

SUMMARY
Tbr1 is a high-confidence autism spectrum disorder (ASD) gene encoding a transcription factor with distinct pre- and postnatal functions. Postnatally, Tbr1 conditional knockout (CKO) mutants and constitutive heterozygotes have immature dendritic spines and reduced synaptic density. Tbr1 regulates expression of several genes that underlie synaptic defects, including a kinesin (Kif1a) and a WNT-signaling ligand (Wnt7b). Furthermore, Tbr1 mutant corticothalamic neurons have reduced thalamic axonal arborization. LiCl and a GSK3β inhibitor, two WNT-signaling agonists, robustly rescue the dendritic spines and the synaptic and axonal defects, suggesting that this could have relevance for therapeutic approaches in some forms of ASD.
Graphical Abstract
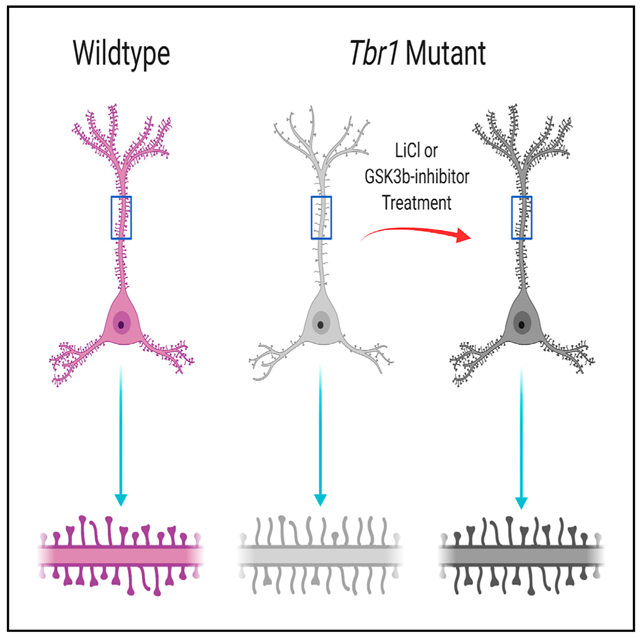
In Brief
Fazel Darbandi et al. demonstrate that TBR1 directly regulates transcriptional circuits in cortical layers 5 and 6, which promote dendritic spine and synaptic density. Enhancing WNT signaling rescues dendritic spine maturation and synaptogenesis defects in Tbr1 mutants. These results provide insights into mechanisms that underlie ASD pathophysiology.
INTRODUCTION
Autism spectrum disorders (ASDs) are defined by deficits in social interaction and abnormalities in language development and repetitive behavior. Considerable genetic and phenotypic heterogeneity has complicated efforts to understand the underlying biology of ASD. However, recent progress in the genomics of ASD has revealed more than 65 high-confidence ASD (hcASD) risk genes (Sanders et al., 2015). Systems analyses suggest that expression of ASD risk genes have important functions in mid-fetal deep-layer cortical excitatory neurons and that disruption may contribute to ASD pathophysiology (Willsey et al., 2013). Among these ASD genes, analysis of the Tbr1 transcription factor (TF) is attractive, as it opens the possibility of defining a transcriptional pathway that includes other ASD genes.
Tbr1 has a central role in the development of mouse early-born excitatory cortical neurons. Tbr1 expression, which begins in newborn neurons, dictates layer 6 identity (Bedogni et al., 2010; Bulfone et al., 1998; Hevner et al., 2001, 2003; McKenna et al., 2011). Using Tbr1layer6 conditional knockouts (CKOs), we recently demonstrated that neonatal Tbr1 function in layer 6 is required for maintaining corticothalamic identity and synaptogenesis (Fazel Darbandi et al., 2018).
Here, we delved deeper into Tbr1’s function in synaptogenesis in several ways. First, we identified convergent synaptic pheno-types in Tbr1layer5 and Tbr1layer6 CKOs and Tbr1constitutive (Tbr1+/−) mutants, including a defect in the formation of mature dendritic spines. Next, we used single-cell RNA sequencing (scRNA-seq) of Tbr1layer5 mutant medial prefrontal cortex (mPFC) neurons and identified Tbr1-regulated genes that impact synapse formation in layer 5, including a kinesin motor protein (Kif1a) and genes in the WNT-signaling pathway (Gsk3β, Ctnnb1, and Wnt7b). We also identified a number of Tbr1-regulated ASD genes in the layer 5 neurons of the mPFC, including Ank2, Ap2s1, Ctnnb1, Dpysl2, Map1a, Rorb, Smarcc2, and Gsk3β. Finally, we found that LiCl, a drug approved by the US Food and Drug Administration, and a GSK3β inhibitor (SB216763; Sigma-Aldrich) that promotes WNT signaling rescue the spine and synaptic defects in adult Tbr1layer5, Tbr1layer6, and Tbr1constitutive (Tbr1+/−) mutants. Lastly, Tbr1layer5 mutants exhibit decreased social interactions with young mice, a phenotype that is rescued with LiCl treatment. The LiCl results suggest an important and novel biological mechanism underlying ASD that may have implications for the treatment of patients with TBR1 mutations and, potentially, other individuals with ASD or related neurodevelopmental disorders.
RESULTS
Tbr1 Regulates Genes Involved in Cytoskeletal Dynamics and Synaptogenesis in Layer 5 Pyramidal Neurons of Neonatal mPFC
In the frontal and motor cortex, Tbr1 is expressed in most excitatory neurons in layers 5 and 6, whereas layer 5 expression in other cortical regions is limited to a minority of neurons (Bulfone et al., 1995). Here, using a floxed allele, we selectively eliminated Tbr1 in cortical layer 5 pyramidal neurons around postnatal day (P)0 using Rbp4-cre ~8 days after Tbr1 expression begins. We refer to these mice as Tbr1layer5 CKOs.
We focused on Tbr1 function in the developing prefrontal cortex (PFC), a region that is implicated in ASD (Willsey et al., 2013). To overcome the limitations caused by cellular heterogeneity of batch RNA sequencing (RNA-seq) (Tbr1 is expressed in ~60% of layer 5 pyramidal neurons at P5 and ~85% at P21; Figures S1A and S1B), we generated scRNA-seq data from fluorescence-activated cell sorting (FACS) of layer 5 neurons isolated from P5 mPFC (Figure 1). We studied the transcriptomic changes from Tbr1wild-type, Tbr1layer5 heterozygous and homozygous CKO cells using the 10X Genomics platform (GenBank GEO: GSE146298).
Figure 1. Tbr1 Regulates Genes that Are Implicated in Controlling the Development of Axons, Synapses, and Dendrites in Layer 5 Pyramidal Neurons of the mPFC.

(A) t-distributed stochastic neighbor embedding (t-SNE) plot displaying 11,070 single neuronal cells from Tbr1layer5 WT (red) and from Tbr1layer5 heterozygous (HET; green), and Tbr1layer5 homozygous (NULL; blue) CKOs. t-SNE was performed after quality control and removal of non-neuronal cell subtypes.
(B) Heatmap of DEX genes (FDR < 0.05) shared between both genotypes (x axis, n = 218) over a randomly selected 1,000 cells from each genotype (y axis, n = 3,000). Genes are ordered by hierarchal clustering within direction of regulation grouping, and the Z score of normalized gene expression data is shown. The genotype for each cell is depicted at the top, and genes with membership in selected enriched GO categories are highlighted at the right.
See also Figures S1-S3.
To identify genotype-dependent gene expression changes, we used a t-distributed stochastic neighbor embedding (t-SNE) dimensionality reduction followed by differential expression (DEX) analysis to identify neuronal cells (Figures 1A and S2). We captured 11,070 cells and 7,174 genes from Tbr1wild-type (n = 1,778 cells), Tbr1layer5 heterozygous (n = 5,357 cells), and Tbr1layer5 homozygous (n = 3,935 cells) mutant mPFCs that were used for downstream analysis (Figure S2). We excluded 873 cells classified as atypical neuronal cells, with lower expression levels of Neurod6 and Nrgn (two excitatory neuronal markers; Figures S3A and S3D) and high levels of housekeeping genes, from DEX analyses (gray cells in Figure 1A). The t-SNE plot demonstrated clear separation between Tbr1wid-type and Tbr1layer5 CKOs (Figure 1A). DEX analysis identified 470 DEX genes when comparing Tbr1layer5 homozygous mutants to Tbr1wild-type (Table S1) and 320 DEX genes when comparing Tbr1ayer5 heterozygous mutants to Tbr1wild-type (Table S2), 218 of which occur in both comparisons (false discovery rate [FDR] ≨ 0.05) (Figure 1B, Table S3). Feature plots showing the expression of layer 5 markers in our scRNA-seq cell population are shown (Figure S3i). Gene Ontology (GO) analysis of DEX genes identified terms including “axon,” “synapse,” “dendrite,” “cell body,” and “neurogenesis” (Figure 1B; Table S4).
To determine whether the changes in gene expression in Tbr1layer5 CKOs are due to direct regulation by TBR1, we used data from TBR1 chromatin immunoprecipitation (ChIP-seq) from P2 wild-type (WT) cortex (Fazel Darbandi et al., 2018). TBR1 binds to the promoters and distal regions of layer 5 DEX genes (within 100 kb) (Figure S1C). This suggests that TBR1 may be involved in controlling the expression by activating or repressing the target genes.
We used in situ hybridization (ISH) to validate the expression of several DEX genes (Table S5). Our scRNA-seq analysis in conjunction with ISH aided in discovering Mgst3, as a new layer 5 marker of prefrontal cortex (Figure S1D). To provide a histological context, we defined laminar boundaries in the prefrontal cortex at P3 using the following probes: Cux2 (layers 2 and 3), Rorb (layer 4), Etv1 (layer 5), Tbr1 (layers 2–3, 5, 6, and 6b), Nr4a2 (subplate; Figure 2i). Cortical layers 2–4 appear as a single layer at this stage (Figures 2A–2F). Expression of Calm2, Kif1a, Mgst3, and Wnt7b was altered as suggested by the scRNA-seq analysis (Figure 2ii; Figure S3ii). Thus, neonatal Tbr1 expression in layer 5 pyramidal neurons directly regulates the expression of genes involved in cytoskeletal dynamics and synapse development.
Figure 2. Tbr1 Regulates Expression in the mPFC.
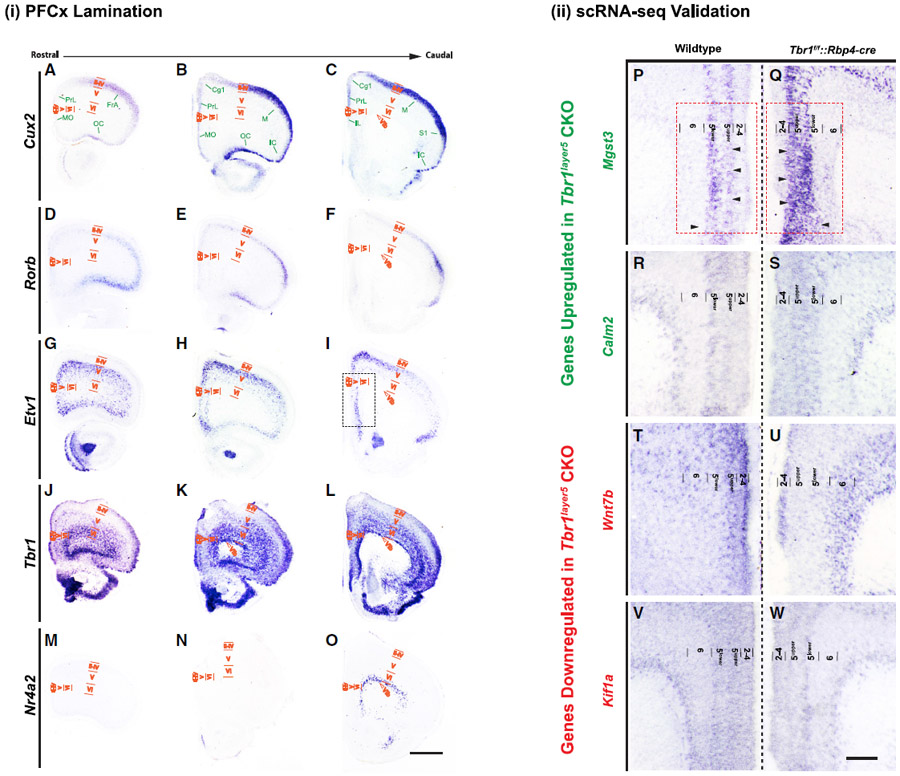
In situ hybridization defines rostral cortical lamination and validates the changes in scRNA-seq expression levels.
(i) PFC lamination. Prefrontal cortical lamination was defined using ISH on coronal sections of neonatal mPFC in WT mice at P3.
(A–O) ISH was performed on rostral, medial, and caudal areas, respectively, using (A–C) Cux2 (layers 2–4); (D–F) Rorb (layer 4); (G–I) Etv1 (layer 5); (J–L) Tbr1 (layers2/3, 5, and 6); and (M–O) Nr4a2 (subplate or layer 6b). Cortical layers in the medial and dorsal regions are labeled. MO, medial orbital cortex; PrL, prelimbic cortex; FrA, frontal association cortex; OC, orbital cortex; Cg1, cingulate cortex area 1; M, motor cortex; S1, primary somatosensory cortex; IC, insular cortex; II-IV, layers 2–4; V, layer 5; VI, layer 6; VIb, subplate. Scale bar, 300 μm.
(ii) scRNA-seq validation. ISH confirms the changes in the transcriptome changes from DEX analysis of scRNA-seq in Tbr1layer5 homozygous mutants.
(P–W) The expression of Mgst3 (P and Q) and Calm2 (R and S) are increased in layer 5upper (Q and S). Tbr1layer5 mutants exhibit reduced expression of Wnt7b (T and U) and Kif1a (V and W) in layer 5 of the mPFC at P3. Only one hemisphere is shown from the ISH images from WT and Tbr1layer5 homozygous CKOs, which are presented as mirror images, to aid in evaluating the changes in laminar gene expression. Color code: downregulated (red) and upregulated (green). Red box shown in (P) and (Q) indicates the region that was dissected for scRNA-seq analyses. Cortical layers 2–4, 5upper, 5lower, 6, and 6b (subplate) are labeled. Scale bar, 100 μm.
See also Figure S3.
Excitatory and Inhibitory Synapses Are Reduced in Tbr1layer5 Mutants
We assessed excitatory synapse numbers on apical dendrites of layer 5 neurons (within layer 2-3) in the mPFC by analyzing VGLUT1+ presynaptic terminals that are apposed to PSD95+ postsynaptic zones at P56 (Figure 3A′) and P21 (Figures S4D-S4F) using immunofluorescence (IF) and confocal microscopy. Inhibitory synaptic density was assessed by counting the overlapping VGAT+ presynaptic inhibitory terminals and Gephyrin+ dendritic postsynaptic zones on the apical dendrites of layer 5 pyramidal neurons (n = 30 dendrites) at P56 (Figure 3D′) and P21 (Figures S4J-S4L). Excitatory and inhibitory synapses were decreased 34% and 42% in Tbr1layer5 heterozygous and 70% and 73% in Tbr1layer5 homozygous mutants at P56, respectively (Figures 3A and 3D). A similar synaptic deficit was also present at P21 (Figures S4G and S4M).
Figure 3. Tbr1 Mutants Have Reduced Excitatory and Inhibitory Synaptic Densities at P56.

IF was used to detect excitatory (i) and inhibitory (ii) synapses onto dendrites of (1) the mPFC of Tbr1wild-type (Rbp4-cre∷tdTomatof/+; red), Tbr1layer5 heterozygous (Tbr1f/+∷Rbp4-cre∷tdTomatof/+; green), and Tbr1layer5 homozygous (Tbr1f/f∷Rbp4-cre∷tdTomatof/+; blue) mutants (n = 30 dendrites), and (2) dendrites of layer 5 neurons from the mPFC of Tbr1wild-type, Tbr1+/− and layer 6 neurons from the SSCx of Tbr1wild-type, Tbr1+/− (n = 15 dendrites).
(A–C′) Excitatory synapses were identified by colocalization of VGLUT1+ boutons and PSD95+ clusters on dendrites of layer 5 pyramidal neurons at P56 (A′ and C′).
(A) Quantification of excitatory synaptic density.
(B) Quantification of the sEPSC frequency from layer 5 neurons at P56 (n = 6/6/6, WT/heterozygous/homozygous cells from two different animals per genotype).
(B′) Sample traces of sEPSC recordings at P56.
(C) Quantification of excitatory synaptic density of Tbr1+/− mutants in cortical layers 5 and 6 at P56.
(D–F′) Inhibitory synapses were identified by co-localizing VGAT+ boutons and Gephyrin+ clusters (D′ and F′).
(D) Quantification of inhibitory synaptic density on dendrites of layer 5 pyramidal neurons at P56.
(E) Quantification of the sIPSC frequency from layer 5 neurons at P56 (n = 7/7/7, WT/heterozygous/homozygous cells from two different animals per genotype). (E′) Sample traces of sIPSC recordings at P56.
(F) Quantification of inhibitory synapse numbers on dendrites of layer 5 and 6 pyramidal neurons of Tbr1+/− mutants at P56.
(iii) In vitro rescue assay was conducted by transfecting Kif1a, Mef2c, Rac3, and Syt4 expression vectors into P0 primary cortical culture from Tbr1wild-type (red) and Tbr1layer5 CKOs (blue) (n = 3 biological replicates).
(G–H′) Excitatory (G) and inhibitory (H) synaptic density was analyzed 14 days post-transfection. Quantification of excitatory (G′) and inhibitory (H′) synaptic density in vitro is indicated. Red box indicates a successful rescue of synaptic density. Two-way ANOVA was used for the statistical analysis of the control, heterozygote, and null. Two-tailed t test with Tukey correction was used for pairwise comparisons. Floating bar graphs represent the minimum-to maximum (min-max) distribution of synaptic density and/or EPSC/IPSC frequency measured from each genotype. Horizontal line in each box denotes the average distribution. Average distribution is numerically indicated in each box (**p < 0.01; ***p < 0.001; ****p < 0.0001). ns, not significant.
See also Figures S4 and S5.
To assess the physiological consequences of the decrease in excitatory and inhibitory synaptic densities, we measured spontaneous excitatory and inhibitory post-synaptic currents (sEPSCs and sIPSCs, respectively) using whole-cell patch clamp on the tdTomato+ layer 5 pyramidal cells in mPFC brain slices at P56 and P21 (Figures 3B′ and 3E′; Figures S4H and S4N). The sEPSC frequency was reduced 25% in Tbr1layer5 heterozygous and 75% in Tbr1layer5 homozygous mutants; furthermore, the frequency of sIPSCs was reduced 30% in Tbr1layer5 heterozygous and 50% in Tbr1layer5 homozygous mutants as compared to cells from Tbr1wild-type mice at P56 (Figures 3B and 3E). Similar decreases were also present at P21 (Figures S4I and S4O). We did not observe changes in the amplitude of sEPSCs and sIPSCs at P21 and P56 (data not shown).
Since most de novo ASD-risk genes are heterozygous, loss-of-function, rare variants, we explored the consequence of constitutive Tbr1 haploinsufficiency on synapse numbers of layer 5 and layer 6 pyramidal neurons using Tbr1+/− mice (Bulfone et al., 1995). We counted excitatory and inhibitory synapse numbers in the mPFC of Tbr1+/−∷Rbp4-cre∷tdTomatof/+ (layer 5 neurons) and the somatosensory cortex (SSCx) of Tbr1+/−∷Ntsr1-cre∷tdTomatof/+ (layer 6 neurons) at P56 (Figure 3, i2 and ii2). Layer 5 excitatory and inhibitory synapse numbers were reduced ~40% and ~35% in the mPFC of Tbr1+/−∷Rbp4-cre∷tdTomatof/+ at P56 (Figures 3C and 3F). Layer 6 neurons in the SSCx of Tbr1+/−∷Ntsr1-cre∷tdTomatof/+ showed ~37% and ~39% decreases in excitatory and inhibitory synaptic densities, respectively (Figures 3C and 3F). Thus, Tbr1 haploinsufficiency results in reduced synaptic density on the excitatory neurons of cortical layers 5 and 6.
Kif1a Expression Restores Normal Synapse Numbers in Tbr1layer5 Mutant Neurons In Vitro
We sought to identify molecular mechanisms underlying the decrease in the excitatory and inhibitory synaptic densities in Tbr1layer5 CKO neurons using the results from the scRNA-seq analysis (Figure 1). We assessed a subset of DEX genes that control synapse biology, including Kif1a (Li et al., 2016), Mef2c (Barbosa et al., 2008), Rac3, and Syt4 (Barber et al., 2009). We examined whether Kif1a, Mef2c, Rac3, and Syt4 could rescue synapse density by expressing them in P0 primary cortical cultures derived from Tbr1wild-type and Tbr1layer5 mutant neurons (n = 3 biological replicates).
After 14 days in vitro, we analyzed the number of excitatory (VGLUT+ presynaptic and PSD95+ postsynaptic) and inhibitory (VGAT+ presynaptic and Gephyrin+ postsynaptic) terminals of Tbr1wlid-type and Tbr1layer5 homozygous mutant neurons (Figure 3, iii). The reduced excitatory and inhibitory synaptic densities onto Tbr1layer5 CKO neurons were recapitulated in vitro (Figures 3G, 3G′, 3H, and 3H′). Only Kif1a rescued the reduction in both excitatory (Figures 3G and 3G′) and inhibitory (Figures 3H and 3H′) synapse numbers. Kif1a, a kinesin motor protein, is implicated in the transport of vesicles for synapse development (Guedes-Dias et al., 2019) and thus may contribute to Tbr1’s function in promoting synapse formation.
Tbr1layer5 CKOs Have Increased Hyperpolarization-Activated Cation Currents (Ihs)
We next examined the intrinsic properties of layer 5 neurons in Tbr1layer5 WT and CKOs using whole-cell patch clamp to measure intrinsic physiological properties of Rbp4-cre∷tdTomato+ neurons of layer 5 in the mPFC (Figure S5A). Resting membrane potential and input resistance were not different between Tbr1wild-type, Tbr1layer5 heterozygotes and homozygotes at P56 (Figures S5B and S5C).
A prominent feature of many layer 5 pyramidal neurons is the presence of an Ih (or h-current) mediated by hyperpolarization-activated cyclic nucleotide–gated HCN channels (Shepherd, 2013). Ih causes a characteristic “sag” and “rebound” in current clamp recordings of responses to steps of hyperpolarizing current. We examined responses to a −200-pA step and found that mPFC layer 5 pyramidal neurons from P56 Tbr1layer5 heterozygotes and homozygotes exhibited a significantly increased “sag and rebound” compared to Tbr1wild-type controls, suggesting increased Ih, while other intrinsic electrophysiological properties were largely unaltered (Figure S5D).
In deep-layer neocortical pyramidal neurons, the presence of Ih shifts the resonant frequency toward higher frequencies (Dembrow et al., 2010). Therefore, to further characterize potential increases in Ih in Tbr1layer5 CKOs, we estimated the resonant frequency by injecting constant current to hold Rbp4-cre+ neurons in current clamp near −70 mV and then introduced a sinusoidal current stimulus with constant amplitude (100 pA, peak to peak) and a frequency that increased linearly from 0 to 20 Hz over 20 s (Figure S5E). Tbr1layer5 heterozygous and homozygous CKOs exhibited an increase in their resonant frequency compared to Tbr1wild-type controls at P56 (Figure S5G).
Lastly, we blocked Ih by bath applying the specific HCN channel antagonist ZD7288 (25 μM; Figure S5F). The resonant frequency was reduced by over 50% in the Tbr1layer5 heterozygous and Tbr1layer5 homozygous CKOs (Figure S5G). Thus, both Tbr1layer5 heterozygotes and homozygotes have an increased Ih in layer 5 pyramidal neurons of the mPFC.
Tbr1 Mutants Have Reduced Mature Dendritic Spine Density
The synaptic deficits described earlier prompted us to investigate the state of dendritic spines in Tbr1layer5 CKOs, Tbr1layer6 CKOs (Fazel Darbandi et al., 2018), and Tbr1+/− mutants. We visualized tdTomato+ spines using Airyscan confocal microscopy to capture 120×-magnification z stack images (using 2× optical zoom) from the dendrites of layer 6 and layer 5 neurons of WT, Tbr1layer5 (Figure 4), Tbr1layer6, and Tbr1+/− mutant neurons at P5, P21, and P60 (Figure S6). We used Imaris software (v.9.2.1) to analyze dendritic spine morphology, density, and distribution.
Figure 4. LiCl Rescues Dendritic Spine Density of Tbr1layer5 CKOs.

(A–L″) In (A)–(L), Rbp4-cre∷tdTomatof/+ allele was used to label the dendrites of layer 5 neurons. Imaris software was used to analyze the dendritic spine density on the apical dendrites of Tbr1layer5 WT and Tbr1layer5 CKO neurons located within layers 2–4 of the mPFC (A′–L′). Changes in the dendritic spine density of layer 5 neurons were examined at P5 (A–D), P21 (E–G), and P60 (H–L). (A″–L″) Merged images.
(M) Quantification of dendritic spine density at P5 and P21. Spine density was improved 24 hr after LiCl treatment at P5 in (C) and (D) and P60 in (K) and (L), compared to the saline-injected control animals in (A) and (B) and in (H) and (J).
(N) Quantification of mature dendritic spines of Tbr1layer5 WT and mutant neurons at P60, 24 h after injection with saline (control) or LiCl. Floating bar graphs represent min-max distribution of the dendritic spine density of layer 5 neurons within layers 2–4 of the mPFC. Horizontal line in each box denotes the average spine density. Average mature dendritic spine density is numerically indicated in each box.
****p < 0.0001. ns, not significant. Scale bar, 8 μm.
See also Figure S6.
There were reductions in the density of mature dendritic spine density in Tbr1 heterozygotes and homozygotes in Tbr1layer5 and Tbr1layer6 CKOs (Figures 4 and S6). Additionally, Tbr1+/− mutants have reduced mature spine density on the dendrites of layer 5 and layer 6 pyramidal neurons (Figure S6). Furthermore, Tbr1 mutant neurons had an increased filamentous spine density (Figure S6). Thus, this defect in mature dendritic spine density may underlie the reduction in synapse numbers in Tbr1 mutants.
Restoring Reduced WNT Signaling in Tbr1 CKOs Rescues Synaptic Deficits
We demonstrated that Tbr1 promotes synaptogenesis onto layer 6 neurons in part via WNT signaling through Wnt7b (Fazel Darbandi et al., 2018). WNT signaling promotes dendrite maturation and synapse formation (Ciani and Salinas, 2005). Here, we found several lines of evidence to further support the role of Tbr1-dependent WNT signaling in synapse development. First, Wnt7b and Ctnnb1 expression was reduced in the mPFC of Tbr1layer5 CKOs (Figures 1 and 2; Tables S1 and S2). Ctnnb1 encodes β-catenin, the critical intracellular transducer of canonical WNT signaling (Budnik and Salinas, 2011). Second, Tbr1layer5 CKOs had increased Gsk3β RNA expression (Figure 1); GSK3β negatively regulates WNT signaling through increasing the destruction of β-catenin (van Noort et al., 2002).
Thus, we tested whether promoting WNT signaling could rescue dendritic spine and synapse phenotypes. Among its several pharmacological effects, there is evidence that LiCl, a WNT-signaling agonist, promotes synapse development (Farooq et al., 2017; Lenox and Wang, 2003; Martin et al., 2018). Thus, we administered LiCl and a GSK3β inhibitor (SB216763, Sigma-Aldrich) to Tbr1 mutants.
LiCl Treatment of Tbr1 Mutants Restores Dendritic Spine Density and Synapse Development
As noted earlier, Tbr1 mutants have a reduced density of mature dendritic spines (Figures 4 and S6). We tested whether promoting WNT signaling by administering LiCl at P5 and P59 could rescue the reduction in mature spine density and synaptogenesis in Tbr1 mutants. We gave a single intraperitoneal (i.p.) injection of 400 mg/kg LiCl; control animals received a single i.p. injection of 4 mL/kg saline. Impressively, LiCl treatment rescued the density of mature dendritic spines within 24 h in Tbr1 mutants; LiCl did not have a clear effect on the density of WT dendritic spines (Figures 4 and S6). These results, in combination with the previously reported evidence that Wnt7b restores synapse numbers on Tbr1layer6 mutant neurons (Fazel Darbandi et al., 2018), led us to test whether LiCl can rescue synapse numbers on adult Tbr1 mutant layer 5 and layer 6 neurons.
We administered LiCl to Tbr1layer5 WT and homozygous CKOs (Figures 5A and 5E), Tbr1layer6 WT and homozygous CKOs (Figures 5B and 5F), and Tbr1+/− mutants (Figure 5). Layer 5 and layer 6 projection neurons were labeled with Rbp4-cre∷tdTomatof/+ and Ntsr1-cre∷tdTomatof/+, respectively. The control and LiCl-treated brains were harvested either 24 h or 4 weeks after injection at P60 (Figures 5 and S7). Confocal images of IF from the mPFC (layer 5) and SSCx (layer 6) showed a nearly complete rescue of synaptic densities, 24 h and 4 weeks after treatment (Figures 5 and S7). LiCl treatment also rescued synaptic densities in the mPFC (layer 5) and SSCx (layer 6) of the constitutive Tbr1+/− mutants (Figure 5).
Figure 5. LiCl Treatment Restores Synapse Numbers and Corticothalamic Axonal Arborization of Tbr1 Mutant Mice.

Excitatory (i) and inhibitory (ii) synaptic densities were quantified at P60 from: (1) apical dendrites of Tbr1layer5CKO and Tbr1layer6CKO mice 4 weeks after P30 injection with saline or LiCl (n = 15 dendrites) and (2) dendrites of layer 5 neurons from the mPFC of Tbr1wiid-type, Tbr1+/− and layer 6 neurons from the SSCx of Tbr1wild-type, Tbr1+/− mice 24 h after injection with saline or LiCl at P59 (n = 15 dendrites).
(A and B) Excitatory synapses were quantified from (A) layer 5 neurons of the mPFC of Tbr1wild-type (green) and Tbr1layer5CKO (orange) mice and (B) layer 6 neurons from the SSCx of Tbr1wild-type (red) and Tbr1layer6CKO (blue) mice at P60, 4 weeks after saline and/or LiCl was administered.
(C and D) Quantification of excitatory synaptic density of (C) layer 5 neurons of the mPFC of Tbr1wild-type (green) and Tbr1+/− (orange) mice and (D) layer 6 neurons from the SSCx of Tbr1wild-type (red) and Tbr1+/− (blue) mice at P60, 24 h after injection with saline or LiCl.
(E and F) Inhibitory synapses were quantified from (E) the mPFC of Tbr1wild-type and Tbr1layer5CKO and (F) the SSCx of Tbr1wild-type and Tbr1layer6CKO mice 4 weeks after saline and/or LiCl was administered at P30.
(G and H) Inhibitory synapses were quantified from (G) layer 5 neurons of the mPFC of Tbr1wild-type and Tbr1+/− and (H) layer 6 neurons of the SSCx of Tbr1wild-type and Tbr1+/− mice at P60 24 h after injection with saline or LiCl. Floating bar graphs represent the min-max distribution of all excitatory and inhibitory synapse numbers measured from each genotype. Horizontal line in each box denotes the average distribution. Average distribution is numerically indicated in each box. Two-tailed t test with Tukey correction was used for pairwise comparisons (***p < 0.001; ****p < 0.0001). ns, not significant.
(I–P) In section iii, corticothalamic axonal arborization in the thalamus is indicated by tdTomato’s endogenous fluorescence of Tbr1layer6 WT (I–L) and Tbr1layer6 homozygous CKO (M–P) mice. The monochrome tdTomato signal (white) is indicated from saline-injected (I and M) mice, 24 h after LiCl injection (J and N), 24 h after GSK3β-inhibitor injection (K and O), and 4 weeks after LiCl injection (L and P). White arrowheads in (M) indicate thalamic regions that have reduced corticothalamic axonal arborization in Tbr1layer6 CKO. Yellow arrowheads in (N)–(P) correspond to improved corticothalamic axonal arborization in Tbr1layer6 CKO at P60 following LiCl treatment after 24 h (N), GSK3β-inhibitor (GSK3βi) treatment after 24 h (O), and LiCl treatment after 4 weeks (P). Yellow box depicts a high magnification of the SSCx, demonstrating that LiCl and GSK3β-inhibitor treatments did not rescue the layer 6 apical dendrite morphogenesis in Tbr1layer6 CKOs. Thalamus, cortex (Cx), and corticothalamic axons (CTAs) are labeled. Scale bars: white, 1 mm; blue, 50 μm.
(iv) Quantification of the tdTomato pixel intensity in the boxed regions in (I) and (M) from saline-injected Tbr1wild-type (WT-Saline) and Tbr1layer6 homozygous mutants (Null-Saline) at P60. tdTomato signal intensity is improved in the thalamus of the Tbr1layer6 homozygous CKO 24 h and 4 weeks after treatment compared to treatment of Tbr1wild-type at 24 h and 4 weeks. Two-tailed t test with Tukey correction was used for pairwise comparisons. Floating bar graphs represent the min-max distribution of tdTomato pixel density measured from region 1 of all genotypes and treatments. Horizontal line in each box denotes the average distribution. Average distribution is numerically indicated in each box (***p < 0.001; ****p < 0.0001).
See also Figures S7, S8, and S9.
Thus, LiCl treatment of Tbr1layer5, Tbr1layer6, and Tbr1+/− mutant mice at P60 rescues both excitatory and inhibitory synaptic deficit in Tbr1 mutant neurons of cortical layers 5 and 6 (Figures 5A-5H). Here, we postulate that Tbr1 mutant neurons are in a “poised” state but are not able to form synapses due to a defect in WNT signaling. Thus, we provide in vivo evidence that augmenting WNT signaling via LiCl treatment is sufficient to restore normal synapse numbers.
GSK3β Inhibitor Restores Defects in Dendritic Spine and Synaptic Density of Tbr1 Mutants
Promoting WNT signaling via LiCl treatment of Tbr1 mutants rescued the defects in mature spine and synaptic density (Figures 5 and S7). Lithium’s best validated mechanisms of action are inhibitory effects on IMP and INPP1, central phosphatases in the phosphoinositide pathway, and on GSK3β, the central kinase in the Wnt/β-catenin and AKT pathways (Lenox and Wang, 2003). To ascertain whether WNT signaling is the main mechanism underlying the defects in dendritic spine and synaptic density of Tbr1 mutants, we used a GSK3β inhibitor (SB216763; Sigma-Aldrich).
A single i.p. injection of GSK3β inhibitor (10 mg/kg) was given to Tbr1 CKOs and WT at P59. Control animals received a single i.p. injection of 4 mL/kg saline at P59. We studied the effects of these treatments on Tbr1layer5 WT and homozygous CKOs (Figure S8i) and on Tbr1layer6 WT and homozygous CKOs (Figure S8ii). Layer 5 and layer 6 projection neurons were labeled with Rbp4-cre∷tdTomatof/+ and Ntsr1-cre∷tdTomatof/+, respectively. The control and GSK3β-inhibitor-treated brains were harvested after 24 h (Figure S8). GSK3β-inhibitor treatment rescued the decrease in mature spine density in Tbr1 CKO mutants (Figures S8C and S8F). Furthermore, IF analysis of excitatory and inhibitory synaptic densities from the Tbr1layer5 CKO mPFC (layer 5; Figures S8A and S8B) and from the Tbr1layer6 CKO SSCx (layer 6; Figures S8D and S8E) showed a nearly complete rescue of synaptic density 24 h after treatment (Figure S8).
Thus, GSK3β inhibitor treatment of Tbr1layer5 and Tbr1layer6 CKO mice at P60 rescues dendritic spine density as well as excitatory and inhibitory synaptic deficit in Tbr1 mutant neurons of cortical layers 5 and 6, respectively (Figure S8). This provides an additional line of evidence that augmenting WNT signaling is a key mechanism in restoring mature dendritic spine and synaptic density in Tbr1 mutants.
LiCl and GSK3β Inhibitor Treatment at P60 Improves Corticothalamic Axonal Arborization in Tbr1layar6 Mutant
Layer 6 corticothalamic neurons extend their axons to the thalamus where they form synapses. Corticothalamic axons enter the thalamus in Tbr1layerr6 CKOs; however, the corticothalamic axonal arborization is reduced in the anteromedial thalamus of Tbr1layer6 CKOs (white arrowheads in Figure 5M) (Fazel Darbandi et al., 2018). Treatment with either LiCl or GSK3β inhibitor rescued this defect after 24 h (yellow arrowheads in Figures 5N and 5O) and 4 weeks (yellow arrowheads in Figure 5P). Quantification of tdTomato pixel intensity in the anteromedial thalamus (boxed region in Figures 5I and 5M) showed a significant increase after treatment (Figure 5, iv). We estimate that LiCl increased the corticothalamic axonal arborization by ~250 μm in 24 h. Axon growth rates in multiple regions of the nervous system and species have been documented to range from 20 to 75 μm/h (equivalent to ~2,000 μm/24 h) (Goldberg, 2003; Lallemend et al., 2012). We postulate that the rescue of the axonal arbors is through enhanced levels of WNT signaling as result of the LiCl or GSK3β inhibitor treatment.
Evidence that WNT Signaling Promotes Synaptogenesis in Tbr1 CKOs through an Autocrine Mechanism
Previously, we demonstrated that restoring in vivo Wnt7b expression in layer 6 pyramidal neurons of Tbr1layer6 CKOs promoted synaptogenesis onto layer 6 neurons (Fazel Darbandi et al., 2018). Here, we have verified this finding and included additional controls (Figures S9A and S9B).
Toward elucidating whether WNT7B functions through autocrine and/or paracrine mechanisms, we used cortical transplantation of Wnt7b-expressing cortical interneurons to study synaptogenesis in Tbr1layer6 CKO and control (WT) mice. We introduced medial ganglionic eminence (MGE)-derived cortical interneurons (MGE donor cells from Nkx2.1-cre∷tdTomatof/+ background) harboring either a Wnt7b expression construct (pLenti-DlxI12b-Wnt7b-GFP) or a control vector (pLenti-DlxI12b-GFP) into deep cortical layers of Tbr1wild-type and Tbr1layer6 CKOs at P1; we analyzed excitatory synaptic density in cortex at P30. We quantified excitatory synapses on apical dendrites of WT and Tbr1layer6 CKOs layer 6 neurons, adjacent to the MGE-transplanted cells (tdTomato+-GFP+) within layer 5 (Figures S9C-S9F). We did not observe a rescue of synapse numbers (Figure S9E). Furthermore, we did not observe an increase of excitatory synapses onto the soma of the transplanted Wnt7b-expressing interneurons (Figure S9F). Thus, this experiment provides evidence that WNT7B promotes synaptogenesis in cortical excitatory neurons through a cell-autonomous autocrine mechanism.
Tbr1layer5 CKOs Exhibit Social Interaction Defects that Are Rescued by LiCl Treatment
We studied motor function, anxiety, and social interaction of Tbr1layer5 mutant mice between P56 and P80. Motor defects were not detected based on speed in an open field or performance on a rotarod (data not shown). To assay social behavior, we measured the time the experimental mouse spent exploring a novel juvenile WT mouse of the same sex. Subsequently, we measured the amount of time the subject mouse spent exploring a novel object. Tbr1layer5 homozygous CKOs exhibited social interaction deficit with a juvenile mouse; we did not observe a social deficit between Tbr1layer5 WT and Tbr1layer5 heterozygous CKOs (data not shown). Loss of Tbr1 in layer 5 neurons did not affect the amount of time Tbr1layer5 CKOs spent exploring a novel object compared to the WT.
The improved synaptic density of Tbr1layer5 CKOs due to LiCl treatment prompted us to assess the impact of LiCl treatment on the social interaction of Tbr1layer5 CKOs. We performed the novel object exploration and social interaction assays at P60 using Tbr1layer5 WT and CKOs that were treated with a single i.p. injection of saline (control) and LiCl (experimental) 4 weeks prior to the behavioral assays. LiCl treatment of Tbr1layer5 homozygous CKOs improved their social interaction deficit with a juvenile mouse (Figure 6A), while LiCl treatment did not affect a novel object assay (Figure 6B). Thus, LiCl rescues defects in dendritic spines, synapse density, and the social behavior of Tbr1layer5 CKOs.
Figure 6. LiCl Treatment Rescues Social Interaction Deficit of Tbr1layer5 Mutants.

(A) Tbr1layer5 homozygous CKOs (blue) showed reduced social interaction with a juvenile mouse at P56–P80. LiCl treatment of Tbr1layer5 CKOs rescued the social deficit phenotype compared to the saline-treated mutants at P56–P80.
(B) LiCl treatment of Tbr1layer5 CKOs did not affect the time spent engaged in novel object exploration compared to the saline-injected control. Floating bar graphs represent the min-max distribution of interaction measured from all genotypes and treatments. Horizontal line in each box denotes the average distribution. Average distribution is numerically indicated in each box.
Two-tailed t test with Tukey correction was used for pairwise comparisons (*p < 0.05).
DISCUSSION
Tbr1 Dosage in Layers 5 and 6 Is Essential for Promoting and Maintaining Dendritic Spine and Synaptic Density
Tbr1 is expressed in post-mitotic excitatory neurons in the neocortex, hippocampus, entorhinal cortex, pallial amygdala, piriform cortex, olfactory bulb, Cajal-Retzius cells, and subplate neurons (Hevner et al., 2001, 2003). Tbr1 is best known for its expression and function in layer 6, where it is required to initiate and then maintain layer 6 identity by repressing markers of layer 5 identity (Fazel Darbandi et al., 2018; McKenna et al., 2011). There is also prominent Tbr1 expression in layer 5 of the rostral cortex, where it is expressed in ~85% of pyramidal neurons (Bulfone et al., 1995).
Here, by deleting Tbr1 late in gestation using a layer-5-specific Cre (Rbp4-Cre), we have investigated the role of Tbr1 in mPFC development. scRNA-seq from FACS-purified layer 5 neurons of Tbr1wild-type and Tbr1layer5 heterozygous and homozygous CKOs demonstrated that Tbr1 deletion in mPFC layer 5 alters the expression of a subset of genes that control synaptogenesis, synaptic maturation, and microtubule assembly (Tables S1 and S2).
The core phenotypes of the Tbr1 CKOs are: (1) reduction in the density of mature dendritic spine density (Figures 4 and S6); (2) increased density of immature filamentous (thin) spines (Figure S6); and (3) reduced density of excitatory and inhibitory synapses (Figures 3 and S4). The dendritic spine defect is apparent at the beginning of synaptogenesis (P5) and is maintained through adolescence and into adulthood (Figures 4 and S6). Notably, the Tbr1 CKOs neurons have an increased Ih. There is evidence that HCN channels, the mediator of Ih, localize to thin spines (Paspalas et al., 2013). Thus, we hypothesize that the increased Ih in Tbr1 CKOs may be attributed to the increased filamentous spine density in Tbr1 CKOs. Support for this notion comes from the observation that layer 5 neurons have an ~2-fold increased density of filamentous spines compared to that of layer 6 neurons (Figures S6E and S6F), which correlates with higher Ih in layer 5 neurons (Shepherd, 2013).
We postulate that the reduced mature spine density is central to the reduction of excitatory synapses and synaptic activity observed in adolescent (P21) and adult (P56) Tbr1layer5 CKOs. In addition, Tbr1layer6 CKOs (Fazel Darbandi et al., 2018), as well as Tbr1+/− constitutive mutants, show defects in dendritic spines and synapses. The fact that we observed defects in dendritic spine and synapse density in Tbr1 heterozygous CKOs and Tbr1+/− constitutive mutants implies that this phenotype could contribute to the behavioral phenotypes in neuropsychiatric disorders such as ASD. This hypothesis is further strengthened by the dendritic spine and synaptic phenotypes in the mPFC, a cortical region with critical functions in cognitive and affective processing.
Molecular Mechanisms Downstream of Tbr1 that Promote Synapse Development
We have evidence that TBR1 controls synaptic development by promoting spine maturation and synaptogenesis through several mechanisms. Tbr1 promotes WNT signaling (discussed more extensively later), and TBR1 directly drives the expression of Cyp26b1, Foxp2, Mef2c, and Wnt7b in layer 6 (Fazel Darbandi et al., 2018), as well as Kif1a and Wnt7b in layer 5. We integrated these findings into a molecular model (Figure 7). The model also postulates how LiCl and GSK3β inhibitor treatments, through promoting WNT signaling, rescues synaptic and axonal phenotypes in Tbr1 mutants (Figure 7).
Figure 7. Model of How Tbr1 Controls Spine Maturation and Synaptogenesis through Promoting WNT Signaling: Links to ASD Pathogenesis.

Schematic representation of how Tbr1 controls spine maturation and synaptogenesis in cortical layers 5 and 6. Tbr1 regulates WNT signaling by promoting Wnt7b and Ctnnb1 and represses Gsk3β expression. LiCl (blue) and GSK3β inhibitor (green) rescues Tbr1 phenotypes through stimulating WNT signaling by inhibiting GSK3β activity. WNT inhibition of GSK3β results in phosphorylation of MAP1A and MAP1B, which promotes microtubule assembly and axonal outgrowth. Tbr1 activates Kif1a, a kinesin motor protein involved in synaptic vesicle trafficking. Furthermore, Tbr1 activates Foxp2 and Mef2c in layer 6 pyramidal neurons. Mef2c promotes the development of excitatory synapses. Lastly, TBR1 promotes expression of Cyp26b1 in layer 6 pyramidal neurons, which controls RA levels and impacts synaptic development. Asterisks indicate hcASD (red, reduced in Tbr1 mutants) and pASD (green, increased in Tbr1 mutants) genes that are involved in these pathways. Cell membrane and nuclear membrane (blue) are indicated. Pathways unique to layer 5 and layer 6 are shown in orange and yellow, respectively. Convergent pathways between layers 5 and 6 are highlighted in blue.
Tbr1 promotes the expression of Foxp2 (a hcASD gene) and Mef2c transcription factors (TFs) in layer 6 (Fazel Darbandi et al., 2018). Mef2c promotes the development of excitatory synapses (Harrington et al., 2016). However, restoring Mef2c expression in Tbr1 mutant neurons failed to rescue their synaptic deficit, suggesting that decreased expression of this TF alone does not underlie the synaptic deficits in Tbr1 mutants.
Tbr1 also promotes the expression of Cyp26b1, a gene encoding a retinoic-acid (RA)-degrading enzyme, in layer 6 pyramidal neurons. Restoring Cyp26b1 expression in primary cortical cultures from Tbr1layer6 CKOs rescued synaptic deficit in vitro (Figures S9E and S9F). RA acts via RARα in synapses to promote protein synthesis (Chen et al., 2014; Chen and Napoli, 2008). This suggests that Tbr1’s control of RA levels, via Cyp26b1, can impact synaptic development (Figure 7).
While these three mechanisms appear to contribute to Tbr1’s orchestration of synapse development, we believe that Tbr1’s control of WNT signaling may be the overriding Tbr1-dependent mechanism (Figure 7).
Tbr1 Promotion of WNT Signaling Drives Dendritic Spine Maturation and Synaptogenesis on Layer 5 and Layer 6 Pyramidal Neurons
WNT signaling is essential in postsynaptic differentiation of excitatory synapses by recruiting NMDA receptors via promoting PSD95 clustering and local activation of CaMKII within dendritic spines (Ciani et al., 2011). Furthermore, CaMKII is required for WNT-mediated spine growth and increased synaptic strength, thus promoting postsynaptic maturation and differentiation (Ciani et al., 2011). Moreover, WNT expression increases microtubule unbundling and stability by signaling through the canonical pathways downstream of GSK3β (Ciani et al., 2004). WNT inhibition of GSK3β results in phosphorylation of microtubule-associated proteins such as MAP1B. This interaction is essential for microtubule assembly, axonal arborization and outgrowth (Ciani et al., 2004).
Transcriptomic and ISH analyses demonstrate that Tbr1 promotes expression of Wnt7b and Ctnnb1 (β-catenin) and represses expression of Gsk3β. Wnt7b encodes a WNT ligand of the canonical WNT signaling pathway (Rosso et al., 2005). Ctnnb1 encodes β-catenin, the central intracellular signaling protein of the canonical WNT signaling pathway (Ciani and Salinas, 2005). GSK3β is a ubiquitously expressed kinase that represses the canonical WNT pathway by targeting β-catenin for ubiquitin-mediated proteasomal degradation (van Noort et al., 2002). Restoring Wnt7b expression rescued the synaptic deficit in Tbr1layer6 mutant neurons in vitro and in vivo (Fazel Darbandi et al., 2018). To test whether Wnt7b is acting through an autocrine or paracrine mechanism, we introduced cortical interneurons ectopically expressing Wnt7b into the deep layers of Tbr1layer6 CKOs. We measured their effect on synapse density onto apical dendrites of WT and Tbr1layer6 CKO layer 6 neurons (Figure S9). Because we did not find a statistically significant increase in synapse density, we surmise that WNT7B primarily promotes synaptogenesis cell autonomously onto layer 6 pyramidal neurons.
Furthermore, restoring Kif1a expression in layer 5 pyramidal neurons rescued synapses in the Tbr1layer5 CKOs in primary cultures of the neonatal cortex. Kif1a is a member of the kinesin family and functions as an anterograde motor protein that controls vesicle delivery in the assembly and function of synapses (Guedes-Dias et al., 2019). GSK3β phosphorylation of kinesins inhibits their activity and thereby reduces anterograde dendritic transport (Gottschalk et al., 2017; Morfini et al., 2002). De novo KIF1A mutations in human have been associated with intellectual disability (Ohba et al., 2015; Yoshikawa et al., 2018) and hereditary spastic paraplegia (Pennings et al., 2020). In Drosophila, loss-of-function mutations in KIF1A homolog Unc-104 causes defects in synaptic transmission by disrupting the formation of mature boutons (Zhang et al., 2017). Thus, the rescue of the dendritic spine and synaptic deficits in Tbr1 mutants via LiCl and GSK3β-in-hibitor treatments could be, in part, attributed to the enhanced activity of KIF1A proteins as a result of reduced GSK3β activity.
LiCl and GSK3β Inhibitor Rescue Defects in Dendritic Spine and Synaptic Density in Tbr1 Mutants
To further explore the hypothesis that reduced WNT signaling in Tbr1 mutants underlies the reduction in synapses, we tested whether a canonical WNT-signaling pathway agonist, LiCl or GSK3β inhibitor, could rescue dendritic spine and synapse defects. Among LiCl’s best validated mechanisms of action is inhibition of GSK3β, a central kinase in the WNT/β-catenin and AKT pathways (Lenox and Wang, 2003).
LiCl or GSK3β-inhibitor treatment (within 24 h) rescued the dendritic spine density of Tbr1 mutant neurons in cortical layers 5 and 6. Furthermore, either LiCl or GSK3β-inhibitor treatment rescued excitatory and inhibitory synapse numbers within 24 h. Remarkably, a single dose of LiCl at P30 led to a sustained rescue of synaptic density, measured 4 weeks after treatment. These results suggest that the Tbr1 mutant’s dendrites have most of the machinery needed to make synapses but have a deficit of the essential signal(s) to initiate synaptogenesis. Once the LiCl- or GSk3β-inhibitor-induced synapses are formed, they appear to be relatively stable.
Corticothalamic axons in the Tbr1layer6 mutants fail to fully arborize within the anterior and anteromedial regions of the thalamus (Fazel Darbandi et al., 2018). This phenotype was also rescued within 24 h of LiCl or GSK3β-inhibitor treatment, suggesting that the reduced WNT signaling underlies the defect of axonal elongation and/or arborization in Tbr1layer6 mutants.
In sum, we postulate that Tbr1 mutant layer 5 and layer 6 cortical neurons have reduced WNT signaling that underlies their defects in dendritic spines, synapses, and axonal arborization. LiCl or GSK3β inhibitor rescues each of these defects, perhaps through promoting WNT signaling.
LiCl Treatment Rescues Social Interaction Deficit in Tbr1layer5 CKOs
We eliminated Tbr1’s function in cortical layer 5 pyramidal neurons. In most cortical areas, a minority of layer 5 neurons express TBR1, whereas in rostral areas, including the PFC, TBR1 is expressed in ~85% of layer 5 excitatory neurons (Figure S1). The PFC has a central function in distributed circuits that control higher cognitive and emotional functions that are disrupted in neuropsychiatric disorders such as ASD. Tbr1layer5 CKOs are viable and fertile, allowing us to study the impact of Tbr1 deletion on the behavior of heterozygous and homozygous CKOs. The Tbr1layer5 CKOs showed no deficit in their motor functions (rotarod and open field) and interest in novel objects. However, Tbr1layer5 homozygous CKOs showed a reduction in social interaction with a juvenile mouse. This phenotype had previously been demonstrated in mice with Tbr1 haploinsufficiency (Huang et al., 2014).
Importantly, treating Tbr1layer5 CKOs with LiCl at P30 rescued the social deficit of Tbr1layer5 CKOs (measured at P56–P80). Thus, perhaps the LiCl-mediated rescue of synaptogenesis may underlie the rescue of the social behavior phenotype. In studies of multiple neuropsychiatric phenotypes, face-valid rodent behavior has, so far, not proven to be a reliable assay for therapeutics development in humans (Sestan and State, 2018). However, the observation here is notable in that it links a risk-specific mutation to an identifiable molecular mechanism and circuit level behavior, offering important traction for future investigations of ASD pathophysiology.
Insights into How Tbr1 May Contribute to ASD Pathogenesis
Co-expression network analysis suggests that the de novo mutations of ASD-risk genes are enriched in excitatory projection neurons of cortical layers 5 and 6 in the PFCs during human mid-fetal development (Willsey et al., 2013), cell types that also express Tbr1. The functions of many ASD-risk genes converge on pathways that control synaptogenesis, synaptic development, and plasticity (Sanders et al., 2015). Thus, in this study, we deleted Tbr1 in the excitatory neurons of mouse layer 5 of the mPFC at a stage similar to human mid-fetal development.
Our single-cell transcriptomic analysis of FACS-purified layer 5 neurons from the mPFC revealed that Tbr1 regulates other ASD genes, including Ank2, Ap2s1, Ctnnb1, Dpysl2, Map1a, Rorb, Smarcc2 (orthologs of high-confidence ASD [hcASD] genes), and Gsk3β (ortholog of a probable ASD [pASD] gene) in either Tbr1layer5 heterozygous or homozygous CKOs. Tbr1layer5 heterozygous and homozygous CKOs demonstrated a decrease in dendritic spines and excitatory and inhibitory synaptic densities and reduced sEPSCs and sIPSCs, phenotypes that are convergent with Tbr1layerr6 CKOs, and constitutive Tbr1+/−. This suggests that decreased TBR1 dosage in human may also impair synaptic development and thereby increase the risk for ASD. While some of the other phenotypes detected in Tbr1layer5 mutants were only present in the homozygotes, including defects in social interaction, these observations could have relevance for ASD, as they denote biological processes that could be altered in Tbr1 heterozygotes.
Tbr1 and Shank3 Mutants Convergently Present Synaptic and Physiological Defects
The complex genetic variation underlying ASD has complicated efforts to understand the mechanism associated with ASD pathology and therapies. A possible solution for such complex diversity is to identify core mechanisms, in which ASD-risk proteins may act convergently on a common pathway (State and Sestan, 2012). Many mutations are thought to predispose to idiopathic ASDs by causing primary impairments in synaptic transmission (Rosti et al., 2014; Sanders et al., 2015).
Reduced or increased Shank expression in Drosophila reduces WNT signaling and excitatory synapses (Harris et al., 2016). In mouse, reduced Shank3 impairs synaptic function by reduction in dendritic arborization, excitatory synaptic density, synaptic transmission, and Ih current (Yi et al., 2016). Similarly, Tbr1 CKOs have evidence for reduced WNT signaling and have reduced mature spine density and excitatory synaptic density (Fazel Darbandi et al., 2018). Likewise, Tbr1 CKOs have abnormal Ih currents in cortical layer 5 (Figure S5) and layer 6 (Fazel Darbandi et al., 2018), although, in Tbr1 CKOs, Ih is increased. TBR1 binds to the Shank1, −2, and −3 loci (P2 TBR1 ChIP-seq data; GEO: GSE119362) (Fazel Darbandi et al., 2018), although there are only subtle changes in Shank RNA expression in the Tbr1 mutants. Thus, synaptic dysfunction and, perhaps, reduced WNT signaling are common features of mouse Tbr1 and Shank3 mutants; these defects may be the core pathophysiology of some forms of ASD.
LiCl as a Therapy for Neurodevelopmental Disorders that Have Reduced Synapse Development
Currently, there are no treatments for ASD that address its core biological defects. The ability to restore synapse numbers following lithium administration in the Tbr1 mutant mice provides an insight to a possible human therapy, especially given that LiCl has a long history of clinical use.
Our study suggests the value of future study of LiCl as a potential treatment for ASD patients with TBR1 mutations. If successful, LiCl could also conceivably prove relevant for ASD syndromes beyond individuals with TBR1 mutations, particularly where reduced synaptic development is a central feature. In a clinical case report, LiCl was reported to reverse clinical regression, stabilize behavioral abnormalities, and restore brain functioning in two SHANK3 patients with ASD (Serret et al., 2015). Additionally, it is plausible that the mechanisms identified here could be relevant for patients with Ank2, Ap2s1, Ctnnb1, Dpysl2, Map1a, Rorb, Smarcc2, and Gsk3β. We also showed that Tbr1layer6 CKOs had arborization defects of their corticothalamic axons that were improved with LiCl, suggesting that LiCl could also improve presynaptic defects. This is consistent with the evidence that WNT signaling positively regulated presynaptic and postsynaptic development (Ahmad-Annuar et al., 2006; Stamatakou and Salinas, 2014).
It is critically important that any hypothesis regarding novel treatments in ASD be subjected to rigorous blinded clinical testing. This is particularly the case for an agent such as LiCl, which has well-known long-term side effects and a narrow therapeutic window. Open-label trials of novel compounds to treat core symptoms in ASD have repeatedly shown promising results early (Choi et al., 2011), only to be followed almost uniformly by negative well-controlled trials. The foregoing consideration of potentially relevant biological mechanisms should not be construed as an immediate clinical recommendation but rather as a justification for additional in-depth and rigorous studies.
Finally, it is remarkable that LiCl in Tbr1 mutant mice restores dendritic spine density, synaptogenesis, and axon arborization. LiCl has a rapid action (24 h); furthermore, the effect of a single dose lasts over 4 weeks. However, there were many features of the Tbr1 mutants that did not appear to be rescued by LiCl, including increased layer 5 and layer 6 filamentous spine density and layer 6 dendritic morphogenesis. Thus, while LiCl may have some promise as a therapy, it is improbable that it would fully rescue normal brain function of ASD patients with TBR1 mutations.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact, Dr. John L. Rubenstein (john.rubenstein@ucsf.edu), without restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures and animal care were approved and performed in accordance with the University of California San Francisco Laboratory Animal Research Center (LARC) guidelines. All strains were maintained on a C57BL/6 background. Animals were housed in a vivarium with a 12hr light, 12hr dark cycle. Postnatally, experimental animals were kept with their littermates. For timed pregnancies, noon on the day of the vaginal plug was counted as embryonic day 0.5.
The Tbr1flox allele was generated by inGenious Targeting Laboratory (Ronkonkoma, NY). LoxP sites were inserted into introns 1 and 3, flanking Tbr1 exons 2 and 3 (Fazel Darbandi et al., 2018). To enable selection of homologous recombinants, the LoxP site in intron 3 was embedded in a neo cassette that was flanked by Flp sites. The neo cassette was removed by mating to a Flp-expressing mouse to generate the Tbr1flox allele. Cre excision removes exons 2 and 3, including the T-box DNA binding region, similar to the constitutive null allele (Bulfone et al., 1998). Rbp4-cre mice (Gensat KL100) were used to delete Tbr1 in layer 5 projection neurons. tdTomatofl/+ (Ai14) mice were crossed with Tbr1f/f mice and used as an endogenous reporter. Tbr1 layer 5 knockout mice (Tbr1layer5 mutant) were generated by crossing Tbr1f/f∷tdTomatof/+ mice with Tbr1f/+∷Rpb4-cre+. The specific gender and age of experimental animals can be found in the Results section and corresponding figure legends.
TRANSGENIC ANIMAL MODELS
The mouse strains used for this research project, B6.FVB(Cg)-Tg(Ntsr1-cre)GN220Gsat/Mmucd, RRID:MMRRC_030648-UCD and B6.FVB(Cg)-Tg(Rbp4-cre)KL100Gsat/Mmucd, RRID:MMRRC_037128-UCD, were obtained from the Mutant Mouse Resource and Research Center (MMRRC) at University of California at Davis, an NIH-funded strain repository, and was donated to the MMRRC by MMRRC at UCD, University of California, Davis. Made from the original strain (MMRRC:032081) donated by Nathaniel Heintz, Ph.D., The Rockefeller University, GENSAT http://gensat.org/index.html and Charles Gerfen, Ph.D., National Institutes of Health, National Institute of Mental Health.
Information about the generation and genotyping of the transgenic lines used in this study can be found in the corresponding original studies: Rbp4-Cre (Gong et al., 2007), lox-STOP-lox-tdTomato (Ai14;(Madisen et al., 2010)). Mice were maintained on C57BL/6J background.
METHOD DETAILS
Genomic DNA extraction and genotyping
Tissue samples were digested in a solution containing 1 mg/mL of proteinase K, 50 mM Tris-HCl pH 8.0, 100 mM EDTA, 100 mM NaCl and 1% SDS. Genomic DNA was extracted using a standard ethanol precipitation protocol. Genotyping was performed with PCR-based assays using purified genomic DNA, and primer-pair combinations flanking the deleted region and detecting Cre and tdTomato alleles.
RNA extraction and cDNA synthesis
Total RNA was extracted from the cortices of wild-type mice at P0 using RNeasy Plus® Mini Kit (QIAGEN) following the manufacturer’s protocol. First strand cDNA was synthesized using Superscript reverse transcriptase II following manufacturer’s protocol (Thermofisher). cDNA library was used as template to clone and generate in situ probes.
Single-Cell RNA-sequencing (scRNA-seq) on FAC-Sorted Cells
Layer specific transcriptome profiling was conducted by using 10X Chromium scRNA-seq on FAC-Sorted cells from medial prefrontal cortex of Tbr1wild-type and Tbr1layer5 heterozygous and homozygous mutants at P5. The medial prefrontal cortex was dissected in HBSS from P5 mice. Cortices were dissociated using a Papain Dissociation System (Worthington Biochemical Corporation) following manufacturer’s protocol. tdTomato+ cells were sorted using BD FACS Aria II Cell Sorter at Center for Advanced Technology (UCSF). Approximately 20,000 tdTomato+ cells were collected from each sample. Following FAC-sorting, the cell suspensions were centrifuged at 300 × g for 5 min. Cells were washed for a total of 3 times with 1 mL 1X PBS supplemented with 0.04% BSA. Following the final wash, the cell pellet was resuspended with 25 μL of 1X PBS supplemented with 0.04% BSA. Cell concentration for each sample was determined using trypan blue and a hemocytometer. We targeted to capture approximately 5000 cells per each genotype to generate scRNA-seq libraries. Single cell RNA-seq was performed using 10X Chromium Single Cell 3′ Reagent Kit v2 following manufacturer’s protocol. Library concentration was assessed with Qubit dsDNA HS Assay Kit following manufacturer’s protocol (Thermofisher). Library fragment size distribution was examined on the Agilent Bioanalyzer 2100 (Agilent Technologies) and Agilent High Sensitivity DNA Kit (Agilent Technologies) following manufacturer’s protocol. Libraries were sequenced on Hiseq4000 at Center for Advanced Technology (UCSF).
Computational Analysis of FAC-Sorted Layer 5 scRNA-Seq data
Read pre-processing
Single cell RNA-sequencing libraries were sequenced on Illumina Hiseq4000 to an average depth of 45K reads per cell. Read quality control, UMI counting, barcode counting, and alignment to the mouse reference genome (mm10) were performed using the “cell-ranger 2.0.1” pipeline provided by the manufacturer.
Filtering and Normalization
The initial dataset contained 17,823 cells with an average of 892 genes per cell. Cells with greater than 30% of mitochondrial genes were removed as this is indicative of poor-quality cells (n = 82). Cells with fewer than 500 or more than 10,000 unique-molecular-identifier (UMI) counts were removed as this often represents sequencing errors (n = 163). Cells with fewer than 500 or more than 3,000 genes were removed based on the distribution (n = 182). Genes which occurred in less than 0.01% of cells were also removed (n = 13065). The remaining 17,396 cells and 14,933 genes were used for downstream analysis. No experimental factors were determined to explain a disproportionate of expression variance using the Single Cell Analysis Toolkit for gene Expression in R (scater; v 1.9.15).
Using the R package Seurat (v 2.3.4), the data was log normalized for each cell by the total expression and scaled to 10,000 transcripts per cell. Variable genes were identified using the FindVariabieGenes() function which calculates the average expression and dispersion for each gene, then bins genes and calculates a z-score for dispersion within each bin. The data was scaled, centered, and regressed on the percent of mitochondrial gene content, number of UMI counts, and the number of genes.
Cell-type Identification and Clustering
TSNE was generated using all principal components accounting for more than 2% of the variance and a clustering resolution of 0.3 which resulted in 12 clusters (average silhouette width 0.16). Three clusters were identified as neuronal cells using known markers Nrgn, Rorb, and Cnih2.
The raw data from the three identified neuronal clusters was retained and filtered again based on the distribution of UMI counts and the number of genes per cell (N = 11,943). We applied more stringent filtering to genes by removing mitochondrial genes, ribosomal genes, pseudogenes, genes that did not occur in 1% of neuronal cells, and genes with a variation below the median variation across all genes (N = 7,174). The data normalized as described above and TSNE was generated using all principal components accounting for more than 4% of the variance and a clustering resolution of 0.3 which resulted in 6 clusters (average silhouette width 0.15). Two clusters were identified as atypical cells due to a reduced expression in excitatory neuronal markers and subsequently removed from downstream DEX analysis.
Differential Gene Expression (DEX) Analysis and Gene Ontology Enrichment
To identify gene signatures of each genotype, we used MAST and the zero-inflated regression (zlm) method to compare raw UMI counts (i.e., non-normalized counts) per gene across the cells in the population (FDR < 0.05). Genes that pass a 0.05 significant threshold are considered as significantly differentially expressed (DEX) genes. Gene Ontology enrichment analysis of common differentially expressed genes was performed using the R package goseq (v 1.34.1) using all expressed genes (N = 7,174) as background.
Data and Code Availability
The data used in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE146298 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146298). In addition, all the scripts that were used for analyzing scRNA-seq data as well as the result files are available on https://github.com/aseveritt/Darbandi_TBR1_L5scRNAseq.
Primary Cell Culture and in vitro Rescue Assay
Primary Cell Culture
Cortex was dissected from P0 Tbr1wild-type and Tbr1layer5 homozygous mutants and dissociated using papain dissociation kit following manufacturer’s protocol (Worthington). A total of 300,000 cells were seeded into tissue culture slides pre-coated with poly-L-lysine (10 mg/ml, Sigma) and then laminin (5 mg/ml, Sigma), and grown in vitro with media containing DMEM-H21 with 5% fetal bovine serum for 3 hr. After the cells recovered, DMEM-H21 media was replaced by Neurobasal medium containing B27 supplement, 25% glucose, and glutamax overnight.
In vitro Rescue Assay
Syt4, Mef2c, Kif1a and Rac3 cDNA was cloned into pcDNA3.1(−) (Thermofisher Scientific). Tbr1layer5 mutant cells were transfected with Syt4, Mef2c, Kif1a, Rac3 expression vectors and Tbr1wild-type were transfected with mock empty vector using Lipofectamine 3000 (Invitrogen) for 6 hr. Following incubation, the media was replaced by Neurobasal medium containing B27 supplement, Penicillin/Streptomycin, 25% glucose, and glutamax. Cultures were grown for 14 days in vitro. After 14 days, cultures were washed 3 times with 0.5 mL 1X PBS for 5 min each and fixed for 15 min with 4% PFA in 1X PBS at RT. Fixed cells were washed 3 times with 0.5 mL 1X PBS and blocked in 1X PBS containing 10% Normal Serum, 0.1% Triton X- 100 and 2% BSA for 1 hr at RT. Primary antibodies including mouse anti-Vglut1 (1:200, Synaptic Systems) and rabbit anti-PSD95 (1:200, Cell Signaling; excitatory synapses), rabbit anti-Vgat (1:500, Synaptic Systems) and mouse anti-gephyrin (1:200, Synaptic Systems; inhibitory synapses) were diluted 1:200 in blocking solution. Cells were stained for excitatory and inhibitory synapses with primary antibodies for 48 hr at 4°C with gentle shaking. On a shaker, the cells were washed 3 times with 0.5 mL 1X PBS for 5 min each and incubated with the secondary antibody for 2 hr (room temperature), washed 3X with 1X PBS, and mounted. This experiment was repeated twice (n = 2).
In vivo Synapse Rescue Assays
We performed in vivo rescue assay of synaptic deficit in Tbr1 mutant mice using three different approaches. First, we directly injected a lentivirus harboring WNT7B. Second, we utilized a transplantation assay to deliver the protein of interest (WNT7B) by introducing MGE progenitor cells, following previously published MGE transplantation assay (Vogt et al., 2015). Lastly, we used a single intraperitoneal injection of LiCl to rescue the decrease in synapse numbers in Tbr1 mutants.
Direct lentiviral injection
In vivo rescue assay was carried out by cloning Wnt7b into a Cre-dependent lentiviral backbone (pLenti-CAG-Flex-IRES-GFP). CAG-Flex-GFP (empty vector) and Wnt7b-IRES-GFP expressing lentivirus (pLenti-CAG-Flex-Wnt7b-IRES-GFP) were generated in HEK293T cells as previously reported (Vogt et al., 2015) using Polyplus jetPRIMEH® transfection reagent following manufacturer’s protocol.
Lentivirus containing CAG-Flex-GFP or Wnt7b-IRES-GFP were injected in the SSCx of Tbr1layer6 wild-type as well as Tbr1layer6 heterozygous and homozygous CKO pups at P1. For injections, a glass micropipette of 50 μm diameter (with a beveled tip) was preloaded with sterile mineral oil and viral suspension was front-loaded into the tip of the needle using a plunger connected to a hydraulic drive (Narishige) that was mounted to a stereotaxic frame. P1 pups from Tbr1layer6 wild-type and Tbr1layer6 heterozygous and homozygous CKOs were anesthetized on ice for 1–2 min before injections. Each pup received 2–3 viral injections (150 nL per site) in the right hemisphere. These sites were about 1 mm apart along the rostral to caudal axis. Viral suspensions were injected into layer 6 of the neonatal SSCx. After injections, pups were put back with the mother to recover after they began to move around on their own. Mice were sacrificed 21 days after injection and transcardially perfused with PBS followed by 4% PFA.
MGE-Derived Interneuron Transplantation Assay
A detailed protocol for the MGE transplantation assay has been previously described (Vogt et al., 2015). First, E13.5 MGEs from Nkx2.1-cre∷tdTomatof/+ embryos were dissected in ice-cold HBSS. Next, cells were mechanically dissociated by repeated pipetting (10–15 times) through a 1000 μL plastic pipette tip in DMEM media that contained 10% fetal bovine serum. Cells were dissociated in DMEM with 10% FBS that was preconditioned in a tissue culture incubator at 37°C and with 5% CO2 to achieve a physiological pH. The cells were then transfected with either DlxI12b-GFP (control) or DlxI12b-Wnt7b-GFP (WNT7B-GFP expressing). Cells were transfected for 30 min at 37°C then pelleted by centrifugation (3 min, 700 × g), and resuspended in 2-3 μL of DMEM, put on ice, and then remaining media containing the transfected MGE cells was removed before loaded into the injection needle. For injections, a glass micropipette of 50 μm diameter (with a beveled tip) was preloaded with sterile mineral oil and cells were front-loaded into the tip of the needle using a plunger connected to a hydraulic drive (Narishige) that was mounted to a stereotaxic frame. Tbr1layer6 Wild-type and Tbr1layer6 homozygous CKO P1 pups were anesthetized on ice for 1-2 min before being placed into a molded surface (modeling clay) for injections. Each pup received 2-3 injections of cells (~100 nL per site) in the right hemisphere. These sites were about 1mm apart along the rostral to caudal axis; cells were injected into layers 5/6 of the neocortex. After injections, pups were put back with the mother to recover after they began to move around on their own. Mice were sacrificed 28 days after transplantation and transcardially perfused with PBS followed by 4% PFA.
Lithium chloride (LiCl) injection
P59 and P30 mice were administered a single intraperitoneal (IP) injection of 400 mg/kg LiCl or saline in a volume of 4 ml/kg (Martin et al., 2018). Treated mice were anesthetized at P60, 24 hr or 4 weeks after LiCl injection with intraperitoneal injection of 100 mg/kg Ketamine containing 15 mg/kg Xylazine. A separate cohort of P58 mice were administered a single IP injection of 400 mg/kg LiCl or saline in a volume of 4 ml/kg. Treated mice were anesthetized 24 hr after LiCl injection with intraperitoneal injection 100 mg/kg Ketamine containing 15 mg/kg Xylazine. All brains were processed at P60. Animals were perfused transcardially with ice-cold 1X PBS and then with 4% PFA in 1X PBS, followed by brain isolation, 1-2 hr post-fixation, cryoprotected in 30% sucrose in PBS, and cut frozen (coronally or sagittally) on a sliding microtome at 40μm for immunohistochemistry.
Histology
For P0 and P3 experiments, neonatal animals were anesthetized on ice. For P21 and P56 experiments, animals were anesthetized with intraperitoneal injection of 100 mg/kg Ketamine containing 15 mg/kg Xylazine. Animals were perfused transcardially with cold PBS and then with 4% PFA in PBS, followed by brain isolation, 1-2 hr post-fixation, cryoprotected in 30% sucrose in PBS, and cut frozen (coronally or sagittally) on a sliding microtome at 40μm for immunohistochemistry or in situ hybridization. All primary and secondary antibodies were diluted in PBS containing 10% Normal Serum, 0.25% Triton X-100 and 2% BSA. The following primary antibodies were used: mouse anti-Vglut1 (1:200, Synaptic Systems), rabbit anti-Vgat (1:500, Synaptic Systems), rabbit anti-PSD95 (1:200, Cell Signaling), mouse anti-gephyrin (1:200, Synaptic Systems). The secondary antibodies for immunofluorescence were Alexa Fluor-conjugated and purchased from Thermofisher. For in vivo synapse immunohistochemistry, a total of n = 30 apical dendrites were counted from each of Tbr1wild-type, Tbr1layer5 heterozygous and Tbr1layer5 homozygous mutants. The coronal sections were pre-treated with pepsin to enhance the staining. Immunofluorescence specimens were counterstained with 1% DAPI to assist the delineation of cortical layers. For in situ hybridization a rostro-caudal coronal series of at least ten sections from n = 2 brains from Tbr1wild-type and Tbr1layer5 heterozygous and homozygous mutants were examined. Anti-sense riboprobes for Calm2, Kif1a, Wnt7b, and Mgst3 were prepared as previously described (Cobos et al., 2005; Fazel Darbandi et al., 2016). We also investigated cortical lamination within rostral cortex including PFCx of wild-type brain at P3 and P21 using anti-sense riboprobes for lamination markers Cux2, Rorb, Etv1, Tbr1 and Nr4a2. ISH was performed using digoxigenin-labeled riboprobes.
Image Acquisition and Analysis
Fluorescent and bright-field images were taken using a Coolsnap camera (Photometrics) mounted on a Nikon Eclipse 80i microscope using NIS Elements acquisition software (Nikon). Confocal imaging experiments were conducted at the Cancer Research Laboratory (CRL) Molecular Imaging Center, supported by Helen Wills Neuroscience Institute at UC Berkeley. Confocal images were acquired using Zeiss LSM 880 with Airyscan with a 63X objective at 1,024 × 1,024 pixels resolution with 2.0X optical zoom using ZEN 2.0 software. Brightness and contrast were adjusted, and images merged using Photoshop or ImageJ software. ImageJ software was used for image processing. For synapse counting (presynaptic and postsynaptic boutons), confocal image stacks (0.4μm step size) were processed with ImageJ software. In brief, background subtraction and smooth filter were applied to each stack. Using a threshold function, each stack was converted into a ‘masks’ image. Furthermore, the channels were co-localized with the Image Calculator plugging. Lastly, the number of co-localizations were counted, and the length of each dendrite was measured in each of the focal plane. Staining for control and mutant were done in parallel as well as the image capturing.
Electrophysiology
Coronal brain slices (250 μm) including medial prefrontal cortex were made from three mice (n = 3) at age p21-28 and at p56-p80. Slicing solution was chilled to 4°C and contained (in mM): 234 sucrose, 26 NaHCO3, 11 glucose, 10 MgSO4, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, bubbled with 5% CO2/ 95% O2. Slices were incubated in artificial cerebrospinal fluid (aCSF) at 32°C for 30 minutes and then at room temperature until recording. aCSF contained (in mM): 123 NaCl, 26 NaHCO3, 11 glucose, 3 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgCl2, also bubbled with 5% CO2/ 95% O2. Neurons were visualized using differential interference contrast or DODT contrast microscopy on an upright microscope (Olympus). Rbp4-cre positive neurons were identified by fluorescent visualization of cre-dependent tdTomato. We obtained somatic whole-cell patch clamp recordings using a Multiclamp 700B (Molecular Devices) amplifier and acquired with pClamp. Patch pipettes (2-5 MΩ tip resistance) were filled with the following (in mM): 130 KGluconate, 10 KCl, 10 HEPES, 10 EGTA, 2 MgCl2, 2 MgATP, 0.3 Na3GTP. All recordings were made at 32-34°C. Series resistance was compensated in all current clamp experiments and monitored throughout recordings. Recordings were discarded if Rs changed by > 25%. For spontaneous EPSC and IPSC recordings cells were held in voltage clamp at −70 mV and +10mV, respectively. In both cases patch pipettes were filled with the following (in mM): 135 Cesium Methanesulfonate, 8 NaCl, 10 HEPES, 0.3 EGTA, 5 QX314, 4 MgATP, 0.3 Na3GTP.
Behavioral Assays
Experiments were conducted during the light cycle (8am to 8pm). Mice were habituated to investigator handling for 1-2min on three consecutive days. On the testing day, mice were transferred to experimental room and allowed to habituate for at least 45 minutes prior to testing. All behavior assays were performed on mice age P56 to P80. We were blind to the genotypes during scoring of videos.
Open-field test
An individual mouse was placed near the wall-side of 50 × 50 cm open-field arena, and the movement of the mouse was recorded by a video camera for 10 min. The recorded video file was analyzed with Any-Maze software (San Diego Instruments). Time in the center of the field (a 25 × 25 cm square) was measured. The open field arena was cleaned with 70% ethanol and wiped with paper towels between each trial.
Elevated plus maze test
An individual mouse was placed at the junction of the open and closed arms, facing the arm opposite to the experimenter, of an apparatus with two open arms without walls (30 × 5 × 0.5 cm) across from each other and perpendicular to two closed arms with walls (30 × 5 × 15 cm) with a center platform (5 × 5 cm), and at a height of 40 cm above the floor. The movement of the mouse was recorded by a video camera for 10 min. The recorded video file was analyzed with Any-Maze software and time in the open arms of the apparatus was measured. The arms of the elevated plus maze apparatus was cleaned with 70% ethanol and wiped with paper towels between each trial.
Rotarod test
The assay consisted of four trials per day over the course of 2 days with the rotarod set to accelerate from 4rpm to 45rpm over 5 minutes. The trial started once five mice were placed on the rotarod rotating at 4rpm in separate partitioned compartments. Each trial ended when a mouse fell off, made three complete revolutions while hanging on, or reached 300 s. Digital videos of the mice on the rotarod were recorded from behind. The rotarod apparatus was cleaned with 70% ethanol and wiped with paper towels between each trial.
Social interaction and novel object task
An individual mouse was allowed to habituate for 5 minutes in their home cage prior to starting the trial. A juvenile (3-4 weeks old) mouse of the same strain and sex was introduced to the home cage. After 5 minutes, the juvenile was removed from the home cage. After a 5 min break a novel object (typically a plastic test tube cap) was introduced into the home cage for five minutes. We scored videos offline, blind to genotype. We measured the number of seconds the mouse spent with its nose in direct contact with the novel object or engaged in social interaction with the juvenile (defined as sniffing, close following, or allo-grooming) in the 300 s following the time the juvenile or object was introduced into the cage. In addition, we noted any aggressive-appearing behaviors toward the juvenile, freezing, and grooming behaviors. We repeated this behavioral assay on adult wild-type and mutant mice that were treated with a single IP injection of LiCl and compared to vehicle treated animals injected with saline.
QUANTIFICATION AND STATISTICAL ANALYSIS
All individual data points are shown as well as mean ± SEM. All statistical analyses were performed using GraphPad Prism 7.0 software. Statistical significance was accepted at the level p < 0.05. We used Student’s t test to compare pairs of groups if data were normally distributed (verified using Lillie test). If more than two groups were compared, we used one-way ANOVA with post hoc tests between groups corrected for multiple comparisons (Holm-Sidak or Tukey). Forthe ISH experiments reported in this paper n = 2 represents two biological replicates for each of the reported genes. We examined the changes in synapse numbers of n = 30 different dendrites from n = 2 animals for each genotype. Whole-cell patch clamp experiments at P21 and P56 were conducted from n = 3 different animals for each age and genotype. Lastly, behavioral analysis was conducted from n = 11/8/9, wild-type/ heterozygous/homozygous animals. The specific n for each experiment as well as the post hoc test, exact F and corrected p values can be found in the Results section.
DATA AND CODE AVAILABILITY
Data and MATLAB analysis scripts are available upon request from the Lead Contact.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Vglutl polyclonal antibody | Synaptic Systems | Cat# 135303 |
| RRID: AB_887875 | ||
| Mouse anti-PSD95 antibody | NeuroMab (UC Davis) | Cat# 75-028 |
| RRID: AB_2307331 | ||
| Rabbit anti-Vgat polyclonal antibody | Synaptic Systems | Cat# 131002 |
| RRID: AB_887871 | ||
| Mouse anti-Gephyrin polyclonal antibody | Synaptic Systems | Cat# 147011 |
| RRID: AB_887717 | ||
| Goat anti-Rabbit IgG Alexa Fluor 488 | Thermofisher Scientific | Cat# A-11008 |
| RRID: AB_143165 | ||
| Goat anti-Mouse Alexa Fluor 647 | Thermofisher Scientific | Cat# A32728 |
| RRID: AB_2633277 | ||
| Bacterial and Virus Strains | ||
| pLenti- CAG- Flex-Wnt7b-IRES-GFP | This paper | N/A |
| pLenti-DlxI12b-Wnt7b-GFP | This Paper | N/A |
| pLenti-DlxI12b-GFP | This Paper | N/A |
| pLenti-CAG-Flex-IRES-GFP | This Paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sucrose | Sigma Aldrich | Cat# S5016 |
| Sodium bicarbonate (NaHCO3) | Sigma Aldrich | Cat# S6014 |
| Glucose | Sigma Aldrich | Cat# G5767 |
| Magnesium sulfate (MgSO4) | Sigma Aldrich | Cat# 230391 |
| Sodium phosphate monobasic monohydrate (NaH2PO4) | Sigma Aldrich | Cat# P9638 |
| Potassium chloride (KCl) | Sigma Aldrich | Cat# P9333 |
| Calcium chloride dehydrate (CaCl2) | Sigma Aldrich | Cat# 223506 |
| Magnesium chloride dexahydrate (MgCl2) | Sigma Aldrich | Cat# M9272 |
| Potassium gluconate (KGluconate) | Sigma Aldrich | Cat# P1847 |
| HEPES | Sigma Aldrich | Cat# H3375 |
| EGTA | Sigma Aldrich | Cat# E4378 |
| Adenosine 5′-triphosphate magnesium salt (Mg-ATP) | Sigma Aldrich | Cat# A9187 |
| Guanosine 5′-triphosphate sodium salt hydrate (Na3GTP) | Sigma Aldrich | Cat# 51120 |
| Cesium Methanesulfonate | Sigma Aldrich | Cat# C1426 |
| Sodium Chloride (NaCl) | Sigma Aldrich | Cat# S9888 |
| QX314 chloride | Tocris | Cat# 2313 |
| ZD7288 | Tocris | Cat# 1000 |
| Critical Commercial Assays | ||
| Bioanalyzer High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| Bioanalyzer RNA 6000 Nano Kit | Agilent | Cat# 5067-1511 |
| Chromium i7 Multiplex Kit, | 10X Genomics | Cat# 120262 |
| Chromium Single Cell 3’ Chip Kit v2 | 10X Genomics | Cat# 120236 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v2 | 10X Genomics | Cat# 120237 |
| Deposited Data | ||
| Tbr1 P5 scRNA-seq Raw and Analyzed Data | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146298 |
| Experimental Models: Cell Lines | ||
| Mouse primary cortical culture | This paper | N/A |
| HEK293 cells | Thermofisher Scientific | Cat# R79007 |
| Experimental Models: Organisms/Strains | ||
| Mouse TBR1 conditional mutant | This paper | N/A |
| Mouse TBR1 constitutive mutant | This paper | N/A |
| Oligonucleotides | ||
| Primer for genotyping flox allele | This Paper | N/A |
| ND.for GAC ACA CAC CCT TCT TCA GTT TAC AGC | ||
| Primer for genotyping flox allele | This Paper | N/A |
| ND.rev CAA GCC CGA CTG CCA ATG TTC TG | ||
| Primer for genotyping Ntsr1-cre allele | This Paper | N/A |
| Ntsr1-cre.for GAC GCC ACG CCC CCC TTA | ||
| Primer for genotyping Ntsr1-cre allele | This Paper | N/A |
| Ntsr1-cre.rev CGG CAA ACG GAC AGA AGC ATT | ||
| Primer for genotyping Rbp4-cre allele | This Paper | N/A |
| Rbp4-cre.for GGG CGG CCT CGG TCC TC | ||
| Primer for genotyping Rbp4-cre allele | This Paper | N/A |
| Rbp4-cre.rev CCC CAG AAA TGC CAG ATT ACG TAT | ||
| Primer for genotyping tdTomato allele | This Paper | N/A |
| tdTomato.for CTG TTC CTG TAC GGC ATG G | ||
| Primer for genotyping tdTomato allele | This Paper | N/A |
| tdTomato.rev GGC ATT AAA GCA GCG TAT CC | ||
| Primer for genotyping constitutive allele | This Paper | N/A |
| LN.for CAT TCA GAG CGA CGC ATC AAA GC | ||
| Primer for genotyping constitutive allele | This Paper | N/A |
| LN.rev CAA GCC CGA CTG CCA ATG TTC TG | ||
| Recombinant DNA | ||
| For complete list of recombinant DNA, please refer to Table S5. | This Paper | N/A |
| pcDNA3.1(−) | Thermofisher | Cat# V79520 |
| Software and Algorithms | ||
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| MiniAnalysis | http://www.synaptosoft.com/MiniAnalysis/ | v6.0.7 |
| GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ | v7.01 |
| MATLAB | https://www.mathworks.com/products/matlab.html | v8.6.0.267246 |
| Clampex and Multiclamp | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite | v10.2 |
| ANY-maze | https://www.stoeltingco.com/any-maze-video-tracking-software-1224.html | v5 |
| Imaris | https://imaris.oxinst.com/downloads | v9.2.1 |
| Single-cell RNA-seq code availability | This Paper | https://github.com/aseveritt/Darbandi_TBR1_L5scRNAseq |
Highlights.
Tbr1 promotes and maintains spine maturation and synaptogenesis through WNT signaling
Promoting WNT signaling rescues dendritic spine and synaptic defects in Tbr1 mutants
TBR1 directly regulates transcriptional circuits that control ASD-risk genes
ACKNOWLEDGMENTS
This work was supported by research grants to J.L.R.R. from the Nina Ireland, NINDS (R01 NS34661 and R01 NS099099), and the Simons Foundation (630332); and to V.S.S. from the NIMH (R01MH100292 and R01MH106507).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.059.
DECLARATION OF INTERESTS
J.L.R.R. is cofounder and stockholder, and currently on the scientific board, of Neurona, a company studying the potential therapeutic use of interneuron transplantation. A.J.W. is a paid consultant for Daiichi Sankyo. M.W.S. is a consultant to BlackThorn and ArRett Pharmaceuticals. All other authors declare no competing interests.
REFERENCES
- Ahmad-Annuar A, Ciani L, Simeonidis I, Herreros J, Fredj NB, Rosso SB, Hall A, Brickley S, and Salinas PC (2006). Signaling across the synapse: a role for Wnt and Dishevelled in presynaptic assembly and neurotransmitter release. J. Cell Biol 174, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber CF, Jorquera RA, Melom JE, and Littleton JT (2009). Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J. Cell Biol 187, 295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AC, Kim M-S, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, and Olson EN (2008). MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. USA 105, 9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RAM, Beyer RP, Bammler TK, Rubenstein JLR, and Hevner RF (2010). Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. USA 107, 13129–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, and Salinas PC (2011). Wnt signaling during synaptic development and plasticity. Curr. Opin. Neurobiol 21, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfone A, Smiga SM, Shimamura K, Peterson A, Puelles L, and Rubenstein JLR (1995). T-brain-1: a homolog of Brachyury whose expression defines molecularly distinct domains within the cerebral cortex. Neuron 15, 63–78. [DOI] [PubMed] [Google Scholar]
- Bulfone A, Wang F, Hevner R, Anderson S, Cutforth T, Chen S, Meneses J, Pedersen R, Axel R, and Rubenstein JLR (1998). An olfactory sensory map develops in the absence of normal projection neurons or GABAergic interneurons. Neuron 21, 1273–1282. [DOI] [PubMed] [Google Scholar]
- Chen N, and Napoli JL (2008). All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 22, 236–245. [DOI] [PubMed] [Google Scholar]
- Chen L, Lau AG, and Sarti F (2014). Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology 78, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, et al. (2011). Pharmacological reversal of synaptic plasticity deficits in the mouse model offragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 1380,106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, and Salinas PC (2005). WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci 6, 351–362. [DOI] [PubMed] [Google Scholar]
- Ciani L, Krylova O, Smalley MJ, Dale TC, and Salinas PC (2004). A divergent canonical WNT-signaling pathway regulates microtubule dynamics: Dishevelled signals locally to stabilize microtubules. J. Cell Biol 164, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani L, Boyle KA, Dickins E, Sahores M, Anane D, Lopes DM, Gibb AJ, and Salinas PC (2011). Wnt7a signaling promotes dendritic spine growth and synaptic strength through Ca2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. USA 108, 10732–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, and Rubenstein JL (2005). Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci 8, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, and Johnston D (2010). Projection-specific neuromodulation of medial prefrontal cortex neurons. J. Neurosci 30, 16922–16937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq M, Kim S, Patel S, Khatri L, Hikima T, Rice ME, and Ziff EB (2017). Lithium increases synaptic GluA2 in hippocampal neurons by elevating the δ-catenin protein. Neuropharmacology 113 (Pt A), 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazel Darbandi S, Poitras L, Monis S, Lindtner S, Yu M, Hatch G, Rubenstein JL, and Ekker M (2016). Functional consequences of I56ii Dlx enhancer deletion in the developing mouse forebrain. Dev. Biol 420, 32–42. [DOI] [PubMed] [Google Scholar]
- Fazel Darbandi S, Robinson Schwartz SE, Qi Q, Catta-Preta R, Pai EL-L, Mandell JD, Everitt A, Rubin A, Krasnoff RA, Katzman S, et al. (2018). Neonatal Tbr1 dosage controls cortical layer 6 connectivity. Neuron 100, 831–845.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL (2003). How does an axon grow? Genes Dev. 17, 941–958. [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, and Gerfen CR (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk MG, Leussis MP, Ruland T, Gjeluci K, Petryshen TL, and Bahn S (2017). Lithium reverses behavioral and axonal transport-related changes associated with ANK3 bipolar disorder gene disruption. Eur. Neuropsychopharmacol 27, 274–288. [DOI] [PubMed] [Google Scholar]
- Guedes-Dias P, Nirschl JJ, Abreu N, Tokito MK, Janke C, Magiera MM, and Holzbaur ELF (2019). Kinesin-3 responds to local microtubule dynamics to target synaptic cargo delivery to the presynapse. Curr. Biol 29, 268–282.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AJ, Raissi A, Rajkovich K, Berto S, Kumar J, Molinaro G, Raduazzo J, Guo Y, Loerwald K, Konopka G, et al. (2016). MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. eLife 5, e20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KP, Akbergenova Y, Cho RW, Baas-Thomas MS, and Littleton JT (2016). Shank modulates postsynaptic Wnt signaling to regulate synaptic development. J. Neurosci 36, 5820–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, and Rubenstein JLR (2001). Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29, 353–366. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Neogi T, Englund C, Daza RAM, and Fink A (2003). Cajal-Retzius cells in the mouse: transcription factors, neurotransmitters, and birthdays suggest a pallial origin. Brain Res. Dev. Brain Res 141, 39–53. [DOI] [PubMed] [Google Scholar]
- Huang T-N, Chuang H-C, Chou W-H, Chen C-Y, Wang H-F, Chou S-J, and Hsueh Y-P (2014). Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat. Neurosci 17, 240–247. [DOI] [PubMed] [Google Scholar]
- Lallemend F, Sterzenbach U, Hadjab-Lallemend S, Aquino JB, Castelo-Branco G, Sinha I, Villaescusa JC, Levanon D, Wang Y, Franck MCM, et al. (2012). Positional differences of axon growth rates between sensory neurons encoded by Runx3. EMBO J. 31, 3718–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenox RH, and Wang L (2003). Molecular basis of lithium action: integration of lithium-responsive signaling and gene expression networks. Mol. Psychiatry 8, 135–144. [DOI] [PubMed] [Google Scholar]
- Li L-B, Lei H, Arey RN, Li P, Liu J, Murphy CT, Xu XZS, and Shen K (2016). The neuronal kinesin UNC-104/KIF1A is a key regulator of synaptic aging and insulin signaling-regulated memory. Curr. Biol 26, 605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P-M, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, lafrati J, Stapornwongkul KS, Dominguez S, Kivimäe S, et al. (2018). DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β-catenin signaling. Mol. Psychiatry 23, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna WL, Betancourt J, Larkin KA, Abrams B, Guo C, Rubenstein JLR, and Chen B (2011). Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J. Neurosci 31, 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, and Brady ST (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Haginoya K, Osaka H, Kubota K, Ishiyama A, Hiraide T, Komaki H, Sasaki M, Miyatake S, Nakashima M, et al. (2015). De novo KIF1A mutations cause intellectual deficit, cerebellar atrophy, lower limb spasticity and visual disturbance. J. Hum. Genet 60, 739–742. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Wang M, and Arnsten AFT (2013). Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb. Cortex 23, 1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings M, Schouten MI, van Gaalen J, Meijer RPP, de Bot ST, Kriek M, Saris CGJ, van den Berg LH, van Es MA, Zuidgeest DMH, et al. (2020). KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. Eur. J. Hum. Genet 28, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, and Salinas PC (2005). Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42. [DOI] [PubMed] [Google Scholar]
- Rosti RO, Sadek AA, Vaux KK, and Gleeson JG (2014). The genetic landscape of autism spectrum disorders. Dev. Med. Child Neurol 56, 12–18. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. ; Autism Sequencing Consortium (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serret S, Thümmler S, Dor E, Vesperini S, Santos A, and Askenazy F (2015). Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: case reports. BMC Psychiatry 15, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestan N, and State MW (2018). Lost in translation: traversing the complex path from genomics to therapeutics in autism spectrum disorder. Neuron 100, 406–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci 14, 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakou E, and Salinas PC (2014). Postsynaptic assembly: a role for Wnt signaling. Dev. Neurobiol 74, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, and Šestan N (2012). Neuroscience. The emerging biology of autism spectrum disorders. Science 337, 1301–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort M, Meeldijk J, van der Zee R, Destree O, and Clevers H (2002). Wnt signaling controls the phosphorylation status of β-catenin. J. Biol. Chem 277, 17901–17905. [DOI] [PubMed] [Google Scholar]
- Vogt D, Wu P-R, Sorrells SF, Arnold C, Alvarez-Buylla A, and Rubenstein JLR (2015). Viral-mediated labeling and transplantation of medial ganglionic eminence (MGE) cells for in vivo studies. J. Vis. Exp 98, 52740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, and Südhof TC (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Kuwahara M, Saigoh K, Ishiura H, Yamagishi Y, Hamano Y, Samukawa M, Suzuki H, Hirano M, Mitsui Y, et al. (2018). The novel de novo mutation of KIF1A gene as the cause for spastic paraplegia 30 in a Japanese case. eNeurologicalSci 14, 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YV, Hannan SB, Kern JV, Stanchev DT, Koç B, Jahn TR, and Rasse TM (2017). The KIF1A homolog Unc-104 is important for spontaneous release, postsynaptic density maturation and perisynaptic scaffold organization. Sci. Rep 7, 38172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE146298 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146298). In addition, all the scripts that were used for analyzing scRNA-seq data as well as the result files are available on https://github.com/aseveritt/Darbandi_TBR1_L5scRNAseq.
Data and MATLAB analysis scripts are available upon request from the Lead Contact.