population
Patients met ‘research criteria for mild cognitive impairment (MCI) and probable Alzheimer dementia, incorporating neurodegeneration biomarkers’ [decreased 18fluorodeoxyglucose uptake in temporoparietal cortex in PET; and/or disproportionate atrophy in medial temporal lobe and medial parietal cortex on magnetic resonance imaging], according to guidelines from the National Institute on Aging-Alzheimer's Association (NIA-AA) (Albert et al., 2011; Jack et al., 2011; McKhann et al., 2011). Written informed consent was obtained from all patients and aged-matched healthy controls (HC) according to the Declaration of Helsinki following ethics approval from the Institutional Review Board at the Houston Methodist Research Institute. Staging of dementia severity was based on the Clinical Dementia Rating Scale (CDR) assessment instrument (Morris, 1993; O'Bryant et al., 2008). Participants with a global CDR score of 0.5 were categorized as MCI (n: 42, M/F: 18/24, mean age: 71.4) and patients with a CDR ≥ 1 were considered to have Alzheimer dementia [n: 46 (24 CDR1 and 22 CDR2 and 3), M/F: 20/26, mean age: 70.0]. Enrolled healthy controls (HC) were required to have a CDR of 0 (n: 41, M/F: 17/24, mean age: 69.3).
Flow cytometry
Multicolour flow cytometry was used to assess the immunophenotype of Tregs. Antibodies against the following molecules were provided by: CD3 BV650 (BD Biosciences), CD8 BV450 (BD Biosciences), CD4 APC-H7 (BD Biosciences), CD25 PerCPCy5.5 (BD Biosciences), CD73 eFluor 450 (eBioscience™), PD-1 BV 650 (Biolegend). Dead cells were stained by LIVE/DEAD® Fixable Blue Dead Cell Stain Kit (Life Technology). For intracellular staining, cells were fixed and permeabilized using the FoxP3/Transcription Factor Staining Buffer Set (eBioscience), and then stained with FoxP3 Alexa Fluor 488 (eBioscience), IL13-PE (eBioscience) and Granzyme B APC (BioLegend). Appropriate isotype controls were used to set the quadrants and to evaluate background staining. Cells were analysed using an LSRII flow cytometer with BD FACSDIVA software.
Immune cell isolation
Mononuclear immune cells were isolated from peripheral blood of participants using Lymphoprep (Stemcell) density gradient centrifugation. Tregs and responders T cells (Tresps) were isolated using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. To increase purity, the positively selected cell fraction containing the CD4+CD25+regulatory T cells was run twice through the MS column. The unlabelled CD4+CD25− cell effluent was collected as the Tresp population.
Treg suppression assays on Tresp proliferation
Isolated Tresps were placed in a 96-well plate at a density of 50 000 cells per well followed by co-culture of corresponding Tregs at a ratio of 1:1 and 2:1 (Tresps:Tregs) in at least triplicates. A CD3/CD28 T-cell stimulation reagent (Miltenyi Biotec) was added to the co-cultures. After 5 days of culture, cells were pulsed with tritiated thymidine (1 Ci/well; Amersham Life Sciences) for 18 h. The cells were harvested and thymidine uptake was measured using a gas-operated-plate reader (Packard Instruments). The percent suppression of proliferation was calculated using the following formula: Percentage suppression = 100 – [(counts per minute of proliferating Tresps in the presence of Tregs/counts per minute of proliferating Tresps in the absence of Tregs) × 100].
Treg suppression assays on IPS-derived pro-inflammatory macrophages
Our lab has recapitulated protocols for the generation of mature monocyte cells from induced pluripotent stem cells. The protocol consists of five sequential steps through which mature monocytes are differentiated from human pluripotent cells in a stepwise manner (Yanagimachi et al., 2013). Mature monocytes were polarized to pro-inflammatory macrophages through treatment with GM-CSF (50 ng/ml) for 7 days, lipopolysaccharide (0.1 ng/ml) and interferon-γ (0.2 ng/ml) for 1 h. These induced pluripotent stem cell-derived macrophages robustly produced proinflammatory IL-6, TNFα and IL-1B cytokines. Tregs from patients and controls were co-cultured with activated macrophages for short (4 h) and longer (24 h) time periods. Cultured media was collected to assess cytokine protein levels via ELISA. Cells were then collected from culture and messenger RNA was isolated for the examination of cytokine transcripts. For IL-13 and CD25 blockade in the co-culture, anti-human IL-13 Ab (MAB2131) and anti-human CD25 Ab (MAB223) were used from R&D Systems. A falcon insert system (Life Science) was utilized to block direct contact between Treg and macrophage populations in the co-culture.
RNA purification, RT-PCR analysis
Using Trizol reagent, followed by Direct-zol RNA MiniPrep Kit (Zymo Research), messenger RNA was extracted from patient immune cell populations and in vitro cell experiments. Quantitative PCR experiments were performed using a One-Step RT-PCR kit with SYBR Green and run on the Bio-Rad iQ5 Multicolor Real-Time PCR Detection Systems. Primers for the study were purchased from BioRad and the relative expression level of each messenger RNA was calculated using the ΔΔCt method with normalization to β-actin and relative to control samples.
Ex vivo Tregs expansion
Bead-selected CD4+CD25high T lymphocytes were suspended at a concentration of 1 × 106 cells/ml in media containing 100 nM of rapamycin (Miltenyi Biotec), 500 IU/ml IL-2 and Dynabeads™ Human Treg Expander (Gibco™) at a 4:1 bead-to-cell ratio. Fresh media containing rapamycin and IL-2 were added to the cells every 2 − 3 days. After 10 days of culture, cells were harvested and washed. The number of expanded Tregs and their suppressive functions were assayed.
Statistical analysis
The minimum sample size was calculated to achieve 80% power with 5% significance level to detect the differences of Treg characteristic and function between varying stages of Alzheimer’s disease. Investigators were blinded to the identity of the groups during outcome assessment. Comparisons were performed using paired or unpaired Student’s t-test (for two groups) or one-way ANOVA (for more than two groups). Correlations were determined using the linear regression. Data were expressed as Mean ± SEM and P-values <0.05 were considered significant.
Data availability
The data collected during this study are available from the corresponding author upon reasonable requests from qualified individuals.
Results
Suppressor T cells’ immunophenotype in Alzheimer’s disease
Single blood samples from 20 Alzheimer patients, 18 MCI and 27 HC were examined by flow cytometry. We applied a gating strategy (Figure 1-A) that allows the identification of the Tregs by an existing established phenotype (CD4+FoxP3+CD25high). The percentage of CD4+FOXP3+CD25high Tregs did not differ among HC, MCI and Alzheimer groups (Figure 1-B). While the mean fluorescent intensity (MFI) of FoxP3 in CD4+FoxP3+CD25high Tregs was comparable among three groups (Figure 1-C), decline in CD25 MFI was identified in Alzheimer group compared with HC (Figure1-D). In evaluating CD8 suppressor T cells, the percentages of CD8+CD25high and CD8+FOXP+ cells were increased in MCI compared with HC and returned to baseline level at Alzheimer dementia stage (Fig. 1E and F).
Figure 1.

Suppressor T cells immunophenotype in Alzheimer disease. (A) Gating strategy to identify CD4 and CD8 suppressor T cells; lymphocytes were delineated by forward/side scatter gating. CD3 were used to identify T cells among the previously selected viable lymphocytes. CD4 T cells and CD8 T cells were identified as uniquely expressing CD4 or CD8 antigens. Tregs were defined as CD4 T cells co-expressing CD25 and FOXP3. The expressions of CD25 and FoxP3 were then determined on CD8 T cells. (B) CD4+FOXP3+ CD25high T-cell percentage (% of total CD4), did not differ among HC, MCI and Alzheimer groups. (C) FoxP3 mean fluorescent intensity in CD4+FOXP3+ CD25high cell population was comparable among the three groups. (D) CD25 mean fluorescent intensity in the CD4+FOXP3+ CD25high cell population was reduced in Alzheimer dementia stage. (E, F) The percentages of CD8+CD25high and CD8+FOXP+ suppressor T cells (% of total CD8) were increased in MCI patients, compared with HCs. CD8+CD25high T-cell population differs significantly between MCI and Alzheimer. P-values are *P < 0.05 and **P < 0.01.
Compromised suppressive function of Tregs on corresponding Tresps in Alzheimer’s disease
CD4+CD25highTregs were positively selected from whole blood based on CD25 expression. Tregs and Tresps were co-cultured with varying numbers of Tregs against the same number of Tresps. First, we evaluated the effects of age and gender on the suppressive function of Tregs. There was no difference in the suppressive function of Tregs among men and women (data not shown). No correlation was found between Treg suppressive activity and the age of our subjects (Figure 2-A). Treg suppressive activity in MCI (n = 29) was comparable to HC (n = 27) at both 1:1 and 2:1 Tresp: Treg ratios (1:1 ratio: HC = 52.8 ± 4.36%, MCI = 47.68 ± 4.40%, P = 0.933; 2:1 ratio, HC = 46.55 ± 3.88%, MCI = 39.71 ± 3.84%, P = 0.890). The suppressive function of Tregs in Alzheimer’s disease patients (n = 23) was reduced compared with both HC (1:1 ratio: Alzheimer disease = 27.45 ± 4.11%, Alzheimer disease versus HC, P = 0.0001; 2:1 ratio Alzheimer’s disease = 23.3 ± 4.96%, Alzheimer versus HC, P = 0.0018) and MCI (1:1 ratio: Alzheimer’s disease versus MCI: P = 0.0034, 2:1 ratio: P = 0.0357) (Figure 2-B).
Figure 2.
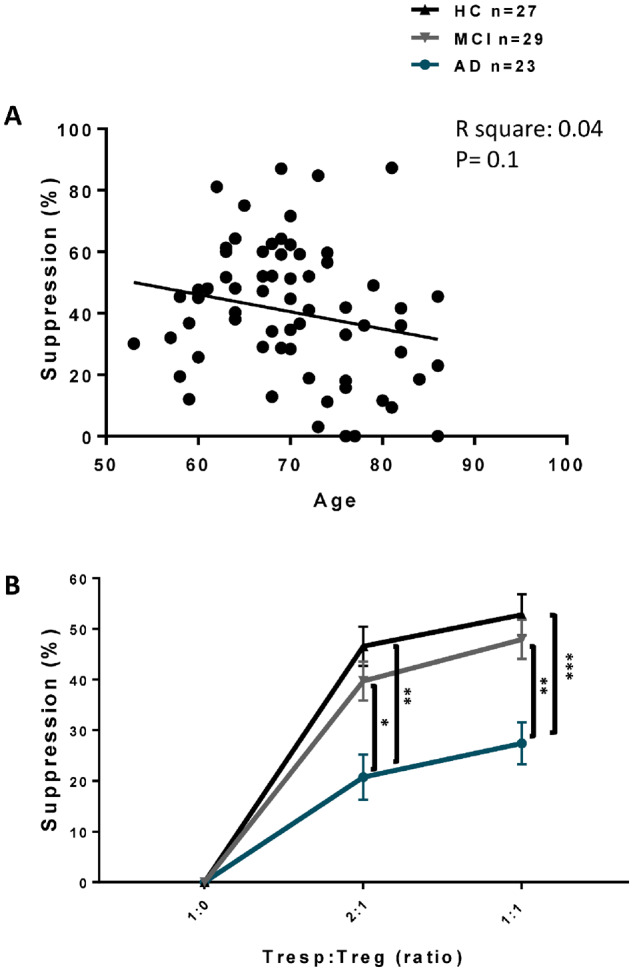
Treg suppressive function on Tresp proliferation. CD4+CD25highTregs were co-cultured with CD4+CD25neg Tresps and proliferation was determined by 3H-thymidine incorporation. (A) No correlation between age and suppressive activity of Tregs on Tresp proliferation (1:1 ratio) was noted. (B) Suppression (%) of Tregs on Tresp proliferation in MCI vs. HC in 1:1 and 2:1 Tresps: Tregs ratios were comparable. Suppression of Tregs on Tresps proliferation in Alzheimer’s disease in both 1:1 and 2:1 Tresps: Tregs ratios were reduced compared with both MCI and HC. P-values are *P < 0.05, **P < 0.01, and ***P < 0.001.
Enhanced immunophenotype and suppressive activity of Tregs on Tresp proliferation following ex vivo expansion
In another experiment, distinct smaller sets of patients and controls were enrolled (10 Alzheimer patients, 10 MCI and 10 HC). CD4+CD25high Tregs were isolated from blood and the suppressive function of Tregs on Tresp proliferation at a 1:1 ratio was assayed. Similar to our initial finding (Figure 2-B), the suppressive function of MCI Tregs (43.45 ± 5.20%) was comparable to HC (47.89 ± 4.23%) (P = 0.983), but the suppressive function of Tregs in Alzheimer’s disease (25.27 ± 4.81%) was decreased compared with HC (P = 0.0130) (Figure 3-A). Tregs from the same individuals were expanded ex vivo in the presence of IL-2, rapamycin and CD3/CD28 beads for 10 days. The expansion rates of Tregs isolated from the HC, MCI and Alzheimer’s disease groups were not different (data not shown). Enhancement in the suppressive function of ex vivo expanded (exp) Tregs on Tresp proliferation was noted in all groups compared with their respective baselines (base) Tregs (exp-HC = 87.7 ± 4.82%, exp vs. base-HC P = 0.0001, exp-MCI = 76.6 ± 4.61%, exp vs. base-MCI P = 0.0002, exp-Alzheimer= 87.98 ± 4.97%, exp vs. base-Alzheimer P < 0.0001). Interestingly, in contrast to baseline Tregs, the suppressive activity of the expanded Tregs became comparable among patients and HC (Figure 3-A). The immunophenotypes of baseline and expanded Tregs were also assayed by flow cytometry. Following expansion, the MFI of CD25 was remarkably (∼20-fold) increased in all three groups (Figure 3-B). The MFI of FoxP3 in expanded Tregs was also elevated (∼1.5 − 2-fold) (Figure 3-C). CD25 and FoxP3 MFIs in expanded Tregs were comparable between patients and controls.
Figure 3.

Treg immunophenotype and suppressive function following ex vivo expansion. (A) In a second group of individuals, suppression (%) of CD4+CD25highTregs on Tresp proliferation (1:1 ratio) at baseline (base) was compromised in Alzheimer patients (n = 10), compared with HC (n = 10). Following 10 days of ex vivo expansion, amplified suppressive function of expanded (exp) Tregs on Tresp proliferation was noted in all groups. CD25 (B) and FoxP3 (C) MFIs in CD4+CD25highTreg population were elevated following ex vivo expansion. P-values are *P< 0.05, **P< 0.01 and ***P< 0.001.
Amplified suppression of Tregs on induced pluripotent stem cell-derived pro-inflammatory macrophages following ex vivo expansion
CD4+CD25highTregs were co-cultured with pro-inflammatory induced pluripotent stem cell-derived macrophages (M1) and the relative changes of IL-6, TNFα and IL1B transcripts (after 4 h) and protein levels in the supernatant (after 24 h) were assayed. At baseline, Tregs from HC attenuated macrophage IL-6 transcript expression by 25% but not significant (P = 0.071) (Figure 4-A). Baseline Tregs of MCI or Alzheimer patients showed no suppression on M1-derived IL-6 transcript or protein (Fig. 4A and B). The Tregs from these same subjects were expanded ex vivo for 10 days and then co-cultured with pro-inflammatory macrophages. A reduction of M1-derived IL-6 transcript and protein expression levels were noted following co-culture with expanded Tregs in all groups (Fig. 4A and B). The co-culture of baseline Tregs with M1 did not attenuate TNFα transcript and protein levels. Expanded Tregs of Alzheimer’s disease, MCI and HC displayed an enhanced capacity to suppress M1-derived TNFα transcript and protein, compared with corresponding baseline Tregs (Fig. 4C and D). Similar to IL6 and TNFa findings, while baseline Tregs did not alter IL1B transcript levels, expanded Tregs displayed an enhanced capacity to suppress M1-derived IL1B transcript in all three groups (Figure 4-E). However, in evaluating IL1B protein, the protein values were too low to be measured reliably (data not shown).
Figure 4.

Suppression of Tregs on induced pluripotent stem cell-derived pro-inflammatory macrophages. Baseline and ex vivo expanded CD4+CD25high Tregs were added to induced pluripotent stem cell-derived pro-inflammatory macrophages (M1) and relative change (% of M1-only) of pro-inflammatory cytokines were measured. (A, B) At baseline, a trend towards decreased macrophage IL6 transcript expression was noted following co-culture with baseline HC Tregs. Expanded Tregs of HC, MCI and Alzheimer’s suppressed M1-IL6 transcript and protein expressions. (C, D) The co-culture of baseline Tregs with M1 did not suppress TNFa transcript and protein levels. Reduction of M1- TNFa transcript and protein expressions were noted following co-culture with expanded Tregs in all three groups. (E) Baseline Tregs did not attenuate IL1B transcript level. Expanded Tregs of Alzheimer disease, MCI and HC displayed an enhanced capacity to suppress M1-derived IL1B transcript. *P < 0.05, **P < 0.01, ***P< 0.001 versus their corresponding baseline Tregs- M1 co-cultures; ##P < 0.01, ###P < 0.001 versus M1-alone cultures.
Up-regulated immunoregulatory gene expressions in expanded Tregs
Messenger RNA was extracted from Tregs of 10 HC, 10 MCI and 10 AD subjects at baseline and following their respective expansions. Transcript levels of immunosuppressive cytokines known to be released from Tregs including TGFβ, IL-10, IL-4 and IL-13, CD25, and immunomodulatory markers that require cell-to-cell proximity or contact [PD1, CTLA4, Granzyme (GZM) A and B, PDL1, PDL2, CD39 and CD73] were examined by quantitative PCR. At baseline, trends towards reduced IL-4, IL-10 and IL-13 transcript expression levels were noted in Alzheimer group. TGFβ expression levels were comparable between Alzheimer, MCI and HC at baseline, but following expansion, the expression levels were down-regulated in all groups. In contrast to IL-4 and IL-10 that did not change following expansion, IL-13 and CD25 transcripts were up-regulated in expanded Tregs (Figure 5-A). The expression levels of PD1, PDL1, PDL2, CTLA4, CD39, CD73 and Granzyme A&B in Tregs were comparable between HC, MCI and AD at baseline. While there were no changes in CD39, PDL1, PDL2 and CTLA4 transcripts following ex vivo expansion, up-regulation of PD1, GZMB and CD73 transcripts in expanded Tregs were observed in all groups. Granzyme A expression levels were down-regulated in expanded Tregs (Figure 5-B).
Figure 5.

Transcript expression profile of immunoregulatory genes in Tregs. Expression profiles of (A) anti-inflammatory cytokines (TGFb, IL4, IL10, IL13), CD25 and (B) proximity or contact-mediated immunoregulatory markers [Granzyme (GZM) A, CD39, PDL1, PDL2, CTLA4, CD73, GZMB, PD1] were evaluated in expanded and baseline Tregs of HC (n = 10), MCI (n = 10) and Alzheimer patients (n = 10). The data were obtained from qPCR analysis when normalized using β-actin. The transcript levels of IL13, CD25 and proximity or contact-mediated immunoregulatory markers (PD1, GZMB and CD73) were up regulated following ex vivo expansion. P-values are *P< 0.05, **P< 0.01 and ***P< 0.001 versus their corresponding baseline Tregs.
The protein expression of genes with upregulated transcript levels following expansion, was further analysed in 5 Alzheimer patients and 5 HCs, using flow cytometer. Higher percentage of CD73+ (Figure 6-A), PD1+ (Figure 6-B) and IL13+ Tregs (Figure 6-C) (% of total CD4+CD25high T cells) were observed following ex vivo expansion in both Alzheimer patients and HCs. In addition to increased frequency, significant enhancement in CD73 (Figure 6-E) and PD1 (Figure 6-F) MFIs were noted in expanded Tregs. The difference between percentage of GZMB+ Treg subpopulation between baseline and expanded Tregs were not different (Figure 6-D), but GZMB MFIs were significantly amplified following expansion (Figure 6-H).
Figure 6.

Protein expression of immunoregulatory genes in Tregs. Using flow cytometer, protein expression of immunoregulatory genes (CD73, PD1, IL13 and GZMB) were analysed in baseline and corresponding ex vivo expanded Tregs of Alzheimer patients (n = 5) and HCs (n = 5). Increase in the percentage of (A) CD73+, (B) PD1+ and (C) IL13+ Tregs (% of total CD4+CD25high Tregs) were noted following ex vivo expansion in both Alzheimer patients and HCs. The change in the percentage of (D) GZMB+Tregs following expansion were not statistically significant. Mean fluorescence intensities (MFIs) of (E) CD73, (F) PD1 and (H) GZMB were amplified in expanded Tregs. The increase in G IL13 MFI in Tregs following expansion was not statistically significant. P-values are *P < 0.05, **P < 0.01 and ***P < 0.001.
Ex vivo expanded Tregs immunomodulation works through a contact-mediated mechanism
Immunoregulatory genes that up-regulated in ex vivo expanded Tregs were further scrutinized as potential candidates for enhanced Treg suppressive function. To test the direct effects of these gene products, expanded Tregs were co-cultured with pro-inflammatory macrophages and the changes in the suppressive function of Tregs on M1-derived IL-6 protein production were measured in the presence of neutralizing anti-IL-13, anti-CD25 or avoiding cell−cell contact using transwells. This assay was replicated with expanded Tregs of total four Alzheimer and four HC individuals. Expanded Alzheimer’s disease and HC Tregs attenuated M1-derived IL-6 protein by 44 and 46%, respectively (Figure 7-A and B). The addition of CD25 or IL-13 neutralizing antibodies to the co-culture did not affect the abilities of expanded Tregs to suppress IL-6 protein production. However, when the expanded Tregs were placed in a transwell, their suppressive function was abrogated in both Alzheimer’s disease and HC groups. As soluble factors, released from expanded Tregs, could still pass through the transwells, these data suggested that expanded Tregs required cell-to-cell contact to exert their suppressive function on pro-inflammatory macrophages.
Figure 7.

The suppressive mechanism of ex vivo expanded Tregs. Expanded Tregs of Alzheimer patients (n = 4) (A) and HC (n = 4) (B) were co-cultured with pro-inflammatory macrophages (M1) and relative fold change of IL6 protein (% of M1-only) were assayed in the presence of IL13 or CD25 neutralizing antibodies (IL13 NA and IL25 NA) or transwells. The absence of direct cell−cell contact by using transwells, blocked suppressive function of ex vivo expanded Tregs. P-values are **P < 0.01.
Discussion
Tregs play a major role in establishing and maintaining immune tolerance. Although several studies evaluated the role of Tregs in Alzheimer animal models (Baruch et al., 2015; Baek et al., 2016; Dansokho et al., 2016), there is still limited knowledge regarding the status of Tregs in Alzheimer patients. This study documented significant alteration in the immunophenotype as well as the suppressive activity of Tregs throughout the course of Alzheimer’s disease. Following ex vivo expansion, the immunoregulatory capacity of Tregs was substantially enhanced in both patients and HC through a cell contact-mediated mechanism.
In analysing the Treg immunophenotype, the percentages of CD4 Tregs were comparable among HC, MCI and Alzheimer groups, confirming the findings of two previous studies (Le Page et al., 2017; Oberstein et al., 2018). However, a recent publication reported reduced levels of Tregs at Alzheimer dementia stage (Ciccocioppo et al., 2019). The major differences between Ciccocioppo et al. and our finding are the larger sample size in the present study and the different strategies of Tregs identification. In further evaluation of Treg population, reduced CD25 surface expression was noted in Alzheimer group. CD25 is crucial for metabolic homeostasis and survival of Tregs (Fan et al., 2018; Toomer et al., 2019). Tregs with lower CD25 expression is previously reported in certain cases of autoimmunity and might have a partial suppressor activity rather than a fully active Tregs (Liu et al., 2006; Ono et al., 2006; Bonelli et al., 2009; Coleman et al., 2012; Prado et al., 2013). CD8+ T cells with immunosuppressive properties have also been described with varying phenotypic definitions (Wang and Alexander, 2009). Although a specific marker for the identification of CD8+ suppressive T cells is still elusive, numerous markers representing subsets of CD8+ Tregs overlap with markers for CD4+ Tregs, particularly the surface CD25 and intracellular FoxP3 markers (Frisullo et al., 2010; Naciute et al., 2017; Yu et al., 2018). In contrast to CD4 suppressor T cell, significant elevation in CD8 suppressor T cells at the MCI stage was noted in our study which could represent an attempt to curtail an ongoing pro-inflammatory immune response.
In the present study, we documented a compromise in Treg suppression on activated Tresp proliferation in Alzheimer’s disease. A failure in Treg immunomodulatory function has been previously reported in other neurodegenerative disorders including amyotrophic lateral sclerosis (ALS) (Beers et al., 2017) and Parkinson disease (Saunders et al., 2012). However, two previous reports in Alzheimer’s disease, did not find a compromise in Treg suppressive activity (Rosenkranz et al., 2007; Saresella et al., 2010). The disagreement between our findings and others might be secondary to different applied methods of Treg isolation and suppression assay. Additionally, in the Rosenkranz et al. study, patients in the Alzheimer group were mainly at a mild stage of the disease (Average MMSE score was 22.6) where Tregs might still be as functional as MCI groups. In the present study, about half of the patients in the Alzheimer group had moderate to severe dementia (CDR 2 and 3). The mechanism of Treg immunomudulatory compromise through neurodegeneration progression is not clear. However, T-cell immunosenesence and a shift of post-thymic differentiation towards the accumulation of effector T lymphocytes other than Tregs (Saresella et al., 2011; Thomas et al., 2020) were reported in Alzheimer patients. It suggests the possibility of decreased proportion of thymus-derived Tregs to peripherally induced Tregs in these patients. Partial immunomodulatory function (Haribhai et al., 2011; Josefowicz et al., 2012) and lack of stability (Zhou et al., 2009; Miyao et al., 2012) have been reported in peripherally induced Tregs, which provide a rationale for impaired Treg population in Alzheimer disease. However, there is a lack of definitive information regarding the proportion of thymus-derived versus peripherally-induced Tregs, differences in suppressive activity of these two subsets and their relative contributions to immune homeostasis in Alzheimer pathology. In a murine model of Alzheimer, Treg elimination caused an up-regulation in the expression of genes related to astrogliosis, microgliosis and neuroinflammation while increasing the amyloid burden in the hippocampus and accelerating the onset of cognitive deficits (Baek et al., 2016; Dansokho et al., 2016). However, these regulatory T cells might have dichotomous effects on the neurodegenerative process by obstructing a selective gateway for immune cell trafficking to the CNS, reducing the recruitment of immunoregulatory cells from the periphery to the CNS and compromising reparative immune responses (Baruch et al., 2015, 2016). In clinical setting of Alzheimer’s disease, the structural integrity of the blood−brain barrier has already broken down (van de Haar et al., 2016a, b) and pro-inflammatory peripheral immune cells have migrated through the activated endothelium by various mechanisms (Zenaro et al., 2017), aggravating Alzheimer’s disease pathology.
For the first time in Alzheimer’s disease, the potential for ex vivo expansion of patients’ Tregs was evaluated. These expanded Tregs expressed 20-fold higher levels of CD25 protein on their cell surfaces. Increased CD25 protein expression plays a critical role in maintaining the survival of the Treg population (Fan et al., 2018). The suppressive activities of these ex vivo expanded Tregs were also examined. Following expansion, the ability of Tregs to suppress Tresp proliferation was significantly increased in both patients and HC. Interestingly, following expansion the immunophenotype and suppressive activity of Tregs from advanced Alzheimer’s disease patients were comparable to those of HC. A similar pattern has been observed following ex vivo expansion of dysfunctional Tregs in other neurological disorders including multiple sclerosis (Lifshitz et al., 2016) and ALS (Alsuliman et al., 2016; Thonhoff et al., 2018).
Innate immune cell populations have now been identified as key players in Alzheimer’s disease pathology. While neuroprotective microglia are important for containment/clearance of Amyloid plaques and maintaining neuronal survival (Daria et al., 2017; Kang et al., 2018; Nordengen et al., 2019), Alzheimer inflammatory microglia (Del-Aguila et al., 2019; Mathys et al., 2019; Gray et al., 2020) may play a detrimental role by eliciting tau pathogenesis and accelerating neurotoxicity (Kitazawa et al., 2011; Kiyota et al., 2012; Tweedie et al., 2012; Ghosh et al., 2013; Zheng et al., 2016; Shi et al., 2017, 2019; Kametani and Hasegawa, 2018). There is increasing evidence that blood−borne macrophages and brain resident microglia orchestrate central nervous system inflammation in Alzheimer pathology (Holmes, 2013; Martin et al., 2017; Sevenich, 2018). We have previously reported the pro-inflammatory phenotype of peripheral monocytes throughout the course of AD (Thome et al., 2018). In the present study, the interplay between Tregs and innate immune cells was investigated. While freshly isolated Tregs showed no effects on activated macrophages, ex vivo expanded Tregs displayed substantially enhanced suppressive activity on pro-inflammatory macrophage-derived cytokine production. Consistent with our findings, a recent report documented that expanded Tregs of healthy donors drive monocyte differentiation towards alternatively activated macrophages that produce lower levels of pro-inflammatory TNF-α and IL-6 and higher levels of anti-inflammatory IL-10 (Romano et al., 2018). In two previous Alzheimer in vivo studies, Treg treatment had contradicting effects on the number and morphology of micrgolia (Dansokho et al., 2016). The immunomodulatory impact of Tregs on microglia phenotype is still elusive. In this regard, further investigation is underway, evaluating the effect of Tregs on Alzheimer pathology, particularly Treg potential effects on microglial immunophenotype shifting.
Tregs have been shown to utilize different regulatory mechanisms to maintain immune homeostasis. The suppressive mechanism of Tregs could be mediated by (i) inhibitory cytokines (TGFβ, IL-10, IL-4, IL-13), (ii) local competition for IL-2 through high CD25 expression and (iii) dependence on proximity or cell-to-cell contact (PD1, CTLA4, GZM A and B, PDL1, PDL2, CD39 and CD73) (Gondek et al., 2005; Sojka et al., 2008; Tang and Bluestone, 2008; Sakaguchi et al., 2009; Ernst et al., 2010; Gubser et al., 2016). In this study, gene expression profiling of expanded Tregs identified upregulation of several genes, including IL-13, CD25, PD1, CD73 and Granzyme B, that could exert suppressive effects through aforementioned modes of action. Further evaluation of these candidate Treg mechanisms of action documented that contact-dependent mechanisms played a major role in modulating pro-inflammatory macrophages. In the other report, physical separation of ex vivo expanded Tregs and Tresps using a Transwell system blocked their suppressive abilities (Rossetti et al., 2015), indicating that cell contact is essential for immunosuppression. Further research is necessary to define more fully the cell contact-mediated pathway involved in expanded Treg immunomodulation.
The major limitation of this study is that patients were enrolled based on 2011 NIA-AA research criteria incorporating biomarkers of neurodegeneration (MRI and fluorodeoxyglucose PET scans). However, as this project started earlier, the patients had not been screened based on 2018 NIA-AA Research Framework, which raises concern about the certainty of the Alzheimer diagnosis in enrolled patients. In conclusion, the immunophenotype and suppressive function of Tregs are compromised in Alzheimer’s disease patients. Accumulating preclinical and clinical evidences suggest that Tregs could be a modifiable therapeutic target. Expanded Tregs are currently being translated into cell therapy for neurodegenerative disorders. Recently, a first-in-human Phase I clinical study of autologous transplantation of expanded normalized Tregs was completed in patients with ALS (Thonhoff et al., 2018). The study demonstrated safety and potential benefit of this treatment strategy, and a Phase II double-blind, placebo-controlled trial is currently underway. The data presented here demonstrate that the ex vivo expansion of Tregs restores their immunophenotype and suppressive function at the Alzheimer’s disease stage. These results provide a rationale for the autologous transfer of expanded Tregs in Alzheimer’s disease patients.
Acknowledgements
Acknowledgement is made to the donors of Alzheimer’s Disease Research, a programme of the BrightFocus Foundation. The authors appreciate the assistance of Victoria Arbones, Katharine G. Dlouhy, Jinghong Wang and Shixiang Wen, of the Houston Methodist Neurological Institute and the Nantz National Alzheimer Center. We are grateful to the Nantz National Alzheimer Center patients and families for making this research possible.
Funding
This research was supported by a BrightFocus Foundation fellowship award (BF-A2019083F) an award from the Houston Methodist Clinician Scientist Recruitment and Retention Program, the Chao Foundation, Harrison Foundation, Moody Foundation and Nantz Houston Methodist Foundation funds.
Competing interests
A.F, D.R.B., W.Z., A.D.T., B.P. and J.R.T. report no disclosures. J.C.M. reports no disclosures relevant to this work, but is on a medical advisory board for G.E. Healthcare, has received research support from AbbVie, ALSA, Aventis, Avanir, Biogen, Eli Lilly, Genentech, Janssen, the Lee Rizzuto Foundation, the National Institute of Aging, and Novartis, and served as an expert consultant in two traumatic brain injury legal cases. S.H.A. reports no disclosures relevant to this work but serves as a scientific consultant to Mitsubishi Tanabe Pharma; served on the speaker's bureau of Avanir; received research support from the ALSA, ALS Finding a Cure and Lee Rizzuto Foundation; and served as an expert consultant in ALS case.
Glossary
- AD =
Alzheimer’s disease
- Base =
baseline
- CDR =
clinical dementia rating scale
- Exp =
expanded
- GZM =
Granzyme
- HC =
healthy control
- MCI =
mild cognitive impairment
- MFI =
mean fluorescent intensity
- NIA-AA =
National Institute on Aging-Alzheimer's Association
- Treg =
regulatory T lymphocyte
- Tresp =
responders T cells
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsuliman A, Appel SH, Beers DR, Basar R, Shaim H, Kaur I, et al. A robust, good manufacturing practice-compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy 2016; 18: 1312–24. [DOI] [PubMed] [Google Scholar]
- Baek H, Ye M, Kang G-H, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer's disease model. Oncotarget 2016; 7: 69347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun 2015; 6: 7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med 2016; 22: 135–7. [DOI] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, et al. ALS patients' regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017; 2: e89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonelli M, Savitskaya A, Steiner C-W, Rath E, Smolen JS, Scheinecker C, et al. Phenotypic and functional analysis of CD4+ CD25- Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol 2009; 182: 1689–95. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo F, Lanuti P, Pierdomenico L, Simeone P, Bologna G, Ercolino E, et al. The Characterization of regulatory T-cell profiles in Alzheimer's disease and multiple sclerosis. Sci Rep 2019; 9: 8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MM, Finlay CM, Moran B, Keane J, Dunne PJ, Mills KHG, et al. The immunoregulatory role of CD4(+) FoxP3(+) CD25(-) regulatory T cells in lungs of mice infected with Bordetella pertussis. FEMS Immunol Med Microbiol 2012; 64: 413–24. [DOI] [PubMed] [Google Scholar]
- Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 2016; 139: 1237–51. [DOI] [PubMed] [Google Scholar]
- Daria A, Colombo A, Llovera G, Hampel H, Willem M, Liesz A, et al. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J 2017; 36: 583–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Aguila JL, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, Fernandez MV, et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 2019; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst PB, Garrison JC, Thompson LF.. Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. Ji 2010; 185: 1993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MY, Low JS, Tanimine N, Finn KK, Priyadharshini B, Germana SK, et al. Differential roles of IL-2 signaling in developing versus mature Tregs. Cell Rep 2018; 25: 1204–13.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti MT, Merlini M, Späni C, Gericke C, Schweizer N, Enzmann G, et al. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer's disease-like cerebral amyloidosis. Brain Behav Immun 2016; 54: 211–25. [DOI] [PubMed] [Google Scholar]
- Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. CD8(+)Foxp3(+) T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol 2010; 71: 437–41. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, et al. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci 2013; 33: 5053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giubilei F, Antonini G, Montesperelli C, Sepe-Monti M, Cannoni S, Pichi A, et al. T cell response to amyloid-beta and to mitochondrial antigens in Alzheimer's disease. Dement Geriatr Cogn Disord 2003; 16: 35–8. [DOI] [PubMed] [Google Scholar]
- Gondek DC, Lu L-F, Quezada SA, Sakaguchi S, Noelle RJ.. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol 2005; 174: 1783–6. [DOI] [PubMed] [Google Scholar]
- Gray SC, Kinghorn KJ, Woodling NS.. Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen Res 2020; 15: 1208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubser C, Schmaler M, Rossi SW, Palmer E.. Monoclonal regulatory T cells provide insights into T cell suppression. Sci Rep 2016; 6: 25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011; 35: 109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Balling R.. The role of regulatory T cells in neurodegenerative diseases. Wires Syst Biol Med 2013; 5: 153–80. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 2013; 5: 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J.. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008; 28: 8354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C. Review: Systemic inflammation and Alzheimer's disease. Neuropathol Appl Neurobiol 2013; 39: 51–68. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu Z, Cao B-B, Qiu Y-H, Peng Y-P.. Treg cells attenuate neuroinflammation and protect neurons in a mouse model of Parkinson's disease. J Neuroimmune Pharmacol 2020; 15: 224–37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 2019; 51: 404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012; 482: 395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kametani F, Hasegawa M.. Reconsideration of amyloid hypothesis and Tau hypothesis in Alzheimer's disease. Front Neurosci 2018; 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Kurti A, Baker KE, Liu C-C, Colonna M, Ulrich JD, et al. Behavioral and transcriptomic analysis of Trem2-null mice: not all knockout mice are created equal. Hum Mol Genet 2018; 27: 211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE-H.. Wild type and P301L mutant Tau promote neuro-inflammation and alpha-Synuclein accumulation in lentiviral gene delivery models. Mol Cell Neurosci 2012; 49: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer's disease model. Ji 2011; 187: 6539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T, et al. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther 2012; 19: 724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page A, Garneau H, Dupuis G, Frost EH, Larbi A, Witkowski JM, et al. Differential phenotypes of myeloid-derived suppressor and T regulatory cells and cytokine levels in amnestic mild cognitive impairment subjects compared to mild Alzheimer diseased patients. Front Immunol 2017; 8, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Selenica M-LB, Reid P, Kraft C, Johnson A, et al. LPS-induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation 2010; 7: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lin J, Wang Z, He S, Ma X, Li D, et al. Oxidized low-density lipoprotein-induced proinflammatory cytokine response in macrophages are suppressed by CD4CD25(+)Foxp3(+) regulatory T cells through downregulating toll like receptor 2-mediated activation of NF-kappaB. Cell Physiol Biochem 2010; 25: 649–56. [DOI] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na S-Y, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci 2013; 33: 17350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz GV, Zhdanov DD, Lokhonina AV, Eliseeva DD, Lyssuck EY, Zavalishin IA, et al. Ex vivo expanded regulatory T cells CD4(+)CD25(+)FoxP3(+)CD127(Low) develop strong immunosuppressive activity in patients with remitting-relapsing multiple sclerosis. Autoimmunity 2016; 49: 388–96. [DOI] [PubMed] [Google Scholar]
- Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006; 203: 1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015; 138: 1738–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry 2018; 8: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Boucher C, Fontaine B, Delarasse C. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer's disease models: effects of aging and amyloid pathology. Aging Cell 2017; 16: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer's disease. Nature 2019; 570: 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT.. Extravascular CD3+ T cells in brains of alzheimer disease patients correlate with Tau but not with amyloid pathology: an immunohistochemical study. Neurodegener Dis 2018; 18: 49–56. [DOI] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 2012; 36: 262–75. [DOI] [PubMed] [Google Scholar]
- Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993; 43: 2412–4. [DOI] [PubMed] [Google Scholar]
- Naciute M, Maciunaite G, Mieliauskaite D, Rugiene R, Zinkeviciene A, Mauricas M, et al. Increased numbers of CD4(+)CD25(+) and CD8(+)CD25(+) T-cells in peripheral blood of patients with rheumatoid arthritis with parvovirus B19 infection. In Vivo 2017; 31: 181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, et al. Tau Oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. JAD 2016; 55: 1083–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordengen K, Kirsebom B-E, Henjum K, Selnes P, Gísladóttir B, Wettergreen M, et al. Glial activation and inflammation along the Alzheimer's disease continuum. J Neuroinflammation 2019; 16: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Bryant SE, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol 2008; 65: 1091–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein TJ, Taha L, Spitzer P, Hellstern J, Herrmann M, Kornhuber J, et al. Imbalance of circulating Th17 and regulatory T cells in Alzheimer's disease: a case control study. Front Immunol 2018; 9: 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeke EB, Okwor I, Uzonna JE.. Regulatory T cells restrain CD4+ T cells from causing unregulated immune activation and hypersensitivity to lipopolysaccharide challenge. Ji 2014; 193: 655–62. [DOI] [PubMed] [Google Scholar]
- Ono M, Shimizu J, Miyachi Y, Sakaguchi S.. Control of autoimmune myocarditis and multiorgan inflammation by glucocorticoid-induced TNF receptor family-related protein(high), Foxp3-expressing CD25+ and CD25- regulatory T cells. J Immunol 2006; 176: 4748–56. [DOI] [PubMed] [Google Scholar]
- Prado C, de Paz B, López P, Gómez J, Rodríguez-Carrio J, Suárez A, et al. Relationship between FOXP3 positive populations and cytokine production in systemic lupus erythematosus. Cytokine 2013; 61: 90–6. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Paolicelli RC.. Microglia-mediated synapse loss in Alzheimer's disease. J Neurosci 2018; 38: 2911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE.. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+CD25+ T cells. J Proteome Res 2009; 8: 3497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano M, Fanelli G, Tan N, Nova-Lamperti E, McGregor R, Lechler RI, et al. Expanded regulatory T cells induce alternatively activated monocytes with a reduced capacity to expand T Helper-17 cells. Front Immunol 2018; 9: 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol 2007; 188: 117–27. [DOI] [PubMed] [Google Scholar]
- Rossetti M, Spreafico R, Saidin S, Chua C, Moshref M, Leong JY, et al. Ex vivo-expanded but not in vitro-induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell-specific demethylated region. Ji 2015; 194: 113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T.. Regulatory T cells: how do they suppress immune responses? Int Immunol 2009; 21: 1105–11. [DOI] [PubMed] [Google Scholar]
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. JAD 2010; 21: 927–38. [DOI] [PubMed] [Google Scholar]
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer's disease. Brain Behav Immun 2011; 25: 539–47. [DOI] [PubMed] [Google Scholar]
- Saunders JAH, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J Neuroimmune Pharmacol 2012; 7: 927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol 2018; 9: 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ; Alzheimer’s Disease Neuroimaging Initiative. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017; 549: 523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manis M, Long J, Wang K, Sullivan PM, Serrano JR, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 2019; 216: 2546–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka DK, Huang YH, Fowell DJ.. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology 2008; 124: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, et al. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol 2011; 178: 2811–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA.. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 2008; 9: 239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Wang W, Su DM.. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing 2020; 17: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome AD Faridar A Beers DR Thonhoff JR Zhao W Wen S, . et al. Functional alterations of myeloid cells during the course of Alzheimer’s disease. Mol Neurodegeneration 2018; 13:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm 2018; 5: e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomer KH, Lui JB, Altman NH, Ban Y, Chen X, Malek TR, et al. Essential and non-overlapping IL-2Ralpha-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat Commun 2019; 10: 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, et al. Tumor necrosis factor-alpha synthesis inhibitor 3,6'-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer's disease. J Neuroinflammation 2012; 9: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 2016. a; 281: 527–35. [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Wong SM, et al. Neurovascular unit impairment in early Alzheimer's disease measured with magnetic resonance imaging. Neurobiol Aging 2016. b; 45: 190–6. [DOI] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer's disease. Nature 2017; 552: 355–61. [DOI] [PubMed] [Google Scholar]
- Wang YM, Alexander SI.. CD8 regulatory T cells: what's old is now new. Immunol Cell Biol 2009; 87: 192–3. [DOI] [PubMed] [Google Scholar]
- Xie L, Choudhury GR, Winters A, Yang S-H, Jin K.. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur J Immunol 2015; 45: 180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagimachi MD, Niwa A, Tanaka T, Honda-Ozaki F, Nishimoto S, Murata Y, et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS One 2013; 8: e59243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ma X, Gong R, Zhu J, Wei L, Yao J, et al. Recent advances in CD8(+) regulatory T cell research. Oncol Lett 2018; 15: 8187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Piacentino G, Constantin G.. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis 2017; 107: 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kong Q, Zhang Z, Ge P, Ba D, He W.. Telomere dysfunction of lymphocytes in patients with Alzheimer disease. Cogn Behav Neurol 2003; 16: 170–6. [DOI] [PubMed] [Google Scholar]
- Zheng C, Zhou XW, Wang JZ.. The dual roles of cytokines in Alzheimer's disease: update on interleukins, TNF-alpha, TGF-beta and IFN-gamma. Transl Neurodegener 2016; 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 2009; 10: 1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data collected during this study are available from the corresponding author upon reasonable requests from qualified individuals.