

Abstract
Parkinson’s disease (PD) is one of the most common movement disorders with loss of dopaminergic neurons and presence of Lewy bodies in certain brain areas. However, it is not clear how Lewy body (inclusion with protein aggregation) formation occurs. Mutations in Leucine-rich repeat kinase 2 (LRRK2) can cause a genetic form of PD and contribute to sporadic PD with the typical Lewy body pathology. Here we used our recently identified LRRK2 GTP-binding inhibitors as pharmacological probes to study the LRRK2-linked ubiquitination and protein aggregation. Pharmacological inhibition of GTP-binding by GTP-binding inhibitors (68 and Fx2149) increased LRRK2-linked ubiquitination predominantly via K27 linkage. Compound 68- or Fx2149 increased G2019S-LRRK2-linked ubiquitinated aggregates, which occurred through the atypical linkage types K27 and K63. Co-expression of K27R and K63R, which prevented ubiquitination via K27 and K63 linkages, reversed the effects of 68 and Fx2149. Moreover, 68 and Fx2149 also promoted G2019S-LRRK2-linked aggresome (Lewy body-like inclusion) formation via K27 and K63 linkages. These findings demonstrate that LRRK2 GTP-binding activity is critical in LRRK2-linked ubiquitination and aggregation formation. These studies provide novel insight into the LRRK2-linked Lewy body-like inclusion formation underlying PD pathogenesis.
Keywords: LRRK2, Parkinson’s disease, GTP-binding inhibitors, ubiquitination, protein aggregation, aggresome
Introduction
Parkinson’s disease (PD) is one of the most common movement disorders with loss of dopaminergic neurons and presence of Lewy bodies in certain brain areas. A Lewy body is composed of abnormal ubiquitin-positive protein aggregation due to dysfunction of the protein degradation system. This includes the dysfunctions in both the ubiquitin-proteasome system (UPS) and the lysosome/autophagy system. Impaired UPS has previously been implicated in protein aggregation in PD pathogenesis (Cook and Petrucelli, 2009; Atkin and Paulson, 2014). Ubiquitination of misfolded proteins typically signals for their processing and degradation by the proteasome or via autophagy, however in some cases can signal for alternative processing (Glickman and Ciechanover, 2002; Hochstrasser, 2009; Pickart, 2000) such as protein aggregation to form inclusions. Lewy bodies in PD brains are ubiquitinated, and these aggregates are believed to represent the neuron’s attempt at protecting itself by sequestering toxic protein aggregates (Hadian et al., 2011; Dantuma and Bott, 2014; Ciechanover and Kwon, 2015).
Several proteins that are involved in PD pathogenesis are directly involved in the UPS, such as Leucine Rich Repeat Kinase 2 (LRRK2) (Skibinski et al., 2014; Smith et al., 2005). Mutations in LRRK2 are among the most common causes of PD and can contribute towards 5–40% of familial PD cases and 1–5% of sporadic PD cases (Zimprich et al., 2004; Mamais et al., 2018; Dzamko et al., 2017; Heckman et al., 2016). LRRK2 undergoes ubiquitination and has implications in PD pathogenesis (Nucifora, Jr. et al., 2016; Smith et al., 2005). In LRRK2 mutation cases, there are ubiquitin positive Lewy bodies and ubiquitin positive neurites in brain regions (Zimprich et al., 2004; Mamais et al., 2018; Dzamko et al., 2017; Heckman et al., 2016). Impaired UPS functions have been observed in LRRK2 PD models and key UPS players have been shown to interact with LRRK2 (Ko et al., 2009). LRRK2 can interact with E3 ubiquitin ligases including Parkin (mutations in which also cause familial PD) and CHIP (Skibinski et al., 2014; Smith et al., 2005; Ko et al., 2009). These interactions influence LRRK2 ubiquitination and degradation, thereby altering disease progression.
LRRK2 protein contains both a kinase and a GTPase domain, and processes kinase, GTP-binding and GTPase activities. Many studies, including our own, suggest a genetic gain-of-function mechanism, potentially involving dysregulation of these enzymatic activities, as an important component of mutant LRRK2-linked pathogenesis (Zimprich et al., 2004; Mamais et al., 2018; Dzamko et al., 2017; Heckman et al., 2016). The most prevalent mutation, G2019S causes about 5% of familial PD, and has been shown to increase the kinase activity of LRRK2 (Zimprich et al., 2004; Mamais et al., 2018; Dzamko et al., 2017; Heckman et al., 2016). A previous study demonstrated that dephosphorylation of LRRK2 leads to an increase in its level of ubiquitination (Zhao et al., 2015), suggesting the LRRK2 kinase activity may play a role in LRRK2 ubiquitination. Thus, we are interested to investigate whether LRRK2 GTP-binding inhibition alters LRRK2-linked ubiquitination and protein aggregation underlying Lewy body formation.
Recently, we have discovered relatively specific LRRK2 GTP-binding inhibitors, 68 and Fx2149, which do not affect LRRK1 or another small GTPase (Li et al., 2014;Li et al., 2015). Moreover, 68 and Fx2149 also decrease phosphorylation of LRRK2, as well as increase neuronal survival both in LRRK2 in vitro and transgenic mouse brain models (Li et al., 2014;Li et al., 2015). In this study, we investigated whether GTP-binding inhibition alters LRRK2-linked ubiquitination and aggregation. We used a combination of immunoprecipitation and immunocytochemistry approaches to monitor these changes both with and without treatment of LRRK2 GTP-binding inhibitors, 68 and Fx2149. Our studies provide novel insight into the roles of LRRK2 GTP-binding activity in protein ubiquitination and Lewy body-like inclusion formation underlying PD pathogenesis.
Materials and Methods
Reagents
Compound 68 was ordered through Chembridge and Fx2149 was designed/synthesized by Dr. Fengtian Xue’s group using 68 as a lead compound (Li et al., 2014;Li et al., 2015). Both compounds were dissolved in 0.1% DMSO for use in biochemical assays. Anti-Flag antibodies, γ-tubulin antibody, anti-Flag-agarose, GTP-agarose, and PR-619 (deubiquitinase inhibitor) were purchased from Sigma (St. Louis, MO, USA). Anti-HA antibody (clone Y-11) was purchased from Santa Cruz (Dallas, TX, USA). Anti-ubiquitin antibody (clone P4D1) was purchased from Covance (Princeton, NJ, USA). Alexa Fluor 568 goat anti-rabbit antibody, LipofectAMINE and Plus transfection reagents, and DMEM cell culture media were from Invitrogen™, and FITC goat anti-mouse antibody was from EMD Millipore (Billerica, MA, USA).
Cell culture and transfection
HEK293T (ATCC, Manassas, VA, USA) were grown in 10% FBS/ 1% penicillin-streptomycin DMEM as described previously (Li et al., 2010). Flag-wild-type and Flag-G2019S LRRK2 plasmids were cloned using a pcDNA3.1 vector backbone as previously described (Ko et al., 2009;Smith et al., 2006). For endogenous ubiquitin studies, LRRK2 constructs were transfected into HEK293T cells using Lipofectamine™ and PLUS™ Reagents (Invitrogen) according to the manufacturer’s protocol for a total of 72 h. For ubiquitin overexpression experiments, both Flag-tagged LRRK2 and HA-tagged ubiquitin plasmids were co-transfected at equal ratios. HA-tagged ubiquitin lysine mutant and lysine-arginine mutant constructs were described previously (Nucifora, Jr. et al., 2016). Compound treatments were administered 24 h prior to cell harvesting or cell fixation.
Ubiquitination assay and western blot analysis
For endogenous ubiquitin experiments, HEK293T cells expressing LRRK2 for 48 h and treated with either vehicle or inhibitors for an additional 24 h were harvested using lysis buffer (Cell Signaling). For PR-619 deubiquitinase inhibitor assay, cells were treated with PR-619 (50 μM) for 30 min prior to harvesting. Lysates were brought to a volume of 500 μg in 500 μL of lysis buffer, and allowed to incubate with anti-Flag agarose for 3 h in 4°C for immunoprecipitation (IP) to pull down LRRK2 variants. Conjugated-agarose was washed three times in cell lysis buffer, once in PBS, and eluted in 2X Loading buffer (Invitrogen) and 3% βME. Samples were denatured for 5 min at 85°C, resolved on 4–12% NuPAGE Bis-Tris gels, and transferred for 3.5 h on PVDF membranes. Membranes were probed for anti-ubiquitin and anti-Flag (LRRK2) controls.
For HA-tagged ubiquitin experiments, HEK293T cells were transfected with LRRK2 and ubiquitin constructs for 48 h and treated with either vehicle or inhibitors for an additional 24 h. Lysates were collected and brought to a volume of 500 μg in 500 μL of lysis buffer and incubated with anti-Flag agarose for 1 h at 4°C. Samples were eluted and western blot procedures were then performed as done with endogenous ubiquitin, however membranes were probed with anti-HA antibody instead.
Quantification of LRRK2 ubiquitination was performed by measuring densities using NIH image J software with experiments repeated 3–5 times. To compare the LRRK2 ubiquitination crossing the blots, we used the WT-LRRK2 ubiquitination alone in each blot as the baseline for ubiquitination (as 1), and any other experimental lanes will be the relative fold increase compared against WT-LRRK2 ubiquitination baseline. Briefly, each blot included a wild type (WT)-LRRK2 baseline ubiquitination control (cells expressing WT-LRRK2 treated with vehicle, or cells expressing WT-LRRK2 and HA-wild type ubiquitin treated with vehicle). The ratio of various LRRK2 ubiquitination in IP sample/input protein, in each experimental lane was measured relative to the control, LRRK2 ubiquitination baseline lane (ratio of WT-LRRK2 ubiquitination in IP sample/input protein).
LRRK2 GTP binding assay and kinase assay
LRRK2 kinase assay was performed as described previously (Li et al., 2014;Li et al., 2015). Cells were transfected with various LRRK2 variants for 36 h, then underwent no-serum starvation for 12 h followed by treatment with 68 or FX 2149 for 1 h. Cell lysates were immunoprecipitated using anti-Flag antibodies followed by immunoblotting assays with anti-phosphorylation LRRK2 antibodies as described previously (Li et al., 2014;Li et al., 2015). GTP-binding assays were performed as described previously (Li et al., 2014;Li et al., 2015). Cell lysates (100 μg protein/per reaction) were treated with vehicle or compounds 68 or Fx2149 for 1 h, then GTP-agarose beads (Sigma) were added for an additional 2 h at 4°C. Beads were washed with cell lysis buffer, then eluted by adding SDS-PAGE loading buffer and heating for 5 min at 72°C. Precipitates were subjected to western blot analysis using anti-Flag antibodies to detect LRRK2 proteins
Mouse and Fx2149 treatment
Normal C57BL/6J mice were injected with Fx2149 at 10 mg/kg intraperitoneally twice daily for three days. The brain homogenates were subjected to LRRK2 IP followed by anti-ubiquitin western blot analysis to detect endogenous LRRK2 ubiquitination. Animal usage and ethics was approved by the Animal Care and Use Committee at the Johns Hopkins University School of Medicine.
LRRK2-linked protein aggregation experiments
HEK293T cells were transfected with Flag-tagged G2019S-LRRK2 constructs for 48 h prior to compound or vehicle treatments for an additional 24 h. In the case of HA-tagged ubiquitin aggregation, these constructs were additionally transfected into cells. At 72 h post-transfection, cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Following blocking with 5% normal goat serum (NGS), cells were co-incubated with primary anti-Flag antibodies and either anti-ubiquitin or anti-HA antibodies for 1 h at 37°C. These cells were stained with fluorescent secondary antibodies FITC (green) anti-mouse and Alexa Fluor 568 (red) anti-rabbit at room temperature for an additional 1 h. Cells were washed with PBS three times and incubated with DAPI nuclear stain prior to imaging. Images were captured on a Zeiss Avixon Camera under a computer controlled fluorescent microscope. The number of cells with at least one LRRK2 aggregate were counted for nine separate fields and divided by the number of total co-transfected cells to determine the percent of cells containing aggregates. This was repeated three times for each condition.
For some cases, cells were co-incubated with primary anti-Flag antibodies and anti-γ-tubulin antibodies (aggresome marker) for 1 h at 37°C, and again stained with fluorescent secondary antibodies at room temperature for 1 h. Images were taken as mentioned during aggregation studies. The number of aggregates positive for both LRRK2 and γ-tubulin were counted for nine separate fields and divided by the number of total LRRK2 expressing cells to determine the percent of cells containing aggregates with γ-tubulin-positive aggresome marker. This was repeated three times for each condition.
Data analysis
Quantitative data represent arithmetic means ± SEM from at least three separate experiments for each condition. Statistically significant differences among groups were analyzed by ANOVA by Sigmastart 3.1 statistical software (Aspire Software International, VA). A p value <0.05 was considered statistically significant.
Results
GTP-binding inhibitors increased LRRK2-linked ubiquitination
Our previous studies reported that 68 and Fx2149 inhibit LRRK2 GTP-binding and reduce LRRK2 kinase activity, measured through the phosphorylation at S935 (Li et al., 2014;Li et al., 2015). Phosphorylation at this residue has been inversely linked to LRRK2 ubiquitination (14)(Meissner et al., 2011). To determine if GTP-binding inhibitors alter LRRK2 ubiquitination, HEK293T cells were transfected with vector, WT-, and G2019S-LRRK2 for 48 hours, then treated with DMSO, compound 68 (100 nM) or Fx2149(100 nM) for 24 hours prior to harvesting. Both 68 and Fx2149 caused a dramatic increase in ubiquitinated WT and mutant G2019S-LRRK2 proteins using a LRRK2 ubiquitin assay (Fig. 1A and B). A greater than 3-fold increase in G2019S-LRRK2 ubiquitination occurred after 68 or Fx 2149 treatment compared to untreated vehicle control (Fig 1B).
Fig. 1. 68 and Fx2149 increased LRRK2 ubiquitination.
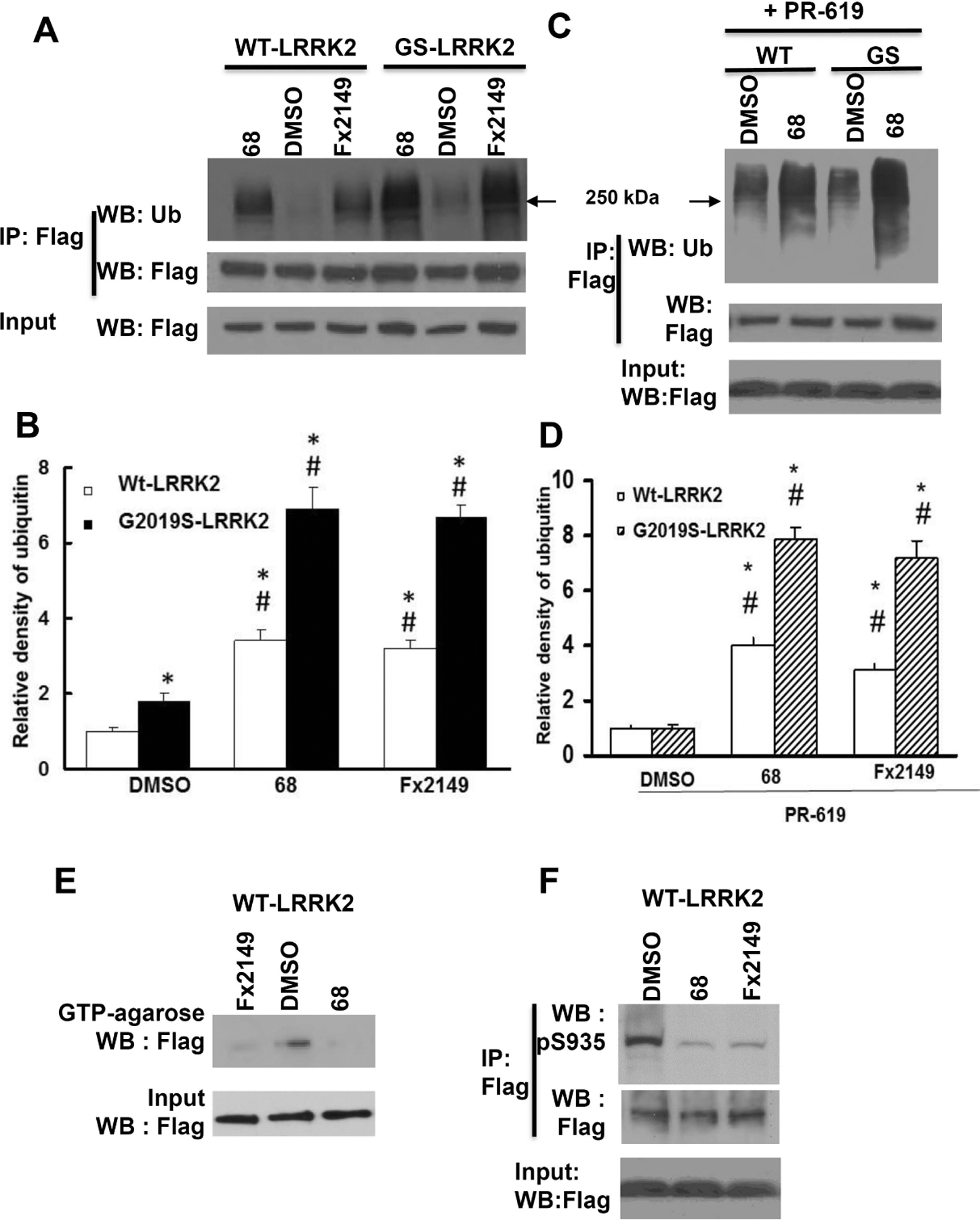
HEK293T cells were transfected with Flag-wild-type (WT)-LRRK2 or Flag-G2019S(GS)-LRRK2 for 48 h, then treated with inhibitors, 0.2% DMSO, 100 nM 68 or 100 nM Fx2149 for an additional 24 h. Cell lysates were harvested and subjected to ubiquitination assays. A, shown are representative blots of three repeated ubiquitination assays. B Quantification of LRRK2 ubiquitination. Arrow: 250 kDa. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle control of each genotype cells. C and D, Cells expressing LRRK2 variants were treated with 68 or Fx2149 for 24 hours, and then with deubiquitinase inhibitor, PR-619 (50 μM) for 30 min prior to lysate collection and in vitro ubiquitin assay. C, Shown are representative blots of three repeated ubiquitination assays. D, Quantification of LRRK2 ubiquitination. E-F, Cells expressing WT-LRRK2 were treated with 68 and Fx2149, and then cell lysates were subjected to LRRK2 GTP binding assays (E and G) and kinase assays (F and G), respectively. G, Quantification of LRRK2 GTP binding and kinase activities. H, Cells were transfected with Flag-WT-LRRK2, K1347A-LRRK2 or D1994N-LRRK2 for 48 hours, then treated with vehicle or 68 for 24 hours. Cell lysates were subjected to ubiquitin assays. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle control of each genotype cells. I, Normal mice (three per group) were treated with vehicle or 10 mg/kg Fx2149 (ip.), twice daily for three days. Mouse brain homogenates were subjected to LRRK2 ubiquitination assays. P<0.05 by student T-test, * vs untreated control mice.
To further validate these results, cells expressing either WT- or G2019S-LRRK2 were treated with 68 or Fx2149 for 24hrs, followed by an additional treatment with deubiquitinase inhibitor, PR-619 (50 μM) for 30 min before harvesting for LRRK2 ubiquitination assays. Blocking deubiquitination with PR-619 exacerbated both WT and G2019S-LRRK2 ubiquitination levels (Fig. 1C and 1D). Most importantly, co-treatment with LRRK2 GTP-binding inhibitor, 68 (100nM) or Fx2149 (100nM) along with PR-619 significantly increased both WT and G2019S-LRRK2 ubiquitination compared with the PR-619 treated cells alone. There was a dramatic increase in PD mutant G2019S-LRRK2 ubiquitination compared with WT-LRRK2 ubiquitination after co-treatment with 68 and PR-619 (Fig 1D). Consistent with our previous findings, 68 and Fx2149 significantly reduced LRRK2 GTP binding and kinase activities (Fig 1E–1G) compared with vehicle treated controls. Compound 68 or Fx2149 had no effect on LRRK2 input levels, demonstrating that the increased ubiquitination levels were not due to an increase in overall protein levels and that the compounds were not affecting LRRK2 clearance. Moreover, we found that non-GTP binding or kinase-dead LRRK2 variants also demonstrated increased ubiquitination with similar levels to 68-treated cells expressing WT-LRRK2 (Fig. 1H). GTP binding inhibitors did not further increase K1347A-LRRK2 or D1994N-LRRK2 ubiquitination. To validate these results in vivo, Fx2149 (which has better brain penetration than 68 (22)) was injected intraperitoneally into normal mice. Fx2149 significantly increased endogenous LRRK2 ubiquitination in mouse brains (Fig. 1I). Taken together, these results indicated that GTP-binding inhibition increased LRRK2 ubiquitination levels.
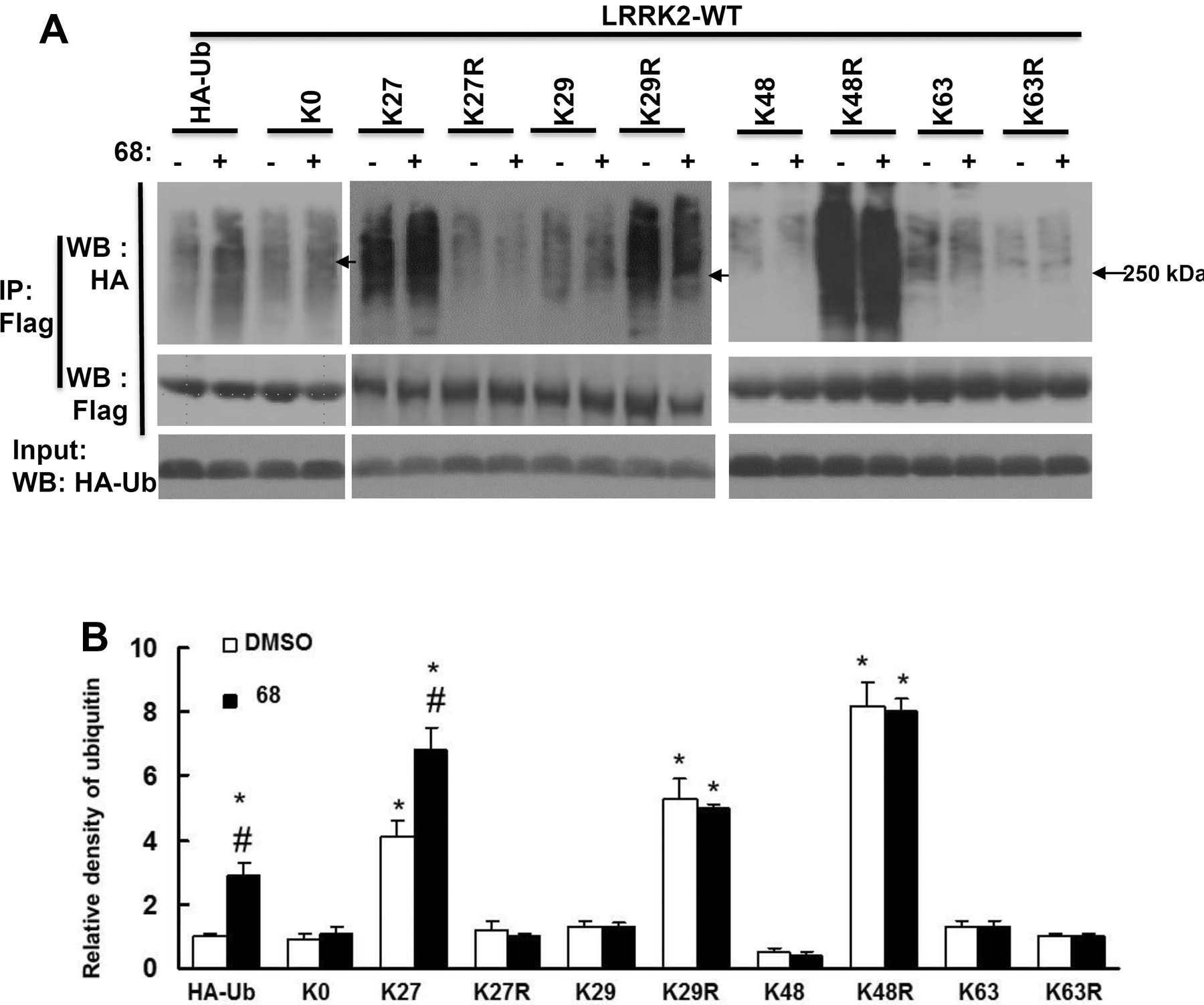
GTP-binding inhibition increased LRRK2 ubiquitination predominantly via K27-linkages
To further assess whether pharmacological GTP-binding inhibition favors specific ubiquitin signals, HEK293T cells were co-transfected with LRRK2 constructs and various HA-tagged ubiquitin constructs for a total of 72 h before subsequent ubiquitination assays. These altered ubiquitination constructs were generated by our group and described previously (Nucifora, Jr. et al., 2016). Briefly, K-ubiquitin alteration constructs (K27, K29, K48, or K63) are only able to be modified by forming chains on each specific lysine residue of ubiquitin (K27, K29, K48, K63 respectively). KR-ubiquitin alteration constructs (K27R, K29R, K48R, K63R) can be modified by all other ubiquitin linkages except K27, K29, K48, or K63 residue respectively, as it is mutated to arginine. K0 construct has no lysine available for modification. HEK293 cells were co-transfected with WT- or G2019S-LRRK2 for 48 hours, and then treated with 0.2% DMSO or 68 (100 nM) for 24h prior to collecting cell lysates for ubiquitination assays.
Co-expression of WT-LRRK2 and K27-, K29-, K63-ubiquitin constructs increased LRRK2 ubiquitination compared with cells expressing LRRK2 and wild type ubiquitin (Fig. 2). Expression of K27-ubiquitin increased the most, with about a 4-fold increase in wild type ubiquitin expressing cells. In contrast, there was no change in cells expressing K0 and K48 constructs. Moreover, treatment of 68 significantly increased the LRRK2 ubiquitination in cells expressing K27 and WT ubiquitin constructs, but did not alter ubiquitination in cells expressing K0, K29, K48 and K63 ubiquitin constructs. Interestingly, co-expression of K27R with LRRK2 significantly reduced the LRRK2 ubiquitination, and reversed the elevated effect of 68, even below the untreated control level. Co-expression of K63R with LRRK2 slightly reduced LRRK2 ubiquitination, but there was no response to 68 treatment. Unexpectedly, there was also a significant increase in LRRK2 ubiquitination in cells expressing K29R and K48R compared with control cells. One explanation is that these constructs could result in preference towards formation of alternative ubiquitination linkages due to blocking the formation of K29- and K48- linked ubiquitination.
Fig. 2. 68 increased WT-LRRK2 ubiquitination predominantly via K27-linkages.
HEK293T cells were co-transfected with Flag-wild-type-LRRK2 and various HA-tagged ubiquitin constructs for 48 h, then treated with 68 (100 nM) for 24 h. Cell lysates were subjected to ubiquitination assays. A, Representative blots from three repeated ubiquitination assays. B, quantification of density of ubiquitination by NIH image J software. Arrow: 250 kDa. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle control of each genotype of cells.
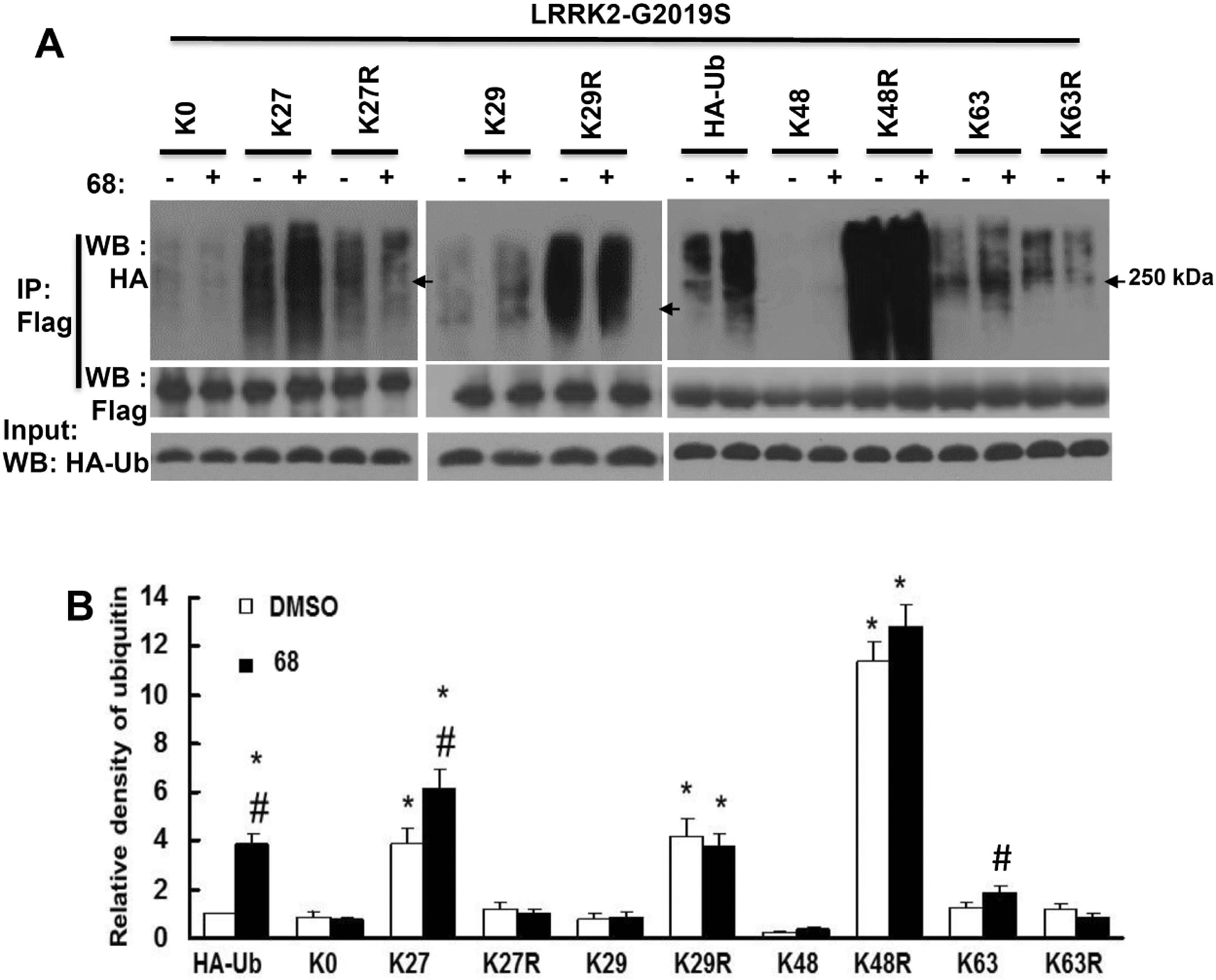
Co-expression of mutant G2019S-LRRK2 with WT-, K27 and K63-ubiquitin constructs also significantly increased ubiquitination but to a higher degree (Fig.3), suggesting that mutant LRRK2 undergoes more ubiquitination than wild type LRRK2. There was no change in cells expressing K0 constructs (Fig. 3). There was significantly decreased ubiquitination during expression of K48-ubiquitin constructs, which is consistent with the notion that K48-ubiquitination results in more proteasome degradation of the targeted protein. Moreover, treatment of 68 further increased the G2019S-LRRK2 ubiquitination in cells expressing K27 and WT ubiquitin constructs, but did not alter ubiquitination in cells expressing K0, K29, K48 and K63 ubiquitin constructs. Co-expression of K27R with G2019S-LRRK2 almost reversed the LRRK2 ubiquitination to the untreated control level. Co-expression of K63R with LRRK2 slightly reduced LRRK2 ubiquitination, but there was no response to 68 treatment. Similar to the co expression of wild type-LRRK2, there was also significantly increased G2019S-LRRK2 ubiquitination in cells expressing K29R and K48R compared with control cells.
Fig. 3. 68 increased G2019S-LRRK2 ubiquitination predominantly via K27-linkages.
HEK293T cells were co-transfected with Flag-G2019S-LRRK2 and various HA-tagged ubiquitin constructs for 48 h, then treated with 68 (100 nM) for 24 h. Cell lysates were subjected to ubiquitination assays. A, Representative blots from three repeated ubiquitination assays. B, quantification of density of ubiquitination by NIH image J software. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle control of each genotype of cells.
68 and Fx2149 increased LRRK2-linked ubiquitinated inclusions
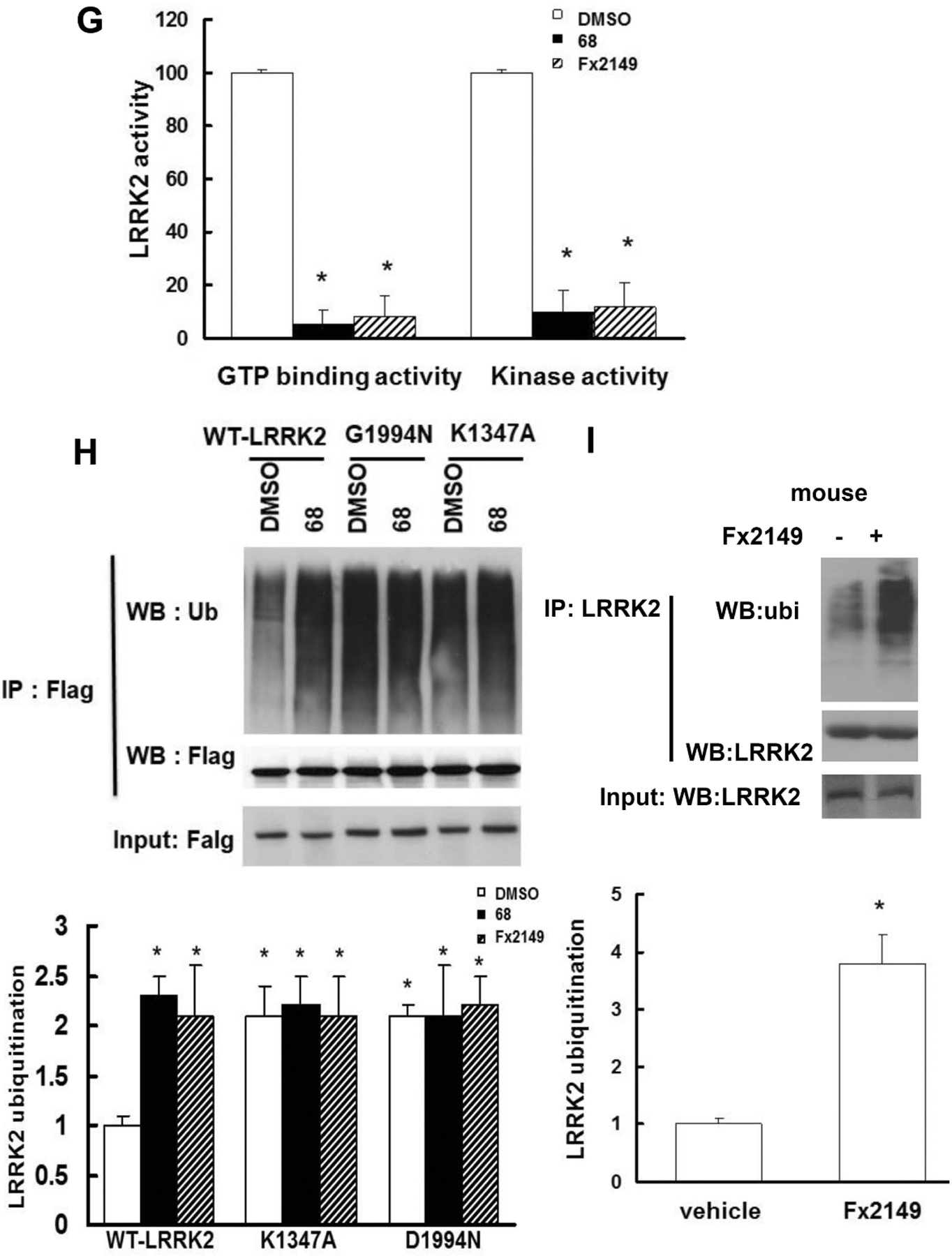
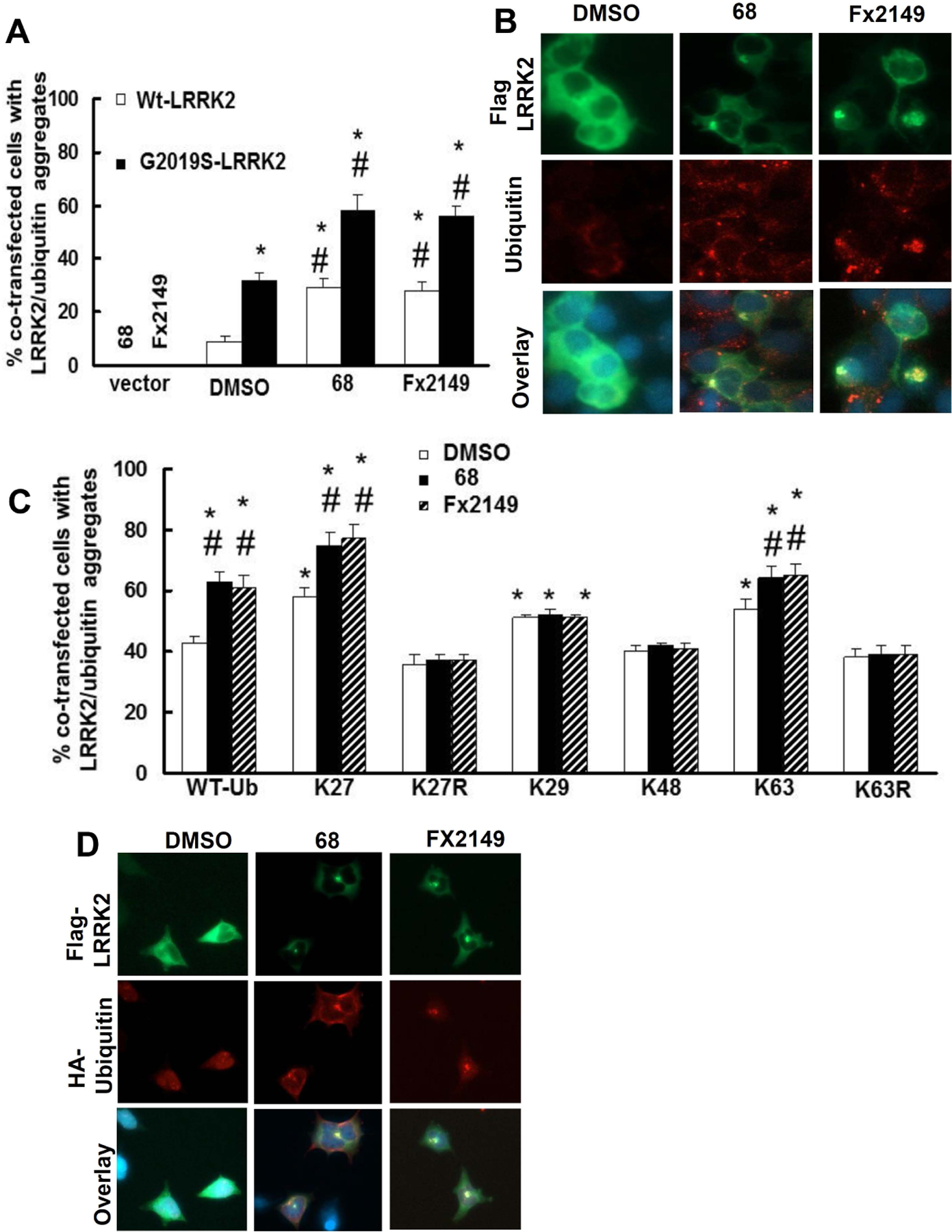
Previous reports have identified that atypical ubiquitin linkages (eg. K27) signal for aggregation of toxic misfolded protein in an attempt to protect the cell (Kalia and Kalia, 2015;Zucchelli et al., 2010;Shaid et al., 2013). Given that GTP-binding inhibitors increased ubiquitin, we were interested in studying whether 68 and Fx2149 alter mutant G2019S-LRRK2-linked aggregation formation. HEK293T cells were transfected with G2019S-LRRK2 for 48 h and treated with 68 or Fx2149 for an additional 24 h prior to detection of LRRK2-linked aggregates via immunostaining. Expression of mutant G2019S-LRRK2 had more ubiquitinated inclusions than cells expressing wild type-LRRK2 (Fig. 4A and 4B). About 90 % of LRRK2-aggreagates were ubiquitin-positive. Moreover, treatment of 68 or Fx2149 significantly further increased the number of ubiquitin-positive LRRK2-aggregates.
Fig. 4. 68 and Fx2149 increase ubiquitin-positive inclusions containing LRRK2.
A and B, HEK293T cells were transfected with WT- or Flag-G2019S-LRRK2 for 48 h, then treated with 0.2%DMSO, 68 (100nM), or Fx2149 (100 nM) for 24 h. Cells were subjected to immunostaining with anti-Flag (Green) and anti-ubiquitin (Red) antibodies. Merged images contain DAPI (blue) nuclear staining. A, Quantification data showed that both 68 and Fx2149 increased ubiquitin positive aggregates. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle treated control of each genotype cells. B. Representative images of LRRK2 transfected cells with ubiquitin positive aggregates. C and E, HEK293T cells were co-transfected with Flag-G2019S-LRRK2 and various HA-ubiquitin constructs for 48 h, and then treated with 0.2%DMSO, 68 (100nM), or Fx2149 (100 nM) for 24 h. Cells were immunostained with anti-Flag (green) and anti-HA(Red) antibodies along with DAPI (blue) nuclear marker. Transfected cells with LRRK2/ubiquitin positive aggregates were recorded. D. Representative images of co-transfected cells with aggregates. P<0.05 by ANOVA, * vs cells expressing G2019S-LRRK2 and wild type ubiquitin treated with 0.2% DMSO; # vs vehicle treated control cells with each ubiquitin construct group.
To determine which ubiquitin-linkage(s) is involved in ubiquitinated inclusion formation, HEK293T cells were co-transfected with G2019S-LRRK2 and various HA-ubiquitin constructs for 48 hours, then treated with 68 or Fx2149 for 24 hours. Cells were stained with anti-Flag and anti-HA antibodies to detect the ubiquitinated positive aggregates. Co-expression of mutant G2019S-LRRK2 with K27-, K29- and K63-ubiquitin constructs further increased the ubiquitinated aggregates compared with cells co-expressing WT-ubiquitin (Fig. 4C and 4D). Moreover, treatment of 68 or Fx2149 further increased the inclusion formation in cells co-expressing G2019S-LRRK2 and WT-, K27-, and K63-ubiquitin constructs compared with untreated co-expressing cells (Fig.4 C and 4D). Co-expression of K27-ubiquitin constructs led to a greater increase in aggregates compared to co-expressing other ubiquitin constructs. A change was not observed in the K29 and K48 conditions between inhibitor-treatment and untreated control groups. Interestingly, co-expression of G2019S-LRRK2 and K27R-, and K63R-ubiquitin constructs reversed 68- or Fx2149- induced inclusion formation (Fig.4 C).
68 and Fx2149 increased LRRK2-linked aggresome formation
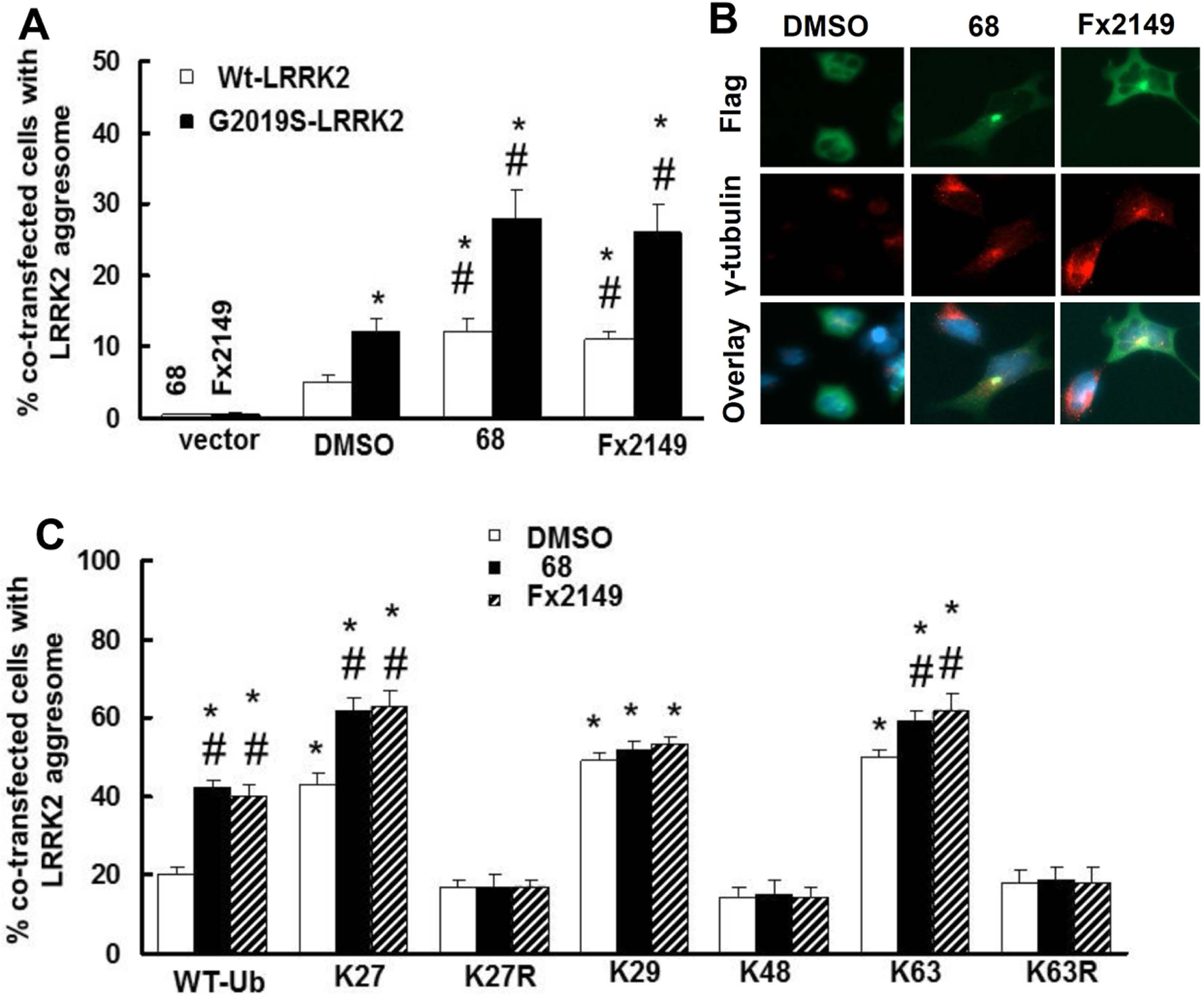
It is believed that the sequestration of toxic proteins is a cell protective response, and that the formation of aggresomes can localize these proteins into a distinct compartment of the cell for further processing. It is suggested that the Lewy body is a type of aggresome. To further assess whether GTP-binding inhibitors alter LRRK2-linked aggresome formation, the aggresomes were detected by using co-immunostaining with anti-Flag-LRRK2 protein and anti- γ-tubulin (an aggresome marker) antibodies (Fig. 5A). HEK293T cells were transfected with G2019S-LRRK2 for 48 h, and left untreated or treated with 68 or Fx2149 for an additional 24 h prior to immunostaining to detect LRRK2-linked aggresomes. Expression of mutant G2019S-LRRK2 had more aggresomes than cells expressing wild type-LRRK2 (Fig. 5A and 5B). Moreover, treatment of 68 or Fx2149 significantly further increased the number of cells with LRRK2-aggresomes.
Fig. 5. 68 and Fx2149 increased LRRK2-aggresomes.
A and B, HEK293T cells were transfected with WT- or Flag-G2019S-LRRK2 for 48 h, then treated with 0.2%DMSO, 68 (100nM), or Fx2149 (100 nM) for 24 h. Cells were subjected to immunostaining with anti-Flag (Green) and γ-tubulin (Red) antibodies. Merged images contain DAPI (blue) nuclear staining. A, Quantification data showed that both 68 and Fx2149 increased LRRK2 aggresomes. P<0.05 by ANOVA, * vs cells expressing WT-LRRK2 treated with 0.2% DMSO; # vs vehicle treated control of each genotype of cells. B. Representative images of LRRK2 transfected cells with ubiquitin positive aggregates. C and D, HEK293T cells were co-transfected with Flag-G2019S-LRRK2 and various HA-ubiquitin constructs for 48 h, and then treated with 0.2%DMSO, 68 (100nM), or Fx2149 (100 nM) for 24 h. Cells were immunostained with anti-Flag (green) and γ-tubulin (Red) antibodies along with DAPI (blue) nuclear marker. Transfected cells with LRRK2/ γ-tubulin (Red) positive aggregates were recorded. P<0.05 by ANOVA, * vs cells expressing G2019S-LRRK2 and wild type ubiquitin treated with 0.2% DMSO; # vs vehicle treated control cells with each ubiquitin construct group.
To determine which ubiquitin-linkage (s) is involved in aggresome formation, HEK293T cells were co-transfected with G2019S-LRRK2 and various HA-ubiquitin constructs for 48 h, then treated with 68 or Fx2149 for 24 h. Cells were stained with anti- γ-tubulin and anti-Flag antibodies to detect LRRK2-aggresome. Co-expression of mutant G2019S-LRRK2 with K27-, K29- and K63-ubiquitin constructs further increased aggresome formation compared with cells co-expressing WT-ubiquitin (Fig. 5C). There was no change when K48 constructs were co-transfected. Moreover, treatment of 68 or Fx2149 further increased the aggresome formation in cells co-expressing G2019S-LRRK2 and WT-, K27-, and K63-ubiquitin constructs compared with untreated co-expressing cells (Fig.5C). Co-expression of K27-ubiquitin constructs increased aggresomes compared with co-expressing other ubiquitin constructs. There was no change in the K29 and K48 co-transfected conditions between inhibitor-treatment and untreated control groups. Interestingly, co-expression of G2019S-LRRK2 and K27R-, and K63R-ubiquitin constructs reversed 68- or Fx2149- induced aggresome formation (Fig.5C).
Discussion
The major findings of this study were that pharmacological inhibition of GTP-binding by 68 or Fx2149 increased LRRK2-linked ubiquitination and protein aggregation. The elevated LRRK2-linked ubiquitination was predominantly through the atypical ubiquitin linkage at the K27 residue. Moreover, 68 and Fx2149 also promote LRRK2-linked aggresome formation via this ubiquitin linkage. These findings demonstrate that LRRK2 GTP-binding activity is critical in LRRK2-linked ubiquitination and protein aggregation.
Ubiquitin is able to signal through a multitude of ways, such as through the formation of additional ubiquitin chains conjugated to specific lysine residues (and one methionine residue) on the ubiquitin protein itself (Atkin and Paulson, 2014;Pickart, 2000;Kulathu and Komander, 2012). Chains on K48 are the most common and most well studied, and target unwanted or misfolded proteins for degradation via the proteasome (Glickman and Ciechanover, 2002; Kulathu and Komander, 2012;Yao, 2010;Michel et al., 2015). K27, K29, and K63 chains still require further investigation, but they have implications related to protein aggregation with an effort by the cell to alternatively process toxic proteins in an attempt to protect itself (Zucchelli et al., 2010;Shaid et al., 2013). Each of these sites is able to be monoubiquitinated, with only one ubiquitin moiety, or polyubiquitinated, with more than one ubiquitin moieties forming branches of varying length. Of these branches, linkages can all be conjugated to the same residue in a homogenous fashion, or display multiple linkage types in a heterogeneous fashion (Kulathu and Komander, 2012;Yao, 2010;Michel et al., 2015). Target proteins can be conjugated to single ubiquitin molecules at multiple sites in a process termed multimonoubiquitination (Kawaguchi, 2013). Polyubiquitination at K27, K29, and K63 is believed to signal for non-proteasomal routes of processing (Nucifora, Jr. et al., 2016;Kulathu and Komander, 2012;Korolchuk et al., 2010;Michel et al., 2015), which may have a role for abnormal proteins not normally handled or recognized by the cell, as well as provide options for cells when the UPS is overwhelmed.
Previously, our group as well as other groups have reported that LRRK2 can be ubiquitinated through K27, K29, K48, and K63 chains (Nucifora, Jr. et al., 2016;Zhao et al., 2015;Tan et al., 2008). Here we found that our recently identified LRRK2 GTP-binding inhibitors, 68 and Fx2149 increased LRRK2 ubiquitination predominantly via K27 linkage. 68 and Fx2149 can reduce LRRK2 phosphorylation via blocking the GTP-binding activity (Li et al., 2014;Li et al., 2015). Consistent with a previous study, dephosphorylation of LRRK2 at S910 and S935 residues upregulate its ubiquitination (Zhao et al., 2015). Our findings indicate that inhibition of GTP-binding and kinase activities of G2019S-LRRK2 promote polyubiquitination at K27 and K63 linkages (a signal for non-proteasomal routes of processing), leading to greater protein aggregation, but not altering LRRK2 degradation. Recent studies provide evidence of consequential aggregation due to K27, K29, and K63-linked ubiquitin signaling, and this is believed to represent a self-protecting mechanism of the cell in sequestering toxic proteins away from the action into an aggresome as they await further processing (Kalia and Kalia, 2015;Arrasate et al., 2004). Our work here demonstrated a role for the LRRK2 GTP-binding domain in its propensity to be ubiquitinated and showed that the non-canonical linkages are involved with mutant LRRK2 inclusion formation and aggresome response. Notably, it is believed that K29-chains can only form as mixed heteroubiquitinated linkages with chains already targeted by K48 ubiquitin (Kulathu and Komander, 2012;Michel et al., 2015). G2019S-LRRK2 mutations cause an impaired and thus overwhelmed UPS (Cook et al., 2012;Park et al., 2016;Bang et al., 2016). A possible reason for the lack of increase in K29 could be due to a disburdening of the previously overwhelmed proteasome pathway through GTP-binding inhibitors. While most of the K48-ubiquitinated LRRK2 resulted in degeneration following inhibitor treatment, this meant less K48-ubiquitinated LRRK2 remained available for further modification via K29-linked ubiquitination.
How a protein’s ubiquitination fate is determined is still unclear, however certain E3 ubiquitin ligases preferentially form lysine-specific chains (Glickman and Ciechanover, 2002; Zucchelli et al., 2010;Komander and Rape, 2012;McDowell and Philpott, 2013). A recent report provides a neuronal protective role for K27 and K29 ubiquitination in LRRK2 through an interaction with WSB1(Nucifora, Jr. et al., 2016). These two types of ubiquitination have signaled for a protective aggregation response in other neurodegenerative disorders, as PD player DJ-1 and HD player huntingtin have both been demonstrated to undergo K27/K29 ubiquitination and subsequent aggregation (Atkin and Paulson, 2014;Dantuma and Bott, 2014;Zucchelli et al., 2010;Arrasate et al., 2004).
Although it is controversial whether protein aggregation is protective for neurons or not, it is generally believed that large aggresomes (such as Lewy bodies) are neuroprotective. Inhibition of LRRK2 GTP-binding activity may lead the cells towards undergoing a self-protection mechanism by sequestering the toxic ubiquitinated mutant proteins into inclusions. Consistent with this idea, treatment of 68 and Fx2149 can protect against G2019S-inducd neurodegeneration in neurons in in vitro and in vivo studies (Li et al., 2014;Li et al., 2015). Recently, LRRK2 demonstrated involvement in selective autophagy, which utilizes ubiquitin-dependent signaling (Park et al., 2016). Moreover, G2019S-LRRK2 caused disruption of the autophagy pathway and subsequent clearance mechanisms, leading to cytotoxicity (Bang et al., 2016). Interestingly, the use of LRRK2 kinase inhibitors caused an increase in protein turnover through proteasome-dependent degradation (Lobbestael et al., 2016). It is likely that both proteasomal and non-proteasomal routes of degradation regain functionality following GTP-binding and kinase inhibition, as the two pathways are tightly regulated and known to complement one another (Cook and Petrucelli, 2009;Korolchuk et al., 2010;Kravtsova-Ivantsiv and Ciechanover, 2012).
In conclusion, our findings demonstrated that LRRK2 GTP-binding inhibitors, 68 and Fx2149 promoted LRRK2 ubiquitination, increased ubiquitinated aggregation, and contributed to an aggresome response. These studies provide novel insight into LRRK2-linked Lewy body-like inclusion formation underlying PD pathogenesis.
Funding:
This work is supported by NIH grants: R01NS093383 and R21NS096620 to Wanli W Smith
Footnotes
Conflict interest: The structure and pharmacological use of compound 68 and Fx 2149 was included in the patent of University of Maryland School of Pharmacy. Wanli W. Smith is an inventor of this patent.
Data availability statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010), 805–810. [DOI] [PubMed] [Google Scholar]
- Atkin G, Paulson H (2014). Ubiquitin pathways in neurodegenerative disease. Frontiers in Molecular Neuroscience, 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang Y, Kim KS, Seol W, Choi HJ (2016). LRRK2 interferes with aggresome formation for autophagic clearance. Molecular and Cellular Neuroscience, 75, 71–80. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Experimental & Molecular Medicine, 47, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Petrucelli L (2009). A critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochimica et Biophysica Acta, 1792(7), 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Stetler C, Petrucelli L (2012). Disruption of protein quality control in Parkinson’s disease. Cold Spring Harbor Perspectives Medicine, 2(5), a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Bott LC (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Frontiers in Molecular Neuroscience, 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Gysbers AM, Bandopadhyay R, Bolliger MF, Uchino A, Zhao Y, … Halliday GM (2017). LRRK2 levels and phosphorylation in Parkinson’s disease brain and cases with restricted Lewy bodies. Movement Disorders, 32(3), 423–432. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological Reviews, 82(2), 373–428. [DOI] [PubMed] [Google Scholar]
- Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M, … Krappmann D (2011). NF-kappaB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-kappaB activation. The Journal of Biological Chemistry, 286(29), 26107–26117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman MG, Soto-Ortolaza AI, Contreras MYS, Murray ME, Pedraza O, Diehl NN, … Ross OA (2016). LRRK2 variation and dementia with Lewy bodies. Parkinsonism Related Disorders, 31, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M (2009). Origin and function of ubiquitin-like proteins. Nature, 458(7237), 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK (2015). alpha-Synuclein and Lewy pathology in Parkinson’s disease. Current Opinion in Neurology, 28(4), 375–381. [DOI] [PubMed] [Google Scholar]
- Kawaguchi K (2013). Role of kinesin-1 in the pathogenesis of SPG10, a rare form of hereditary spastic paraplegia. The Neuroscientist, 19(4):336–344. [DOI] [PubMed] [Google Scholar]
- Ko HS, Bailey R, Smith WW, Liu Z, Shin JH, Lee YI, … Dawson VL (2009). CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proceedings of the National Academic of Sciences of the United States of America, 106(8), 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M, (2012). The ubiquitin code. Annual Review of Biochemistry, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Menzies FM, Rubinsztein DC (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Letters, 584(7), 1393–1398. [DOI] [PubMed] [Google Scholar]
- Kravtsova-Ivantsiv Y, Ciechanover A (2012). Non-canonical ubiquitin-based signals for proteasomal degradation. Journal of Cell Science, 125(Pt 3), 539–548. [DOI] [PubMed] [Google Scholar]
- Kulathu Y, Komander D (2012). Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nature Reviews. Molecular Cell Biology, 13(8), 508–523. [DOI] [PubMed] [Google Scholar]
- Li T, He X, Thomas JM, Yang D, Zhong S, Xue F, Smith WW (2015). A novel GTP-binding inhibitor, FX2149, attenuates LRRK2 toxicity in Parkinson’s disease models. PLoS One, 10(3), e0122461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Yang D, Zhong S, Thomas JM, Xue F, Liu J, … Smith WW (2014). Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Human Molecular Genetics, 23(23), 6212–6222. [DOI] [PubMed] [Google Scholar]
- Li X, Moore DJ, Xiong Y, Dawson TM, Dawson VL (2010). Reevaluation of phosphorylation sites in the Parkinson disease-associated leucine-rich repeat kinase 2. Journal of Cell Science, 285(38), 29569–29576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobbestael E, Civiero L, De WT, Taymans JM, Greggio E, Baekelandt V (2016). Pharmacological LRRK2 kinase inhibition induces LRRK2 protein destabilization and proteasomal degradation. Scientific Reports, 6, 33897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamais A, Manzoni C, Nazish I, Arber C, Sonustun B, Wray S, … Bandopadhyay R (2018). Analysis of macroautophagy related proteins in G2019S LRRK2 Parkinson’s disease brains with Lewy body pathology. Brain Research, 1701, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell GS, Philpott A (2013). Non-canonical ubiquitylation: mechanisms and consequences. The International Journal of Biochemistry & Cell Biology, 45(8), 1833–1842. [DOI] [PubMed] [Google Scholar]
- Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, … Bezard E (2011). Priorities in Parkinson’s disease research. Nature Reviews. Drug Discovery, 10(5), 377–393. [DOI] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, … Komander D (2015). Assembly and specific recognition of k29- and k33-linked polyubiquitin. Molecular Cell, 58(1), 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora FC Jr., Nucifora LG, Ng CH, Arbez N, Guo Y, Roby E, … Ross CA (2016). Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nature Communication, 7, 11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Han S, Choi I, Kim B, Park SP, Joe EH, Suh YH (2016). Interplay between Leucine-Rich Repeat Kinase 2 (LRRK2) and p62/SQSTM-1 in Selective Autophagy. PLoS One, 11(9), e0163029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2000). Ubiquitin in chains. Trends in Biochemical Science, 25(11), 544–548. [DOI] [PubMed] [Google Scholar]
- Shaid S, Brandts CH, Serve H, Dikic I (2013). Ubiquitination and selective autophagy. Cell Death and Differetiation, 20(1), 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S (2014). Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. The Journal of Neuroscience, 34(2), 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA (2006). Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nature Neuroscience, 9(10), 1231–1233. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, … Ross CA (2005). Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proceedings of the National Academic of Sciences of the United States of America, 102(51), 18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, … Lim KL (2008). Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Human Molecular Genetics, 17(3), 431–439. [DOI] [PubMed] [Google Scholar]
- Yao TP (2010). The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes & Cancer, 1(7), 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Molitor TP, Langston JW, Nichols RJ (2015). LRRK2 dephosphorylation increases its ubiquitination. The Biochemical Journal, 469(1), 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, … Gasser T (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44(4), 601–607. [DOI] [PubMed] [Google Scholar]
- Zucchelli S, Codrich M, Marcuzzi F, Pinto M, Vilotti S, Biagioli M, … Gustincich S (2010). TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Human Molecular Genetics, [DOI] [PubMed] [Google Scholar]