

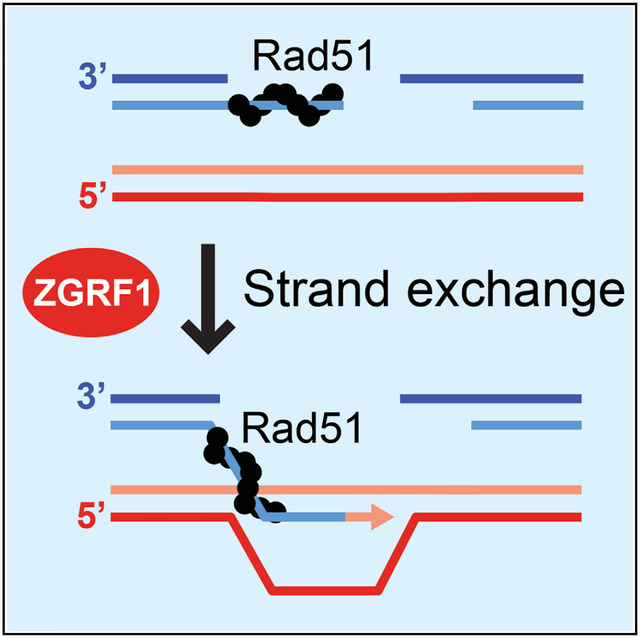
SUMMARY
Replication-blocking DNA lesions are particularly toxic to proliferating cells because they can lead to chromosome mis-segregation if not repaired prior to mitosis. In this study, we report that ZGRF1 null cells accumulate chromosome aberrations following replication perturbation and show sensitivity to two potent replication-blocking anticancer drugs: mitomycin C and camptothecin. Moreover, ZGRF1 null cells are defective in catalyzing DNA damage-induced sister chromatid exchange despite accumulating excessive FANCD2, RAD51, and γ-H2AX foci upon induction of interstrand DNA crosslinks. Consistent with a direct role in promoting recombinational DNA repair, we show that ZGRF1 is a 5′-to-3′ helicase that catalyzes D-loop dissociation and Holliday junction branch migration. Moreover, ZGRF1 physically interacts with RAD51 and stimulates strand exchange catalyzed by RAD51-RAD54. On the basis of these data, we propose that ZGRF1 promotes repair of replication-blocking DNA lesions through stimulation of homologous recombination.
Graphical Abstract
In Brief
DNA helicases are important for DNA repair processes. Here, Brannvoll et al. show that ZGRF1 is a 5′-to-3′ DNA helicase that promotes the resolution of replication-blocking DNA lesions by homologous recombination. ZGRF1 is recruited to sites of DNA damage and directly stimulates the RAD51 recombinase.
INTRODUCTION
Helicases play important roles in DNA replication, transcription, and repair because of their ability to remodel nucleic acid structures. Helicases use the energy from ATP hydrolysis to translocate along DNA or RNA in the 3′-to-5′ or 5′-to-3′ direction, which can lead to strand separation in duplex DNA or in RNA:DNA hybrids. This activity can also melt secondary structures in single-stranded DNA (ssDNA) or RNA molecules. The human genome is predicted to encode more than 95 helicases, some of which are associated with human diseases (Uchiumi et al., 2015; Umate et al., 2011).
DNA interstrand crosslinks (ICLs) represent one of the most genotoxic DNA lesions, because they block DNA replication and, as a consequence, prevent chromosome segregation in mitosis (Chan et al., 2018). ICLs arise spontaneously at a low frequency in human cells because of aldehydes, nitrous acid, and other reactive chemicals produced by normal cellular metabolism (reviewed in Lopez-Martinez et al., 2016). Notably, rapidly dividing cancer cells are hypersensitive to ICL-inducing drugs such as mitomycin C (MMC), cisplatin, and oxaliplatin, which are used as cancer therapeutic agents. ICLs are repaired by the Fanconi anemia (FA) pathway during S phase when an X-shaped DNA structure is generated around the lesion via replication fork convergence or single-fork traverse of the ICL (Huang et al., 2013; Zhang et al., 2015). ICL repair via the FA pathway is initiated upon lesion recognition of the ICL by the UHRF1 and UHRF2 proteins (Motnenko et al., 2018) and the FANCM-MHF1-MHF2-FAAP24 complex, which recruit the FANCI-FANCD2 (FANCI-D2) heterodimer and the FA core complex to chromatin, respectively. The FA core complex is an E3 ubiquitin ligase that monoubiquitylates FANCI-D2 to facilitate recruitment of SLX4/FANCP and subsequently the association of DNA endonucleases MUS81, SLX1, FAN1, and XPF/ERCC4/FANCQ. At the X-shaped DNA structures, these endonucleases cleave one of the parental DNA strands on each side of the ICL, generating a DNA break across from the “unhooked” ICL adduct on the other parental strand. Replication of the ICL-containing strand is completed by translesion synthesis (TLS), and this strand then serves as a template for repair of the DNA double-strand break (DSB) remaining on the other strand by homologous recombination (HR). Finally, the ICL is removed by nucleotide excision repair to restore DNA integrity (reviewed in Ceccaldi et al., 2016).
The HR step of ICL repair is catalyzed by the RAD51 recombinase, which is loaded by BRCA2/FANCD1 onto 3′ single-stranded overhangs generated as a result of DSB end resection (Symington, 2016). RAD51 catalyzes invasion of the 3′ single-stranded end into the sister duplex, where it primes DNA synthesis, leading to an extended D-loop. The D-loop can be resolved by synthesis-dependent strand annealing (SDSA), which leads exclusively to non-crossover (NCO) recombination products, or by classical DSB repair (DSBR), which leads to the formation of a double-Holliday junction (dHJ) that can be resolved into either NCO or crossover (CO) recombination products (reviewed in Zhao et al., 2019). The FANCM translocase promotes SDSA by disassembling D-loops before they are converted into dHJs (Deans and West, 2009; Gari et al., 2008). SDSA is thought to be the preferred pathway for replication-coupled DSBR in mitotically growing cells (Larocque and Jasin, 2010; Zapotoczny and Sekelsky, 2017), because this will prevent loss of heterozygosity arising when CO recombination occurs between homologous chromosomes.
The FANCM-MHF1-MHF2 complex is evolutionarily conserved in eukaryotes, with Mph1 being the homolog of FANCM in the budding yeast Saccharomyces cerevisiae. A fourth subunit of the yeast complex, Mte1 (Mph1-associated telomere maintenance protein 1), was recently identified as a regulator of Mph1 activity (Silva et al., 2016; Xue et al., 2016; Yimit et al., 2016). Mte1 binds with high affinity to branched DNA molecules such as D-loops, Y-structures, and Holliday junctions (HJs). Mte1 interacts with Mph1 via its C terminus and selectively regulates the different activities of Mph1. Mte1 stimulates the replication fork regression and branch migration activities of Mph1, but D-loop dissociation by Mph1 is inhibited. In vivo, Mte1 promotes mitotic CO recombination and protects cells against genotoxic agents that cause replication stress. The Mte1-Mph1 complex also co-localizes with Rad52 recombination foci (Silva et al., 2016; Xue et al., 2016). Similarly, Dbl2/Mte1 in the fission yeast Schizosaccharomyces pombe co-localizes with Rad22/Rad52 and Fml1/Mph1, and dbl2Δ mutant cells are sensitive to replication stress (Yu et al., 2013). Human ZGRF1 (zinc finger GRF-type containing 1, C4orf21) shares homology with Mte1/Dbl2 in its N-terminal DUF2439 domain. The C terminus of ZGRF1 is not conserved in Mte1/Dbl2 and is predicted to encode a (GRF-type) Zn finger DNA binding domain and a helicase domain (Figure 1A). Human ZGRF1 is largely uncharacterized, but genome-wide small interfering RNA (siRNA) knockdown screens have suggested a role for ZGRF1 as a regulator of HR and ICL repair (Adamson et al., 2012; Smogorzewska et al., 2010).
Figure 1. ZGRF1 Is Important for DNA Interstrand Crosslink Repair.
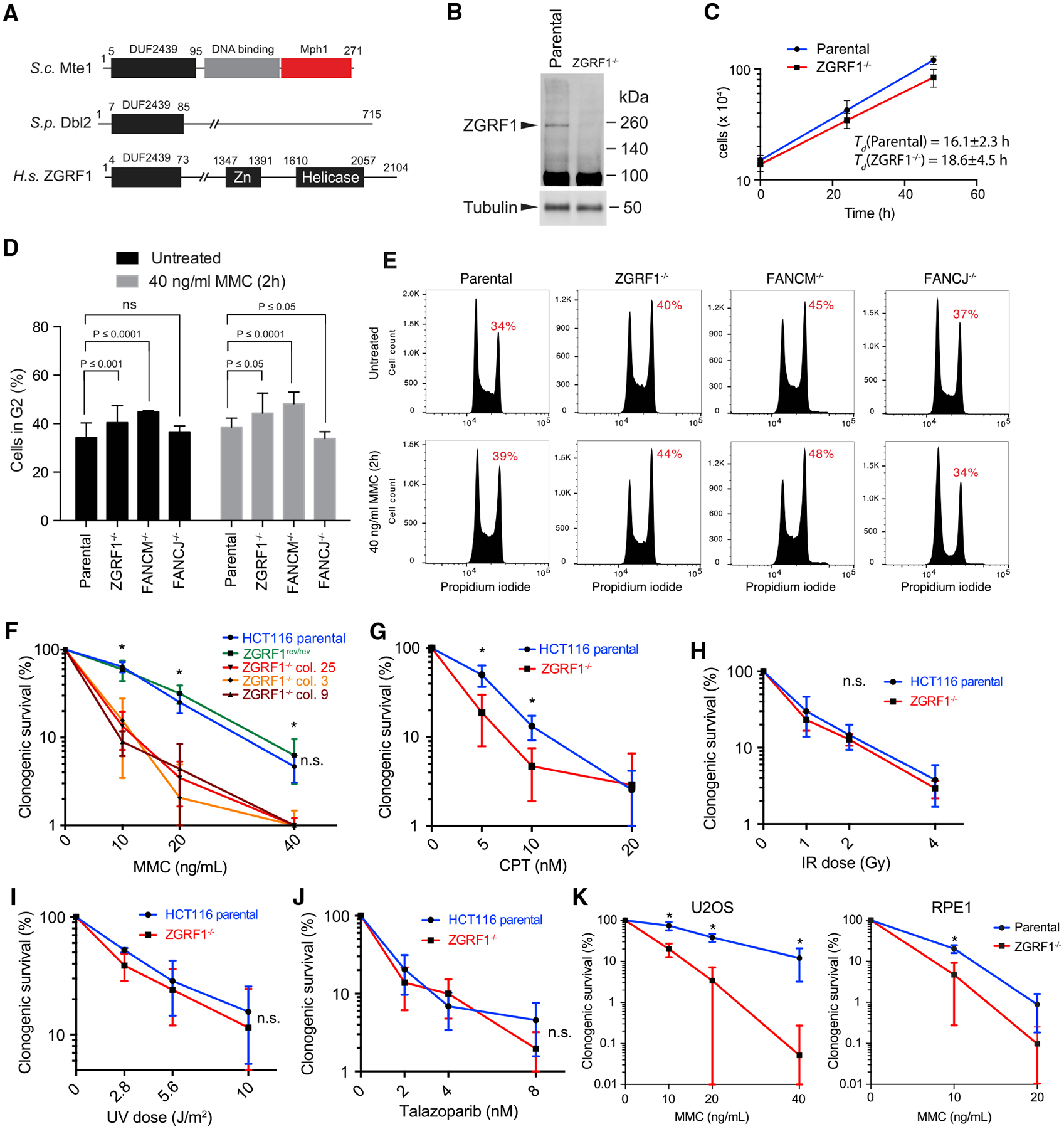
(A) Domain organization of ZGRF1. The DUF2439 domain is conserved between S. cerevisiae Mte1, S. pombe Dbl2, and human ZGRF1. The DNA binding and Mph1 interaction domains are indicated for Mte1. The putative DNA binding Zn finger and helicase domains are indicated for ZGRF1.
(B) Western blot of ZGRF1 in HCT116 parental and ZGRF1−/− cell lines.
(C) ZGRF1−/− cells exhibit slow growth. HCT116 parental and ZGRF1−/− cells were cultured for 48 h, and cell density was determined at 24 h intervals. Error bars indicate SD (n = 5).
(D) ZGRF1−/− cells accumulate in G2. Quantification of G2 accumulation in HCT116 parental, ZGRF1−/−, FANCM −/−, and FANCJ−/− cells in unperturbed condition and in response to MMC treatment. Means from three independent experiments are plotted as bars. Error bars represent 95% confidence intervals. p values were calculated using two-way ANOVA on the basis of three independent biological replicates.
(E) Representative cell cycle profiles of propidium iodide stained cells, either untreated or treated with 40 ng/mL MMC for 2 h followed by recovery for 24 h.
(F) Colony formation assay of HCT116 parental, ZGRF1rev/rev, and three independent ZGRF1−/− cell lines treated with the indicated doses of MMC for 24 h. The graph of the parental cell line is statistically different from each of the knockout cell lines (p < 0.05, t test) but not significantly different from ZGRF1rev/rev (n.s.).
(G–J) Colony formation assays of cells treated with the indicated doses of CPT (G) for 24 h, ionizing radiation (X-rays) (H), ultraviolet radiation (UV-C) (I), or the poly (ADP-ribose) polymerase (PARP) inhibitor talazoparib (J).
(K) Colony formation assay of U2OS and RPE-1 parental and ZGRF1−/− cell lines treated with the indicated doses of MMC for 24 h.
n ≥ 3 for each cell line. All graphs show the mean with 95% confidence interval. Statistical significance was calculated using unpaired t tests without assuming consistent SD. *p < 0.05. n.s., no significant difference.
To investigate the roles of ZGRF1 in human cells, we examined the consequences of genetic deletion of ZGRF1, and we characterized the purified recombinant protein biochemically. We demonstrate that human ZGRF1-knockout cells are sensitive to the DNA interstrand crosslinking agent MMC and the topoisomerase I (TOP1) inhibitor camptothecin (CPT). Furthermore, ZGRF1-deficient cells exhibit elevated levels of chromosome aberrations after MMC treatment, reduced sister-chromatid exchange (SCE) following MMC or CPT exposure, and a defect in gene conversion. Furthermore, we show that the ZGRF1 protein is a 5′-to-3′ helicase with the ability to remodel DNA molecules, interact with RAD51, and stimulate RAD51-RAD54-catalyzed DNA strand exchange. We conclude that ZGRF1 is a helicase that facilitates repair of severe replication-blocking lesions by HR.
RESULTS
ZGRF1−/− Cells Are Sensitive to DNA Interstrand Crosslinks and Covalent Protein-DNA Complexes
To investigate the role of ZGRF1 in DNA repair, we created a homozygous knockout of ZGRF1 in the colon cancer cell line HCT116 using CRISPR-Cas9 (Figure S1). Immunoblotting confirmed that no full-length ZGRF1 protein is produced in the knockout cell lines (Figure 1B). ZGRF1−/− cells show a slightly slower growth rate than parental cells (Figure 1C) and increased accumulation in G2/M, which is similar to what is seen in FANCM−/− cells (Figures 1D and 1E). By contrast, G2-M checkpoint defective FANCJ−/− cells fail to arrest in G2/M upon MMC treatment (Figures 1D and 1E) (Yu et al., 2003). We examined the sensitivity of ZGRF1−/− cells to a range of DNA-damaging agents using a clonogenic survival assay. Consistent with results from a previous RNAi screen (Smogorzewska et al., 2010), ZGRF1−/− cells exhibited sensitivity to the DNA crosslinking agent MMC (Figure 1F). ZGRF1−/− cells also exhibited mild sensitivity to the TOP1 inhibitor CPT (Figure 1G), but not to ionizing radiation (Figure 1H), ultraviolet radiation (Figure 1I), the poly (ADP-ribose) polymerase (PARP) inhibitor talazoparib (Figure 1J), or hydroxyurea (HU; Figure S4D). These data suggest that ZGRF1 is critical for cellular responses to DNA replication-blocking lesions and most notably to ICLs. To confirm that the MMC sensitivity of ZGRF1−/− cells is due to ablation of ZGRF1, we reverted the mutant alleles back to wild-type (WT) using CRISPR-Cas9 and confirmed that this fully rescued the MMC sensitivity of the mutant cell line (Figure 1F).
To analyze whether the DNA damage sensitivity of ZGRF1−/− cells extends beyond the HCT116 cell line, we used CRISPR-Cas9 to target the ZGRF1 gene in the human osteosarcoma U2OS cell line. Exposure of U2OS parental and ZGRF1−/− cells to increasing concentrations of MMC revealed a sensitivity of the mutant cell line comparable with that of the ZGRF1−/− HCT116 cell line (Figure 1K). Similarly, we showed that knockout of ZGRF1 in the untransformed primary epithelial cell line RPE-1 causes MMC sensitivity (Figure 1K). These results indicate that MMC sensitivity is a general phenotype associated with ZGRF1−/− cells and is not linked to a specific state of transformation.
To further characterize the consequences of MMC exposure at the chromosomal level, we examined metaphase spreads for chromosomal aberrations. Treatment of parental cells with 20 ng/mL MMC for 24 h resulted in a small increase in chromosomal aberrations (0.56 versus 0.12 aberrations per metaphase spread in untreated cells). The types of chromosomal aberrations observed in parental cells were usually confined to small gaps or breaks on chromosome arms (Figures 2A and 2B). ZGRF1−/− cells exhibited very few chromosomal aberrations when untreated, which was comparable with parental cells (0.13 and 0.12 aberrations per metaphase spread, respectively). However, they exhibited a significantly higher proportion of metaphase spreads with aberrations when treated with MMC (1.16 aberrations per metaphase spread in ZGRF1−/− cells versus 0.56 aberrations per metaphase spread in parental cells; Figures 2A–2C). Moreover, the types of aberrations were altered, with ZGRF1−/− cells harboring larger gaps on chromosome arms, chromosome arm fusions, and, in rare cases, radial chromosomes (Figure 2C). Taken together, these data indicate a role for ZGRF1 in the repair of replication-blocking DNA damage such as those caused by ICLs and covalent protein-DNA complexes.
Figure 2. ZGRF1 Contributes to the FA Pathway.
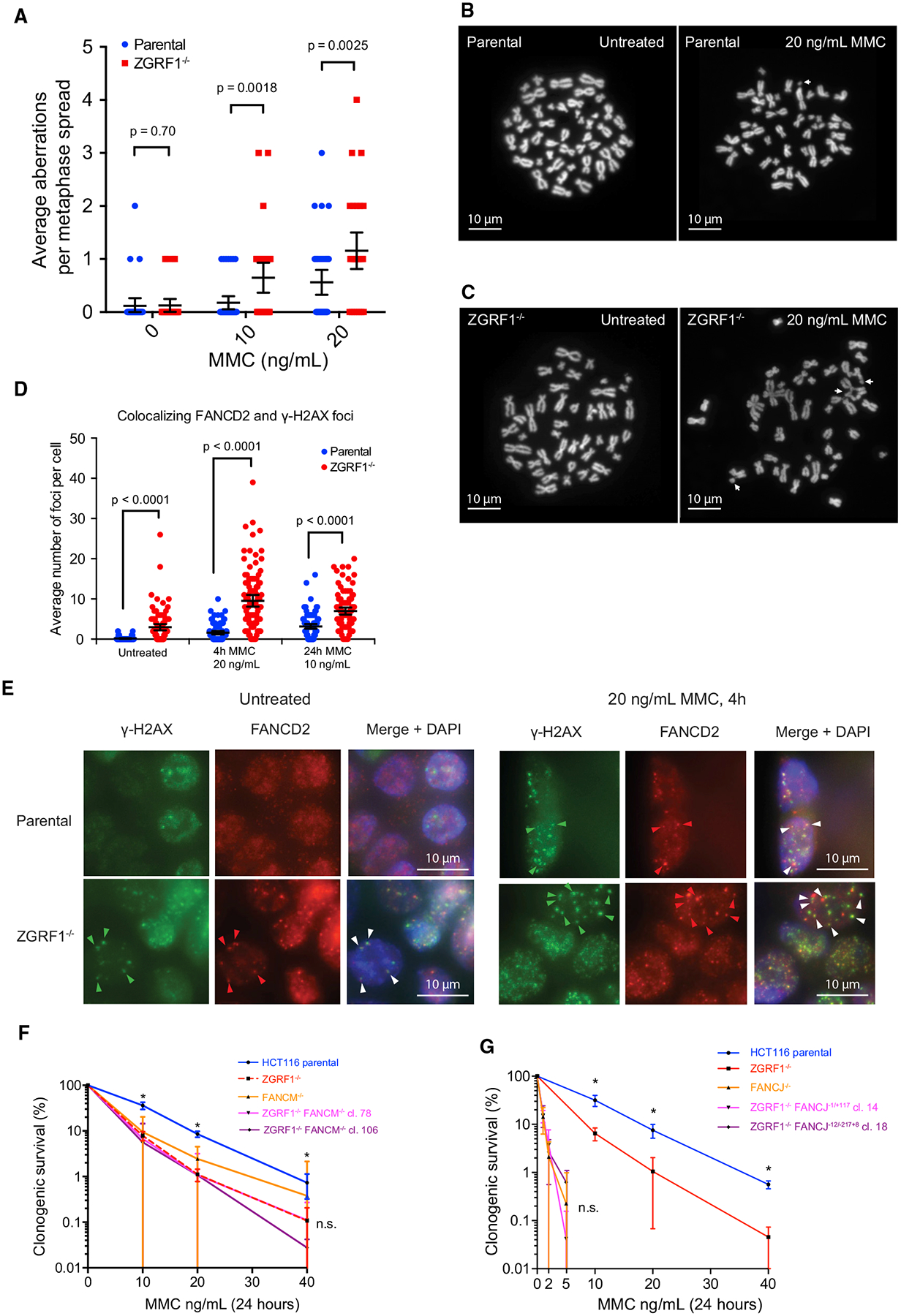
(A) Quantification of chromosomal aberrations in HCT116 parental and ZGRF1−/− cells. Means with 95% confidence intervals are plotted. p values were calculated using Mann-Whitney U tests. N ≥ 32 spreads per condition.
(B and C) Examples of metaphase spreads from parental cells (B) or ZGRF1−/− cells (C) untreated or treated with 20 ng/mL MMC for 24 h. White arrows mark chromosomal aberrations. The images shown highlight the types of aberrations scored rather than being representative of the number of aberrations seen per spread.
(D) ZGRF1−/− cells show a higher frequency of co-localizing γ-H2AX and FANCD2 foci under both unperturbed conditions and when treated with MMC. Quantification of co-localizing γ-H2AX and FANCD2 foci under unperturbed conditions and with two different treatments with MMC is shown. Error bars show 95% confidence intervals. p values were calculated using Mann-Whitney U tests. N > 100 cells for each condition.
(E) Representative images of HCT116 parental and ZGRF−/− cells under unperturbed conditions or after treatment with 20 ng/mL MMC for 4 h. Arrows mark co-localizing foci for one cell, where the pixel intensity of FANCD2 foci was a minimum of 6,000 arbitrary units higher than background. Cells with very low or absent γ-H2AX foci were not scored, as this indicated inadequate immunostaining.
(F) Colony formation assay of HCT116 parental and ZGRF1−/− and FANCM−/− single- and double-mutant cell lines treated with the indicated doses of MMC for 24
h. The graph of the parental cell line is statistically different from each of the knockout cell lines (p < 0.05, t test), but the knockout cell lines are not significantly different from each other (n.s.).
(G) Colony formation assay of HCT116 parental and ZGRF1−/− and FANCJ single- and double-mutant cell lines treated with the indicated doses of MMC for 24 h. The graph of the parental cell line is statistically different from the ZGRF1-knockout cell line (p < 0.05, t test). The FANCJ single- and double-mutant cell lines are not significantly different from each other (n.s.).
Graphs in (F) and (G) show the mean with 95% confidence interval. Statistical significance was calculated using unpaired t tests without assuming consistent SD. *p < 0.05. n.s., no significant difference. n ≥ 3 for each cell line.
ZGRF1 Contributes to the FA Pathway for Surviving ICLs
An important pathway for ICL repair in S phase is the FA pathway. To examine the relationship between ZGRF1 and the FA pathway, we performed immunofluorescence imaging of γ-H2AX, a marker of both stalled and collapsed replication forks (Sirbu et al., 2011; Ward and Chen, 2001), and FANCD2, a key FA protein that is monoubiquitylated in response to ICLs and forms nuclear foci at sites of DNA damage (Garcia-Higuera et al., 2001; Knipscheer et al., 2009). Therefore, co-localizing γ-H2AX and FANCD2 foci likely mark sites of active repair by the FA pathway. Interestingly, even under unperturbed conditions, ZGRF1−/− cells exhibited increased levels of co-localizing foci compared with parental cells (Figures 2D and 2E). Following treatment with MMC, these co-localizing foci accumulated to a higher level in ZGRF1−/− cells (Figures 2D and 2E). Taken together, these data indicate that ZGRF1−/− cells accumulate DNA lesions that are recognized by the FA pathway. Moreover, it suggests that ZGRF1 is not required for the activation of the FA pathway and likely acts downstream of FANCI-D2 monoubiquitylation (Garcia-Higuera et al., 2001).
To substantiate this conclusion, we performed epistasis analysis of ZGRF1 with FANCM and FANCJ. Two independent double-mutant cell lines were constructed for each pair of genes by CRISPR-Cas9 and confirmed by sequencing and western blotting (Figures S2 and S3). The ZGRF1 and FANCM single and double mutants were significantly more sensitive to MMC than the parental HCT116 cell line, but not significantly different from each other, suggesting that ZGRF1−/− and FANCM−/− are epistatic for MMC sensitivity (Figure 2F). We were unable to generate null alleles of FANCJ on both chromosomes in the ZGRF1−/− background, while we could successfully generate FANCJ null alleles in the parental background. Editing of the second FANCJ allele in the ZGRF1−/− background repeatedly resulted in in-frame insertions and deletions, suggesting that ZGRF1−/− is synthetic lethal with FANCJ−/−. Nevertheless, clonogenic survival of ZGRF1−/− cells possibly containing hypomorphic mutant alleles of FANCJ showed that ZGRF1−/− cells containing FANCJ indels are significantly more sensitive to MMC than ZGRF1−/− alone, and the double mutants exhibit MMC sensitivity comparable with the FANCJ−/− single mutant (Figure 2G). Taken together, this analysis suggests that ZGRF1 acts in the same pathway as FANCM. Furthermore, it is likely that FANCJ is critical for survival in the absence of ZGRF1, suggesting some functional redundancy between these two proteins.
MMC Induces Nuclear FANCD2-Co-localizing ZGRF1 Foci during S Phase
To examine the localization of ZGRF1 during ICL repair, we tagged endogenous ZGRF1 with a tandem Venus-eYFP tag (2xYFP) at its C terminus. Expression of full-length ZGRF1–2xYFP was confirmed by western blotting (Figure S4A). Live-cell microscopy showed that ZGRF1–2xYFP localizes to the nucleus but with a large cell-to-cell variation in the expression level (Figure S4B). In contrast, cells synchronized at the G1-S transition with thymidine displayed a more uniform and higher level of ZGRF1 (Figure S4C), suggesting that ZGRF1 abundance is cell cycle regulated and peaks in S phase. To examine the localization of ZGRF1 during ICL repair, cells were arrested at the G1-S transition with thymidine and treated with MMC for 4 h before being released into S phase in fresh medium containing Hoechst for nuclear staining. Fluorescence microscopy of cells before and after release into S phase showed that MMC treatment induces the formation of nuclear ZGRF1 foci in S phase (Figures 3A and 3B). To test if ZGRF1 foci colocalize with components of the FA pathway, we introduced mCherry-FANCD2 into the ZGRF1–2xYFP cell line by random integration (Motnenko et al., 2018) and selected clones that express mCherry-FANCD2 at levels similar to or slightly lower than endogenous FANCD2 to avoid any toxic effects that overexpression may cause (Figure S4E). Live-cell imaging of this cell line showed that the majority (78%–82%, 992 of 1,216 foci and 1523 of 1,948 foci, respectively) of both spontaneous and MMC-induced ZGRF1 foci colocalize with FANCD2 (Figures 3C and 3D). Taken together, these data suggest a direct role for ZGRF1 in the response of the FA pathway to collisions between replication forks and ICLs during S phase.
Figure 3. ZGRF1 Colocalizes with FANCD2 at DNA Damage-Induced Foci.
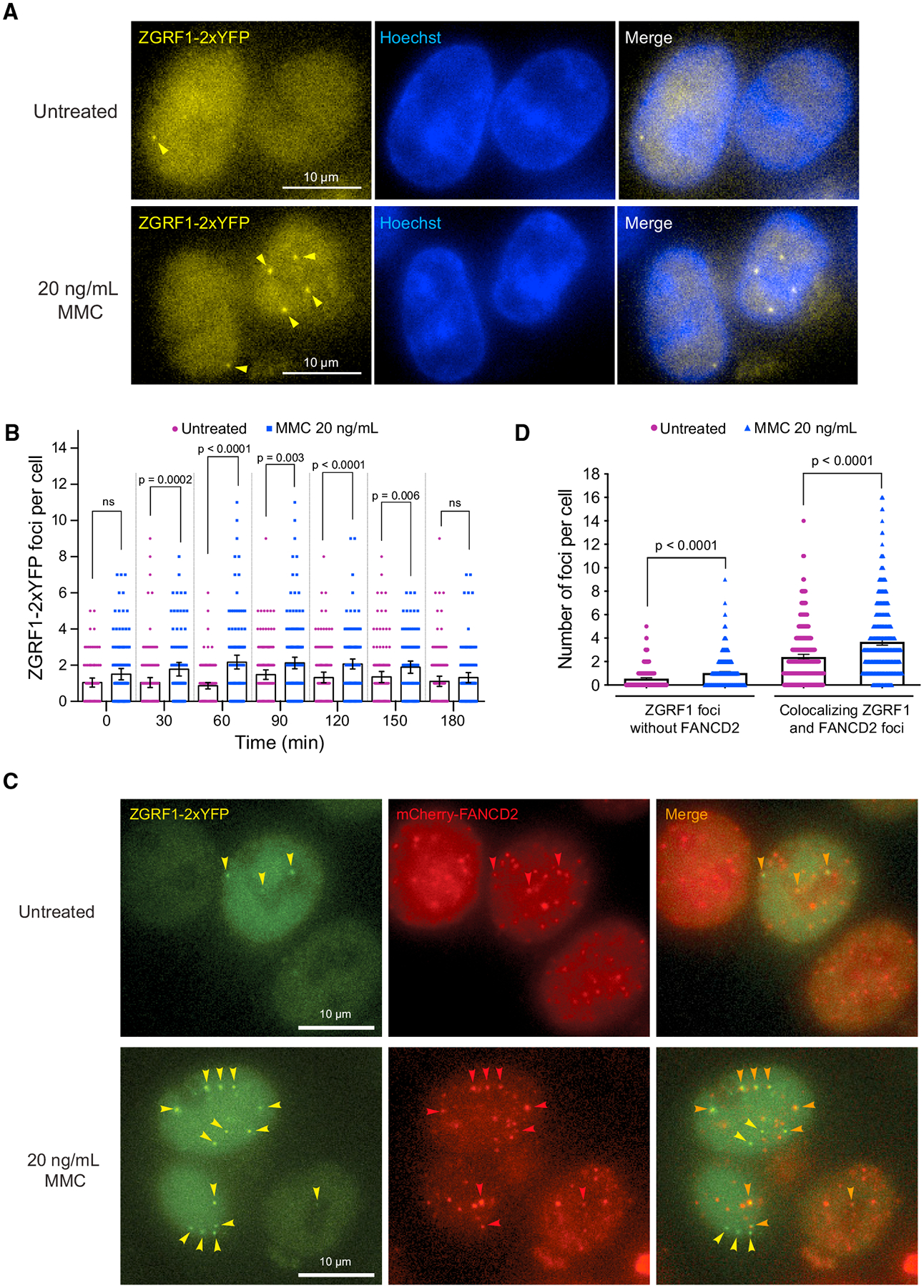
(A) ZGRF1 localizes to nuclear foci during ICL repair. Cells expressing ZGRF1–2xYFP from the endogenous promoter were synchronized at the G1/S border by treatment with 2 mM thymidine for 18 h before release into S phase in Leibovitz’s L-15 medium containing 0.4 μM Hoechst 33258. Four hours before release, 20 ng/mL of MMC or vehicle was added to the cultures. Arrows indicate ZGRF1 foci.
(B) Quantification of ZGRF1 foci after MMC treatment. Quantification of the experiment in (A). Error bars show 95% confidence intervals. p values were calculated using Mann-Whitney U tests. N = 90–160 cells for each condition.
(C) Colocalization of ZGRF1 and FANCD2. Cells expressing ZGRF1–2xYFP from the endogenous promoter and ectopically integrated mCherry-FANCD2 were synchronized in S phase with 2 mM thymidine 18 h prior to microscopy. Four hours before microscopy, 20 ng/mL MMC or vehicle was added to the culture. Yellow arrows mark ZGRF1 foci, red arrows mark FANCD2 foci, and orange arrows mark the co-localizing foci.
(D) Quantification of co-localizing FANCD2 and ZGRF1 foci in the experiment reported in (C).
Error bars show 95% confidence intervals. p values were calculated using Mann-Whitney U tests. N > 400 cells for each condition.
ZGRF1 Is Required for HR Repair at Replication-Blocking Lesions
Several lines of evidence suggest a role for ZGRF1 in promoting HR. In addition to the observed CPT sensitivity (Figure 1G), knockdown of ZGRF1 was shown in a genome-wide screen to cause reduced recombination in a green fluorescent protein (GFP) reporter assay (Adamson et al., 2012). In S. cerevisiae, deletion of the ZGRF1-related gene MTE1 causes a reduction in the rate of CO versus NCO products resulting from recombinational repair of a DSB (Silva et al., 2016). To examine if ZGRF1 regulates HR, we first validated the impact of ZGRF1 on HR in the DR-GFP recombination assay (Figure 4A), where a site-specific DSB induced by the I-SceI meganuclease is repaired by gene conversion to produce a functional GFP gene (Pierce et al., 2001), which is scored by flow cytometry. After knockout of ZGRF1 by CRISPR-Cas9 (Figures S4F and S4G), we observed a 7-fold reduction in the I-SceI-induced DR-GFP recombination frequency relative to the parental control (Figure 4B).
Figure 4. ZGRF1 Contributes to Homologous Recombination.
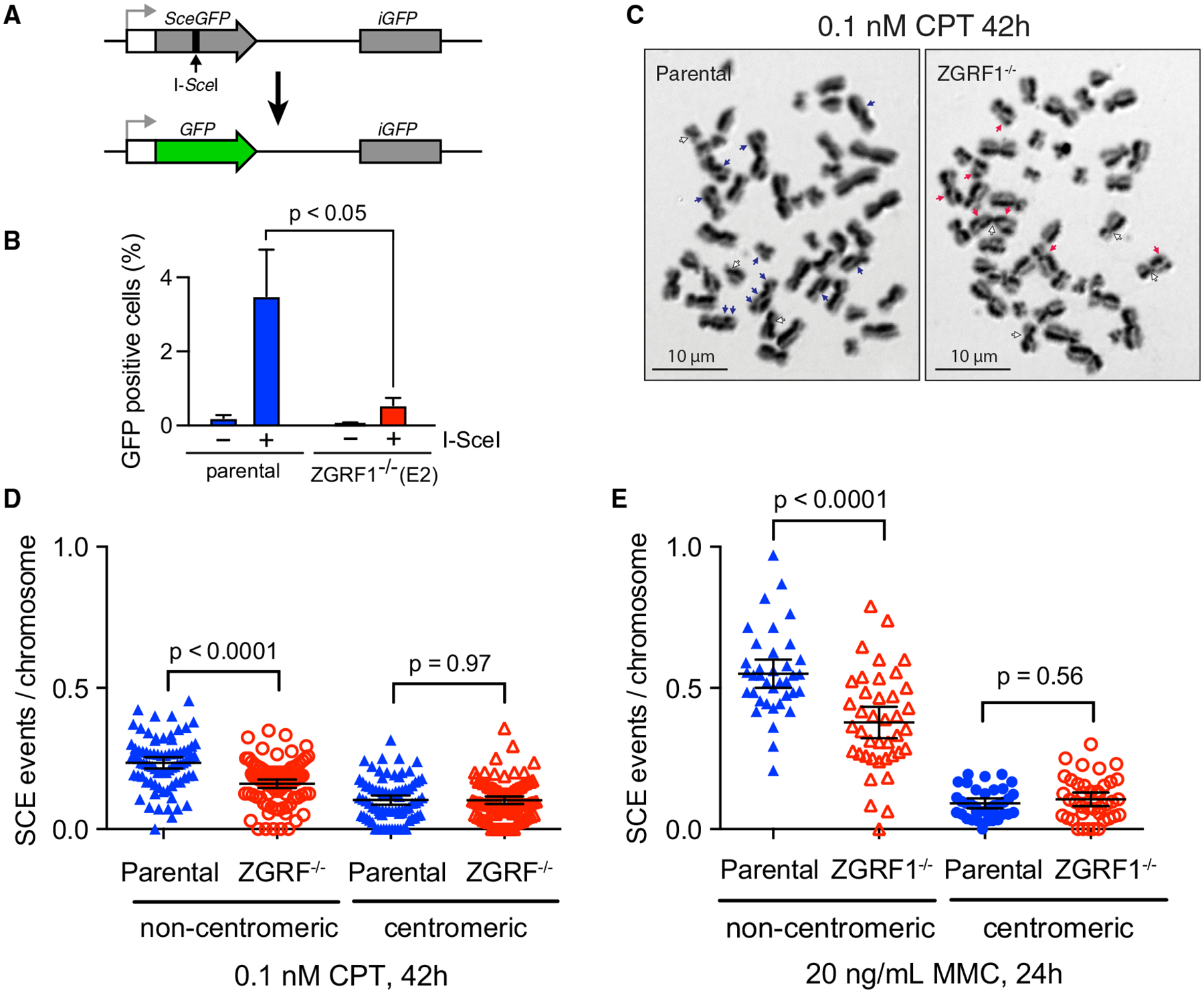
(A) Schematic illustration of the DR-GFP assay. A recognition site of the I-SceI meganuclease has been integrated into the open reading frame (ORF) of the GFP gene (SceGFP), thereby disrupting the ORF, and a truncated GFP gene (iGFP) fragment with the correct ORF sequence is placed downstream in the construct. Repair of the cleaved I-SceI site by gene conversion using the downstream iGFP as a template results in a functional GFP gene that is measured using flow cytometry. White box, promoter; gray arrow, transcription start site.
(B) ZGRF1 promotes gene conversion. The percentage of GFP-positive cells with (+) or without (−) I-SceI expression is shown for U2OS DR-GFP parental and ZGRF1−/− cells (p < 0.05, multiple t test). Error bars indicate SEM (n = 4).
(C) Representative images of sister chromatid exchange events (SCEs) in metaphase spreads in cells treated with 0.1 nM CPT for 42 h. Blue and red arrows mark non-centromeric exchanges for HCT116 parental and ZGRF1−/− cells, respectively, while white arrows mark centromeric exchanges.
(D and E) ZGRF1−/− cells show a decrease in the number of non-centromeric SCEs per chromosome compared with HCT116 parental cells. Quantifications of SCE frequencies in HCT116 parental and ZGRF1−/− cells treated with 0.1 nM CPT for 42 h or 20 ng/mL MMC for 24 h are shown. Two independent experiments were performed. N ≥ 35 metaphase spreads and 674 chromosomes per condition.
Error bars show 95% confidence intervals. p values were calculated using Mann-Whitney U tests.
To test if loss of ZGRF1 also affects recombination induced by replication-blocking lesions, we measured the frequency of SCEs, which represent CO events during HR. For this, we analyzed parental and ZGRF1−/− cells mock-treated or treated with either MMC or CPT, which leads to SCEs that can be visualized in metaphase spreads (Simpson and Sale, 2006). SCEs that occurred across the centromere (centromeric) or across the chromosome arm (non-centromeric) were scored separately (Figures 4C–4E), because a centromeric SCE is indistinguishable from a twisting of the chromosome arms during chromosome spreading. Although there was no significant difference between ZGRF1−/− and parental cells in the levels of centromeric SCEs, we did observe a significantly lower level of non-centromeric SCE events in ZGRF1−/− cells following treatment with either MMC or CPT (Figures 4C–4E). These data suggest that ZGRF1 is required to promote HR repair following DNA replication-blocking lesions.
ZGRF1 Exhibits 5′-to-3′ Helicase Activity
To link the phenotypic characterization of ZGRF1−/− cells to the function of the ZGRF1 protein, we expressed ZGRF1 in insect cells and purified the protein to near homogeneity along with a ZGRF1-K1660A variant in which the Walker A motif is mutated (Figure 5A). We observed that WT ZGRF1, but not ZGRF1-K1660A, exhibited DNA-dependent ATPase activity upon the addition of single- or double-stranded DNA (dsDNA), or replicative form I DNA (RF1) (Figure 5B). To test if ZGRF1 possesses helicase activity, we incubated ZGRF1 with helicase substrates that harbor a 15 bp duplex region and a 45 nt 5′ or 3′ single-stranded overhang (Figures 5C and 5D). ZGRF1, but not ZGRF1-K1660A, efficiently dissociated the substrate with the 5′ single-stranded overhang (Figure 5C) but showed no activity on the substrate with the 3′ overhang (Figure 5D). This indicates that ZGRF1 is an ATP hydrolysis-dependent helicase with a 5′-to-3′ polarity of translocation on ssDNA.
Figure 5. Human ZGRF1 Is a 5′-to-3′ Helicase that Directly Stimulates RAD51-Catalyzed Strand Exchange.
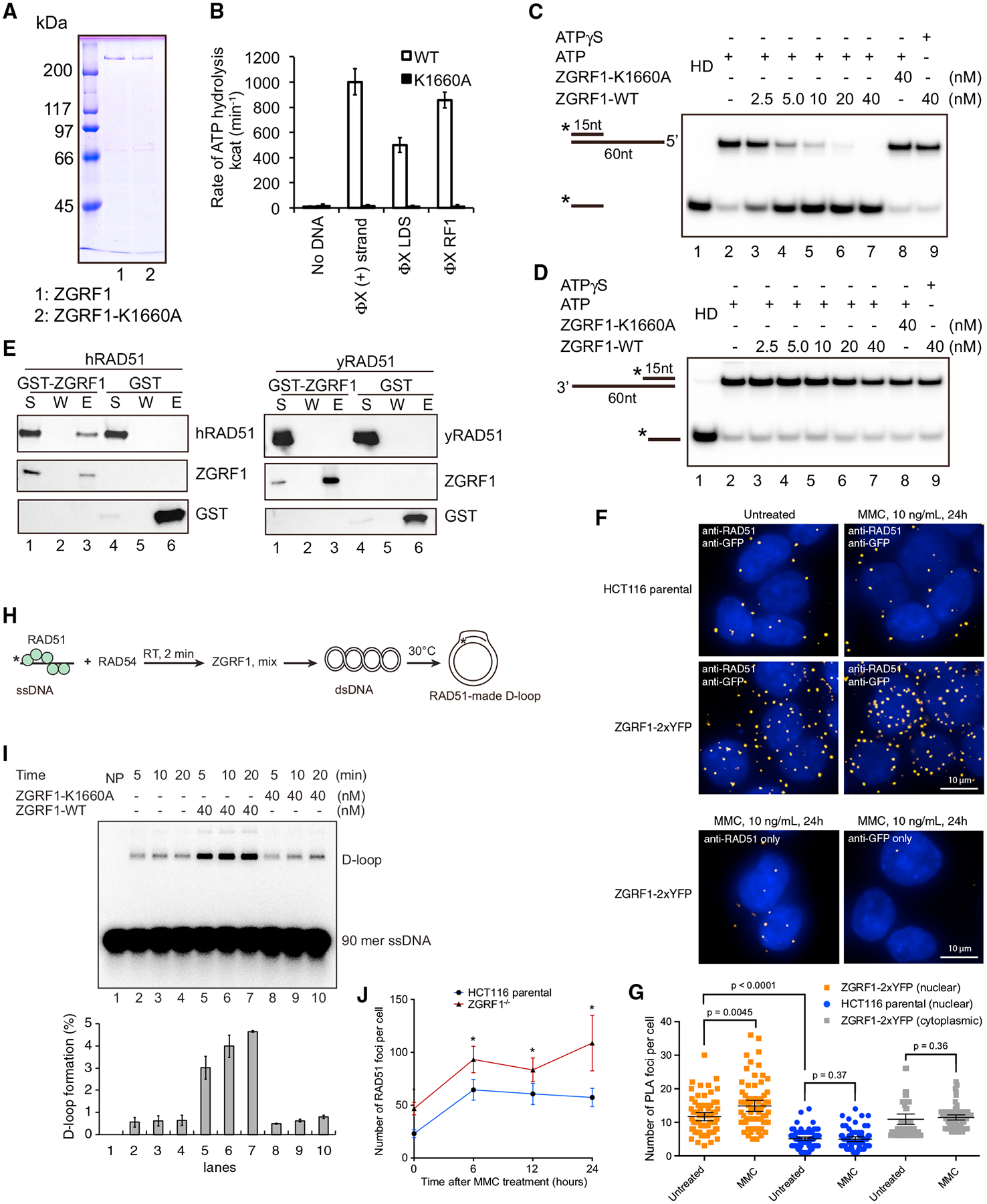
(A) SDS-PAGE analysis of purified ZGRF1 and its helicase dead mutant ZGRF1-K1660A.
(B) ATP hydrolysis by ZGRF1 (wild-type or K1660A mutant) with ϕX174 viral (+) strand, linear dsDNA (LDS), or replicative form I DNA (RF1).
(C and D) ZGRF1 or ZGRF1-K1660A was incubated with DNA substrates harboring either a 5′ (C) or a 3′ (D) ssDNA overhang and either ATP or ATP-γ-S at 30°C for 5 min in the presence of trap oligos to prevent the spontaneous re-annealing of the separated strands. HD, heat denatured.
(E) ZGRF1 interacts with human but not yeast RAD51. GST-ZGRF1 or GST (0.3 μg) was incubated with human or yeast RAD51 (0.5 mg) on Glutathione Sepharose 4B resin. The supernatant (S), wash (W), and SDS eluate (E) were resolved using SDS-PAGE followed by western blotting. GST-ZGRF1 or GST was detected using α-GST-HRP antibody, while human or yeast RAD51 was detected using α-hRAD51 or α-yRad51 antibodies.
(F) ZGRF1 and RAD51 interact in vivo. The proximity ligation assay (PLA) was used to examine the interaction between ZGRF1 and RAD51 in HCT116 cells. Primary antibodies were directed against GFP (to detect ZGRF1–2xYFP) and RAD51, and protein interactions were visualized as fluorescent foci inside the nucleus marked by DAPI staining. HCT116 parental cells that do not contain ZGRF1 tagged with YFP were used as a negative control as well as other negative controls, where either of the primary antibodies was omitted.
(G) Quantification of PLA in (F). Graph shows quantification of nuclear and cytoplasmic PLA foci for 71–85 cells for each condition. p values were calculated using Mann-Whitney U tests. Error bars represent mean with 95% confidence intervals.
(H) Experimental setup for examining RAD51-catalyzed strand exchange. Asterisk denotes 5′−32P radiolabel.
(I) ZGRF1 stimulates RAD51-RAD54-mediated strand exchange. Time course analysis of strand exchange catalyzed by RAD51 and RAD54, in the absence or presence of ZGRF1 or its helicase dead mutant ZGRF1-K1660A was shown. NP, no protein. The D-loop product was quantified, and data are means ± SD (n = 3).
(J) ZGRF1 acts downstream of RAD51 focus formation. HCT116 parental or ZGRF1−/− cells were treated with 1 mg/mL MMC for 1 h and allowed to recover in fresh medium for the indicated amount of time before fixation and immunostaining for RAD51. N ≥ 40 cells for each data point.
Graphs show the mean with 95% confidence intervals (*p < 0.05, multiple t test).
ZGRF1 Stimulates RAD51-RAD54 Catalyzed Strand Exchange
To investigate how ZGRF1 collaborates with other genome maintenance proteins during ICL repair, we first tested the physical interaction of ZGRF1 with a number of candidate proteins by co-affinity precipitation. This mini-screen revealed putative interactions with FANCM, RAD51, and Polδ (Figures 5E, S5A, and S5B), but not with Polε, PCNA, RPA, or yeast Rad51 (Figures S5C and 5E). Because of the potential role of ZGRF1 in HR, we decided to focus on the RAD51 interaction. To analyze whether ZGRF1 and RAD51 interact in situ, we turned to proximity ligation in the ZGRF1–2xYFP cell line using antibodies against YFP and RAD51 (Söderberg et al., 2006). Using this assay, we observed increased proximity ligation specifically in the nucleus of oligo-conjugated antibodies directed to ZGRF1–2xYFP and RAD51 after MMC treatment (Figures 5F and 5G). The MMC-induced increase was not observed in the cytoplasm or in the parental cell line or if either of the anti-YFP or anti-RAD51 antibodies were omitted. We therefore conclude that RAD51 and ZGRF1 physically interact in the context of ICL repair.
HR is critically dependent on RAD51 to catalyze strand exchange between the recombining DNA molecules. To test if the ability of ZGRF1 to promote recombination in vivo (Figures 4A–4E) reflects a direct stimulation of RAD51, we examined RAD51-catalyzed strand exchange in vitro by measuring the invasion of a 90-mer single-stranded oligonucleotide into a homologous double-stranded target to form a D-loop structure in the presence of the RAD54 translocase (Figure 5H) (Mazina and Mazin, 2004). ZGRF1 was introduced into the reaction only after RAD51-ssDNA nucleoprotein filament formation, and just before the addition of dsDNA to initiate the D-loop formation (Figure 5H). We observed a 4-fold stimulation of RAD51-RAD54-catalyzed strand exchange by WT ZGRF1, but not by the ATPase dead ZGRF1-K1660A mutant (Figure 5I). In comparison, ZGRF1 did not stimulate strand exchange catalyzed by yeast Rad51-Rad54 (Figures S5D and S5E), suggesting that the stimulation depends on the direct physical interaction with human RAD51. Taken together, these analyses suggest that ZGRF1 promotes recombination by directly stimulating RAD51 strand exchange activity.
To distinguish whether ZGRF1 promotes recombination by facilitating the loading of RAD51 at ICLs or primarily through stimulation of RAD51 after its binding to ssDNA, we quantified RAD51 foci in HCT116 parental and ZGRF1−/− cells after a challenge with 1 mg/mL MMC for 1 h. This treatment led to a dramatic increase in RAD51 foci in both cell lines (Figure 5J), but the ZGRF1−/− cells accumulated RAD51 foci to higher levels, suggesting that ZGRF1 is not required to load RAD51 at sites of DNA damage, but rather it promotes recombinational repair after RAD51 recruitment.
ZGRF1 Catalyzes Branch Migration and D-Loop Dissociation
The purified yeast Mte1-Mph1-MHF complex can remodel a number of DNA substrates representing DNA repair and replication intermediates (Mitchel et al., 2013; Prakash et al., 2005, 2009; Silva et al., 2016; Xue et al., 2014, 2016; Zheng et al., 2011). To test if ZGRF1 has similar activities, we first examined DNA branch migration using a mobile HJ (MHJ; Figure 6A). Conversion of this substrate into duplex DNA indicates that ZGRF1 can catalyze HJ branch migration in a manner dependent on ATP hydrolysis.
Figure 6. ZGRF1 Has Branch Migration and D-Loop Dissociation Activity.
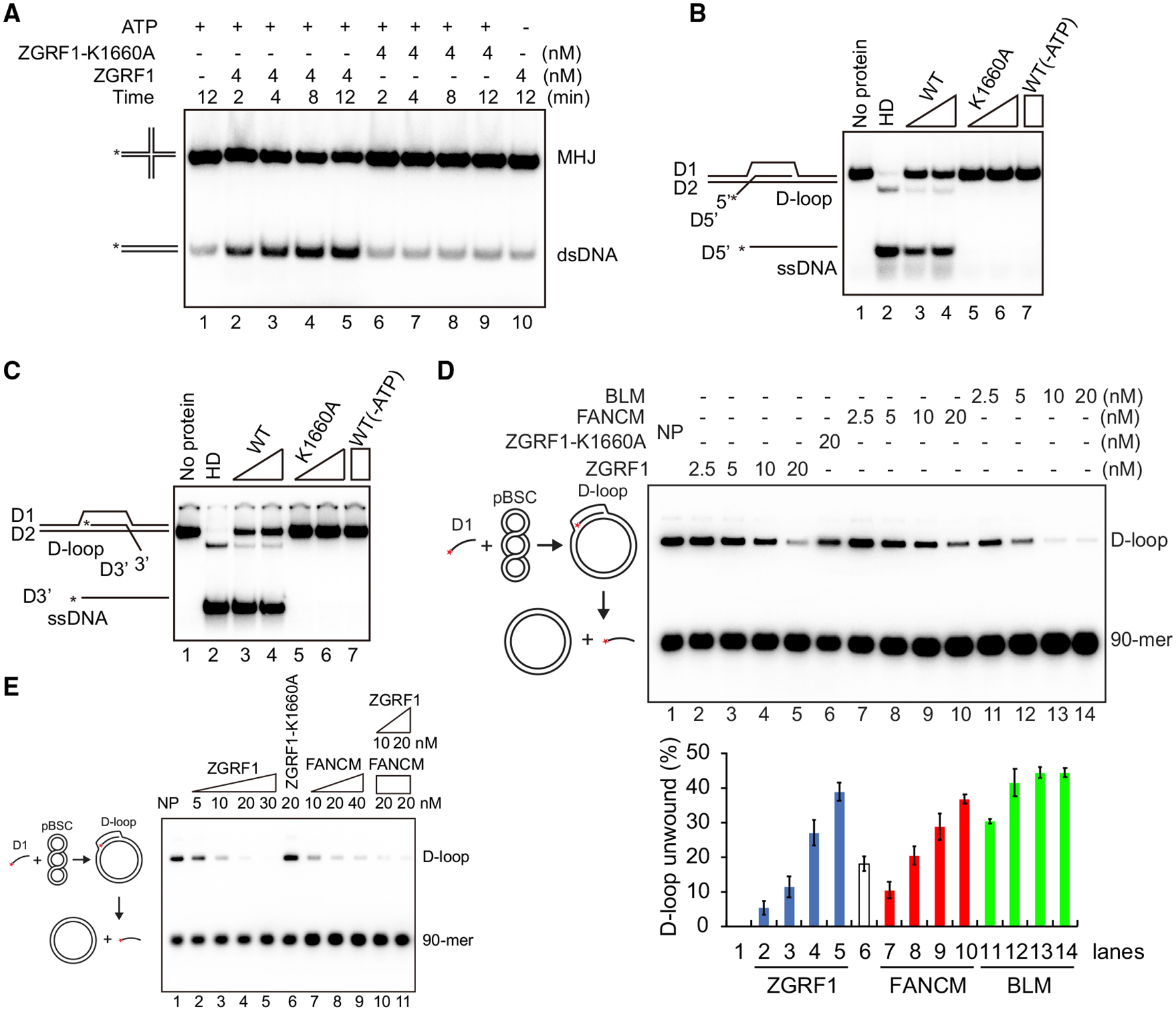
(A) ZGRF1 has branch migration activity. ZGRF1 or ZGRF1-K1660A was incubated with the movable Holliday junction (MHJ) at 37°C for the indicated times with or without ATP being present.
(B and C) D-loop dissociation by ZGRF1. ZGRF1 (WT) or ZGRF1-K1660A (K1660A) was incubated with D-loop structures with a 5′ (B) or 3′ (C) ssDNA overhang at 37°C for 10 min.
(D) Comparison of extended D-loop dissociation activity of ZGRF1, BLM, and FANCM. The deproteinized Rad51-made D-loops (~2.2 nM) were incubated with ZGRF1 (2.5–20 nM), ZGRF1-K1660A (20 nM), FANCM (2.5–20 nM), or BLM (2.5–20 nM) at 37°C for 10 min. The reaction products were resolved in 0.9% agarose gels. NP, no protein. The percentage of D-loop unwound was quantified, and data are means ± SD (n = 3).
(E) ZGRF1 and FANCM independently catalyze dissociation of extended Rad51 made D-loops. Rad51-made D-loops were first deproteinized with SDS and Proteinase K and partially purified. ZGRF1, ZGRF1-K1660A and/or FANCM was incubated with the D-loops at 37°C for 10 min. NP, no protein.
Next, we examined D-loops, which are also important intermediates of HR. To test if ZGRF1 can dissociate D-loops, we assembled 30 bp D-loops with protruding 30 nt 3′ or 5′ overhangs. Incubation of ZGRF1 with any of these substrates led to the release of the free radiolabeled oligonucleotide in a manner dependent on ATP hydrolysis (Figures 6B and 6C), indicating that ZGRF1 can dissociate D-loops irrespective of the orientation of their overhangs. We also tested a more physiological D-loop substrate generated by Rad51-catalyzed strand invasion (Figure 6D). After deproteinization and purification, addition of ZGRF1 to the D-loop led to the efficient dissociation of the labeled invading DNA strand (Figure 6D). Side-by-side comparison of this activity with BLM and FANCM indicated that relative efficiency of D-loop dissociation was greatest with BLM, and that ZGRF1 and FANCM were similar (Figure 6D). As ZGRF1 and FANCM have the potential to interact physically (Figure S5B), we tested if the two proteins would synergize in D-loop dissociation, but no obvious functional interaction was observed (Figure 6E). Further comparison of the joint D-loop dissociation efficiency of FANCM and ZGRF1 showed that a combination of both enzymes neither stimulated nor inhibited the reaction efficiency (Figure S6A), indicating that FANCM and ZGRF1 can dissociate D-loops independently of each other in vitro. Finally, to test if ZGRF1 can dissociate DNA flap structures, we incubated a half D-loop structure with ZGRF1. This substrate was dissociated by ZGRF1 in a manner dependent on ATP hydrolysis (Figures S6B) and more efficiently than FANCM (Figures S6C), indicating that ZGRF1 could play a role in processing these types of intermediates during DNA repair.
Taken together, the biochemical activities of ZGRF1 are consistent with an in vivo role of ZGRF1 in promoting HR at replication-blocking lesions.
DISCUSSION
On the basis of our biochemical, genetic, and cellular analyses, we propose that ZGRF1 may contribute to several aspects of recombinational DNA repair. First, our finding that ZGRF1 physically interacts with RAD51 and stimulates RAD51-RAD54-catalyzed strand exchange indicates that ZGRF1 could play a pre-synaptic role in repair of ICLs and other replication-blocking lesions such as CPT-induced TOP1-DNA covalent intermediates. Second, ZGRF1 may also play a post-synaptic role through its ability to catalyze D-loop dissociation and HJ branch migration. Notably, ZGRF1 can dissociate D-loops even without a free single-stranded tail (Figures 6D and 6E), indicating that ZGRF1 can use the displaced strand of the D-loop as an entry point to remodel the D-loop. Using the 5′-to-3′ directionality of the ZGRF1 helicase, it may catalyze the partial dissociation of the 3′ end of the extended strand from the D-loop, allowing it to re-anneal to the other side of the DSB, which could stimulate the formation of CO products that we observe in the SCE assay (Figures 4C–4E). The epistatic relationship between FANCM and ZGRF1 for resistance to MMC (Figure 2F) suggests that the two proteins act in an interdependent manner to perform the same function in ICL repair. This would be consistent with the observed physical interaction between the two proteins (Figure S5B). Although FANCM and ZGRF1 appear to dissociate D-loops independently in vitro (Figure 6E), it is possible that they may work together in this capacity in vivo. The apparent synthetic lethality between FANCJ and ZGRF1 (Figure 2G) suggests that the two proteins provide a redundant function to ICL repair. This redundant function is unlikely to be related to the checkpoint function of FANCJ (Yu et al., 2003), because ZGRF1 appears to have a functional G2-M checkpoint (Figure 1E). Instead, it may be the 5′-to-3′ helicase activity that is essential and provided by either FANCJ or ZGRF1. In summary, we favor a model in which ZGRF1 promotes ICL repair by facilitating HR through stimulation of RAD51-catalyzed strand exchange and possibly also by dissociation of D-loops and HJ branch migration. A similar contribution to repair may be operating, when replication forks converge on a CPT-trapped TOP1-DNA cleavage complex or replication forks collapse at the DNA nick stabilized by TOP1 trapping.
The lack of sensitivity of ZGRF1−/− cells to PARP inhibition is reminiscent of that reported for some FA patient cell lines (FA-A, FA-L, FA-D2, FA-I, FA-J), and contrasts with sensitivity of FA-D1 (BRCA2) and FA-P cells (Kim et al., 2013). Interestingly, ZGRF1 depletion and mutation of BRCA1 were reported to cause a synthetic sensitization to PARP inhibitors (Zimmermann et al., 2018), suggesting that ZGRF1 could be inhibited to eliminate BRCA-deficient cancer cells. Perhaps in the absence of BRCA1 or BRCA2 to catalyze recombinational bypass of the trapped PARP, cells become reliant on ZGRF1 to promote template switching. The lack of sensitivity of ZGRF1−/− cells to HU is similar to that reported for other ICL repair factors such as FANCJ, FANCA, BRCA1, and BRCA2 (Chen et al., 2016; Peng et al., 2018; Schlacher et al., 2011, 2012; Xu et al., 2017). In contrast, FANCD2 and RAD51 are required for survival after HU treatment (Chen et al., 2016). A more detailed understanding of the coordination of ZGRF1 repair activities with that of other repair factors awaits further epistasis analysis as well as dissection of its interaction with other proteins.
ZGRF1 forms nuclear foci in response to MMC that mostly (78%–82%; Figure 3D) colocalize with FANCD2, indicating that ZGRF1 recognizes DNA lesions repaired by the FA pathway. However, the number of ZGRF1 foci at any given time is low (typically 2–5 foci; Figure 3) compared with the number of co-localizing γ-H2AX and FANCD2 foci (typically 9–11 foci; Figure 2D) observed after the same dose of MMC. The lack of ZGRF1 at some FANCD2 foci could have several explanations. First, ZGRF1 may only recognize a subset of the DNA lesions that are recognized by FANCD2. Second, some ZGRF1 foci may fall below our detection limit, because ZGRF1 foci are generally fainter than FANCD2 foci. Third, ZGRF1 may only be recruited to FANCD2 foci during the recombination step of ICL repair. Relating to the latter point, it should be noted that some ZGRF1 foci appear to localize at the periphery of the nucleolus, which may be related to a previous report that recombinational repair at the rDNA takes place only after relocalization of the DNA lesion to the periphery of the nucleolus (van Sluis and McStay, 2015). It will therefore be important in future studies to dissect the spatiotemporal relationship between ZGRF1 foci and other repair factors.
To date, there are no reports of mutations in ZGRF1 associated with cancer or FA. However, genome-wide association studies have linked rare missense mutations in ZGRF1 to childhood apraxia of speech and obesity (Gao et al., 2015; Peter et al., 2016). It is worth noting that several other neurological disorders are caused by mutation in DNA repair genes such as ataxia-telangiectasia (ATM) (Savitsky et al., 1995), oculomotor apraxia (APTX) (Ahel et al., 2006), spinocerebellar ataxia with axonal neuropathy (TDP1) (Takashima et al., 2002), and motor neuron disease (SETX) (Hirano et al., 2011), but the role of ZGRF1 in human pathology awaits future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Michael Lisby (mlisby@bio.ku.dk).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not apply code. Original/source data for Figures 1B, 2A–2E, 3A–3D, 4C–4E, 5C–5G, 5I, 5J, 6A–6E, S2C, S3C, S4A–S4C, S4E, S4F, S5A–S5C, S5E, and S6A–S6C are available through Mendeley (https://data.mendeley.com/datasets/d532xvdrn2/draft?a=cdd41557-9d54-48fc-bc37-1e63b2be48bb).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human cell lines U2OS (female), HCT116 (male) and hTERT RPE-1 (female) were a gift from Jakob Nilsson (University of Copenhagen). Source and identifier (if applicable) of cell lines used in this study are also listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Raw imaging and western data | This paper | https://data.mendeley.com/datasets/d532xvdrn2/draft?a=cdd41557-9d54-48fc-bc37-1e63b2be48bb |
| Antibodies | ||
| Rabbit polyclonal anti-ZGRF1 | This paper | Lot no. 28429 |
| Rat anti-tubulin | Abcam | Cat#ab6160; RRID:AB_305328 |
| Swine anti-rabbit | Dako | Cat#P0217; RRID:AB_2728719 |
| Rabbit anti-rat | Dako | Cat#P0450; RRID:AB_2630354 |
| Mouse anti-GFP | Roche Diagnostics | Cat#11814460001; RRID:AB_390913 |
| Rabbit anti-mouse | Dako | Cat#P0161; RRID:AB_2687969 |
| Mouse anti-FANCM | Novusbio | Cat#NBP2–50418; RRID:AB_2716711 |
| Rabbit anti-FANCJ | Novusbio | Cat#NB100–416; RRID:AB_2066307 |
| Rabbit anti-FANCD2 | Novusbio | Cat#NB100–182SS; RRID:AB_1108397 |
| Mouse anti-γH2AX | Millipore | Cat#05–636; RRID:AB_309864 |
| Alexa Fluor 568 goat anti-rabbit IgG | Life Technologies | Cat#A-11011; RRID:AB_143157 |
| Alexa Fluor 488 goat anti-mouse IgG | Life Technologies | Cat#A-21121; RRID:AB_2535764 |
| Rabbit anti-RAD51 | Bio Academia | Cat#70–001; RRID:AB_2177110 |
| Anti-GST-HRP | New England Biolabs | Cat#E2624S |
| Rabbit anti-human RAD51 | Santa Cruz Biotech | Cat#sc-8349; RRID:AB_2253533 |
| Anti-yeast Rad51 | Lab raised | N/A |
| Mouse anti-myc-HRP | Thermo Fisher Scientific | Cat#R951 −25; RRID:AB_2556561 |
| Mouse anti-FLAG-HRP | Sigma | Cat#A8592; RRID:AB_439702 |
| Rat anti-HA-HRP | Roche | Cat#12013819001; RRID:AB_390917 |
| Anti-human PCNA | Lab raised | N/A |
| Rabbit anti-human RPA70 | Abcam | Cat#ab79398; RRID:AB_1603759 |
| Rabbit anti-mouse IgG-HRP | Pierce | Cat#31450; RRID:AB_228427 |
| Goat anti-rabbit IgG-HRP | Sigma | Cat#A6154; RRID:AB_258284 |
| Goat anti-rat IgG-HRP | Santa Cruz Biotech | Cat#Sc-2032; RRID:AB_631755 |
| Bacterial and Virus Strains | ||
| E. coli strain DH5α | Lifetechnologies | Cat#18258012 |
| E. coli strain BL21(DE3) | New England Biolabs | Cat#C2527I |
| E. coli strain DH10Bac | Invitrogen | Cat#10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Complete protease inhibitor cocktail | Roche | Cat#25735720 |
| Dynabeads Protein G | Life Technologies | Cat#10004D |
| GFP-Trap® M beads | Chromotek | Cat# gtm-20 |
| Mitomycin C | Sigma | Cat#M7949 |
| Talazoparib | SMS-Gruppen Denmark | Cat#S7048 |
| Hydroxyurea | Sigma | Cat#H8627 |
| KaryoMAX colcemid | Life Technologies | Cat#15212–012 |
| BrdU | Sigma | Cat#B5002 |
| Camptothecin | Merck | Cat#C9911 |
| Critical Commercial Assays | ||
| Guide-it sgRNA Screening Kit | Takara Bio | Cat#632639 |
| Factor Xa Cleavage Capture Kit | Merck | Cat#69037 |
| DPX Mountant for histology | Sigma | Cat#06522 |
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat#DUO92002 |
| Duolink In Situ PLA Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat#DUO92004 |
| Duolink In Situ Detection Reagents Orange | Sigma-Aldrich | Cat#DUO92007 |
| Experimental Models: Cell Lines | ||
| HCT116 | (Brattain et al., 1981) | N/A |
| U2OS | (Pontén and Saksela, 1967) | N/A |
| RPE-1 | (Jiang et al., 1999) | N/A |
| Oligonucleotides | ||
| Primers and oligonucleotides, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| pX458 | (Ran et al., 2013) | Addgene Plasmid #48138 |
| pBluescript SK+ | Fermentas GmbH, Germany | (Short et al., 1988) |
| pAB_KO1 | This paper | N/A |
| pAB_KO_rev | This paper | N/A |
| pAB_KO_rev3 | This paper | N/A |
| pX461 | (Ran et al., 2013) | Addgene Plasmid #48140 |
| pAB_2xYFP_A | This paper | N/A |
| pAB_2xYFP_B | This paper | N/A |
| pAK_ZGRF1–2xYFP | This paper | N/A |
| pCre-GFP | (Williams et al., 2015) | N/A |
| pKSV1 | This paper | N/A |
| pKSV2 | This paper | N/A |
| pKSV15 | (Motnenko et al., 2018) | N/A |
| pGEX-3X-ZGRF1-N | This paper | N/A |
| pCS2-mRFP | (Sartori et al., 2007) | N/A |
| pCBA-I-SceI | (Richardson et al., 1998) | N/A |
| Software and Algorithms | ||
| TIDE (https://tide.nki.nl) | N/A | (Brinkman et al., 2014) |
| FlowJo v10.4.1 | FlowJo, LLC | RRID:SCR_008520 |
| Volocity v5.4 | PerkinElmer | RRID:SCR_002668 |
| SoftWoRx 7.0.0 | Applied Precision | N/A |
| Prism 8 | GraphPad software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_000306 |
Cell culture
Cell lines used in this study are listed in Table S1. Human colorectal carcinoma HCT116 cells were cultured at 37°C, 5% CO2 in McCoy’s 5A (Modified) medium (Life Technologies, cat. no. 26600023), containing 10% heat-inactivated FBS (GIBCO by Life Technologies, cat. no. 10500–064), and 10,000 units/mL of penicillin and 10,000 μg/mL of streptomycin (GIBCO by Life Technologies, cat. no. 15140–122). Conditioned medium was made by culturing 80% confluent HCT116 cells for 24 h in the above-mentioned medium, aspirating the medium and centrifuging it at high speed to remove any cells from the suspension.
Human osteosarcoma U2OS and hTERT-immortalized retinal pigment epithelial (hTERT RPE-1) cells were cultured at 37°C, 5% CO2 in complete medium containing DMEM, high glucose, GlutaMAX Supplement, and pyruvate (ThermoFisher Scientific, cat. no. 31966–021), 10% heat-inactivated FBS (GIBCO by Life Technologies, cat. no. 10500–064), and 10,000 units/mL of penicillin and 10,000 μg/mL of streptomycin (GIBCO by Life Technologies, cat. no. 15140–122). Conditioned medium was made by culturing 20% confluent U2OS cells for 48 h (so the cells would be 80% confluent after 48 h) in the above-mentioned medium, aspirating the medium and centrifuging it at high speed to remove any cells from the suspension. The conditioned medium was then diluted in complete medium (3:2).
METHOD DETAILS
Design of gRNA
To generate a knockout of ZGRF1, a guide RNA (gRNA) was designed to target the second exon of ZGRF1 (Figure S1A) and have the fewest off-target hits. Target sequence (including the PAM sequence): AAGTCAAAAGTGTGGCAAGATGG. The designed gRNA was tested for Cas9 cleavage efficiency of HCT116 genomic DNA in vitro using a Guide-it sgRNA Screening Kit (Takara Bio, cat. no. 632639) according to the manufacturer’s instructions. Single-cell sorted clones were screened for insertions and deletions (indels) by sequencing and TIDE (Tracking of Indels by Decomposition) analysis (Brinkman et al., 2014). Multiple rounds of sequential transfection yielded several independent ZGRF1−/− clones (Figures S1B–S1D). To tag ZGRF1 with 2xYFP, two guides were designed: Guide A 2xYFP targeting CACTTGTTATTGAAGTATACAGG and Guide B 2xYFP targeting TGTCTCCAGAACTCATCTGTTGG (PAM sequences underlined). ZGRF1 was knocked out in the U2OS and RPE-1 cell lines essentially as described for the HCT116 cell line. The guides designed to knock out FANCM (Figure S2) and FANCJ (Figure S3) were CGGGACAAGCTCCTCTAGAAAGG and TGTTTGTTGGAGAGTCCCACAGG (PAM sequences underlined), respectively.
CRISPR/Cas9 plasmids and cloning
Plasmids used in this study are listed in Table S2. Designed gRNAs were cloned into the CRISPR-Cas9 plasmid pX458 (Addgene plasmid #48138) (Ran et al., 2013) for knocking out ZGRF1, FANCM and FANCJ or into pX461 (Addgene plasmid #48140) for tagging ZGRF1 with 2xYFP. Briefly, pX458 or pX461 was digested with BbsI, dephosphorylated and gel purified. Two 5′-phosphorylated oligonucleotides designed to contain the Cas9 targeting sequence (excluding the PAM sequence), and complementary overhangs to BbsI digested pX458 or pX461 when annealed together, were synthesized (TAG Copenhagen). A guanine (G) was added to the 5′ end of the gRNA as the U6 RNA polymerase III promoter used to express the gRNA prefers this nucleotide as the first base of its transcript (Ran et al., 2013). Guide primers were mixed in equimolar ratio and annealed using a PCR machine by heating at 95°C for 5 min and ramping down to 25°C at a rate of 5°C/min. Annealed primers were ligated into BbsI-digested, dephosphorylated pX458 or pX461 and transformed into DH5α E. coli. DNA from ampicillin-resistant clones was screened by restriction digest and sequenced at the gRNA locus to verify sequence integrity. Guide primers ZGRF1_KO_guide1_FW and ZGRF1_KO_guide1_RV for ZGRF1 knockout were ligated into pX458 to generate plasmid pAB_KO1. Guide primers ZGRF1_rev_gRNA_FW and ZGRF1_rev_gRNA_RV for ZGRF1−/− reversion were ligated into pX458 to generate plasmid pAB_KO_rev. Guide primers ZGRF1_rev_gRNA3_FW and ZGRF1_rev_gRNA3_RV for ZGRF1rev/−* reversion were ligated into pX458 to generate plasmid pAB_KO_rev3. GuideA_ZGRF1_FW and GuideA_ZGRF1_RV were ligated into pX461 to generate plasmid pAB_2xYFP_A for 2xYFP tagging. GuideB_ZGRF1_FW and GuideB_ZGRF1_RV were ligated into pX461 to generate plasmid pAB_2xYFP_B for 2xYFP tagging. Guide primers FANCJ_KO_guide1_FW and FANCJ_KO_guide1_RV for FANCJ knockout and guide primers FANCM_KO_guide1_FW and FANCM_KO_guide1_RV for FANCM knockout were ligated into pX458 to generate plasmid pKSV1 and pKSV2, respectively. Oligonucleotides used in this study are listed in Table S3.
Construction of ZGRF1–2xYFP donor template
A 1052 bp 5′ homology arm placed right before the stop codon of ZGRF1 was amplified by PCR using the primers 5′arm_FW and 5′arm_RV adding a 5′ KpnI and a 3′ SalI restriction site. A 872 bp 3′ homology arm placed downstream of the ZGRF1 stop codon was amplified by PCR using the primers 3′arm_FW and 3′arm_RV adding a 5′ BamHI and a 3′ NotI restriction site. A 728 bp fragment encoding Venus YFP (excluding stop codon) with a 5′ SalI and 3′ EcoRI restriction site and a 720 bp fragment encoding EYFP with a 5′ EcoRI and a 3′ BamHI restriction site were synthesized by GeneArt Services at Thermo Fisher Scientific. The four fragments were cloned into pBluescript SK+ starting with insertion of the homology arms followed by a three-point ligation of the backbone and the two YFP’s. Subsequently, the BamHI restriction site was used to introduce a loxP-flanked blasticidin S resistance (BSR) cassette (Arakawa et al., 2001) downstream of the EYFP to create the final plasmid pAK_ZGRF1–2xYFP for tagging ZGRF1 C-terminally with Venus and EYFP (2xYFP).
Transfection of HCT116 cells
Unless otherwise stated, HCT116 cells were transfected using FuGENE 6 Transfection Reagent (Promega, cat. no. E2311) according to the manufacturer’s instructions scaled for cell culture plates with Ø = 10 cm and using a FuGENE 6 Transfection Reagent:DNA ratio of 3:1. For tagging of ZGRF1 with 2xYFP, cells were co-transfected with equal moles of pAB_2xYFP_A, pAB_2xYFP_B and linearized pAK_ZGRF1–2xYFP. To remove the blasticidin resistance cassette, ZGRF1 2xYFP tagged cells were transfected with pCre-GFP and single cell FACS sorted. It was verified that cells became sensitive to blastidicin (5 μg/mL).
For reverting ZGRF1−/− or ZGRF1rev/−* cells, cells were co-transfected with 3.125 μg pAB_KO_rev and 1.875 μg ZGRF1_rev_ssDNA_template_2 or 3.125 μg pAB_KO_rev3 and 1.875 mg ZGRF1_rev_ssDNA_template_2, respectively, using Lipofectamine 3000 Reagent (Invitrogen, cat. no. L3000–015) according to the manufacturer’s instruction scaled for 6-well plate wells. The reverted ZGRF1 allele contained a diagnostic BamHI restriction site for screening candidate clones.
To generate FANCJ mutant and FANCM−/− clones, Lipofectamine 3000 Reagent (Invitrogen, cat. no. L3000–015) was used according to the manufacturer’s instruction scaled for a 6-well plate. Parental and ZGRF1−/− HCT116 cells were transfected with either pKSV1 (FANCJ-KO-gRNA) or pKSV2 (FANCM-KO-gRNA).
mCherry-FANCD2-PuroR expressing cells were obtained by random integration using FuGENE 6 transfection reagent according to the manufacturer’s instructions scaled for a 6-well plate. HCT116 ZGRF12xYFP/WT cells were transfected with plasmid pKSV15 with a Reagent:DNA ratio of 3:1. 24 h after transfection, cells were treated with 0.8 μg/mL puromycin dihydrochloride (Thermo Fisher Scientific, cat. no. A1113803) for 3 days in order to select for puromycin resistant cells.
Transfection of U2OS and hTERT RPE-1 cells
U2OS and hTERT RPE-1 cells were transfected using FuGENE 6 Transfection Reagent according to the manufacturer’s instructions scaled for cell culture plates with Ø = 10 cm and using a FuGENE 6 Transfection Reagent:DNA ratio of 3:1.
Fluorescence activated cell sorting
48 h after transfection, GFP-positive cells were sorted using a BD FACSJazz Cell Sorter (BD Biosciences, cat. no. 655490). Cells were trypsinized, suspended in growth medium, and then filtered (50 μm; BD Biosciences, cat no. 340629) before sorting. For ZGRF1, FANCM and FANCJ knockout, single cells were sorted into 96-well plates containing 40% conditioned media and 60% fresh media. For the ZGRF1 2xYFP tagging, 100 cells were sorted into each well. 5 days later, selection of ZGRF1 2xYFP colonies was performed using 5 μg/mL blasticidin (GIBCO by Life Technologies, cat. no. A11139–03).
Genotyping of FACS-sorted clones
10 days after cell sorting, genomic DNA was extracted from single colonies. Briefly, cells were gently washed with PBS and 30 μL QuickExtract DNA Extraction Solution (Lucigen, cat. no. QE09050) was added to each well. The plate was incubated at 70°C for 30 min, and 1 μL of each well was used for subsequent PCRs.
A 346 bp region containing the ZGRF1 Cas9 target site was amplified using primers ZGRF1_KO_IDAA_FW and ZGRF1_KO_IDAA_RV. The resulting product was gel purified and Sanger sequenced (Eurofins Genomics). Sequences were analyzed for indels using the online tool TIDE (Tracking of Indels by Decomposition) (Brinkman et al., 2014).
Clones containing indels were validated by fragment length analysis by capillary electrophoresis of FAM-labeled PCR products spanning the Cas9 targeting site. PCR products were created using a tri-primer PCR strategy as described previously (Yang et al., 2015). Primer molar ratio used for ZGRF1_KO_IDAA_FAMFW:FamF:ZGRF1_KO_IDAA_RV was 1:10:10.
Analysis of fragment length was carried out by Eurofins Genomics using size standard ROX500 and filter set ABI-D. For each analysis, a parental HCT116 sample was included for reference.
The strategy to verify FANCJ and FANCM knockout was similar to the description above. A 776 bp region containing the FANCJ Cas9 target site was amplified using the primers, FANCJ_KO_700_FW and FANCJ_KO_700_RV and a 714 bp region containing the FANCM Cas9 target site was amplified using the primers FANCM_KO_700_FW and FANCM_KO_700_RV. The resulting PCR products were sequenced and analyzed for indels using the online tool TIDE.
To verify integration of 2xYFP downstream of ZGRF1, a 1180 bp PCR fragment was generated using the primers ZGRF1_outsite_FW2 (binds genomic DNA upstream the ZGRF1 5′ homology arm) and ZGRF1_YFP_RV (binds in YFP).
Generation of anti-ZGRF1 antibodies
To raise a rabbit antibody against a 102 amino acid N-terminal fragment of ZGRF1, a 306 bp region encoding the ZGRF1 DUF2439 domain was amplified from cDNA by PCR using primers ZGRF1-N-F and ZGRF1-N-R, adding a 5′ BamHI and a 3′ XmaI restriction site, and cloned into pGEX-3X (GE Healthcare, cat. no. 28954654) to generate pGEX-3X-ZGRF1-N.
For expression and purification of GST tagged ZGRF1 DUF2439 and untagged ZGRF1 DUF2439, pGEX-3X-ZGRF1-N was transformed into E. coli BL21(DE3). The bacteria were then grown at 37°C to OD600 = 0.7 followed by 4 h of expression induction with 100 μM IPTG (Sigma, cat. no. I5502). The culture was harvested at 4000 × g for 10 min followed by washing and re-suspension in ice-cold RecBuffer (50 mM Piperazine-HCl (Sigma, cat. no. P45907) pH 9.8, 500 mM NaCl, 1 mM DTT (VWR, cat. no. 82021–254), 1 mM EDTA, 0.5 mM PMSF (Roche, cat. no. 10837091001), complete protease inhibitor cocktail (Roche, cat. no. 25735720)). 0.4 mg/mL lysozyme (Sigma, cat. no. L7651) was added and the cells were sonicated for 3 × 30 s in a sonicator (Hielscher, UP200S, point tip max). To complete cell lysis, the solution was supplemented with 1% Triton X-100 (Sigma, cat. no. T8787) and rocked 30 min at 4°C before it was spun at 16,000 × g for 10 min. Whole cell extract was then incubated with Glutathione Sepharose 4B (GE Healthcare, cat. no. 17-0756-01) while rocking at 4°C for 30 min and the protein bound resin was washed with cold RecBuffer supplemented by 0.1% Triton X-100. To purify GST tagged protein, resin was transferred to a Mobicol “classic” column (MoBiTec, cat. no M1002) with a 10 μm pore size filter attached (MoBiTec, cat. no. M2210) and the protein was eluted with Elution Buffer (50 mM Tris-HCl pH 8.0, 10 mM reduced glutathione (AppliChem, cat. no. A2084), 150 mM NaCl, 1 mM DTT, 1 mM EDTA). To purify untagged ZGRF1 DUF2439, the protein bound resin was equilibrated with Cleavage Buffer (PBS, 150 mM NaCl, 1 mM CaCl2, pH 7.5) and incubated with Factor Xa protease (Merck, cat. no. 69037) for 24 h at 25°C. The concentration of NaCl was then increased to approx. 700 mM to release untagged ZGRF1 DUF2439 from the resin before the GST-bound resin was removed using centrifugal columns (Merck, cat. no. UFC40SV25). Finally, Factor Xa was removed using the Factor Xa Cleavage Capture Kit (Merck, cat. no. 69037) according to the manufacturer’s instructions.
Polyclonal antibodies against the ZGRF1 DUF2439 domain were raised in rabbits by BioGenes GmbH. GST tagged ZGRF1 DUF2439 was used for immunization and untagged ZGRF1 DUF2439 was used for subsequent purification of the serum to remove antibodies not specific for the DUF2439 domain.
Western blotting
For all western blots in this study, cells were grown to 80% confluency. For visualization of secondary antibodies, ECL mixture (GE Healthcare, cat. no. RPN2232) was prepared and added to the membrane followed by imaging of chemiluminescence (ImageQuant LAS 4000).
For western blotting of ZGRF1: Cells were washed once with PBS, and cells isolated using cell scrapers. Cells were centrifuged at 250 × g for 5 min at 4°C, supernatant was removed, and cells were re-suspended in 800 mL ice-cold PBS. This was followed by centrifugation at 2500 × g for 30 s at 4°C and re-suspension in 1.8 mL NP-40 buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl pH 8.0, 1 mM PMSF, complete protease inhibitor cocktail). Samples were lysed by syringing through a 23G needle 10 times on ice and centrifuged at 13000 × g for 20 min at 4°C. The supernatant was used for pulldown of ZGRF1 using Dynabeads Protein G (Life Technologies, cat. no. 10004D) according to the manufacturer’s instructions. To each sample of beads, 2.42 μg anti-ZGRF1 rabbit antibody (BioGenes GmbH, lot no. 28429) and 1 μg rat anti-tubulin antibody (Abcam, cat. no ab6160) were bound. PBS with 0.1% Tween-20 was used as washing buffer. Samples were eluted in 1X SDS sample buffer (2X SDS sample buffer (125 mM Tris-HCl pH 6.8, 5% SDS, 0.004% Bromophenol Blue, 10% beta-mercaptoethanol, 20% glycerol) diluted 1:2 in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl pH 8.0, 1 mM PMSF, complete protease inhibitor cocktail)) at 70°C with shaking at 1000 RPM for 10 min. Eluted samples were boiled, run on an SDS-PAGE gel until the 40 kDa marker ran out of the gel, and transferred to a 0.45 μm ethanol activated PVDF membrane at 40 mA for 18 h at 4°C in an ice bucket. The membrane was cut at 60 kDa to separate tubulin from ZGRF1 and blocked with 5% skim milk in TBST for 1 h at room temperature with gentle shaking followed by 3 washes with TBST for 10 min. Each membrane was incubated with their respective primary antibodies at 4°C overnight (1:1000 rabbit anti-ZGRF1 (BioGenes GmbH, lot no. 28429) in 5% BSA in TBST or 1:20.000 rat anti-tubulin (Abcam, cat. no ab6160) in 5% milk in TBST). Membranes were washed 3 times with TBST for 10 min and incubated with respective secondary antibodies (1:1000 swine anti-rabbit (Dako, cat no. P0217) for ZGRF1 and 1:2000 rabbit anti-rat (Dako, cat. no. P0450) for tubulin) in 5% skim milk in TBST for 1 h followed by another 3 washes for 10 min in TBST.
For western blotting of GFP: After syringing and centrifugation, the supernatant was used for pull-down of GFP using GFP-Trap® M beads (Chromotek, cat. no. gtm-20) according to the manufacturer’s instructions. PBS was used as washing buffer. Samples were eluted in 2X SDS sample buffer at 95°C with shaking at 1000 RPM for 10 min. Eluted samples were boiled, run on an SDS-PAGE gel, transferred to a 0.45 μm nitrocellulose membrane and blocked as described in the section above. The membrane was incubated with primary antibody in 5% BSA in PBST at 4°C overnight (1:1000 mouse anti-GFP (Roche Diagnostics, cat. no. 11814460001)). The membrane was washed 3 times with TBST for 10 min and incubated with secondary antibody (1:1000 rabbit anti-mouse (Dako, cat. no. P0161)) in 5% skim milk in TBST for 1 h followed by another 3 washes for 10 min in TBST.
For western blotting of FANCM, FANCJ and FANCD2: The appropriate cell lines were harvested by trypsinization. 10 million cells of each cell line were spun down at 250 × g for 5 min at 4°C. The pellet was resuspended in 1 mL ice cold PBS and washed by spinning at 1300 × g for 30 s at 4°C. This was followed by resuspending in 800 μL ice cold PBS and centrifugation at 1300 × g for 30 s at 4°C. The samples were then resuspended in 200 μL RIPA buffer and lysed by syringing through a 23G needle 10 times on ice and centrifuged at 20.000 × g for 20 min at 4°C. Supernatant was transferred to a new tube and 5x SDS-PAGE loading dye (with 2-mercaptoethanol) was added. Samples were boiled for 5 min at 95°C and run on a precast 4%–20% SDS-PAGE gel (BIO-RAD, cat. no. #456–1093) until the 15 kDa ladder band had run out of the gel. Proteins were transferred to a 0.45 μm nitrocellulose membrane at 40 mA for 18 h at 4°C in an ice bucket with magnetic stirring. After transfer, the membrane was blocked with 5% skim milk in TBST for 1 h at room temperature with gentle shaking followed by 3 washes with TBST for 10 min. The membrane was cut between 70 and 100 kDa to separate Tubulin from FANCM, FANCJ or FANCD2 and then each membrane were incubated with their respective primary antibody in 5% skim milk in TBST at room temperature for 1 h or for 18 h at 4°C (1:500 Mouse anti-FANCM (Novusbio, NBP2–50418), 1:500 Rabbit anti-FANCJ (Novusbio, NB100–416), 1:2000 Rabbit anti-FANCD2 (Novusbio, NB100–182SS) or 1:5000 Rat anti-Tubulin). Membranes were washed 3 times with TBST for 5 min and incubated with respective secondary antibodies (1:1000 swine anti-rabbit for FANCJ and FANCD21:1000, rabbit anti-mouse for FANCM and 1:2000 rabbit anti-rat for tubulin) in 5% skim milk in TBST for 1 h followed by another 3 washes for 5 min in TBST.
Cell cycle analysis
Parental HCT116 and knockout cell lines were seeded in 10 mL growth media in cell culture plates with Ø = 10 cm at low confluency (0.2 × 106 cells). The cells were allowed to adhere for 48 h and then treated with 40 ng/mL MMC (Sigma, cat. no. M7949) for 2 h. Following treatment, the cells were washed with PBS 3 times and incubated in fresh growth media for additional 24 h. Samples were harvested by trypsinization, spun down at 250 × g for 5 minutes and resuspended in 1 mL PBS. Immediately before fixation in 70% ice cold ethanol, the cells were filtered (50 μm; BD Biosciences, cat no. 340629) to obtain a mono-dispersed cell suspension. The fixed cells were stained with propidium iodide as described in Pozarowski and Darzynkiewicz (2004). The cell cycle distribution was analyzed with a BD FACSJazz Cell Sorter (BD Biosciences, cat. no. 655490).
Colony formation assays
HCT116 parental and ZGRF1−/− cells were plated in duplicate and then treated with the indicated doses of DNA-damaging agents. For the epistasis analysis with FANCM−/− cells and FANCJ mutant cells, all cells were seeded in 40% conditioned media. Cells treated with CPT (Merck, cat. no. C9911) or MMC were washed with fresh media 24h after drug addition. Talazoparib-treated samples (SMS-Gruppen Denmark, cat. no. S7048) were grown in the presence of the drug continuously until fixation. UV samples were treated with a 254 nm UV-C irradiator fitted with G15T8 15W germicidal UV tubes, while IR samples were treated with an X-ray irradiator (Faxitron, cat. no. CP-160). Cells were grown for 10–12 days after treatment, fixed with methanol:acetic acid (3:1) and stained with crystal violet in methanol. The surviving fraction at each drug concentration was calculated by normalizing to the plating efficiency of untreated samples. To measure HU sensitivity, HCT116 parental and ZGRF1−/− cells were plated in duplicate and allowed to adhere overnight followed by treatment with the indicated doses of hydroxyurea (Sigma, cat. no. H8627). The samples were grown in the presence of the drug continuously until fixation 8 days after HU was added.
U2OS and RPE-1 parental and ZGRF1−/− cells were plated in duplicate, treated with the indicated doses of DNA-damaging agent (MMC) and washed with fresh medium 24 h after drug addition. Cells were grown for 12 and 9 days (U2OS and RPE-1 cells, respectively) after treatment, fixed with methanol:acetic acid (3:1) and stained with crystal violet in methanol. Colonies were manually counted and the surviving fraction at each drug concentration was calculated by normalizing to the plating efficiency of untreated samples.
DR-GFP recombination assay
U2OS cells with the integrated DR-GFP assay were co-transfected with an I-SceI expression vector (pCBA-I-SceI) together with a vector expressing monomeric red fluorescent protein (pCS2-mRFP) in 1:3 ratio to mark the I-SceI-positive cells (Pierce et al., 2001) or with pCS2-mRFP alone as a negative control. Cells were harvested two days after transfection and subjected to flow cytometric analysis to examine recombination induced by double-strand breakage. The mRFP-positive sub-populations of cells were analyzed for HR efficiency to circumvent possible differences in transfection efficiencies. Fluorescence-activated cell sorting data were analyzed with FlowJo software to reveal the percentage of GFP-positive cells relative to the number of transfected cells (mRFP positive).
Metaphase spreads and chromosomal aberrations
Cells were grown in normal growth media or in media containing MMC for 24 h before harvest. 4 h before harvest, cells were treated with KaryoMAX colcemid solution in PBS (GIBCO by Life Technologies, cat. no. 15212–012) at a final concentration of 0.1 μg/mL. Mitotic shake-off was performed and cells were centrifuged at 500 × g for 3 min, washed once with PBS and once with 75 mM KCl. Cells were centrifuged at 500 × g for 5 min followed by resuspension in 75 mM KCl, incubated on ice for 10 min and centrifuged at 1000 × g for 10 min at 4°C. Cell pellets were washed with, and finally re-suspended in ice-cold methanol:acetic acid (3:1) and placed at −20°C for a minimum of 30 min. Spreads were performed by releasing a 10 μL drop from a height of 30 cm onto a slide that had been soaked for a minimum of 3 h in methanol:acetic acid (3:1) in a Coplin jar. After drying, DAPI mounting media (4% n-propyl gallate, 80% glycerol, 1.5 μg/mL DAPI) was added to the slides and a coverslip was applied.
Metaphase spreads were imaged using a microscope (AxioImager Z1; Carl Zeiss) equipped with a 100 × objective lens (Plan Apochromat, NA 1.4; Carl Zeiss), a cooled CCD camera (Orca-ER; Hamamatsu Photonics), differential interference contrast (DIC), and an illumination source (HXP120C; Carl Zeiss). Images were acquired and analyzed to score for chromosomal aberrations using Volocity (PerkinElmer) software. Images were taken at room temperature with 10 optical sections separated by 0.3 μm.
A chromosome arm gap was defined as having a minimum length equivalent to that of the width of the chromosome arm.
Sister chromatid exchange assay
Cells were seeded at low density and allowed to grow for 24 h before BrdU (Sigma, cat. no. B5002) addition to a final concentration of 10 μM. CPT and MMC was added at the indicated concentrations and for the indicated time before mitotic shake-off. 42 h after BrdU addition (two HCT116 doubling times) metaphase spreads were performed as described above. After spreading, dried slides were aged at 65°C for 1 h and stained with 20 μg/mL Hoechst 33258 (Life Technologies, cat. no. C10639) in dH2O for 30 min. This was followed by washing 3 times with dH2O and slides being placed with the chromosomes facing upward in a tray with just enough 0.1 M phosphate buffer (pH 6.8) to cover the slides. Slides were exposed to 254 nm UV light for 12.5 min in a 254 nm UV Stratalinker 1800 (Stratagene, cat. no. 400072) fitted with five G8T5 8W germicidal UV tubes, followed by another 3 washes in dH2O. Slides were then incubated with pre-heated 2X saline-sodium citrate (SSC) buffer in a Coplin jar at 65°C for 1 h followed by 3 washes in dH2O. Slides were stained by immersion in a 1:20 solution of Giemsa stain (1g Giemsa powder dissolved in 66 mL glycerol and 66 mL absolute ethanol) in 0.1 M phosphate buffer pH 6.8 for 4 min. Slides were washed in running dH2O and dried with filter paper. Mounting medium (DPX Mountant for histology, Sigma, cat. no. 06522) was added and a coverslip affixed.
Image capture and analysis was performed using the same microscope and software as described for metaphase spreads.
Fluorescence microscopy and image analysis
For immunofluorescence microscopy, sterile coverslips were placed into the wells of a 24-well plate and cells were seeded at low density in media with or without MMC. For cells being treated less than 24 h, media was changed to MMC-containing media at the indicated time. 24 h after seeding, cells were washed twice with PBS and fixed in 3% paraformaldehyde in PBS (adjusted to pH 7.4) for 10 min at room temperature. Cells were washed 3 times with ice-cold PBS, permeabilized by incubating 10 min with 0.1% Triton X-100 in PBS, washed 3 times for 5 min in PBS and blocked with 3% BSA (Merck, cat. no. A4503) and 22.52 mg/mL glycine in PBST (PBS+ 0.1% Tween 20) for 60 min. Cells were washed twice with PBS and incubated with primary antibodies (1:2000 anti-FANCD2 (Novus Biologicals, cat. no. NB100–182SS) and 1:500 anti-γH2AX (Millipore, cat. no. 05–636)) in 3% BSA in PBST overnight at 4°C. The following day cells were washed 3 times with PBS for 5 min. Cells were incubated with secondary antibodies (1:500 Alexa Fluor 568 goat anti-rabbit IgG (Life Technologies, cat no. A-11011) and 1:500 Alexa Fluor 488 goat anti-mouse IgG (Life Technologies, cat. no. A-21121)) in 3% BSA in PBST (Merck, cat. no. A4503) for 1 h at room temperature in the dark and then washed 3 times with PBS for 5 min. Samples were counterstained using DAPI mounting media (4% n-propyl gallate, 80% glycerol, 1.5 μg/mL DAPI).
For live cell imaging of ZGRF1–2xYFP, 104 cells were seeded per well in an 8 well μ-Slide (Ibidi) in 200 μL of McCoy’s 5A (Modified) medium, containing 10% heat-inactivated FBS and 10,000 units/mL of penicillin and 10,000 μg/mL of streptomycin, and cultured at 37°C, 5% CO2 for 3 days. Cells were synchronized before S phase by adding 2 mM thymidine (Sigma, T1895–1G) 18 h prior to microscopy. Four hours before microscopy, 20 ng/mL MMC was added to the culture. Thirty minutes before release into S phase, 0.4 μM Hoechst 33258 (Sigma, B2883) was added to the media. Cells were release into S phase in Leibovitz’s L-15 medium without phenol red (GIBCO, cat. no. 21083027) containing 10% fetal calf serum and 0.4 μM Hoechst 33258.
For live cell imaging of ZGRF1–2xYFP and mCherry-FANCD2 (cl 72, 87, 132 and 140), 104 cells were seeded per well in an 8 well μ-Slide (Ibidi) in 200 μL of growth media cultured at 37μC, 5% CO2 for 3 days. Cells were synchronized before S phase with 2 mM thymidine added 18 h prior to microscopy. Four hours before microscopy, 20 ng/mL MMC was added to the culture. Cells were release into S-phase in Leibovitz’s L-15 medium without phenol red containing 10% fetal calf serum.
Fluorescence was detected and imaged using a microscope (DeltaVision Elite; Applied Precision) equipped with a 100 × objective lens with a numerical aperture of 1.35 (U-PLAN S-APO, NA 1.4; Olympus), a cooled EMCCD camera (Evolve 512; Photometrics), and a solid-state illumination source (Insight; Applied Precision, Inc). Immunofluorescence images were taken at room temperature with 10 optical sections separated by 0.33 μm. Live cell images of ZGRF1–2xYFP and ZGRF1–2xYFP mCherry-FANCD2 were taken at 37°C with 8 and 15 optical sections respectively, separated by 0.5 μm
Images were acquired with softWoRx (Applied Precision) software. Processing and quantitative measurements of fluorescence intensities were performed with Volocity software (PerkinElmer).
RAD51 immunostaining and image analysis
Sterile coverslips were placed into the wells of a 24-well plate and cells were seeded at low density in fresh media. The following day, MMC was added at a concentration of 1000 ng/mL. After 1 h incubation, cells were washed twice with fresh warm medium and allowed to recover in fresh, warm medium for the indicated amount of time before pre-extraction and fixation.
To pre-extract cytosolic proteins, media was removed, and cells were washed twice with PBS. Cells were then incubated with 0.1% Triton X-100 in 20 mM PIPES pH 6.8, 1 mM MgCl2, 10 mM EGTA in ddH2O for 60 s followed by fixation in 4% formaldehyde and 0.1% Triton X-100 in 20 mM PIPES pH 6.8, 1 mM MgCl2, 10 mM EGTA in ddH2O for 15 minutes. Following fixation, coverslips were rinsed 4 times with PBS for 5 minutes and saturated overnight by incubation with 3% BSA, 0.5% Triton X-100 in PBS at 4°C.
The following day, cells were washed once with PBS for 5 min and incubated with blocking and aldehyde quenching solution (3% BSA, 22.52 mg/mL glycine in PBST) for 30 min.
Cells were then washed once in PBST and incubated with 1:1000 anti-RAD51 (Bio Academia, cat. no. 70–001) in 3% BSA in PBST for 90 min at room temperature on a rocking table. This was followed by three washes in PBST for 5 minutes and incubation with 1:500 Alexa Fluor 568 goat anti-rabbit IgG (Life Technologies, cat no. A-11011) in 3% BSA in PBST for 45 min (in the dark) at room temperature on a rocking table. Following a further 3 washes with PBST, samples were counterstained using DAPI mounting media (4% n-propyl gallate, 80% glycerol, 1.5 μg/mL DAPI) and coverslips were sealed to microscopy slides using clear nail polish. Slides were kept at 4°C overnight and imaged the next day.
Fluorescence was detected and images were acquired as described under ‘Fluorescence microscopy and image analysis’. Images were taken at room temperature with 10 optical sections separated by 0.6 μm and the same laser intensity and exposure time was used for all images in the same channel. Processing and quantitative measurements of fluorescence intensities were performed with Volocity software (PerkinElmer). Foci were quantified using a custom measurement protocol in Volocity with foci for each cell being automatically counted if they lay within the nucleus (as defined by DAPI staining), exceeded a minimum spot intensity of 400% and were the brightest spots within a radius of 0.4 μm.
Expression and purification of human ZGRF1
The human ZGRF1 cDNA (C4orf21-pCMV6) was purchased from Origene. The ZGRF1 protein coding sequence was introduced into the pENTR D-TOPO vector (Invitrogen) and then transferred into the pDEST20 vector to generate GST-(TEV)-ZGRF1-(His)6. A bacmid was prepared in the E. coli strain DH10Bac (Invitrogen) and used to generate a baculovirus in Sf9 insect cells (Invitrogen). Hi5 insect cells (Invitrogen) were used for ZGRF1 expression. All the purification steps were conducted at 0–4°C. Briefly, a cell pellet (8 g, from ~600 mL culture) was suspended in 100 mL of K buffer (20 mM KH2PO4, pH 7.4, 10% glycerol, 0.5 μM EDTA, 0.01% Igepal, and 1 mM DTT) containing a cocktail of protease inhibitors (aprotinin, chymostatin, leupeptin, and pepstatin A at 5 mg/mL each, and 1 mM phenyl-methylsulfonyl fluoride) and 500 mM KCl. Cells were disrupted by sonication, and the crude lysate was subject to ultracentrifugation (100,000 g for 90 min). The clarified lysate was incubated with 2 mL Glutathione Sepharose 4B resin (GE Healthcare) for 1.5 h. After washing the resin three times with 50 mL K buffer containing 500 mM KCl, ZGRF1 was eluted with K buffer with 500 mM KCl and 15 mM reduced glutathione. The elution was incubated with 0.5 mL Ni-NTA resin (QIAGEN) for 2.5 h. After washing the resin three times with 50 mL K buffer containing 500 mM KCl, once with 25 mL K buffer containing 500 mM KCl and 15 mM imidazole, ZGRF1 was eluted with three aliquots of 0.6 mL K buffer containing 500 mM KCl and 200 mM imidazole. The eluate was dialyzed for 12 h against K buffer containing 500 mM KCl. Purified ZGRF1 was stored at −80°C in 5 μL aliquots. The yield of highly purified protein was ~100 μg. Expression and purification of ZGRF1-K1660 followed the same procedures, and with a similar overall yield of highly purified protein.
Expression and purification of other proteins
FANCM, FANCM-K117R, BLM, RPA, PCNA, RAD51, RAD54, Mph1, yRad51, yRad54, human and yeast Polδ and human Polε were purified to near homogeneity using previously described or similar procedures (Sigurdsson et al., 2002; Singh et al., 2010; Van Komen et al., 2002; Wilson et al., 2013; Xue et al., 2013, 2016).
ATPase assay
ZGRF1 or ZGRF1-K1660A (6 nM) was incubated with or without ϕX174 viral (+) strand, ϕX174 linear dsDNA, ϕX174 RF1 (30 μM nucleotides or base pairs) in 10 μL buffer (30 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 1 mM DTT, 100 μg/mL BSA, 50 mM KCl) containing 0.15 mM ATP and 25 nCi [γ−32P]-ATP at 37°C for 10 or 20 min. The reaction was terminated by adding an equal volume of 0.5 M EDTA, followed by thin layer chromatography and phosphorimaging analysis (Xue et al., 2014).
DNA unwinding assay
DNA substrates with a 45nt 5′ or 3′ ssDNA overhang and a 15bp duplex region were prepared by annealing the corresponding oligonucleotides (with one of the oligonucleotides being labeled with 32P) listed in Table S3. In the unwinding reaction, ZGRF1 or ZGRF1-K1660A (2.5, 5.0, 10, 20, 40 nM) was incubated with 10 nM substrate in reaction buffer (35 mM Tris-Cl, pH 7.5, 1 mM DTT, 3 mM ATP, 2 mM MgCl2, 60 mM KCl, and 100 nM of “trap” DNA (the unlabeled form of the oligonucleotide that harbors the 32P label in the substrate) at 30°C for 5 min. Reaction mixtures were deproteinized by treatment with SDS (0.1%) and proteinase K (0.5 mg/mL) for 10 min at 30°C and then resolved in a 15% polyacrylamide gel in TAE buffer (40 mM Tris, 20 mM acetate acid and 1 mM EDTA) at 4°C. Gels were dried onto Whatman DE81 paper (Whatman International Limited) and analyzed in a Personal Molecular Imager FX PhosphorImager (Bio-Rad).
Human RAD51-mediated D-loop formation assay
The D-loop formation assay (Figures 5H and 5I) mediated by human RAD51 and RAD54 was modified as described (Raynard and Sung, 2009). Briefly, human RAD51 (216 nM) was first incubated with 32P-labeled ssDNA (90-mer, 750 nM nucleotides) at 37°C for 10 min, in a buffer containing 35 mM Tris-HCl, pH 7.5, 2 mM ATP, 100 μg/mL BSA, 1 mM DTT, 20 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 15 mM phosphocreatine and 30 units/mL of creatine phosphokinase, to assemble the RAD51-ssDNA presynaptic filament. RAD54 (90 nM) was then added to the reaction and incubated at room temperature for 2 min. Finally, ZGRF1, or ZGRF1-K1660A (40 nM), or buffer control was added to the reaction followed by addition of pBluescript supercoiled DNA (48 μM base pairs). The reaction was then carried out at 30°C for 5, 10 and 20 min, deproteinized and reaction products were resolved on a 0.9% agarose gel in TBE buffer (45 mM Tris-borate, 1 mM EDTA, pH 8.0) at room temperature. Gels were dried and subject to phosphorimaging analysis. The experiment examining the effect of ZGRF1 on yeast Rad51-Rad54-mediated D-loop formation was carried out in a similar way except the reaction time at 30°C for the final step was 4, 8, 12 min (Figures S5D and S5E).
Holliday junction branch migration assay
The movable Holliday junction (MHJ) substrate was prepared as described (Xue et al., 2014). The indicated concentration of ZGRF1 or ZGRF1-K1660A was incubated with the MHJ (5 nM) in reaction buffer (25 mM Tris–HCl, pH 7.5, 1 mM DTT, 100 μg/mL BSA, 50 mM KCl, 1 mM MgCl2, 2 mM ATP, 15 mM phosphocreatine and 30 units/mL of creatine phosphokinase). The reaction was incubated at 37°C and terminated at the indicated times by treatment with SDS (0.5% final) and proteinase K (0.5 mg/mL) for 5 min at 37°C. Reaction mixtures were resolved in an 8% polyacrylamide gel in TBE buffer (45 mM Tris-borate, 1 mM EDTA, pH 8.0) at 4°C. Gels were dried and subject to phosphorimaging analysis.
D-loop dissociation assay
D-loop substrates with either a 5′ or 3′ invading strand were made by hybridizing oligonucleotides listed in Table S3. The top, unpaired ssDNA region in these substrates bears no homology to the bottom DNA strand that is hybridized to a complementary oligo-nucleotide. These substrates (5 nM) were incubated with ZGRF1 or ZGRF1-K1660A (10 and 20 nM) in reaction buffer (25 mM Tris-Cl, pH 7.5, 1 mM DTT, 2 mM ATP, 2 mM MgCl2, 50 mM KCl) at 37°C for 10 min. Reaction mixtures were deproteinized before being resolved in 7% polyacrylamide gel in TAE buffer at 4°C and analyzed, as above.
The Rad51-mediated D-loop reaction (250 μL) was performed with a 32P-labeled 90-mer oligonucleotide as described (Xue et al., 2016). The reaction was deproteinized with SDS and proteinase K. After an extraction with phenol-chloroform-isoamyl alcohol (25:24:1), the buffer was exchanged with buffer H (35 mM Tris-HCl pH 7.5, 1 mM DTT, 9.3 mM MgCl2, and 30 mM KCl) using a Zeba Spin-desalting Column (Thermo Scientific). The deproteinized Rad51-made D-loops (~2.2 nM) were incubated with ZGRF1 (5–30 nM), ZGRF1-K1660A (20 nM), or FANCM (10–40 nM) in reaction buffer (25 mM Tris-Cl, pH 7.5, 1 mM DTT, 2 mM ATP, 2 mM MgCl2, 50 mM KCl) at 37°C for 10 min. Reaction mixtures were deproteinized before being resolved in 0.9% agarose gels in TBE buffer at 4°C and analyzed, as above.
The 5′ DNA flap substrate that resembles a branch migratable D-loop structure was constructed as described (Schwab et al., 2015) using the oligonucleotides (XX1, XX2, D5′F; Table S3). ZGRF1, ZGRF1-K1660A (2 nM), FANCM or FANCM-K117R (4 nM) was incubated with the substrate (5 nM) in reaction buffer (25 mM Tris-Cl, pH 7.5, 1 mM DTT, 1 mM ATP, 2 mM MgCl2, 50 mM KCl) at 30°C for the indicated time. Reaction mixtures were deproteinized before being resolved in 7% polyacrylamide gels in TAE buffer at 4°C and analyzed, as above.
Proximity ligation assay
Sterile coverslips were placed into the wells of a 24-well plate and 40.000 cells were seeded per well in fresh media. The following day, media was replaced with either fresh media or media containing 10 ng/mL MMC. After 24 h treatment, coverslips were washed twice with PBS and fixed in 3% paraformaldehyde in PBS (adjusted to pH 7.4) for 15 min at room temperature. Cells were washed twice with PBS for 3 min, permeabilized by the addition of 0.1% Triton X-100 in 20 mM glycine for 10 min and once again washed twice with PBS.
Blocking, overnight primary antibody incubation, PLA probe incubation, probe ligation, probe amplification, final wash and slide mounting were all performed using reagents and protocol as recommended by the manufacturer (Duolink PLA Technology, Merck) with reagent volumes scaled up for 24-well plates. Primary antibodies used were 1:2000 mouse anti-GFP (Roche Diagnostics, cat. no. 11814460001) and 1:2000 rabbit anti-RAD51 (Bio Academia, cat. no. 70–001). PLA probes used were Duolink In Situ PLA Probe Anti-Rabbit PLUS (Sigma-Aldrich, cat. no. DUO92002) and Duolink In Situ PLA Probe Anti-Mouse MINUS (Sigma-Aldrich, cat. no. DUO92004). Ligation and amplification were performed using the Duolink In Situ Detection Reagents Orange (Sigma-Aldrich, cat. no. DUO92007).
Fluorescence was detected and images were acquired as described under ‘Fluorescence microscopy and image analysis’. Images were taken at room temperature with 10 optical sections separated by 0.6 μm and the same laser intensity and exposure time was used for all images in the same channel. Amplified PLA product was visualized in the TRITC channel. Processing and quantitative measurements of fluorescence intensities were performed with Volocity software (PerkinElmer). Only foci that overlapped the DAPI staining of each nucleus were counted.
Co-affinity precipitation of ZGRF1 interactors
GST-tagged ZGRF1 or GST alone (0.3 μg) was incubated with human or budding yeast RAD51 (0.5 mg, Figure 5E), or human or budding yeast Polδ (0.5 μg, Figure S5A), or human Flag-FANCM (0.25 μg, Figure S5B), or human Polε or PCNA, RPA (0.5 μg each, Figure S5C) in 30 μL K buffer (20 mM KH2PO4, pH 7.4, 10% glycerol, 0.5 mM EDTA, 0.01% Igepal, and 1 mM DTT) with 80 mM KCl for 30 min at 4°C. The reaction mixture was incubated with 10 μL of Glutathione Sepharose 4B resin (GE Healthcare Life Sciences) for 30 min at 4°C. After washing the resin three times with 200 μL of K buffer with 80 mM KCl, bound proteins were eluted with 20 μL of 2% SDS. Twenty percent of the supernatant (S) and SDS elution (E) fractions, and 2% of the wash (W) fraction were analyzed by 4%–20% gradient SDS-PAGE, transferred to Nitrocellulose membrane (BioRad). Blots were probed with the following antibodies: GST-ZGRF1 and GST (NEB, E2624S), human RAD51 (Santa Cruz Biotech, sc-8349), yeast Rad51 (lab raised antibody), human Myc-Polδ3 subunit (Thermo Fisher Scientific, R951–25), yeast Flag-Polδ3 subunit (Sigma, A8592), FANCM-Flag (Sigma, A8592), human HA-Polε1 subunit (Roche, 12013819001), human PCNA (lab raised antibody), human RPA70 subunit (Abcam, ab79398). If needed, the blots were incubated with HRP-conjugated secondary antibodies (Pierce 31450 for rabbit anti-mouse IgG-HRP; Sigma A6154 for goat anti-rabbit IgG-HRP; Santa Cruz Biotech Sc-2032 for goat anti-rat IgG-HRP) before visualization of protein signals using the ECL kit (Thermo Scientific Pierce).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were completed using Prism 8 (GraphPad). The applied statistical tests and number of biological replicates are indicated in the figure legends. No statistical methods or criteria were used to estimate sample size or to include/exclude samples. Multiple clones and multiple cell lines were analyzed to confirm results were not caused by clonal variations. Unless otherwise stated, all experiments were performed at least twice and representative experiments are shown.
Supplementary Material
Highlights.
The human ZGRF1 gene encodes a 5′-to-3′ DNA helicase
ZGRF1 null cells are sensitive to mitomycin C and camptothecin
ZGRF1 promotes homologous recombination and sister chromatid exchange
ZGRF1 physically interacts with RAD51 and stimulates strand exchange by RAD51-RAD54
ACKNOWLEDGMENTS
This work was supported by the Danish Council for Independent Research (FSS) and the Villum Foundation to A.K.W.S. and M.L.; the Danish National Research Foundation (DNRF115) to A.B., H.W.M., M.L., and I.D.H.; the Dagmar Marshall Foundation to A.B.; and the Novo Nordisk Foundation to K.S.V. and M.L. P.S. and X.X. were supported by National Institutes of Health grants R21 ES029513 and R21 ES028792, and P.S. also received support from grant R35 CA241801. We would like to thank Martin Cohn and Jakob Nilsson for sharing reagents and Julien Duxin for comments on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107849.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, and Elledge SJ (2012). A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol 14, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, and West SC (2006). The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443, 713–716. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Lodygin D, and Buerstedde JM (2001). Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattain MG, Fine WD, Khaled FM, Thompson J, and Brattain DE (1981). Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res 41, 1751–1756. [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- Chan YW, Fugger K, and West SC (2018). Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol 20, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bosques L, Sung P, and Kupfer GM (2016). A novel role for nonubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 35, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, and West SC (2009). FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 36, 943–953. [DOI] [PubMed] [Google Scholar]
- Gao C, Wang N, Guo X, Ziegler JT, Taylor KD, Xiang AH, Hai Y, Kridel SJ, Nadler JL, Kandeel F, et al. (2015). A comprehensive analysis of common and rare variants to identify adiposity loci in Hispanic Americans: the IRAS Family Study (IRASFS). PLoS ONE 10, e0134649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, and D’Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 7, 249–262. [DOI] [PubMed] [Google Scholar]
- Gari K, Décaillet C, Delannoy M, Wu L, and Constantinou A (2008). Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. U S A 105, 16107–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Quinzii CM, Mitsumoto H, Hays AP, Roberts JK, Richard P, and Rowland LP (2011). Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler 12, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, and Chiu CP (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet 21, 111–114. [DOI] [PubMed] [Google Scholar]
- Kim Y, Spitz GS, Veturi U, Lach FP, Auerbach AD, and Smogorzewska A (2013). Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, and Walter JC (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque JR, and Jasin M (2010). Mechanisms of recombination between diverged sequences in wild-type and BLM-deficient mouse and human cells. Mol. Cell. Biol 30, 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Martinez D, Liang CC, and Cohn MA (2016). Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell. Mol. Life Sci 73, 3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina OM, and Mazin AV (2004). Human Rad54 protein stimulates DNA strand exchange activity of hRad51 protein in the presence of Ca2+. J. Biol. Chem 279, 52042–52051. [DOI] [PubMed] [Google Scholar]
- Mitchel K, Lehner K, and Jinks-Robertson S (2013). Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9, e1003340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motnenko A, Liang CC, Yang D, Lopez-Martinez D, Yoshikawa Y, Zhan B, Ward KE, Tian J, Haas W, Spingardi P, et al. (2018). Identification of UHRF2 as a novel DNA interstrand crosslink sensor protein. PLoS Genet 14, e1007643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Cong K, Panzarino NJ, Nayak S, Calvo J, Deng B, Zhu LJ, Morocz M, Hegedus L, Haracska L, and Cantor SB (2018). Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep 24, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B, Wijsman EM, Nato AQ Jr., Matsushita MM, Chapman KL, Stanaway IB, Wolff J, Oda K, Gabo VB, and Raskind WH; University of Washington Center for Mendelian Genomics (2016). Genetic candidate variants in two multigenerational families with childhood apraxia of speech. PLoS ONE 11, e0153864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, and Jasin M (2001). Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 15, 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontén J, and Saksela E (1967). Two established in vitro cell lines from human mesenchymal tumours. Int. J. Cancer 2, 434–447. [DOI] [PubMed] [Google Scholar]
- Pozarowski P, and Darzynkiewicz Z (2004). Analysis of cell cycle by flow cytometry. Methods Mol. Biol 281, 301–311. [DOI] [PubMed] [Google Scholar]
- Prakash R, Krejci L, Van Komen S, Anke Schürer K, Kramer W, and Sung P (2005). Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3′ to 5′ DNA helicase. J. Biol. Chem 280, 7854–7860. [DOI] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, and Ira G (2009). Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynard S, and Sung P (2009). Assay for human Rad51-mediated DNA displacement loop formation. Cold Spring Harb. Protoc 2009, pdb.prot5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Moynahan ME, and Jasin M (1998). Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12, 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, and Jackson SP (2007). Human CtIP promotes DNA end resection. Nature 450, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 60, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short JM, Fernandez JM, Sorge JA, and Huse WD (1988). Lambda ZAP: a bacteriophage lambda expression vector with in vivo excision properties. Nucleic Acids Res 16, 7583–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Van Komen S, Petukhova G, and Sung P (2002). Homologous DNA pairing by human recombination factors Rad51 and Rad54. J. Biol. Chem 277, 42790–42794. [DOI] [PubMed] [Google Scholar]
- Silva S, Altmannova V, Luke-Glaser S, Henriksen P, Gallina I, Yang X, Choudhary C, Luke B, Krejci L, and Lisby M (2016). Mte1 interacts with Mph1 and promotes crossover recombination and telomere maintenance. Genes Dev 30, 700–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LJ, and Sale JE (2006). Sister chromatid exchange assay In Sub-cellular Biochemistry, Buerstedde J-M and Takeda S, eds. (New York: Springer; ), pp. 399–403. [DOI] [PubMed] [Google Scholar]
- Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, Sachpatzidis A, Wahengbam K, Pierce AJ, Xiong Y, et al. (2010). MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol. Cell 37, 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP, and Elledge SJ (2010). A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, and Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Symington LS (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, et al. (2002). Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet 32, 267–272. [DOI] [PubMed] [Google Scholar]
- Uchiumi F, Seki M, and Furuichi Y (2015). Helicases and human diseases. Front. Genet 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umate P, Tuteja N, and Tuteja R (2011). Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol 4, 118–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Komen S, Petukhova G, Sigurdsson S, and Sung P (2002). Functional cross-talk among Rad51, Rad54, and replication protein A in heteroduplex DNA joint formation. J. Biol. Chem 277, 43578–43587. [DOI] [PubMed] [Google Scholar]
- van Sluis M, and McStay B (2015). A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev 29, 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, and Chen J (2001). Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem 276, 47759–47762. [DOI] [PubMed] [Google Scholar]
- Williams MR, DeSpenza T Jr., Li M, Gulledge AT, and Luikart BW (2015). Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J. Neurosci 35, 943–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, and Ira G (2013). Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ning S, Wei Z, Xu R, Xu X, Xing M, Guo R, and Xu D (2017). 53BP1 and BRCA1 control pathway choice for stalled replication restart. eLife 6, e30523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Raynard S, Busygina V, Singh AK, and Sung P (2013). Role of replication protein A in double Holliday junction dissolution mediated by the BLM-Topo IIIα-RMI1-RMI2 protein complex. J. Biol. Chem 288, 14221–14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Choi K, Bonner J, Chiba T, Kwon Y, Xu Y, Sanchez H, Wyman C, Niu H, Zhao X, and Sung P (2014). Restriction of replication fork regression activities by a conserved SMC complex. Mol. Cell 56, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Papusha A, Choi K, Bonner JN, Kumar S, Niu H, Kaur H, Zheng XF, Donnianni RA, Lu L, et al. (2016). Differential regulation of the anti-crossover and replication fork regression activities of Mph1 by Mte1. Genes Dev 30, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Steentoft C, Hauge C, Hansen L, Thomsen AL, Niola F, Vester-Christensen MB, Frödin M, Clausen H, Wandall HH, and Bennett EP (2015). Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res 43, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimit A, Kim T, Anand RP, Meister S, Ou J, Haber JE, Zhang Z, and Brown GW (2016). MTE1 functions with MPH1 in double-strand break repair. Genetics 203, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, and Chen J (2003). The BRCT domain is a phospho-protein binding domain. Science 302, 639–642. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ren JY, Zhang JM, Suo F, Fang XF, Wu F, and Du LL (2013). A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair (Amst.) 12, 433–443. [DOI] [PubMed] [Google Scholar]
- Zapotoczny G, and Sekelsky J (2017). Human cell assays for synthesis-dependent strand annealing and crossing over during double-strand break repair. G3 (Bethesda) 7, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, and Walter JC (2015). DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol 22, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wiese C, Kwon Y, Hromas R, and Sung P (2019). The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu. Rev. Biochem 88, 221–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XF, Prakash R, Saro D, Longerich S, Niu H, and Sung P (2011). Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst.) 10, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Ž, Muir M, Fluteau A, Aregger M, McEwan A, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not apply code. Original/source data for Figures 1B, 2A–2E, 3A–3D, 4C–4E, 5C–5G, 5I, 5J, 6A–6E, S2C, S3C, S4A–S4C, S4E, S4F, S5A–S5C, S5E, and S6A–S6C are available through Mendeley (https://data.mendeley.com/datasets/d532xvdrn2/draft?a=cdd41557-9d54-48fc-bc37-1e63b2be48bb).