

Abstract
Clinical trials aimed at improving results of hematopoietic cell transplantation (HCT) by adjuvant cell-based interventions in children have been limited by small numbers and pediatric-specific features. The need for a larger number of pediatric HCT centers to participate in trials has resulted in a demand for harmonization of disease-specific clinical trials and immune-monitoring. Thus far, most phase I/II trials select different end points evaluated at disparate time points, making inter-study comparisons difficult and, sometimes, impossible. In this review, we discuss the various aspects that are important to consider for harmonizing clinical trial design as well as the critical elements for standardized (immune)-monitoring protocols in cell-based intervention trials in the context of HCT. Comparison data from trials applying harmonized trial design will lead to optimized immunotherapeutic treatment protocols to maximize clinical efficacy while minimizing toxicity.
Keywords: cell based therapy, clinical trial design, hematopoietic cell transplantation, immune monitoring, pediatric cancer
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is a potentially curative treatment option for a variety of malignant and non-malignant diseases. Treatment-related complications (graft-versus-host disease [GvHD] and viral reactivation) and relapse unfortunately remain unwanted sequelae of the procedure. Multiple studies aim to improve the safety and efficacy of HCT mainly by enhancing engraftment or the use of innovative immunotherapies, including combination cell therapy and antibody approaches.
These trials bring an important set of data to light, but most single-center phase I/II cell therapy trials select different end points evaluated at disparate time points, making inter-study comparisons difficult and, sometimes impossible. The need for a larger number of pediatric HCT centers to participate in trials, applying harmonized end points, has brought out the demand for international collaborative groups. Although the goal of many early-phase trials is to identify a maximal tolerated dose, it is also desirable that as much efficacy information as possible is obtained from these trials. In particular, this is true for studies in pediatric patients in whom the relatively small numbers and pediatric-specific variables further complicate side-by-side study comparisons.
Better understanding of the mechanisms and biology of immune reconstitution after HCT and adjuvant cell-based intervention will provide us with clues for the further optimization of immunotherapeutic treatment protocols with the goal of reaching optimal clinical efficacy while minimizing toxicity. In this context, harmonizing designs of disease-specific clinical trials and immune-monitoring for additional immune therapeutic strategies in HCT will facilitate comparisons between early-phase trials enabling optimized dosing regimens and immune monitoring tools for “head-to-head” phase III trials.
Standard protocols in diagnostic immunology laboratories are continuously advancing, but the challenge remains to perform highly sophisticated techniques in a standardized manner and in validated settings for multi-center studies. Direct comparisons are often limited because of confounding factors, such as the immune status of the patient and parameters such as age, genetics and underlying disease. Population-specific traits require further investigation before such a protocol can be applied to a heterogeneous population. For pediatric patients in particular, the immune status, including the presence of immune (effector) cells before and during therapy, is generally undetermined. Hence, the effect size of immune parameters in patients treated with immune-based therapies is often unknown, which may hamper power calculations for the required numbers of patients in future trials. In addition, the acquisition and handling of patient samples requires specific logistics (documented in standard operating procedures [SOPs]) in terms of minimal sample type and volume to acquire sufficient cells for analyses (eg, shortly after HCT) or cell fragility during assay handling. As such, even marginal differences in sample preparation and bio-banking may limit comparison of results generated from different centers.
Harmonizing immune monitoring and clinical trial design
Many diseases, including cancer, are associated with alterations in numbers and function of immune cells within the peripheral circulation and especially at sites of tumor progression [1]. Such immune (response) signatures could serve as biomarkers or as surrogate end points when evaluating treatment responses.Despite the considerable progress in the development of immune-monitoring methodologies, the remaining challenge is how to correlate changes in immune parameters with clinical end points. In malignant disorders, this correlation is further complicated by the complexity of interactions (if known) between the host immune system and the tumor micro-environment. Recent progress in our understanding of the cellular and molecular pathways involved in the immune response has facilitated the selection of relevant immune end points. Also, impressive technological advances in methods that enable multiplex profiling of immune phenotypes, definition of regulatory cell subsets, identification of critical signaling molecules and recognition of biologically important targets have increased our knowledge of potential immune biomarkers that may correlate with patient outcomes [2]. However, for children this information is largely lacking, which hampers the development of optimal immunotherapeutic strategies for this population. Leveraging advances in multiplex technologies (eg, genetics, immune-phenotyping and protein assays) may nevertheless provide us with more insight into the immune status before (in case of reduced intensity condition) and after allo-HCT over time, “mechanisms of action” and immunobiology of post-HCT/adjuvant cell therapies.
In the setting of allo-HCT, critical variables in immune reconstitution are strongly associated with the development of life-threatening complications such as viral reactivation, GvHD and relapse [3] (recently reviewed in de Koning et al. [4]).The failure or success of novel immune approaches to circumvent these complications is also highly affected by the immune status. Hence, the design and the evaluation of studies evaluating the efficacy of novel immune therapies must be standardized for multiple single-center trials. By harmonizing clinical trial design, immune-based therapies can be compared in a more standardized way, enabling us to gain more insights regarding the mechanisms of action as well as the immunobiology of novel therapeutics or combination treatment regimens. Nevertheless, the markers and phenotypes studied in one setting may not be considered relevant in another, supporting the definition of a set of general recommended protocols and a set of add-on trial-specific parameters (Table 1).
Table 1.
General parameters that could be included in harmonized immune monitoring protocols across most studies/centers and advanced parameters that may be of value in specific studies and that can only be performed in specialized immunology labs or analyzed in a central laboratory.
| General | Advanced | ||
|---|---|---|---|
| Genetics | HLA, KIR | Fc⬜R | |
| Cell phenotyping | T | CD45RO/RA, CD3, CD4, CD8, CD27, γδ | Intracellular cytokines |
| Treg | CD45, CD4, CD25, CD127, FoxP3 | after PMA/ionomycin stimulation | |
| B | CD45, CD19, CD38, CD27, IgM/G/D, CD21 | Specific TCR by multimer approach | |
| NK/NKT | CD45, CD3, CD56,TCRα24/β11) | ||
| DC/mono | CD11c, HLA-DR, CD14, CD16, CD1c, CD141, CD303 | ||
| Secretome | – | Multiplex panel (eg, IL-7, ST2,TNF-α, IL-6, HGF, IL-2R, IL-8, GM-CSF, etc) | |
| Cell function | – | NK cell lyses | |
| T-cell proliferation on antigens and mitogens | |||
| B-cell maturation | |||
| PK | ATG, Campath (if part of conditioning) | Trial drug | |
| MRD | qPCR (targets expressed, flow cytometry) | Next-generation sequencing | |
| Viral load | CMV, EBV, HV6, adenovirus | – | |
| GvHD | Clinical staging | ST2 | |
To achieve these goals, trial design should include the following consensus end points (Figure 1) described in SOPs:
Disease/complication-specific markers for phase I/II studies: for example, minimal residual disease (MRD), GvHD and viral load after cell therapy assessments at standard time points.
Standardized sampling and monitoring of immunological markers in accredited quality controlled laboratories.
Bio-banking of samples (eg, plasma, cells, tissue, bone marrow and DNA) at defined time points in a standardized and comprehensively documented manner.
Statistical considerations and use of bioinformatics platforms.
Figure 1.
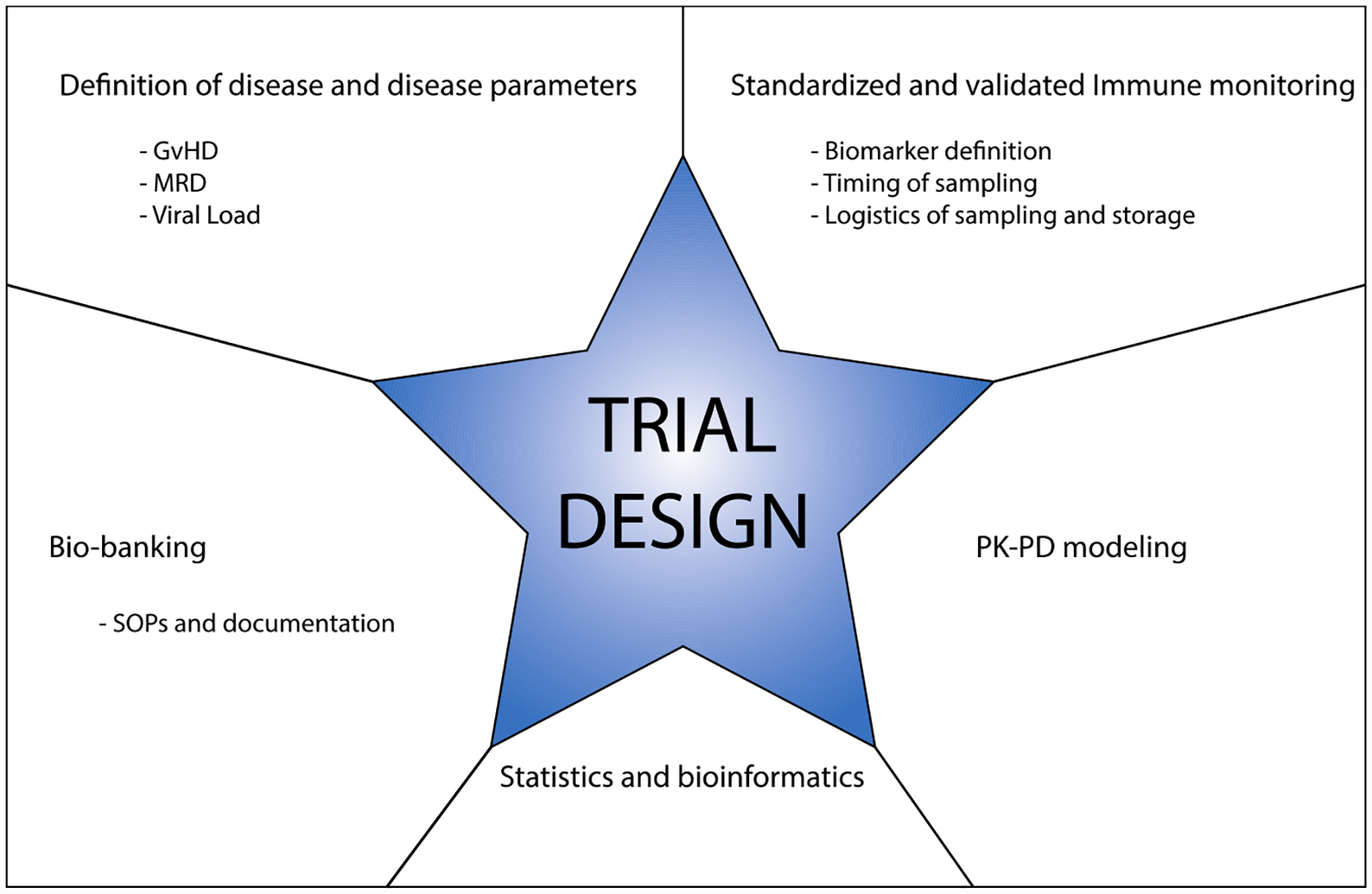
Relevant parameters for immune monitoring to be considered for harmonization early in the process of multi-center trial design.
Genetics
Various genetic variants and polymorphisms in the immune system have been identified to have impact on survival. Non-permissive human leukocyte antigen (HLA)-DPB1 mismatches increased mortality after myelo-ablative unrelated allogeneic HCT [5].Also patients receiving haplo-identical, T-cell–depleted HSC transplant with KIR ligand incompatibility had a significantly higher probability of overall and disease-free survival compared with those without KIR ligand incompatibility [6]. Moreover, patients with relapsed or refractory neuroblastoma have a better response to the additional immune therapy anti-GD2 immunocytokine hu14.18/IL-2 if they have a KIR-HLA ligand mismatch, even in an autologous setting [7]. Irrespective of HCT, the failure or success of antibody therapy (discrimination of responders versus non-responders) may further depend on polymorphisms in FcγR genes (ie, FcgRIIa-H131R and FcgRIIIa-V158) as was shown for rituximab in non-Hodgkin lymphoma patients [8] and in the colorectal and breast cancer settings [9–11]. In patients receiving adjuvant interferon (IFN)-γ therapy, HLA polymorphisms (eg, HLA-Cw6, HLA-B44) were associated with better overall survival in patients with melanoma [12]. Hence, knowledge of these and undoubtedly numerous other genetic markers may therefore affect the efficacy of cell therapies. HLA-KIR typing is widely available in transplantation studies and should be part of a general immune monitoring platform.When antibody therapy is included, it is advised to include genotyping of FcγR types.
Immune cell phenotyping
When developing immunotherapeutic interventions, two major questions that arise are (i) how many cells within each leukocyte subset are present in patients at different disease stages and during different treatment regimens, and (ii) what is the functional response of these cells to the immunotherapeutic treatment?
Flow cytometry is often available for comprehensive immune phenotyping usually in accredited laboratories within transplant centers. Markers identifying the most common leukocyte subsets are broadly used and can therefore be considered as a “standard” panel: CD45 (lymphocytes), CD3 (T cells), CD19 (B cells) and CD16/CD56 (natural killer [NK]) cells. In some studies, this panel has been extended to identify the differentiation and activation state of subsets of T (T-helper, regulatory T cells), B and NK(T) cells, as well as cells from the myeloid lineage (monocytes, dendritic cell subsets) (reviewed in de Koning et al. [4].).These studies have shown that patients with low numbers of CD4 + T cells, NK and NKT cells in the first weeks after HCT show a higher susceptibility to complications associated with lower overall chances of survival [3,13,14].
Furthermore, data on immune reconstitution are important because the success of additional immunotherapies may be largely dependent on the presence of particular cell types. It is believed that antibody-dependent cell-mediated cytotoxicity (ADCC) is a primary effector mechanism responsible for the clinical efficacy of antibody treatment [15]. The efficacy of antibody therapy may thus be determined by the presence and functionality of cells that mediate ADCC, for example, NK cells and neutrophils. Circulating levels of these leukocyte subsets are, however, not definitively related to treatment response.
Other subsets may also be suitable as biomarkers to predict clinical efficacy. For example, monocyte activation and changes in HLA-DR expression may precede clinical detection of infections that may help to identify high-risk HCT patients at an early stage [16,17].Although the predictive value of dendritic cell subsets after HCT remains to be determined, levels of CD1c/BDCA-1 + myeloid dendritic cells (DC) were a predictive biomarker in adults for response to sunitinib [18]. In another study, high baseline frequencies of peripheral blood DC correlated with a clinical response to high-dose interleukin (IL)-2 [19].Together, these data emphasize the importance of antigen-presenting cells in endogenous and therapy-induced anti-tumor immunity, arguably warranting the incorporation of DC markers in immune-monitoring panels.
However, not only the presence of particular cell types but also the ratio between immune stimulatory and regulatory cells (eg, within the tumor microenvironment, such as myeloid or lymphoid suppressor cells), may be a better predictor of response to immune therapies. Elevated levels of IL10-dependentT-regulatory type 1 (Tr1) cells were detected in the peripheral blood of advanced-stage cancer patients compared with patients with early-stage disease or healthy donors [20]. In contrast to CD4+CD25hiFoxp3+ naturalTreg (nTreg), these Tr1 cells exhibit a CD4+CD25−Foxp3low/neg phenotype and suppress target cells through IL-10 and transforming growth factor (TGF)-β secretion as opposed to contact-dependent suppression. Hence, monitoring of Tr1 rather than nTreg may be more relevant in the cancer setting [21].
The presence of tumor antigen–specific T cells as determined by tetramer analyses, or the recently developed combination of ultraviolet-induced peptide exchange and peptide-major histocompatibility complex (MHC) combinatorial coding, may further provide information on the success of immune therapy to expand pre-existing T cells or broaden the T-cell repertoire against tumor and/or virus-associated antigens [22]. This presence of antigen-specificT cells in peripheral blood, however, does not necessarily correlate with clinical response, and some would argue that the cytokine profile secreted by tumor-infiltrating (or delayed-type hypersensitivity [DTH]) T cells may be a more important indicator of clinical response [23].
Taken together, a variety of specialized subsets may have potential as predictive markers for clinical efficacy, but they require more sophisticated staining protocols, making it less broadly applicable when developing harmonized panels for multiple clinical trials. In some cases, this may bring out the requirement for centralized analyses of bio-banked material. Harmonization of standardized panels has been shown to be possible in the ONE Study [24], in which a flow cytometry panel yielded acceptable variability in a standardized assay performed on samples from transplant patients at multiple international sites. In this study, whole-blood analysis was performed requiring fresh samples and relatively large amounts of antibodies. A reduction in variability may also be achieved by the introduction of single cell mass cytometry, which may explain the biological action and mechanisms of separate cells in more detail [25]. It is important to realize that trials that use whole-blood assays may produce different percentages of cell subsets when compared with studies that use peripheral blood mononuclear cells (PBMC).The same is true when comparing freshly isolated PBMCs with bio-banked material, which has been subjected to freeze/thaw procedures that affect expression levels of various markers. Even when the same samples are collected, variations can be introduced by the selection of antibody clones, combination of clones and fluorochromes, choice of flow cytometer, calibration/compensation settings, staining procedures (ie, buffers, incubation time, fixation methods) and the gating strategies. In summary, minimizing the variability in sample handling and the pre-analysis phase is critical for standardization.
Secretome
Measuring the production of cytokines, chemokines and growth factors, as well as their profile (ie, the secretome) represents an integral part of immunomonitoring during immunotherapeutic treatments [26].These biomarkers may distinguish diverse disease/response patterns, identify surrogate markers of efficacy and provide more insight into the therapeutic mode of action. Peripheral blood is often the only source for protein analysis, which may lack sensitivity to reflect local responses in affected tissues. Proteins, such as IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), hepatocyte growth factor (HGF), ST2 (suppressor of tumor-genicity) and soluble IL-2a have been suggested as potential biomarkers for GvHD, whereas increased levels of TNF-α and IL-6 are associated with robust immune responses to viral reactivation (reviewed in de Koning et al. [4].).
Commonly used methods to identify these markers include antibody-based enzyme-linked immunosorbent assays or multiplex platforms, such as protein micro-arrays, high-performance liquid chromatography, electro-chemiluminescence and bead-based multiplex immunoassays [27].Again, different technologies and reagents (eg, antibodies and recombinants for standard curves) may lead to different concentrations and dramatic variability in results depending on how the pre-analytic samples are handled (eg, differences in processing and storage). Cytokine levels differ considerably between serum and plasma samples obtained from the same donor because of release of platelet-associated molecules into serum [28]. Moreover, the type of anticoagulant used in plasma isolation also influences cytokine levels and should at least be reported in publications, which is not always the case. More worrisome is the sensitivity of these markers for time-and/or temperature-sensitive changes. Some markers are markedly affected when blood samples are kept at room temperature for more than 4 h before serum or plasma separation [27]. In addition, storing samples for more than 2 years introduces fluctuations in cytokine levels, and repetitive freeze/thaw cycles influence individual markers to different degrees. Furthermore, heterophylic antibody binding (eg, caused by the presence of rheumatoid factor) can give rise to false-positive results, necessitating appropriate sample pretreatment to prevent this from occurring.These phenomena at least underscore the need for extensive documentation with respect to all biomarker analysis before any conclusions can be made when comparing patient cohorts treated at multiple sites. Nevertheless, biomarker profiles may have diagnostic/prognostic value and should be included in a standardized way when developing immune-monitoring end points for phase I/II and III studies. Given the lack of validated panels and specificity/sensitivity for certain clinical complications, as well as the critical requirements for sampling and storage, it is too early to implement multiplex assays in general immune monitoring. However, we encourage consortia such as the Westhafen Intercontinental Group (WIG) to apply harmonized, standardized logistics and (centralized) analyses to facilitate biomarker identification across different treatment strategies.
Functional assays
Functional assays provide additional insights into the capacity of the patient’s immune cells (either host- or donor-derived) to proliferate and function, but data after HCT are largely lacking. Discouraging factors may be the complexity of techniques and the considerable differences between NK cell functionality among individuals. It has been postulated that NK cells derived from cancer patients show lower levels of activation markers and are therefore less able to kill tumor cell lines in vitro [29].The same holds true forT-cell subsets. The proliferative capacity of T cells can be analyzed by means of a standardized re-stimulation protocol with mitogens and T-cell–specific stimuli. Moreover, the ability of tumor antigen-specificT cells to produce the effector cytokine IFN-γ rather than only IL-5 may better correlate with clinical responses than simply the detection of theseT cells ex vivo [23]. Previous studies with adoptively transferred virus-specificT cells provide proof of concept for the relationship between T-cell proliferation, expression of activation markers and cytokine production and the clinical efficacy [30,31]. Here again, protocols may largely differ in different centers and, although conclusions should essentially be similar, the extent or relative functionality of the immune cells of interest may differ. Performing functional tests is definitely possible with the use of properly bio-banked samples, but results may not be concordant when using freshly isolated samples. In addition, the time elapsed before functional assays are per formed and the duration of the experiment may be critical variables to be considered when attempting to draw valid conclusions.
Monitoring (clinical) effect: MRD, viral load
In early-phase clinical trials, sometimes conclusive or objective clinical efficacy end points are not available; however, surrogate end points may hold valuable information (eg, MRD, viral-load and GvHD bio-markers). For patients with a malignancy, monitoring the impact of immune therapy on tumor burden and viral load is an important “effect-of-therapy” tool, and various methods have been developed over the last decade to evaluate this kind of response. Monitoring for leukemia MRD after immune therapy (including HCT) relies on different techniques, including (i) chimerism assays in which the ratio between donor- and recipient-derived cells can be assessed with the use of fluorescence in situ hybridization (FISH) or polymerase chain reaction (PCR) and enables the detection of cells of donor origin independently from known blast characteristics or specific markers (this technique has a sensitivity limit in the range of 1%). (ii) Cytogenetics studies in which abnormal karyotypes can be followed with the use of FISH or reverse transcriptase (RT)-PCR assessments also have the problem of sensitivity, and, with this technique, only a known clonal aberration is followed longitudinally, thus missing the possible development of sub-clonal evolutions. (iii) MRD monitoring can also be applied to specific molecular targets, which can be followed through RT-PCR. However, one drawback is that targets may not be identifiable for all patients (eg, in AML, only 40% to 50% of patients have an identifiable target). Using multi-parameter flow cytometry, however, aids in the identification of a panel of altered antigens characteristic of leukemia blasts, which is estimated to be possible for approximately 80% of patients. Although both techniques have a high sensitivity (between 10−4 and 10−5) [32,33], neither can precisely take into account and describe the evolution of genetic modifications that a blast clone may develop during and after treatment. Novel, more sensitive and accurate tools that do have this ability, such as the use of next-generation sequencing (NGS) techniques, have been developed [34]. NGS is capable of describing the whole genomic repertoire of the cells of interest, in a single reaction. NGS has the capacity to identify the presence of recurrent disease early and has the potential to detect clonal evolution. Implementing and developing these more specific and accurate tools are important to (i) study the effect of therapy, (ii) characterize leukemia cells isolated at diagnosis, after cell therapy and at relapse and (iii) detect new targets. Therefore,NGS offers the potential to better understand the relationship between leukemia clonal evolution and response to chemo/immunotherapy, as well as tumor immune escape mechanisms. Ultimately, this technology potentially facilitates the development of individualized, targeted therapeutics for leukemia patients that may be efficacious with less toxicity. Besides synchronizing the definitions and detection techniques, inter-trial comparative MRD analyses would benefit from agreements in sampling at the same time intervals.
Pharmacokinetics-pharmacodynamics and drug-effect information
Drugs given as part of the conditioning regimen as well as after HCT will influence both short-term and long-term immune reconstitution and may therefore have an unknown effect on a cell-based therapeutic. In the context of HCT, predictable immune reconstitution would be important when studying the effects of a cell therapy. Therefore, it is important to understand the influence of drugs used before and after HCT on immune reconstitution. In pediatrics and certainly in pediatric HCT, the use of unlicensed and off-label drugs is a common occurrence with an incidence ranging from 18% to 60% [35].To better understand the effects of transplant-related drugs on immune reconstitution, it is important to obtain critical and comprehensive pharmacokinetic (PK) and pharmacodynamic (PD) information. Individualization of drug dosing on the basis of PK/PD modeling and the use of immune reconstitution–related biomarkers holds promise for improved HCT outcomes by decreasing transplantation-related toxicity (eg, GvHD, viral reactivation) and relapse rates. A recent PK-PD study demonstrating the impact of variable exposures on immune reconstitution was conducted in pediatric patients receiving ATG. This study determined that a higher exposure to ATG after HCT, which was observed primarily in patients with a higher body weight, resulted in delayed or absent immune reconstitution, leading to poorer overall survival rates [3].This study concluded that by individualizing the dose of ATG, immune reconstitution may become more predictable, which is of utmost importance when developing cell therapy trials and a harmonized clinical trial design for pediatric patients after HCT in particular.
Bioinformatics platforms
The above-mentioned aspects of standardizing trial design and immune/response monitoring will generate large volumes of data in a relatively limited number of patients. Therefore, partners in bio-informatics must develop tools to deliver immune, PK, MRD-monitoring and bioinformatics pipelines to facilitate the identification of immune parameters and bio-markers associated with response to therapy. Bio-marker identification in clinical trials (especially in patients with rare diseases) is generally complicated by small patient numbers. However, it has been shown that parameters, which differ with disease activity and/or time, can be powerful markers even when detected in a small number of patients [36]. A prerequisite for this type of analysis is the follow-up of these markers in an unrelated validation cohort. Furthermore, such a platform requires “state-of-the-art” bio-monitoring and bio-banking and the establishment of quality controls between laboratories.
Conclusions
Immune-based therapies in the context of HCT are being developed for a variety of indications to improve outcomes for diseases with a clear unmet need, for example, chemo-resistant cancer and the prevention and/or treatment of viral reactivations after HCT. In the majority of these primarily phase I/II trials, various end points are chosen at different time points, thereby making inter-study comparison difficult and sometimes impossible. To facilitate comparisons between studies, the development of a standard panel of immunological and genetic tools measured at standardized time points as well as a panel of disease-specific readouts is needed. In addition to reaching a consensus on “clinical trial design,” setting up an international, technical and bio-informatics platform is of the utmost importance. Ensuring the use of “state-of-the-art” bio-monitoring and bio-banking as well as developing standardized quality controls between laboratories, and developing bio-informatics pipelines are required for harmonization. Committees such as the Westhafen Intercontinental Group are expected to take the initiative in collaboration with European and American working committees to develop a consensus on harmonized clinical trial design for cell therapies. Moreover, the development of a “standard” and “international” consensus may ultimately make it easier for groups to obtain ethical approvals, thus facilitating the development of harmonized cell therapy trials worldwide.
Footnotes
Disclosure of interests: The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
References
- [1].Butterfield LH, Vujanovic L, Pardee AD. Approaches to immunologic monitoring of clinical trials In: The tumor immunoenvironment. Dordrecht: Springer Netherlands; 2013. p. 663–94. doi: 10.1007/978-94-007-6217-6_29. [DOI] [Google Scholar]
- [2].Whiteside TL. Immune responses to cancer: are they potential biomarkers of prognosis? Front Oncol 2013;3:107. doi: 10.3389/fonc.2013.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Admiraal R, van Kesteren C, Jol-van der Zijde CM, Lankester AC, Bierings MB, Egberts TCG, et al. Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haemopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol 2015;2:e194–203. doi: 10.1016/S2352-3026(15)00045-9. [DOI] [PubMed] [Google Scholar]
- [4].de Koning C, Plantinga M, Besseling P, Boelens JJ, Nierkens S. Immune-reconstitution after allogeneic hematopoietic cell transplantation in children. Biol Blood Marrow Transplant 2015; doi: 10.1016/j.bbmt.2015.08.028. [DOI] [PubMed] [Google Scholar]
- [5].Pidala J, Lee SJ, Ahn KW, Spellman S, Wang H-L, Aljurf M, et al. Nonpermissive HLA-DPB1 mismatch increases mortality after myeloablative unrelated allogeneic hematopoietic cell transplantation. Blood 2014;124:2596–606. doi: 10.1182/blood-2014-05-576041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood 2003;102:814–19. doi: 10.1182/blood-2003-01-0091. [DOI] [PubMed] [Google Scholar]
- [7].Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcγ receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res 2010;70:9554–61. doi: 10.1158/0008-5472.CAN-10-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood 2002;99:754–8. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- [9].Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- [10].Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol 2007;25:3712–18. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- [11].Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- [12].Wang E, Zhao Y, Monaco A, Uccellini L, Kirkwood JM, Spyropoulou-Vlachou M, et al. A multi-factorial genetic model for prognostic assessment of high risk melanoma patients receiving adjuvant interferon. PLoS ONE 2012;7:e40805. doi: 10.1371/journal.pone.0040805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bartelink IH, Belitser SV, Knibbe CAJ, Danhof M, de Pagter AJ, Egberts TCG, et al. Immune reconstitution kinetics as an early predictor for mortality using various hematopoietic stem cell sources in children. Biol Blood MarrowTransplant 2013;19:305–13. doi: 10.1016/j.bbmt.2012.10.010. [DOI] [PubMed] [Google Scholar]
- [14].Rubio M-T, Moreira-Teixeira L, Bachy E, Bouillié M, Milpied P, Coman T, et al. Early posttransplantation donor-derived invariant natural killer T-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood 2012;120:2144–54. doi: 10.1182/blood-2012-01-404673. [DOI] [PubMed] [Google Scholar]
- [15].Chester C, Marabelle A, Houot R, Kohrt HE. Dual antibody therapy to harness the innate anti-tumor immune response to enhance antibody targeting of tumors. Curr Opin Immunol 2015;33C:1–8. doi: 10.1016/j.coi.2014.12.010. [DOI] [PubMed] [Google Scholar]
- [16].Döring M, Rohrer KM, Erbacher A, Gieseke F, Schwarze C-P, Bader P, et al. Human leukocyte antigen DR surface expression on CD14+ monocytes during adverse events after hematopoietic stem cell transplantation. Ann Hematol 2015;94:265–73. doi: 10.1007/s00277-014-2185-y. [DOI] [PubMed] [Google Scholar]
- [17].Döring M, Cabanillas Stanchi KM, Haufe S, Erbacher A, Bader P, Handgretinger R, et al. Patterns of monocyte subpopulations and their surface expression of HLA-DR during adverse events after hematopoietic stem cell transplantation. Ann Hematol 2015;94:825–36. doi: 10.1007/s00277-014-2287-6. [DOI] [PubMed] [Google Scholar]
- [18].van Cruijsen H, van der Veldt AAM, Vroling L, Oosterhoff D, Broxterman HJ, Scheper RJ, et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res 2008;14:5884–92. doi: 10.1158/1078-0432.CCR-08-0656. [DOI] [PubMed] [Google Scholar]
- [19].Finkelstein SE, Carey T, Fricke I, Yu D, Goetz D, Gratz M, et al. Changes in dendritic cell phenotype after a new high-dose weekly schedule of interleukin-2 therapy for kidney cancer and melanoma. J Immunother 2010;33:817–27. doi: 10.1097/CJI.0b013e3181ecccad. [DOI] [PubMed] [Google Scholar]
- [20].Bergmann C, Strauss L, Wang Y, Szczepanski MJ, Lang S, Johnson JT, et al. T regulatory type 1 cells in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res 2008;14:3706–15. doi: 10.1158/1078-0432.CCR-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Whiteside TL, Schuler P, Schilling B. Induced and natural regulatory T cells in human cancer. Expert Opin Biol Ther 2012;12:1383–97. doi: 10.1517/14712598.2012.707184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti–CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 2014;6: doi: 10.1126/scitranslmed.3008918.254ra128. [DOI] [PubMed] [Google Scholar]
- [23].Aarntzen EHJG, Figdor CG, Adema GJ, Punt CJA, De Vries IJM. Dendritic cell vaccination and immune monitoring. Cancer Immunol Immunother 2008;57:1559–68. doi: 10.1007/s00262-008-0553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Streitz M, Miloud T, Kapinsky M, Reed MR, Magari R, Geissler EK, et al. Standardization of whole blood immune phenotype monitoring for clinical trials: panels and methods from the ONE study. Transplant Res 2013;2(1):17. doi: 10.1186/2047-1440-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bendall SC, Simonds EF, Qiu P, Amir E-AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Clay TM, Hobeika AC, Mosca PJ, Lyerly HK, Morse MA. Assays for monitoring cellular immune responses to active immunotherapy of cancer. Clin Cancer Res 2001;7: 1127–35. [PubMed] [Google Scholar]
- [27].Keustermans GCE, Hoeks SBE, Meerding JM, Prakken BJ, de Jager W. Cytokine assays: an assessment of the preparation and treatment of blood and tissue samples. Methods 2013;61:10–17. doi: 10.1016/j.ymeth.2013.04.005. [DOI] [PubMed] [Google Scholar]
- [28].de Jager W, Bourcier K, Rijkers GT, Prakken BJ, Seyfert-Margolis V. Prerequisites for cytokine measurements in clinical trials with multiplex immunoassays. BMC Immunol 2009;10:52. doi: 10.1186/1471-2172-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mamessier E, Pradel LC, Thibult M-L, Drevet C, Zouine A, Jacquemier J, et al. Peripheral blood NK cells from breast cancer patients are tumor-induced composite subsets. J Immunol 2013;190:2424–36. doi: 10.4049/jimmunol.1200140. [DOI] [PubMed] [Google Scholar]
- [30].Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM, et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010;116:4360–7. doi: 10.1182/blood-2010-01-262089. [DOI] [PubMed] [Google Scholar]
- [31].Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood 2013;121: 5113–23. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for “prime time?”. Hematology 2014;2014:222–33. doi: 10.1182/asheducation-2014.1.222. [DOI] [PubMed] [Google Scholar]
- [33].Jaso JM, Wang SA, Jorgensen JL, Lin P. Multi-color flow cytometric immunophenotyping for detection of minimal residual disease in AML: past, present and future. Bone Marrow Transplant 2014;49:1129–38. doi: 10.1038/bmt.2014.99. [DOI] [PubMed] [Google Scholar]
- [34].Pulsipher MA, Carlson C, Langholz B, Wall DA, Schultz KR, Bunin N, et al. IgH-V(D)J NGS-MRD measurement pre- and early post- allo-transplant defines very low and very high risk ALL patients. Blood 2015;125(22):3501–8. doi: 10.1182/blood-2014-12-615757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lindell Osuagwu L, Korhonen MJ, Saano S, Helin Tanninen M, Naaranlahti T, Kokki H. Off-label and unlicensed drug prescribing in three paediatric wards in Finland and review of the international literature. J Clin Pharm Ther 2009;34:277–87. doi: 10.1111/j.1365-2710.2008.01005.x. [DOI] [PubMed] [Google Scholar]
- [36].Bellutti Enders F, vanWijk F, Scholman R, Hofer M, Prakken BJ, van Royen Kerkhof A, et al. CXCL10, TNFR2 and Galectin-9 correlate with disease activity in Juvenile Dermatomyositis. Arthritis Rheum 2014;66(8):2281–9. doi: 10.1002/art.38676. [DOI] [PubMed] [Google Scholar]