

In situ vaccine–based combinatorial therapy induced potent tumor-specific immunity against tumor recurrence and metastasis.
Abstract
Application of cancer vaccines is limited due to their systemic immunotoxicity and inability to satisfy all the steps, including loading of tumor antigens, draining of antigens to lymph nodes (LNs), internalization of antigens by dendritic cells (DCs), DC maturation, and cross-presentation of antigens for T cell activation. Here, we present a combinatorial therapy, based on a α-cyclodextrin (CD)–based gel system, DOX/ICG/CpG-P-ss-M/CD, fabricated by encapsulating doxorubicin (DOX) and the photothermal agent indocyanine green (ICG). Upon irradiation, the gel system exhibited heat-responsive release of DOX and vaccine-like nanoparticles, CpG-P-ss-M, along with chemotherapy- and phototherapy-generated abundant tumor-specific antigen storage in situ. The released CpG-P-ss-M acted as a carrier adsorbed and delivered antigens to LNs, promoting the uptake of antigens by DCs and DC maturation. Notably, combined with PD-L1 blocking, the therapy effectively inhibited primary tumor growth and induced tumor-specific immune response against tumor recurrence and metastasis.
INTRODUCTION
Tumor metastasis and recurrence are the cause of over 90% of human cancer-related deaths (1). Nanoscale drug delivery systems (DDSs) with chemotherapy, photothermal, and photodynamic therapeutic approaches have been extensively applied in treating primary tumors (2). Unfortunately, they have not been successfully used for treating metastasis and tumor relapse, in part due to their poorer enhanced permeability and retention (EPR) effect (1), increased heterogeneity (3, 4), and resistance development to chemotherapy (5, 6).
Recent studies (7–9) have shown that enhancing patients’ antitumor immunity is the best approach to prevent and control tumor metastasis. Cancer immunotherapeutic approaches, such as cancer vaccines (10, 11), checkpoint blockade therapies (12, 13), and cell-based therapies [such as dendritic cell (DC)–based T cell adoptive transfer] (14–16), have shown promise. Thus, introducing immunotherapeutic strategies with DDS appears to be a feasible approach to counter metastasis and tumor recurrence.
Lymph nodes (LNs) contain phagocytotically active DCs (17, 18), particularly CD8+ DCs, that are efficient in cross-presenting tumor antigens (19, 20) and thus serve as an optimized target site in DDS-based vaccine strategy for inducing antitumor immunity. An efficient tumor vaccine must overcome many barriers and ensure a cascade of five critical events: loading tumor-specific antigens by DDS, draining to LNs, internalization by DCs, DC maturation for costimulatory molecule expression, and presenting peptide–MHC-I complexes to T cells—in short, the LDIMP cascade. Thus, the overall probability (R) to induce potent antitumor immune response is a multiplying product of efficiencies of each step (RL, RD, RI, RM, and RP).
It is therefore critical to maximize efficiencies of all the five events without allowing any event close to zero. Unfortunately, most DDSs that co-deliver antigens and adjuvants have shown disappointing results in clinical trials due to either low RD or RI, directly leading to low RM and further reduplicative administrations, off-target–induced immunotoxicity, immune tolerance, and high costs (10, 21, 22). While designing a vaccine that could effectively target LNs and simultaneously enhance antigen internalization by DCs, we face an inherent dilemma: Development of nanoparticles with characteristics that facilitate internalization by DCs, such as large size, positive charge, and low PEGylation, hinders LN drainage (23, 24).
Traditional cancer vaccines that used one or more model antigens remain unsuccessful, mainly due to lack of antigen specificity induced by tumor heterogeneity and progression (14, 21, 25). Because chemotherapy could further up-regulate the expression of programmed death ligand 1 (PD-L1) in the malignant tumor cells, which is originally overexpressed and induces adaptive immune resistance (26, 27), anti–PD-L1 antibody treatment was also included in this combinatorial therapy to block PD-L1–dependent immune evasion.
Here, to control tumor progression, metastasis, and recurrence, we have prepared previously unidentified α-cyclodextrin (CD)–based gel systems using supramolecular host-guest interactions, by combining a chemotherapeutic drug, doxorubicin (DOX), a model chemotherapeutic agent for favoring immunogenic cell death (28, 29); a photothermal agent, indocyanine green (ICG), which is widely exploited for photothermal therapy of solid tumors (25, 30); and an immunomodulator, CpG, a potent adjuvant that induces DC maturation to effectively integrate chemotherapy and photothermal therapy with immunotherapy. We show that this approach is efficient in achieving all the stages of the LDIMP cascade (Fig. 1).
Fig. 1. LDIMP process.
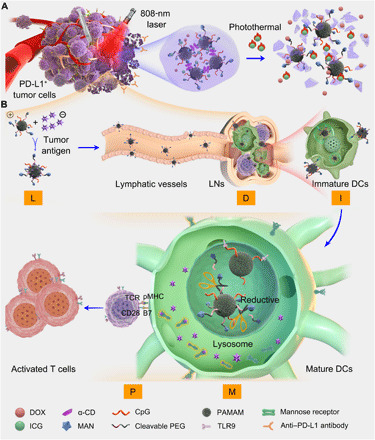
(A) Fabrication of the integrated regimen and the release process of CpG-P-ss-M. (B) Simplified mechanism of CpG-P-ss-M–mediated DC maturation for cancer immunotherapy. Letters LDIMP in orange frame represent loading tumor-specific antigens by DDS, draining to LNs, internalization by DCs, DC maturation for costimulatory molecule expression, and presenting peptide–MHC-I complexes to T cells, respectively.
The small-sized dendritic polyamidoamine (PAMAM) dendrimer (about 5 nm) was co-decorated with reductive-cleavable 4-aminophenyl-α-d-mannopyranoside (MAN)–polyethylene glycol (PEG) chains and CpG to obtain CpG-P-ss-M. The thermal-responsive gel system was achieved by mixing α-CD and CpG-P-ss-M under ultrasonication, which also coencapsulated DOX and ICG simultaneously. As shown in Fig. 1A, upon 808-nm laser irradiation, ICG generated heat and induced apoptosis of tumor cells. Furthermore, hyperthermia partially destroyed the gel, releasing DOX, apoptotic tumor antigens, and CpG-P-ss-M. Released DOX, in turn, caused continuous direct cytotoxicity against primary tumors, especially deep inside the tumor site where laser irradiation efficiency is poor. CpG-P-ss-M went through all the stages of LDIMP journey: loading antigens via electrostatic adsorption from apoptotic tumor cells, easily draining to LNs due to their neutral charge and small size, MAN-triggered active internalization by DCs in LNs (23, 31, 32), promoting DC maturation by CpG exposure intracellularly in response to reductive conditions, and presenting antigens to T cells, resulting in strong antitumor immune response (Fig. 1B).
RESULTS
Synthesis and characterization of CpG-conjugated nanoparticles
To prepare MAN-modified reductive-responsive PAMAM nanoparticles (P-ss-M), the linker carrying disulfide bond was introduced into the PEG segment (fig. S1A). The following MAN modification was obtained via amino group reacting with the N-hydroxysuccinimide (NHS)–functionalized PEG segment (NHS-PEG-ss-COOH or NHS-PEG-NHS) (fig. S1, B and C).
The reductive-nonresponsive MAN unmodified nanoparticles, PP (PAMAM-PEG), as parallel controls, were successfully synthesized in the absence of disulfide bond or MAN group (fig. S2, A and B). The successful synthesis of PM (MAN modified control) and P-ss-M was confirmed by 1H nuclear magnetic resonance (NMR) spectrum (fig. S2, C and D) and matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF-MS) analyses (fig. S3, A to C). To amplify the immunotherapeutic effect, PAMAM was covalently coupled with the agonist of Toll-like receptor 9 (TLR9), CpG, via amidation reaction between C-terminal CpG with primary amino groups of PAMAM. This chemical conjugation, in contrast to electrostatic adsorption followed in other formulations, better maintained the stability and integrity of the nanosystem during lymphatic circulation, ensuring sufficient uptake of CpG by DCs. Agarose gel electrophoresis confirmed the structural integrity of CpG-conjugated nanoparticles without unremoved free CpG (fig. S3D). The zeta potential was reduced after PEG conjugation, suggesting that the neutralization of amidogen occurred (fig. S3E).
Dynamic laser scattering (DLS) revealed that the hydrodynamic diameters of CpG-conjugated PP, PM, and P-ss-M nanoparticles were 22.1 ± 2.7, 24.6 ± 4.2, and 22.1 ± 4.6 nm, respectively. Transmission electronic microscopy (TEM) further confirmed these well-distributed nanoparticles with spherical morphology (Fig. 2, A to C). After incubating P-ss-M nanoparticles in 10 mM glutathione to mimic intracellular reductive condition of endosome or lysosome, the MALDI-TOF-MS analysis showed the peak at 13,754, close to the molecular weight of PAMAM (MW = 14,215 Da), indicating efficient reductive-responsive cleavage of PEG from PAMAM. In contrast, negligible molecular weight changes were observed for PP and PM (fig. S4, A to C).
Fig. 2. CpG-P-ss-M elicits antigen-specific CD8+ CTL response by improving the LDIMP process.
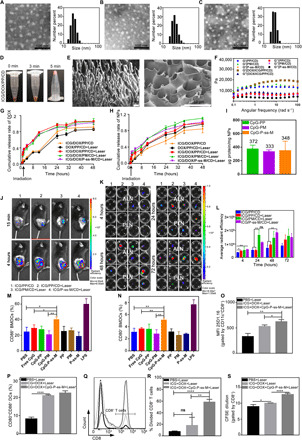
(A to C) TEM images and DLS analysis of CpG-PP (A), CpG-PM (B), and CpG-P-ss-M (C). (D) Photograph of gelation process after ultrasonication. (E) Scanning electron microscopy images of PP/CD gel. (F) Frequency-dependent rheological properties of gels. (G and H) Cumulative release rate of DOX (G) and nanoparticles (NPs) (H) upon irradiation. (I) Quantification of proteins captured by nanoparticles. (J) Live images of melanoma-bearing mice after intratumoral administration. Red circles, location of subiliac LNs. (K) Ex vivo imaging of popliteal and axillary LNs collected at different times after administration. (L) Semiquantitative data of fluorescence signal from popliteal, sciatic, axillary, and accessory axillary LNs. (M and N) Percentages of CD86+ (M) and CD80+ (N) BMDCs gated by CD11c+. (O) Mean fluorecent intensity (MFI) of 25D1.16 signal gated by CD11c+CD8+ and (P) percentages of CD80+CD86+ gated by CD11c+ in BMDCs/B16-OVA multicellular spheroid cocultured system. (Q to S) Percentages of CD8+CTLs (Q and R) and CFSE dilution (S) in CD8+ T/BMDCs/B16-OVA multicellular spheroid cocultured system. *P < 0.05, **P < 0.01, ****P < 0.0001. ns, not significant. Error bars represent mean ± SD; n = 3. Photo credit (D): Lin Qin, West China School of Pharmacy, Sichuan University.
Thermal-triggered drug release gel system
Gel fabrication was performed by the host-guest interaction between α-CD with PEG chains (33, 34). Briefly, PEG chains penetrated into the cavities of α-CD to form polypseudorotaxanes, which then transformed into supramolecular hydrogels via hydrogen bonds between neighboring α-CDs.
Proper gelation time for convenient administration was optimized at different concentrations of PP by measuring rheological properties along with time (fig. S4, D to F). In addition, PP (20 mg ml−1) was selected for further experiments. As shown in Fig. 2D, the mixture solution of drugs (ICG and DOX), PP, and α-CD turned to be viscous and turbid 3 min after ultrasonication (1 min, 300 W) and rapidly turned into gel with an interconnected porous, multilayered network structure (Fig. 2E). Drug-loaded gels, DOX/ICG/PP/CD, displayed rheological properties similar to PP/CD, indicating that neither drug incorporation nor MAN modification affected the mechanical properties of the gel (Fig. 2F).
The thermal-triggered drug release of the gel was evaluated under 808-nm laser irradiation 1 hour after gelation. Within a short time, hyperthermia markedly triggered the release of free DOX from the gels loaded with ICG after laser treatment (Fig. 2G and fig. S5A). Differently, nanoparticles exhibited time-dependent release pattern (Fig. 2H) due to gradual dissociation of the gel. This sustained release of nanoparticles from the gel enables sufficient exposure time to grab more tumor antigens in the tumor microenvironment.
Improving LDIMP process
The tumor antigen loading capacity of CpG-P-ss-M was determined by quantifying the total amount of tumor cell lysates captured by CpG-P-ss-M (Fig. 2I). The increased size (fig. S5B), change in zeta potential (fig. S5C), and silver staining image of SDS-PAGE (polyacrylamide gel electrophoresis) gel of CpG-P-ss-M/protein (fig. S5D) further confirmed that CpG-P-ss-M successfully grabbed tumor antigens. Next, we investigated whether nanoparticles migrate to and accumulated in the LNs once they were released from the gel using in vivo imaging system (IVIS) spectrum.
All the PEGylated nanoparticles and α-CD rapidly fabricated into the drug-loaded gel in situ and showed good retention without laser irradiation (Fig. 2J and fig. S6, A to C) in vivo. Notable accumulation of nanoparticles in the draining LNs (DLNs), subiliac LNs, was observed in the laser irradiation groups, especially in the group treated with MAN-modified nanoparticles (Fig. 2J), suggesting the LN targeting and accumulating capacity of MAN-modified nanoparticles.
The optimal size (20 to 30 nm) of the nanoparticles plays an important role in LN migration (23, 24). With an increase in time, complicated bioenvironments affected gel stability, particularly degradation of α-CD by amylase, and caused time-dependent release of nanoparticles without laser irradiation, consistent with the release properties in vitro (Fig. 2H) and similar intensity in DLNs (fig. S6D). To evaluate the distribution of P-ss-M in systemic LNs, the major LNs (popliteal, sciatic, axillary, and accessory axillary LNs) were collected for ex vivo imaging (Fig. 2K) and quantitative analysis (Fig. 2L). The fluorescent intensity of MAN-decorated nanoparticles was much higher than that of unmodified ones after injection for 4, 24, and 48 hours (Fig. 2L), validating the prominent DC targeting capacity of MAN in LNs, consistent with the ex vivo imaging results of popliteal and axillary LNs (Fig. 2K).
The cellular uptake of P-ss-M was investigated by flow cytometry and laser confocal scanning microscopy (LCSM). The fluorescent signal of MAN-decorated nanoparticles significantly increased and was much stronger than that of the control group PP by bone marrow–derived DCs (BMDCs) and DC 2.4 cells (fig. S7, A to E), suggesting that MAN efficiently promoted the cellular uptake through receptor-mediated endocytosis, consistent with overexpression of mannose receptors on DCs (32, 35, 36). The detachment of reductive-responsive cleavable fragments from P-ss-M released mannose receptors back to the plasma, which could “shuttle” another CpG-P-ss-M nanoparticle, resulting in enhanced internalization of P-ss-M, compared with PM and PP. Because CpG is a short single-stranded synthetic DNA fragment, its application is limited by rapid extracellular and enzymatic degradation and poor uptake by DCs in both blood and lymphatic circulation. Free CpG showed negligible effect in promoting DC maturation; in contrast, the CpG-P-ss-M group had significantly increased percentages of CD80+ and CD86+ DCs (Fig. 2, M and N, and fig. S7, F and G), suggesting that reductive-responsive exposure of CpG binds with TLR9 and induces DC maturation efficiently.
We further investigated the tumor antigen cross-presenting ability of CD8+ BMDCs (19, 20, 31) by staining ovalbumin I (OVAI)–peptide–MHC class I complexes, H-2Kb–SIINFEKL, with 25D1.16 antibody. OVAI peptide presentation was significantly higher in the group treated with ICG + DOX + CpG-P-ss-M + Laser, followed by ICG + DOX + Laser (Fig. 2O and fig. S8A). However, the percentages of CD80+ and CD86+ DCs were not significantly different between the above two groups (Fig. 2P and fig. S8B), which could be attributed to excessive antigen exposure, consistent with previous findings (8, 25, 37).
Consistent with this increased cross-presentation, upon coculturing with BMDCs and B16-OVA multicellular spheroids, OVAI-specific, CFSE [5(6)-carboxyfluorescein diacetate N-succinimidyl ester]–labeled CD8+ T cells proliferated efficiently in the ICG + DOX + CpG-P-ss-M + Laser group when compared to the other groups (Fig. 2, Q to S), suggesting efficient cross-presentation in the CpG-P-ss-M groups. Collectively, these results indicate that CpG-P-ss-M elicits antigen-specific CD8+ cytotoxic T lymphocyte (CTL) response by improving the LDIMP process.
Evaluation of antitumor effects with combinatorial therapy
Up-regulation of PD-1/PD-L1 in cancer is one of the major impediments for effective tumor therapy (12, 13, 26). It is convincible that, in addition to inducing DC maturation via CpG, PD-L1 blockade could synergistically enhance antitumor immunity (13, 25). Here, we tested this possibility by carrying out the experiment depicted in Fig. 3A. Slightly increased body weight (Fig. 3B) and the absence of cytotoxicity in all major organs (38) relative to those in the saline group (fig. S9) suggested limited systemic toxicity of the treatments.
Fig. 3. Antitumor effects in the bilateral B16F10 tumor model.
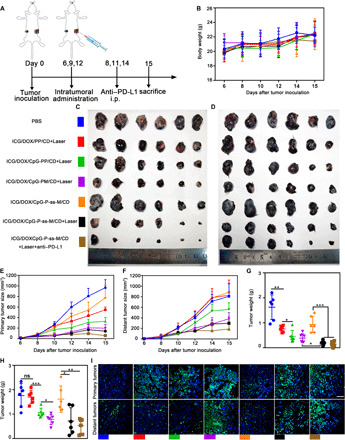
(A) Schematic depicting the experimental approach. (B) Body weight of B16F10 tumor–bearing mice (n = 6). (C and D) Representative images of primary (C) and distant tumors (D) after treatment. (E and F) Primary (E) and distant (F) tumor size curves (n = 6). (G and H) Weight of primary (G) and distant tumors (H). (I) TUNEL staining of primary and distant tumors; scale bar, 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001. Error bars represent mean ± SD. Photo credit (C and D): Lin Qin, West China School of Pharmacy, Sichuan University.
In contrast to ICG/DOX/PP/CD + Laser, all the CpG-loaded groups plus laser irradiation inhibited tumor growth, suggesting the development of productive antitumor capacity in these groups, owing to immunomodulatory CpG effects and the release of immunogenic antigens from dying and apoptotic tumor cells (Fig. 3, C, E, and G). Thus, the regression of distant (left) tumors revealed that antitumor immunity was successfully generated in response to primary tumor treatment. MAN-decorated groups presented a significantly enhanced inhibition activity (Fig. 3, D, F, and H), suggesting that LN targeting and active MAN-mediated internalization of P-ss-M by DCs are critical for DC maturation, antigen presentation, and cytotoxic T cell activation. Notably, the combination of PD-L1 blockade exhibited the strongest antitumor effect against both primary and metastatic tumors, where tumor sizes in this group were significantly lower than in any other groups. In addition, the remaining small tumors showed the signs of regression, such as severe apoptosis and poor proliferative index determined by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) (Fig. 3I) and hematoxylin and eosin (H&E) staining (fig. S9).
To see whether the antitumor response observed in the bilateral tumor model is due to antigen-specific antitumor immunity, the underlying treatment-triggered immune response was investigated in detail. The frequencies of mature DCs in spleens (Fig. 4, A to D) and LNs (Fig. 4, E and F) of the ICG/DOX/CpG-P-ss-M/CD + Laser group were significantly higher than those in other groups, suggesting that the uptake of CpG-P-ss-M by DCs and subsequent CpG-mediated DC maturation is necessary for inducing antitumor immunity.
Fig. 4. Combinatorial therapy promoted DC maturation and induced potent CD8+ T cell response in vivo.
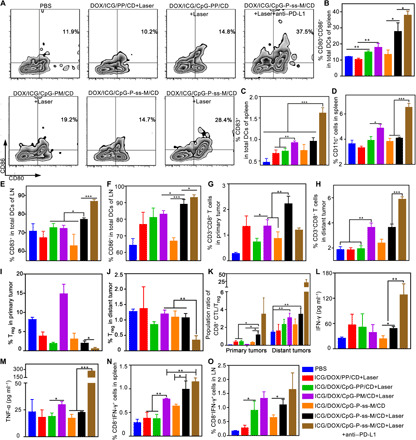
(A to C) Percentages of CD80+ cells, CD86+ cells (A and B), and CD83+ cells (C) gated on CD11c+ cells in spleens. (D) Percentages of CD11c+ cells in the spleen. (E and F) Percentages of CD83+ (E) and CD86+ cells (F) gated on CD11c+ cells in LNs. (G and H) Percentages of CD3+CD8+ T cells in the primary (G) and distant tumors (H). (I and J) Percentages of Tregs (CD3+CD4+Foxp3+) in the primary (I) and distant tumors (J). (K) Ratio of CD8+ T cells:Foxp3+ Tregs in primary and distant tumors. (L and M) IFN-γ (L) and TNF-α (M) levels in serum, determined by enzyme-linked immunosorbent assay (ELISA). (N and O) IFN-γ production by antigen-specific CD8+ CTL cells in the spleens (N) and LNs (O). *P < 0.05, **P < 0.01, ***P < 0.001. Error bars represent mean ± SD (n = 3 to 4).
In contrast, ICG/DOX/CpG-PP/CD + Laser failed to up-regulate DC maturation markers, indicating that antigen delivery alone is bad at stimulating DC maturation in vivo. DC frequencies observed in the primary and distant tumors of different treatments (fig. S10, A and B) correlated with the frequencies of tumor-infiltrating CD8+ T cells (Fig. 4, G and H), indicating the essential role of DC maturation for antitumor immunity generation.
Combination of PD-L1 blockade therapy resulted in the strongest antitumor immune response. The highest frequency of distant tumor-infiltrating CTLs (5.87%) (Fig. 4H) and increased costimulatory molecule–expressing DC population (Fig. 4, A to F) in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 group suggests the synergistic effect of PD-L1 blockade therapy. The frequencies of suppressive regulatory T cells (Tregs) (CD3+CD4+Foxp3+) from this group significantly decreased to 0.474% in the primary tumors and to 0.338% in the distant tumors, which are 4.0- and 3.1-fold lower in the primary and distant tumors, respectively, relative to ICG/DOX/CpG-P-ss-M/CD + Laser (Fig. 4, I and J).
Furthermore, an increase in the CD8+ CTL/Treg ratio (Fig. 4K) and serum proinflammatory cytokine [interferon-γ (IFN-γ) and tumor necrosis factor–α (TNF-α)] levels (Fig. 4, L and M) was observed in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 group. Actually, antigen adsorption in vivo is difficult to measure and the amount of tumor-specific proteins could be affected by nontumor proteins. To address this concern, antigen-specific immune response induced by combinatorial therapy was directly assessed by the frequencies of IFN-γ–secreting CD8+ CTLs in spleens and LNs of treated mice. Consistent with DC maturation and CD8+ CTL proliferation data, IFN-γ–secreting CD8+ CTL frequencies were highest in the PD-L1 blockade combinatorial therapy, followed by the MAN-decorated nanoparticle group (Fig. 4, N and O, and fig. S10C), indicating the successful tumor-specific antigen adsorption and delivery of LNs in vivo. Together, these results suggest that MAN decoration, CpG, and PD-L1 blockade play critical roles in the generation of antigen-specific antitumor immunity in situ.
Inhibition of postoperative tumor metastasis
Antimetastatic effect was evaluated using a postoperative B16-OVA tumor model (Fig. 5A). The median survival time of mice receiving ICG/DOX/CpG-P-ss-M/CD + Laser and ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 therapy was considerably prolonged to 51 and 52 days, respectively, when compared to ICG/DOX/CpG-P-ss-M/CD (37 days)–treated and saline (23 days)–treated groups (Fig. 5B), suggesting that the combination of chemotherapy, photothermal therapy, and immunotherapy prevents tumor recurrence and prolongs survival time.
Fig. 5. Combinatorial therapy suppresses postoperative tumor relapse.
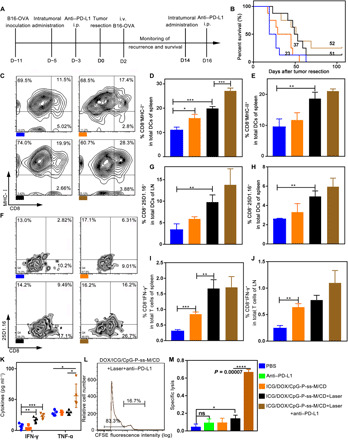
(A) Schematic diagram of the postoperative model and the administration schedule. i.p. intraperitoneal; i.v., intravenous. (B) Survival curve of each group using the log-rank (Mantel-Cox) test (n = 8). (C to E) Percentages of CD8+MHC-I+ cells (C and D) and CD8+MHC-II+ cells (E) in total CD11c+ DCs in spleens (n = 3). (F to H) The percentage of CD8+ 25D1.16+ gated on CD11c+ in LNs (F and G) and spleens (H) was detected by flow cytometry (n = 3). (I and J) Percentages of CD8+IFN-γ+ cells in spleen (I) and LN (J) (n = 3). (K) IFN-γ and TNF-α levels in serum measured by ELISA kit (n = 4). (L and M) In vivo CTL response (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars represent mean ± SD.
Next, the antigen-presenting ability of mature DCs was determined in vivo. The frequencies of mature CD8+ DCs (MHC-I+ and MHC-II+ expressing) in spleens were markedly higher (Fig. 5, C to E, and fig. S10D): 19.9% and 27.1% in groups treated with ICG/DOX/CpG-P-ss-M/CD + Laser and ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 (Fig. 5D), respectively. The up-regulated peptide–MHC-I complexes on CD8+ DCs receiving combinatorial therapy without or with PD-L1 blockade in spleens and LNs over 1.9- and 2.9-fold higher than those of the saline group (Fig. 5, F to H, and fig. S10E) demonstrate enhanced DC maturation and superior tumor antigen cross-presentation in these groups.
Consistently, these groups contained higher frequencies of OVA-specific IFN-γ–secreting CTLs in both spleens and LNs (Fig. 5, I and J), followed by ICG/DOX/CpG-P-ss-M/CD treatment. Similarly, highest levels of IFN-γ and TNF-α were observed in the serum of mice treated with ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 (Fig. 5K). Together, combinatorial therapy markedly promoted DC maturation and antigen cross-presentation in vivo, resulting in robust antitumor immune response.
We next examined the killing capacity of antigen-specific CTLs. OVAI-pulsed target cells were significantly killed in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 group—about 66% lysis as compared to 4.2% in the saline group (P = 0.00002) (Fig. 5, L and M); however, mice treated with anti–PD-L1 antibody alone did not exhibit any appreciable OVAI-specific lysis. These results confirmed a synergistic role for multifunctional CpG-P-ss-M nanoparticles and PD-L1 blockade therapy, resulting in efficient suppression of postoperative tumor relapse and prolonged median survival time due to improved LDIMP process, a long sought-after goal in the therapy of tumor patient.
Long-term immune response
Long-term memory is essential for preventing tumor recurrence and metastasis (21, 25, 39, 40). Encouraged by antigen-specific CD8+ CTL–mediated tumor suppression in the above studies, the memory response induced in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1–treated group was evaluated using resectional lung metastasis and rechallenge tumor models (Fig. 6A).
Fig. 6. Long-term immune response induced by combinatorial therapy.
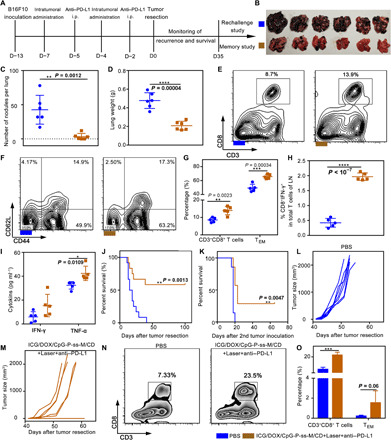
(A) Male C57BL/6 mice bearing melanoma received resection after two treatments. All mice were randomly divided into three parts. Memory study in the first cohort of mice. (B and C) Macroscopic tumor nodules on the lung surface (n = 6). (D) Weight of lung tissues (n = 6). (E to G) TEM (CD44+CD62L− gated by CD3+CD8+) in spleens were detected by flow cytometry (n = 5). (H) Detection of tumor antigen–specific CTLs by intracellular IFN-γ staining in LNs (n = 5). (I) IFN-γ and TNF-α levels in the serum (n = 5). (J) Survival curve of (i) phosphate-buffered saline (PBS) and (ii) ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 groups (n = 12) in the second cohort. (K to O) Rechallenge study in the last cohort. (K) Survival curve of (i) naïve mice, as age-matched control + rechallenge, and (ii) ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 + rechallenge (n = 7). (L and M) Individual contralateral tumor growth curves of two groups. (N and O) Frequencies of CD3+CD8+ T cells and TEM in the LNs (n = 4). Data in (J) and (K) were analyzed using the log-rank (Mantel-Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars represent mean ± SD. Photo credit (B): Lin Qin, West China School of Pharmacy, Sichuan University.
Long-term antitumor immunity generated in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1–treated group was confirmed using the first cohort by transferring the splenocytes from the treated or age-matched unchallenged naïve mice to naïve young mice (Fig. 6A). The transferred mice were challenged with the double dose of tumor cells intravenously on day 35 after surgery. All the challenged mice were monitored up to 18 days. The transferred splenocytes from the treated mice suppressed tumor establishment in the lung of native recipient mice (Fig. 6, B to D). Consistent with this, the frequencies of effector memory T cell (TEM) (CD3+CD8+CD44+ CD62L−) subset (in spleen), IFN-γ–secreting CTLs (in LNs), and serum levels of their cytokines, IFN-γ and TNF-α, increased significantly in the group treated with ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1 when compared to other groups (Fig. 6, E to I). These results suggest that functional memory cells were generated in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1–treated group and were responsible for the observed long-term protection.
In the second cohort, the tumor recurrence and longest survival time were monitored, while the other cohort was used for rechallenge study. The median survival time in both cohorts of the saline-treated group was 17 days (Fig. 6, J and K). Unexpectedly, over 50% survival rate was found in the ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1–treated group even after 100 days (Fig. 6J), suggesting that combinatorial therapy–induced antitumor immunity hinders tumor recurrence and prolongs survival time.
The last cohort of mice was used for rechallenge tumor model. On day 35 after primary tumor resection, the mice were challenged with B16F10 cells on the contralateral side to mimic metastasis. Because almost all mice died from tumor recurrence and invisible metastasis in the saline group, age-matched naïve mice were used as the control group. The analysis of contralateral tumor volume growth curves and CD8+ CTLs and TEM frequencies suggested that combinatorial therapy significantly hindered tumor growth and increased survival rate by inducing robust antitumor response (Fig. 6, K to O). Collectively, these results provided convincing evidence that combinatorial therapy generated a potent long-term memory that prevented tumor recurrence and metastasis.
DISCUSSION
The in situ vaccine-like combinatorial therapy tested here may address several critical issues and afford superior tumor protection. In this work, we have described a versatile gel system integrated with chemotherapy, photothermal therapy, and immunotherapy. This combinatorial therapeutic platform offers key advantages over other immunotherapy strategies that are currently being tested in terms of generating robust tumor-specific immunity and suppressing established primary tumors and tumor metastasis effectively.
First, the hyperthermia and release of DOX-induced immunogenic cell death appeared to generate both tumor-associated antigens and mutated neoantigens, resulting in the escape of immune tolerance and production of unique antigen-specific antitumor immunity in any given patient. Thus, this therapy could be used to treat large population with a specific tumor type and a broad spectrum of different solid tumors. In addition, this combinatorial therapy does not involve complex processes such as characterizing tumor antigens and then loading antigen in vitro, and huge cost associated with these processes. Reagents used in this combinatorial therapy, such as DOX, ICG, and anti–PD-L1 antibody, without any extra modifications are all U.S. Food and Drug Administration–approved agents. Thus, this therapy is cost effective and easy to practice in the clinic. Second, this gel system acts like vaccine in situ—inside the tumor, the continuous release of DOX results in the sustained release of tumor-specific antigens, thereby generating an in situ “antigen reservoir.” Furthermore, the released nanoparticles carry tumor antigens and immune modulators and induce the maturation of DCs for cross-presentation efficiently.
Furthermore, as a result of local action, immunotoxicity in systemic tissues is very limited. Last, the reduced Treg frequencies and the elevated antigen-specific IFN-γ+ CD8+ CTLs in the spleens and LNs of mice treated with combinatorial therapy suggest that our regimen improves LDIMP cascade and blocks negative signals delivered by PD-L1 receptors on tumor cells and DCs. Thus, combinatorial therapy reverses suppressive tumor microenvironment and generates robust tumor-specific immunity, suppressing primary, recurrent, and metastatic tumors. Application of this integrated system in the clinic would therefore provide a safer, feasible, and cost-effective treatment for a range of tumor types in a broad population.
MATERIALS AND METHODS
Materials
PAMAM dendrimers (ethylenediamine core, G4, MW = 14214.7 Da) was obtained from Sigma-Aldrich Co. Ltd. (USA). DOX hydrochloride was purchased from Beijing Huafeng United Technology Co. Ltd. (Beijing, China). ICG was purchased from Dalian Meilun Biotech Co. Ltd. (Dalian, China). NHS-PEG5000-NHS, mPEG5000-NHS, and HO-PEG5000-NHS were synthesized by and purchased from JenKem Technology Co. Ltd. (Beijing, China). CpG 1826 (5′-TCCATGACGTTCCTGACGTT-3′) with C-terminal modification and other peptides was synthesized by and bought from Sangon Biotech Co. Ltd. (Shanghai, China). Anti–PD-L1 antibody was obtained from Selleck Chemicals. α-CD was bought from Shanghai Aladdin Biochemical Technology Co. Ltd. (Shanghai, China). MAN and other chemical agents were purchased from J&K Scientific Ltd. (Beijing, China).
Cell lines and animals
Murine melanoma B16F10 cell line and mouse BMDC line DC 2.4 were purchased from Chinese Academy of Sciences Cell Bank (Shanghai, China). B16-OVA, OVA-transfected B16F10, was a gift from Y. Ma of the Shenzhen Institutes of Advanced Technology of the Chinese Academy of Science. These cells were cultured according to American Type Culture Collection–recommended procedures. BMDCs were generated from the bone marrow of male C57BL/6 mice according to a typical method (41). Male C57BL/6 mice (6 to 8 weeks, 18 to 20 g) were obtained from Dashuo Biotechnology Co. Ltd. (Chengdu, China). OT-I mice containing transgenic insert for mouse Tcra-V2 and Tcrb-V5 genes were bought from The Jackson Laboratory (USA). All animal experiments were performed under the guidelines evaluated and approved by the ethics committee of Sichuan University.
Synthesis and characterization of CpG-PP, CpG-PM, and CpG-P-ss-M
We first prepared MAN-PEG5000-NHS and MAN-PEG5000-CH2CH2-S-S-CH2CH2-COOH. MAN (2.7 mg; 1 equiv.) and NHS-PEG5000-NHS (50 mg; 1 equiv.) were dissolved in 4 ml of methanol, and 4 μl of triethylamine was added. The mixture was stirred for 24 hours in the protection of argon. Purified MAN-PEG5000-NHS was obtained by washing in dichloromethane, then precipitating with diethyl ether, and lastly drying in vacuum pressure. NHS-PEG5000-OH (100 mg), 3,3′-dithiodipropionic acid (3.56 mg), and DMAP (4-dimethylaminopyridine) (24 μg) were stirred in three-necked flask, dichloromethane as solvent, with argon protection. Then, 8.25 mg of DCC (dicyclohexylcarbodiimide) dissolved in dichloromethane was slowly dropped into the flask under ice bath condition, and the mixture was proceeded further with stirring for 24 hours at 25°C. MAN (5.4 mg) was dissolved in methanol and added in the above flask for another 24 hours. Last, MAN-PEG5000-CH2CH2-S-S-CH2CH2-COOH power was purified by dialysis (cutoff molecular weight, 1000 Da) three times in dimethyl sulfoxide and three times in ultrapure water to remove unreacted MAN and other reagents and dried by lyophilization. The products’ structure was confirmed by 1H NMR spectrum using Varian Mercury400 (Varian Inc. Palo Alto, USA). PP (PAMAM-PEG), PM (PAMAM-PEG-MAN), and P-ss-M (PAMAM-PEG-CH2CH2-S-S-CH2CH2-MAN) were obtained by connecting the amidogen of PAMAM and the glycol-N-hydroxysuccinimide ester or carboxyl groups of PEG via amide bond (─CO─NH─). These nanoparticles were purified by dialysis (cutoff molecular weight, 7000 Da) and lyophilization. MALDI-TOF-MS and 1H NMR spectrum were used to determine the product. CpG was then decorated onto these nanoparticles via amide bond and quantified by measuring absorption at 270 nm. The sizes and zeta potential of all the nanoparticles were characterized by DLS (Malvern ZetaSizer Nano ZS), and their morphologies were captured by TEM (H-600, Hitachi, Japan).
Formulation procedure
To obtain drug-loaded gels, 5 mg of α-CD was mixed with 50 μl of mixture solution (nanoparticles, 20 mg ml−1; DOX, 0.4 mg ml−1; ICG, 0.1 mg ml−1) by vortex. The mixture solution rapidly transformed into the gel state after ultrasonication (300 W, 1 min). Before the formation of the gel, it was administered by intratumoral injection and the administration dosages of DOX, ICG, and CpG were 1, 0.25, and 0.5 mg kg−1, respectively. The anti–PD-L1 antibody was intraperitoneally administered 48 hours later after intratumoral injection, and the dosage was 5 mg kg−1.
Heat-trigged drug release from gel in vitro
The gelation time of different concentrations and gel properties after loading drugs were characterized by rheological properties (elastic modulus, G′, and viscous modulus, G″). To evaluate the release manner of DOX and nanoparticles, DOX-loaded gels were irradiated using 808-nm laser (1 W/cm2, 3 min). Released DOX- and Cy5.5-labeled nanoparticles from the gel were collected in the supernatant and measured at an excitation/emission wavelength of 470/590 nm and 650/710 nm (SPARK 10M, Tecan, Switzerland), respectively.
Cellular uptake in vitro
DC 2.4, B16F10 cells, and BMDCs were seeded in 12-well plates at a density of 5 × 104 per well and cultured for 24 hours. Cy5.5-labeled PP, PM, and P-ss-M in fetal bovine serum–free medium were added into the well and further incubated for 1, 2, and 4 hours, respectively. The concentration of nanoparticles was 0.05 mg ml−1. The fluorescence of Cy5.5 was measured by flow cytometry (FACSVerse, BD, USA) and captured using a confocal laser microscope (A1R+, Nikon, Japan).
DC maturation and cross-presentation of tumor antigens in vitro
DC 2.4 cells and BMDCs in 12-well plates were stimulated with CpG-PP, CpG-PM, and CpG-P-ss-M for 24 hours, with LPS (lipopolysaccharide; 2 μg ml−1) as positive control. Mature DCs expressed maturation markers (CD80 and CD86), which were analyzed by flow cytometry. The B16-OVA multicellular spheroids were prepared as described previously (30). The formulation [CpG-P-ss-M (0.1 mg ml−1), DOX (20 μg ml−1), and ICG (5 μg ml−1)] was added into wells and further incubated for 1 hour. After laser treatment (1 W/cm2, 3 min), 3 × 104 BMDCs per well were added and coincubated for another 24 hours. These cells were collected and stained, and the percentage of CD11c+CD8+25D1.16+ cells was counted by flow cytometry.
Antigen absorption by CpG-P-ss-M
Tumor cell lysate was extracted from B16-OVA cells using the Protein Extraction Kit. CpG-P-ss-M and control nanoparticles were incubated with cell lysates ex vivo for 1 hour at 37°C. Excess free protein was removed by gel filtration using Sepharose CL-4B (Sigma-Aldrich), and nanoparticles/proteins, representing nanoparticle-absorbing proteins, were collected and concentrated via ultrafiltration. Then, the total protein amount and the change of size and zeta potential were tested to confirm the antigen adsorption by CpG-P-ss-M.
T cell proliferation in vitro
B16-OVA multicellular spheroids were incubated with different formulations [CpG-P-ss-M (0.1 mg ml−1), DOX (20 μg ml−1), and ICG (5 μg ml−1)] and then received irradiation (1 W/cm2, 3 min). BMDCs were added into each well and incubated for 2 days. CD8 T cells isolated from the splenocytes of OT-I mice, which are restricted to recognize ovalbumin peptide residues 257 to 264 in the context of H-2Kb, were labeled CFSE and then added into each well. After incubating for 2 days, all cells were collected and stained with CD8-antigen presenting cells (APC). T cell proliferation was evaluated by quantification of CD8+ T cells and CFSE dilution.
In vivo distribution of P-ss-M
Melanoma-bearing mice were intratumorally injected with ICG-loaded gel formed with Cy5.5-labeled nanoparticles (50 mg kg −1) and CD (250 mg kg −1). The fluorescence signal was captured with an IVIS Spectrum system (Caliper, MA, USA) at 4, 24, 48, and 72 hours. LNs (including subiliac, popliteal, sciatic, axillary, and accessory axillary LNs), tumors, and major organs were isolated for ex vivo imaging at each time point.
Photothermal effect in vivo
To examine gel formation in vivo, 50 μl of phosphate-buffered saline (PBS), free ICG, ICG/PP/CD, ICG/PM/CD, and ICG/P-ss-M/CD were intratumorally administrated at an ICG concentration of 0.25 mg kg−1. The 808-nm laser (1 W/cm2, 3 min) was irradiated, where formulations were injected 1 hour ago. The temperature elevation was recorded with an infrared camera (Fotric 220, ZXF, USA).
In vivo antitumor evaluation
On day 0, 6- to 8-week-old male C57BL/6 mice were inoculated subcutaneously with 5 × 105 B16F10 cells on their lower back and 2.5 × 105 B16F10 cells in the contralateral side. On day 6, mice were randomly divided into seven groups and intratumorally injected with different formulations every 3 days as follows: (i) PBS, (ii) DOX/ICG/PP/CD + Laser, (iii) DOX/ICG/CpG-PP/CD + Laser, (iv) DOX/ICG/CpG-PM/CD + Laser, (v) DOX/ICG/CpG-P-ss-M/CD, (vi) DOX/ICG/CpG-P-ss-M/CD + Laser, and (vii) DOX/ICG/CpG-P-ss-M/CD + Laser + anti–PD-L1. Tumor size (length × width2 × 0.5) and animal weight were monitored every 2 days. At the end point, spleens and LNs were harvested for DC maturation study by counting the percentage of CD11c+CD80+, CD11c+CD83+, and CD11c+CD86+ using flow cytometry. The productions of cytokines in serum, such as IFN-γ and TNF-α, were tested with an enzyme-linked immunosorbent assay (ELISA) kit under instruction. The percentage of CD3+CD8+, CD3+CD4+, and CD3+CD4+Foxp3+ cells of tumor cells was counted. Histological analysis on major organs and tumors was performed by H&E staining and TUNEL staining.
Postoperative tumor recurrence model
On day −11, mice were inoculated with 106 B16-OVA cells in the lower back. On day −5, mice were randomly divided into four groups and treated with different formulations as follows: (i) PBS, (ii) DOX/ICG/CpG-P-ss-M/CD, (iii) DOX/ICG/CpG-P-ss-M/CD + Laser, and (iv) DOX/ICG/CpG-P-ss-M/CD + Laser + anti–PD-L1 with dosing regimen as mentioned above for one time. On day 0, mice underwent tumor resection surgery and were intravenously injected with 2 × 105 B16-OVA cells. Mice received postoperative treatments once on day 14. The first therapeutics was intended to prime and induce antitumor immunity, providing sufficient amount of tumor antigens, and to reduce the volume of bigger tumors, which is often used in the clinic before resection. Similarly, the second therapeutics after tumor resection is intended to suppress the invasive recurrence to prolong the median survival time of treated mice and to allow metastasis to occur, boosting the primary antitumor immunity generated with the first treatment.
We isolated spleens and LNs from each group to test DC maturation by detecting the percentages of CD11c+CD8+MHC-I+ and CD11c+CD8+MHC-II+ cells and cross-presentation in vivo by comparing the percentage of CD11c+CD8+25D1.16+ cells using flow cytometry. Recurrence and survival time, which were determined by animal committee (tumor volume, ~2000 mm3), were further monitored.
Detection of tumor antigen–specific CTLs
Spleens and LNs were collected from B16F10 and B16-OVA models. We added specific peptides to restimulate IFN-γ production and brefeldin A, an intracellular protein transport inhibitor aiming to inhibit IFN-γ excretion into the prepared single-cell suspension for 4-hour incubation. The OVA257–264 peptide (SIINFEKL) was for the B16-OVA model, and gp10025–33 (EGSRNQDWL) and TRP-2181–188 (VYDFFVWL) were both for the B16F10 model. Then, following the regular surface and intracellular staining procedure, the percentage of CD8+ T cells expressing IFN-γ was determined by flow cytometry.
In vivo cytotoxicity T cell assay
Melanoma-bearing mice, which were inoculated subcutaneously with 1 × 106 B16-OVA cells, received two treatments at a 3-day interval. Seven days after the last treatment, splenocytes from naïve mice were pulsed with the OVA257–264 peptide SIINFEKL and the irrelevant lung carcinoma H-2Kb peptide Mut1 FEQNTAQP for 2 hours at 37°C. After washing three times with PBS, OVA257–264 peptide–pulsed and Mut1-pulsed splenocytes were incubated with high (4 μM, CFSEhigh)– and low (0.5 μM, CFSElow)–concentration CFSE (Invitrogen) for 15 min. Equal amount of CFSEhigh- and CFSElow-labeled splenocytes was intravenously injected into immunized mice and naïve mice as control. Splenocytes were collected 16 hours later, and residual CFSEhigh and CFSElow target cells remaining in the recipients’ spleens were analyzed by flow cytometry. Percentage of specific lysis = [1 − (ratio of CFSEhigh/CFSElow obtained from immunized mice)/(ratio of CFSEhigh/CFSElow obtained from naive mice)] × 100%.
Tumor rechallenge study
To prove the long-term immune memory by combinatorial therapy, male C57BL/6 mice bearing melanoma underwent tumor resection surgery after two treatments with PBS or ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1. Thirty-five days later, a cohort of the above mice and naïve mice, as age-matched control, was inoculated subcutaneously with 5 × 105 B16F10 cells in the contralateral side. The recurrence, protection against contralateral tumor challenge, and the longest survival time were monitored. At the end point, which was determined by the animal committee (tumor volume, ~2000 mm3), splenocytes were isolated and the percentage of TEM (CD3+CD4+CD44+CD62L−) was detected by flow cytometry.
Adoptive transfer of tumor immunity
Another tumor model, lung metastasis, was also performed to show the cytotoxicity of memory T cells. In parallel with rechallenge study, 35 days after primary tumor resection surgery, splenocytes isolated from these two groups—(i) naïve mice, without any challenge and as age-matched control, and (ii) the remaining mice treated with ICG/DOX/CpG-P-ss-M/CD + Laser + anti–PD-L1—were intravenously injected into naïve mice followed by lung metastasis tumor challenge intravenously 1 day later. At the end point, macroscopic tumor nodules on the whole surface of lung tissues were counted. TEM of LNs and spleen were counted using flow cytometry.
Statistical analysis
The results were expressed as SD. Data were analyzed using two-tailed Student’s t test for a two-group comparison. The survival comparison was determined using the log-rank (Mantel-Cox) test. Differences with P < 0.05, P < 0.01, P < 0.001, P < 0.0001, and not significant were considered statistically significant and were labeled with *, **, ***, ****, and ns, respectively.
Supplementary Material
Acknowledgments
We thank C. Li (Analytical and Testing Center, Sichuan University) for assistance with images and X. Sun (West China School of Pharmacy, Sichuan University) for manuscript modification. Funding: This study was supported by the National Natural Science Foundation of China (nos. 81961138009 and 51973135), the 111 Project (no. B18035), and the “Fundamentals of Research Funds” for the Central Universities (nos. SCU2017A001 and 2018SCUH0024). J.C. and N.A.P.’s experiments and studies at The University of Texas at Austin were supported by funds from the Institute of Biomaterials, Drug Delivery and Regenerative Medicine. Author contributions: H.G. and J.C. designed the project with assistance by N.A.P. and C.S.U. K.S. and J.C. performed the material synthesis experiments. K.S. performed in vitro cell experiments. L.Q. performed all animal in vivo experiments and analyzed all data. F.T., Z.Y., T.L., Y.W., and C.H. assisted with the in vivo experiments. L.Q., K.S., and H.G. wrote the manuscript. N.A.P. and C.S.U. guided manuscript modification. H.G. supervised the antitumor evaluation of the combinatorial therapy. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eabb3116/DC1
REFERENCES AND NOTES
- 1.Schroeder A., Heller D. A., Winslow M. M., Dahlman J. E., Pratt G. W., Langer R., Jacks T., Anderson D. G., Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 12, 39–50 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Li C., Wang J., Wang Y., Gao H., Wei G., Huang Y., Yu H., Gan Y., Wang Y., Mei L., Chen H., Hu H., Zhang Z., Jin Y., Recent progress in drug delivery. Acta Pharm. Sin. B 9, 1145–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fidler I. J., Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 38, 2651–2660 (1978). [PubMed] [Google Scholar]
- 4.Suzuki H., Freije D., Nusskern D. R., Okami K., Cairns P., Sidransky D., Isaacs W. B., Bova G. S., Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 58, 204–209 (1998). [PubMed] [Google Scholar]
- 5.Coley H. M., Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 34, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Real P. J., Sierra A., De Juan A., Segovia J. C., Lopez-Vega J. M., Fernandez-Luna J. L., Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21, 7611–7618 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Mellman I., Coukos G., Dranoff G., Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka K., Banchereau J., Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 12, 265–277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg S. A., Yang J. C., Restifo N. P., Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 10, 909–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollingsworth R. E., Jansen K., Turning the corner on therapeutic cancer vaccines. npj Vaccines 4, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keskin D. B., Anandappa A. J., Sun J., Tirosh I., Mathewson N. D., Li S., Oliveira G., Giobbie-Hurder A., Felt K., Gjini E., Shukla S. A., Hu Z., Li L., Le P. M., Allesøe R. L., Richman A. R., Kowalczyk M. S., Abdelrahman S., Geduldig J. E., Charbonneau S., Pelton K., Iorgulescu J. B., Elagina L., Zhang W., Olive O., Cluskey C. M., Olsen L. R., Stevens J., Lane W. J., Salazar A. M., Daley H., Wen P. Y., Chiocca E. A., Harden M., Lennon N. J., Gabriel S., Getz G., Lander E. S., Regev A., Ritz J., Neuberg D., Rodig S. J., Ligon K. L., Suvà M. L., Wucherpfennig K. W., Hacohen N., Fritsch E. F., Livak K. J., Ott P. A., Wu C. J., Reardon D. A., Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cogdill A. P., Andrews M. C., Wargo J. A., Hallmarks of response to immune checkpoint blockade. Br. J. Cancer 117, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardoll D. M., The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carreno B. M., Magrini V., Becker-Hapak M., Kaabinejadian S., Hundal J., Petti A. A., Ly A., Lie W.-R., Hildebrand W. H., Mardis E. R., Linette G. P., A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golubovskaya V., CAR-T cell therapy: From the bench to the bedside. Cancer 9, 150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hee Lee J., Park M.-S., Hwang J.-E., Cho S.-H., Bae W.-K., Shim H.-J., Kim D.-E., Chung I.-J., Dendritic cell-based immunotherapy for colon cancer using an HLA-A*0201-restricted cytotoxic T-lymphocyte epitope from tumor-associated antigen 90K. Cell. Mol. Immunol. 10, 275–282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sainte-Marie G., The lymph node revisited: Development, morphology, functioning, and role in triggering primary immune responses. Anat. Rec. 293, 320–337 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Randolph G. J., Angeli V., Swartz M. A., Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5, 617–628 (2005). [DOI] [PubMed] [Google Scholar]
- 19.den Haan J. M., Lehar S. M., Bevan M. J., CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192, 1685–1696 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudziak D., Kamphorst A. O., Heidkamp G. F., Buchholz V. R., Trumpfheller C., Yamazaki S., Cheong C., Liu K., Lee H.-W., Park C. G., Steinman R. M., Nussenzweig M. C., Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acharya A. P., Sinha M., Ratay M. L., Ding X., Balmert S. C., Workman C. J., Wang Y., Vignali D. A. A., Little S. R., Localized multi-component delivery platform generates local and systemic anti-tumor immunity. Adv. Funct. Mater. 27, 1604366 (2017). [Google Scholar]
- 23.Jiang H., Wang Q., Sun X., Lymph node targeting strategies to improve vaccination efficacy. J. Control. Release 267, 47–56 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Trevaskis N. L., Kaminskas L. M., Porter C. J. H., From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 14, 781–803 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Wang T., Wang D., Yu H., Feng B., Zhou F., Zhang H., Zhou L., Jiao S., Li Y., A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 9, 1532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou W., Wolchok J. D., Chen L., PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okazaki T., Honjo T., The PD-1-PD-L1 pathway in immunological tolerance. Trends Immunol. 27, 195–201 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Green D. R., Ferguson T., Zitvogel L., Kroemer G., Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J.-L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., Zitvogel L., Kroemer G., Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Hu C., Cun X., Ruan S., Liu R., Xiao W., Yang X., Yang Y., Yang C., Gao H., Enzyme-triggered size shrink and laser-enhanced NO release nanoparticles for deep tumor penetration and combination therapy. Biomaterials 168, 64–75 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Cruz L. J., Rosalia R. A., Kleinovink J. W., Rueda F., Löwik C. W. G. M., Ossendorp F., Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: A comparative study. J. Control. Release 192, 209–218 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Engering A. J., Cella M., Fluitsma D., Brockhaus M., Hoefsmit E. C., Lanzavecchia A., Pieters J., The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur. J. Immunol. 27, 2417–2425 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Li J., Loh X. J., Cyclodextrin-based supramolecular architectures: Syntheses, structures, and applications for drug and gene delivery. Adv. Drug Deliv. Rev. 60, 1000–1017 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Wang J., Williamson G. S., Yang H., Branched polyrotaxane hydrogels consisting of alpha-cyclodextrin and low-molecular-weight four-arm polyethylene glycol and the utility of their thixotropic property for controlled drug release. Colloids Surf. B Biointerfaces 165, 144–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C., Liu P., Zhuang Y., Li P., Jiang B., Pan H., Liu L., Cai L., Ma Y., Lymphatic-targeted cationic liposomes: A robust vaccine adjuvant for promoting long-term immunological memory. Vaccine 32, 5475–5483 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Yang R., Xu J., Xu L., Sun X., Chen Q., Zhao Y., Peng R., Liu Z., Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano 12, 5121–5129 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Pan Y., Li X., Kang T., Meng H., Chen Z., Yang L., Wu Y., Wei Y., Gou M., Efficient delivery of antigen to DCs using yeast-derived microparticles. Sci. Rep. 5, 10687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umeshappa C. S., Singha S., Blanco J., Shao K., Nanjundappa R. H., Yamanouchi J., Parés A., Serra P., Yang Y., Santamaria P., Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines. Nat. Commun. 10, 2150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheever M. A., Thompson D. B., Klarnet J. P., Greenberg P. D., Antigen-driven long term-cultured T cells proliferate in vivo, distribute widely, mediate specific tumor therapy, and persist long-term as functional memory T cells. J. Exp. Med. 163, 1100–1112 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enamorado M., Iborra S., Priego E., Cueto F. J., Quintana J. A., Martínez-Cano S., Mejías-Pérez E., Esteban M., Melero I., Hidalgo A., Sancho D., Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 8, 16073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lutz M. B., Kukutsch N., Ogilvie A. L., Rössner S., Koch F., Romani N., Schuler G., An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223, 77–92 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/36/eabb3116/DC1