

Abstract
Glycopeptides identified by tandem mass spectrometry rely on the identification of the peptide backbone sequence and the attached glycan(s) by the incomplete fragmentation of both moieties. This may lead to ambiguous identifications where multiple structures could explain the same spectrum equally well due to missing information in the mass spectrum or incorrect precursor mass determination. To date, approaches to solving these problems have been limited, and few inroads have been made to address these issues. We present a technique to address some of these challenges and demonstrate it on previously published data sets. We use a linear modeling approach to learn the influence of the glycan composition on the retention time of a glycopeptide and use these models to validate glycopeptides within the same experiment, detecting over 400 incorrect cases during the MS/MS search and correcting 75 cases that could not be identified based on mass alone. We make this technique available as a command line executable program, written in Python and C, freely available at https://github.com/mobiusklein/glycresoft in source form, with precompiled binaries for Windows.
Keywords: glycoproteomics, software, retention time, bioinformatics, C18 chromatography, metallic cation adduction, ambiguity, chromatogram extraction, chromatogram scoring
Graphical Abstract

INTRODUCTION
Glycosylation is one of the most abundant and diverse co- and post-translational protein modifications in nature.1 The majority of cell surface and secreted proteins are believed to be glycosylated,2 which leads to glycosylation being involved in a wide range of biological processes.1 Glycans come in several varieties with two major groups, N-linked glycans and O-linked glycans, each having distinct structural motifs both within and between groups.3,4 The same diversity that makes glycosylation a flexible functional component also makes it more difficult to study.4,5 Mass spectrometry (MS) is one of the most powerful tools for the study of many post-translational modifications, including glycosylation,6 particularly when coupled to separation techniques like liquid chromatography (LC). Different strategies have been proposed for enriching and separating glycopeptides prior to analysis6,7 as well.
Glycopeptide identification from tandem MS is a challenging task, requiring that we identify both the peptide and the attached glycan or glycans from incomplete, often mixed, fragmentation. The completeness of the glycan fragmentation depends heavily on the glycan’s size, the peptide sequence, the precursor charge, and the activation energy used in the case of collisional dissociation.8 This makes it difficult to fully characterize a glycan’s structure or composition, requiring that we control the false discovery rate (FDR) for both the peptide and the glycan components.9 In practice, this involves modeling how much evidence is present in the observed peptide+Y ions for a given glycan compared with an empirical null model. To reliably fragment as many different glycopeptides as possible, this requires using multiple collision energies,8,9 at the cost of longer tandem MS acquisition times and relying on instrument vendor-specific settings. We propose that retention time can be used to provide an orthogonal means of measuring the glycan composition that can augment evidence from fragmentation alone.
It has been well established in the literature that the addition of each monosaccharide induces some shift in the retention time of glycans and glycopeptides,10–13 and a computational method used to propagate identifications using monosaccharide differences that exploited this information has been described. Recently, Ang et al.14 published an extension to SSRCalc relating this phenomenon to shifts in hydrophobicity relative to the deglycosylated peptide analyzed in the same LC–MS run. SRRCalc’s model is tied to the chromatographic conditions and instrumentation modeled in ref 14. This may be prohibitive for groups using different chromatographic techniques, and if deglycosylated peptides are not present, then they must first be predicted and then used as a starting point for extrapolation, compounding the prediction error.
Glycans are made up of a wide array of monomers sharing a few common masses. These masses combined in varying proportions with common chemical shifts can reach similar sums that are difficult to differentiate on intact mass alone. We list a few relevant pairs of ambiguous combinations in Table 1. We note that other than NeuAc to 2 Fuc, all of these substitutions induce errors on the scale of hundredths of daltons or smaller. Such small errors may still pass a high-mass-accuracy error tolerance on the parts-per-million scale for a sufficiently large glycopeptide and, with the exception of those cases that eliminate NeuAc or NeuGc, cannot be reliably detected in a collisional dissociation tandem mass spectrum. Previous work has described some of these more common errors and others,15,16 whereas others have observed other shifts without connecting them to their substitution effects.17 We claim that the correct glycan can be determined by the retention time, letting us measure how often they corrupt glycopeptide identifications.
Table 1.
Common Glycan Composition-Specific Ambiguous Substitutions Discussed in This Studya
| component | mass (Da) | substitution | mass (Da) | error (Da) | note |
|---|---|---|---|---|---|
| NeuAc 1 | 291.0954 | Fuc 2 | 292.115 | 1.02 | monoisotopic peak error |
| NeuAc 1 NH3 1 | 308.121 | Hex 1 Fuc 1 | 308.110 | 0.011 | ammonium adduct |
| SO3 1 HexNAc 2 | 486.115 | Hex 3 | 486.158 | 0.0429 | sulfate substituent group |
| HexNAc 2 Fuc 1 NeuAc 2 | 1134.407 | Hex 7 | 1134.369 | 0.037 | |
| NeuAc 1 Hex 1 | 453.148 | NeuGc 1 Fuc 1 | 453.148 | 0.0 |
Because we cannot assume a chromatographic configuration or sample preparation to produce a broadly applicable method, we cannot suggest a general reusable model. We therefore propose a linear modeling approach that may work well with a modest amount of data. To demonstrate this, we apply this technique to two N-glycopeptide-focused case studies, one with a relatively simple proteome but a complex glycome and another with a complex proteome and a restricted glycome. We demonstrate how this technique may be used to detect deviation from expected patterns and correct common errors in glycan assignment and to identify glycoforms with insufficient glycan fragmentation to accept in isolation. We also include an implementation of this approach with the GlycReSoft version 0.4 suite of tools18 for postsearch model fitting and scoring. Although the work here used this search engine, the retention time modeling procedure does not require it and may be applied to any glycopeptide identifications written in a suitably formatted comma-separated text file. Data are available via ProteomeXchange with the identifier PXD018208.19
METHODS
Mass Spectrometry Data Acquisition
This study analyzed two previously published data sets taken from PXD00349820 and PXD009654.21 The data used from PXD003498 were acquired using a Waters NanoAcquity nanoflow chromatograph (Waters, Milford, MA) mounted with a Waters Xbridge reversed-phase column (150 μm × 100 mm) packed with 1.7 μm BEH C18 resin and a Waters trap column (180 μm × 20 mm) packed with 5 μm symmetry C18 stationary phase attached to a Q-Exactive Plus mass spectrometer (ThermoFisher Scientific, San Jose, CA). The data from PXD009654 were acquired using the Ultimate 3000 system (Thermo, San Jose, CA) for separation. The LC–MS/MS system contained a 4 cm C18 capillary trap column (200 μm i.d., C18 AQ beads (5 μm, 120 Å)) and a 12 cm C18 capillary analysis column (75 μm i.d., C18 AQ beads (3 μm, 120 Å)) attached to a Q-Exactive mass spectrometer (Thermo). For further details, please see the original publications.
Mass Spectrum Preprocessing
We downloaded six Thermo.raw data files for PXD003498 corresponding to Phil-82 and Phil-BS tryptic glycopeptide data and six case/control glycopeptide data-dependent acquisition Thermo.raw files from PXD009654.21 We processed the profile mass spectra using GlycReSoft’s deconvoluter, using no background reduction and averaging each MS1 spectrum with the preceding and following MS1 scan and using a glycopeptide averagine H15.75C10.93S0.02O6.47N1.65 for MS1 and the Senko peptide averagine22 for MSn spectra.
Glycopeptide Database Definition
For PXD003498, we created a glycan composition database using 3–10 Hex, 2–9 HexNAc, 0–4 Fuc, 0–5 NeuAc, and 0–1 sulfate, subject to the constraints that NeuAc < HexNA − 1 and Fuc < HexNAc. We combined this with a database of 10 protein sequences of IAV hemagglutinin proteins from Philippines 1982 and 1982-BS strains as well as reference sequences for other IAV proteins, adding constant carbamidomethylation on cysteine and variable deamidation on asparagine. For PXD009654, we created an N-glycan database of 448 compositions through biosynthetic simulation,23 which we combined with the UniProt Human Reference Proteome UP000005640,24 adding constant carbamidomethylation on cysteine and variable oxidation on methionine.
Glycopeptide Identification
We used a variant of GlycReSoft using a scoring model and a multipart FDR estimation strategy similar to pGlyco29 to identify glycopeptides in each data set. PXD009654 was originally acquired with stepped collision energy, making it an appropriate approach. Although PXD003498 was acquired with a single collision energy, we required that both the peptide and the glycan components be accurately identified to make our downstream model fitting more accurate.
We modified the glycan scoring step to allow the addition of a glycan modification, which may or may not persist through fragmentation. For PXD009654, we considered only +NH3 (ammonium adduct, +17.026), whereas for PXD003498 we allowed +NH3, +Na–H (sodium adduct, +21.98), and +K–H (potassium adduct, +37.95). Ammonium adducts would be reduced to proton adducts by collisional dissociation, but metallic cation adducts could persist through fragmentation. We also performed a search of PXD009654 without ammonium adduction as a control. We additionally modified the total score to have a slight bias toward better precursor mass accuracy.
Retention Time Modeling
We defined the retention time (RT) of a glycopeptide according to the apex of its chromatographic peak. We applied a Gaussian smoothing with σ = 1 to further reduce the jaggedness of the observed chromatographic peak following spectrum-level averaging. We used only the most intense chromatographic peak when multiple peaks were resolved.
We selected glycopeptides that were identified unadducted with an MS1 score greater than 018 and that passed a 1% Joint FDR threshold for modeling from PXD003498, and we modeled retention time using a weighted linear model RTi ~ I(Peptidei) + Hexi + HexNAci + Fuci + NeuAci + sulfatei, using for each peptide as well as across peptides within a single sample. We treated Peptide as a group intercept term.
We evaluated a within-peptide group glycoform based on the difference between its predicted and observed retention times , where St is the survival function of the t distribution with the predicting model’s degrees of freedom. This produces a score between 0 and 1, where 1 reflects a perfect match between the observed and predicted retention times, whereas 0 reflects an improbable divergence from model expectations.
For PXD009654, we applied the same criteria as for PXD003498 for filtering identified glycopeptides. Because fewer glycoforms per peptide were identified, we pooled all samples’ identifications together, and we modeled the retention time using a weighted linear model RTi ≈ I(Peptidei) + I(Samplei) + Hexi + HexNAci + Fuci + NeuAci, using . We treated both Peptide and Sample as group intercept terms. Because of the extreme differences in gradient conditions between the early and late stages of each LC–MS run, we split the glycopeptides in the data set into two subgroups, those eluting before 200 min and those eluting after 200 min. We additionally constrained the training data to require that at least two distinct glycoforms were identified for each peptide backbone in the training set. We fit a preliminary model on all of the filtered data, then scored its training data, discarding examples that scored <0.01 to reduce the influence of outliers on the parameter estimation. We carried out further analyses using the filtered model.
Incorrect Identification Detection
For each model fit, we scored each chromatogram within its training set, labeling those cases that scored below 0.1 as outliers for further analysis. We labeled these observations as a monoisotopic mass error (converting 1 NeuAc to 2 Fuc), an ammonium adduct (converting 1 NeuAc to 1 Fuc 1 Hex), an in-source fragmentation event, a chromatographic peak error, a glycan composition assignment error, or a lack of peptide sequence intercept. A monoisotopic mass error converting 1 NeuAc to 2 Fuc may occur frequently as a result of poor isotopic pattern quality during deconvolution in one or more charge states. We assigned the monoisotopic mass error label when replacing 2 Fuc with 1 NeuAc, which produced an improved model score. The same approach is sufficient for the ammonium adduct case, replacing 1 Fuc and 1 Hex with 1 NeuAc in the glycan composition, requiring a difference of 2 between Hex and HexNAc in the original glycan composition. A peptide sequence that was not part of the model training data does not have a peptide-specific intercept to use as a point of reference directly using the algorithm used here. We labeled an observation as in-source fragmentation where a smaller glycan composition eluted at an unexpected time and could be nested within the chromatographic peak of a larger glycan composition for the same peptide sequence. The remaining two labels, chromatographic peak error and glycan composition assignment error, could be assessed only by manual validation.
Unassigned MS1-RT Identification
We extracted all chromatograms from the deconvoluted experimental signal and searched for δ monosaccharide offsets from observed glycopeptides and scored those matches. We marked glycopeptides that were not part of the training examples as potentially novel and collected all MS2 spectra associated with them, which were used for spectral similarity comparisons between the identified glycopeptide and the RT predicted case.
RESULTS
Glycopeptides Identified
For PXD003498, we identified 209 distinct glycopeptides passing the FDR, MS1, and adduction filters across all six samples from hemagglutinin and neuraminidase, spanning 18 distinct peptide sequences with variable deamidation, of which 37 were sulfated. For PXD009654, we identified 472 distinct glycopeptides passing the filters across all six samples observed during all but the first gradient stage, shown in Table 2. In total there were 1512 glycopeptides identified, including replicates, of which 395 were identified with ammonium adduction based upon precursor mass accuracy.
Table 2.
Number of Glycopeptides Identified at Distinct Gradient Stages in PXD009654a
| gradient stage | time interval (min) | glycopeptide count |
|---|---|---|
| 3–6% | 0–5 | 0 |
| 6–35% | 5–170 | 266 |
| 35–45% | 170–195 | 92 |
| 45–90% | 195–210 | 34 |
| 90% | 210–220 | 63 |
Most glycopeptides eluted at between 6 and 45% hydrophobic gradient conditions, whereas ~12% eluted under 90% hydrophobic conditions, showing distinctly different chromatographic behavior.
We assigned multiple adduction types to the same glycopeptide in PXD003498, which do not alter the retention time of the analyte, as shown in Figure 1a. These also influence the fragments produced, as shown in Figure 2a. This implies that events like ammonium adduction or metallic cation adduction do not alter the retention time under conditions like those used in PXD003498. We were also able to assign ammonium adduction in PXD009654, as shown in Figure 2b.
Figure 1.
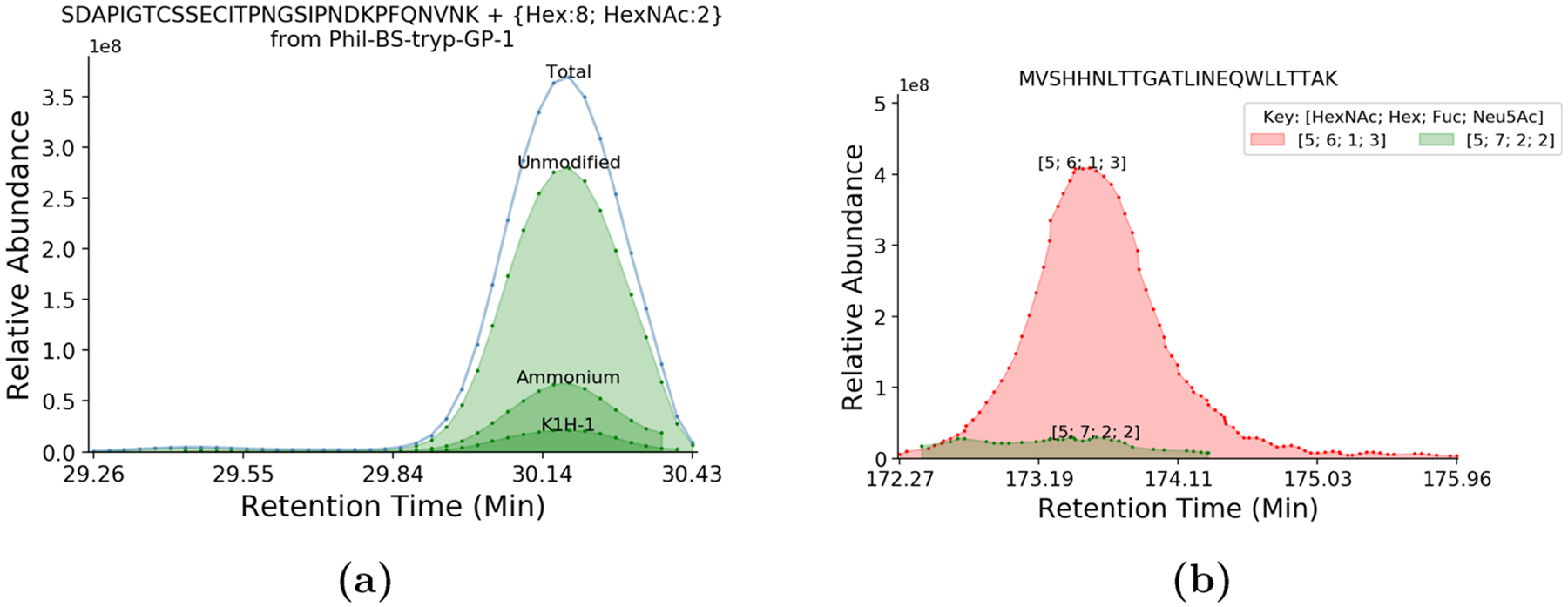
(a) Extracted ion chromatogram for SDAPIGTCSSECITPNGSIPNDKPFQNVNK glycosylated with Hex8HexNAc2, segregated by adduct type. We show that adducted analyte coelutes simultaneously with its normal protonated form and that some adducts are capable of altering the fragmentation pattern of a glycopeptide. (b) Extracted ion chromatograms for MVSHHNLTTGATLINEQWLLTTAK glycosylated with Fuc1Hex6HexNAc5NeuAc3 and putatively Fuc2Hex7HexNAc5NeuAc2. These two signals are completely overlapped in time. Assuming glycoform retention time shifts are driven by the glycan, we would expect these two compositions to elute further apart. This suggests that Fuc2Hex7HexNAc5NeuAc2 is actually an NH +4 adduct of Fuc1Hex6HexNAc5NeuAc3.
Figure 2.
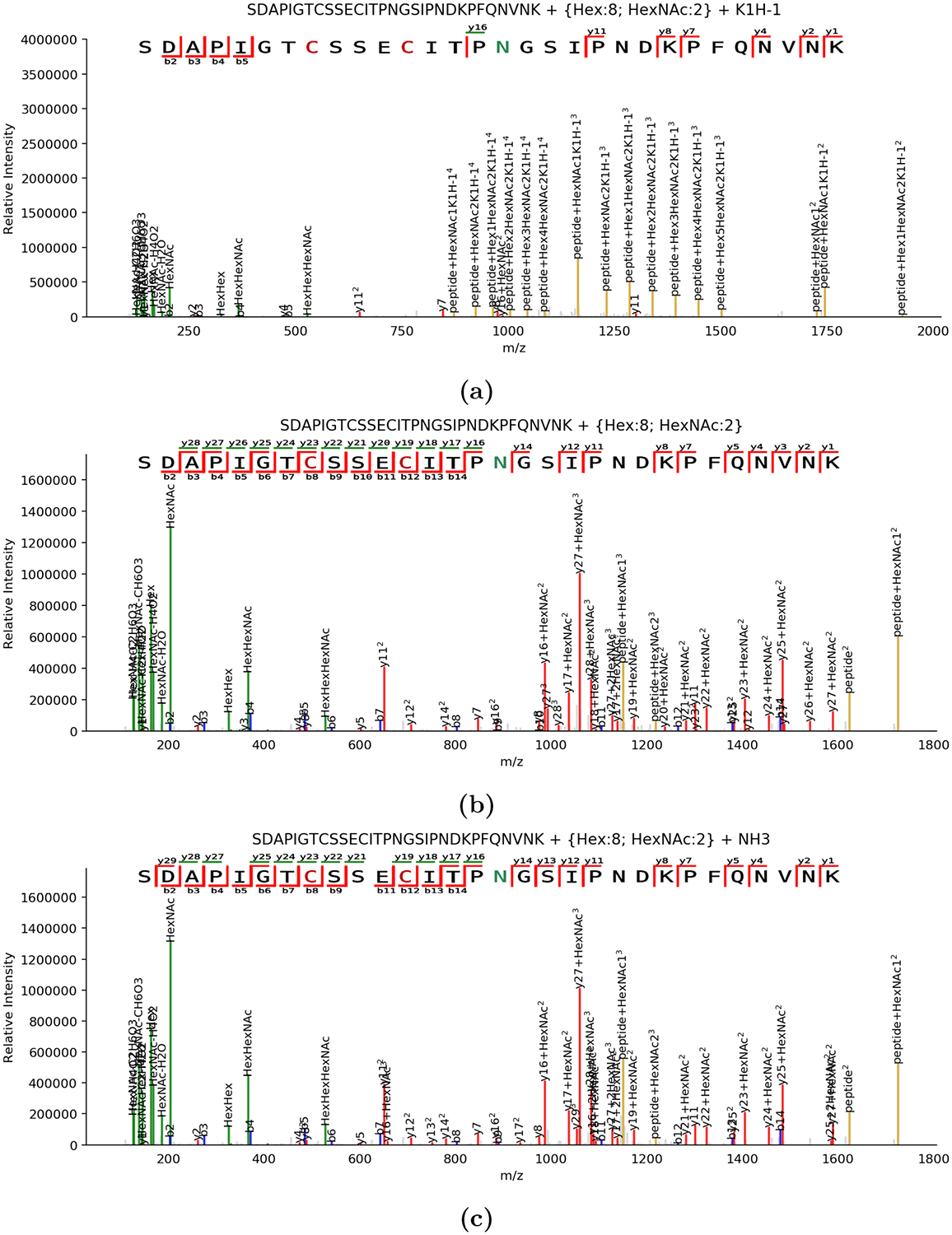
Set of annotated spectra for the same precursor in three different adduction states. All spectra shown are derived from 4+ precursors. (a) MS2 spectrum for SDAPIGTCSSECITPNGSIPNDKPFQNVNK glycosylated with Hex8HexNAc2 carrying a potassium adduct. In particular, we note that peptide+HexNAc+2 is present both with and without potassium adduction, whereas most other peptide+Y ions are only observed adducted. It is arguable that the potassium adduct may also associate with the peptide sequence at E or D, but insufficient fragmentation is available to confirm this. (b) MS2 spectrum for the same glycopeptide without any nonproton adducts. It has substantially more abundant peptide backbone fragmentation than the potassium adducted form in panel a, with fewer glycan-dominated fragments. (c) MS2 spectrum for the same glycopeptide matched to an ammoniated precursor, which has high similarity with the native spectrum (cosine similarity of 0.86).
Retention Time Modeling
For each sample in PXD003498, we estimated a sample-wide model, and a peptide specific model for each peptide that had more than 30 glycoforms, shown in Table 3. The two peptide specific models performed well, producing an R2 of 0.98 for both NGTYDHDVYR and NCTLIDALLGDPHCDGFQNEK. The experimental versus predicted retention times are shown in Figure 3a,b, respectively. The mean score of the cross-peptide model on its training data was 0.897, and the mean score for the peptide specific models were 0.961 for NGTYDHDVYR and 0.942 for NCTLIDALLGDPHCDGFQNEK.
Table 3.
Parameter Estimates (in min) for the Linear Model Fitted from Phil-BS-tryp-GP-1.raw of PXD003498a
| feature name | cross-peptide | NGTYDHDVYR | NCTLIDALLGDPHCDGFQNEK |
|---|---|---|---|
| QVIVDSGNR | 16.49 | - | - |
| NGTYDHDVYR | 16.67 | 16.19 | - |
| NGTYDHDVYRDEALN(d)NR | 20.89 | - | - |
| NGTYDHDVYRDEALNNR | 20.76 | - | - |
| GSNNSFFSR | 22.78 | - | - |
| YPVLNVTMPNN(d)GK | 32.83 | - | - |
| TITNDQIEVTNATELVQSSSTGR | 33.58 | - | - |
| SDAPIGTCSSECITPNGSIPNDKPFQNVNK | 31.09 | - | - |
| SDAPIGTCSSECITPNGSIPN(d)DKPFQNVNK | 31.66 | - | - |
| ILDGKNCTLIDALLGDPHCDGFQNEK | 42.47 | - | - |
| NCTLIDALLGDPHCDGFQNEK | 42.68 | - | 42.81 |
| NCTLIDALLGDPHCDGFQN(d)EK | 42.99 | - | - |
| Fuc | −0.13 | −0.08 | −0.16 |
| Hex | −0.09 | −0.10 | −0.10 |
| HexNAc | −0.06 | 0.05 | −0.09 |
| NeuAc | 0.60 | 0.59 | 0.0 |
| sulfate | 1.14 | 0.66 | 1.32 |
| count | 105 | 38 | 35 |
Parameter estimates for sulfate between the three models are substantial, suggesting that there is a physical difference underlying its behavior. We also observed a slight shift in the retention time apex caused by deamidation, denoted with a (d), although the start of a deamidated analyte is masked by the much more abundant native analyte’s ending.
Figure 3.

We show the accuracy of the within-peptide predictive models for PXD003498 compared with its reference data and use it to predict the retention times of theoretical assignments based on the MS1 mass match alone. The R2 shown in both plots is calculated from the fitted points only. We observed that in both cases, the model was able to predict unsulfated and singly sulfated cases accurately, but it did not work accurately on structures matched with two sulfates. The two-sulfate cases were all lower in abundance and had worse chromatographic peak quality than the singly sulfated cases, which, in turn, were lower in abundance than the unsulfated cases.
We fit a model across all samples in PXD009654, including all peptides eluting in the retention time intervals 0–200 (“early”) and 200–220 (“late”). We used 953 initial and 828 outlier-filtered observations for the early model and 222 initial and 220 outlier-filtered observations in the late model, with the parameters shown in Table 4. The R2 values were 0.999 and 0.996 for the early and late models, respectively. The mean score was 0.659 for the early model and 0.899 for the late model. We fit a model over the early time segment in the search without ammonium adducts (“uncorrected early”), with an R2 of 0.999 and with a mean score 0.493. The parameters are also shown in Table 4.
Table 4.
Parameter Estimates (in minutes) for PXD009654 across All Samples Segregated by Gradient Phase with Peptide- and Sample-Specific Coefficients Omitted for Brevitya
| feature name | early | late | uncorrected early | averaged δ early |
|---|---|---|---|---|
| Hex | −0.01 | 0.10 | 4.22 | |
| HexNAc | −0.07 | −0.27 | −2.59 | |
| Fuc | −0.47 | −0.09 | 0.04 | −0.38 |
| NeuAc | 6.53 | 2.39 | 4.75 | 6.31 |
There are significant differences in the parameters estimated for Fuc and NeuAc between the two conditions. Because of the magnitude of these differences, it is possible that they have overwhelmed the weight of the other monosaccharide parameters. The parameter estimates may be further confounded by the enrichment for sialylated glycopeptides used in this study. In the “uncorrected early” case, we see that the parameter values for Hex and NeuAc are pulled closer together to satisfy the ammonium adduct cases, and the HexNAc parameter must scale to compensate. For validation, for each distinct peptide sequence in each sample in the early partition, we computed the retention time apex difference between a glycan composition and a matched glycopeptides with the addition of a single monosaccharide for NeuAc and Fuc, shown in the “averaged δ early” column. We observe values much closer to the “early” model’s estimates than “uncorrected early”.
Incorrect Identification Detection
In the early partition, we evaluated the 953 identifications with the filtered model and found 160 observations with a score below 0.1, and 14 out of 206 were in the late partition. In the 160 cases in the early partition, we labeled 21 ammonium adduct errors, 43 deconvolution errors, 20 lack of peptide sequence intercept cases, 5 in-source fragmentation cases, and 70 unresolved cases for manual inspection. In the 14 cases in the late partition, we labeled 3 ammonium adduct errors and 11 deconvolution errors. Had no ammonium adduct correction been performed, there would be 407 additional ammonium adduct errors to contend with, accounting for 21.6% of the identifications having incorrect glycan assignments, or 24.3% of all identifications would be incorrect and accurately classified according to the model and the rules we specified. The fractions of identifications labeled are shown in Figure 4.
Figure 4.
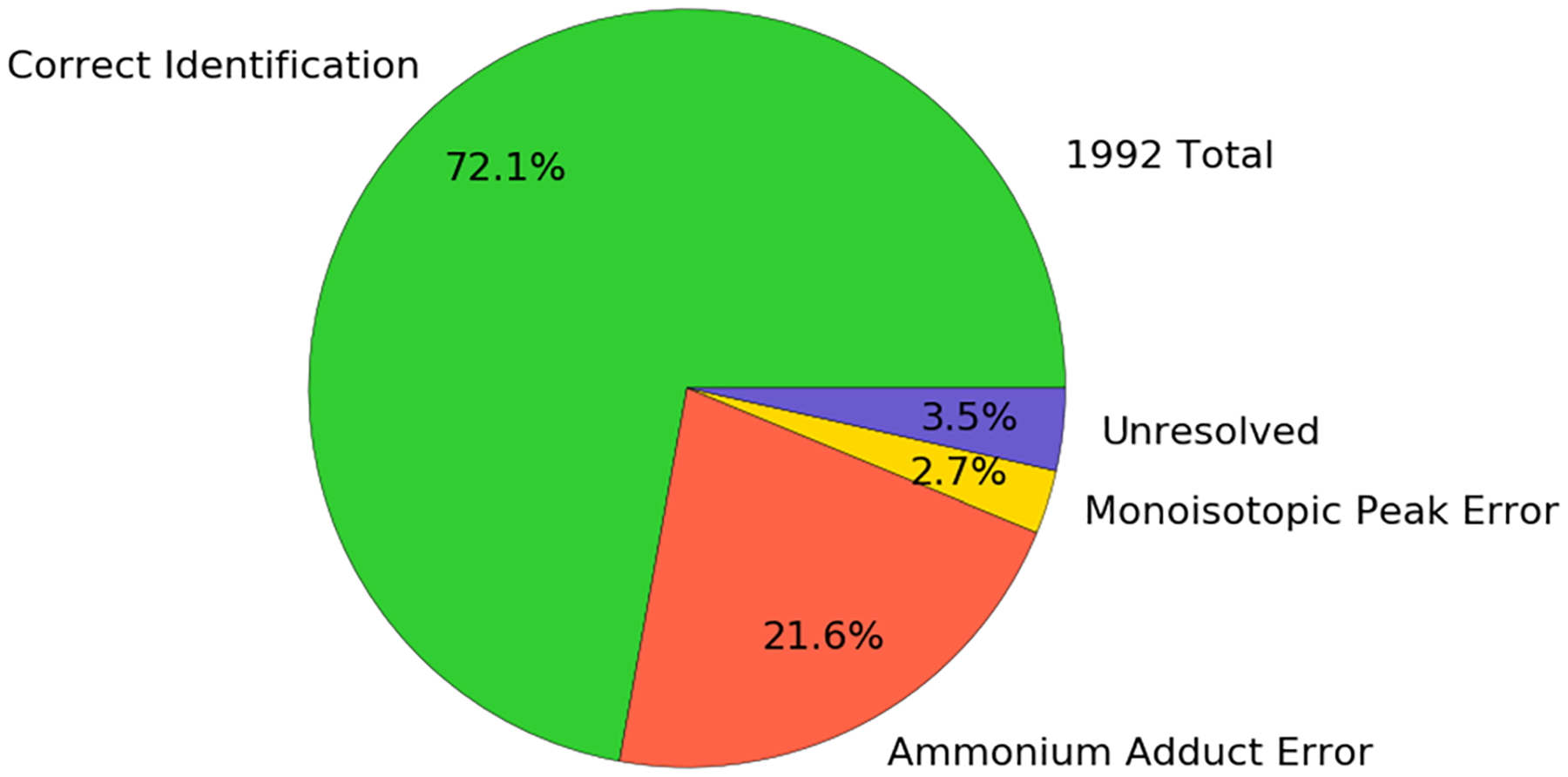
Pie chart describing the fraction of identifications that would be labeled as correct identifications, incorrect, or unresolved from PXD009654. The ammonium adducted fraction would be assigned a different glycan composition if ammonium adduction was not considered in the search, although the correct model estimates could be used to detect them.
No database identifications from PXD003498 fell below the threshold of 0.1.
Unassigned MS1-RT Identification
We extracted all deconvoluted chromatographic features and found 174 possible unassigned analytes that could be matched to δ monosaccharides mass shifts of database glycopeptides in Phil-BS-tryp-GP-1.raw of PXD003498. After filtering by model score, we found that 105 of these passed a score threshold of 0.75, and 72 passed a threshold of 0.9. The full distribution of scores is shown in Figure 5a. Several of these were linked with supporting MS/MS that did not pass the glycan FDR but had high-quality peptide backbone fragmentation to validate them.
Figure 5.

(a) Histogram of model scores for unidentified analytes assigned a peptide backbone and glycan composition relative to a database identified glycopeptide. (b) Spectrum from an unassigned precursor, not accepted initially because of insufficient glycan fragmentation but supported with a retention time model score of 0.95.
We found 152 possible unassigned analytes that could be matched in the early partition of PXD009654, but few had supporting MS/MS, and there was no correlation with the model score. Furthermore, there was little correlation between the model score and the prediction validation. There were 26 possible cases in the late partition, with similar concerns.
DISCUSSION
Glycopeptide identification relies on the often incomplete fragmentation of both the peptide and the glycan, and the range of precursor ion microheterogeneity may be too large to reliably cover within a single data-dependent acquisition LC–MS/MS. The nature of the collisional dissociation of the glycan component also prevents pairs of nearly isobaric but structurally distinct glycans from being discriminated, leading to distorted or implausible results. We sought to overcome these challenges to extract biologically meaningful insight from glycoproteomics. The chromatographic retention time of glycopeptides carries some structured information describing the glycan attached to a peptide, relative to other glycans attached to the same peptide, which can be used orthogonally to resolve the identity of a glycopeptide. A computational method that can leverage retention time information would allow us to address some of these shortcomings.
Our linear modeling approach learned the per-monosaccharide retention time shifts caused by a glycan relative to some peptide-specific theoretical baseline. We showed that such a model could be learned from individual LC–MS/MS experiments or averaged across multiple experiments using the same chromatographic conditions. We demonstrated its efficacy in predicting the retention times of correctly and incorrectly assigned glycopeptides, with and without chemical adduction. This approach allows an approximate retention time prediction for cases without the deglycosylated peptide present, as in ref 14. Sulfated N-glycans can be discriminated from nonsulfated forms of the same glycan by the retention time shift caused by the sulfate modification and would otherwise be identified as hybrid or high mannose structures, possibly with improbable fucosylation caused by the substitution rules described in Table 1. This may also be useful when analyzing proteoglycan glycosylation including glycosaminoglycans and keratan sulfate precursors.23 Our approach worked well for a single sulfate group but did not extrapolate two sulfates accurately, as shown in Figures 3a,b, suggesting that there may be other factors at work, such as peptide–glycan interactions, although no two-sulfate case was identified by MS/MS. We did not look for sulfated glycans in PXD009654.
In the uncorrected early case, the model had a lower average score for its training data, and its parameters did not reflect the observed differences when considering single monosaccharide differences between two glycans on the same backbone, as shown in Table 4. We could not compute an average estimate for Hex or HexNAc because these monosaccharides are readily contaminated by uncorrected ammonium adduction. Even so, the uncorrected early model did not reproduce its training data as well as the early model did, with an average score of 0.56 compared with the corrected model at 0.65. This could be explained by overfitting, so we used a hierarchical three-fold cross-validation, holding replicate representation balanced over each split and all replicate- and peptide-level parameters equal, and computed the average score on the test set for each model. The average of these averages came to 0.56 for the uncorrected early model and 0.59 for the corrected model. The difference between these two values is smaller than the full data set average, but combined with the other evidence, we believe this makes a reasonable case for the corrected model.
The demonstration of cation adduction in the course of normal glycoproteomics analysis is a challenge for many existing tools. Several studies9,17,20,21,25,26 have been published with compositions resembling uncorrected ammonium adducts, like the one described in Figure 1b, or have had ammonium adduction detected in their data by the authors or others. We demonstrate here a technique that can identify them and possibly correct them. Methods to reduce the abundances of adducted glycopeptide ions in LC–MS data have not yet been described. Other metallic cation adducts may also be present, including sodium, calcium, and iron, as shown in Figure S-1. It can be difficult to deconvolute ammonium and sodium adduction because their isotopic patterns overlap, as may other metallic cation pairs such as calcium and potassium. These adducts can also induce significant changes in the fragmentation process, as shown in Figures 2a and Figure S-2, contrasting the fragmentation of a single glycopeptide in its native state with an ammonium adduct and a potassium adduct. Although we did not actively look for metallic cation adduction in PXD009654, it is present, as shown in Figure S-2a and by the MS/MS of the sodium and potassium adducted species. This technique may also be similarly used for deconvolution artifacts, even when the correct glycan has fucosylation.16 These deconvolution error cases may be more difficult to quantify because they may reflect an issue with the raw precursor data itself rather than the interpretation. This is of particular note in ref 21, where Hex7HexNAc5NeuAc2Fuc2 at HPTR N126 is reported as differentially expressed but coelutes exactly with Hex6HexNAc5NeuAc3Fuc1 at the same site, suggesting that it is instead an ammonium adduct of the smaller glycan, as shown in Figure 1b. The early model fit to this data would give the Hex7HexNAc5NeuAc2Fuc2 a score close to 0, whereas it would evaluate Hex6HexNAc5NeuAc3Fuc1 at 0.97 on 20160301_Serum_SA_glycopeptide_HCC_1.raw. Similarly, Hex5HexNAc4NeuAc1Fuc3 is reported to be differentially expressed but could also be the result of a deconvolution artifact. Using the same sample from ref 21, the early model scores the artifact composition close to zero, but the “corrected” composition score is 0.27.
Although accounting for adduction during the search disambiguated the majority of cases, it would be desirable to eliminate these adducts experimentally. MS/MS spectra are often acquired on adducted forms of previously identified ions, not identifying new structures. These adducts split the signal of each structure across multiple ions, increasing the quantification error and increasing the complexity of MS1 spectra, reducing our ability to observe other, low abundance signals. Finally, accounting for them dramatically increases search times because each spectrum must be considered with all adducts, increasing the number of structures to match against each spectrum.
Our method performed well on PXD003498, where there were many glycoforms to learn parameters on a single peptide backbone sequence. This compounded with the large range of microheterogeneity, which made this modeling approach useful. The comparative lack of microheterogeneity to exploit in the majority of the peptides covered in PXD009654, along with the larger variability in outcome caused by averaging over multiple samples and the parameter magnitude, contributed to the poorer performance in the de novo prediction. In general, basing an identification on the retention time and accurate mass alone is tenuous and requires that other supporting information be considered, similar to cases like cofragmented data-independent-acquisition spectra. The scoring function itself could also benefit from a better representation of the scale of the observed error in the training data, which might also help when parameter magnitudes are large.
This problem may be modeled in many different ways. A linear mixed effects model may be more appropriate for learning across peptide and sample groups than a classical linear model, because these represent group-like structures. It might also be possible to achieve better parameter estimates by learning relative differences among all glycoforms of the same peptide, normalizing away the peptide-specific term, followed by a back-calculation to estimate the theoretical starting point for each peptide group. Such a recalibration procedure may be useful for projecting either modeling approach onto data from peptides not in the model but governed by the same chromatographic conditions. Our strategy may also be useful when implementing a match-between-runs strategy for glycopeptide quantification or an invariant to preserve during the LC–MS alignment of enriched glycopeptides, a specialized subproblem that is not well studied.27 Likewise, our results from PXD009654 may have been improved by first aligning all six samples to reduce the between-sample errors shown in Figure S-5, but we did not want to distort the within-sample retention times. Our approach did not require using deglycosylated peptides and used either the earliest or the latest eluting glycoform as a proxy for the base peptide. If we had included the deglycosylated peptide, then the parameter estimates for each peptide-specific intercept would have been more reliable, and the fitting of peptide-specific models would therefore have been more stable14 potentially required fewer distinct glycoforms. Our method should be applicable without modification of data including deglycosylated peptides, encoded as having a glycan with zero of each monosaccharide.
Previous publications have described similar retention time shifts for glycans and glycopeptides.10,11 The fact that our observations, those found in ref 14, and others do not match is likely due to differences in the chromatographic conditions and instrumentation. Even between our two case studies we see substantial differences in monosaccharide-induced retention time shifts. This further suggests that unless standardized methods and equipment come into use, appropriate models may be difficult to find. Indeed, ref 21 notes that using deglycopeptides to provide anchor points for validating glycopeptides,12 which indirectly exploited the phenomenon studied here and in ref 14, failed when using longer chromatography columns. A normalization approach similar to spiked in iRT peptides28 might be able to help to get accurate parameter estimates, but drastic differences in gradient conditions may require that there be many peptide backbones, and the broad range of glycoforms to be sampled at each point might make this impractical in complex samples.
CONCLUSIONS
We present a computational approach for the orthogonal validation of N-glycopeptide identifications by retention time, even for some cases where inadequate glycan fragmentation is observed, and show common cases where errors occur if a technique like this is not used. We demonstrated these techniques on published data sets, showing cases where they may have impacted the results of the studies from which these data sets were drawn. This method does not require the addition of any new reagents and may be estimated from any identified glycosite with sufficient complexity and coverage. We implement this postprocessing step as a library and tool in the GlycReSoft collection, which are freely available at https://github.com/mobiusklein/glycresoft.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by NIH grant U01CA221234.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00051.
Figure S-1. MS spectrum of SDAPIGTCSSECITPNGS-IPNDKPFQNVNK{Hex:8; HexNAc:2} showing multiple metallic cation adducted species alongside the native precursor. Figure S-2. Metallic cation adduction in PXD009654 MSn spectra of MVSHHN(N-glycosylation)LTTGATLINEQWLLTTAK{Hex:6; Hex-NAc:5; NeuAc:3}. Figure S-3. Distribution of pairwise retention time differences for single monosaccharides in PXD009654. Figure S-4. Distribution of pairwise monosaccharide retention time offsets when ammonium adducts are not considered. Figure S-5. Dispersion within glycoform between runs. Table S-1. Columns required by the retention time modeling tool “glycresoft analyze retention-time fit-glycopeptide-retention-time” (PDF) Glycopeptide chromatogram evaluations done on glycopeptides identified from PXD009654’s “early” partition. G glycopeptide chromatogram evaluations done on glycopeptides identified from PXD009654’s “late” partition (ZIP)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00051
The authors declare no competing financial interest.
Contributor Information
Joshua Klein, Program for Bioinformatics, Boston University, Boston, Massachusetts 02215, United States;.
Joseph Zaia, Program for Bioinformatics and Department of Biochemistry, Boston University, Boston, Massachusetts 02215, United States;.
REFERENCES
- (1).Varki A Biological roles of glycans. Glycobiology 2017, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Apweiler R On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta, Gen. Subj 1999, 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- (3).Stanley P; Schachter H; Taniguchi N Essentials of Glycobiology; Cold Spring Harbor Laboratory Press, 2009. [PubMed] [Google Scholar]
- (4).Cummings RD The repertoire of glycan determinants in the human glycome. Mol. BioSyst 2009, 5, 1087–104. [DOI] [PubMed] [Google Scholar]
- (5).Moremen KW; Tiemeyer M; Nairn AV Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kim U; Oh MJ; Lee J; Hwang HY; An HJ MS-based technologies for the study of site-specific glycosylation. Mass Spectrom. Lett 2018, 8, 69–78. [Google Scholar]
- (7).Ongay S; Boichenko A; Govorukhina N; Bischoff R Glycopeptide enrichment and separation for protein glycosylation analysis. J. Sep. Sci 2012, 35, 2341–2372. [DOI] [PubMed] [Google Scholar]
- (8).Hinneburg H; Stavenhagen K; Schweiger-Hufnagel U; Pengelley S; Jabs W; Seeberger PH; Silva DV; Wuhrer M; Kolarich D The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. J. Am. Soc. Mass Spectrom 2016, 27, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu M-Q; Zeng W-F; Fang P; Cao W-Q; Liu C; Yan G-Q; Zhang Y; Peng C; Wu J-Q; Zhang X-J; et al. pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun 2017, 8, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang B; Tsybovsky Y; Palczewski K; Chance MR Reliable determination of site-specific in vivo protein N-glycosylation based on collision-induced MS/MS and chromatographic retention time. J. Am. Soc. Mass Spectrom 2014, 25, 729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hu Y; Shihab T; Zhou S; Wooding K; Mechref Y LC-MS/MS of permethylated N-glycans derived from model and human blood serum glycoproteins. Electrophoresis 2016, 37, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cheng K; Chen R; Seebun D; Ye M; Figeys D; Zou H Large-scale characterization of intact N-glycopeptides using an automated glycoproteomic method. J. Proteomics 2014, 110, 145–154. [DOI] [PubMed] [Google Scholar]
- (13).Choo MS; Wan C; Rudd PM; Nguyen-Khuong T GlycopeptideGraphMS: Improved Glycopeptide Detection and Identification by Exploiting Graph Theoretical Patterns in Mass and Retention Time. Anal. Chem 2019, 91, 7236–7244. [DOI] [PubMed] [Google Scholar]
- (14).Ang E; Neustaeter H; Spicer V; Perreault H; Krokhin O Retention Time Prediction for Glycopeptides in Reversed-Phase Chromatography for Glycoproteomic Applications. Anal. Chem 2019, 91, 13360–13366. [DOI] [PubMed] [Google Scholar]
- (15).Darula Z; Medzihradszky KF Carbamidomethylation Side Reactions May Lead to Glycan Misassignments in Glycopeptide Analysis. Anal. Chem 2015, 87, 6297–6302. [DOI] [PubMed] [Google Scholar]
- (16).Lee LY; Moh ES; Parker BL; Bern M; Packer NH; Thaysen-Andersen M Toward Automated N-Glycopeptide Identification in Glycoproteomics. J. Proteome Res 2016, 15, 3904–3915. [DOI] [PubMed] [Google Scholar]
- (17).Toghi Eshghi S; Shah P; Yang W; Li X; Zhang H GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem 2015, 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Klein J; Carvalho L; Zaia J Application of network smoothing to glycan LC-MS profiling. Bioinformatics 2018, 34, 3511–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vizcaíno JA; Csordas A; Del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu QW; Wang R; Hermjakob H 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Khatri K; Klein JA; White MR; Grant OC; Leymarie N; Woods RJ; Hartshorn KL; Zaia J Integrated Omics and Computational Glycobiology Reveal Structural Basis for Influenza A Virus Glycan Microheterogeneity and Host Interactions. Mol. Cell. Proteomics 2016, 15, 1895–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Qin H; Dong X; Mao J; Chen Y; Dong M; Wang L; Guo Z; Liang X; Ye M Highly Efficient Analysis of Glycoprotein Sialylation in Human Serum by Simultaneous Quantification of Glycosites and Site-Specific Glycoforms. J. Proteome Res 2019, 18, 3439–3446. [DOI] [PubMed] [Google Scholar]
- (22).Senko MW; Beu SC; McLaffertycor FW Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom 1995, 6, 229–233. [DOI] [PubMed] [Google Scholar]
- (23).Klein J; Zaia J glypy: An Open Source Glycoinformatics Library. J. Proteome Res 2019, 18, 3532–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hu Y; Shah P; Clark DJ; Ao M; Zhang H Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Anal. Chem 2018, 90, 8065–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bollineni RC; Koehler CJ; Gislefoss RE; Anonsen JH; Thiede B Large-scale intact glycopeptide identification by Mascot database search. Sci. Rep 2018, 8, 2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mayampurath A; Song E; Mathur A; Yu C-Y; Hammoud Z; Mechref Y; Tang H Label-free glycopeptide quantification for biomarker discovery in human sera. J. Proteome Res 2014, 13, 4821–32. [DOI] [PubMed] [Google Scholar]
- (28).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.