

SUMMARY
Defective cholesterol efflux pathways in mice promote the expansion of hematopoietic stem and progenitor cells and a bias toward the myeloid lineage, as observed in chronic myelomonocytic leukemia (CMML). Here, we identify 5 somatic missense mutations in ABCA1 in 26 patients with CMML. These mutations confer a proliferative advantage to monocytic leukemia cell lines in vitro. In vivo inactivation of ABCA1 or expression of ABCA1 mutants in hematopoietic cells in the setting of Tet2 loss demonstrates a myelosuppressive function of ABCA1. Mechanistically, ABCA1 mutations impair the tumor-suppressor functions of WT ABCA1 in myeloproliferative neoplasms by increasing the IL-3Rβ signaling via MAPK and JAK2 and subsequent metabolic reprogramming. Overexpression of a human apolipoprotein A-1 transgene dampens myeloproliferation. These findings identify somatic mutations in ABCA1 that subvert its anti-proliferative and cholesterol efflux functions and permit the progression of myeloid neoplasms. Therapeutic increases in HDL bypass these defects and restore normal hematopoiesis.
In Brief
Viaud et al. show that ABCA1 mutants identified in CMML patients diminish the tumor-suppressor functions of ABCA1 and cooperate with Tet2 loss to confer the hypersensitivity of myeloid progenitors to IL-3 receptor β canonical signaling, which can be prevented by raising HDL levels.
Graphical Abstract
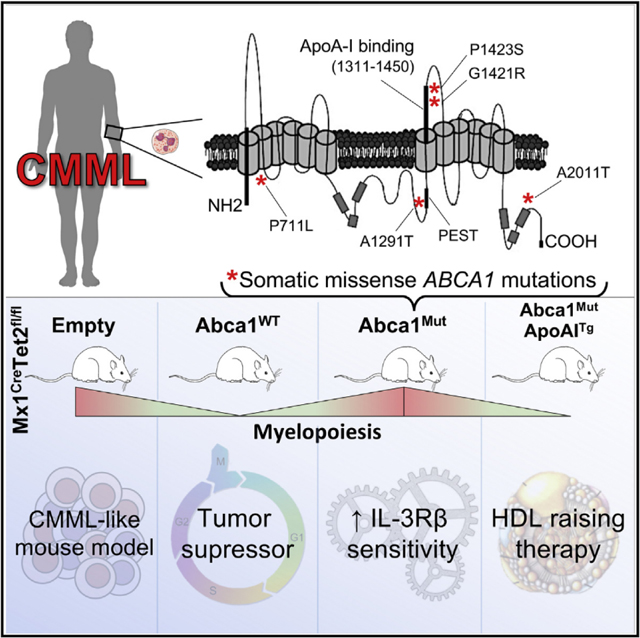
INTRODUCTION
Most human adult cancers develop through a multistep acquisition of a wide range of somatic mutations that initiate or maintain self-renewal of the malignant clone. The elucidation of the somatic mutational landscape of many solid tumors and hematologic malignancies has occurred in the last decade (Sjöblom et al., 2006; Ley et al., 2013). These mutations are referenced in the Catalog of Somatic Mutations In Cancer (COSMIC) and The Cancer Genome Atlas (TCGA) and may provide potential insights into the mechanisms underlying cancer. In myeloid malignancies, hematopoietic stem and progenitor cells (HSPCs) acquire specific combinations of leukemia disease alleles required to promote hematopoietic transformation (Shih et al., 2015; Kunimoto et al., 2018). Recent studies have shown that mutations in a small number of genes, including loss-of-function mutations in ten-eleven translocation 2 (TET2) are common in the elderly and provide a proliferative advantage to hematopoietic stem cells (HSCs), giving rise to clonal hematopoiesis (Busque et al., 2012). Clonal hematopoiesis mutations are associated with an ~10-fold increase in the risk of developing a hematological malignancy, including myeloproliferative disorders and leukemias and a 2- to 3-fold risk of developing atherosclerotic cardiovascular disease (CVD).
Increased high-density lipoprotein (HDL) levels are well known to be associated with a reduced risk of CVD. A recent meta-analysis of randomized controlled trials of lipid-altering therapies revealed that for every 10-mg/dL increase in the plasma HDL-cholesterol level among trial participants, there was a 36% lower risk of cancer incidence during >625,000 person-years of follow- up and >8,000 incident cancers (Jafri et al., 2010). While not establishing causation, this association suggests that HDL may be linked to tumor cell biology in humans. The ability of HDL and its apolipoproteins to promote the efflux of cholesterol from cells depends in part on the ATP-binding cassette transporters ABCA1 and ABCG1, but it can also be mediated by scavenger receptor B1 and passive efflux pathways (Tall and Yvan-Charvet, .The reduced expression of cholesterol efflux mediators and the increased levels of cellular cholesterol have been associated with different solid tumors; however, it is unclear whether these are secondary changes or have a role in promoting cell proliferation (Bovenga et al., 2015; Clendening and Penn, 2012; Dang, 2012; Lin and Gustafsson, 2015; Mullen et al., 2016). Mice with defective cholesterol efflux in hematopoietic cells develop progressive myeloid expansion with an underlying dramatic HSPC expansion in the bone marrow (BM), an enhanced interleukin-3-granulocyte-macrophage colony-stimulating factor (IL-3-GM-CSF) signaling pathway, and marked extramedullary hematopoiesis (Murphy et al., 2011; Wang et al., 2014; Westerterp et al., 2012; Yvan-Charvet et al., 2010). We also demonstrated that HDL-raising therapies could limit Mpl-W515Land Flt3-ITD-driven myeloproliferative disorders (Gautier et al., 2013).
While these findings suggest a potential role for cholesterol efflux pathways in modulating the development of myeloid malignancies and leukemias, they have not established a causative role of specific somatic mutations in cholesterol efflux genes in these disorders. Chronic myelomonocytic leukemia (CMML) is typically a disease of the elderly with few treatment options. Recent studies in CMML patients have shown changes reminiscent of those observed in mice with defective cholesterol efflux in hematopoietic cells, including: (1) frequently mutated tumor-suppressor genes encoding regulators of GM-CSF signaling (RAS, CBL), (2) hypersensitivity of myeloid progenitors to GM-CSF, and (3) a proportion of “classical” CD14+CD16− monocytes >94% (Itzykson et al., 2017). In addition, these patients often have mutations in genes associated with clonal hematopoiesis, including TET2 and ASXL1. In the present study, we have used high-throughput sequencing to identify mutations in ABCA1 in CMML patient samples. Further studies in a mouse model of myeloproliferative neoplasms driven by hematopoietic Tet2 deficiency have shown that these somatic mutations abrogate the tumor-suppressor function of WT ABCA1, resulting in the failure to suppress canonical IL3-receptor β (IL-3Rβ) signaling-driven myelopoiesis. The loss of the myelosuppressive function of ABCA1 mutants can be overcome by raising HDL levels through the overexpression of the human apolipoprotein A-1 (apoA-1) transgene.
RESULTS
Identification of ABCA1 Somatic Mutations in CMML
Sequencing of full-length ABCA1, ABCG1, and NR1H2/3 (liver X receptors [LXRs]) in 26 CMML samples revealed a somatic mutational frequency of 19% of samples for ABCA1 (n = 5) and 0% for ABCG1 and NR1H2/3. All of the mutations were somatic missense mutations, with only 1 mutation observed in each patient sample (Figures 1A and S1 A; Table S1). The identity of the paired samples was verified by Sequenom single-nucleotide polymorphism (SNP) genotyping, demonstrating that the likelihood of a match occurring by chance was <1 × 10−13 (data not shown). These ABCA1 mutations occur in evolutionarily conserved regions (Figures 1B and 1C). The ABCA1 mutations have not been previously described, even though different ABCA1 mutations have been identified in Tangier disease (Brunham et al., 2006; Sjöblom et al., 2006). Sequencing of other genes implicated in the pathogenesis of CMML in the same samples revealed that ABCA1 mutations co-existed with known oncogenic mutations in JAK2, Flt3, and N-Ras (Emanuel, 2008). We noted that (1) of the 4 genes sequenced, somatic non-synonymous mutations were found in only 2 of the 4 genes and (2) the somatic nonsynonymous mutation rate for ABCA1 was higher than the expected background silent mutation rate and higher than expected by chance alone by binomial tests (p = 3.6 × 10−10 for ABCA1), suggesting that mutations in ABCA1 do not represent passenger gene effects.
Figure 1. Identification of Loss of Function ABCA1 Mutants in CMML.

Forward (upper trace) and reverse (lower trace) sequence traces of ABCA1 gene demonstrating a heterozygous cytosine-to-thymine substitution (arrows) present in myeloid cell DNA from patients with chronic myelomonocytic leukemia (CMML). The mutation is not present in buccal DNA from the same patient (upper trace).
(A and B) DNA sequence (A) and protein translation (B) for both the wild-type (WT) and mutant ABCA1 alleles. The mutations result in amino acid substitution at codons 711, 1,291, 1,421,1,423, and 2,011.
(C ) Representative 3D structure of ABCA1 transporter. The asterisks represent localizations ABCA1 mutants.
(D and E) [3H]-Thymidine proliferation assays (Pulsed for 2h) were performed in HEK293 cells transiently transfected with plasmid constructs expressing ABCA1-WT and ABCA1 mutants or empty vector (D), or in THP-1 monocytic leukemia cells transduced for 72 h with lentiviral particles expressing ABCA1-WT, ABCA1 mutants or empty vector (E).
(F) THP-1 cells transduced for 72 h with ABCA1 mutants exhibit growth advantage over a 7-day period compared with ABCA1-WT.
(G and H) Expression of phosphoSTAT5 (G) and BODIPY staining (H) determined by flow cytometry in these cells.
(I) Confocal images of lipid raft staining in THP-1 cells transduced for 72 h with empty, ABCA1-WT, and ABCA1 mutants.
Values are mean ±SEMs of at least 3 experiments performed in triplicate. *P < 0.05 ABCA1-WT versus empty control. #P < 0.05 ABCA1 mutants versus empty control. §p < 0.05 versus ABCA1-WT.
Functional Analysis of ABCA1 Mutations In Vitro
Given the key role of the ABCA1-dependent cholesterol efflux pathway in controlling myeloid expansion (Tall and Yvan-Charvet, 2015), we sought to test whether ABCA1 CMML mutations affect cellular proliferation. We used site-directed mutagenesis to introduce each of these 5 somatic mutations individually into the ABCA1 cDNA. To compare the ability of ABCA1 mutants to control proliferation, we transiently transfected HEK293 cells with the ABCA1 cDNAs. Overexpression of wild-type (WT)-ABCA1 resulted in an ~1.7-fold decrease in cell proliferation compared with empty vector-transfected cells (Figure 1D). All of the mutants located in either the N- and C-terminal regions (P711L and A2011T), the PEST sequence (A1291T), or the apoA-1 binding region (G1421R and P1423S) exhibited a significant reduction in anti-proliferative activity (Figures 1C and 1D). We also tested the relevance of these ABCA1 mutations in human THP-1 monocytic leukemia cell lines that express endogenous ABCA1 (Figure S1B). ABCA1-P711L, ABCA1-A1291T, ABCA1-G1421R, ABCA1-P1423S, and ABCA1-A2011T displayed reductions in anti-proliferative activity, compared to ABCA1-WT in lentivirus-transduced cells (Figure 1E), which is consistent with observations in HEK293 cells. Although the proliferation rate of THP1 cells was slowed down by transient transfection, we showed a growth advantage of all mutations over a culture period of 1 week compared to WT-ABCA1 expression (Figure 1F). Stable cell lines expressing ABCA1-G1421R and ABCA1-A2011T (Figure S1C) with a normal proliferation rate showed growth and proliferative advantages compared to ABCA1-WT (Figures S1D and S1E). We also observed an activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway in ABCA1 mutant-transduced cells compared to WT-ABCA1, as illustrated by the higher levels of pSTAT5 quantified by flow cytometry (Figure 1G). Because the assessment of cholesterol efflux capacity is not practical in suspension cells, we next tested the dependence of ABCA1 mutations on their efflux capacity in differentiated THP-1 macrophages. In this setting, 3 of the 5 ABCA1 mutations (ABCA1-A1291T, ABCA1-G1421R, and ABCA1-P1423S) showed a decrease in the ability of apoA-1 to promote cholesterol efflux compared with WT-ABCA1 (Figure S1F). Nevertheless, quantification of BODIPY (bore-dipyrromethene)-neutral lipid staining revealed higher neutral lipid accumulation in ABCA1 mutant-transduced cells compared to WT-ABCA1 (Figure 1H). The quantification of cellular cholesterol content confirmed these findings (Figure S1G). The increased proliferation of these cells was also associated with increased cholera toxin subunit B (CTx-B) staining of ABCA1 mutant-transduced cells compared to WT-ABCA1 at the cell surface (ABCA1-A1291T, ABCA1-G1421R, and ABCA1 -P1423S) or in intracellular endosomal-like structure (ABCA1-P711L and ABCA1-A2011T) (Figure 1l), suggesting the increased formation of cholesterol-rich lipid raft or perturbed intracellular cholesterol trafficking (Dietrich et al., 2001).
ABCA1 Mutants Associated with CMML Fail to Suppress Myelopoiesis In Vivo
Previous studies have suggested that the loss of ABCA1 function alone is insufficient to promote prominent myelopoiesis in hypercholesterolemic mice (Yvan-Charvet et al., 2010). We hypothesized that the proliferative effects of ABCA1 mutants observed in CMML may become more evident when combined with other CMML mutant alleles. Tet2 inactivation through loss-of-function mutation is commonly found in CMML (Bowman and Levine, 2017; Solary et al., 2014). Therefore, to assess the in vivo effects of ABCA 7 mutants, BM cells from WT or Mx1-Cre+Tet2fl/fl mice (i.e., mice bearing the conditional Tet2 allele and the interferon-inducible Cre transgene) were transduced with pLKO-Puro-GFP lentiviral vectors containing WT-ABCA1 or ABCA1 mutants and transplanted into lethally irradiated C57BL76J mice (Figure 2A). Animals were analyzed 5 weeks after BM reconstitution (TO) and at the indicated time point following polyinosinic:polycytidylic acid (PIPC) injection (Figure 2A). Consistent with earlier works (Moran-Crusio et al., 2011; Quivoron et al., 2011), we observed the loss of Tet2 expression in the BM of WT recipient mice transplanted with Mx1-Cre+Tet2fl/fl BM compared to Mx1-Cre+ BM (Figure S2A), and ablation of the gene was paralleled by a significant reduction in hydroxylation of 5-methylcyto- sine (5hmC) in a pool of peripheral blood cells, which reflect the enzymatic activity of TET2 (Figure S2B). Quantification of Abca1 mRNA expression confirmed similar levels of overexpression of ABCA1-WT and mutants in the BM of Mx1-Cre+Tet2fl/fl recipients (Figure 2B) and controls (data not shown). Despite similar leukocyte counts (Figure S2C), the overexpression of WT-ABCA1 on aTet2-deficient background caused a marked reduction in myeloid cells (Gr-1highCD11bhigh) in blood over time (Figure 2C), reflecting mainly lower inflammatory Ly6Chl monocyte and neutrophil counts (Figures 2D and S2D). In contrast, ABCA1 mutant-expressing animals on a Tet2-deficient background exhibited higher peripheral myeloid cells (both monocytes and neutrophils) compared to ABCA1-WT-transduced animals (Figures 2C, 2D, and S2D). These effects were not observed when ABCA1-WT or mutants were transduced on a WT background (Figures S2E and S2F). T and B cell numbers and hematocrit and platelet counts were normal on both backgrounds (data not shown). These data indicate that ABCA1 mutants impede the protective effect of ABCA1-WT in preventing myeloid expansion on a Tet2-deficient background. The 5 ABCA1 mutations identified in CMML patients were found to be loss-of-function mutations, as demonstrated by their failure to suppress blood leukocyte counts in the setting of Tet2 deficiency.
Figure 2. Loss of Functional ABCA1 Reduces Tumor Suppression in Myelomonocytic Leukemia Induced by Tet2 Loss.

(A) Experimental overview. BM from Mx1-Cre+ or Mx1-Cre+Tet2fl/fl mice were transduced with lentiviral particles expressing ABCA1-WT, ABCA1 mutants, or empty vector before bone marrow transplantation (BMT) into lethally irradiated WT mice, and after a 5-week recovery period, the mice were injected with poly(l:C) and analyzed over a 12-week-period.
(B) Modulation of Abca1 mRNA expression levels in the BM of the aforementioned mouse models.
(C) Quantification of the percentage of peripheral blood myeloid cells determined by hematology cell counter over the course of 12 weeks after poly (l:C) injection in recipient mice transplanted with empty, ABCA1-WT, or ABCA1 mutants expressing Mx1-Cre+Tet2fl/fl BM.
(D) Peripheral blood myeloid subsets (CD115+Ly6Chl and CD115+Ly6Clo monocytes and CD115-Ly6Chl neutrophils) were also quantified in these mice at the indicated time point.
(E) Representative spleen (upper panel) and hematoxylin and eosin (H&E) staining of paraffin-embedded spleen sections from recipient mice transplanted with control or M×1-Cre+Tet2fl/fl BM expressing empty, ABCA1-WT, or ABCA1 mutants (lower panel). Original magnification × 200. Arrows indicate extensive cellular infiltrate.
(F) Quantification of spleen weight of these mice.
(G and H) Representative dot plot (G) and quantification (H) of CD11 b+ Grl+ myeloid cells determined by flow cytometry in the spleens of recipient mice transplanted with control or Mx1-Cre+Tet2fl/fl BM expressing empty, ABCA1-WT, or ABCA1 mutants.
The results are means ± SEMs of 5–9 animals per group. ND, not detectable. *p < 0.05 versus empty control on a Tet2-deficient background. §p < 0.05 versus ABCA1-WT. #p < 0.05 and ##p < 0.001 versus Mx1-Cre+ controls.
ABCA1 Mutants Fail to Prevent CMML-Associated Extramedullary Hematopoiesis and Splenomegaly
The overexpression of ABCA1-WT suppressed the splenomegaly of animals transplanted with Tet2-deficient BM (Figures 2E and 2F). Despite a variability within the groups, this was statistically not observed in ABCA1 mutant-transduced animals on a Tet2-deficient background (Figure 2F). Although the median survival of Tet2-deficient mice was estimated to be 560 days (Kunmoto et al., 2018), the Kaplan-Meier survival curve indicated that the survival of ABCA1-WT-transduced animals on aTet2-deficient background is significantly better compared to empty vector-transduced animals and ABCA1 mutant-transduced animals over a 140-day period (Figure S2G). Pathological examination revealed a robust infiltration of myeloid cells, including significant destruction of normal spleen architecture in ABCA1 mutant-transduced animals on a Tet2-deficient background compared to ABCA1-WT transduced animals, similar to what was observed in empty vector-transduced animals (Figure 2E). Flow analysis of the spleen confirmed an increased proportion and number of CD11 b+Grl+ myeloid cells in ABCA1 mutant-transduced animals on a Tet2-deficient background, and to some extent, on a WT background, compared to ABCA1-WT-transduced animals (Figures 2G, 2H, and S2H). An increased percentage of HSPCs (LSK cells [Lineage Sca1+ c-Kit+]) was also observed in the spleens of ABCA1 mutant-transduced animals compared to ABCA1-WT-transduced animals (Figure S2I). Thus, unlike WT ABCA1, specific ABCA1 mutants associated with human CMML were unable to limit the increased extramedullary hematopoiesis and splenomegaly that are classical features of CMML.
ABCA1 Mutants Fail to Suppress Expansion and Myeloid Bias of Tet2-Deficient HSPCs
Tet2 loss or defective cholesterol efflux pathways leads to BM HSPCs and differentiation toward a myeloid lineage fate in vivo (Moran-Crusio et al., 2011; Quivoron et al., 2011; Yvan-Charvet et al., 2010). Analysis of the BM HSPCs showed a reduction in the LSK cells in ABCA1-WT-transduced animals on a Tet2-deficient background compared to empty control-transduced animals. This effect was lost in ABCA1 mutant-transduced animals (Figure 3A). Although ABCA1 mutants barely altered the percentage of BM megakaryocyte-erythrocyte progenitors (MEPs; Lineage−Sca1− c-Kit+CD34lowFcyRlow) (Figures S3A and S3B), the granulocyte-monocyte progenitors (GMPs; Lineage−Seal−c-Kit+CD34hiFcγRhi) and the common myeloid progenitors (CMPs; Lineage Seal c-Kit+CD34hiFcγRlow) were significantly increased in ABCA1 mutant-transduced BM compared to ABCA1-WT-transduced BM both on WT (Figures S3A and S3B) and Tet2-deficient backgrounds (Figure 3B). To determine whether ABCA1 mutants engage HSPCs to differentiate into the myelomonocytic lineages, BM cells from ABCA1 mutant-transduced animals were cultured ex vivo in the presence of IL-3 and GM-CSF. BM cultures showed that ABCA1 mutants disengaged the suppressive effects of ABCA1 on HSPC expansion (Figure 3C) and myeloid lineage expansion (Figure 3D) in theTet2-deficient background. These findings suggest that in contrast to ABCA1-WT, ABCA1 mutants allow Tet2-deficient HSPCs to proliferate and to favor myelopoiesis as a result of increased IL-3-GM-CSF signaling. Of note, in secondary transplants (Figure 3E), ABCA1 mutant-transduced animals had a significantly higher percentage of peripheral Gr-1highCD11bhigh myeloid cells compared ABCA1-WT-transduced animals (Figure 3F) and failed to suppress splenomegaly (Figure S3C) or splenic cholesterol accumulation (Figure S3D). These findings confirm that ABCA1 inactivation not only confers a significant competitive advantage to Tet2-deficient FISPCs in vivo but also enhances a TET2-induced transplantable myeloproliferative disorder.
Figure 3. ABCA1 Mutants Support Tet2-Deficient HSPC Expansion and Myeloid Lineage Commitment and Spreads CMML-like Disease in Serial BM Transplantation.

(A and B) Quantification of hematopoietic stem (A) and progenitor (B) cells in the BM of recipient mice transplanted with control or Mx1-Cre+Tet2fl/fl BM expressing empty, ABCA1-WT, or ABCA1 mutants. Lineage(Lin)− Sca1−c-Kit+ LSK cells are hematopoietic stem and progenitor cells (HSPCs); Lin_Sca1− c-Kit+CD34hiFcγRhi are granulocyte-monocyte progenitors (GMPs); and Lin−Sca1−c-Kit+CD34hiFcγRlow are common myeloid progenitors (CMPs). The results are the means ± SEMs of 5–9 animals per group.
(C and D) The quantification of hematopoietic progenitors (C) and myeloid cells (D) in BM cultures isolated from Mx1-Cre+Tet2fl/fl BM expressing empty, ABCA1-WT, or ABCA1 mutants and grown ex vivo for 72 h in liquid culture in the presence or absence of 6 ng/mL IL-3 and 2 ng/mL GM-CSF. The results are the means ± SEMs of experiments performed in triplicate.
(E) Experimental overview. BM from WT and ABCA1 mutant-transduced animals on aTet2-deficient background were serially transplanted into lethally irradiated WT mice and analyzed 7 weeks later.
(F) Quantification of the percentage of peripheral blood CD11bhi,Gr-1hi myeloid cells was determined by flow cytometry at the end of the study period. The results are means ± SEMs.
*p < 0.05 versus empty control transduced animals on a Tet2-deficient background. §p < 0.05 versus ABCA1-WT. #p < 0.05 and ##p < 0.001 versus Mx1-Cre+ controls.
ABCA1 Deficiency Cooperates with Tet2 Loss to Propagate Myeloid Transformation
In parallel, we crossed Mx1-Cre+Tet2fl/fl mice (Moran-Crusio et al., 2011; Quivoron et al., 2011) to Abca1fl/fl mice (Yvan-Charvet et al., 2010) to generate Mx1-Cre+Tet2fl/flAbca1fl/fl (referred to subsequently as DKOAHSC [double knockout, DKO]) mice. BM cells from these mice were subsequently transplanted into lethally irradiated C57BL/6J mice (Figure 4A). Animals were analyzed 5 weeks after BM reconstitution (TO) and at the indicated time point following PIPC injection (Figure 4A). We confirmed the excision of both Tet2 and Abcal mRNA expression in the BM of Mx1-Cre+Tet2fl/fl mice and Mx1-Cre+Abca1fl/fl mice, respectively (Figure 4B). DKOAHSC mice also underwent a marked increase in peripheral myeloid cells compared to single Abcal or Tet2-deficient mice (Figures 4C, 4D, and S4A). These findings show a cooperative effect between ABCA1 and Tet2 mutations on myeloid expansion. In addition, Abcal deficiency exacerbated the splenomegaly of Tet2-deficient animals, while having no effect on its own (Figures 4E and 4F). DKOΔHSC mice also exhibited higher myeloid cell infiltration compared to single-KO mice after pathological examination (Figure 4E), with an increased percentage and number of eosinophil, neutrophil, monocyte, and red pulp macrophages (RPMs) determined by flow cytometry (Figures 4G, S4B, and S4C). Thus, the complete deletion of ABCA1 in Tet2-deficient mice increased spleen size and enhanced extramedullary myelopoiesis. Consistently, DKOΔHSC mice showed an increased frequency of LSK cells compared with WT or Tet2-deficient mice (Figure 4H). There was also an additive effect ABCA1 and TET2 deficiency in promoting BM myeloid progenitor expansion (Figure 4l). These data indicate that concurrent ABCA1 deficiency and Tet2 loss in hematopoietic cells exacerbated myeloid transformation, supporting the finding that ABCA1 mutations identified in CMML patients were loss-of-function mutations.
Figure 4. ABCA1 Invalidation Propagates Myelopoiesis and Accelerates Extramedullary Hematopoiesis on a Tet2-Deficient Background.

(A) Experimental overview. BM from Mx1-Cre+, Mx1-Cre+Abca1fl/fl, Mx1-Cre+Tet2fl/fl, and Mx1-Cre+Tet2,l/flAbca1fl/,fl mice were transplanted into lethally irradiated WT mice, and after a 5-week recovery period, the mice were injected with poly(l:C) and analyzed over a 20-week period.
(B) Modulation of Abca1 and Tet2 mRNA expression levels in the BM of the aforementioned mouse models.
(C) Quantification of the percentage of peripheral blood myeloid cells determined by hematology cell counter over the course of 20 weeks after poly (I:C) injection in recipient mice transplanted with the BM from Mx1-Cre+, Mx1-Cre+Abca1fl/fl, Mx-Cre+Tet2fl/fl, and Mx1-Cre+Tetfl/fl Abca1fl/fl mice.
(D) Peripheral blood myeloid subsets (CD115+Ly6Chi and CD115+Ly6Clo monocytes and CD115−Ly6Chi neutrophils) were also quantified in these mice at the indicated time point
(E) Representative H&E staining of paraffin-embedded spleen sections from these mice. Original magnification × 200. Arrows indicate extensive cellular infiltrate.
(F and G) Quantification of spleen weight (F) and myeloid subsets (eosinophils, neutrophils, monocytes, and red pulp macrophages [RPMs] in the spleens (G) of recipient mice transplanted with the BM from Mx1-Cre+Abca1fl/fl, Mx1-Cre+Tet2, and Mx1-Cre+Tet2fl/fl Abca1fl/fl mice.
(H and I) Quantification of hematopoietic stem (H) and progenitor (MEPs); Lin−Sca1−c-kit+CD34hiFcγRhi are GMP; and Lin−Sca1−c-kit+CD34hi FcγRlow are CMPs.
The results are the mean ± SEMs of 5–7 animals per groups ND, not detectable. #p < 0.05 and ##p < 0.001 versus Mx1-Cre+ controls.
Cholesterol Accumulation Links ABCA1 Mutants and Tet2 Loss to IL3-Rβ Signaling Hypersensitivity
We next sought to identify the mechanisms responsible for the lack of tumor-suppressor function of ABCA1 mutants in Tet2-deficient BM cells. Increased cholesterol accumulation and reduced expression of ABCA1 have been repeatedly observed in cancer cells (Bovenga et al., 2015; Lin and Gustafsson, 2015). Thus, taking advantage of publicly available gene expression datasets (Kunimoto et al., 2018), we interrogated whether Tet2-deficient LSK, CMP, and GMPs cells could transcriptionally regulate cholesterol metabolic pathways. We did not observe a major transcriptional regulation of the genes involved in cholesterol metabolism (<10% overall changes), including LXR target genes and ABCA1 in Tet2-deficient hematopoietic progenitors (Figure S5A). The functional behavior of these cells was next assessed by quantifying the neutral lipid accumulation in single-KO and DKOΔHSC HSPCs by flow cytometry using BODIPY staining. An increase in cellular neutral lipid content in single-KO and DKOΔHSC HSPCs and committed myeloid progenitors (i.e., GMP and CMP) was observed compared to controls (Figures 5A and 5B). The combined deficiency of Tet2 and Abca1 also led to a cumulative BODIPY-neutral lipid staining per cell (expressed as mean fluorescence intensity [MFI]) in myeloid progenitors compared to single-KO cells (Figure 5B) but not in HSPCs (Figure 5A). Consistently, the overexpression of WT-ABCA1 reduced BODIPY staining inTet2-deficientCMP progenitors, but not in Tet2-deficient HSPCs (Figures 5C and 5D). However, ABCA1 mutants failed to reduce BODIPY staining on a Tet2-deficient background (Figures 5C and 5D) and to some extent on a control background (i.e., ABCA1-A1291T, G1421H, and P1423S) (Figure S5B). The anti-tumor activity of ABCA1 could be linked to the inhibition of sterol regulatory element- binding protein-2 (SREBP-2) and cholesterol biosynthesis target genes, which has recently been linked to the reduced expression of cell growth-facilitating mevalonate products in a solid tumor (Moon et al., 2019). However, the accumulation of cholesterol in ABCA1 mutant-transduced Tet2-deficient BM cells rather was associated with the reduced expression of genes in the mevalonate pathway (Figure 5E), which is a well-known feedback regulatory mechanism by which sterols accumulating in the endoplasmic reticulum (ER) limit the processing and expression of the target genes of SREBP-2 (Brown and Goldstein, 2009; Spann and Glass, 2013).
Figure 5. Cholesterol Accumulation Couples ABCA1 Invalidation and Tet2 Deficiency to IL-3 Receptor β Signaling Hypersensitivity.

(A-D) Quantification of BODIPY staining by flow cytometry expressed as mean fluorescence intensity (MFI) as a surrogate of cellular cholesterol neutral lipid per cell (A-D) in BM hematopoietic stem (A and C) and progenitor (B and D) cells (i.e., LSKs, MEPs, CMPs, and GMPs) of recipient mice transplanted with Mx1-Cre+, Mx1-Cre+Abca1fl/fl, Mx1-Cre+Tet2fl/fl,and Mx1-Cre+Tet2fl/,lAbca1fl/fl BM (A and B) or control and Mx1-Cre+Tet2fl/fl BM expressing empty, ABCA1-WT, or ABCA1 mutants (C and D). Results are means ± SEMs of 5–9 animals per group.
(E) mRNA expression of SREBP-2 and cholesterol biosynthesis target genes (Hmgcr, Mvk, Idi1, Sc4mol, and Dhcr24) from empty, WT, and ABCA1 mutant-transduced BM on a Tet2-deficient background isolated at the end of the study period. The expression of mRNA was normalized to m36B4. mRNA levels were expressed as percentage over WT whole BM cells.
(F) Proliferation rates were determined after 2 h [3H]-thymidine pulse labeling in BM cells from empty, ABCA1-WT, and ABCA1 mutant-transduced animals on a Tet2-deficient background that were grown for 72 h in liquid culture in the presence or absence of 6 ng/mL IL-3 and 2 ng/mL GM-CSF and the indicated chemical compounds. Results are means ± SEMs of cultures from at least 3 independent mice.
*p < 0.05 versus empty control-transduced animals on a Tet2-deficient background. §p < 0.05 versus ABCA1-WT. #p < 0.05 and ##p < 0.001 versus Mx1-Cre+ controls.
Given the activation of the IL3-Rβ canonical pathway in myeloid cells with a defective cholesterol efflux pathway (Yvan-Charvet et al., 2010) and the hypersensitivity of Tet2-deficient myeloid cells to GM-CSF (Kunimoto et al., 2018), we next assessed whether the myelosuppressive function of ABCA1 on a Tet2-deficient background could be attributed to its role in removing excess cellular cholesterol in committed myeloid progenitors. Excess cellular cholesterol was removed by treatment with cyclodextrin in ABCA1 mutant-transduced Tet2-deficient BM cells cultured in the presence or absence of IL-3 and GM- CSF. We validated our ex vivo BM culture proliferation assay by showing that inhibition of the IL-3Rβsignaling pathway using the IL-3Rβ blocking antibody prevented BM proliferation in IL-3 and GM-CSF treatment (Figure 5F). We next showed that both the IL-3Rβ blocking antibody and cyclodextrin prevented the proliferation of ABCA1 mutant-transduced Tet2-deficient BM cells, similar to the effect of ABCA1-WT overexpression (Figure 5F), without affecting cell viability (data not shown). Consistently, western blot analysis showed higher levels of pErk1/2 or pJak2 in ABCA1 mutant-transduced Tet2-deficient BM cells compared to ABCA1-WT-transduced cells (Figure S5C). mRNA expression analysis also revealed higher cyclin D1 and PU.1 gene expression in ABCA1 mutant-transduced Tet2-deficient BM cells (Figure S5D). These genes are IL-3R3 signaling targets involved in proliferation and myeloid lineage commitment. In contrast, we did not observe changes in the expression of the key transcription factor CCAAT/enhancer-binding protein alpha (C/EBPα) or in the negative regulators of RAS signaling, including dual-specificity phosphatase 1 (DUSP1) or sprouty-related, EEVH1 domain-containing protein 1 (Spredl) (Kunimoto et al., 2018). These findings suggest that the tumor-suppressor function of ABCA1 relies on its capacity to modulate cholesterol-dependent IL-3R3 signaling and downstream mitogen-activated protein kinase (MAPK) and JAK2 signaling. We and others recently showed that reduced autophagy and enhanced mitochondrial reactive oxygen species (ROS) production were 2 major metabolic events promoting HSPC expansion and myeloid commitment downstream of the IL-3R3 signaling pathway (Lum et al., 2005; Sarrazy et al., 2016). Thus, we estimated autophagic activity using the Cyto-ID probe by flow cytometry. Reduced autophagic activity was observed in Tet2- and Abca1-deficient myeloid progenitors (Figure S5E) and in ABCA1 mutant-transduced CMP and GMP cells compared to ABCA1-WT-transduced BM both on WT and Tet2-deficient cells (Figure S5F). Finally, suppression of mitochondrial ROS with tempol prevented the basal proliferation and IL-3-GM-CSF hypersensitivity of ABCA1 mutant-transduced Tet2-deficient BM cells (Figure 5F). These data confirm that ABCA1 mutations impair the myelosuppressive function of ABCA1 by increasing IL-3-GM-CSF hypersensitivity and downstream metabolic sequelae in Tet2-deficient myeloid progenitors.
Increased HDL Reverses Increased Myelopoiesis and Splenomegaly Caused by ABCA1 Mutants
Given the ability of increased HDL to suppress HSPC myeloid lineage commitment and rescue the myeloproliferative disorder of mice with defective cholesterol efflux (Yvan-Charvet et al., 2010), we hypothesized that raising HDL would alleviate some of the myeloproliferative phenotypes of ABCA1 mutant-transduced Tet2-deficient animals. First, HDL treatment ex vivo reduced the proliferation rates of ABCA1 mutant-transduced Tet2-deficient BM cells (Figure 6A). This was not the case with low-density lipoprotein (LDL) treatment (data not shown). HDL treatment also reduced BODIPY cholesterol staining (Figure 6B) of ABCA1 mutant-transduced Tet2-deficient BM cells. We next explored the efficacy of the apoA-1 transgene to increase HDL levels in vivo (Figures 6C and 6D). Transduction of ABCA1-A1291T, G1421R, and A2011T was selected as the proof of concept and compared to BM transduced with empty vector or ABCA1-WT. Similar to ABCA1-WT-transduced BM cells, the human apoA-1 transgene prevented the peripheral myeloid expansion of mice transplanted with ABCA1 mutant-transduced Tet2-deficient BM (Figure 6E) and also abolished their splenomegaly (Figure 6F). These data demonstrate that increased apoA-1 production and increased HDL show robust preclinical efficacy in myeloproliferative neoplasms driven by ABCA1 and TET2 mutants.
Figure 6. HDL Overcome Loss-of-Function ABCA1 Mutants and Limit the Myeloproliferative Disorder Induced by These Mutants in Tet2-Deficient Mice.

(A) Proliferation rates were determined after 2 h [3H]-thymidine pulse labeling in BM cells from empty, ABCA1-WT, and ABCA1 mutant-transduced animals on a Tet2-deficient background that were grown for 72 h in liquid culture in the presence or absence of 50 (μg/mL polyethylene glycol (PEG)-HDL.
(B) The quantification of BODIPY staining was determined by flow cytometry in these cells and expressed as the MFI. The results are means ± SEMs of cultures from at least 3 independent mice.
(C and D) Levels of human apoA-1 (hApoA-1) (C) and plasma HDL-cholesterol levels (D) were determined in WT or apoA-1 transgenic recipient mice transplanted with Mx1-Cre+Tet2fl/fl BM transduced with lentiviral particles expressing ABCA1-WT or ABCA1 mutants or empty vector.
(E and F) Peripheral blood CD11b+Grl+ myeloid subsets (E) and spleen weight (F) were quantified at the end of the study period (i.e., 7 weeks post-poly(l:C) injection that followed a 5-week recovery period post-BMT) in WT or apoA-1 transgenic recipient mice transplanted with Mx1-Cre+Tet2fl/fl BM transduced with lentiviral particles expressing ABCA1-WT or ABCA1 mutants or empty vector. Results are means ± SEMs of 4–5 animals per group.
*p < 0.05 versus empty control-transduced animals on a Tet2-deficient background. §p < 0.05 versus ABCA1-WT. #p < 0.05 versus non-HDL-treated conditions or transduced animals on a non-ApoA1Tg background.
DISCUSSION
Here, we identified recurrent somatic mutations in the cholesterol exporter ABCA1 in CMML patients. The expression of ABCA1 mutants in vitro confers a growth and proliferative advantage in vitro compared to the expression of WT-ABCA1. The relevance of these mutations is underscored by the loss of the tumor-suppressor function of WT-ABCA1 in the context of Tet2 deficiency. ABCA1 deficiency also accelerated the myeloproliferative disorder of Tet2-deficient BM transplanted mice, as illustrated by increased myelopoiesis and extramedullary hematopoiesis. The myelosuppressive function of ABCA1 was further illustrated by the failure of ABCA1 mutants to limit a fully penetrant myeloproliferative disorder after serial transplantation. The underlying mechanism involves the accumulation of cellular cholesterol in myeloid progenitors, resulting in IL-3Rβ signaling hypersensitivity with sustained ERK1/2 and JAK2 signaling and subsequent metabolic perturbations (e.g., reduced autophagy, enhanced mitochondrial ROS formation). In addition, the overexpression of the WT human apoA-1 transgene rescued the loss of the myelosuppressive function of ABCA1 mutants. These findings provide further evidence that these effects were caused by defective cholesterol efflux and suggest a therapeutic application of HDL-raising therapies for CMML patients bearing ABCA1 mutants.
The classical hallmarks of cancer are intimately intertwined with an assortment of metabolic processes that a tumor cell effectively hijacks to facilitate malignant transformation (Boroughs and DeBerardinis, 2015; Hanahan and Weinberg, 2011; Kroemer and Pouyssegur, 2008). Several studies, largely performed in vitro, suggest that altered cholesterol metabolism (i.e., enhanced cholesterol synthesis and reduced cholesterol efflux) is linked to the cellular transformation that is most likely to provide the essential building blocks required to maintain their aberrant survival and growth (Bovenga et al., 2015; Clendening and Penn, 2012; Dang, 2012; Lin and Gustafsson, 2015; Mullen et al., 2016). This synergistic metabolic regulation is a highly cooperative process that is thought to involve a transcriptional response of non-mutant genes (i.e., “cooperation response genes”) induced by oncogenic mutations, including the downregulation of the cholesterol exporter ABCA1 (Hirsch et al., 2010; McMurray et al., 2008). Conversely, short amphipathic a-helical peptides, the presumed mimetics of apoA-1 (the major apolipoprotein constitutive of HDL), inhibited ovarian tumor development (Su et al., 2010), synthetic HDL nanoparticles inhibited B cell lymphoma xenografts (Yang et al., 2013), and injection of human apoA-1 promoted tumor and métastasés regression in mice inoculated with malignant melanoma cells (Zamanian-Daryoush et al., 2013). These findings indicate decreased ABCA1 efflux activity in a variety of solid tumors that may have pathophysiological importance. However, efforts to identify causal somatic mutations in genes governing cholesterol efflux pathways have been limited so far to human colon cancer samples (Sjöblom et al., 2006; Smith and Land, 2012). Similar to solid tumors, early studies have associated hematological malignancy, especially chronic myelocytic leukemia, with perturbed cholesterol metabolism (Dessì et al., 1995; Gilbert and Ginsberg, 1983). Hypercholesterolemia and defective hematopoietic cholesterol efflux pathways control many aspects of HSPC homeostasis in mice, including their proliferation and expansion, their myeloid lineage commitment, and their mobilization from the BM (Lacy et al., 2019; Soehnlein and Swirski, 2013; Tall and Yvan-Charvet, 2015). We also demonstrated that the overexpression of the human apoA-1 transgene prevented the myeloprolifération of mice bearing the Mpl-W515L or Flt3-ITD oncogenes (Gautier et al., 2013). The discovery of ABCA1 mutants in CMML samples with loss of myelosuppressive activity in cell culture and in vivo provides causal evidence of the role of ABCA1 efflux activity in myeloproliferative disorders.
Functional studies have suggested co-occurrence of specific combinations of leukemia disease alleles to drive myeloid transformation (Kunimoto et al., 2018; Shih et al., 2015). These mutations include genes that control signal transduction (e.g., somatic RAS mutations) and metabolic pathways (e.g., somatic isocitrate dehydrogenase [IDH] mutations) and those that regulate epigenetic modifications. One of the epigenetic modifiers most commonly mutated in myeloid malignancies is TET2 (Itzykson et al., 2017), which catalyzes the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, leading to DNA demethylation (Tahiliani et al., 2009). These mutations are thought to largely arise spontaneously as a result of replicative stress in aging HSCs providing the first-step acquisition of clonal expansion and a bias toward the myeloid lineage (Moran-Crusio et al., 2011; Quivoron et al., 2011), but they are not sufficient to induce myeloid transformation and IL-3Rβ hypersensitivity (Itzykson et al., 2017). Our findings suggest that these ABCA1 mutants, due to their loss of myelosuppressive functions, could play a pivotal role in providing a second hit, which cooperates with CH alleles to drive myeloid transformation.
Somatic mutations in ABCA1 have not previously been described in leukemia; however, loss-of-function mutations in the ABCA1 gene have previously been characterized in Tangier disease, a rare autosomal-recessive disorder (Kang et al., 2010). Apart from mutations located in the apoA-1 binding pocket, loss-of-function mutations have been located in the island C-terminal regions or the PEST sequence, involving several mechanisms such as mislocalization of ABCA1 away from the cell surface, impaired dimerization, ER exit failure, or protein interaction-dependent destabilization and degradation, all leading to impaired cholesterol efflux (Kang et al., 2010). Thus, the proliferative advantage of some ABCA1 mutants (ABCA1-A1291T, ABCA1-G1421R, and ABCA1-P1423S) over empty control could be attributed to a change in the dimerization with endogenous ABCA1, as previously observed in Tangier disease (i.e., dominant-negative effect) (Brunham et al., 2006; Kang et al., 2010). Engineered mutations in ABCA1 have also revealed the additional regulation of receptor tyrosine kinase signaling by lipid-dependent and -independent export activity (Vaughan et al., 2009; Tang et al., 2009). Thus, future studies are necessary to pinpoint the molecular mechanism at the origin of the impaired ABCA1 efflux activity or signal transduction and subsequent myelosuppressive functions of the newly identified ABCA1 mutants.
Monocytosis and IL-3Rβ hypersensitivity are hallmarks of CMML (Itzykson et al., 2017), and we previously showed that the accumulation of plasma membrane cholesterol in myeloid progenitors with defective cholesterol efflux pathways is the culprit of enhanced IL-3R0 signaling (Yvan-Charvet et al., 2010). However, ABCA1 anti-tumor activity in murine hepatocellular carcinomas driven by p53 loss has recently been attributed to a distinct mechanism involving the inhibition of SREBP-2 maturation with the consequent reduced expression of cell growth-facilitating mevalonate products, which are intermediates in the cholesterol biosynthesis pathway (Moon et al., 2019). This mechanism is inconsistent with our data in the hematopoietic context, since reduced expression of SREBP-2 and cholesterol biosynthesis target genes were observed in ABCA1 mutant-transduced leukemic cells, and the loss of the anti-proliferative activity of ABCA1 mutants was overcome by HDL or cyclodextrin-mediated cholesterol efflux in cell culture and by increased HDL levels in vivo. HDL and cyclodextrin promote cholesterol efflux by non-ABCA1 pathways, which should deplete cellular and ER cholesterol (Tall and Yvan-Charvet, 2015), leading to increased SREBP2 processing and increased cholesterol biosynthesis (Brown and Goldstein, 2009; Spann and Glass, 2013). Moreover, the overexpression of cholesterol efflux-promoting genes leads to the increased expression of SREBP-2 target genes (Wang et al., 2006). The reason for the apparent discrepancy between these studies is unknown, but it could be related to the specific mutant alleles and tissue context being evaluated (Lee et al., 2013). The present study rather shows that the IL-3R3 hypersensitivity observed in the presence of ABCA1 mutants metabolically reprograms myeloid cells downstream of receptor tyrosine kinase signaling activation, which is the culprit of mitochondrial ROS production, known to drive hyperproliferation and leukemic hematopoiesis (Vander Heiden et al., 2009; Abdel-Wahab and Levine, 2010).
Treatment for CMML remains challenging, since there are few validated therapeutic targets, and conventional cytotoxic drugs have limited activity. The only curative option to date is allogenic stem cell transplantation. However, most patients are diagnosed at an advanced age and are not candidates for stem cell transplantation (Itzykson et al., 2017). Our data indicate that cooperative epigenetic remodeling by TET2 loss and ABCA1 mutants supports myeloid transformation, which may provide a rationale for mechanism-based therapy. Given the emerging association between HDL and cancer (Pirro et al., 2018), the identification of ABCA1 mutations in CMML provides a potential target for therapeutic intervention with HDL-raising therapies such as infusions of reconstituted HDL, which are in phase III clinical trials for acute coronary syndrome. This could be repurposed for the treatment of myeloid malignancies with genetic inactivation of ABCA1 function.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfiled by the Lead Contact, Dr. Laurent Yvan-Charvet (yvancharvet@unice.fr).
There are restrictions to the availability of plasmid generated and stable cell lines due to the lack of an external centralized repository for its distribution and our need to maintain the stock. We are glad to share repository with reasonable compensation by requestor for its processing and shipping.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary CMML patient samples
All samples were collected on MSKCC protocol 06–107. Protocol 06–107 was most recently approved by the Human Subjects Protection Committee at the Memorial Sloan Kettering Cancer Center on July 09, 2019 and is fully HIPAA compliant. Peripheral blood and/or bone marrow samples were collected from 26 patients (men and women) with CMML; informed consent was obtained from all patients included in this study. Matched normal tissue in the form of a buccal swab was available for all patients.
Mice
WT, Mx1-Cre+ (B6.Cg-Tg(Mx1-cre)1Cgn/J), Tet2fl/fl (B6;129S-Tet2tm1.1laai/J), Abca1fl/fl (B6.129S6-Abca1tm1Jp/J) and human apoA-1 transgenic male mice (B6.Tg(ApoA1)1Rub/J), were obtained from the Jackson Laboratory between the age of 6 and 10 weeks. Human apoA-1 transgenic mice were selected based on the human apoA-1 levels in the range of 150–300mg/dL (ELISA do not detect mouse apoA-1) as previously described (Rubin et al., 1991; Yvan-Charvet et al., 2010). Mx1-Cre+ Tet2fl/fl mice, Mx1-Cre+ Abcalfl/fl mice and Mx1-Cre+ Tet2fl/flAbcalfl/fl littermate mice were used for this study. Animal procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at Mediterranean Center of Molecular Medicine (C3M). Mice were maintained on a 12h light/12hdarkness lighting schedule. Animals had ad libitum access to both food and water.
Cell Line
THP-1 cells (human acute monocytic leukemia cell line, TIB-202, ATCC) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% C02.
HEK293 cells (human embryonic kidney, CRL-1573, ATCC) were grown in DMEM containing 10% FBS and cultured at 37°C in 5% CO2.
METHOD DETAILS
Genetic analysis of primary patient samples
Genomic DNA was extracted from viably frozen peripheral blood granulocytes and buccal swabs. High-throughput DNA sequence analysis was used to screen for mutations in ABCA1, ABCG1, NR1H2, and NR1H3. All DNA samples were whole genome amplified using 029 polymerase to ensure sufficient material was available for sequence analysis. M13-appended gene-specific primers were designed to amplify and sequence all coding exons of all isoforms of the above-mentioned genes. Primer sequences and the genomic coordinates of all amplicons sequenced are included in Table S1. Bidirectional sequence traces were analyzed for missense and nonsense mutations using Mutation Surveyor (Softgenetics, Inc., State College, PA), and all traces were reviewed manually and with Mutation Surveyor for the presence of frameshift mutations. Mutations were annotated according to the predicted effects on coding sequence using NM_005502.2, NM_004915.3, NM_007121.4, and NM_001130101.1 as the reference sequence for ABCA1, ABCG1, NR1H2, and NR1H3 respectively. Non-synonymous mutations were first compared to published SNP data (dbSNP, https://www.ncbi.nlm.nih.gov/projects/SNP) such that previously annotated SNPs were not considered pathogenic mutations. Missense mutations not in the published SNP database were annotated as somatic mutations based on either on reported data demonstrating these are somatic mutations or sequence analysis of that demonstrated these mutations were present in tumor and not in matched normal DNA. All somatic mutations were validated by resequencing non-amplified source DNA for the particular amplicon where the mutation was noted. Genomic DNA from paired samples was verified to belong to the same patient by genotyping of the specimens for 42 highly polymorphic single-nucleotide polymorphisms using mass-spectrometry based genotyping as described previously. In order to determine whether the genes in which non-synonymous mutations were identified were mutated at a rate higher than expected by chance alone, we first calculated the rate of non-synonymous mutations in the sequenced genes. We then performed binomial test in R (http://www.r-project.org/) to compare the rate of non-synonymous mutations in the genes identified in this study with the expected background rate of 0.22 – 2.5 × 10−6 synonymous mutations identified in several prior large-scale sequencing studies2–10 as well as the expected ratio of silent:non-silent mutations (0.31–0.41) from the same studies.
Plasmids
Mouse ABCA1 cDNA, with a homology of 97% to human ABCA1 cDNA, was used to generate P711L, A1291T, G1421R, P1423Sand A2011T mutant cDNAs and cloned into pLKO lentiviral vectors to genetically perturb cells by lentiviral infection and avoid cross reactivity.
Mice and treatments
Bone marrow (BM) transplantation into lethally irradiated WT recipients and serial BM transplantation studies were performed as previously described (Yvan-Charvet et al., 2010). After 5 weeks of reconstitution, mice were i.p injected with poly:IC (250 μg/injection with a cumulative dose of 750 μg/mice, Invivogen) to induce gene deletion/recombination. Mice were used between 3 and 5 months after the injections of poly:IC depending of the experiment to allow the development of myeloproliferative neoplasms (Moran-Crusio et al., 2011).
Lentiviral BM transplantation
The lentiviral BM transplant assay was performed as previously described (Gautier et al., 2013). In brief, Mx1-Cre+ and Mx1-Cre+ Tet2fl/fl mice were injected with 5-fluorouracile (3mg/mice of 5-FU, F6627, Sigma) 3 days before the experiment to enrich HSPCs within the BM. Control, ABCA1-WT and ABCA1 mutant lentiviral particles (pLKO lentiviral vector containing a MSCV-IRES-EGFP sequence, Genecust) were tittered and used to transduce Mx1-Cre+ or Mx1-Cre+ Tet2fl/fl cells. BM cells were cultured for 24 h in transplantation media (RPMI + 10% FBS + 6 ng/ml IL-3 (Corning), 10 ng/ml IL-6, and 10 ng/ml stem cell factor (Milteny Biotech)) and treated with lentiviral particles (MOI of 5 in the presence of polybrene (Sigma)). After washing, the cells were used for BM transplantation into lethally irradiated WT or human apoA-1 transgenic recipient mice as indicated in the figure legends. The transduction efficiency ranged from 70%−90% in LSK cells before implantation as previously described (Gough and Raines, 2003; Pikman et al., 2006; Westerterp et al., 2012). After 5 weeks of reconstitution, mice were i.p injected with poly:IC (250 μg/ injection with a cumulative dose of 750 ng/mice, Invivogen) to induce gene deletion/recombination. Mice were used between 3 and 5 months after the injections of poly:IC depending of the experiment.
White blood cell counts-
Leukocytes, differential blood counts, platelets and erythrocytes were quantified from whole blood using a hematology cell counter (HEMAVET® 950).
Histopathology
Mice were euthanized and tissues were harvested and fixed in 4% paraformaldehyde. Spleen was serially paraffin sectioned using a Microm HM340E microtome (Microm Microtech, Francheville France) and stained with H&E for morphological analysis as previously described (Yvan-Charvet et al., 2010).
HEK293 cell transfection and culture
HEK293 cells at a density of 106 cells/well were transiently transfected with similar amounts of control empty vector (pcDNA 3.1+), ABCA1-WT or mutant cDNA using LipofectAMINE 2000 according to the manufacturer’s instructions (Invitrogen). Then, cells were incubated for different times in DMEM containing 10% FBS before treatments as indicated in the figure legends.
Human THP-1 monocytic leukemia cells and treatments
Non-adherent THP-1 monocytes were transduced at MOI of 5 with control, ABCA1-WT and ABCA1 mutant lentiviral particles (pLKO lentiviral vector containing a CMV promoter, Genecust) in the presence of polybrene (Sigma) and used 3 days later for experiments as described in the figure legends. THP1 cells were treated for 16 hours with puromycin 24 hours after transfection to improve the transient transduction/transfection efficiency up to 60% to 80% (data not shown), which slowed down the proliferation rate of these cells. In some experiments, stable overexpressing ABCA1-WT and ABCA1 mutants THP-1 macrophages were generated after lentiviral transduction and GFP selection of a puromycin resistant pLKO vector containing ABCA1 gene.
BM harvest and treatment
Briefly, femurs were flushed with ice-cold PBS and centrifugated for 5min at 1,000rpm to extract BM cells. After red blood cell lysis, over 90% of BM cells were CD45-positive cells of hematopoietic origin (Westerterp et al., 2012). Primary BM cells were resuspended in IMDM (GIBCO) containing 10% FCS (STEMCELL Technologies) and cultured for 1 h in tissue culture flasks to remove adherent cells, including macrophages. The transduction rate of control, ABCA1-WT and ABCA1 mutant lentiviral particles was determined after BM transplantation as described above. Suspended cells were then normalized to the same concentration and cultured for 72 h in the presence of 6 ng/mL IL-3 and 2ng/mL GM-CSF (R&D Systems). In some experiments, the cyclodextrin (Sigma) was used at the final concentration of 5 mM, tempol (EMD Millipore) at 4 mM and anti-IL3Rp AF549 antibody (R&D Systems) at 50 ng/mL.
[3H]-Thymidine proliferation assay
For proliferation assays, cells were pulsed for 2 h with 2 μCi/ml [3H]-thymidine, and the radioactivity incorporated into the cells was determined by standard procedures using a liquid scintillation counter.
Isotopic cholesterol efflux assay
THP-1 monocytes were treated with 100 nmol/L PMA (Phorbol myristate acetate) for 24 hour to facilitate differentiation into macrophages and cultured for 24 h in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) containing 2 μCi/ml of [3H]-cholesterol. Cholesterol efflux was performed for 6 h in 0.2% BSA DMEM containing 15 ng/mL apoA-l. The cholesterol efflux was expressed as the percentage of the radioactivity released from the cells in the medium relative to the total radioactivity in cells plus medium (Yvan-Charvet et al., 2010).
Cellular and tissue cholesterol content
Total lipids were extracted with chloroform/methanol from total cell lysates. Cholesterol mass in cells was determined using colorimetric kits (Wako Chemicals).
Flow cytometry analysis
BM cells, peripheral blood and splenocytes were collected from leg bones, blood and spleen cells after manual flushing or grinding, lysis to remove red blood cells and filtering through a 40-μm cell strainer as previously described (Yvan-Charvet et al., 2010). For peripheral blood leukocytes analysis, 100 [iL of blood were collected into EDTA tubes before red blood cell lysis and filtration. Freshly isolated BM, spleen and blood cells were stained with the appropriate antibodies for 30 min on ice. Cellular cholesterol content was quantified using the Bodipy-cholesterol probe (Life Technologies). Phosphoflow staining was performed according to the manufacturer’s instruction (BD Biosciences). HSPC and hematopoietic progenitor subsets and myeloid cell populations were analyzed by flow cytometry using an LSR Fortessa (Becton Dickinson) or sorted with a FACSAria II instrument (Becton Dickinson). All gating strategies are depicted in the Figures. Data were analyzed with FlowJo software (Tree Star).
Cholera Toxin staining
After a wash in complete growth medium, THP-1 transduced monocytes were stained 15min at 37°C, in 1 μg/ml working solution of Cholera Toxin Subunit B, Alexa Fluor 594 conjugate (Invitrogen, C34777). Cells were then stained with 1ng/ml working solution of DAPI (4’,6-diamidino-2-phenylindole) and washed 3 times in PBS1X. Immunostaining of cells was read on a Nikon Confocal A1R microscope.
Antibodies
TCR-β (H57–597), F4/80 (BM8), CD2 (RM2–5), CD3e (145–2C11), CD4 (GK1.5), CD8b (53–6.7), CD19 (eBio1D3), CD45R (B220, RA3–6B2), Gr-1 (Ly6G, RB6–8C5), Cd11b (Mac1, M1/70), Ter119 (Ly76) and NK1.1 (Ly53, PK136)-FITC were all from eBioscience and used for lineage determination. c-Kit (CD117, ACK2)-APCeFluor780 from eBioscience, Sca-1-Pacific blue from Biolegend, FcgRII/lll-PE (CD 16/32, 2.4G2), CD34 (RAM34)-AlexaFluor 647, CD135 (Flt3, A2F10)-PE, CD150 (Slamfl, TC15–12F12.2)-PECy7 were from Biolegend and used to quantify HSPCs and progenitor subsets. Peripheral leukocytes were stained with CD115 (AFS98)-APC, CD45 (30-F11)-APCCy7 and Ly6C/G or Gr-1 (RB6–8C5)-PercPCy5.5 from eBioscience and BD Biosciences, respectively.
RNA analysis
Total RNA extraction, cDNA synthesis and real-time PCR were performed as described previously (Yvan-Charvet et al., 2010). m36B4 RNA expression was used to account for variability in the initial quantities of mRNA.
Analysis of DNA hydroxymethylation
Global 5-hmC levels in genomic DNA isolated from blood cells were evaluated by ELISA, following manufacturer’s instructions (MethylFlash Global DNA Hydroxymethylation ELISA Easy Kit).
Western Blotting
The expression of ABCA1, TET2, phospho JAK2 and ERK were measured in BM cell by western blot analysis. Briefly, cell extracts were electrophoresed on 4%−20% gradient SDS-PAGE gels and transferred to 0.22-fim nitrocellulose membranes. The membrane was blocked in Tris-buffered saline, 0.1 % Tween20 containing 5%(w/v) nonfat milk (TBST-nfm) at room temperature (RT) for 1 h and then incubated with the primary antibody (TET2, phospho JAK2 and ERK antibodies from Cell Signaling and ABACI antibody from Abeam) in TBST-nfm at RT for4h, followed by incubation with the appropriate secondary antibody coupled to horseradish peroxidase. Proteins were detected by ECL chemiluminescence (Pierce). Intensity of each protein strips was quantified using ImageJ software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are shown as mean ± SEM. Statistical significance was performed using two-tailed parametric Student’s t test or by one-way analysis of variance (ANOVA, 4-group comparisons) with a Bonferroni multiple comparison post-test according to the dataset (GraphPad software, San Diego, CA). Results were considered as statistically significant when p < 0.05. All the statistical details of experiment can be found in the figure legends.
DATA AND CODE AVAILABILITY
The datasets supporting the current study have not been deposited in a public repository because of a pending patent at the time of submission but are available from the corresponding author on request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-IL3R antibody | R&D Systems | Cat# AF549; RRID:AB_355432 |
| TCR-β (H57–597)-FITC | eBioscience | Cat# 11–5961-82; RRID:AB_465323 |
| F4/80 (BM8)-FITC | eBioscience | Cat# 11–4801-82; RRID:AB_465323 |
| CD2 (RM2–5)-FITC | eBioscience | Cat# 11–0021-82; RRID:AB_464873 |
| CD3e (145–2C11)-FITC | eBioscience | Cat# 11–0031 −81; RRID:AB_464881 |
| CD4 (GK1.5)-FITC | eBioscience | Cat# 11–0041-82; RRID:AB_464892 |
| CD8b (Ly-3)-FITC | BioLegend | Cat# 126606; RRID:AB_961295 |
| CD19 (eBiol D3)-FITC | eBioscience | Cat# 11–0193-82; RRID:AB_657666 |
| CD45R (B220, RA3–6B2)-FITC | eBioscience | Cat# 14–0452-82; RRID:AB_467254 |
| Gr-1 (Ly6G, RB6–8C5)-FITC | eBioscience | Cat# 11–5931-82; RRID:AB_465314 |
| Cd11b (Mac1, M1/70)-FITC | eBioscience | Cat# 11–0112-41; RRID:AB_11042156 |
| Ter119 (Ly76)-FITC | eBioscience | Cat# 11–5921-82; RRID:AB_465311 |
| NK1.1 (Ly53, PK136)-FITC | eBioscience | Cat# 11–5941-82; RRID:AB_465318 |
| c-Kit (CD117, ACK2)-APCeFluor780 | eBioscience | Cat# 47–1172-82; RRID:AB_1582226 |
| Sea-1 -Pacific blue | Biolegend | Cat# 108119; RRID:AB_493274 |
| FcgRII/III-PE (CD16/32, 2.4G2) | Biolegend | Cat# 156606; RRID:AB_2800704 |
| CD34 (RAM34)-AlexaFluor 647 | Biolegend | Cat# 119314; RRID:AB_604089 |
| CD135 (Flt3, A2F10)-PE | Biolegend | Cat# 135305; RRID:AB_1877218 |
| CD150 (Slamfl, TC15–12F12.2)-PECy7 | Biolegend | Cat# 115913; RRID:AB_439796 |
| CD115 (AFS98)-APC | Biolegend | Cat# 135509; RRID:AB_2085222 |
| CD45 (30-F11)-APCCy7 | Biolegend | Cat# 103115; RRID:AB_312980 |
| Ly6C/G or Gr-1 (RB6–8C5)-PercPCy5.5 | BD Bioscience | Cat# 552093; RRID:AB_394334 |
| ABCA1 Antibody | Abeam | Cat# ab18180; RRID:AB_444302 |
| TET2 Antibody | Cell Signaling | Cat# 4501 OS; RRID:AB_2799277 |
| Phospho-Jak2 (Tyr1008) (D4A8) Rabbit mAb | Cell Signaling | Cat# 8082; RRID:AB_10949104 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat# 4377; RRID:AB_331775 |
| Bacterial and Virus Strains | ||
| pLKO lentiviral | Genecust | N/A |
| Biological Samples | ||
| Peripheral blood | MSKCC | HSPCMSKCC06–107 |
| Bone marrow | MSKCC | HSPCMSKCC06–107 |
| Buccal swab | MSKCC | HSPCMSKCC06–107 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| poly:IC | Invivogen | tlrl-pic-5 |
| 5-fluorouracile | Sigma | F6627 |
| IL-6, Mouse | Milteny Biotech | 130–094-065 |
| IL-3 Culture Supplement, Mouse | Corning | 354040 |
| Stem Cell Factor, Mouse | Milteny Biotech | 130–094-079 |
| polybrene | Sigma | TR-1003 |
| 4% paraformaldehyde | Sigma | 1004968350 |
| LipofectAMINE 2000 | Invitrogen | 11668030 |
| GM- CSF | R&D Systems | 415-ML-005 |
| Methyl-β-cyclodextrin | Sigma | C4555 |
| Tempol | EMD Millipore | 581500, CAS 2226–96-2 |
| Thymidine, [Methyl-3H] | PerkinElmer | NET027W001 MC |
| Phorbol myristate acetate | Sigma | P8139 |
| Apolipoprotein A-l from human plasma | Sigma | A0722 |
| Bodipy-cholesterol probe | Life Technologies | D3922 |
| Fixation Buffer (BD Cytofix) | BD Bioscience | 554655 |
| Perm Buffer III | BD Bioscience | BD Bioscience |
| Cholera Toxin Subunit B, Alexa Fluor 594 conjugate | Invitrogen | C34777 |
| DAPI (4’,6-diamidino-2-phenylindole) | ThermoFisher Scientific | D1306 |
| Critical Commercial Assays | ||
| Total Cholesterol colorimetric kits | Wako Chemicals | 999–02601 |
| MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Colorimetric) | Epigentek | P-1032–96 |
| Experimental Models: Cell Lines | ||
| HEK293 cells (human embryonic kidney) | ATCC | CRL-1573 |
| THP-1 monocytes (human acute monocytic leukemia cell line) | ATCC | TIB-202 |
| Experimental Models: Organisms/Strains | ||
| WT mice | Jackson Laboratory | SN000664 |
| Mx1-Cre mice (B6.Cg-Tg(Mx1-cre)1Cgn/J) | Jackson Laboratory | SN003556 |
| Abca1fl/fl mice (B6.129S6-Abca1tm1 Jp/J) | Jackson Laboratory | SN028266 |
| Tet2fl/fl (B6; 129S-Tet2tm 1.1 laai/J) | Jackson Laboratory | SN017573 |
| Human apoA-1 transgenic (B6.Tg(ApoA1)1Rub/J) | Jackson Laboratory | SN 001927 |
| Oligonucleotides | ||
| See Table S1 for genetic analysis of primary patient samples | N/A | N/A |
| $Software and Algorithms | ||
| FlowJo software | Tree Star | https://www.flowjo.com |
| Other | ||
| 4–20% Criterion Tris-HCI Protein Gel, 12+2 well, 45 μl | BioRad | 3450032 |
| Fetal Bovine Serum, Heat Inactivated | Fischer scientific | 12350273 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFischer | 15140122 |
| RMP11640, no glutamine | ThermoFischer | 31870074 |
| L-Glutamine (200 mM) | ThermoFischer | 25030024 |
Highlights.
ABCA1 somatic mutations were identified in CMML patients
ABCA1 mutations fail to repress myeloproliferative neoplasms in Tet2-deficient mice
ABCA1 mutations sustain IL-3Rβ signaling-driven myelopoiesis in Tet2-deficient HSPCs
Overexpression of apoA-1 overcomes ABCA1/TET2 co-mutant myeloproliferative neoplasms
ACKNOWLEDGMENTS
We thank Dr. Frédéric Larbret for assistance with flow cytometry and Dr. Véronique Corcelle for assistance in animal facilities. A PACA region PhD fellowship and the La Ligue Nationale Contre le Cancer support M.V. This work was supported by the Inserm Atip-Avenir program, the European Marie Curie Career Integration Grant (CIG-630926), and the Fondation ARC (ARC-R14027AA-RAC14007AAA) to L.Y.-C. This work was supported in part by NIH grants HL107653 and HL137663 and the Leducq Foundation to A.R.T., and by MSKCC Support Grant/Core Grant P30 CA008748.
DECLARATION OF INTERESTS
R.L.L. is on the supervisory board of QIAGEN and is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, each of which include an equity interest. He receives research support from and has consulted for Celgene and Roche, has received research support from Prelude Therapeutics, and has consulted for Astellas, Incyte, Janssen, Morphosys, and Novartis. He has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews. The authors have filed a patent, EB19024, on the use of HDL-raising therapies in the treatment of myeloproliferative neoplasms.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.Org/10.1016/j.celrep.2020.02.056.
REFERENCES
- Abdel-Wahab O, and Levine RL (2010). Metabolism and the leukemic stem cell. J. Exp. Med. 207, 677–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, and DeBerardinis RJ (2015). Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovenga F, Sabbà C, and Moschetta A. (2015). Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 21, 517–526. [DOI] [PubMed] [Google Scholar]
- Bowman RL, and Levine RL (2017). TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Med. 7, a026518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (2009). Cholesterol feedback: from Schoen- heimer’s bottle to Scap’s MELADL. J. Lipid Res. 50 (Suppl), S15–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham LR, Singaraja RR, and Hayden MR (2006). Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu. Rev. Nutr. 26,105–129. [DOI] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 44, 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clendening JW, and Penn LZ (2012). Targeting tumor cell metabolism with statins. Oncogene 31, 4967–4978. [DOI] [PubMed] [Google Scholar]
- Dang CV (2012). Links between metabolism and cancer. Genes Dev. 26, 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessì S, Batetta B, Spano O, Sanna F, Tonello M, Giacchino M, Tessitore L, Costelli P, Baccino FM, Madon E, et al. (1995). Clinical remission is associated with restoration of normal high-density lipoprotein cholesterol levels in children with malignancies. Clin. Sci. (Lond.) 89, 505–510. [DOI] [PubMed] [Google Scholar]
- Dietrich C, Volovyk ZN, Levi M, Thompson NL, and Jacobson K. (2001). Partitioning of Thy-1, GM1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl. Acad. Sci. USA 98, 10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuel PD (2008). Juvenile myelomonocytic leukemia and chronic myelo- monocytic leukemia. Leukemia 22, 1335–1342. [DOI] [PubMed] [Google Scholar]
- Gautier EL, Westerterp M, Bhagwat N, Cremers S, Shih A, Abdel-Wahab O, Lütjohann D, Randolph GJ, Levine RL, Tall AR, and Yvan-Char- vet L. (2013). HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J. Exp. Med. 210, 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert HS, and Ginsberg H. (1983). Hypocholesterolemia as a manifestation of disease activity in chronic myelocytic leukemia. Cancer 51,1428–1433. [DOI] [PubMed] [Google Scholar]
- Gough PJ, and Raines EW (2003). Gene therapy of apolipoprotein E-deficient mice using a novel macrophage-specific retroviral vector. Blood 101, 485–491. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hirsch HA, lliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, Tsichlis PN, Shirley Liu X, and Struhl K. (2010). A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell 17, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson R, Duchmann M, Lucas N, and Solary E. (2017). CMML: clinical and molecular aspects. Int. J. Hematol. 105, 711–719. [DOI] [PubMed] [Google Scholar]
- Jafri H, Alsheikh-Ali AA, and Karas RH (2010). Baseline and on-treatment high-density lipoprotein cholesterol and the risk of cancer in randomized controlled trials of lipid-altering therapy. J. Am. Coll. Cardiol. 55, 2846–2854. [DOI] [PubMed] [Google Scholar]
- Kang MH, Singaraja R, and Hayden MR (2010). Adenosine-triphosphate- binding cassette transporter-1 trafficking and function. Trends Cardiovasc. Med. 20, 41–49. [DOI] [PubMed] [Google Scholar]
- Kroemer G, and Pouyssegur J. (2008). Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482. [DOI] [PubMed] [Google Scholar]
- Kunimoto H, Meydan C, Nazir A, Whitfield J, Shank K, Rapaport F, Maher R, Pronier E, Meyer SC, Garrett-Bakelman FE, et al. (2018). Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell 33, 44–59.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy M, Atzler D, Liu R, de Winther M, Weber C, and Lutgens E. (2019). Interactions between dyslipidemia and the immune system and their relevance as putative therapeutic targets in atherosclerosis. Pharmacol. Ther. 193, 50–62. [DOI] [PubMed] [Google Scholar]
- Lee BH, Taylor MG, Robinet P, Smith JD, Schweitzer J, Sehayek E, Falzarano SM, Magi-Galluzzi C, Klein EA, and Ting AH (2013). Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Cancer Res. 73, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoad- ley K, Triche TJ Jr., Laird PW, Baty JD, et al. ; Cancer Genome Atlas Research Network (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-Y, and Gustafsson J-A (2015). Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 15, 216–224. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, and Thompson CB (2005). Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248. [DOI] [PubMed] [Google Scholar]
- McMurray HR, Sampson ER, Compitello G, Kinsey C, Newman L, Smith B, Chen S-R, Klebanov L, Salzman P, Yakovlev A, and Land H. (2008). Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 453, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S-H, Huang C-H, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JP, Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp-Hill R, et al. (2019). p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasantha kumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20,11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen PJ, Yu R, Longo J, Archer MC, and Penn LZ (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 76, 718–731. [DOI] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo C-L, Wang M, Sanson M, Abramowicz S, Welch C, et al. (2011). ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Invest. 121, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, et al. (2006). MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3, e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirro M, Ricciuti B, Rader DJ, Catapano AL, Sahebkar A, and Banach M. (2018). High density lipoprotein cholesterol and cancer: marker or causative? Prog. Lipid Res. 71, 54–69. [DOI] [PubMed] [Google Scholar]
- Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern M-H, et al. (2011). TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38. [DOI] [PubMed] [Google Scholar]
- Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, and Clift SM (1991). Inhibition of early atherogenesis in transgenic mice by human apolipo-protein Al. Nature 353, 265–267. [DOI] [PubMed] [Google Scholar]
- Sarrazy V, Viaud M, Westerterp M, Ivanov S, Giorgetti-Peraldi S, Guinamard R, Gautier EL, Thorp EB, De Vivo DC, and Yvan-Charvet L. (2016). Disruption of Glutl in Hematopoietic Stem Cells Prevents Myelopoiesis and Enhanced Glucose Flux in Atheromatous Plaques of ApoE(−/−) Mice. Circ. Res. 118, 1062–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony- Debre I, Viale A, Socci N, Sun Y, et al. (2015). Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274. [DOI] [PubMed] [Google Scholar]
- Smith B, and Land H. (2012). Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2, 580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehnlein O, and Swirski FK (2013). Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol. Metab. 24,129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solary E, Bernard OA, Tefferi A, Fuks F, and Vainchenker W. (2014). The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 28, 485–496. [DOI] [PubMed] [Google Scholar]
- Spann NJ, and Glass CK (2013). Sterols and oxysterols in immune cell function. Nat. Immunol. 14, 893–900. [DOI] [PubMed] [Google Scholar]
- Su F, Kozak KR, Imaizumi S, Gao F, Amneus MW, Grijalva V, Ng C, Wagner A, Hough G, Farias-Eisner G, et al. (2010). Apolipoprotein A-l (apoA-l) and apoA-l mimetic peptides inhibit tumor development in a mouse model of ovarian cancer. Proc. Natl. Acad. Sci. USA 107, 19997–20002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, and Yvan-Charvet L. (2015). Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Liu Y, Kessler PS, Vaughan AM, and Oram JF (2009). The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J. Biol. Chem. 284, 32336–32343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324,1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AM, Tang C, and Oram JF (2009). ABCA1 mutants reveal an interdependency between lipid export function, apoA-l binding activity, and Janus kinase 2 activation. J. Lipid Res. 50, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Ranalletta M, Matsuura F, Peng F, and Tall AR (2006). LXR- induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler. Thromb. Vase. Biol. 26, 1310–1316. [DOI] [PubMed] [Google Scholar]
- Wang M, Subramanian M, Abramowicz S, Murphy AJ, Gonen A, Witztum J, Welch C, Tabas I, Westerterp M, and Tall AR (2014). Interleukin-3/granulocyte macrophage colony-stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler. Thromb. Vase. Biol. 34,976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, and Yvan-Charvet L. (2012). Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 11, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Damiano MG, Zhang H, Tripathy S, Luthi AJ, Rink JS, Ugolkov AV, Singh ATK, Dave SS, Gordon LI, and Thaxton CS (2013). Biomimetic, synthetic HDL nanostructures for lymphoma. Proc. Natl. Acad. Sci. USA 7 70, 2511–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, and Tall AR (2010). ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328,1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Lindner D, Tallant TC, Wang Z, Buffa J, Klipfell E, Parker Y, Hatala D, Parsons-Wingerter P, Rayman P, et al. (2013). The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J. Biol. Chem. 288, 21237–21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository because of a pending patent at the time of submission but are available from the corresponding author on request.