

Abstract
Cyclin-dependent kinases 4 and 6 (CDK4/6) have emerged as important therapeutic targets. Pharmacological inhibitors of these kinases function to inhibit cell cycle progression and exert other important effects on the tumor and host environment. Due to their impact on the cell cycle, CDK4/6 inhibitors (CDK4/6i) have been hypothesized to antagonize the anti-tumor effects of cytotoxic chemotherapy in tumors that are CDK4/6 dependent. However, there are multiple preclinical studies that illustrate potent cooperation between CDK4/6i and chemotherapy. Furthermore, the combination of CDK4/6i and chemotherapy is being tested in clinical trials to both enhance anti-tumor efficacy and limit toxicity. Exploitation of the non-canonical effects of CDK4/6i could also provide an impetus for future studies in combination with chemotherapy. Thus, while seemingly mutually exclusive mechanisms are at play, the combination of CDK4/6 inhibition and chemotherapy could exemplify rational medicine.
CDK4/6 in cell-cycle progression:
Cyclin dependent kinases (CDKs) are serine/threonine kinases that regulate the sequential progression of the cell cycle in eukaryotic organisms. The molecular functions of these kinases in different phases of the cell cycle have been well characterized (1, 2). The cell cycle machinery in higher eukaryotes is tightly regulated by the presence of more than 10 proteins in the CDK family that can have overlapping and distinct functions (2). Cell-cycle initiation occurs in G1 phase, which is conventionally governed by the activation of CDK4 and CDK6 kinases that are downstream of mitogenic signals (3–5). The catalytic activity of CDK4 and CDK6 is positively regulated by the binding of D-type cyclins (D1, D2 and D3). Expression of D-type cyclins is induced in response to mitogenic stimuli and remains high as the cells progress to the G1/S phase boundary (6). Therefore, unlike other cyclins and CDKs that are regulated by other components of the cell-cycle machinery, the expression of D-type cyclins --and by extension CDK4/6 associated kinase activity--largely depend on mitogenic signaling pathways (7, 8). Transcription of D-type cyclins is intimately linked to multiple pathways that coalesce to lead to the accumulation of transcripts (7, 9, 10). Mitogenic signaling pathways also regulate the stability and localization of these proteins (11, 12). Importantly, a host of growth inhibitory mechanisms also impact CDK4/6 activity, including the induction of endogenous CDK4/6-specific inhibitors with specific stresses (e.g. CDKN2A which encodes p16INK4A), and active mechanisms of cyclin D1 degradation (13, 14). Thus, CDK4/6 activity acts as a sensor linking multiple-signaling pathways to the initiation of the cell cycle (15–17).
CDK4/6 regulates the cell cycle through phosphorylation of key substrates. Unlike the prototypical CDK1 and CDK2, which can phosphorylate many substrates, CDK4/6 has a very limited repertoire of targets (18). CDK4 and CDK6 selectively phosphorylate the RB tumor suppressor protein and additional members of the RB family (18–21). RB-family proteins function as transcriptional co-repressors and limit the expression of E2F target genes that include multiple genes required for cell cycle progression, DNA replication, and mitotic progression (22, 23). The phosphorylation of RB, which is initiated by CDK4 or CDK6 serves to limit transcriptional repression and enable progression through latter phases of the cell cycle defining the canonical CDK4/6-RB pathway (Fig 1A).
Figure 1. Different cell cycle states in cancer.
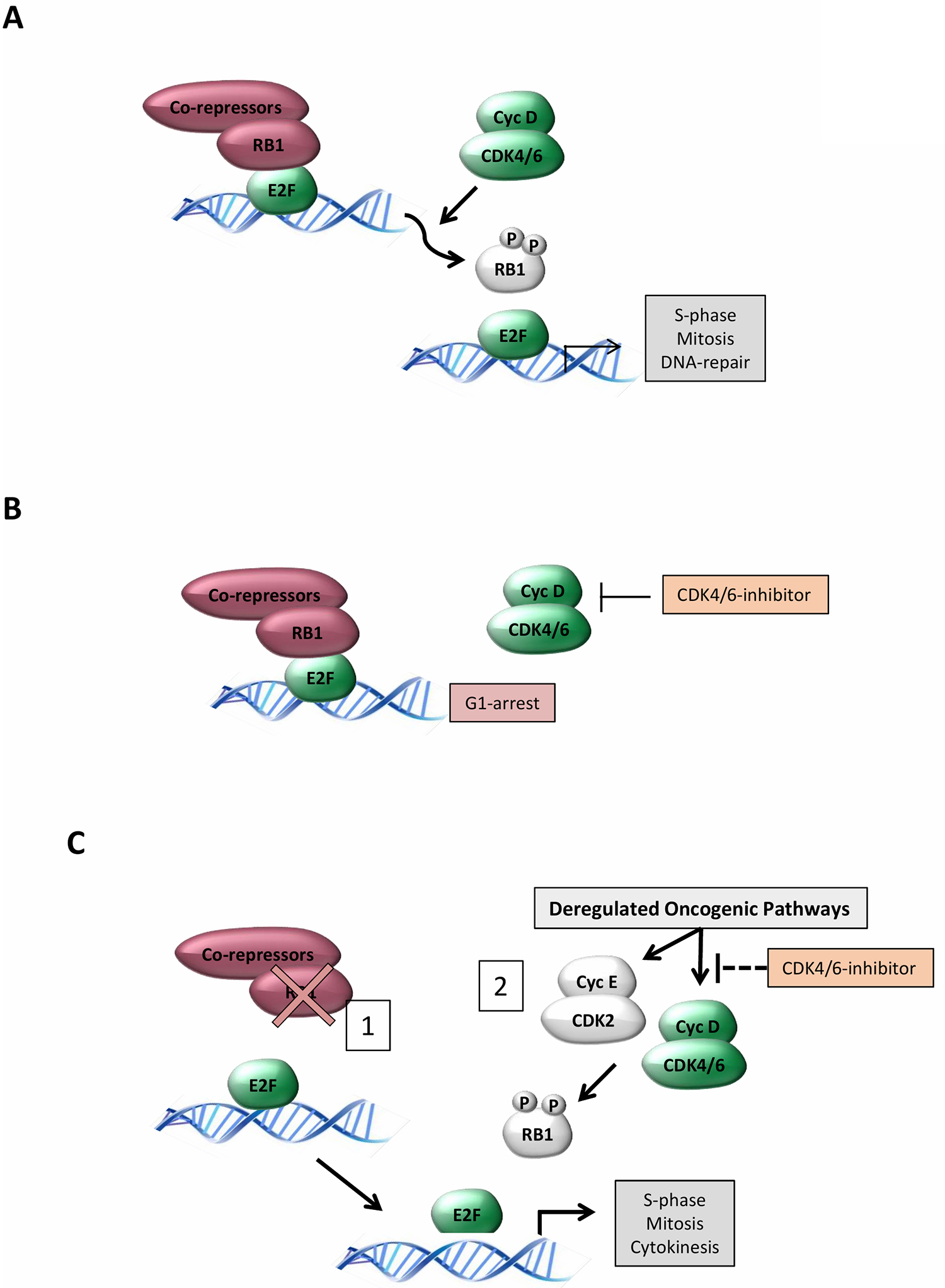
(A) The Canonical G1/S regulatory circuit: CDK4/6 kinase activity is stimulated down-stream of mitogenic/oncogenic signals to initiate the phosphorylation of RB and related proteins. Phosphorylation facilitates the de-repression of E2F-family of transcription factors that drive the expression of many genes required for DNA replication, mitosis, and cell division. (B) CDK4/6 dependent cells: In cells, tumors, or tissues that are dependent on CDK4/6 activity treatment with pharmacological inhibitors yields the robust activation of RB. This event limits other CDK activities and represses the expression of essential genes for cellular division resulting in a G1/G0 like arrest. (C) CDK4/6-independent cells: There are clearly two distinct states that yieldCDK4/6 independent proliferation. (1) Loss of RB as occurs in a subset of human tumors removes the down-stream target and as such inhibition of CDK4/6 has minimal efficacy in controlling cell cycle. (2) through various mechanisms RB phosphorylation can remain during pharmacological inhibition. This cell cycle plasticity can be generated through either CDK4/6 or CDK2 complexes and is prevalent in a number of tumor types that retain the RB tumor suppressor.
The requirement for CDK4/6 in cell division has been interrogated utilizing multiple approaches and has illustrated important features of the cell cycle. The inhibition of CDK4/6 by the expression of endogenous inhibitors (e.g. p16INK4A) potently arrests cells that contain a functional RB protein and subsequently limits gene expression controlled by RB/E2F (Fig 1A). Multiple experimental methods (e.g. antibody injection, RNAi, etc.) have further suggested that D-type cyclins and/or CDK4/6 activity are generally important for progression from G1/S in normal cells as well as multiple cancer models (24). These findings contrast with studies in mouse models that clearly demonstrate that the cell cycle can proceed with genetic deletion of CDK4 and 6 or deletion of all D-type cyclins (25, 26). In this context, adaptation occurs in many tissues by enabling CDK2 or CDK1 activity to drive cell cycle entry. However, genetic suppression of CDK4/6 activity can limit or block tumor development in select models (27–30). This was clearly shown in the context of HER2-driven breast cancer where CDK4/6 activity is required both for tumor etiology and maintenance (31).
Pharmacological inhibitors of CDK4/6—mechanisms of action and resistance:
Due to the function of CDK4/6 in coordinating cell division, pharmacological inhibitors have been developed as anti-cancer drugs. There are five selective CDK4/6 inhibitors (CDK4/6i); palbociclib (PD0332991), ribociclib (LEE011), abemaciclib (LY2835219), trilaciclib, (G1T28) and lerociclib (G1T38) (32–38). Currently, three of these drugs are FDA-approved for the treatment of ER+ metastatic breast cancer based on multiple randomized clinical trials (palbociclib, ribociclib, abemaciclib). While all of these compounds are selective for CDK4/6, palbociclib, ribociclib, abemaciclib, and lerociclib are formulated for oral long-term dosing. Trilaciclib was formulated specifically for intravenous delivery and short half-life with the intended goal of preventing chemotherapy-induced host toxicities. Consistent with their mechanism of action, all CDK4/6i have cytostatic activity that is associated with RB-dependent suppression of the G1/S transition (32, 36). Pharmacological CDK4/6i mimic the effect of RB activation (Fig 1B) and suppress the expression of genes that are conventionally regulated by the E2F-family of transcription factors (39, 40). Since many of these genes are involved in core functions of DNA replication and mitotic progression, and are considered essential for proliferation, the magnitude of transcriptional repression downstream from CDK4/6 inhibition is critical for cytostatic activity.
Multiple determinants of response to CDK4/6 inhibition are being elucidated through both preclinical investigation and the analysis of clinical specimens (Fig 1C). This work has illustrated that there are multiple cell cycle related alterations present in models or tumors (e.g. RB loss or overexpression of cyclin E) that are associated with resistance to CDK4/6 inhibitors (41–44) (Fig 1C). Conversely, a number of oncogenic signaling pathways (e.g. RAS/MAPK, PTEN/PI3K, or HIPPO) have emerged as contributing to resistance (45–47). These derangements enable escape from CDK4/6 inhibition by facilitating the inactivation/phosphorylation of RB even in the presence of the pharmacological CDK4/6i. This is believed to occur due to “plasticity”, which is associated with either incomplete inhibition of CDK4/6 or the ability of CDK2 to initiate the phosphorylation of RB. While inhibition of CDK4/6 can arrest cells, dual inhibition of CDK4 and CDK2 has been shown to be required for durable responses in preclinical models (48). In addition, the hyperactivation of CDK2 kinase in breast cancer cells due to overexpression of cyclin E1/cyclin E2 drives resistance to CDK4/6i (49). Conversely, RNAi mediated knockdown of CCNE1 or CDK2 along with CDK4/6 inhibition can reverse acquired resistance to CDK4/6 inhibition in select models (50–52). Thus, the level of CDK2 activity during response is an important determinant and potential biomarker for the efficacy of CDK4/6i.
The spectrum of CDK4/6i tumor sensitivities and theoretical intersection with chemotherapy:
From preclinical and clinical studies, emerging data indicate that there are tumors that are sensitive to CDK4/6i, but that many of these sensitive tumors develop adaptive resistance mechanisms. In these contexts, CDK4/6i combination therapies can enhance the efficacy and durability of the tumor response. In contrast, there are a subset of cancers that are intrinsically CDK4/6-independent (e.g. as a consequence of RB loss) (33, 53). The consequences of the addition of CDK4/6i to chemotherapy must be considered within this framework. In patients with CDK4/6-independent tumors, the anticipated clinical benefit would be to protect normal cells from chemotherapy as the normal cells are sensitive to CDK4/6 inhibition and the tumor cells are insensitive to CDK4/6 inhibition (Fig 2A). In patients with CDK4/6-dependent tumors, there may be opportunities to enhance anti-tumor efficacy; however, there is a theoretical risk that CDK4/6 inhibition in this setting may antagonize the intended cytotoxicity of the chemotherapy (Fig 2B). Preclinical and clinical data suggest that the risk of chemotherapy antagonism by CDK4/6i is not as well understood as initially thought. In addition, the molecular determinants of CDK4/6-independence and -dependence are complex, such that it can be difficult to identify those tumors that truly rely upon CDK4/6 for proliferation. We discuss these approaches using the “theoretical” binary tumor classification of CDK4/6-independent and -dependent and acknowledge that the spectrum of dependence may actually be continuous.
Figure 2. Differential response to CDK4/6 inhibition determines mode of interaction with chemotherapy.
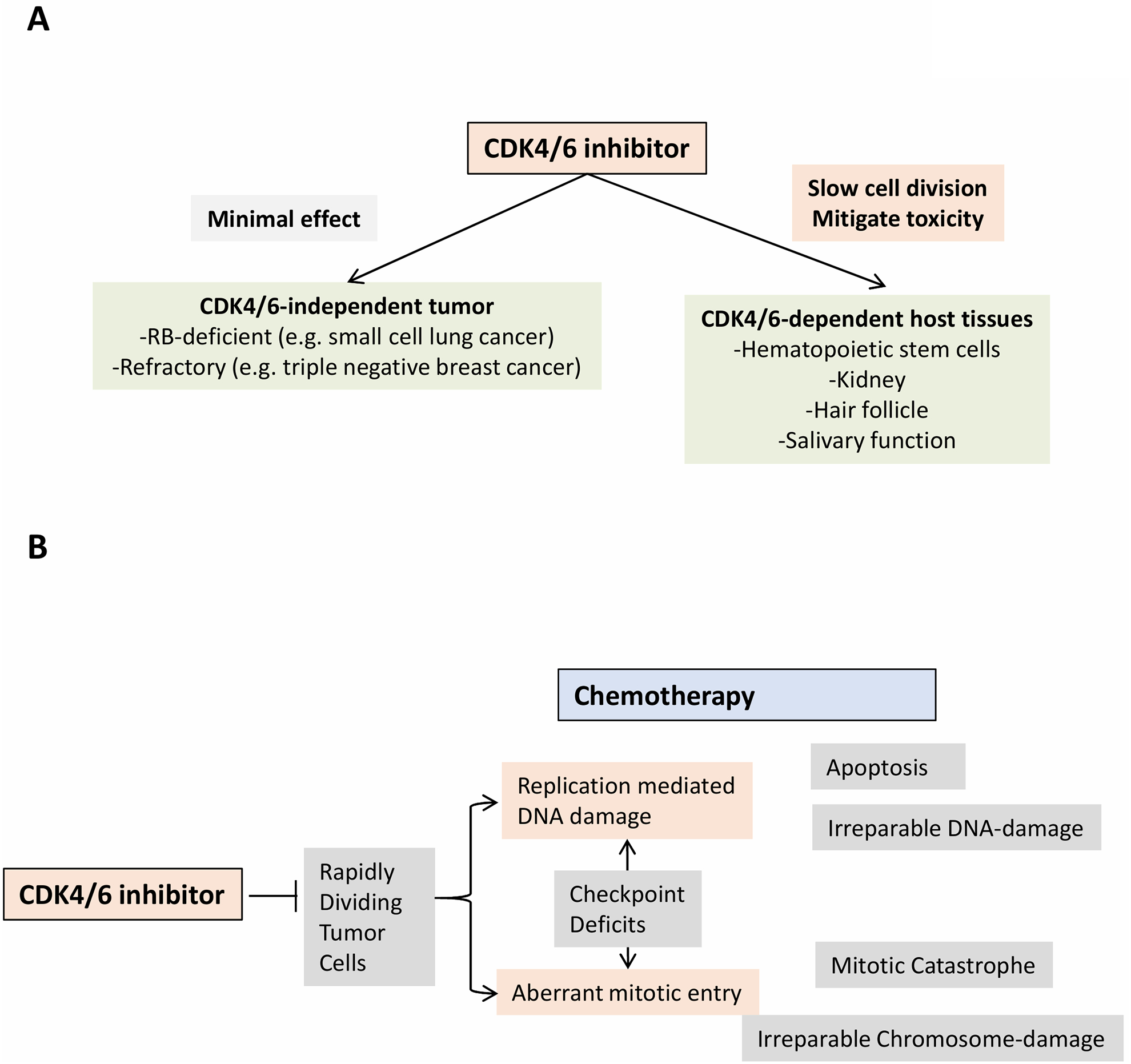
(A) CDK4/6 independent tumors: In CDK4/6 independent tumor models, by definition the pharmacological inhibitors will have minimal effect. Small cell lung cancer tumors exhibit near universal loss of the RB tumor suppressor, while triple negative breast cancer exhibits multiple mechanisms that render CDK4/6 independence. In contrast, the host-tissues will be responsive to the CDK4/6 inhibitor and the reduced cell division could be expected to mitigate the toxicity associated with chemotherapy in specific tissue settings. (B) Conceptual frame-work for antagonism: Chemotherapy represents a diverse class of drugs that exploit the rapid division of tumor cells that impinges on DNA replication or mitotic division. This sensitivity is often enhanced as a result of cell cycle checkpoint deficits in tumor cells. Ultimately, apoptosis, necrosis, or mitotic catastrophe lead to cytotoxic activity. Since CDK4/6 inhibitors can slow cell cycle progression they could negatively impact on the efficacy of chemotherapy.
CDK4/6-independent Tumors - host protection:
The use of CDK4/6i to arrest cells in the G1 phase in cancer patients who are being treated with chemotherapy may not seem to be intuitive. However, this biological phenomenon can be exploited to prevent chemotherapy-induced cellular damage of normal cells that harbor an intact RB pathway (54, 55) (Fig 2A). One of the common side effects of chemotherapy is myelosuppression, that can lead to the exhaustion of hematopoietic stem and progenitor cells (HSPCs) (56–58).
Trilaciclib (G1T28) has been developed to specifically prevent chemotherapy-induced myelosuppression (38). Trilaciclib maintains a selective and reversible G1-arrest in the RB proficient HPSCs and prevents or mitigates the acute and long-term hematopoietic toxicity of the cytotoxic chemotherapeutic agent, 5-fluorouracil, when administered concurrently (38, 55). Similarly, the cytostatic effect of CDK4/6i can prevent or mitigate the hematopoietic toxicity of ionizing radiation by preventing HSPCs from immediately entering the cell cycle when the cells sense the radiation-induced DNA damage (54).
To translate these findings to the clinic, three randomized, placebo-controlled, double-blind clinical trials designed to evaluate the myelopreservation effects of trilaciclib versus placebo in combination with chemotherapy have been completed in small cell lung carcinoma (SCLC) (59–62)(Table 1). SCLC was chosen as the first clinical setting to test the myelopreservation benefit of trilaciclib because: (1) standard of care chemotherapy regimens are myelosuppressive, (2) SCLC replicates independently of CDK4/6 due to obligate loss of RB (63), thereby minimizing theoretical concerns related to chemotherapy antagonism, and (3) SCLC treated in the 1st line setting is a chemosensitive tumor which provided an optimal background upon which to demonstrate that trilaciclib does not antagonize chemotherapy efficacy. In all three studies, trilaciclib demonstrated consistent clinical benefit across myelosuppression endpoints, including highly statistically significant improvements for both primary endpoints: the duration of severe neutropenia [SN] in Cycle 1 (a surrogate for febrile neutropenia and infections) and the percentage of patients with SN. Furthermore, integrated analysis from the three studies demonstrated statistically significant improvement across multiple hematopoietic lineages; including neutrophils (duration of SN in Cycle 1, percentage of patients with SN), red blood cells (RBC) (percentage of patients with Grade 3 or 4 anemia, percentage of patients receiving RBC transfusions on or after 5 weeks, the rate of RBC transfusions on or after 5 weeks), and platelets (percentage of patients with Grade 3 or 4 thrombocytopenia) (59). Consistent with the improvement in chemotherapy safety, patient reported outcome (PRO) measures demonstrated an improved experience for patients receiving trilaciclib, including improved measures of fatigue (59). Importantly, the addition of trilaciclib to chemotherapy did not have an adverse effect on anti-tumor efficacy (60–62).
Table 1:
Clinical studies of CDK4/6 inhibitors with chemotherapy
| Study Title | CDK4/6 Inhibitor | Chemotherapy | Phase | Description | NCT# |
|---|---|---|---|---|---|
| Phase 2 Study of Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib in Patients With Untreated Extensive Stage Small Cell Lung Cancer | Trilaciclib | Carboplatin, Etoposide, and Atezolizumab | 2 | This is a study to investigate the potential clinical benefit of trilaciclib (G1T28) in preserving the bone marrow and the immune system, and enhancing antitumor efficacy when administered with carboplatin, etoposide, and atezolizumab (E/P/A) therapy in first line treatment for patients with newly diagnosed extensive-stage SCLC. | NCT03041311 |
| Phase 2 Study of the Safety, Efficacy, and Pharmacokinetics of G1T28 in Patients With Metastatic Triple Negative Breast Cancer Receiving Gemcitabine and Carboplatin Chemotherapy | Trilaciclib | Gemcitabine and Carboplatin | 2 | his is a study to investigate the potential clinical benefit of trilaciclib (G1T28) in preserving the bone marrow and the immune system, and enhancing chemotherapy antitumor efficacy when administered prior to carboplatin and gemcitabine (GC therapy) for patients with metastatic triple negative breast cancer. | NCT02978716 |
| Phase 1b/2a Safety and Pharmacokinetic Study of G1T28 in Patients With Previously Treated Extensive Stage Small Cell Lung Cancer (SCLC) Receiving Topotecan Chemotherapy | Trilaciclib | Topotecan | 1/2 | This is a study to investigate the potential clinical benefit of trilaciclib (G1T28) in preserving the bone marrow and the immune system, and enhancing chemotherapy antitumor efficacy when administered prior to topotecan in patients previously treated for extensive-stage SCLC. | NCT02514447 |
| Phase 1b/2a Safety and Pharmacokinetic Study of G1T28 in Patients With Extensive Stage Small Cell Lung Cancer (SCLC) Receiving Etoposide and Carboplatin | Trilaciclib | Combination and Etoposide | 1/2 | This is a study to investigate the potential clinical benefit of trilaciclib (G1T28) in preserving the bone marrow and the immune system, and enhancing chemotherapy antitumor efficacy when administered prior to carboplatin and etoposide in first line treatment for patients with newly diagnosed extensive-stage SCLC. | NCT02499770 |
| A Phase 1 Study Of Palbociclib, A CDK 4/6 Inhibitor, In Combination With Chemotherapy In Children With Relapsed Acute Lymphoblastic Leukemia (ALL) Or Lymphoblastic Lymphoma (LL) | Palbociclib | Cytarabine, Methotrexate, Hydrocortisone, Doxorubicin, Prednisolone, Vincristine, Pegaspargase, Prednisone | 1 | AINV18P1 is a Phase 1 study where palbociclib will be administrated in combination with a standard re-induction platform in pediatric relapsed Acute Lymphoblastic Leukemia (ALL) and lymphoblastic lymphoma (LL). | NCT03792256 |
| A Phase I Study of the CDK4/6 Inhibitor PD-0332991, 5-Fluorouracil, and Oxaliplatin in Patients With Advanced Solid Tumor Malignancies | Palbociclib | 5-Fluorouracil and Oxaliplatin | 1 | The purpose of this study is to test the safety and effectiveness of a new combination of drugs, palbociclib and 5-Fluorouracil and Oxaliplatin for patients with advanced solid tumor malignancies. | NCT01522989 |
| Phase 1B Study of PD-0332991 in Combination With T-DM1 in the Treatment of Patients With Advanced HER2 (Human Epidermal Growth Factor Receptor 2)-Positive Breast Cancer | Palbociclib | T-DM1 | 1 | This is a phase 1B inter-patient dose escalation study of PD-0332991 in combination with T--DM1 in patients with recurrent or metastatic HER2-positive breast cancer after prior trastuzumab or other HER2-directed therapies. | NCT01976169 |
| A Phase 1 Trial of PD0332991 and Paclitaxel in Patients With Rb-Expressing Advanced Breast Cancer | Palbociclib | Paclitaxel | 1 | This study is a phase I, single arm, open-label trial of PD0332991 in combination with Paclitaxel in patients with Rb-expressing metastatic breast cancer. | NCT01320592 |
| An Open-Label Phase 1B Study of Palbociclib (Oral CDK4/6 Inhibitor) Plus Abraxane (Nab-Paclitaxel) In Patients with Metastatic Pancreatic Ductal Adenocarcinoma | Palbociclib | Nab-Paclitaxel | 1 | This is a Phase 1, open label, multi center, multiple dose, dose escalation, safety, pharmacokinetic and pharmacodynamic study of palbociclib in combination with nab-P, in sequential cohorts of adult patients with mPDAC, with MTD expansion cohort(s). | NCT02501902 |
| A Phase 1 Study of Palbociclib in Combination With Cisplatin or Carboplatin in Advanced Solid Malignancies | Palbociclib | Carboplatin or Cisplatin | 1 | This phase I trial studies the side effects and best dose of palbociclib with cisplatin or carboplatin in treating patients with solid tumors that have spread to other places and usually cannot be cured or controlled with treatment. | NCT02897375 |
| A Phase I Study of the CDK4/6 Inhibitor PD-0332991, 5-Fluorouracil, and Oxaliplatin in Patients With Advanced Solid Tumor Malignancies | Palbociclib | 5FU Oxaliplatin | 1 | The purpose of this study is to test the safety and effectiveness of a new combination of drugs, PD-0332991 and 5-Fluorouracil and Oxaliplatin for patients with advanced solid tumor malignancies. | NCT01522989 |
| A Phase 1b/2 Study of the Oral CDK4/6 Inhibitor LEE011 (Ribociclib) in Combination With Docetaxel Plus Prednisone in Metastatic Castration Resistant Prostate Cancer | Ribociclib | Docetaxel, Ribociclib, Prednisone, Filgrastim | 1/2 | his is a Phase Ib/II open label clinical trial in patients with metastatic castration resistant prostate cancer. The objective of the phase Ib portion of the study is to establish the maximum tolerated dose (MTD) and dose limiting toxicities (DLT) of docetaxel (75 mg/m2 IV q21 days) and prednisone (5mg orally BID) in combination with ribociclib in escalating oral daily doses in patients with metastatic CRPC with prior resistance to abiraterone and/or enzalutamide who have not undergone prior chemotherapy for metastatic disease. | NCT02494921 |
| A Phase I Study of CDK4/6 Inhibitor LEE011 Combined With Gemcitabine in Patients With Advanced Solid Tumors or Lymphoma | Ribociclib | Gemcitabine | 1 | This phase I trial studies the side effects and best dose of ribociclib and gemcitabine hydrochloride in treating patients with solid tumors or lymphoma that have spread to other places in the body and usually cannot be cured or controlled with treatment | NCT02414724 |
| Phase I Study of CDK4/6 Inhibitor Ribociclib (LEE011) Combined With Gemcitabine in Patients With Advanced Solid Tumors | Ribociclib | Gemcitabine | 1 | This phase I trial studies ribociclib and gemcitabine hydrochloride in treating patients with solid tumors that have spread to other places in the body. | NCT03237390 |
| Study of Ribociclib With Everolimus + Exemestane in HR+ HER2- Locally Advanced/Metastatic Breast Cancer Post Progression on CDK 4/6 Inhibitor. (TRINITI-1) | Ribociclib | Everolimus Exemestane | 1/2 | The purpose of this study is determine if the triplet combination of ribociclib, everolimus and exemastane is effective in the treatment of locally advanced/metastatic breast cancer following treatment with a CDK 4/6 inhibitor | NCT02732119 |
| Phase Ib Trial of LEE011 With Everolimus (RAD001) and Exemestane in the Treatment of Hormone Receptor Positive HER2 Negative Advanced Breast Cancer | Ribociclib | Everolimus Exemestane | 1 | This study evealuates the safety and tolerability of the triplet combination of LEE011 + everolimus + exemestane in patients naïve or refractory to CDK4/6 inhibitor-based therapy, and the safety and tolerability of the doublet combination of LEE011 + exemestane in patients refractory to CDK4/6 inhibitor-based therapy. | NCT01857193 |
| An Open-Label, Phase Ib/II Clinical Trial Of Cdk 4/6 Inhibitor, Ribociclib (Lee011), In Combination With Trastuzumab Or T-Dm1 For Advanced/Metastatic Her2-Positive Breast Cancer | Ribociclib | T-DM1 Trastuzumab Fulvestrant | 1/2 | This study tests the combination of ribociclib in combination with T-DM1, trastuzumab, or trastuzumab plus fulvestrant in patients with advanced/metastatic Her2+ breast cancer. | NCT02657343 |
| A Phase I Trial of Ribocilcib (LEE011) and Weekly Paclitaxel in Patients With Rb+ Advanced Breast Cancer | Ribociclib | Paclitaxel | 1 | This is a Phase I study to assess the safety and MTD of paclitaxel + ribociclib (LEE011) in patients with Rb+, advanced breast cancer. Dose escalation will be performed using standard 3 + 3 dosing strategy. | NCT02599363 |
| A Phase Ib/II Study of LEE011 and Chemoembolization In Patients With Advanced Hepatocellular Carcinoma | Ribociclib | TACE | 2 | The purpose of this study is determine whether the combination therapy with LEE011 and chemoembolization in patients with locally advanced Hepatocellular Carcinoma not amenable to curative therapies will provide greater efficacy than chemoembolization alone with a tolerable safety profile. | NCT02524119 |
| A Phase 1b/2 Study of the Oral CDK4/6 Inhibitor LEE011 (Ribociclib) in Combination With Docetaxel Plus Prednisone in Metastatic Castration Resistant Prostate Cancer | Ribociclib | Docetaxel Prednisone | 1/2 | This is an open-label study of ribociclib (dosed at the RP2D) in combination with docetaxel and prednisone to determine the efficacy and safety of the treatment combination in patients with metastatic castration resistant prostate cancer. | NCT02494921 |
| A Phase 1b Study of LY2835219 in Combination With Multiple Single Agent Options for Patients With Stage IV NSCLC | Abemaciclib | Pemetrexed Gemcitabine | 1 | The main purpose of this study is to evaluate the safety and tolerability of abemaciclib in combination with another anti-cancer drug in participants with NSCLC that is advanced or has spread to other parts of the body (stage IV). | NCT02079636 |
| A Phase 2 Study of Abemaciclib in Patients With Brain Metastases Secondary to Hormone Receptor Positive Breast Cancer, Non-small Cell Lung Cancer, or Melanoma | Abemaciclib | Pemetrexed Gemcitabine | 2 | The main purpose of this study is to evaluate the safety and effectiveness of the study drug known as abemaciclib in participants with hormone receptor positive breast cancer, non-small cell lung cancer (NSCLC), or melanoma that has spread to the brain. Some cohorts allow concurrent pemetrexed and/or gemcitabine. | NCT02308020 |
| A Phase II Trial Program Exploring The Integration Of Novel HER2-targeted Tyrosine Kinase Inhibitor Pyrotinib and CDK4/6 Inhibitor SHR6390 Into Current Chemotherapy/Endocrine Therapy Regimes For Prior Trastuzumab-treated Advanced HER2-positive Breast Cancer | SHR6390 | Capecitabine | 2 | Patients previously failing transtuzumab therapy are randomized to Pyrotinib and SHR6390 plus capcitabine, letrozole, or placebo. | NCT04095390 |
Similar to SCLC, triple negative breast cancer (TNBC) is thought to be a mostly CDK4/6-independent tumor, based on both tumor genetics and the relatively poor response of such tumors to CDK4/6i therapy in preclinical studies (64, 65). Trilaciclib has been tested in combination with gemcitabine and carboplatin in patients with metastatic TNBC (66). In this study the addition of trilaciclib to chemotherapy generally did not improve myelosuppression endpoints; however, there were positive trends for RBC and platelet measures, and patients in the trilaciclib arms received significantly more chemotherapy than the control. In contrast, the anti-tumor efficacy results demonstrated a clinically meaningful survival benefit in both combination groups compared to the chemotherapy alone control group. Median progression-free survival was 5.7 months (95% CI: 3.4–9.2) for the control group compared to 9.4 (6.1–13.0; hazard ratio [HR]: 0.60, p=0.13) and 7.3 (6.2–12.19; HR:0.59, p=0.12) for the two trilaciclib groups. Median overall survival was 12.6 months (6.3–15.6) for the chemotherapy control group compared to 20.1 (10.2-not reached; HR: 0.33, p=0.028) and 17.8 (12.9-not reached; HR: 0.34, p=0.0023) for the two trilaciclib + chemotherapy groups. While patients receiving trilaciclib received more chemotherapy, it is unlikely that this can explain the magnitude of survival benefit achieved with transient CDK4/6 inhibition. Instead, an alternative mechanism of action related to enhanced anti-tumor immunity is more likely as discussed in more detail below.
Antagonism of chemotherapy-mediated cytotoxicity in CDK4/6-dependent pre-clinical models:
Based on the intra-cellular targets of many chemotherapeutic agents, it is evident that dividing cells are more chemo-sensitive than arrested cells which underlies the therapeutic index of such agents (67). Considering the mechanism of action of CDK4/6i in inhibiting cell division through activation of the RB pathway, it is hypothesized that concurrent CDK4/6 inhibition may antagonize the cytotoxic effects of chemotherapeutic agents in tumors that are CDK4/6 dependent (Fig 2B). Indeed, a number of preclinical reports have described the antagonistic effects of combining a CDK4/6i with chemotherapy (50, 68–70) (Table 2). In breast cancer cell lines, xenografts, and GEMM models, treatment with CDK4/6i can limit the acute induction of tumor-specific toxicity with taxanes, anthracyclines, and platinum-agents (68, 70–72). These effects are RB-dependent and link the antagonism of chemotherapy cytotoxicity with the cell cycle pause induced by CDK4/6 inhibition (68, 70, 71). In considering these data, it is important to appreciate that most studies utilized conditions where CDK4/6 inhibition elicits profound cell cycle inhibition and that effects measured on antagonism were relatively short-term; however, it should not be discounted that such antagonism could be clinically relevant and caution should be taken to evaluate whether these effects will be seen in specific clinical settings.
Table 2:
Preclinical Studies Demonstrating Some Evidence of Antagonism Between CDK4/6 Inhibitors and Chemotherapy
| Author | Tumor Type | Context | Chemo | Outcome |
|---|---|---|---|---|
| Franco et al. Oncotarget 2014 | Human PDA | In vitro | Gemcitabine 5FU |
|
| McClendon et al. Cell Cycle 2012 | Human TNBC | In vitro In vivo |
Doxorubicin |
|
| Dean et al. JBC 2012 | Human TNBC | In vitro In vivo |
Doxorubicin Paclitaxel Radiation |
|
| Roberts et al. JCI 2012 | TNBC HER2 | In vitro In vivo |
Carboplatin Doxorubicin, Etoposide Camptothecin,Paclitaxel |
|
| Konecny et al. Clin Ca Re. 2011 | Human Ovarian | In vitro | Carboplatin |
|
| Cretella et al. Scientific Reports 2019 | Human TNBC | In vitro | Paclitaxel |
|
Potential cooperation between CDK4/6i and chemotherapy in cancer therapy in preclinical models:
In contrast to the above reports, a number of preclinical studies suggest that the combination of chemotherapy and CDK4/6 inhibition can have cooperative anti-tumor effects; similar observations are beginning to emerge from clinical studies. Palbociclib, ribociclib, and abemaciclib have been shown to enhance, rather than antagonize, chemotherapy cytotoxicity when combined with camptothecin, carboplatin, cisplatin, docetaxel, doxorubicin, 5FU, gemcitabine, irinotecan, paclitaxel, and temozolomide (73–86). These effects were shown in RB-proficient in vitro and in vivo models of non-small cell lung carcinoma (NSCLC), ovarian cancer, gastric cancer, TNBC, atypical teratoid rhabdoid tumors, Ewing sarcoma, pancreatic cancer and glioblastoma using both sequential and concurrent dosing schedules (73–86) (Table 3).
Table 3:
Preclinical Studies Demonstrating Enhanced Efficacy with CDK4/6 Inhibitors + Chemotherapy Treatment
| Author | Inhibitor | Tumor Type | Context | Chemotherapy | Outcome |
|---|---|---|---|---|---|
| Hamilton et al. Molecules 2014 | Palbociclib | SCLC | In vitro | Camptothecins |
|
| Gelbert et al. Inv New Drug 2014 | Abemaciclib | Lung cancer | In vivo | Gemcitabine |
|
| Hashizume et al. Neuro-Oncology 2016 | Palbociclib | ATRT and glioblastoma | In vitro In vivo |
Radiation |
|
| O’Brien et al. MCT 2018 | Abemaciclib | TNBC | In vitro In vivo |
Docetaxel, Carboplatin |
|
| Iyengar et al. Oncotarget 2018 | Ribociclib | Ovarian Ca | In vitro In vivo |
Cisplatin |
|
| Dowless et al. Clin Ca Re 2018 | Abemaciclib | Ewing sarcoma | In vitro In vivo |
Doxorubicin, Etoposide, Cisplatin, Temozolomide Irinotecan |
|
| Chou et al. Gut 2018 | Palbociclib | Pancreatic Cancer | In vitro In vivo |
Gemcitabine Paclitaxel |
|
| Raub et al. Drug Metab Dispos 2015 | Abemaciclib | Glioblastoma | In vivo | Temozolomide |
|
| Wang et al. Int J Mol Med. 2018 | Palbociclib | Gastric Ca | In vitro | 5FU |
|
| Gao et al. Cell Oncol. 2017 | Palbociclib | Ovarian | In vitro | Paclitaxel |
|
| Zhang et al. Cancer Biology & Therapy. 2013 | CINK4 | NSCLC | In Vitro | Paclitaxel |
|
| Cao et al. Oncogene 2019 | Palbociclib | Squam Lung | In vitro In vivo |
Paclitaxel |
|
| Kumarasamy et al. Oncogene 2019 | Palbociclib, Ribociclib | PDAC | In vitro In vivo |
Gemcitabine Docetaxel |
|
| Salvador-Barbero et al. Cancer cell 2020 | Palbociclib | PDAC | In vitro In vivo |
Taxol |
|
Ca, cancer; NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer; Squam lung, squamous lung cancer; TNBC, triple negative breast cancer; PDAC, Pancreatic ductal adenocarcinoma
While the CDK4/6-RB-E2F axis is responsible for controlling expression of genes required for cell cycle progression, DNA replication, and mitotic progression (23, 39), the unexpected observations of cooperation described above may be due to other less well understood mechanisms. One mechanistic explanation of the enhanced, rather than antagonistic activity of combination CDK4/6i plus chemotherapy regimens, has been the reduced expression of specific E2F-regulated genes, whose products are targeted by chemotherapy (Fig. 3A). Palbociclib treatment reduces thymidylate synthase (TS; 5FU target), Topoisomerase 1, and Topoisomerase 2 alpha expression. These effects on gene expression could potentially enhance the response to select chemotherapies by limiting the threshold needed for efficacy of chemotherapy (70, 78, 87). Similarly, E2F regulates the expression of multiple genes required for DNA damage repair and thus would limit the ability of tumor cells to recover from chemotherapy-mediated damage. Consistent with the impact on DNA-repair machinery, it has been shown that CDK4/6i can cooperate with PARP inhibitors ostensibly by limiting the ability of damaged cells to carry out HR-mediated repair (88) (Fig. 3A). Conversely, Gao et al demonstrated a role for MDR (multidrug resistance; P-glycoprotein) in paclitaxel resistance, an effect counteracted by both CDK4 siRNA and palbociclib treatment (76). Finally, multiple studies have demonstrated that CDK4/6 inhibition can enhance chemotherapy-induced apoptosis (76, 80, 86), and that CDK4/6 are upstream regulators of transcription factors that control global gene expression leading to changes in metabolism, DNA repair and cell plasticity, all of which can render a cancer cell more susceptible to chemotherapy cytotoxicity (89). Collectively, these results suggest that the net effect of concomitant CDK4/6 inhibition during chemotherapy exposure in patients with CDK4/6-dependent tumors will provide cooperation rather than antagonism (Table 3).
Figure 3. Cellular response to CDK4/6 inhibition in combination with chemotherapy.
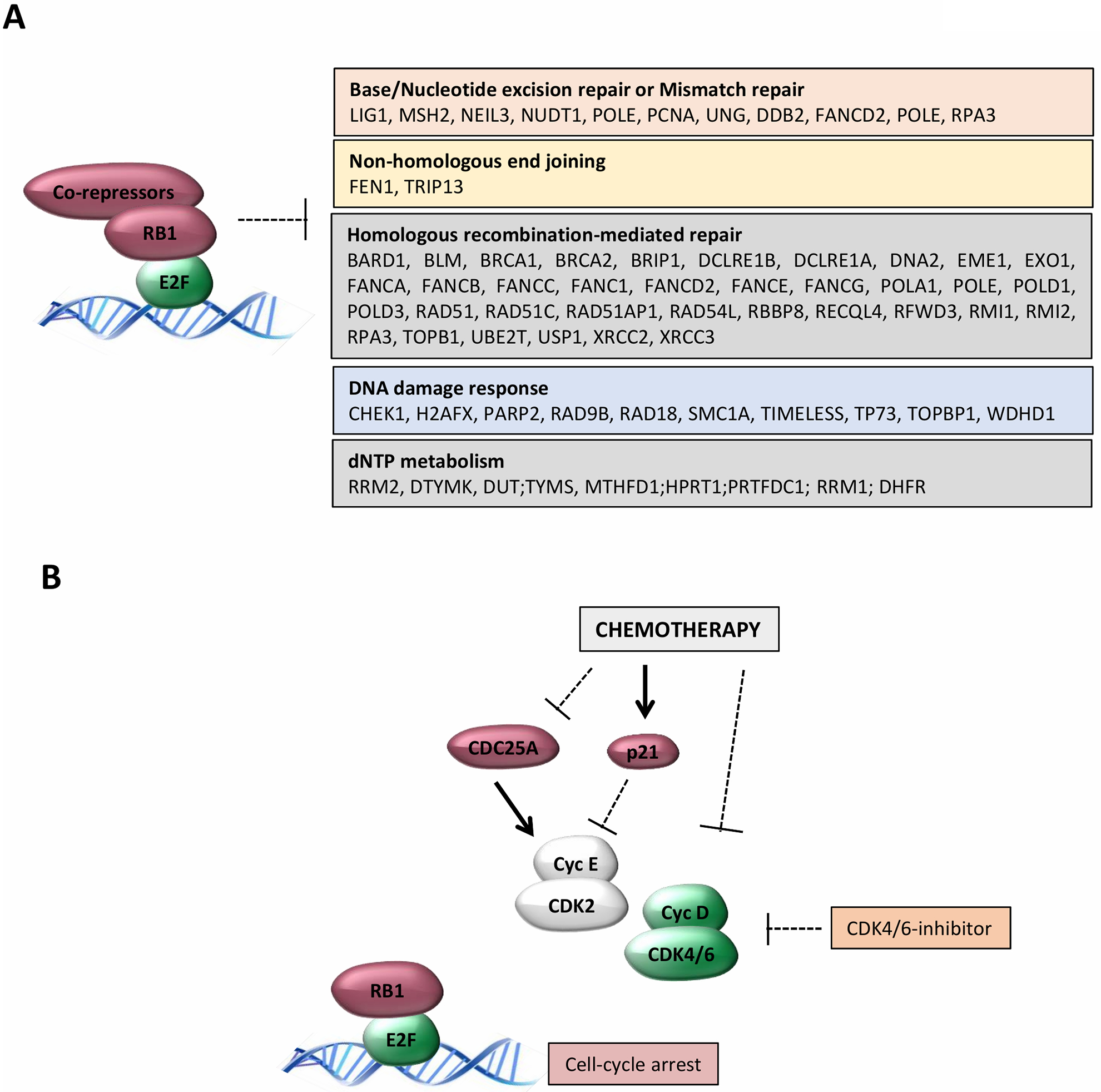
(A) Downstream from the activation of the RB-pathway are the repression of multiple genes involved in different pathways that could impinge on different features of response to distinct chemotherapeutic agents. (B) There are multiple mechanisms through which chemotherapy impinges on the activity of CDK2 and CDK4/6 complexes that would be expected to enhance the efficacy of pharmacological CDK4/6 inhibitors. For example, CDC25A is rapidly degraded or the CDK2-inhibitor P21CIP1 is rapidly induced as a specific response to chemotherapy. Conversely, degradation of cyclin D1 and inhibition of CDK4/6 complexes is a consequence of different chemotherapy agents that induce S-phase block.
Strategies for incorporating CDK4/6i into chemotherapy regimens:
Based on the available preclinical and clinical data, there are several therapeutic strategies by which CDK4/6i can be incorporated into standard chemotherapy regimens to provide therapeutic benefit to patients:
Protection of Normal Tissues:
As described above, protection of HSPCs to reduce dose-limiting myelosuppression has been demonstrated preclinically and clinically (38, 60–62, 68, 90). While myelosuppression is recognized as a common complication of chemotherapy, damage to other normal tissues including the gastrointestinal track, kidney and hair follicles also occurs. CDK4/6 inhibition has been shown to ameliorate kidney injury in preclinical models following both cisplatin treatment and acute renal ischemia, and to provide intestinal radioprotection (39, 91, 92). Additionally, alopecia, while not life threatening, is one of the most distressing side effects of chemotherapy. Similar to other tissues, transient CDK4/6 inhibition has been shown to protect hair follicles from taxane-induced damage in preclinical models (93). While clinical benefit for this approach has been shown in a CDK4/6-independent setting (SCLC), the question remains whether it can be employed in CDK4/6-dependent tumors without antagonizing chemotherapy anti-tumor efficacy. In vivo evaluation of trilaciclib with chemotherapy in CDK4/6 sensitive breast cancer models has not shown antagonism at the tumor level, and with some models, the combination shows enhanced anti-tumor efficacy (94). Interestingly, subset analysis of patients in the trilaciclib metastatic TNBC (mTNBC) study using PAM50 and other molecular stratification approaches revealed no antagonism, and demonstrated improved PFS and OS across all groups (55, 66, 95, 96).
Concurrent interactions, maintenance therapy, and staggered strategies to enhance anti-tumor efficacy:
Since the mechanisms of CDK4/6i and chemotherapy action are distinct, there could be drug interactions that would enhance the efficacy of each class of agent. Chemotherapy is well known to impact CDK-biology at multiple points that would be expected to enhance the cytostatic response to CDK4/6 inhibition (73). For example, chemotherapy can impact CDK2 activity via the induction of the endogenous CDK inhibitor p21 or loss of the CDC25a protein phosphatase which would yield increased the inhibitory phosphorylation on CDK2 (Fig. 3B). This cooperation has been illustrated in models of pancreatic cancer and other tumor models that do not show robust response to CDK4/6i (97, 98). Regarding resistance to chemotherapy, as discussed above, CDK4/6i can impact the expression of genes associated with DNA-repair and dNTP-metabolism. These effects could be broadly relevant to chemotherapy that induces DNA damage or is associated with nucleotide metabolism or function (Fig. 3A). Whether these interactions manifest clinically remains unclear, although multiple clinical trials are interrogating CDK4/6i and chemotherapy combinations where there is clear overlap in the treatment.
Given the canonical action of chemotherapy and CDK4/6i, it is appealing to separate/stagger the dosing of the chemotherapy and the CDK4/6i. In this context, the chemotherapy can have the desired impact of killing the tumor cells, while the CDK4/6i prevents the expansion of cells that are not killed by the chemotherapy (99). In ovarian cancer models, following the release from cisplatin-mediated S-phase arrest, tumor cells undergo normal cell-cycle progression and proliferation, which is significantly blocked by CDK4/6 inhibition, indicating a positive interaction (81). Similarly, the targeted microtubule poison trastuzumab-emtansine (T-DM1) displayed a cooperative anti-tumor effect where CDK4/6i could block the recovery of residual cells following T-DM1 treatment (100). Presently there are a number of clinical trials that explore the interaction of CDK4/6 inhibition and chemotherapy using dosing strategies to enhance durability of response (Table 1). In one of the first reported trials, the combination of paclitaxel with palbociclib appeared to have efficacy in heavily pre-treated breast cancer (101).
As CDK4/6i are not associated with cumulative toxicity, which is a common feature of chemotherapy, they can be given for long periods of time. Therefore, chronic administration of a CDK4/6i in the maintenance treatment setting, after the tumor has been de-bulked and chemotherapy discontinued could lead to improved patient outcomes by delaying tumor progression and or allowing the host immune system to eliminate the residual disease (81).
Enhancing the response to immunotherapy:
There is significant preclinical data demonstrating that CDK4/6i can enhance immune checkpoint inhibitor (ICI) efficacy through enhanced T-cell activation, increased antigen presentation, increased expression of PD-L1, and reduced T-cell exclusion and immune evasion gene signature (89, 102, 103) In the clinic, chemotherapy has successfully been used to enhance ICI efficacy (104–109) through induction of immunogenic cell death, enhancement of immunosurveillance and T cell activity, and reduction of immunosuppressive cell types (110–113). Despite these benefits, chemotherapy induced myelosuppression and immunosuppression may limit the full benefit of combinatorial treatments with ICIs. Given that intratumor immune cells are highly proliferative, one strategy to further enhance chemotherapy/ICI combinations is through transient CDK4/6 inhibition during chemotherapy exposure. Trilaciclib has been shown to favorably alter the tumor immune microenvironment through transient T cell inhibition (114, 115). Treatment of immunocompetent tumor models with trilaciclib plus chemotherapy/ICI combinations significantly improved anti-tumor efficacy and survival compared to chemotherapy/ICI combinations (114, 115).
Summary and under-explored areas:
In summary, while it was originally hypothesized that CDK4/6i combined with chemotherapy could potentially result in antagonistic anti-tumor effects (at least in CDK4/6-dependent tumors), emerging data suggest that these agents can be safely combined using different clinical strategies to enhance anti-tumor efficacy and/or reduce chemotherapy-induced toxicity. There are a number of unanswered questions whose answers could help guide implementation of these strategies in the clinic. Understanding which of these clinical strategies would be best employed in tumors that are “truly” CDK4/6-dependent is still needed. However, identifying “truly” CDK4/6-dependent tumors remains elusive as predictive biomarkers have not been validated in clinical practice. Identification and validation of such biomarkers would allow clinical testing of CDK4/6i + chemotherapy combinations in a homogeneous CDK4/6-dependent tumor population to definitively determine whether CDK4/6 inhibition during chemotherapy exposure interferes with the intended anti-tumor efficacy of chemotherapy. Additionally, understanding the contribution of anti-tumor effects arising from cell cycle inhibition in tumor cells versus non-tumor cells (e.g. cancer associated fibroblasts and immune cells) and through non-canonical biological processes controlled by CDK4/6 will further aid the rational design of novel CDK4/6i plus chemotherapy and/or immunotherapy regimens.
Acknowledgements:
The authors acknowledge their colleagues at Roswell Park and G1 Therapeutics for thoughtful discussion and review of the manuscript. This work was supported by grants AKW and ESK from the NIH CA211878 and CA247362.
Footnotes
CONFLICT OF INTEREST STATEMENT: At the time of the initiation of this manuscript PJR was an employee of G1 Therapeutics which is involved in the clinical development of CDK4/6 inhibitors. The other authors have no potential conflicts of interest to report.
References
- 1.Beach D, Durkacz B, Nurse P. Functionally homologous cell cycle control genes in budding and fission yeast. Nature. 1982;300(5894):706–9. [DOI] [PubMed] [Google Scholar]
- 2.Nurse P, Thuriaux P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics. 1980;96(3):627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker SJ, Reddy EP. CDK4: A Key Player in the Cell Cycle, Development, and Cancer. Genes Cancer. 2012;3(11–12):658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. 2013;5(3):a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagano M, Theodoras AM, Tam SW, Draetta GF. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 1994;8(14):1627–39. [DOI] [PubMed] [Google Scholar]
- 7.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270(40):23589–97. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20(5):187–90. [DOI] [PubMed] [Google Scholar]
- 9.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65(4):701–13. [DOI] [PubMed] [Google Scholar]
- 10.Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, et al. Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol. 1998;18(9):5609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1(3):226–31. [DOI] [PubMed] [Google Scholar]
- 12.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11(8):957–72. [DOI] [PubMed] [Google Scholar]
- 13.Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102(1):55–66. [DOI] [PubMed] [Google Scholar]
- 14.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704–7. [DOI] [PubMed] [Google Scholar]
- 15.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joyce D, Bouzahzah B, Fu M, Albanese C, D’Amico M, Steer J, et al. Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-kappaB-dependent pathway. J Biol Chem. 1999;274(36):25245–9. [DOI] [PubMed] [Google Scholar]
- 17.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14(24):3102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323–34. [DOI] [PubMed] [Google Scholar]
- 19.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7(3):331–42. [DOI] [PubMed] [Google Scholar]
- 20.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14(3):2077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farkas T, Hansen K, Holm K, Lukas J, Bartek J. Distinct phosphorylation events regulate p130- and p107-mediated repression of E2F-4. J Biol Chem. 2002;277(30):26741–52. [DOI] [PubMed] [Google Scholar]
- 22.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16(2):245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knudsen ES, Witkiewicz AK. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer. 2017;3(1):39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493–504. [DOI] [PubMed] [Google Scholar]
- 26.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118(4):477–91. [DOI] [PubMed] [Google Scholar]
- 27.Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448(7155):811–5. [DOI] [PubMed] [Google Scholar]
- 28.Malumbres M, Barbacid M. Is Cyclin D1-CDK4 kinase a bona fide cancer target? Cancer Cell. 2006;9(1):2–4. [DOI] [PubMed] [Google Scholar]
- 29.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–21. [DOI] [PubMed] [Google Scholar]
- 30.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9(1):13–22. [DOI] [PubMed] [Google Scholar]
- 31.Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9(1):23–32. [DOI] [PubMed] [Google Scholar]
- 32.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38. [PubMed] [Google Scholar]
- 33.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin Cancer Res. 2017;23(13):3251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corona SP, Generali D. Abemaciclib: a CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des Devel Ther. 2018;12:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investigational new drugs. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bisi JE, Sorrentino JA, Jordan JL, Darr DD, Roberts PJ, Tavares FX, et al. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget. 2017;8(26):42343–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bisi JE, Sorrentino JA, Roberts PJ, Tavares FX, Strum JC. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol Cancer Ther. 2016;15(5):783–93. [DOI] [PubMed] [Google Scholar]
- 39.DiRocco DP, Bisi J, Roberts P, Strum J, Wong KK, Sharpless N, et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. American journal of physiology Renal physiology. 2014;306(4):F379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knudsen ES, Witkiewicz AK. Defining the transcriptional and biological response to CDK4/6 inhibition in relation to ER+/HER2- breast cancer. Oncotarget. 2016;7(43):69111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018–32. [DOI] [PubMed] [Google Scholar]
- 42.Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011;17(6):1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, Dunn GP, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A. 2010;107(25):11501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108(5):1744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Leeuw R, McNair C, Schiewer MJ, Neupane NP, Brand LJ, Augello MA, et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin Cancer Res. 2018;24(17):4201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornell L, Wander SA, Visal T, Wagle N, Shapiro GI. MicroRNA-Mediated Suppression of the TGF-beta Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019;26(10):2667–80 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nature communications. 2019;10(1):1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel P, Tsiperson V, Gottesman SRS, Somma J, Blain SW. Dual Inhibition of CDK4 and CDK2 via Targeting p27 Tyrosine Phosphorylation Induces a Potent and Durable Response in Breast Cancer Cells. Mol Cancer Res. 2018;16(3):361–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Portman N, Alexandrou S, Carson E, Wang S, Lim E, Caldon CE. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr Relat Cancer. 2019;26(1):R15–R30. [DOI] [PubMed] [Google Scholar]
- 50.Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knudsen ES, Kumarasamy V, Ruiz A, Sivinski J, Chung S, Grant A, et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016;76(8):2301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–4. [DOI] [PubMed] [Google Scholar]
- 54.Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. The Journal of clinical investigation. 2010;120(7):2528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asghar US, Barr AR, Cutts R, Beaney M, Babina I, Sampath D, et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin Cancer Res. 2017;23(18):5561–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gardner RV, Lerner C, Astle CM, Harrison DE. Assessing permanent damage to primitive hematopoietic stem cells after chemotherapy using the competitive repopulation assay. Cancer Chemother Pharmacol. 1993;32(6):450–4. [DOI] [PubMed] [Google Scholar]
- 57.Gardner RV. Long term hematopoietic damage after chemotherapy and cytokine. Front Biosci. 1999;4:e47–57. [DOI] [PubMed] [Google Scholar]
- 58.Mauch P, Constine L, Greenberger J, Knospe W, Sullivan J, Liesveld JL, et al. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol Biol Phys. 1995;31(5):1319–39. [DOI] [PubMed] [Google Scholar]
- 59.Weiss JSK, Gwaltney C, Daniel D, Adler S, Wo0lfe S, Malik RK, Morris SR, Antal JM, Andric Z. Positive effects of trilaciclib on patient myelosuppression-related symptoms and fucntioning: results from three phase 2 randomized, double-blind, placebo-controlled small cell lung cancer trials. Supportive Care in Cancer. 2019;27:S274. [Google Scholar]
- 60.Weiss JM, Csoszi T, Maglakelidze M, Hoyer RJ, Beck JT, Domine Gomez M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30(10):1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hart LL, Andric ZG, Hussein MA, Ferrarotto R, Beck JT, Subramanian J, et al. Effect of trilaciclib, a CDK 4/6 inhibitor, on myelosuppression in patients with previously treated extensive-stage small cell lung cancer receiving topotecan. Journal of Clinical Oncology. 2019;37(15_suppl):8505-. [Google Scholar]
- 62.Daniel D, Kuchava V, Bondarenko I, Ivashchuk O, Spigel D, Dasgupta A, et al. 1742PDTrilaciclib (T) decreases myelosuppression in extensive-stage small cell lung cancer (ES-SCLC) patients receiving first-line chemotherapy plus atezolizumab. Annals of Oncology. 2019;30(Supplement_5). [Google Scholar]
- 63.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6(7):740–53. [DOI] [PubMed] [Google Scholar]
- 65.Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014;16(3):207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan AR, Wright GS, Thummala AR, Danso MA, Popovic L, Pluard TJ, et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2019;20(11):1587–601. [DOI] [PubMed] [Google Scholar]
- 67.Gudkov AV, Komarova EA. Radioprotection: smart games with death. The Journal of clinical investigation. 2010;120(7):2270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104(6):476–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, et al. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle. 2012;11(14):2747–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dean JL, McClendon AK, Knudsen ES. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J Biol Chem. 2012;287(34):29075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witkiewicz AK, Chung S, Brough R, Vail P, Franco J, Lord CJ, et al. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell reports. 2018;22(5):1185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jin D, Tran N, Thomas N, Tran DD. Combining CDK4/6 inhibitors ribociclib and palbociclib with cytotoxic agents does not enhance cytotoxicity. PLoS One. 2019;14(10):e0223555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao J, Zhu Z, Wang H, Nichols TC, Lui GYL, Deng S, et al. Combining CDK4/6 inhibition with taxanes enhances anti-tumor efficacy by sustained impairment of pRB-E2F pathways in squamous cell lung cancer. Oncogene. 2019;38(21):4125–41. [DOI] [PubMed] [Google Scholar]
- 74.Chou A, Froio D, Nagrial AM, Parkin A, Murphy KJ, Chin VT, et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut. 2018;67(12):2142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dowless M, Lowery CD, Shackleford T, Renschler M, Stephens J, Flack R, et al. Abemaciclib Is Active in Preclinical Models of Ewing Sarcoma via Multipronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin Cancer Res. 2018;24(23):6028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao Y, Shen J, Choy E, Mankin H, Hornicek F, Duan Z. Inhibition of CDK4 sensitizes multidrug resistant ovarian cancer cells to paclitaxel by increasing apoptosiss. Cell Oncol (Dordr). 2017;40(3):209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investigational new drugs. 2014;32(5):825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamilton G, Klameth L, Rath B, Thalhammer T. Synergism of cyclin-dependent kinase inhibitors with camptothecin derivatives in small cell lung cancer cell lines. Molecules. 2014;19(2):2077–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hashizume R, Zhang A, Mueller S, Prados MD, Lulla RR, Goldman S, et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol. 2016;18(11):1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120(5):1095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iyengar M, O’Hayer P, Cole A, Sebastian T, Yang K, Coffman L, et al. CDK4/6 inhibition as maintenance and combination therapy for high grade serous ovarian cancer. Oncotarget. 2018;9(21):15658–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Brien N, Conklin D, Beckmann R, Luo T, Chau K, Thomas J, et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol Cancer Ther. 2018;17(5):897–907. [DOI] [PubMed] [Google Scholar]
- 83.Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab Dispos. 2015;43(9):1360–71. [DOI] [PubMed] [Google Scholar]
- 84.Wang D, Sun Y, Li W, Ye F, Zhang Y, Guo Y, et al. Antiproliferative effects of the CDK6 inhibitor PD0332991 and its effect on signaling networks in gastric cancer cells. Int J Mol Med. 2018;41(5):2473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Zhou L, Zhao S, Dicker DT, El-Deiry WS. The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell cycle. 2017;16(12):1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang XH, Cheng Y, Shin JY, Kim JO, Oh JE, Kang JH. A CDK4/6 inhibitor enhances cytotoxicity of paclitaxel in lung adenocarcinoma cells harboring mutant KRAS as well as wild-type KRAS. Cancer Biol Ther. 2013;14(7):597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Min A, Kim JE, Kim YJ, Lim JM, Kim S, Kim JW, et al. Cyclin E overexpression confers resistance to the CDK4/6 specific inhibitor palbociclib in gastric cancer cells. Cancer Lett. 2018;430:123–32. [DOI] [PubMed] [Google Scholar]
- 88.Yi J, Liu C, Tao Z, Wang M, Jia Y, Sang X, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine. 2019;43:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell. 2018;34(1):9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He S, Roberts PJ, Sorrentino JA, Bisi JE, Storrie-White H, Tiessen RG, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017;9(387). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pabla N, Gibson AA, Buege M, Ong SS, Li L, Hu S, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A. 2015;112(16):5231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei L, Leibowitz BJ, Wang X, Epperly M, Greenberger J, Zhang L, et al. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. The Journal of clinical investigation. 2016;126(11):4076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Purba TS, Ng’andu K, Brunken L, Smart E, Mitchell E, Hassan N, et al. CDK4/6 inhibition mitigates stem cell damage in a novel model for taxane-induced alopecia. EMBO Mol Med. 2019;11(10):e11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sorrentino JA BJ, Thompson D, Lai AY, Hall CR, Strum JC, Roberts PJ. Trilaciclib, a Cdk4/6 Inhibitor, does not impair the efficacy of chemotherapy in Cdk4/6-dependent tumor models. European Journal of Cancer. 2018;103:e23–e147. [Google Scholar]
- 95.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11(6):e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prat A, Bianchini G, Thomas M, Belousov A, Cheang MC, Koehler A, et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin Cancer Res. 2014;20(2):511–21. [DOI] [PubMed] [Google Scholar]
- 97.Kumarasamy V, Ruiz A, Nambiar R, Witkiewicz AK, Knudsen ES. Chemotherapy impacts on the cellular response to CDK4/6 inhibition: distinct mechanisms of interaction and efficacy in models of pancreatic cancer. Oncogene. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Salvador-Barbero B, Alvarez-Fernandez M, Zapatero-Solana E, El Bakkali A, Menendez MDC, Lopez-Casas PP, et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell. 2020;37(3):340–53 e6. [DOI] [PubMed] [Google Scholar]
- 99.Cretella D, Fumarola C, Bonelli M, Alfieri R, La Monica S, Digiacomo G, et al. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci Rep. 2019;9(1):13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Witkiewicz AK, Cox D, Knudsen ES. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes Cancer. 2014;5(7–8):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clark AS, McAndrew NP, Troxel A, Feldman M, Lal P, Rosen M, et al. Combination Paclitaxel and Palbociclib: Results of a Phase I Trial in Advanced Breast Cancer. Clin Cancer Res. 2019. [DOI] [PubMed] [Google Scholar]
- 102.Knudsen ES, Pruitt SC, Hershberger PA, Witkiewicz AK, Goodrich DW. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer. 2019;5(5):308–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018;8(2):216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378(22):2078–92. [DOI] [PubMed] [Google Scholar]
- 105.Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018. [DOI] [PubMed] [Google Scholar]
- 106.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2040–51. [DOI] [PubMed] [Google Scholar]
- 108.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375(19):1823–33. [DOI] [PubMed] [Google Scholar]
- 109.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379(22):2108–21. [DOI] [PubMed] [Google Scholar]
- 110.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155(4):1063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44(2):343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–21. [DOI] [PubMed] [Google Scholar]
- 113.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73. [DOI] [PubMed] [Google Scholar]
- 114.Lai AY, Sorrentino JA, Strum JC, Roberts PJ. Transient exposure of trilaciclib, a CDK4/6 inhibitor, modulates gene expression in tumor immune infiltrates to promote a pro-inflammatory tumor microenvironment. Cancer Research. 2018;78(13 Supplement):1752-. [Google Scholar]
- 115.Sorrentino JA, Lai AY, Strum JC, Roberts PJ. Trilaciclib (G1T28), a CDK4/6 inhibitor, enhances the efficacy of combination chemotherapy and immune checkpoint inhibitor treatment in preclinical models. Cancer Research. 2017;77(13 Supplement):5628-.28904063 [Google Scholar]