

Abstract
Cofilins are small protein of the actin depolymerizing family. Actin polymerization/depolymerization is central to a number of critical cellular physiological tasks making cofilin a key protein for several physiological functions of the cell. Cofilin activity is mainly regulated by phosphorylation on serine residue 3 making this post-translational modification key to the regulation of myofilament integrity. In fact, in this form, the protein segregates in myocardial aggregates in human idiopathic dilated cardiomyopathy. Since myofilament network is an early target of oxidative stress we investigated the molecular changes induced by oxidation on cofilin isoforms and their interplay with the protein phosphorylation state to get insight on whether/how those changes may predispose to early protein aggregation. Using different and complementary approaches we characterized the aggregation properties of cofilin-2 and its phosphomimetic variant (S3D) in response to oxidative stress in silico, in vitro and on isolated cardiomyocytes.
We found that the phosphorylated (inactive) form of cofilin-2 is mechanistically linked to the formation of an extended network of fibrillar structures induced by oxidative stress via the formation of a disulfide bond between Cys39 and Cys80. Such phosphorylation-dependent effect is likely controlled by changes in the hydrogen bonding network involving Cys39. We found that the sulfide ion inhibits the formation of such structures. This might represent the mechanism for the protective effect of the therapeutic agent Na2S on ischemic injury.
Keywords: Cofilin, Redox properties, Amyloid, Phosphorylation, Cysteine, Sulfide
Graphical abstract
Highlights
-
•
Cofilin changes the hydrogen bonding network involving Cys39 in its phosphorylated form.
-
•
This phosphorylation-dependent effect of oxidation promotes the formation of a disulfide bond between Cys39 and Cys80.
-
•
The formation of the Cys39 and Cys80 disulfide bond favors the formation of fibrillar aggregates.
-
•
The sulfide ion inhibits the formation of such structures.
Abbreviations
- ADF
Actin Depolymerizing Factor
- AFM
Atomic Force Microscopy
- Arg
Arginine
- CD
Circular Dichroism
- CV
Cyclic Voltammetry
- Cys
Cysteine
- E°
standard reduction potential
- ATR-FTIR Spectroscopy
Attenuated Total Reflectance - Fourier Transform InfraRed Spectroscopy
- Glu
Glutamine
- IAA
Iodoacetamide
- IF
ImmunoFluorescence
- Leu
Leucine
- MD
Molecular Dynamics
- MS/MS
Tandem Mass Spectrometry
- PTM
Post-Translational Modification
- ROS
Reactive Oxygen Species
- S3D
Phosphomimetic cofilin mutant (serine 3 substituted by an aspartic acid)
- S–S
intramolecular disulfide bridge
- TEM
Transmission Electron Microscopy
- ThT
Thioflavin-T
- TMT
Tandem Mass Tags
- V
Volts
1. Introduction
Proteins of the actin depolymerizing factor (ADF)/cofilin family are small proteins involved in a number of physiological processes [1,2]. Their main function is the depolymerization, severing and treadmilling of actin filaments. Through this primary function, they play numerous roles in the cell including maintaining sarcomere's integrity for proper myocyte contractility [[3], [4], [5], [6], [7], [8], [9], [10], [11], [12]]. Owing to their diverse cellular functions, abnormalities in proteins of this family are linked to a number of diseases, along with those collectively known as proteinopathies, characterized by protein precipitation in insoluble aggregates. Due to its recognized involvement in brain proteinopathies, most studies focused on the structure, function and regulation of the ubiquitous isoform cofilin-1. However, aggregates of the muscle isoform cofilin-2, precipitating with its substrate actin, have been described also in the skeletal muscle and the heart [4,[13], [14], [15], [16], [17], [18]].
Cofilin activities and functions are mainly regulated by phosphorylation at serine-3 (Ser3) [[19], [20], [21]] and changes in this post-translational modification (PTM) are associated with proteinopathies both in the brain and in the heart [1,2,13,14]. However, other PTMs such as oxidation are also recognized pathogenic mechanism. Notably, the intertwining between phosphorylation and redox signaling emerged as a fundamental mechanism underlying cofilin changes in response to cellular environmental disorders and stress [6,8,9,22].
While Ser are substrates for phosphorylation, at the molecular level, cysteine residues (Cys) are key to the modifications induced by redox changes. Cys are particularly susceptible to oxidative stress forming covalent bonds within and between proteins. The number and position of the Cys would determine the consequences of oxidative stress on the protein's molecular structure and function and, consequently, on the cell. Interestingly, cofilin-1 contains four Cys (Cys39, Cys80, Cys139, Cys147), whereas the muscle isoform, cofilin-2, only bears two (Cys39 and Cys80). The presence of pairs of Cys allows the formation of intra- and intermolecular disulfide bridges. While both isoforms can form protein aggregates [13,14,[16], [17], [18],[23], [24], [25], [26]], whether oxidative stress would differently affect the isoforms propensity to aggregate based on the Cys content is unknown. Furthermore cofilin-1 aggregates following oxidative stress seem to contain only unphosphorylated protein [14,16], while the muscle aggregates are formed prevalently by phosphorylated cofilin-2 [13].
Such differences in the Cys distribution and phosphorylation state between cofilin isoforms aggregates led us to hypothesize that oxidative stress may have different effects on molecular changes or types of aggregation between the two isoforms. It also prompted us to investigate the interplay between phosphorylation and oxidation in the different mechanism by which oxidative stress promotes aggregation of cofilin-2. Specifically, we investigated how oxidative stress affects the molecular structure of the un/phosphorylated forms of cofilin-2, its interplay with phosphorylation in Ser3 and if/how this interaction fosters protein aggregation and its functional consequences on cells. To this purpose, we used the cofilin-2 phosphomimetic mutant S3D, in which the serine residue is substituted by an aspartic acid residue, which is known to well mimic the effect of phosphorylation.
Finally, since sulfide ion is a known reducing agent and has been proposed as therapeutic agent to limit ischemic damage by suppressing cofilin-2 [27,28] we explored the molecular mechanisms by which sulfide ion affects the propensity of cofilin-2 to aggregation.
2. Materials and methods
A detailed description of the materials and experimental procedures is reported in SI 1.
No safety hazards were encountered.
3. Results
3.1. Cofilin-2 can be oxidized to form a disulfide bridge between Cys39 and Cys80 and is facilitated by phosphorylation
Oxidative stress is known to promote cofilin-1 aggregation, which is proposed to be strongly dependent upon the formation of an intramolecular disulfide bridge between the two internal Cys39-80 [[29], [30], [31]]. These two residues are also present in the muscle isoform (cofilin-2). Instead, two external Cys residues (Cys139 and Cys147) are absent in cofilin-2 and it is unknown whether this would produce different aggregation response under oxidative stress. Therefore, we investigated the oxidation process of the two Cys featured by cofilin-2 and establish whether they can form an intramolecular disulfide bridge (S–S). For this, we used an electrochemical approach which allows a direct detection of the redox properties. Since Ser-3 phosphorylation regulates the structure and function of cofilin isoforms and is associated with actin bundles aggregation [13,17,18,26,27], we tested WT and the phosphomimetic mutant where Ser in position 3 is replaced with Asp to inhibit the binding to actin mimicking the inactive phosphorylated form of cofilin (S3D hereafter).
We recorded a well-shaped cyclic voltammetry (CV) response (a signal consisting of well-defined and resolved peaks) consisting of a cathodic peak and the corresponding anodic counterpart on both the proteins at high scan rate (Fig. 1), indicating a reversible oxidation/reduction process. The standard reduction potential E° was calculated as the semi-sum of the cathodic and anodic peak potentials and referred to SHE. We obtained E°WT = -0.168 V and E°S3D = -0.207 V (pH = 7, T = 20 °C). We observed a marked dependence of the electrochemical signals on the potential scan rate v, on the starting potential and on the delay time for both forms (details about the electrochemical behavior in SI2 and Fig. SI 2.1- Fig. SI 2.4). The E° values of both WT and S3D showed a linear pH-dependence (SI2, Fig. SI 2.5) in the pH range 5–10, consistent with a single 2e−/2H+ mechanism as requested by the formation of a disulfide bridge. Control experiments performed after Cys alkylation showed that no electrochemical response occurs for the S-alkylated cofilin-2. Moreover, the E° values are similar to those obtained for disulfide reduction in other proteins [32,33]. Therefore, the observed electrochemical response can be confidently assigned to the formation of an intramolecular disulfide bridge between Cys39 and Cys80.
Fig. 1.

Direct oxidation/reduction of cofilin cysteines. Cyclic voltammogram at high scan rate (2 Vs−1) for cofilin-2 WT (orange line) and S3D (blue line) immobilized on anionic SAM of MUA/MU. Potentials are vs. SHE. Potential scans started from E = +0.2 V. Delay time: 50 s; Electrolyte solution: 10 mM sodium perchlorate and 5 mM buffer phosphate at pH 7. T = 293 K. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
S3D is characterized by a markedly more negative E°' value and by a significantly higher increase in current as a function of the delay time compared to WT (SI2, Fig. SI 2.4). Overall, the voltammetric results indicate that Ser3 phosphorylation facilitates the formation of Cys39-80 S–S bridge under (electrochemical) oxidative stimuli.
To explore the possibility that Cys residues could yield the disulfide bridge by chemical oxidation (we chosen hydrogen peroxide, the least reactive ROS), we performed the CV measurements in cathodic (reductive) scan on WT and S3D cofilin-2 treated with 50 μm H2O2 for 1 h. For the WT, we did not observe any signal in cathodic scan with a delay time = 0 s, i.e. without electrochemically pre-oxidizing the adsorbed protein (SI2, Fig. SI 2.6A). Conversely, the S3D mutant showed an intense, well-defined cathodic signal (SI2, Fig. SI 2.6B) also without any delay time. Shape and intensity of this signal remained unchanged regardless of the delay time value.
These results provide further evidence that phosphorylation of cofilin facilitates the formation of the disulfide bridge upon (mild) chemical oxidation.
3.2. Effects of chemical oxidation by H2O2 on the cys residues of cofilin-2 WT and S3D are shown by Tandem Mass Tags spectrometry
Oxidative stress causes cardiodepression by changing the expression or phosphorylation of proteins [29]. Thus, here we further verified the differential susceptibility to oxidation modification between the phosphorylated and non-phosphorylated form of cofilin-2 using Mass Spectrometry. Oxidative modification of Cys is one of the major PTM involved in ROS-mediated cellular signaling. These modifications include sulfenic acid (Cys-SOH), sulfonic acid (CysSO3H), and inter- and intra-disulfide adduct formations, among others [30]. The most direct and sensitive method for determining the presence of such structural changes is the measurement of the mass of the oxidized cysteine residue. For this reason, we investigate the effects of chemical oxidation by H2O2 on the Cys residues of cofilin-2 WT and S3D using ‘Tandem Mass Tags’ (iodoTMT) spectrometry (SI3) [31]. The analysis of the MS/MS spectrum of the two peptides 34AVLFCLSDDKR46 and 73LLPLNDCR82 (peptides containing the two cysteines Cys39 and Cys80 that are obtained by tryptic digestion) upon addition of iodoacetamide (IAA) allows highlighting their oxidation state as schematically represented in SI3. IodoTMT is based on the use of IAA to add a carbamidomethylation to the reduced Cys residue (SH-) resulting in an increase in mass of about 57 Da (SI3 A and B). Oxidized cysteines are not reactive and, consequently, do not carry the modification (SI3 A and C).
The MS/MS spectra of WT H2O2-treated cofilin-2 show that in both peptides the Cys are alkylated (data not shown), indicating that even upon chemical oxidation with H2O2 the Cys are still reduced. Conversely, S3D cofilin-2 treated with H2O2 produces peptides with Cys oxidized to sulfonic acid (RSO3H; +47 Da) or thiosulfonic acid (RS2O2H; +63 Da), as shown by the MS/MS spectra for the AVLFCLSDDKR fragment containing Cys 39 reported in SI 4 [30]. The two oxidized forms are degradation products of disulfide bridge [30]. The data resulting from iodoTMT spectrometry therefore support the electrochemical results, indicating that, in the phosphorylated form, Cys 39 and Cys 80 are more prone to change their oxidation state to form a disulfide bridge (-S-S-).
3.3. In silico studies on cofilin-2 in unphosphorylated and phosphorylated form by molecular dynamics (MD) simulations
Protein phosphorylated state favors changes induced by oxidation [29]. To understand the molecular and structural basis of the phosphorylation-dependent oxidation of cofilin-2, we performed in silico studies on the unphosphorylated and phosphorylated form of cofilin-2 by molecular dynamics (MD) simulations, paying particular attention to the environment surrounding the cysteine residues. Overall, MD simulations did not show major structural changes upon Ser3 phosphorylation (SI 5). Rearrangements close to Cys39 and Cys80 were explored by mapping the occupancy percentage of other residues around 5 Å from the S atom of the Cys residues and analyzing the H-bonds involving their side chains along the whole sampling obtained for both forms. The conformation and dynamics around Cys80 remain almost unaltered upon phosphorylation, while the dynamics of the surroundings of Cys39 changes appreciably (Fig. 2). In particular, the main H-bond partners of the –SH group of Cys39 are Leu40 and Arg81 in the unphosphorylated form and change to Glu50 in the phosphorylated one [32]. In the latter, Cys39 becomes a hydrogen bond donor, resulting in a partially negative charge on the sulfur and decreasing of the thiol pKa. In this way, the Cys is more easily oxidizable [[33], [34], [35], [36], [37]]. This mechanism is known to underly the physiological redox regulation of a number of proteins in the body [34,37,38].
Fig. 2.

Effect of phosphorylation on the hydrogen bonds network of cys39 and cys80 by MD simulation. (A) percentage occupancy around the sulfur atom of Cys39 or Cys80 along the whole trajectory, in gray scale. In black we have 100% of occupancy, in white 0%. For clarity reason, we have used the same structure to draw the data. (B) Hydrogen bonds persistence of every couple, as mean ± max/min values. The first number of the couple identifies the donor, the second one identifies the acceptor.
3.4. Cofilin-2 forms amyloid structures upon aerobic aging or in the presence of H2O2
Phosphorylation of cofilin-2 is involved in formation of pathological aggregates in the heart and favors the formation of disulfide bridges under mild oxidative (H2O2) conditions [13,14,39,40]. Here we investigated whether the presence of S–S bridge is the mechanisms that mediates oxidative stress induced aggregation of cofilin-2. The possibility of forming amyloid structures is a critical aspect of this problem. For this reason, we used a variety of techniques (Circular Dichroism, Thioflavin-T (ThT) fluorescence emission and Fourier-Transform InfraRed Spectroscopy FTIR) able to directly identify the presence of the amyloid β-sheet. To this purpose, we investigated the progressive appearance of amyloid structures under unstressed conditions and upon incubation with H2O2 (H2O2/protein molar ratio = 10).
Circular dichroism (CD): The far-UV spectra mainly depend on secondary structure content and are indicative of the presence of α-helix and β-sheet structures or other conformation. The CD spectra of cofilin-2 WT and S3D (Fig. 4) are typical of proteins featuring both α-helices and β-sheet rich regions (involving indeed in both proteins about 30% and 20% of the amino acid residues, respectively). In fact, a minimum at 208 nm, a well-shaped shoulder at about 222 nm and a very shallow shoulder at about 230 nm are observed (Fig. 3) [41]. Far-UV CD spectra of untreated cofilin-2 WT (Fig. 3A) and S3D (Fig. 3B) and cofilin-2 WT treated with H2O2 (Fig. 3C) showed quite similar time dependence. In fact, aging of these samples led to a slow, but progressive decrease in the intensity of the CD signals suggesting the formation of protein aggregates. Moreover, all these samples, regardless of whether or not they were exposed to H2O2, featured a peak forming around 215 nm, which increases with time and is well clean-cut after 3 days (Fig. 3A, B and C). This signal can be confidently associated with the formation of β-sheets, typical of amyloid structures [42,43]. After three days, the proteins form amorphous aggregates. Instead, the far-UV spectra of cofilin-2 S3D treated with H2O2 (Fig. 3D) shows, already from the first day, a more intense band at 215 nm. This signal becomes predominant after three days (Fig. 3D) and is accompanied by the formation of distinctly fibrillary aggregates.
Fig. 4.

Effect of oxidative stress on cofilin amyloid formation shown by ThT fluorescence emission. (A) Plot of the time course of ThT fluorescence intensity for: ■ cofilin-2 WT, ▲cofilin-2 WT treated with H2O2, ♦ cofilin-2 S3D, ● cofilin-2 S3D treated with H2O2. Protein concentration 5 μM, 10:1 H2O2/protein molar ratio. The plot shows the mean value of 8 replicates, the error bar corresponds to the standard deviation. (B) Plot of the time course of ThT fluorescence intensity for WT (■) and S3D (♦) exposed to H2O2 (10:1 H2O2/protein molar ratio, protein concentration 5 μM) for 2 h, then treated with Na2S at 2:1 M ratio with the protein. As references, the same samples treated with H2O2 but not with Na2S are also reported: (▲) cofilin-2 WT, (●) S3D. The plot shows the mean value of 8 replicates, the error bar corresponds to the standard deviation.
Fig. 3.

Effect of oxidative stress on the cofilin secondary structure shown by Far-UV CD. Far-UV CD spectra of cofilin-2 WT (A, C) and S3D (B, D) as a function of time (t = 0 blue, t = 1 day orange, t = 2 days gray, t = 3 days yellow), samples untreated (A, B) and treated with H2O2 (C, D). H2O2/protein molar ratio = 10. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
ThT fluorescence emission: To confirm the formation of amyloid structures, fluorescence emission measurements were performed on samples of WT and S3D cofilin-2 in the presence of ThT, a fluorescent dye used to visualize amyloid structures [[44], [45], [46]]. Under the same experimental conditions used for CD measurements, we invariably observed the time dependent appearance of an emission band at 482 nm, diagnostic for the binding of ThT to amyloid structures (Fig. 4A). The signal intensity, however, decreases after >80 h (Fig. 4A). The time dependences of ThT fluorescence emission for the different samples (WT and S3D, with and without H2O2) are rather similar. This lack of differences is not surprising since the ThT emission is known to be dependent on type and dimension of β-aggregates (oligomers or fibrils) [46,47].
Infrared spectroscopy: The band of amide I vibration in proteins is very sensitive to the secondary structure of the backbone [48,49]. In fact, the α-helix is characterized by an amide I band falling near 1650 cm−1, while that of the (amyloid) β-sheet structures shifts to about 1630 cm−1. ATR-FTIR spectra realized on cofilin-2 WT and S3D, show a broad band at 1649 nm which can be confidently assigned to the α-helices of the protein with a minor contribution of the β-sheet structures (SI 6). For both forms, this band moves to 1638 nm (SI 6A and 6B, blue and red lines) over time. Upon treatment with H2O2, the WT protein shifts again to 1638 nm (SI 6A, green line), while S3D band moves up to 1629 nm (SI 6B, green line). The broad band at 1638 nm could arise to the overlapping of a band due to α-helices or random coil and a more pronounced contribution of β-sheets. For S3D, however, the contribution due to the β-sheets structures is remarkably enhanced. Therefore, the WT and S3D aggregates that are formed over time (including WT treated with H2O2) appear to be only partially composed of amyloid structures. Conversely, the sharp band at 1629 nm suggest that the aggregates formed by S3D treated with H2O2 predominantly contains amyloid structures, in agreement with the CD measurements.
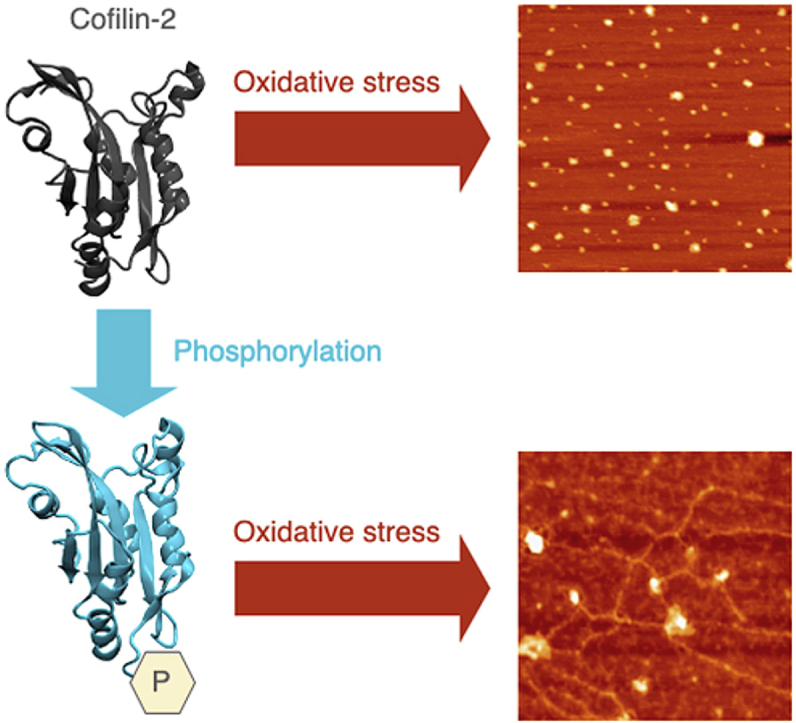
3.5. Phosphorylation drives the morphology of protein aggregates upon exposure to H2O2
The morphology of the protein aggregates can be very different and plays a key role in several degenerative diseases, therefore we followed the aggregation process of cofilin-2 WT and S3D over time both in the absence and presence of H2O2 by Atomic Force Microscopy (AFM), a technique that allows to observe directly the morphology of the aggregate protein structures that are formed. The images where taken on freshly cleaved atomic-flat surface of clinochlore, a phyllosilicate which presents physico-chemical properties suitable for studying the adsorption of proteins and protein aggregates [50,51]. In fact, the surface of this specific substrate simultaneously exposes regions of the TOT layer (TOT layer stands for tetrahedral-octahedral-tetrahedral layer) with negative surface potential and regions with positive surface potential, belonging to the brucite-like layer (bright areas) [51,52]. We observed aggregates of untreated WT and S3D cofilin-2 deposited on the positively charged, hydrophobic, bright areas (SI 7A, C) as isolated protein structures of pseudo-circular/elliptical shape (red arrows) together with a prevailing population of structured globular aggregates of varying morphology (white arrows). The approximate minimum dimensions of the isolated pseudo-circular structures for both cofilin-2 WT and S3D are below 1 nm in height and <15 nm in width. Instead, fewer interactions were seen on the negatively charged, hydrophilic surfaces. Exposure of WT cofilin-2 to H2O2 (10:1 H2O2/protein molar ratio) for three days resulted in the deposition of globular aggregates preferentially on the positively charged (hydrophobic) areas that were almost completely saturated (SI 7B). Several structures, however, were present also on the negatively charged (hydrophilic) areas. The aggregates adsorbed on the positively charged (hydrophobic) areas were densely packed to form a monolayer. Moreover, we observed punctual pseudo-circular/elliptic structures adsorbed even onto the monolayer itself. The average height of the monolayer is about 1.5 nm. The smallest observed structures have an average height of less than one nm and a width of less than 10 nm. The negatively charged (hydrophilic) areas, instead, showed isolated adsorbed structures with a pseudo circular/elliptical geometry and size similar to the punctual aggregates adsorbed on the positively charged (hydrophobic) areas.
S3D shows a different behavior upon exposure to H2O2 (SI 7D): well defined filamentous (fibrillar) structures completely covered the positively charged (hydrophobic) areas, while no adsorbed aggregate was found on the negatively charged (hydrophilic) surfaces. The filamentous structures have the following approximate dimensions: height about 2 nm and average length of 110 nm. In several cases the fibrils overlap and intertwine.
Since bare mica is unable to immobilize cofilin-2 and its aggregates, we performed further AFM measurements using Mg(II)-treated mica. This processing is known to activate the surface of the mica to the adsorption of biopolymers and can be successfully used for negatively charged proteins. Under this experimental condition we confirmed the morphological characteristics of the cofilin-2 aggregates with and without H2O2 although aggregates were less densely populated (Fig. 5, SI 7 and SI 8) [47,[53], [54], [55]]. Overall, purified cofilin-2 forms aggregates over time under aerobic and oxidative (H2O2) conditions, but, primarily, oxidative stress induces the formation of amyloid fibrillary structures only for the phosphorylated cofilin-2.
Fig. 5.

Effect of oxidative stress on cofilin aggregation imaged by Atomic Force Microscopy. Morphological AFM images of the aggregates formed after 3 days on mica surfaces treated with Mg(II) by cofilin-2: WT, WT exposed 2 h to 50 μM H2O2, WT exposed 2 h to 50 μM H2O2 and then treated with 10 μM sulfide ion, S3D, S3D exposed 2 h to 50 μM H2O2 and S3D exposed 2 h to 50 μM H2O2 and then treated with 10 μM sulfide ion after three days. Protein concentration about 5 μM.
3.6. The susceptibility of phosphocofilin-2 to form amyloid β-sheet aggregates under oxidative stress is prevented by sulfide ion
Sulfide has been proposed as a therapeutic agent in ischemia associated with cofilin-2 dyshomeostasis [27,28]. Here, we tested whether the mechanisms for this effect could be related to the ability to directly affect cofilin-2 aggregation processes with ThT fluorescence, AFM and ATR-FTIR. Fluorescence emission measurements of ThT (emission to 482 nm) were carried out on cofilin-2 WT and S3D samples exposed to H2O2 and then treated with Na2S. Fig. 4B shows the change in ThT fluorescence with time with and without treatment with Na2S. The time dependence of the fluorescence intensity of WT was not affected by the presence of sulfide (Fig. 4B) while ThT fluorescence emission of S3D in the presence of sulfide was significantly lowered throughout the whole range of the investigated times.
Similar to the results of ThT fluorescence, AFM measurements showed that the morphologies of the aggregates of WT cofilin-2 do not change over time in the presence of sulfide (Fig. 5), whereas strong differences were observed for the S3D mutant (Fig. 5). In fact, the fibrillary structures characteristic of S3D exposed to H2O2 (Fig. 5, SI 7, SI 8 and SI 9) disappeared with sulfide and were substituted by globular aggregates (Fig. 5).
Using ATR-FTIR, regardless of the treatment with sulfide, the spectra of WT samples exposed to H2O2 for 2 h showed an enlarged amide I band at about 1638 nm (SI 10). Untreated S3D samples, instead, showed a narrow band at 1629 nm (see above), but after exposition to sulfide the amide I band shifts to 1640 nm and broadens, resulting very similar to that observed for WT.
These results suggest that, for S3D, the treatment with sulfide decreases the amyloid β-sheet in cofilin-2 aggregates.
3.7. Oxidative stress induces cofilin aggregation and sarcomeric disarray in cardiomyocytes
Cofilin-2 precipitates in cofilin-actin rods in cardiomyocytes from human failing hearts and in mouse models of cofilin-2 knock-down. Here we tested whether the oxidative cofilin-2 structural changes induced by H2O2 observed in vitro and in silico are relevant also in the more complex cellular environment of ex vivo isolated cardiomyocytes recapitulating the accumulation of stress fibers observed in the mouse model [13]. To this end, we exposed normal C57Bl/6 WT mice cardiomyocytes to 10 μM H2O2 treatment and imaged them by transmission electron microscopy (TEM) and immunofluorescence (IF). Concentrations of H2O2 (>0.1 μM) are considered supra-physiological and known to induce oxidative stress and cell damage [56] and a range of 0.1–50 μM H2O2 has been shown to induce loss of high energy phosphate [57].
The exposed cells displayed signs of misfolded protein aggregate accumulation together with sarcomeric addensations and sarcomeric disarray (Fig. 6A), similar to what was found in patients with nemaline cardiomyopathy and mice models of cofilin-2 deletion [13].
Fig. 6.

Effect of oxidative stress on isolated cardiomyocytes morphology. (A) TEM images of C57Bl/6 WT cardiomyocytes exposed to normal media showed intact sarcomeric structure and mitochondrial morphology (upper panels). Following 1 h exposure to 10 μM H2O2 cells showed sarcomeric disarray, addensations (arrow) (lower left panel) and mitophagy (lower right panel). (B) Immunofluorescence images. Upper panels show C57Bl/6 WT isolated cardiomyocytes cultures in normal media or H2O2 for 15 or 30 min. Images show cofilin-2 redistribution to the sarcolemma after 15 min, cofilin aggregation (arrows) and pre-amyloid oligomers (PAO) accumulation (arrowheads) in the treated cells after 30 min. Cofilin-2 loses the sarcomeric distribution in the treated cells and redistributes in the sarcolemma. In blue: DAPI staining of the cell nucleus. In red A11 stained PAO; In green cofilin-2. Bottom panel: negative control. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Known changes described in cardiomyocytes following oxidative stress such as mitophagy were also evident in the images. Disarray was confirmed by IF as soon as 15 min after exposure, and positive staining for oligomeric aggregates was evident after 30 min of H2O2 exposure (Fig. 6B) supporting the susceptibility of cofilin-2 to oxidative stress in intact cells.
4. Discussion
A number of physiological functions in the cell depend on the dynamic organization of the actin cytoskeleton, which, in striated muscle cells - including cardiomyocytes - is also essential for contractility. The rearrangement of actin cytoskeleton is primarily mediated by cofilin that, because of its critical functions, is tightly regulated by PTM. Phosphorylation is the main PTM regulating normal cofilin activity, yet, other PTM, such as oxidation, may affect cofilin function leading, to myocardial damage.
The cardiodepressive effect of oxidative stress has been attributed, for the most part, to the effect on expression or phosphorylation of Ca2+ handling proteins, however, oxidative stress was also shown to induce changes in the phosphorylation status of the sarcomeric proteins [29,58,59], although oxidant agents, such as HNO have been shown to also have a cardioprotective effect via oxidation of both Ca2+ handling [60] and sarcomeric proteins [61]. Instead the effect of oxidative stress hasn't been shown for the sarcomeric regulatory proteins. Since cofilin activity is, itself, regulated by phosphorylation, oxidative stress may mediate the abnormal function of cofilin and, consequently, of the thin filaments, found in degenerative diseases.
Abnormalities of cofilin have been, in fact, shown to participate in the pathogenesis of degenerative diseases of the brain, skeletal and cardiac muscle and, most recently in myocardial ischemia [13,14,17,18,23]. Importantly, those studies identified cofilin-2 as a critical target of a therapeutic agent (sulfide) for this disease [27,28]. Thus, here we investigated the chemical consequences of changes in the redox equilibrium on cofilin-2, its phosphorylation state, how those changes may predispose to protein aggregation observed in diseases, and if oxidation of cofilin-2 represents the mechanism of protection of sulfide.
4.1. Molecular response of cofilin to oxidative stress
Although oxidation is possible for unphosphorylated cofilin, we found that oxidative stress affects the phosphorylated protein to a greater extent than the unphosphorylated counterpart.
In cofilin-1, oxidation has been shown to determine the formation of disulfide bridges responsible for losing its actin-depolymerizing ability [39,62]. Instead no data are available for the muscle isoform, cofilin-2. This is important, because cofilin isoforms differ in Cys content underlying potential different influence of redox imbalance on protein structure and function and/or susceptibility to stress. Cofilin-1 contains four Cys paired in the external or internal structure of the protein (Cys39 and Cys80 buried internally, Cys137 and 139 exposed on the surface), while only the two internal Cys are present in cofilin-2.
Since cysteines are unique amino acids for their property to undergo reversible redox reactions as part of their normal function, their oxidation might cause the formation of cross-linked bonds (Cys-S--S-Cys) with the aim to provide structural stability to the proteins or change their structure or activity. Disulfide formation indeed requires the proximity of two Cys, but also chemical conditions favouring this process, like modifications of the thiol group acidity [34,38]. ROS species, in particular H2O2, are among factors responsible for Cys oxidation [63,64].
Our results provide evidence that, similar to what was obtained for cofilin-1 [39,62], a putative disulfide bridge forms under oxidative stress in cofilin-2 [33,65]. To verify this, we repeated the measurements with proteins treated with an alkylating agent. Cys alkylation, confirmed by mass spectroscopy, abrogated altogether the redox electrochemical response of the protein, supporting the fact that Cys are prone to a (partially) reversible oxidation.
4.2. Phosphorylated cofilin-2 is more prone to oxidation and to form intramolecular disulphide bridges
Under proper conditions, both unphosphorylated (WT) and phosphorylated (phosphomimetic S3D) cofilin-2 can be oxidized, in a partially reversible manner, as shown in our CV measurements. However, their ability and rate of formation of the disulfide bridge appear different. The electrochemical investigation indicates that the oxidation of the Cys is rather slow for both WT and S3D. This process, however, results remarkably slower for WT as shown by the plot of the peak currents vs. delay (pre-oxidation) time and the larger peak-to-peak separation (ΔEp = 130 mV for WT and ΔEp = 56 mV for S3D) which indicates that the electron transfer rate is much faster for S3D [66]. In addition, S3D oxidation is thermodynamically favored. In fact, this mutant showed an E°' value more negative than WT (E°WT = -0.168 V and E°S3D = -0.207 V), according to its overall higher tendency to convert to the oxidized non-native form.
4.3. Changes in H-bond network of Cys39 account for the phosphorylated cofilin-2 permissive oxidation
Interestingly oxidation of the Cys residues and the formation of the disulfide bridge only occurred for the phosphomimetic form of cofilin-2 when oxidation was promoted by exposure to H2O2 as shown in our MS experiments.
Our in silico modeling allowed us to elucidate the chemical basis of the oxidation process. Inspection of the three-dimensional structure of cofilin-l and -2 sampled with MD simulations showed that the mean distance between the sulfur atoms of the two internal Cys is large (about 1 nm) and the percentage occupancy of spaces compatible with the formation of the disulfide bridge is rather limited, reasonably accounting for the slow oxidation processes.
The in silico simulations provided an explanation for the selective oxidation of the S3D form. Phosphorylation on Ser-3 leaves the environment of the Cys80 is almost unaltered, while it changes the H-binding partner of the internal Cys39 from the Leu40 and Arg81 to Glu50 making Cys39 much more prone to oxidation than the corresponding residue in WT, in which Cys39 acts as an acceptor of the H-bond, a mechanism described also for other proteins [34,67] and causing the formation of intramolecular disulfide bridges.
4.4. Phosphorylation favors the formation of amyloid β-sheets
The above conformational changes in phosphorylated cofilin-2 in response to redox state may predispose to protein aggregation in diseases associated with oxidative stress. While we were not able to correlate oxidation with the kinetic of formation of amyloid β-sheets in the protein aggregates using ThT binding, CD and ATR-FTIR measurements demonstrated the appearance fibrillar content in the S3D treated with H2O2.
For both phosphorylated and unphosphorylated species, treated or untreated with H2O2, we invariably found evidence of the formation of amyloid β-sheets and the time course of the monitored signal suggested that a seeding mechanism was followed. Nevertheless, the variability of the replicates hampered the understanding of the mechanism by which phosphorylation affects cofilin-2 oxidation by H2O2. This shortcoming was previously reported for several amyloid β-aggregation phenomena and it was suggested to be an effect of the stochasticity of the seeding process [68].
In support of the CD and ATR-FTIR measurements, the AFM results showed that S3D treated with H2O2 forms elongated fibrillar structure, confirming that phosphorylated cofilin-2 can give rise to amyloid-like fibrils. The AFM results also indicate a higher affinity of the misfolded cofilin for positively charged hydrophobic surfaces. This suggests the possibility that misfolded proteins have higher propensity to adhere to the hydrophobic lipid bilayer of the cell membrane favouring the seeding of misfolded proteins released from dying cells. Thus, released aggregates may more easily deposit over a resting cardiomyocyte forming circular species and, over time, transmembrane channels. On the other hand, within the cell, the preferred hydrophobicity may favour the adhesion of misfolded proteins to intracellular membranes (ER, Golgi, lysosomes), lipid rich structures (lipid bodies), lipid metabolism organelles (mitochondria) or cell membrane for exocytosis favouring cell damage and further extracellular damping.
4.5. Oxidized cofilin-2 induces aggregates
The exposure of cardiomyocytes to oxidative stress supported the biological relevance of the oxidation-induced aggregation of cofilin-2. Exposure to supra-physiological concentrations of H2O2 induced the aggregation of cofilin and sarcomeric disarray. The increased oxidation observed after phosphorylation could diminish the pool of cofilin-2 by targeting cofilin-2 to degradation or by removing it from the phosphorylation/dephosphorylation cycle, as oxidized cofilin-2 cannot react with LIMK1 [62,69]. These events could act as triggers and form the basis for a feedback mechanism, in which an initial small direct and/or indirect formation of aggregates leads to substantial protein dishomeastasis and burden on the protein quality control (PQC) system. Scheme 1 summarizes the effect of aerobic aging and H2O2 reaction on cofilin-2 WT and its S3D mutant which can lead accumulation of misfolded protein aggregate of different morphologies in cardiomyocytes.
Scheme 1.

Scheme depicting the effect of aerobic conditions and H2O2reaction on cofilin-2 WT and its S3D mutant. Purified cofilin aggregates over time. Cofilin unphosphorylated and phosphorylated are represented by their isosurfaces of electrostatic potential (in blue: positive potential, in red: negative). Under oxidative stress the phosphomimetic form of cofilin progresses towards the formation of amyloid fibers, whereas the WT maintains the aggregation path found under aerobic conditions. Sulfide prevents the formation of both large aggregates as well as amyloid fibers in the WT and S3D.
4.6. Sulfide is effective in inhibiting the aggregation of phosphorylated cofilin-2
The sulphide ion strongly affects the aggregation process of S3D and therefore, reasonably, of phosphorylated cofilin-2. In particular, sulphide depresses the formation of β-sheet structures. Given the key role played by Cys39 in the protein aggregation, the possibility of a direct interaction between this amino acid residue and the sulphide ion cannot be excluded. Our findings may also provide a chemical explanation for the previously described cofilin-2 mediated protective effect of treatment with sodium sulfide (Na2S) against ischemic and inflammatory injury [27,28]. In addition to the proposed mechanism of the induction of microRNA (miR-21) suppressing cofilin-2 [27], it is also possible that sulfide would induce Cys S-sulfhydration of unbound cysteine residues [70] breaking the disulfide bond. Thus, sulfide may exert a dual mechanism by breaking cofilin-2 disulfide bond and occupying the free Cys residues thereby changing the microenvironment around the Cys39. This mechanism may also be invoked to explain the reduction in the inflammatory response by mediating T-cell activation and migration.
5. Conclusions
Our findings support the importance of cofilin-2 in proteinopathies. The susceptibility of the phosphorylated cofilin-2 to oxidative stress may identify a possible trigger that initiate the aggregation cascade in non-ischemic and ischemic cardiomyopathies as well as in the aging organs where oxidative stress is elevated. Finally, the Cys39 intramolecular interaction and the formation of the disulfide bridge may represent the mechanism mediating the susceptibility of the inactive cofilin to oxidative stress and explain the therapeutic effect of sulfide. Our findings underly the importance of preventing aggregation for the development of proteinopathies and the potential use of sulfide donors as therapeutic agents in ischemic and non-ischemic diseases in the brain, skeletal muscle and heart as well as aging.
6. LIMITATIONS and STUDY in PERSPECTIVE
Our study was mainly in silico and in vitro. Although some measurements made in cardiomyocytes result meaningful, in vitro cellular experiments with the phosphorylated form of cofilin-2 as well as in vivo experiments are required for the translational relevance. Thus, future studies would focus on testing the mechanism of formation of fibrillar structures in vivo, to understand how mitochondria are involved in the processes of formation of globular and fibrillar amyloid structures, to verify the possibility that the aggregation process, under physiological or pathological conditions, can also involve other proteins.
Although our results suggest that the two internal Cys residues (present in both cofilin isoforms) are sufficient to destabilize the proteins structure under oxidative stress and potentially facilitate protein aggregation, it still remains to understand the role of phosphorylation in oxidation induced aggregation in cofilin-1 vs cofilin-2 that differs in terms of phosphorylated proteins content.
Fundings
This work was supported by the American Heart Association (grant AHA 14IRG18980028) and MUSC (discretional funds) to FdM and by the University of Modena and Reggio Emilia (grant UNIMORE FAR2020) to MB.
Declaration of competing interest
No conflicts of interest were disclosed.
Acknowledgements
An appreciated technical support was provided by Centro Interdipartimentale Grandi Strumenti (CIGS) of Università di Modena e Reggio Emilia and its staff.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101691.
Contributor Information
Marco Borsari, Email: marco.borsari@unimore.it.
Federica del Monte, Email: delmonte@musc.edu.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hild G., Kalmar L., Kardos R., Nyitrai M., Bugyi B. The other side of the coin: functional and structural versatility of ADF/cofilins. Eur. J. Cell Biol. 2014;93(5–6):238–251. doi: 10.1016/j.ejcb.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Van Troys M., Huyck L., Leyman S., Dhaese S., Vandekerkhove J., Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur. J. Cell Biol. 2008;87(8–9):649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Wiggan O., Schroder B., Krapf D., Bamburg J.R., DeLuca J.G. Cofilin regulates nuclear architecture through a myosin-II dependent mechanotransduction module. Sci. Rep. 2017;7:40953. doi: 10.1038/srep40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munsie L.N., Desmond C.R., Truant R. Cofilin nuclear-cytoplasmic shuttling affects cofilin-actin rod formation during stress. J. Cell Sci. 2012;125(Pt 17):3977–3988. doi: 10.1242/jcs.097667. [DOI] [PubMed] [Google Scholar]
- 5.Dopie J., Skarp K.P., Rajakyla E.K., Tanhuanpaa K., Vartiainen M.K. Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. U. S. A. 2012;109(9):E544–E552. doi: 10.1073/pnas.1118880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chua B.T., Volbracht C., Tan K.O., Li R., Yu V.C., Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat. Cell Biol. 2003;5(12):1083–1089. doi: 10.1038/ncb1070. [DOI] [PubMed] [Google Scholar]
- 7.Xiang S.Y., Ouyang K., Yung B.S., Miyamoto S., Smrcka A.V., Chen J., Heller Brown J., PLCepsilon PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 2013;6(306):ra108. doi: 10.1126/scisignal.2004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Posadas I., Perez-Martinez F.C., Guerra J., Sanchez-Verdu P., Cena V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J. Neurochem. 2012;120(4):515–527. doi: 10.1111/j.1471-4159.2011.07599.x. [DOI] [PubMed] [Google Scholar]
- 9.Li G.B., Cheng Q., Liu L., Zhou T., Shan C.Y., Hu X.Y., Zhou J., Liu E.H., Li P., Gao N. Mitochondrial translocation of cofilin is required for allyl isothiocyanate-mediated cell death via ROCK1/PTEN/PI3K signaling pathway. Cell Commun. Signal. 2013;11:50. doi: 10.1186/1478-811X-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C.K., Park H.J., So H.H., Kim H.J., Lee K.S., Choi W.S., Lee H.M., Won K.J., Yoon T.J., Park T.K., Kim B. Proteomic profiling and identification of cofilin responding to oxidative stress in vascular smooth muscle. Proteomics. 2006;6(24):6455–6475. doi: 10.1002/pmic.200600124. [DOI] [PubMed] [Google Scholar]
- 11.Kotiadis V.N., Leadsham J.E., Bastow E.L., Gheeraert A., Whybrew J.M., Bard M., Lappalainen P., Gourlay C.W. Identification of new surfaces of cofilin that link mitochondrial function to the control of multi-drug resistance. J. Cell Sci. 2012;125(Pt 9):2288–2299. doi: 10.1242/jcs.099390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li G., Zhou J., Budhraja A., Hu X., Chen Y., Cheng Q., Liu L., Zhou T., Li P., Liu E., Gao N. Mitochondrial translocation and interaction of cofilin and Drp1 are required for erucin-induced mitochondrial fission and apoptosis. Oncotarget. 2015;6(3):1834–1849. doi: 10.18632/oncotarget.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian K., Gianni D., Balla C., Assenza G.E., Joshi M., Semigran M.J., Macgillivray T.E., Van Eyk J.E., Agnetti G., Paolocci N., Bamburg J.R., Agrawal P.B., Del Monte F. Cofilin-2 phosphorylation and sequestration in myocardial aggregates: novel pathogenetic mechanisms for idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2015;65(12):1199–1214. doi: 10.1016/j.jacc.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minamide L.S., Striegl A.M., Boyle J.A., Meberg P.J., Bamburg J.R. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2000;2(9):628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 15.Vandebrouck A., Domazetovska A., Mokbel N., Cooper S.T., Ilkovski B., North K.N. In vitro analysis of rod composition and actin dynamics in inherited myopathies. J. Neuropathol. Exp. Neurol. 2010;69(5):429–441. doi: 10.1097/NEN.0b013e3181d892c6. [DOI] [PubMed] [Google Scholar]
- 16.Bamburg J.R., Bernstein B.W. Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton (Hoboken) 2016;73(9):477–497. doi: 10.1002/cm.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agrawal P.B., Greenleaf R.S., Tomczak K.K., Lehtokari V.L., Wallgren-Pettersson C., Wallefeld W., Laing N.G., Darras B.T., Maciver S.K., Dormitzer P.R., Beggs A.H. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am. J. Hum. Genet. 2007;80(1):162–167. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal P.B., Joshi M., Savic T., Chen Z., Beggs A.H. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 2012;21(10):2341–2356. doi: 10.1093/hmg/dds053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan T.E., Lockerbie R.O., Minamide L.S., Browning M.D., Bamburg J.R. Isolation and characterization of a regulated form of actin depolymerizing factor. J. Cell Biol. 1993;122(3):623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agnew B.J., Minamide L.S., Bamburg J.R. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 1995;270(29):17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 21.Bamburg J.R. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 22.Wabnitz G.H., Goursot C., Jahraus B., Kirchgessner H., Hellwig A., Klemke M., Konstandin M.H., Samstag Y. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis. 2010;1:e58. doi: 10.1038/cddis.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein B.W., Shaw A.E., Minamide L.S., Pak C.W., Bamburg J.R. Incorporation of cofilin into rods depends on disulfide intermolecular bonds: implications for actin regulation and neurodegenerative disease. J. Neurosci. 2012;32(19):6670–6681. doi: 10.1523/JNEUROSCI.6020-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfannstiel J., Cyrklaff M., Habermann A., Stoeva S., Griffiths G., Shoeman R., Faulstich H. Human cofilin forms oligomers exhibiting actin bundling activity. J. Biol. Chem. 2001;276(52):49476–49484. doi: 10.1074/jbc.M104760200. [DOI] [PubMed] [Google Scholar]
- 25.Ockeloen C.W., Gilhuis H.J., Pfundt R., Kamsteeg E.J., Agrawal P.B., Beggs A.H., Dara Hama-Amin A., Diekstra A., Knoers N.V., Lammens M., van Alfen N. Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul. Disord. : NMD. 2012;22(7):632–639. doi: 10.1016/j.nmd.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agrawal P.B., Strickland C.D., Midgett C., Morales A., Newburger D.E., Poulos M.A., Tomczak K.K., Ryan M.M., Iannaccone S.T., Crawford T.O., Laing N.G., Beggs A.H. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations. Ann. Neurol. 2004;56(1):86–96. doi: 10.1002/ana.20157. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen K., D D., Das A., He J., Toldo S., Abbate A., Salloum F.N. Hydrogen sulfide attenuates ischemic heart failure by suppressing pro-apoptotic cofilin-2. Circulation. 2014;130:A16666. [Google Scholar]
- 28.Chau Vh D.T., Romeo F., Balan B., Cain C., Toldo S., Tang D., Shah K., Kasirajan V., Salloum F.N. vol. 136. 2017. p. A16744. (Cardiac Expression Profiles of Cofilin-2 and H2S-Producing Enzyme 3-Mercaptopyruvate Sulfurtransferase in Patients with End-Stage Ischemic Cardiomyopathy). [Google Scholar]
- 29.Steinberg S.F. Oxidative stress and sarcomeric proteins. Circ. Res. 2013;112(2):393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H.J., Ha S., Lee H.Y., Lee K.J. ROSics: chemistry and proteomics of cysteine modifications in redox biology. Mass Spectrom. Rev. 2015;34(2):184–208. doi: 10.1002/mas.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao C., Behring J.B., Shao D., Sverdlov A.L., Whelan S.A., Elezaby A., Yin X., Siwik D.A., Seta F., Costello C.E., Cohen R.A., Matsui R., Colucci W.S., McComb M.E., Bachschmid M.M. Overexpression of catalase diminishes oxidative cysteine modifications of cardiac proteins. PloS One. 2015;10(12) doi: 10.1371/journal.pone.0144025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou P., Tian F., Lv F., Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins. 2009;76(1):151–163. doi: 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- 33.Johnson D.L., Polyak S.W., Wallace J.C., Martin L.L. Probing the stability of the disulfide radical intermediate of thioredoxin using direct electrochemistry. Lett. Pept. Sci. 2003;10(5–6):495–500. [Google Scholar]
- 34.Witt A.C., Lakshminarasimhan M., Remington B.C., Hasim S., Pozharski E., Wilson M.A. Cysteine pKa depression by a protonated glutamic acid in human DJ-1. Biochemistry. 2008;47(28):7430–7440. doi: 10.1021/bi800282d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazmanian K., Sargsyan K., Grauffel C., Dudev T., Lim C. Preferred hydrogen-bonding partners of cysteine: implications for regulating cys functions. J. Phys. Chem. B. 2016;120(39):10288–10296. doi: 10.1021/acs.jpcb.6b08109. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez R., Riddle M., Woo J., Momand J. Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 2008;17(3):473–481. doi: 10.1110/ps.073252408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobbs G.A., Mitchell L.E., Arrington M.E., Gunawardena H.P., DeCristo M.J., Loeser R.F., Chen X., Cox A.D., Campbell S.L. Redox regulation of Rac1 by thiol oxidation. Free Radic. Biol. Med. 2015;79:237–250. doi: 10.1016/j.freeradbiomed.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madzelan P., Labunska T., Wilson M.A. Influence of peptide dipoles and hydrogen bonds on reactive cysteine pKa values in fission yeast DJ-1. FEBS J. 2012;279(22):4111–4120. doi: 10.1111/febs.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samstag Y., John I., Wabnitz G.H. Cofilin: a redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol. Rev. 2013;256(1):30–47. doi: 10.1111/imr.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bamburg J.R., Bernstein B.W. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol Rep. 2010;2:62. doi: 10.3410/B2-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manavalan P., J J.W. Sensitivity of circular dichroism to protein tertiary structure class. Nature. 1983;305:831–832. [Google Scholar]
- 42.Srisailam S., Kumar T.K., Rajalingam D., Kathir K.M., Sheu H.S., Jan F.J., Chao P.C., Yu C. Amyloid-like fibril formation in an all beta-barrel protein. Partially structured intermediate state(s) is a precursor for fibril formation. J. Biol. Chem. 2003;278(20):17701–17709. doi: 10.1074/jbc.M300336200. [DOI] [PubMed] [Google Scholar]
- 43.Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006;1(6):2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisert R., Felau L., Brown L.R. Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Anal. Biochem. 2006;353(1):144–146. doi: 10.1016/j.ab.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 45.Biancalana M., Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta. 2010;1804(7):1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maezawa I., Hong H.S., Liu R., Wu C.Y., Cheng R.H., Kung M.P., Kung H.F., Lam K.S., Oddo S., Laferla F.M., Jin L.W. Congo red and thioflavin-T analogs detect Abeta oligomers. J. Neurochem. 2008;104(2):457–468. doi: 10.1111/j.1471-4159.2007.04972.x. [DOI] [PubMed] [Google Scholar]
- 47.Walsh D.M., Lomakin A., Benedek G.B., Condron M.M., Teplow D.B. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997;272(35):22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 48.Barth A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta. 2007;1767(9):1073–1101. doi: 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Sarroukh R., Goormaghtigh E., Ruysschaert J.M., Raussens V. ATR-FTIR: a "rejuvenated" tool to investigate amyloid proteins. Biochim. Biophys. Acta. 2013;1828(10):2328–2338. doi: 10.1016/j.bbamem.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 50.Moro D., Ulian G., Valdre G. Single molecule investigation of glycine-chlorite interaction by cross-correlated scanning probe microscopy and quantum mechanics simulations. Langmuir. 2015;31(15):4453–4463. doi: 10.1021/acs.langmuir.5b00161. [DOI] [PubMed] [Google Scholar]
- 51.Valdrè G., Tosoni S., Moro D. Zeolitic-type Brønsted-Lowry sites distribution imaged on clinochlore. Am. Mineral. 2011;96(10):1461–1466. [Google Scholar]
- 52.Valdre’ G M.D., Brigatti M.F. Crystallographic features and cleavage nanomorphology of chlinochlore: specific applications. Clay Clay Miner. 2009;57(2):183–193. [Google Scholar]
- 53.Bernini F., Malferrari D., Pignataro M., Bortolotti C.A., Di Rocco G., Lancellotti L., Brigatti M.F., Kayed R., Borsari M., Del Monte F., Castellini E. Pre-amyloid oligomers budding:a metastatic mechanism of proteotoxicity. Sci. Rep. 2016;6:35865. doi: 10.1038/srep35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harper J.D., Lieber C.M., Lansbury P.T., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-beta protein. Chem. Biol. 1997;4(12):951–959. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 55.Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997;4(2):119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 56.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janero D.R., Hreniuk D., Sharif H.M. Hydrogen peroxide-induced oxidative stress to the mammalian heart-muscle cell (cardiomyocyte): nonperoxidative purine and pyrimidine nucleotide depletion. J. Cell. Physiol. 1993;155(3):494–504. doi: 10.1002/jcp.1041550308. [DOI] [PubMed] [Google Scholar]
- 58.Canton M., Menazza S., Sheeran F.L., Polverino de Laureto P., Di Lisa F., Pepe S. Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 2011;57(3):300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 59.Canton M., Skyschally A., Menabo R., Boengler K., Gres P., Schulz R., Haude M., Erbel R., Di Lisa F., Heusch G. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur. Heart J. 2006;27(7):875–881. doi: 10.1093/eurheartj/ehi751. [DOI] [PubMed] [Google Scholar]
- 60.Keceli G., Majumdar A., Thorpe C.N., Jun S., Tocchetti C.G., Lee D.I., Mahaney J.E., Paolocci N., Toscano J.P. Nitroxyl (HNO) targets phospholamban cysteines 41 and 46 to enhance cardiac function. J. Gen. Physiol. 2019;151(6):758–770. doi: 10.1085/jgp.201812208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao W.D., Murray C.I., Tian Y., Zhong X., DuMond J.F., Shen X., Stanley B.A., Foster D.B., Wink D.A., King S.B., Van Eyk J.E., Paolocci N. Nitroxyl-mediated disulfide bond formation between cardiac myofilament cysteines enhances contractile function. Circ. Res. 2012;111(8):1002–1011. doi: 10.1161/CIRCRESAHA.112.270827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klemke M., Wabnitz G.H., Funke F., Funk B., Kirchgessner H., Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. 2008;29(3):404–413. doi: 10.1016/j.immuni.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 63.De Pinto V., Reina S., Gupta A., Messina A., Mahalakshmi R. Role of cysteines in mammalian VDAC isoforms' function. Biochim. Biophys. Acta. 2016;1857(8):1219–1227. doi: 10.1016/j.bbabio.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bachi A., Dalle-Donne I., Scaloni A. Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chem. Rev. 2013;113(1):596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- 65.Chobot S.E., Hernandez H.H., Drennan C.L., Elliott S.J. Direct electrochemical characterization of archaeal thioredoxins. Angew Chem. Int. Ed. Engl. 2007;46(22):4145–4147. doi: 10.1002/anie.200604620. [DOI] [PubMed] [Google Scholar]
- 66.Laviron E. Influence of the adsorption of the depolarizer or of a product of the electrochemical reaction on polarographic currents: XVII. Theoretical study of a reversible surface reaction followed by a first order chemical reaction in linear potential sweep voltammetry. J. Electroanal. Chem. Interfacial Electrochem. 1972;35(1):333–342. [Google Scholar]
- 67.Roos G., Foloppe N., Messens J. Understanding the pK(a) of redox cysteines: the key role of hydrogen bonding. Antioxidants Redox Signal. 2013;18(1):94–127. doi: 10.1089/ars.2012.4521. [DOI] [PubMed] [Google Scholar]
- 68.Hortschansky P., Schroeckh V., Christopeit T., Zandomeneghi G., Fandrich M. The aggregation kinetics of Alzheimer's beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 2005;14(7):1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernstein B.W., Bamburg J.R. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20(4):187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Filipovic M.R. Persulfidation (S-sulfhydration) and H2S. Handb. Exp. Pharmacol. 2015;230:29–59. doi: 10.1007/978-3-319-18144-8_2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.