

Abstract
Peroxynitrite (PNT) is a highly reactive oxidant that plays a key role in the destruction of foreign pathogens by specific phagocytic immune cells such as macrophages. However, when its production is dysregulated, this oxidant can contribute to cardiovascular disease, neurological diseases, and cancer. To facilitate the detection of PNT in living cells, we designed and synthesized a fluorescent sensor termed PS3 that accumulates in membranes of the endoplasmic reticulum (ER). This subcellular targeting enhances the proximity of PS3 to the phagosome of macrophages where PNT is generated. When PS3-treated macrophages are stimulated with 10 μm opsonized tentagel microspheres, antibody-dependent cellular phagocytosis (ADCP) of these particles results in production of endogenous PNT, oxidative cleavage of the fluorescence-quenching phenolic side chain of PS3, and increased fluorescence that can be detected by confocal laser scanning microscopy, flow cytometry, and other assays. We describe methods for the synthesis of PS3 and evaluation of its photophysical properties, selectivity, and reactivity. We further report differential production of PNT during ADCP by the phagocytic cell lines RAW 264.7, J774A.1, and THP-1, as detected by confocal microscopy and changes in fluorescence intensity on 96-well plates. This approach may be useful for identification of modulators of PNT and related studies of ADCP.
1. Introduction
1.1. Fluorescent sensors of cellular peroxynitrite (PNT)
Biochemical processes generate a wide variety of reactive small molecules. These include reactive oxygen and nitrogen species (ROS and RNS) that play complex roles in biological systems. Some of these species, such as RNS peroxynitrite (ONO2−, PNT), are biosynthesized by specific cell types through tightly regulated pathways. An example includes host defense involving antibody-dependent cellular phagocytosis (ADCP). In this process, some phagocytic cells such as macrophages engulf foreign cells or particles, trigger the formation of PNT in the phagosome, and harness the reactivity of PNT to destroy pathogens. This RNS can be thought of as a naturally occurring binary chemical weapon formed by the rapid reaction of nitric oxide (•NO), produced by nitric oxide synthases, with superoxide (O2•−), generated by NADPH oxidases, among other pathways. In cells, PNT is so reactive that its half-life is only about 10 milliseconds (Bartesaghi & Radi, 2018; Szabo, Ischiropoulos, & Radi, 2007) During this short time, PNT can diffuse across cell membranes in its even more reactive protonated form (ONO2H, pKa=6.8), but it can generally only travel up to about 20μm before participating in a chemical reaction. In vivo, a predominant and rapid initial reaction is with CO2, present at a concentration of about 1mM, to form nitrosoperoxycarbonate (ONO2CO2−). However, reaction with thiol and selenol peroxidases, some transition metal centers, homolysis into hydroxyl radical (HO•) and nitrogen dioxide (•NO2), and oxidation of a wide variety of substrates can also occur (Bartesaghi & Radi, 2018; Ferrer-Sueta et al., 2018). Substrates directly oxidized by PNT in cells include protein cysteine, methionine, and tryptophan residues, lipids, and nucleic acids. PNT does not directly react with tyrosine, but its reaction with CO2, some transition metals, and proton-catalyzed homolysis, leads to other species that oxidize tyrosine to tyrosine radicals. These radicals react with •NO2 to form 3-nitrotyrosine (Alvarez & Radi, 2003; Ferrer-Sueta et al., 2018; Ferrer-Sueta & Radi, 2009; Pacher, Beckman, & Liaudet, 2007). This modification can alter protein function, kill pathogens, contribute to inflammation, and is linked to pathology of cardiovascular disease, neurode-generative diseases, and cancer (Beckman, Beckman, Chen, Marshall, & Freeman, 1990; Ferrer-Sueta & Radi, 2009; Pacher et al., 2007; Prolo, Alvarez, & Radi, 2014; Radi, Beckman, Bush, & Freeman, 1991; Szabo et al., 2007). Because PNT is associated with both normal biology and human pathology, methods for its detection, analysis, and modulation in biological systems are of substantial interest.
The direct detection of endogenous PNT generated by cells is challenging due to its fleeting nature. Consequently, some of the first methods for its detection were indirect, such as analysis of nitrotyrosine residues of modified proteins (Ferrer-Sueta et al., 2018; Ischiropoulosd et al., 1995; Wardman, 2007), or via low-throughput electrochemical approaches (Vasilescu, Gheorghiu, & Peteu, 2017). More recent methods are predominantly based on the development of fluorescent sensors compatible with living cells (Chan, Dodani, & Chang, 2012; Chen, Ren, Wright, & Ai, 2013; Fernandez & Vendrell, 2016; Prolo, Rios, Piacenza,Álvarez, & Radi, 2018). One such strategy utilizes small pro-fluorophores that react with peroxynitrite or its secondary oxidants to generate fluorescent products. Sensors of this type can become fluorescent upon nitration of phenols (Ueno, Urano, Kojima, & Nagano, 2006), oxidation of chalcogenides (e.g., Se or Te containing compounds) (Wang, Yu, Li, Sun, & Han, 2013; Yu, Li, Wang, & Han, 2013) oxidation of boronates or boronic acids (Murfin et al., 2019; Prolo et al., 2015; Rios et al., 2016; Weber, Mackenzie, Bull, & James, 2018; Yu, Song, Li, Wang, & Han, 2012; Zielonka et al., 2012; Zielonka, Sikora, Joseph, & Kalyanaraman, 2010), and through a variety of other oxidative cleavage and related reactions (Knewtson, Rane, & Peterson, 2018; Peng et al., 2014; Zhou, Kwon, Kim, Ryu, & Yoon, 2015). Reaction-based luminescent probes of PNT and related species have been recently reviewed (Bezner, Ryan, & Lippert, 2020). In many cases, high selectivity for PNT can be achieved in the presence of other reactive species such as nitric oxide, superoxide, hydroxy radical, hypochorite, and peroxides. However, for some sensors, high selectivity requires inclusion of DMSO as a scavenger of hydroxyl radicals or hypochlorite. Despite this progress, it remains challenging to achieve the combination of high cellular permeability, favorable selectivity and reactivity profiles, and low cytotoxicity needed to detect endogenous peroxynitrite produced during physiological processes such as antibody-dependent cellular phagocytosis (Gul & van Egmond, 2015; Hendriks, Choi, de Bruyn, Wiersma, & Bremer, 2017; Kamen et al., 2019). Although a wide variety of fluorescent sensors of PNT have been described, to our knowledge the only highly selective commercially available sensor is NiSPY-3 (Ueno et al., 2006). Hydroxyphenylfluorescein (HPF) (Setsukinai, Urano, Kakinuma, Majima, & Nagano, 2003) is sold as a sensor of PNT, but this compound additionally reacts with hydroxyl radical, complicating its use for studies of PNT (Price, Reiners, Santiago, & Kessel, 2009; Setsukinai et al., 2003). The structures of these compounds, and two other more recently developed oxidatively-cleavable fluorescent sensors of peroxynitrite (HKGreen3A and HKGreen4A), are shown in Fig. 1.
Fig. 1.
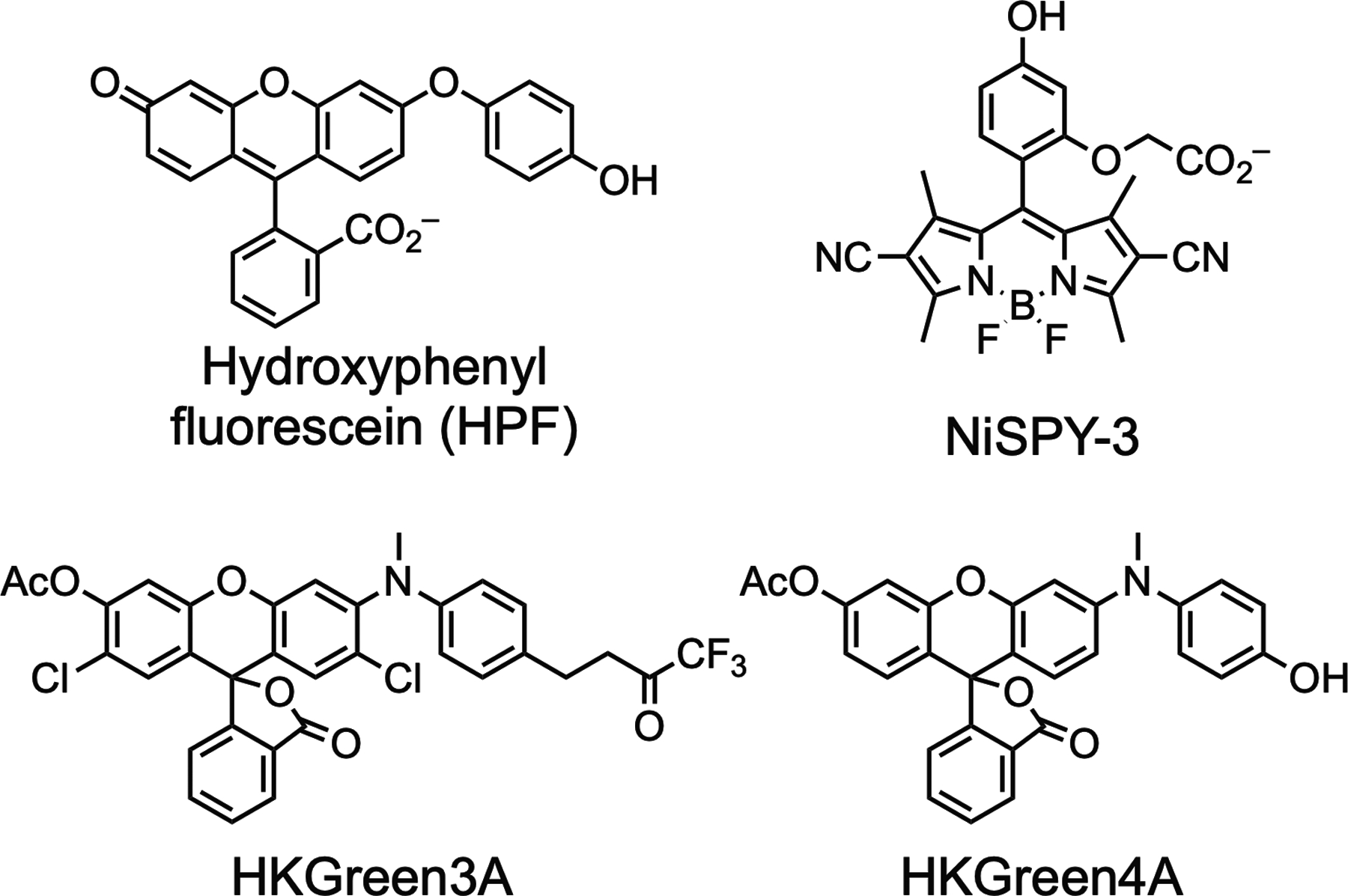
Structures of the sensors HPF, NiSPY-3, HKGreen3A, and HKGreen4A. Oxidation results in cleavage of the phenol side chains of HPF and HKGreen4A, or the trifluoromethylketone side chain of HKGreen3A, to provide a fluorescent product. Nitration of the phenol of NiSPY-3, indirectly mediated by PNT, generates a fluorescent product.
1.2. Subcellular targeting of sensors for detection of endogenous peroxynitrite
Targeting of small molecules to specific subcellular compartments is a promising but relatively unexplored strategy for enhancing specificity and biological activity (Rajendran, Knolker, & Simons, 2010). This approach has been used to design fluorescent sensors of PNT targeted to mitochondria ( Jia et al., 2016; Zhang et al., 2015; Zhu et al., 2018) and lysosomes (Guo et al., 2018). As an alternative subcellular targeting approach, we previously reported the synthesis of two fluorescent chemotypes, fluorinated hydrophobic rhodols (Meinig, Fu, & Peterson, 2015) and N-alkylresorufamines (Phaniraj, Gao, Rane, & Peterson, 2016), that preferentially localize in the endoplasmic reticulum (ER) in living cells. This critical organelle is involved in many biological processes, including the biosynthesis and post-translational modification of secreted and cell surface proteins, and plays important roles in human disease (Wang & Kaufman, 2016). The ER is defined by a complex series of dense tubular membranes that have an extensive surface area approximately 30 times larger than the cellular plasma membrane (Nixon-Abell et al., 2016). Based on their high hydrophobicity, and our studies of structure-activity relationships, we predict that fluorinated hydrophobic rhodols and related compounds preferentially insert into membranes of the ER, but the molecular basis for this selectivity remains to be elucidated. A wide variety of other ER-targeted fluorophores (Colston, Horobin, Rashid-Doubell, Pediani, & Johal, 2003; Fujisawa, Tamura, Yasueda, Kuwata, & Hamachi, 2018; Li et al., 2018; Lin, Buccella, & Lippard, 2013; McDonald et al., 2016), including derivatives of glibenclamide that associate with this organelle by binding ER-localized sulfonylurea receptors (Smith, Taneja, Mankouri, & Sivaprasadarao, 2007), have also been described. Other ER-targeted fluorescent sensors of hydrogen sulfide (Zhang et al., 2019), changes in pH (Ghule et al., 2015), hydrogen peroxide (Gao, Tian, Zhang, Jing, & Zhang, 2017), and temperature have been previously reported (Arai, Lee, Zhai, Suzuki, & Chang, 2014).
To detect PNT during ADCP by innate immune cells, we investigated targeting of fluorescent sensors to the extensive membranes of the ER. Using a fluorinated hydrophobic rhodol as an ER-targeting motif, and incorporating a fluorescence-quenching oxidatively-cleavable phenol side chain similar to other sensors of PNT such as HPF (Setsukinai et al., 2003), HKGreen4 (Peng et al., 2014), and HKYellow (Peng et al., 2016), we designed and optimized peroxynitrite sensor 3 (PS3) (Knewtson et al., 2018). As shown in Fig. 2, this sensor uses a 2,6-dimethyl phenol as a cleavable side chain that extensively quenches its fluorescence, increases its association with ER membranes, and enhances its reactivity with PNT. Reaction with this oxidant yields a highly fluorescent rhodol (1) and 2,6-dimethylbenzoquinone. We previously reported (Knewtson et al., 2018) the use of this approach to detect PNT upon phagocytosis of opsonized (antibody-bound) tentagel microspheres (beads) by the mouse macrophage cell line RAW 264.7. This remarkably mild method for activating production of PNT does not require additional stimulation with agents such as lipopolysaccharide, but the polar polyethylene glycol that is grafted to the polystyrene matrix of tentagel beads may enhance immunostimulatory responses compared with other types of opsonized beads.
Fig. 2.
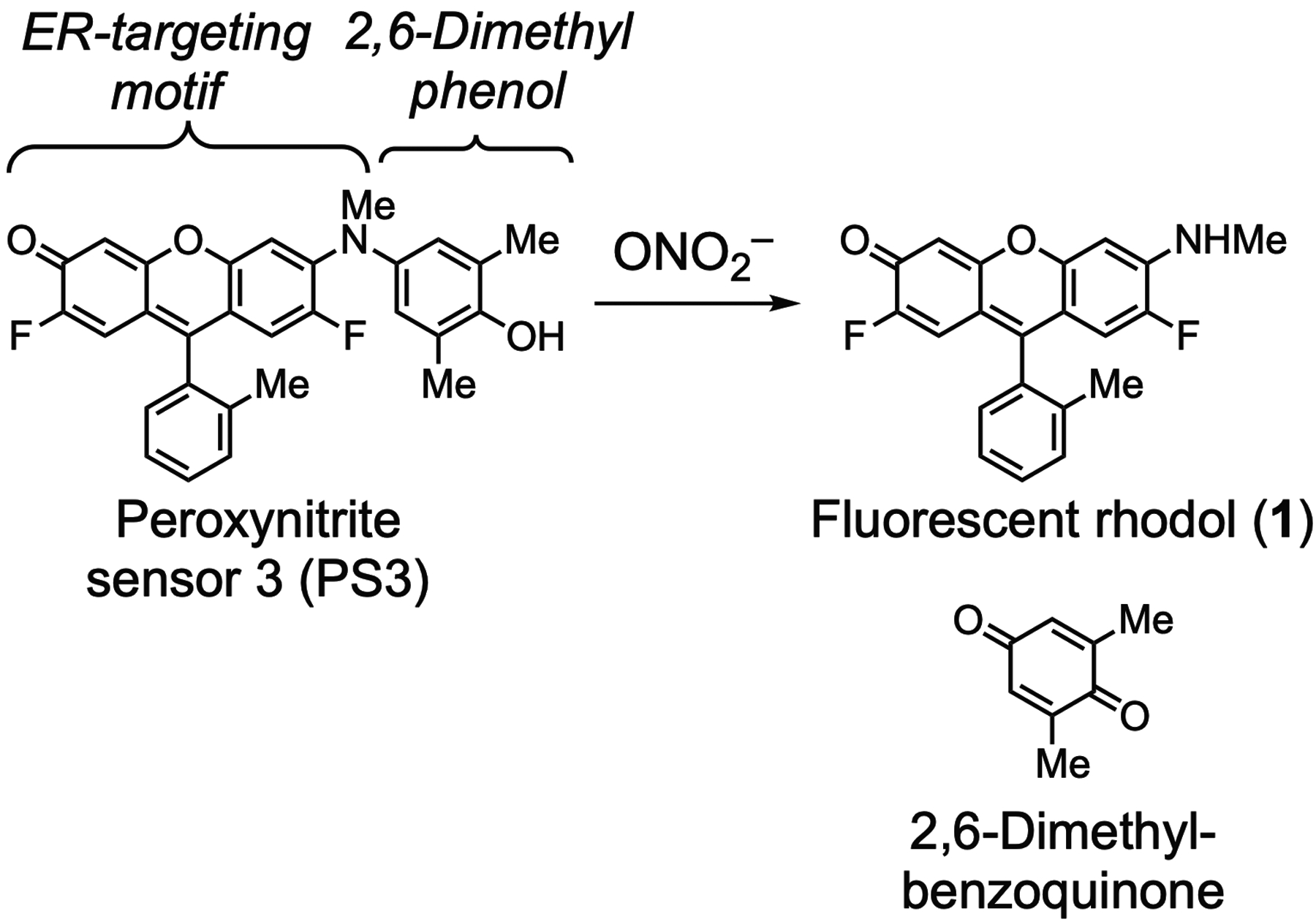
Structures of peroxynitrite sensor 3 (PS3) and products of reaction with peroxynitrite.
To trigger ADCP, we used the bead-based assay illustrated in Fig. 3. In this assay, monosized aminotentagel microspheres (10 μm diameter) provided chemically well-defined particles that can be readily modified and analyzed by confocal microscopy and flow cytometry. These microspheres were derivatized with amine-reactive N-succinimidyl N-(2,4-dinitrophenyl)-6-aminocaproate (DNP-NHS) to allow opsonization with commercially available rabbit anti-DNP polyclonal antibodies. Addition of these antibody-bound beads to the mouse RAW 264.7 macrophage cell line results in recognition of the beads by Fc receptors on cell surfaces, phagocytosis, and production of PNT in the phagosome. By accumulating in the proximal membranes of the endoplasmic reticulum, PS3 achieves a high effective concentration that allows it to readily react with this oxidant to form a fluorescent rhodol product (1). Both PS3 and the fluorescent rhodol 1 can be observed by confocal laser scanning microscopy to predominantly colocalize in the ER with the probe ER tracker blue white DPX (Knewtson et al., 2018). However, this fluorescent rhodol also rapidly exchanges between cells, and it distributes throughout cell culture media, offering another approach for detection by changes in the fluorescence intensity of media. In this chapter, we present methods for the synthesis of PS3 and immunostimulatory tentagel beads for analysis of production of PNT during ADCP. Comparison of PS3 with the commercially available PNT sensors HPF (Setsukinai et al., 2003) and NiSPY-3 (Ueno et al., 2006), by confocal laser scanning microscopy revealed that only PS3 was sufficiently sensitive to detect PNT during ADCP under the conditions investigated here. We further show that PNT can be detected by PS3 during ADCP using a simple high-throughput method of analysis of changes in the fluorescence intensity of media seeded with cells and cultured on 96-well plates.
Fig. 3.
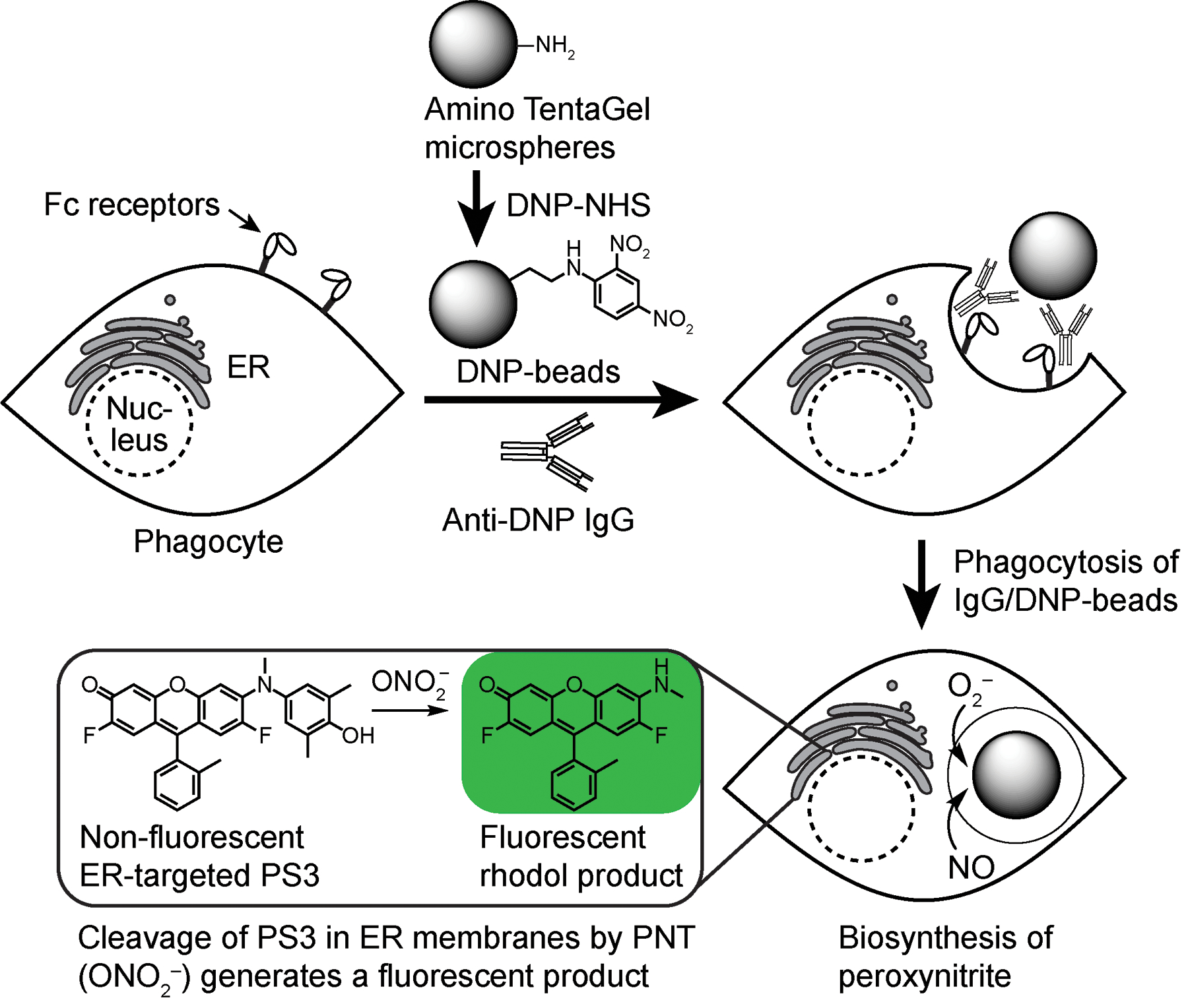
Assay for detection of endogenous PNT triggered by ADCP. Phagocytosis of opsonized tentagel beads results in biosynthesis of PNT and conversion of non-fluorescent PS3 into a fluorescent rhodol (1). This fluorescent product remains predominantly associated with membranes of the ER, but it also rapidly exchanges between cells in a population, and it additionally diffuses into cell culture media to facilitate detection.
2. Synthesis of peroxynitrite sensor 3 (PS3)
We previously reported the synthesis of the fluorophore Pennsylvania Green (Mottram, Boonyarattanakalin, Kovel, & Peterson, 2006). This analogue of fluorescein was designed as a hybrid of the related fluorophores Oregon Green (Sun, Gee, Klaubert, & Haugland, 1997) and Tokyo Green (Urano et al., 2005). The spectral properties of Pennsylvania Green (Ex. 494nm; Em. 514nm; QY=0.91; ε494nm,pH 7.4 =82,000M−1cm−1) are similar to fluorescein, but the fluorine substituents increase the acidity of the phenol (pKa=4.8), enhancing its fluorescence in acidic cellular environments (Mottram et al., 2006). Additionally, by replacing the carboxylate of fluorescein with a methyl group, the hydrophobicity of Pennsylvania Green is substantially increased, facilitating passive diffusion across cellular membranes (Mottram, Maddox, Schwab, Beaufils, & Peterson, 2007). As shown in Figs. 4 and 5, Pennsylvania Green and 2,6-dimethyl-4-nitrophenol (2) were used as building blocks to synthesize PS3. The nitrophenol 2 was protected with a MOM group to afford 3, which was reduced with H2 and catalytic Pd/C to yield aniline 4. Reductive amination of 4 with paraformaldehyde and sodium borohydride yielded the N-methyl aniline 5. Buchwald-Hartwig amination of 5 with the triflate (Tf ) derivative of Pennsylvania Green (6, Fig. 5) provided the methoxymethyl ether (MOM)-protected derivative 7. Deprotection with trifluoroacetic acid (TFA) afforded PS3 as a light-sensitive magenta solid.
Fig. 4.
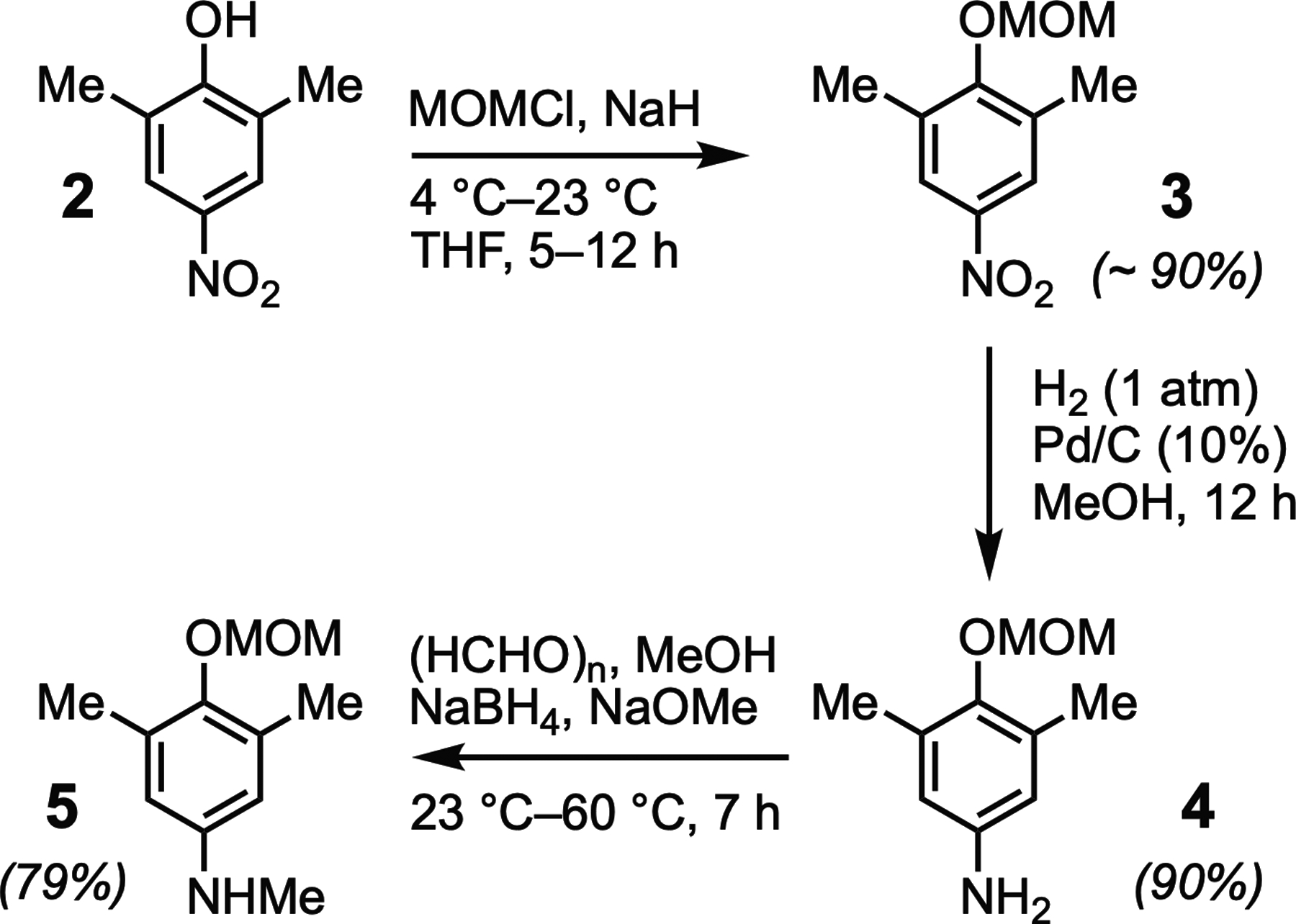
Synthesis of 4-(methoxymethoxy)-N,3,5-trimethylaniline (5) from 2,6-dimethyl-4-nitrophenol (2).
Fig. 5.
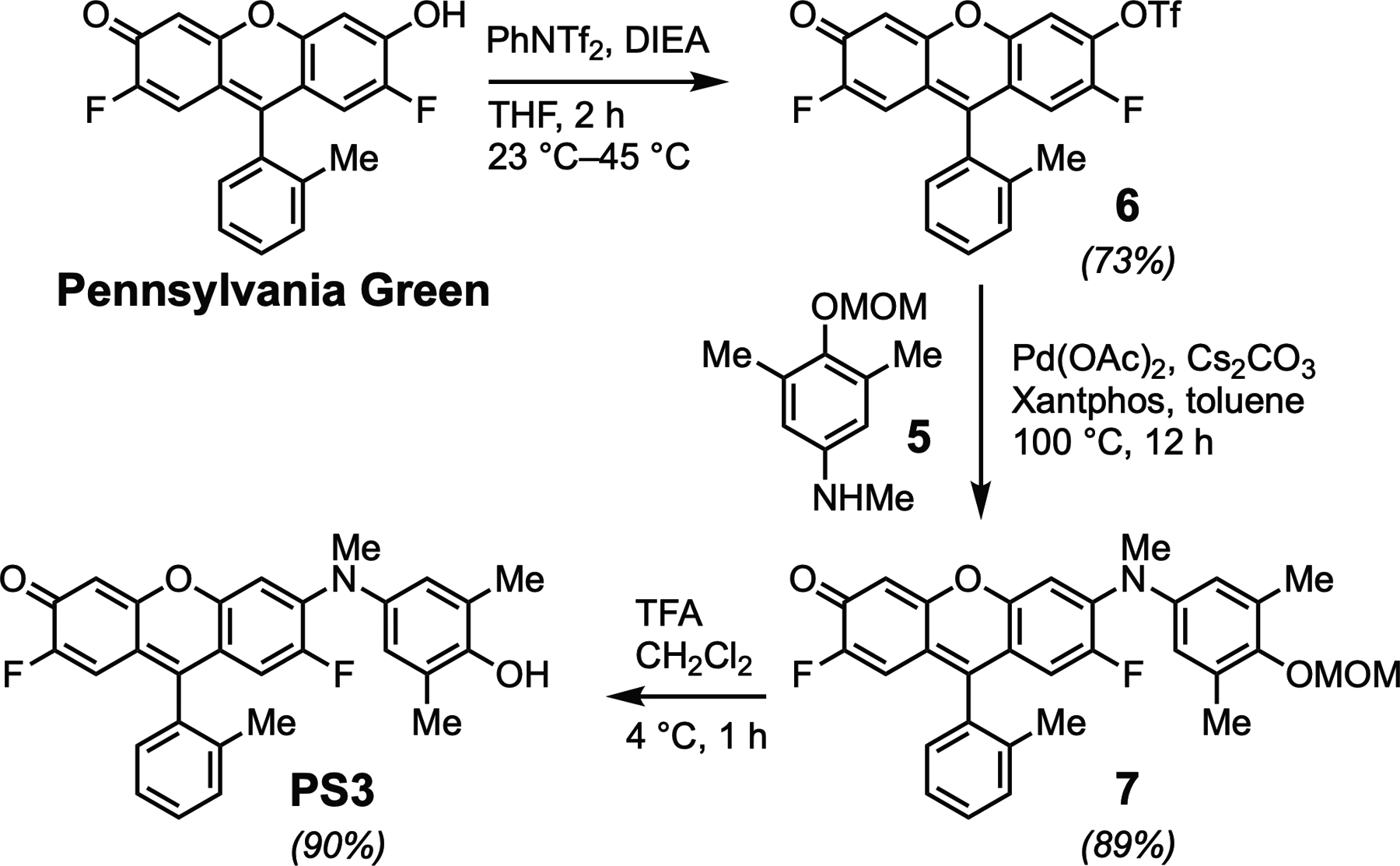
Synthesis of PS3 from Pennsylvania Green.
2.1. Materials
2,6-Dimethyl-4-nitrophenol (Sigma Aldrich, #132713).
Sodium hydride (Sigma Aldrich, #452912).
Methyl chloromethyl ether (TCI America, #M0680).
10% Palladium on carbon (wet support, Sigma Aldrich, #520829).
Sodium methoxide (Sigma Aldrich, #164992).
Paraformaldehyde (Sigma Aldrich, #P6148).
Sodium borohydride (Sigma Aldrich, #71320).
N-Phenyl-bis(trifluoromethanesulfoimide) (Oakwood chemicals, #093934).
N,N-Diisopropylethylamine (Sigma Aldrich, #550043).
Palladium (II) acetate (Sigma Aldrich, #520764).
Xantphos (Sigma Aldrich, #526460).
Cesium carbonate (Sigma Aldrich, #554855).
Trifluoroacetic acid (TCI America, #T0431).
Silica gel (Silicycle, #R12030B).
RediSepRf Gold 50g C18 column (Teledyne ISCO, #69-2230-336).
Teledyne ISCO Combiflash EZ Prep.
2.1.1. 2-(Methoxymethoxy)-1,3-dimethyl-5-nitrobenzene (3)
Dry a single-necked round bottom flask (50mL) equipped with a magnetic stir bar in an oven (150°C) for 1h. Seal this flask with a rubber septum after removing it from the oven. Evacuate the flask under high vacuum for 5min and flush with dry argon. Repeat at least 3 times, allowing the flask to return to ambient temperature.
Transfer 2,6-dimethyl-4-nitrophenol (2, 500mg, 3mmol, 1 equiv.) to the dry round bottom flask and add anhydrous THF (12mL, 0.25mM) under dry argon.
Cool the reaction mixture to 4°C by placing the flask in a water ice bath.
Add sodium hydride (196mg, 1.5 equiv.) to the reaction mixture in several portions with stirring over 10–15min. Stir this suspension at 4°C for an additional 10min. Note: Addition of sodium hydride results in a slight exotherm; avoid adding this reagent in a single portion.
Add methyl chloromethyl ether (340μL, 1.5 equiv.) dropwise to the reaction mixture at 4°C. Stir the reaction mixture for an additional 10–15min.
Remove the ice bath, allow the reaction mixture to warm to ambient temperature, and stir for 5h. Note: The mixture remains in suspension through the course of the reaction.
Monitor the conversion of 2 to 3 by TLC. Tip: Draw an aliquot (~100 μL) of the reaction mixture using a syringe fitted with a long needle. Dilute with dichloromethane (~400 μL) containing a drop of methanol for analysis by TLC.
After 2 is converted to 3, cool the reaction mixture 4°C with a water-ice bath. Add water dropwise under constant stirring until the mixture ceases effervescing, indicating that excess sodium hydride is quenched.
Dilute the quenched reaction mixture with diethyl ether (~100mL), and transfer to a separatory funnel (250mL).
Extract the upper organic layer twice with water (~50mL) and remove and discard the lower aqueous layer after each extraction. Wash the organic layer with saturated aqueous sodium chloride (~50mL). Tip: Diethyl ether is very volatile, and it is important to occasionally vent the pressure generated during the extraction.
Transfer the upper organic layer to an Erlenmeyer flask (250mL). Dry over anhydrous magnesium sulfate to remove residual moisture.
Remove the magnesium sulfate by filtration. Evaporate the solvent under vacuum to isolate the crude product. This crude product (ca. 90% yield) can be carried to the next step without further purification.
2.1.2. 4-(Methoxymethoxy)-3,5-dimethylaniline (4)
Caution: This reaction uses highly flammable hydrogen and palladium on carbon. Use appropriate personal protective equipment (PPE) and safety measures. Waste palladium on carbon should not be allowed to dry out completely and should be stored under water prior to disposal.
To a single necked round bottom flask (100mL) equipped with a magnetic stir bar, add palladium on carbon (10%, Pd/C) wetted with water (50%, 1.14g, 0.2 equiv.). Rapidly add methanol (10mL). Note: This commercially available form of palladium on carbon is easier to handle compared with dry palladium on carbon. To maximize safety, the fume hood should not house other flammable material.
In a separate flask, prepare (2-(methoxymethoxy)-1,3-dimethyl-5-nitrobenzene (3, 600mg, 2.84mmol, 1 equiv.) in methanol (20mL). Transfer this solution to the flask containing Pd/C.
Seal the reaction flask with a rubber septum, partially evacuate under vacuum, and flush with hydrogen from a balloon fitted with a syringe and needle. Tip: Evacuate the flask using a vacuum line fitted with a needle until you see a gentle boil of the solvent. Gently introduce hydrogen from a filled balloon. Prepare additional hydrogen balloons for use if the first one becomes deflated.
Stir the reaction mixture at ambient temperature under 1atm of hydrogen and monitor the progress of the reaction by TLC (eluent: 25% ethyl acetate in hexane). Conversion of starting material to product is usually observed within 1–2h.
Use a Büchner filtration assembly to remove the Pd/C and work up the reaction. Load a Bucher funnel with a celite plug (2″ depth) and wet with methanol. Load the crude reaction mixture on the celite plug and filter under vacuum. Wash the celite plug with copious amounts of methanol and allow to remain wet to minimize fire hazard. Note: Post filtration, the wet celite should be immediately disposed of in a container of water and should always be completely immersed in water.
Concentrate the filtrate to dryness and purify the crude product by silica gel flash chromatography (eluent: 100:0 hexane/ethyl acetate to 40:60 hexane/ethyl acetate, 90% yield).
2.1.3. 4-(Methoxymethoxy)-N,3,5-trimethylaniline (5)
Weigh 4-(methoxymethoxy)-3,5-dimethylaniline (4, 514mg, 2.84mmol, 1 equiv.) in a three-necked round bottom flask (100mL) equipped with a reflux condenser and a magnetic stir bar.
Seal the flask using a rubber septum, evacuate under vacuum, and flush three times with dry argon.
Introduce anhydrous methanol (14mL, ~0.2mM) into the flask using a syringe. Stir to effect complete dissolution of the starting material.
Add sodium methoxide (767mg, 5.0 equiv.). Note: A slight exotherm may be observed, and a cold-water bath can be used to equilibrate to ambient temperature.
Add paraformaldehyde (119mg, 1.4 equiv.). Stir at ambient temperature for 5h under Ar.
Add sodium borohydride (107mg, 1 equiv.) at ambient temperature. Transfer the reaction flask to an oil bath preheated to 55°C. Stir the reaction at 55°C for an additional 5h. Monitor reaction progress by TLC (eluent: ethyl acetate/hexane (6:4)).
Upon completion, dilute the reaction mixture with ethyl acetate (100mL) and extract twice with aq. KOH (1N,~25mL).
Separate the upper organic layers, and dry over anhydrous magnesium sulfate.
Remove the magnesium sulfate by filtration. Isolate the crude product by removal of solvent.
Purify the product by silica gel flash chromatography (100:0 hexane/ ethyl acetate to 40:60 hexane/ethyl acetate, 79% yield).
2.1.4. 2,7-Difluoro-3-oxo-9-(o-tolyl)-3H-xanthen-6-yl trifluoromethanesulfonate (6)
Dry a single-necked round bottom flask (25mL) equipped with a magnetic stir bar in an oven (150°C) for 1h. Seal this flask with a rubber septum after removing it from the oven. Evacuate the flask under high vacuum (5min) and flush with dry argon. Repeat at least three times, allowing the flask to return to ambient temperature.
Quickly transfer Pennsylvania Green (490mg, 1.44mmol, 1 equiv.) to the dry flask. Note: Pennsylvania Green is commercially available from AK Scientific (AMTGC257) or can be prepared from 2,7-difluoro-3,6-dihydroxy-9H-xanthen-9-one, available through an improved synthesis (Woydziak, Fu, & Peterson, 2012), as previously reported (Mottram et al., 2006).
Quickly transfer N-phenyl-bis(trifluoromethanesulfoimide) (770mg, 2.2mmol, 1.5 equiv.) to the dry flask.
Add anhydrous THF (10mL, ~0.15mM) via syringe under a positive pressure of argon. Note: The orange red slurry of Pennsylvania Green is only partially soluble at this concentration.
Add N,N-diisopropylethylamine (0.64mL, 3.6mmol, 2.5 equiv.) dropwise by syringe with constant stirring for 5min. The mixture may clarify, but it will remain orange-red throughout the course of the reaction.
Transfer this round bottom flask to an oil bath preheated to 45°C. Continuously maintain the reaction under a positive pressure of dry argon.
Monitor the progress of the reaction by TLC. Remove a small aliquot from the reaction flask for comparison with the reactants/reagents. Tip: Remove the reaction flask from the oil bath, allow to cool to ambient temperature while stirring, draw ca. 100 μL of the reaction mixture via syringe, dilute this sample with dichloromethane (~400 μL), and analyze immediately.
Conversion of Pennsylvania Green to product can be observed after ~2h. Allow the reaction mixture to cool to ambient temperature and concentrate to near dryness with a rotary evaporator. Note: This process removes only the volatile solvent and a small amount of residual N,N-diisopropylethylamine.
Dissolve the crude product in a minimum amount of dichloromethane. Load on the top of a silica gel column and purify the product by flash chromatography (eluent: 100:0 hexanes/ethyl acetate to 70:30 hexanes/ethyl acetate). After removal of solvents, store the yellow foamy solid in the dark at −20°C (73% yield).
2.1.5. 2,7-Difluoro-6-((4-(methoxymethoxy)-3,5-dimethylphenyl) (methyl)amino)-9-(o-tolyl)-3H-xanthen-3-one (7)
The following reaction is preferably conducted in a glove box, but it can be performed in a fume hood if the entire assembly is maintained under an atmosphere of dry Ar at all times. This Buchwald-Hartwig amination reaction is sensitive to both oxygen and moisture.
Dry a three necked round bottom flask equipped with a reflux condenser and magnetic stir bar in an oven for ca. 1h and allow to cool in a dry desiccator. Transfer the reaction flask into a glove box and weigh all reagents in this flask in the glove box in the following order: aniline 5, palladium(II) acetate, Xantphos, triflate 6, and cesium carbonate.
To the reaction flask, add 4-(methoxymethoxy)-N,3,5-trimethylaniline 5, (12.5mg, 63.8 μmol, 1.2 equiv.), palladium (II) acetate (1.2mg, 5.3 μmol, 0.1 equiv.), Xantphos (4.6mg, 8.0 μmol, 0.15 equiv.), 2,7-difluoro-3-oxo-9-(o-tolyl)-3H-xanthen-6-yl trifluoromethanesulfonate 6, (25mg, 53 μmol, 1 equiv.), and cesium carbonate (43.3mg, 132.9 μmol, 2.5 equiv.).
Add anhydrous toluene (1mL, 0.05mM) to the reaction mixture and seal the flask with a rubber septum. Remove the flask from the glove box.
Attach a dry reflux condenser to the flask and transfer the reaction flask to an oil bath preheated to 100°C. Heat the reaction mixture at 100°C under Ar for 12–16h. Note: The color of the reaction mixture changes from yellow to red upon heating.
Remove and analyze an aliquot of the reaction mixture by TLC (eluent: ethyl acetate/hexane (6:4)). Monitor reaction progress by comparison to the limiting reagent 6. Note: minimize introduction of oxygen and moisture during this process by obtaining TLC samples as infrequently as possible.
When the starting material is converted to product by TLC, remove the reaction mixture from the oil bath and allow to cool to ambient temperature.
Pipet the crude mixture onto a silica gel column prewetted with hexane. Dilute any residual crude product remaining in the reaction flask with a minimal amount of dichloromethane and transfer to the column.
Purify the product by flash chromatography (100:0 hexane/ethyl acetate to 20:80 hexane/ethyl acetate). After removal of solvents, the pure product is obtained as a dark red solid (89% yield).
2.1.6. 2,7-Difluoro-6-((4-hydroxy-3,5-dimethylphenyl) (methyl)amino)-9-(o-tolyl)-3H-xanthen-3-one (PS3)
Weigh 2,7-difluoro-6-((4-(methoxymethoxy)-3,5-dimethylphenyl) (methyl)amino)-9-(o-tolyl)-3H-xanthen-3-one (7, 13mg, 25.2 μmol, 1 equiv.) into a single necked round bottom flask equipped with a magnetic stir bar under argon. Cover the flask with aluminum foil to protect from light.
Add dichloromethane (0.5mL, 0.05mM) and cool to 4°C with a water ice bath.
Add trifluoroacetic acid dropwise to the reaction mixture at 4°C with stirring to a final concentration of 30% v/v with respect to dichloromethane (0.15mL). The color of the reaction mixture will change from red to magenta upon introduction of TFA.
Stir the reaction mixture at 4°C for ~1h. Analyze the course of the reaction by TLC.
Concentrate the crude reaction mixture to dryness using a rotary evaporator. Remove residual trifluoroacetic acid azeotropically using toluene. Tip: For azeotropic removal of trifluoroacetic acid, add 10–20 times excess toluene by volume to the concentrated crude mixture. Concentrate using a rotary evaporator. Repeat this process 3–5 times.
Dissolve the crude mixture in a minimum quantity of dimethyl sulfoxide (~1mL) and purify by reverse phase flash chromatography eluting with a gradient of water and acetonitrile (both containing 0.1% v/v trifluoroacetic acid). Note: If available, this can be facilitated by use of a Teledyne Isco combiflash automated purifier using a 60g Redisep Gold column with a gradient of 90:10 water/acetonitrile to 10:90 water/acetonitrile over 16min.
Concentrate the fractions containing the product using a rotary evaporator to remove acetonitrile. Remove the residual water by lyophilization in a foil-wrapped flask to obtain the pure product as a magenta powder. Store this product in the dark at −20°C (90% yield). Note: PS3 is prone to photodegradation and exposure to light must be minimized during the purification and concentration step. Keep solutions of PS3 away from ambient light at all times to avoid photooxidation, which can contribute to background fluorescence.
If needed as a control compound, the synthesis of rhodol 1 has been previously reported (Knewtson et al., 2018).
3. Photophysical properties, reactivity, and selectivity of PS3
Absorbance and fluorescence emission spectra of PS3, and rhodol 1, its product of cleavage by PNT, are shown in Fig. 6. Other photophysical data, such as the molar absorptivity of PS3 in DMSO (ε525nm =29,200M−1 cm−1), have been previously reported (Knewtson et al., 2018). Using the Beer-Lambert Law (A= εcl), and this value of ε, concentrations of stock solutions of this sensor in DMSO can be readily determined by absorbance spectroscopy. In 1-octanol, a mimic of cellular membranes, PS3 exhibits an absorbance λmax of 526nm and a fluorescence emission λmax of 538nm. However, under these conditions pure PS3 is essentially non-fluorescent with a very low quantum yield (Φ=0.0004). In contrast, the maximal absorbance of rhodol 1 is blue-shifted to 515nm and its fluorescence emission at 536nm is increased by 1800-fold (Φ=0.72) compared to PS3. Changes in fluorescence emission spectra when PS3 is treated with pure PNT to form 1 are shown in Fig. 6. We previously reported (Knewtson et al., 2018) that under pseudo-first order conditions the half-life of PS3 treated with the PNT generator SIN1 is 49s.
Fig. 6.
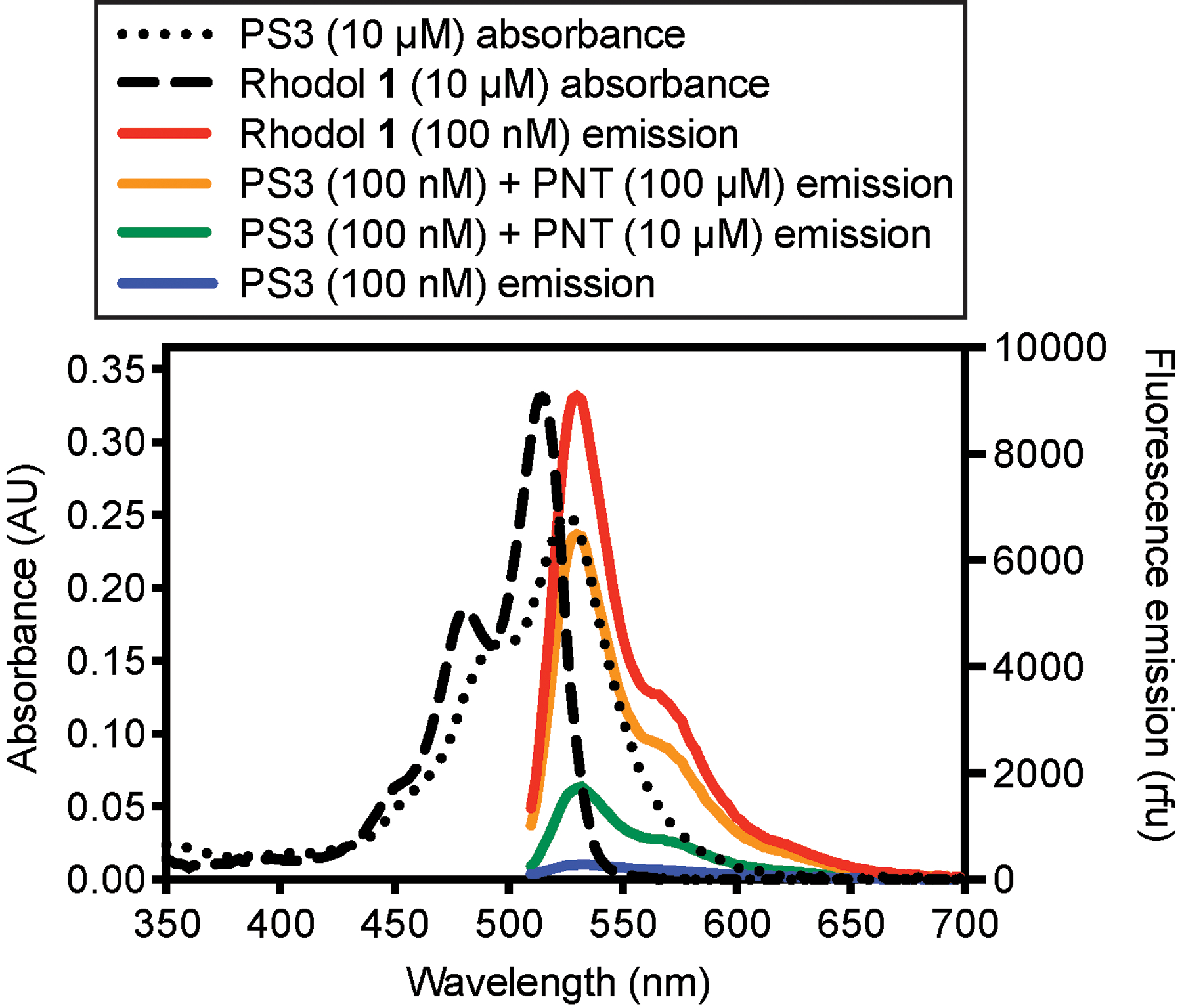
Absorbance and emission spectra of PS3 and rhodol 1 in the membrane-mimetic solvent 1-octanol. Fluorescence emission spectra were acquired by excitation from 474 to 490nm. For reaction with PNT, solutions of PS3 (100nM, 1mL, 0.4% DMSO) were shaken in centrifuge tubes (30°C, 1h) to promote oxidative cleavage.
The selectivity of reaction of PS3 with PNT, sodium hypochlorite (NaOCl), and hydrogen peroxide (H2O2) was analyzed by analytical reverse-phase HPLC (Fig. 7). The biological oxidants NaOCl and H2O2 were investigated because they have been shown to be produced by neutrophils to kill pathogens. These phagocytic innate immune cells generate hypochlorous acid by promoting reaction of hydrogen peroxide with chlo-ride, catalyzed by the enzyme myeloperoxidase (MPO) (Degrossoli et al., 2018; Winterbourn & Kettle, 2013). To evaluate selectivity, PS3 in PBS (10μM) was allowed to react with 10-fold or 100-fold excess oxidant (23°C, 1h). This reaction mixture was then immediately analyzed by analytical reverse-phase HPLC, and peaks were identified by comparison with unreacted PS3 and rhodol 1. As shown in Fig. 7, PS3 (10μM) reacted cleanly with PNT (1mM) to form 1 in >99% yield, whereas very little reaction with H2O2 (1mM) was observed under these conditions (1% yield). Treatment with this high concentration of sodium hypochlorite (1mM) promoted some conversion of PS3 to 1 (17% yield), but at a lower concentration of these oxidants (100μM), PS3 was over 10-fold more selective for PNT (45% yield) compared with NaOCl (3% yield). These results support our previous (Knewtson et al., 2018) studies that demonstrated selectivity of PS3 for reaction with PNT compared with NaOCl, H2O2, hydroxyl radical, alkyl peroxides, superoxide, and nitric oxide. Additionally, we previously demonstrated (Knewtson et al., 2018) that the limit of detection of PNT by PS3 using a Perkin Elmer LS55 fluorescence spectrometer is 40±10nM, a concentration where a twofold change in fluorescence can be measured.
Fig. 7.
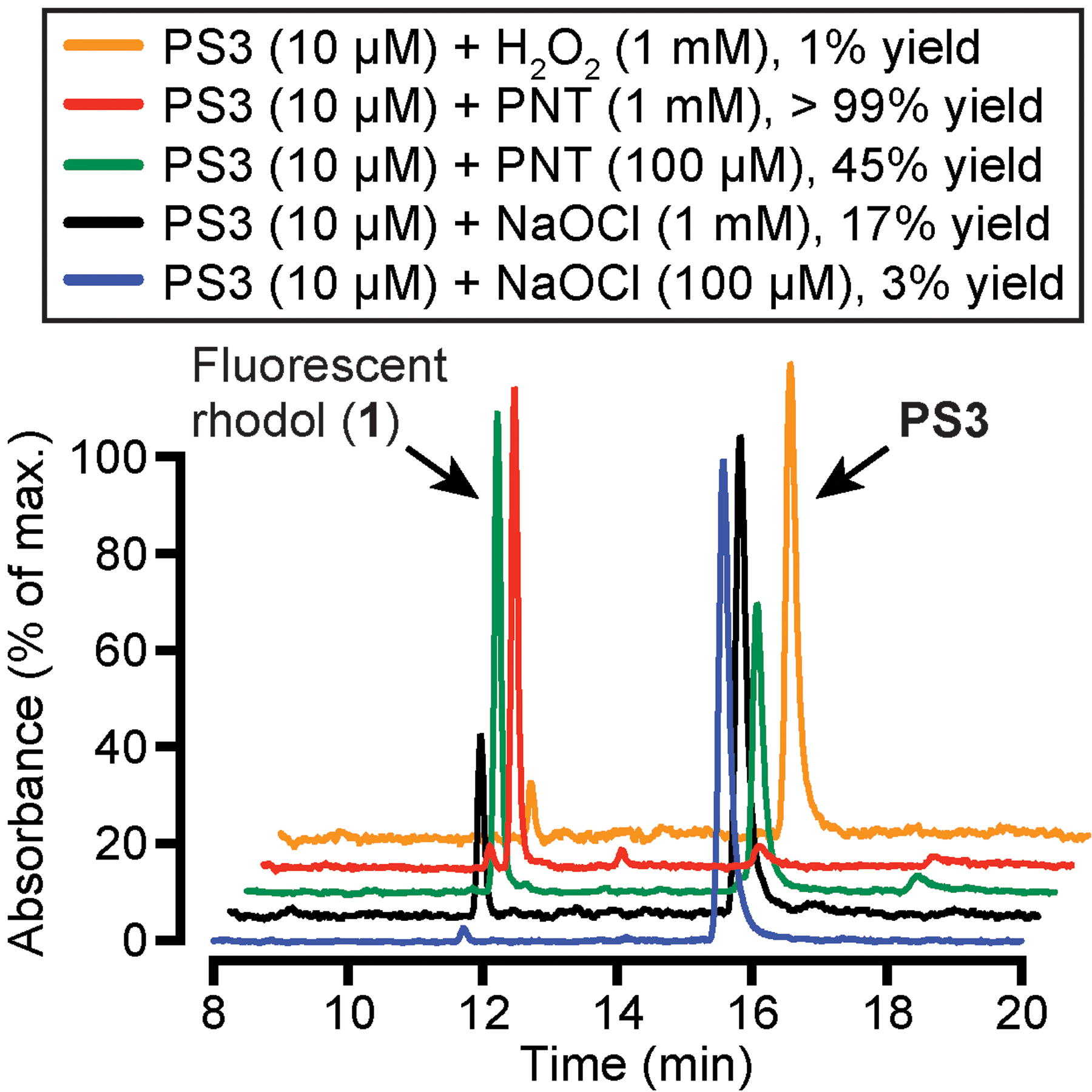
Analysis of selectivity of oxidation of PS3 by analytical reverse-phase HPLC. PS3 was allowed to react with PNT, NaOCl, and H2O2 for 1h at 23°C in PBS (pH 7.4, 0.4% DMSO). Retention times: 1 = 11.7min; PS3= 15.6min. Yields were measured by integration of peaks. The order of the stacked plots (top to bottom) matches the key above.
3.1. Optical spectroscopy
3.1.1. Materials
Seal-Rite 1.5mL graduated microcentrifuge tube (USA Scientific, 1615-5500).
CytoOne 96 well plate (USA Scientific, CC7682-7596).
Fluotrac 200 96-well flat bottom black plate (USA Scientific, 5665-5096).
Peroxynitrous acid sodium salt (PNT, Cayman chemicals, 81565).
Aqueous NaOH (0.3M).
Molecular biology grade DMSO (Sigma Aldrich, D8418).
1-Octanol (Sigma Aldrich, 293245).
3.1.2. Determination of molar extinction coefficients of fluorophores
Carefully weigh three separate portions of at least 1mg of dry fluorophore powder that has been dried under high vacuum for at least 24 h to remove all traces of moisture.
Add DMSO to generate master stock solutions of known concentration in the mM range based on molecular weight. Prepare serial dilutions of these solutions in DMSO to provide six stock solutions of different concentrations in the micromolar range (0.5–15μM).
Record absorbance spectra of these μM stock solutions with a spectrophotometer.
Determine the molar extinction coefficient (ε) by plotting absorbance λmax versus concentration. Fit the data using linear least squares (including a zero intercept) to determine the slope, which corresponds to ε.
The following values were previously measured (Knewtson et al., 2018) in DMSO: PS3, ε525nm =29,200M−1 cm−1; rhodol 1, ε517nm =30,900M−1 cm−1.
3.1.3. Determination of the quantum yields of PS3 and rhodol 1 relative to rhodamine 6G in 1-octanol
Serially dilute absorbance-normalized 10mM DMSO stock solutions of each fluorophore with DMSO to provide five stock solutions (0.25–2mM).
For absorbance measurements, prepare additional dilutions in 1-octanol in triplicate (1:500, 0.5–4μM for PS3, rhodol 1, and rhodamine 6G). Transfer solutions to a semi-micro quartz cuvette (Sigma Aldrich, Z27667–7) for analysis on an Agilent 8453 spectrophotometer or similar instrument.
For fluorescence emission measurements, prepare additional dilutions in 1-octanol in triplicate (1:500, 0.5–4μM for PS3; 2–10nM for rhodol 1 and rhodamine 6G). Transfer solutions to a Quartz SUPRASIL Macro/Semi-micro cell (PerkinElmer B0631132) for analysis on a Perkin Elmer LS-55 spectrophotometer or similar instrument.
Generate absorbance and fluorescence emission spectra (Ex. 488nm ±5nm).
Plot the integrated fluorescence emission at each concentration against the absorbance measured at 488nm. For rhodol 1 and rhodamine 6G, absorbance values for the low concentrations used for fluorescence studies were determined by extrapolation of absorbance data made at higher concentrations. Fit the data using linear least squares, including a zero intercept (GraphPad Prism 7), to determine the slope, which is proportional to the quantum yield. Calculate the quantum yield as: Φx =Φst • Gradx/Gradst, where Φx =quantum yield of the unknown, Φst =quantum yield of the standard, Grad =slope of the best linear fit.
The quantum yield of rhodamine 6G in octanol (Φ=0.96) (Magde, Wong, & Seybold, 2002) was used as the standard. Because the fluorophores were all analyzed in the same solvent, there is no need to correct for differences in refractive index.
The following values were previously measured (Knewtson et al., 2018) in 1-octanol: PS3, Φ=0.0004; rhodol 1, Φ=0.72.
3.1.4. Analysis of reactivity of PS3 with PNT by fluorescence spectroscopy
To prepare an absorbance-normalized concentrated stock solution of PS3 in DMSO, weigh PS3 (~1mg) in a microcentrifuge tube (1.5mL). Add molecular biology grade DMSO (100μL). Sonicate to ensure complete dissolution (at least 5min).
Determine the concentration of this solution by absorbance spectroscopy and the Beer-Lambert law (PS3 ε525nm =29,200M−1 cm−1). Dilute to a final concentration of 10mM by addition of molecular biology grade DMSO using the dilution equation C1V1 = C2V2, where C = concentration and V = volume. This DMSO stock solution can also be used for the biological assays described later in this chapter. However, solutions of PS3 should be used immediately after preparation from dry powder to minimize background fluorescence.
For reactivity studies in octanol, serially dilute this 10mM DMSO stock solution with 1-octanol to yield a 10μM solution (1mL).
Acquire absorbance spectra at 10μM. For the data shown in Fig. 6, samples were loaded on a clear flat-bottom 96-well plate and analyzed with a BMG Labtech ClarioStar Plus plate reader.
Serially dilute the 10μM stock solution of PS3 and rhodol 1 with 1-octanol to yield 100nM solutions (2mL each). Add DMSO to standardize the final concentration at 0.4%.
Cool commercially available peroxynitrous acid sodium salt (provided in 0.3M aq. NaOH) on ice. Dilute to a final concentration of 10mM using freshly prepared aq. 0.3M NaOH. Determine the concentration of PNT by absorbance spectroscopy using the Beer-Lambert law (PNT ε302nm =1670M−1 cm−1).
Aliquot a 100nM solution of PS3 into 1.5mL microcentrifuge tubes. For reactivity studies, add sodium peroxynitrous acid (10mM) to a final [10μM] and [100μM].
Shake these mixtures (200 rpm) in an incubator (30°C, 1h).
Acquire fluorescence emission spectra. For the data shown in Fig. 6, samples were loaded on a Fluotrac 200 96-well flat bottom black plate and analyzed with a BMG Labtech ClarioStar Plus plate reader (Ex. 482nm±8nm, Em. 510–700nm).
3.1.5. Analysis of reactivity of PS3 with oxidants by HPLC
3.1.5.1. Materials
Hydrogen peroxide (30% in water, 9.8M, Sigma Aldrich, H3410).
Sodium hypochlorite solution (4.00–4.99% available chlorine, 0.53M, Sigma Aldrich, 239,305).
PBS (pH 7.2, MilliporeSigma, 79,378).
Prepare an absorbance-normalized 10mM stock solution of PS3 in DMSO as previously described.
Prepare an absorbance-normalized 10mM stock solution of PNT in 0.3M aq. NaOH as previously described.
Prepare a 10mM stock solution of hydrogen peroxide in PBS (10μL of 30% H2O2 diluted with 990μL PBS to [100mM], followed by a further 10-fold dilution in PBS).
Prepare a 10mM solution of sodium hypochlorite (190μL of aq. NaOCl diluted with 810μL PBS to [100mM], followed by a further 10-fold dilution in PBS).
For reaction with 1mM oxidants, pipet 180μL of the 10mM solution of PS3 into three HPLC vials. Add 20μL of 10mM PNT, hydrogen peroxide, or sodium hypochlorite.
For reaction with 100μM oxidants, pipet 199μL of 10mM PS3 into two HPLC vials. Add 1μL of 10mM PNT or sodium hypochlorite to obtain a final concentration of 100μM of each oxidant.
Cap the vial, gently swirl the solution, and allow the reaction to proceed for 1h at 23°C.
Analyze the conversion of PS3 to 1 by reverse-phase HPLC at 23°C. For the data shown in Fig. 7, samples were analyzed with a Agilent 1220 instrument fitted with a Hamilton PRP-1 column (7μM, 4.1mm×25cm) by elution with 90:10 water: acetonitrile to 10:90 water: acetonitrile over 25min (mobile phases contain 0.1% TFA).
4. Detection of peroxynitrite resulting from antibody-dependent cellular phagocytosis (ADCP) of opsonized tentagel microspheres
When treated with opsonized 10μm amino-tentagel microspheres, the mouse RAW 264.7 macrophage cell line undergoes ADCP, as observed by microscopy (Fig. 8). To opsonize, these beads were acylated with the NHS ester of 2,4-dinitrophenyl aminohexanoic acid (DNP) and treated with rabbit polyclonal anti-DNP IgG. Under the conditions shown in Fig. 8, ca. 10% of these beads were observed to be phagocytosed by DIC and confocal laser scanning microscopy. To detect PNT generated during ADCP, cells were treated with PS3 at a final concentration of 10 μM. Here, we describe comparison of PS3 with commercially available PNT sensors, hydroxyphenyl fluorescein (HPF) and NiSPY-3, under the same conditions. Confocal fluorescence micrographs of RAW 264.7 macrophages treated with these sensors and IgG/DNP-beads revealed that enhanced cellular fluorescence resulting from ADCP was only observed in cells treated with PS3 (Fig. 8, compare panel B with D and F). In contrast to NiSPY-3, HPF increased the fluorescence of IgG/DNP-beads both in the presence and absence of cells. However, when phagocytosed, the fluorescence of HPF-bound beads was partially quenched (Fig. 8, panel D), presumably due to the sensitivity of this fluorescein-derived fluorophore to the acidic conditions of the phagosome.
Fig. 8.
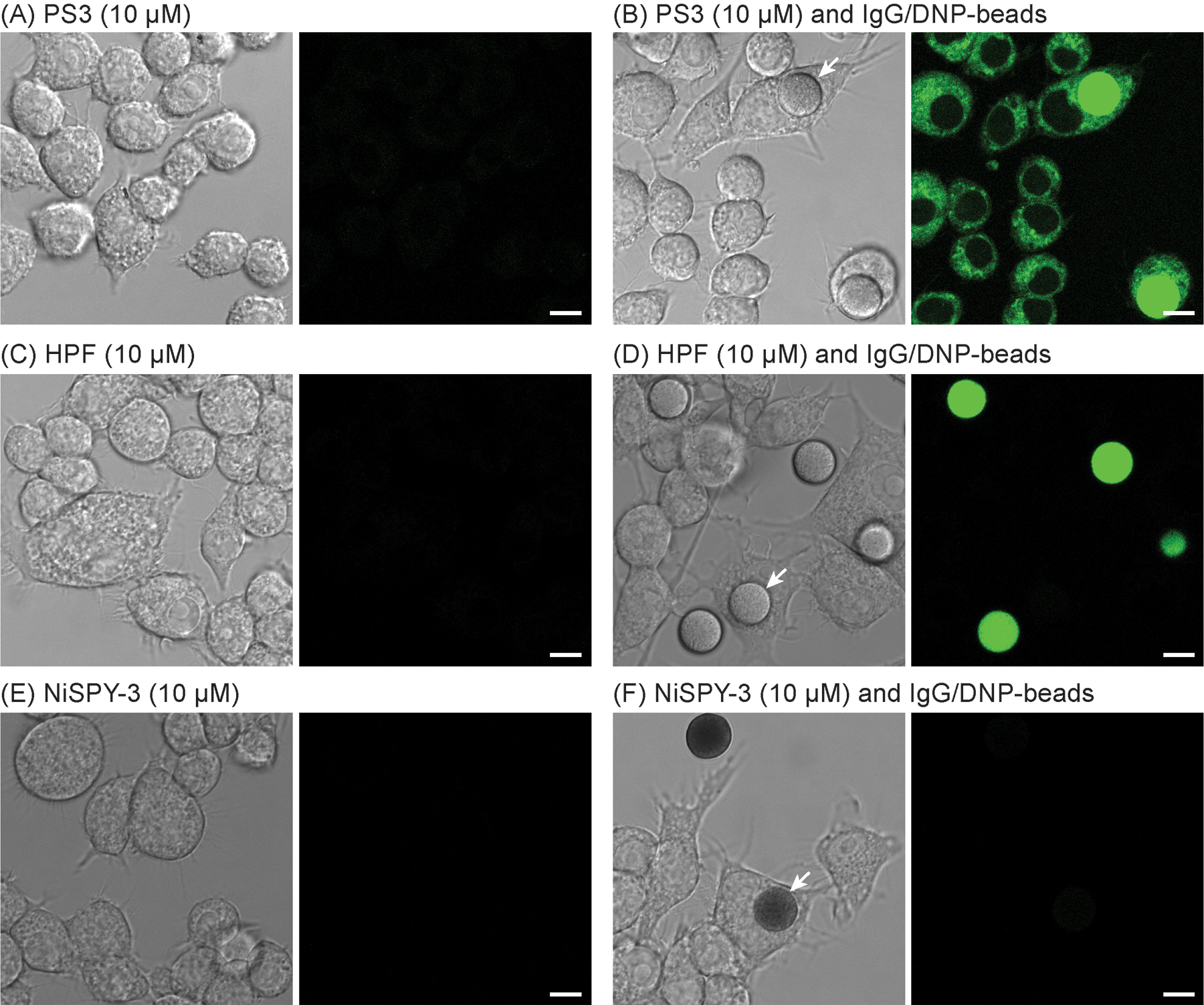
DIC (left) and confocal fluorescence (right, Ex. 488nm, Em. 500–600nm) micrographs of living RAW 264.7 cells treated (4 h, 10μM) with PS3 (A and B, 0.2% DMSO), HPF (C and D, 0.2% DMF), and NiSPY-3 (E and F, 0.2% DMSO) in the absence (A, C and E) and presence (B, D and F) of IgG/DNP-beads (100,000/mL). White arrows point at representative phagocytosed beads. Increased cellular fluorescence upon ADCP was only observed with PS3 (B). Images were acquired with a Leica SP8 microscope (63× objective). Scale bar=8μm.
To investigate the response of PS3 in other cell lines, we imaged mouse J774A.1 macrophages, human THP-1 monocytes, and human Jurkat lymphocytes after treatment with IgG/DNP-beads (Fig. 9). Similar to RAW 264.7 cells, J774A.1 cells become highly fluorescent upon ADCP. In contrast, little response was observed with non-differentiated THP-1 monocytes, although extensive phagocytosis of beads was observed. As expected, in non-phagocytic Jurkat lymphocytes, no enhancement of fluorescence was detected, providing a useful negative control for this assay.
Fig. 9.
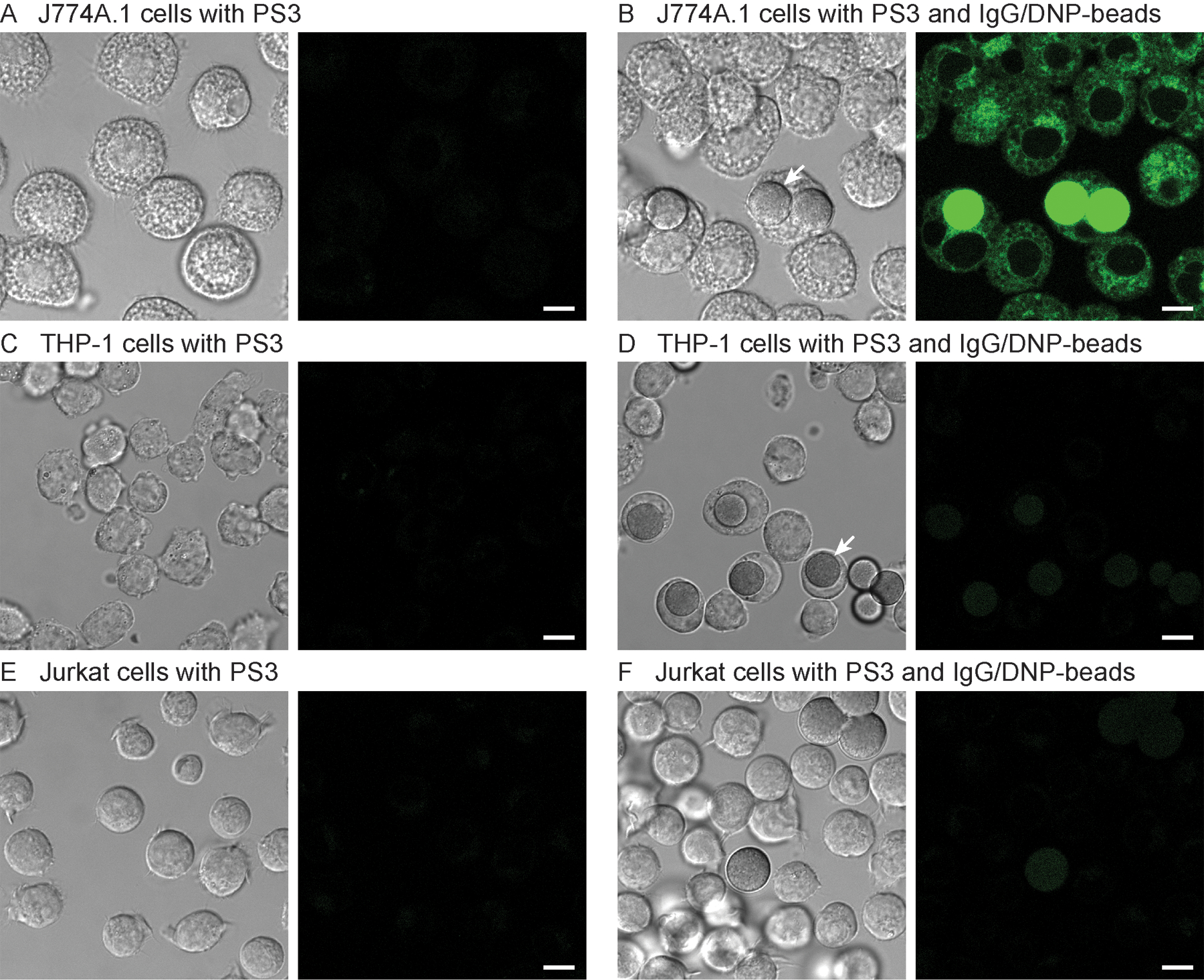
DIC (left) and confocal fluorescence (right, Ex. 488nm, Em. 500–600nm) micrographs of living J774A.1 (A and B), THP-1 (C and D), and Jurkat (E and F) cells treated with PS3 (10μM, 0.2% DMSO) for 4h in the absence (A, C and E) and presence (B, D and F) of IgG/DNP-beads (100,000/mL). White arrows point at representative phagocytosed beads. Images were acquired with a Leica SP8 microscope (63× objective). Scale bar=8μm.
We previously described (Knewtson et al., 2018) the detection of PNT during ADCP using the cell line RAW 264.7 by confocal microscopy and flow cytometry. Although these methods of analysis are effective, they are relatively low throughput, and flow cytometry required differential fluorescent labeling of cells and tentagel beads to effectively distinguish these species. To provide a simpler and higher-throughput alternative, we report here a method for the analysis of fluorescence intensity of cell culture media containing cells on 96-well plates. To validate this method, the four cell lines studied by confocal microscopy were treated with PS3 and stimulated with IgG/DNP-beads. As shown in Fig. 10, when cultured on 96-well plates, ADCP triggers substantial (>10-fold) increases in fluorescence in both the RAW 264.7 and J774.A1 cell lines, whereas a lesser threefold to fourfold (statistically significant) enhancement in the fluorescence of THP-1 cells was observed. After treatment for 4h, controls that omit the IgG or the DNP-beads showed no effect, but DNP-beads alone were slightly stimulatory after 24h and enhanced fluorescence by twofold in the most responsive RAW 264.7 cell line. In all cases, the non-phagocytic Jurkat cell line showed no response as expected. Additional control experiments where the media was separated from the cells indicated that diffusion of rhodol 1 into the media represents a major contributor to the fluorescent signal detected with this assay approach (data not shown). Consistent with this notion, to detect PNT by confocal microscopy, flow cytometry, or changes in fluorescent intensity of the medium, cells treated with PS3 were not subjected to any additional washing steps. When these cells are washed with cell culture medium, the fluorescent rhodol 1 rapidly effluxes from ER membranes, leading to loss of cellular fluorescence. Comparison of the data for the THP-1 cell line (Fig. 10) with the confocal micrographs of THP-1 cells (Fig. 9) indicates that this plate reader-based assay is more sensitive than confocal microscopy for detection of PNT.
Fig. 10.
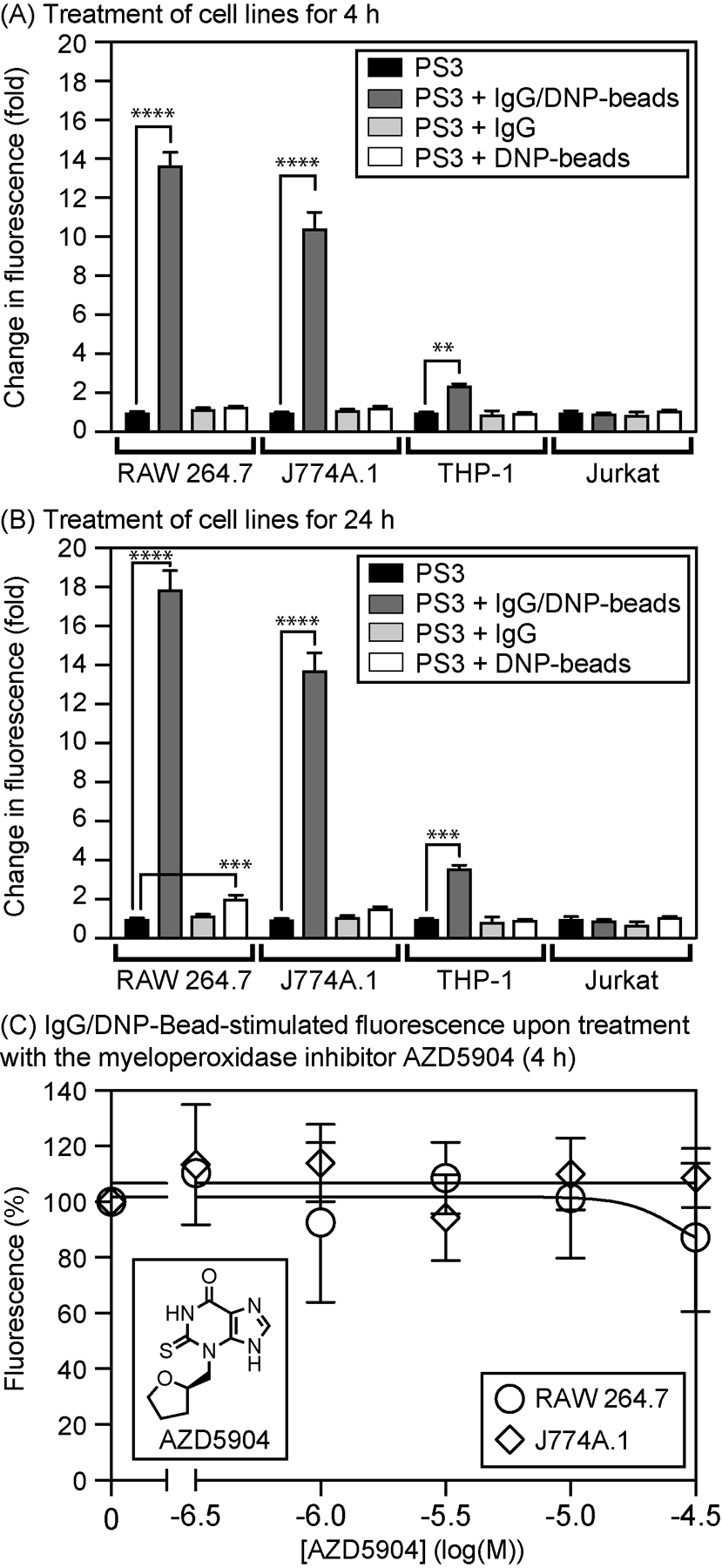
Changes in fluorescence of complete media seeded with different cell lines on a 96-well plate. Cells were treated with PS3 (10μM), IgG/DNP-beads (200,000/mL), DNP-beads alone (200,000/mL), or IgG alone (2μg/mL) for 4h (A) or 24h (B) at 37°C in a CO2 (5%) incubator. In (C), the myeloperoxidase inhibitor AZD5904 was additionally added to RAW 264.7 or J774A.1 cells with PS3 and IgG/DNP-beads (4h). Error bars represent SD (N ≥3). Statistical significance analyzed by one-way ANOVA (GraphPad Prism 8): ****, P <0.0001; ***; P <0.001; **, P <0.01. Samples were analyzed with a BMG LabTech Clariostar Plus plate reader (Ex. 480±7.5nm /Em. 520±7.5nm).
Because neutrophils are known to produce hypochlorite to kill pathogens, and PS3 shows some reactivity with hypochlorite (Fig. 7), we used this sensitive plate reader-based assay to evaluate the effect of AZD5904, a potent inhibitor of the HOCl-generating enzyme myeloperoxidase. This myeloperoxidase inhibitor has a reported IC50 of 140nM and has been shown to reduce HOCl in PMA-stimulated human neutrophils by >90% when added at 1μM (Tiden et al., 2011). As shown in Fig. 10, AZD5904 did not appreciably affect fluorescence from PS3 stimulated by IgG/DNP-beads in RAW 264.7 and J774A.1 at concentrations of up to 100μM, providing additional evidence that oxidation of PS3 by hypochlorite is not observed during ADCP by RAW 264.7 or J774A.1 cells.
4.1. Preparation of opsonized tentagel microspheres
4.1.1. Materials
TentaGel M NH2 microspheres (10μm, monosized, Rapp Polymere, M30102).
PBS (pH 7.2, MilliporeSigma, 79378).
N-Succinimidyl N-(2,4-dinitrophenyl)-6-aminocaproate (DNP-NHS, MilliporeSigma, 55472).
DMSO, suitable for cell culture (MilliporeSigma, D2438).
Personal microcentrifuge (USA Scientific Plastics, 2631-006).
Ethanol, 200 proof.
Rabbit polyclonal anti-DNP IgG antibody (Vector Laboratories, SP-0603).
Swell tentagel beads (15mg) by suspending in PBS (1mL, 1h) at ambient temperature.
Prepare a stock solution of DNP-NHS in DMSO (5mg, 253μL, 50mM). Note: DNP-NHS requires sonication for full dissolution at this concentration. Use this solution immediately after preparation and do not store for future use.
After swelling of the beads for 1h, pellet by centrifugation with a personal microcentifuge (30s). Discard the supernatant.
Suspend the bead pellet in PBS (300μL).
Add DNP-NHS (5μL, 50mM) to acylate amino groups on beads. Shake at ambient temperature for 1h.
Wash beads with EtOH (800μL), followed by PBS (2×1mL).
Suspend the washed beads in PBS (200μL). To include a unopsonized DNP-bead negative control experiment, remove a portion of the DNP-bead suspension (50μL). To prepare opsonized beads, add rabbit Anti-DNP IgG (1mg/mL, 50μL) at a final concentration of 200μg/mL. Shake the solution at ambient temperature for 1h.
Measure the density of beads by flow cytometry or other method (10mg of tentagel beads typically yields 15–20 million beads/mL). For the data shown in Fig. 10, a final concentration of 200,000 beads/mL was added to each well of a 96-well plate.
For the IgG alone experiment shown in Fig. 10, a final concentration of 2μg/mL IgG was used.
4.2. Culture of RAW 264.7, J774A.1, THP-1, and Jurkat cells
4.2.1. Materials
RAW 264.7 cell line (ATCC TIB-71, BSL 2).
J774A.1 cell line (ATCC TIB-67, BSL 1).
THP-1 cell line (ATCC TIB-202, BSL 1).
Jurkat cell line (ATCC TIB-152, BSL 1).
RPMI medium (Sigma Aldrich R8758).
Dulbecco’s Modified Eagle F-12 medium (Sigma Aldrich D6429).
Fetal bovine serum, (FBS, Life Technologies, 26140079).
Penicillin-streptomycin, (Sigma Aldrich, P4333).
Corning T-75 flask (CytoOne CC7682-4175).
Cell scraper (CytoOne CC7600-0220).
Conical bottom centrifuge tube (15mL, Thermo Fisher Scientific 339652).
4.3. Culture of suspension cell lines (THP-1 and Jurkat)
To revive THP-1 and Jurkat cells from frozen liquid N2 stocks, warm RPMI-1640 supplemented with FBS (10%, complete media) to 37°C in a water bath.
Thaw the capped frozen vial containing cells in a water bath at 37°C. Note: Only a partial thaw is sufficient, and a small lump of ice should remain in the vial. Limit the toxicity of the DMSO (5%) cryoprotectant by minimizing the time the cells are placed in the water bath (<1 min).
Move the vial, the media bottle, and an empty T-75 flask to a biosafety cabinet. Sterilize all items by spraying with 70% ethanol.
By adding warm complete media (~5mL) to the vial in several aliquots, transfer the contents to a 15mL conical vial. Centrifuge at 2000rpm for 2min at ambient temperature. Remove the supernatant by aspiration. Resuspend the remaining cell pellet in complete media (~1mL).
Pipet warm complete media (15mL) into the T-75 flask. Transfer the re-suspended cells into the T-75 flask.
Culture the cells in a vented T-75 flask in a humidified incubator (37°C, 5% CO2). Tip: Expansion from a frozen stock may require up to 7 days.
Monitor the density of the cell culture using an inverted microscope. Analyze cell density by cell counting, flow cytometry, or a similar method.
Passage (subculture) when the cell density reaches 8×105 cells/mL by transferring 10mL of the cell culture to a 15Ml conical tube. Centrifuge at 2000rpm for 2min. Remove the supernatant and resuspend the cell pellet in 1mL of fresh complete media. Transfer the cell suspension to a T-75 flask and dilute with fresh complete media to 2–4 105 cells/mL (THP-1) or 1×105 cells/mL (Jurkat). Cells can alternatively be maintained by adding fresh complete media (13 mL) to a portion of the cell culture (~2 mL) every 2–3 days. Do not allow the cell density to exceed 1×106 cells/mL (THP-1) or 3×106 cells/mL (Jurkat).
4.4. Culture of adherent cell lines (RAW 264.7 and J774A.1)
To revive RAW 264.7 and J774A.1 from frozen liquid N2 stocks, warm Dulbecco’s Modified Eagle F-12 media supplemented with FBS (10%) and penicillin-streptomycin (1%, complete media). Using this media, thaw, resuspend, and transfer cells to a T-75 flask and incubator as previously described for suspension cell lines. These cells become adherent in ca. 6h.
After 3–4 days, discard the media by aspiration. Replace with fresh complete media (15mL). Tip: When removing the media, orient the flask vertically to avoid damaging the cells.
When cells reach high confluency (>90%), use single swift strokes with a sterile cell scraper from top to bottom or side to side to suspend the cells (avoid dragging the scraper along the adherent surface of the flask to minimize cellular damage).
Aspirate 5mL of media, transfer this cell suspension to a 15mL conical centrifuge tube, and centrifuge at 2000rpm for 2min.
Remove the supernatant and resuspend the cell pellet in 1mL fresh complete media. Gently aspirate using a pipette and transfer 100μL of this suspension to a fresh T-75 flask containing 15mL of media. Incubate at 37°C (5% CO2).
Replace medium every 2 to 3 days.
4.5. Preparation of solutions of PS3 for bioassays
Transfer a small amount (e.g., 0.2mg) of dry PS3 powder to a microcentrifuge tube (1.5mL). Add molecular biology grade DMSO (10 μL) to generate a concentrated stock solution (>10mM).
Measure the concentration of this stock solution by UV–vis spectroscopy by applying the Beer-Lambert law (A= εcl, ε525 nm =29,210M−1 cm−1). Dilute with molecular biology grade DMSO to obtain a final concentration of 10mM.
Serially dilute this DMSO stock by 1000-fold in 10-fold increments using appropriate media to a final concentration of 10 μM (0.1% DMSO, v/v).
Wrap PS3 in foil to protect from light and use immediately after dissolution. Extended exposure of solutions of PS3 to ambient light can lead to photooxidation and result in elevated background fluorescence.
4.6. Preparation of cell lines for imaging
For adherent cell lines (RAW 264.7 and J774A.1), grow to >90% confluency in a T-75 flask. Use a cell scraper to dislodge the cells and measure the cell density using a flow cytometer or other method.
For suspension cell lines (THP-1 and Jurkat), grow to 8×105 cells/mL.
Transfer cells to a 15mL conical tube and centrifuge at 2000 rpm for 2min.
Remove the supernatant and resuspend the cell pellet in fresh complete media at a final concentration of 3×105 cells/mL.
For adherent cell lines (RAW 264.7 and J774A.1), pipet this cell suspension onto an 8-well ibidi μ-slide (300 μL/well) and incubate at 37°C for 16h (5% CO2). Carefully aspirate the media and wash the wells with PBS (300 μL/well). Add pre-prepared solutions (300 μL/well) of PS3, beads, and/or other agents in complete media.
For suspension cell lines (THP-1 and Jurkat), treat 1mL aliquots of the cell culture with PS3, beads, and/or other agents and pipet this cell suspension onto an 8-well ibidi μ-slide (300 μL/well).
Ensure that solutions of beads are mixed thoroughly before dispensing as the beads tend to sediment. After addition of beads to cells, pipet very gently to avoid damage.
Return the 8-well ibidi μ-slide to the CO2 incubator at 37°C for the treatment period.
Image the cells by confocal laser scanning microscopy (Ex. 488nm; Em. 500–600nm) without any additional washing steps.
4.7. Preparation of cells for analysis of fluorescence intensity on 96-well plates
Grow cells in complete media (15mL) in a vented T-75 flask at 37°C (5% CO2).
For adherent cell lines (RAW 264.7 and J774A.1), when >90% confluency is reached, use a sterile cell scraper to suspend the cells.
For suspension cell lines (THP-1 and Jurkat), grow to 8×105 cells/mL.
Determine the density using cell counting, flow cytometry, or a similar method.
Transfer the cell suspension to a 15mL conical tube and centrifuge at 2000rpm for 2min.
Remove the supernatant and resuspend cells in fresh complete media. Dilute in complete media to 2×105 cells/mL in a sufficient volume needed for assays.
For adherent cell lines (RAW 264.7 and J774A.1), dispense 200μL of this cell suspension into each assay well of a 96 well plate. Cultivate for 16–20h prior to treatment.
For suspension cell lines (THP-1 and Jurkat), prepare samples in 650μL of complete media for assays in triplicate.
On the day of the assay, prepare a fresh 10mM DMSO stock solution of PS3 from dry powder and dilute to 10μM in complete media (final [DMSO]=0.1%). Note: for a 96-well plate, prepare 220μL per well to dispense 200μL to each well.
For wells containing DNP-beads (negative control) and IgG/DNP-beads (positive control), add the appropriate volume of concentrated bead stock solutions (15–20M beads/mL) to afford a final concentration of 200,000 beads/mL. For wells containing IgG alone (negative control), add anti-DNP (200μg/mL) to a final concentration of 2μg/mL.
For adherent cell lines (RAW 264.7 and J774A.1), carefully aspirate the media from the assay plate and replace with complete treatment media (200μL). Mix thoroughly prior to dispensing into wells as the beads tend to sediment.
Incubate at 37°C (5% CO2) for 4h.
Remove plate and read fluorescence. A strong signal can be obtained with excitation at 480nm and emission at 525nm as is typically used for fluorescein. Note: If appropriate, instrument gain should be calibrated from wells containing opsonized beads, as these wells will exhibit the highest fluorescence.
Return plate to incubator and read after treatment for 24h.
Decontaminate biohazardous waste. For solids, autoclave. For liquids, treat with 10% bleach for >10min.
5. Future perspectives
pNitric oxide, superoxide, and PNT have been described metaphorically in the literature as the good, the bad, and the ugly (Beckman & Koppenol, 1996). PNT is considered ugly because it is highly toxic and rapidly reacts to form a wide variety of oxidized products. It additionally promotes nitration of tyrosine residues of biologically critical proteins, leading to their functional inactivation. However, this exceptional reactivity is beneficial when production of PNT is tightly controlled by phagocytic cells such as macrophages to kill foreign pathogens. Because PNT is so reactive, it is difficult to detect the transient production of this RNS during natural processes such as ADCP. To facilitate detection of endogenous PNT, we designed and synthesized PS3 as a fluorescent sensor that accumulates in membranes of the endoplasmic reticulum. This subcellular targeting approach enhances the proximity of this sensor to intracellular sites of generation of PNT such as the phagosome of macrophages. In contrast to the commercially available fluorescent sensors HPF and NiSPY-3, PS3 can uniquely detect PNT upon ADCP when phagocytic cell lines are stimulated with opsonized 10μm tentagel beads. This approach can be used to investigate cellular production of PNT by confocal microscopy and flow cytometry (Knewtson et al., 2018). We additionally describe here its use in simpler and higher-throughput assays of fluorescence intensity of cells in culture on microtiter plates. This approach has potential for phenotypic drug discovery of modulators of production of PNT and for related studies of ADCP.
Acknowledgments
We thank the NIH (R01-CA211720) and The Ohio State University Comprehensive Cancer Center (OSUCCC) for financial support.
References
- Alvarez B, & Radi R (2003). Peroxynitrite reactivity with amino acids and proteins. Amino Acids, 25(3–4), 295–311. 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- Arai S, Lee SC, Zhai D, Suzuki M, & Chang YT (2014). A molecular fluorescent probe for targeted visualization of temperature at the endoplasmic reticulum. Scientific Reports, 4, 6701 10.1038/srep06701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi S, & Radi R (2018). Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biology, 14, 618–625. 10.1016/j.redox.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, & Freeman BA (1990). Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Academy of Sciences of the United States of America, 87(4), 1620 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, & Koppenol WH (1996). Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. American Journal of Physiology. Cell Physiology, 271(5 Pt. 1), C1424–C1437. 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bezner BJ, Ryan LS, & Lippert AR (2020). Reaction-based luminescent probes for reactive sulfur, oxygen, and nitrogen species: Analytical techniques and recent progress. Analytical Chemistry, 92(1), 309–326. 10.1021/acs.analchem.9b04990. [DOI] [PubMed] [Google Scholar]
- Chan J, Dodani SC, & Chang CJ (2012). Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nature Chemistry, 4(12), 973–984. 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Ren W, Wright QE, & Ai HW (2013). Genetically encoded fluorescent probe for the selective detection of peroxynitrite. Journal of the American Chemical Society, 135(40), 14940–14943. 10.1021/ja408011q. [DOI] [PubMed] [Google Scholar]
- Colston J, Horobin R, Rashid-Doubell F, Pediani J, & Johal KK (2003). Why fluorescent probes for endoplasmic reticulum are selective: An experimental and QSAR-modelling study. Biotechnic & Histochemistry, 78(6), 323–332. 10.1080/10520290310001646659. [DOI] [PubMed] [Google Scholar]
- Degrossoli A, Muller A, Xie K, Schneider JF, Bader V, Winklhofer KF, et al. (2018). Neutrophil-generated HOCl leads to non-specific thiol oxidation in phagocytized bacteria. Elife, 7, pii: e32288 10.7554/eLife.32288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, & Vendrell M (2016). Smart fluorescent probes for imaging macrophage activity. Chemical Society Reviews, 45(5), 1182–1196. 10.1039/c5cs00567a. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Campolo N, Trujillo M, Bartesaghi S, Carballal S, Romero N, et al. (2018). Biochemistry of Peroxynitrite and protein tyrosine nitration. Chemical Reviews, 118(3), 1338–1408. 10.1021/acs.chemrev.7b00568. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G, & Radi R (2009). Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chemical Biology, 4(3), 161–177. 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- Fujisawa A, Tamura T, Yasueda Y, Kuwata K, & Hamachi I (2018). Chemical profiling of the endoplasmic reticulum proteome using designer labeling reagents. Journal of the American Chemical Society, 140(49), 17060–17070. 10.1021/jacs.8b08606. [DOI] [PubMed] [Google Scholar]
- Gao CC, Tian Y, Zhang RB, Jing J, & Zhang XL (2017). Endoplasmic reticulum-directed ratiometric fluorescent probe for quantitive detection of basal H2O2. Analytical Chemistry, 89(23), 12945–12950. 10.1021/acs.analchem.7b03809. [DOI] [PubMed] [Google Scholar]
- Ghule NV, Bhosale RS, Kharat K, Puyad AL, Bhosale SV, & Bhosale SV (2015). A Naphthalenediimide-based fluorescent sensor for detecting the pH within the rough endoplasmic reticulum of living cells. ChemPlusChem, 80(3), 485–489. 10.1002/cplu.201402307. [DOI] [PubMed] [Google Scholar]
- Gul N, & van Egmond M (2015). Antibody-dependent phagocytosis of tumor cells by macrophages: A potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Research, 75(23), 5008–5013. 10.1158/0008-5472.CAN-15-1330. [DOI] [PubMed] [Google Scholar]
- Guo BP, Jing J, Nie LX, Xin FY, Gao CC, Yang W, et al. (2018). A lysosome targetable versatile fluorescent probe for imaging viscosity and peroxynitrite with different fluorescence signals in living cells. Journal of Materials Chemistry B, 6(4), 580–585. 10.1039/c7tb02615c. [DOI] [PubMed] [Google Scholar]
- Hendriks D, Choi G, de Bruyn M, Wiersma VR, & Bremer E (2017). Antibody-based Cancer therapy: Successful agents and novel approaches. International Review of Cell and Molecular Biology, 331, 289–383. 10.1016/bs.ircmb.2016.10.002. [DOI] [PubMed] [Google Scholar]
- Ischiropoulosd H, Beckman JS, Crow JP, Ye YZ, Royall JA, & Kooy NW (1995). Detection of peroxynitrite. Methods, 7(1), 109–115. 10.1006/meth.1995.1016. [DOI] [Google Scholar]
- Jia X, Chen Q, Yang Y, Tang Y, Wang R, Xu Y, et al. (2016). FRET-based Mito-specific fluorescent probe for ratiometric detection and imaging of endogenous peroxynitrite: Dyad of Cy3 and Cy5. Journal of the American Chemical Society, 138(34), 10778–10781. 10.1021/jacs.6b06398. [DOI] [PubMed] [Google Scholar]
- Kamen L, Myneni S, Langsdorf C, Kho E, Ordonia B, Thakurta T, et al. (2019). A novel method for determining antibody-dependent cellular phagocytosis. Journal of Immunological Methods, 468, 55–60. 10.1016/j.jim.2019.03.001. [DOI] [PubMed] [Google Scholar]
- Knewtson KE, Rane D, & Peterson BR (2018). Targeting fluorescent sensors to endoplasmic reticulum membranes enables detection of peroxynitrite during cellular phagocytosis. ACS Chemical Biology, 13(9), 2595–2602. 10.1021/acschembio.8b00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Zhou DY, Li Y, Liu HW, Wu P, Ou-Yang J, et al. (2018). Efficient two-photon fluorescent probe for imaging of nitric oxide during endoplasmic reticulum stress. ACS Sensors, 3(11), 2311–2319. 10.1021/acssensors.8b00567. [DOI] [PubMed] [Google Scholar]
- Lin W, Buccella D, & Lippard SJ (2013). Visualization of peroxynitrite-induced changes of labile Zn2+ in the endoplasmic reticulum with benzoresorufin-based fluorescent probes. Journal of the American Chemical Society, 135(36), 13512–13520. 10.1021/ja4059487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde D, Wong R, & Seybold PG (2002). Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: Improved absolute standards for quantum yields. Photochemistry and Photobiology, 75(4), 327–334. . [DOI] [PubMed] [Google Scholar]
- McDonald L, Liu B, Taraboletti A, Whiddon K, Shriver LP, Konopka M, et al. (2016). Fluorescent flavonoids for endoplasmic reticulum cell imaging. Journal of Materials Chemistry B, 4(48), 7902–7908. 10.1039/c6tb02456d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinig JM, Fu L, & Peterson BR (2015). Synthesis of fluorophores that target small molecules to the endoplasmic reticulum of living mammalian cells. Angewandte Chemie, International Edition, 54(33), 9696–9699. 10.1002/anie.201504156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottram LF, Boonyarattanakalin S, Kovel RE, & Peterson BR (2006). The Pennsylvania green fluorophore: A hybrid of Oregon green and Tokyo green for the construction of hydrophobic and pH-insensitive molecular probes. Organic Letters, 8(4), 581–584. 10.1021/ol052655g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottram LF, Maddox E, Schwab M, Beaufils F, & Peterson BR (2007). A concise synthesis of the Pennsylvania green fluorophore and labeling of intracellular targets with O6-benzylguanine derivatives. Organic Letters, 9(19), 3741–3744. 10.1021/ol7015093. [DOI] [PubMed] [Google Scholar]
- Murfin LC, Weber M, Park SJ, Kim WT, Lopez-Alled CM, McMullin CL, et al. (2019). Azulene-derived fluorescent probe for bioimaging: Detection of reactive oxygen and nitrogen species by two-photon microscopy. Journal of the American Chemical Society, 141(49), 19389–19396. 10.1021/jacs.9b09813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon-Abell J, Obara CJ, Weigel AV, Li D, Legant WR, Xu CS, et al. (2016). Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science, 354(6311), aaf3928 10.1126/science.aaf3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, & Liaudet L (2007). Nitric oxide and peroxynitrite in health and disease. Physiological Reviews, 87(1), 315–424. 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Chen XM, Gao L, Zhang T, Wang W, Shen JG, et al. (2016). A rationally designed rhodamine-based fluorescent probe for molecular imaging of peroxynitrite in live cells and tissues. Chemical Science, 7(8), 5407–5413. 10.1039/c6sc00012f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Wong N-K, Chen X, Chan Y-K, Ho DH-H, Sun Z, et al. (2014). Molecular imaging of Peroxynitrite with HKGreen-4 in live cells and tissues. Journal of the American Chemical Society, 136(33), 11728–11734. 10.1021/ja504624q. [DOI] [PubMed] [Google Scholar]
- Phaniraj S, Gao Z, Rane D, & Peterson BR (2016). Hydrophobic resorufamine derivatives: Potent and selective red fluorescent probes of the endoplasmic reticulum of mammalian cells. Dyes and Pigments, 135, 127–133. 10.1016/j.dyepig.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M, Reiners JJ, Santiago AM, & Kessel D (2009). Monitoring singlet oxygen and hydroxyl radical formation with fluorescent probes during photodynamic therapy. Photochemistry and Photobiology, 85(5), 1177–1181. 10.1111/j.1751-1097.2009.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo C, Alvarez MN, & Radi R (2014). Peroxynitrite, a potent macrophage-derived oxidizing cytotoxin to combat invading pathogens. BioFactors, 40(2), 215–225. 10.1002/biof.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo C, Alvarez MN, Rios N, Peluffo G, Radi R, & Romero N (2015). Nitric oxide diffusion to red blood cells limits extracellular, but not intraphagosomal, peroxynitrite formation by macrophages. Free Radical Biology & Medicine, 87, 346–355. 10.1016/j.freeradbiomed.2015.06.027. [DOI] [PubMed] [Google Scholar]
- Prolo C, Rios N, Piacenza L,Álvarez MN, & Radi R (2018). Fluorescence and chemiluminescence approaches for peroxynitrite detection. Free Radical Biology & Medicine, 128, 59–68. 10.1016/j.freeradbiomed.2018.02.017. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, & Freeman BA (1991). Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Archives of Biochemistry and Biophysics, 288(2), 481–487. 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Knolker HJ, & Simons K (2010). Subcellular targeting strategies for drug design and delivery. Nature Reviews. Drug Discovery, 9(1), 29–42. 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- Rios N, Piacenza L, Trujillo M, Martinez A, Demicheli V, Prolo C, et al. (2016). Sensitive detection and estimation of cell-derived peroxynitrite fluxes using fluorescein-boronate. Free Radical Biology & Medicine, 101, 284–295. 10.1016/j.freeradbiomed.2016.08.033. [DOI] [PubMed] [Google Scholar]
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ, & Nagano T (2003). Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. The Journal of Biological Chemistry, 278(5), 3170–3175. 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Taneja TK, Mankouri J, & Sivaprasadarao A (2007). Molecular cell biology of KATP channels: Implications for neonatal diabetes. Expert Reviews in Molecular Medicine, 9(21), 1–17. 10.1017/S1462399407000403. [DOI] [PubMed] [Google Scholar]
- Sun WC, Gee KR, Klaubert DH, & Haugland RP (1997). Synthesis of fluorinated fluoresceins. The Journal of Organic Chemistry, 62(19), 6469–6475. Retrieved from <Go to ISI>://A1997XX49000011. [Google Scholar]
- Szabo C, Ischiropoulos H, & Radi R (2007). Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nature Reviews. Drug Discovery, 6(8), 662–680. 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Tiden AK, Sjogren T, Svensson M, Bernlind A, Senthilmohan R, Auchere F, et al. (2011). 2-Thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. The Journal of Biological Chemistry, 286(43), 37578–37589. 10.1074/jbc.M111.266981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno T, Urano Y, Kojima H, & Nagano T (2006). Mechanism-based molecular design of highly selective fluorescence probes for nitrative stress. Journal of the American Chemical Society, 128(33), 10640–10641. 10.1021/ja061972v. [DOI] [PubMed] [Google Scholar]
- Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, & Nagano T (2005). Evolution of fluorescein as a platform for finely tunable fluorescence probes. Journal of the American Chemical Society, 127(13), 4888–4894. 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- Vasilescu A, Gheorghiu M, & Peteu S (2017). Nanomaterial-based electrochemical sensors and optical probes for detection and imaging of peroxynitrite: A review. Microchimica Acta, 184, 649–675. 10.1007/s00604-017-2093-7. [DOI] [Google Scholar]
- Wang M, & Kaufman RJ (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature, 529(7586), 326–335. 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- Wang B, Yu F, Li P, Sun X, & Han K (2013). A BODIPY fluorescence probe modulated by selenoxide spirocyclization reaction for peroxynitrite detection and imaging in living cells. Dyes and Pigments, 96(2), 383–390. 10.1016/j.dyepig.2012.09.006. [DOI] [Google Scholar]
- Wardman P (2007). Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radical Biology & Medicine, 43(7), 995–1022. 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Weber M, Mackenzie AB, Bull SD, & James TD (2018). Fluorescence-based tool to detect endogenous peroxynitrite in M1-polarized murine J774.2 macrophages. Analytical Chemistry, 90(17), 10621–10627. 10.1021/acs.analchem.8b03035. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, & Kettle AJ (2013). Redox reactions and microbial killing in the neutrophil phagosome. Antioxidants & Redox Signaling, 18(6), 642–660. 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- Woydziak ZR, Fu L, & Peterson BR (2012). Synthesis of fluorinated benzophenones, xanthones, acridones, and thioxanthones by iterative nucleophilic aromatic substitution. The Journal of Organic Chemistry, 77(1), 473–481. 10.1021/jo202062f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Li P, Wang B, & Han K (2013). Reversible near-infrared fluorescent probe introducing tellurium to mimetic glutathione peroxidase for monitoring the redox cycles between peroxynitrite and glutathione in vivo. Journal of the American Chemical Society, 135(20), 7674–7680. 10.1021/ja401360a. [DOI] [PubMed] [Google Scholar]
- Yu F, Song P, Li P, Wang B, & Han K (2012). A fluorescent probe directly detect peroxynitrite based on boronate oxidation and its applications for fluorescence imaging in living cells. Analyst, 137(16), 3740–3749. 10.1039/C2AN35246J. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chen J, Xiong H, Zhang Y, Chen W, Sheng J, et al. (2019). An endoplasmic reticulum-targetable fluorescent probe for highly selective detection of hydrogen sulfide. Organic & Biomolecular Chemistry, 17(6), 1436–1441. 10.1039/c8ob02998a. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu J, Sun YQ, Huo Y, Li Y, Liu W, et al. (2015). A mitochondriatargetable fluorescent probe for peroxynitrite: Fast response and high selectivity. Chemical Communications, 51(13), 2721–2724. 10.1039/c4cc09122a. [DOI] [PubMed] [Google Scholar]
- Zhou X, Kwon Y, Kim G, Ryu JH, & Yoon J (2015). A ratiometric fluorescent probe based on a coumarin-hemicyanine scaffold for sensitive and selective detection of endogenous peroxynitrite. Biosensors & Bioelectronics, 64, 285–291. 10.1016/j.bios.2014.08.089. [DOI] [PubMed] [Google Scholar]
- Zhu BC, Zhang M, Wu L, Zhao ZY, Liu CY, Wang ZK, et al. (2018). A highly specific far-red fluorescent probe for imaging endogenous peroxynitrite in the mitochondria of living cells. Sensors and Actuators B: Chemical, 257, 436–441. 10.1016/j.snb.2017.10.170. [DOI] [Google Scholar]
- Zielonka J, Sikora A, Joseph J, & Kalyanaraman B (2010). Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: Direct reaction with boronate-based fluorescent probe. The Journal of Biological Chemistry, 285(19), 14210–14216. 10.1074/jbc.M110.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J, Zielonka M, Sikora A, Adamus J, Joseph J, Hardy M, et al. (2012). Global profiling of reactive oxygen and nitrogen species in biological systems: High-throughput real-time analyses. The Journal of Biological Chemistry, 287(5), 2984–2995. 10.1074/jbc.M111.309062. [DOI] [PMC free article] [PubMed] [Google Scholar]