

Abstract
N6-methyladenosine (m6A) is the most abundant post-transcriptional modification and involves a series of important biological processes. Therefore, accurate detection of the m6A site is very important for revealing its biological functions and impacts on diseases. Although both experimental and computational methods have been proposed for identifying m6A sites, few of them are able to detect m6A sites in different tissues. With the consideration of the spatial specificity of m6A modification, it is necessary to develop methods able to detect the m6A site in different tissues. In this work, by using the convolutional neural network (CNN), we proposed a new method, called im6A-TS-CNN, that can identify m6A sites in brain, liver, kidney, heart, and testis of Homo sapiens, Mus musculus, and Rattus norvegicus. In im6A-TS-CNN, the samples were encoded by using the one-hot encoding scheme. The results from both a 5-fold cross-validation test and independent dataset test demonstrate that im6A-TS-CNN is better than the existing method for the same purpose. The command-line version of im6A-TS-CNN is available at https://github.com/liukeweiaway/DeepM6A_cnn.
Keywords: m6A, convolution neural network, one-hot encoding, spatial specificity of gene expression
Graphical Abstract
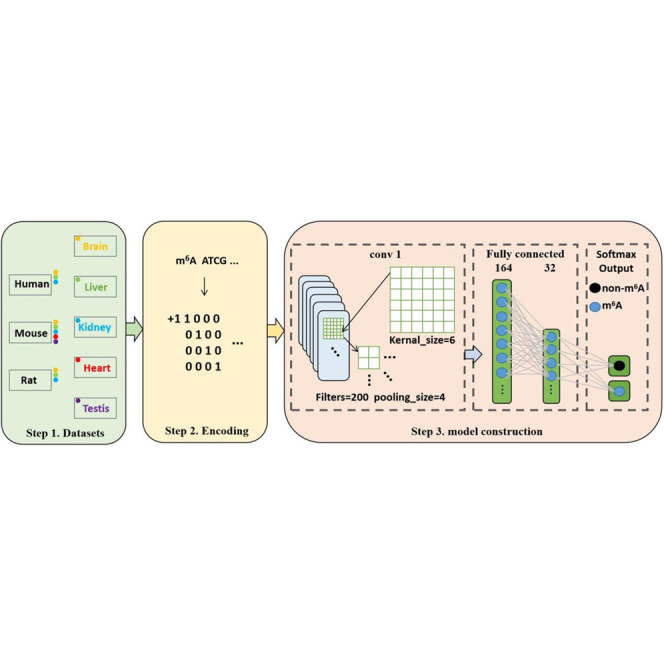
With the consideration of the spatial specificity of m6A modification, it is necessary to develop methods able to detect m6A in different tissues. With the use of a convolutional neural network, Liu and colleagues proposed a computational method that can identify m6A sites in multiple tissues of Homo sapiens, Mus musculus, and Rattus norvegicus.
Introduction
As a common and abundant of RNA post-transcriptional modification (PTM), N6-methyladenosine (m6A) modification plays an important role in almost all processes of cell cycles, such as affecting translation efficiency,1 cell development,2 cell viability,3 etc. m6A is catalyzed by a methyltransferase complex containing METTL3, METTL14, and WTAP. As a kind of dynamic PTM, m6A can be erased by the demethylases FTO and ALKBH5.4 Recently, more and more studies have revealed that m6A is closely correlated with diseases, such as obesity,5 thyroid tumor,6 prostate cancer,7 zika virus,8 and acute myelogenous leukemia.9 However, our knowledge about the functions of m6A modifications is still unintelligible. To deepen our understanding on the functions of m6A, the key step is to know the precise position of m6A in transcriptomes.
There are two main ways to identify m6A sites. One way is using experimental methods, such as Methylated RNA Immunoprecipitation (MeRIP),10 m6A sequencing (m6A-seq)11, photo-crosslinking-assisted (PA)-m6A-seq,12 and m6A-crosslinking immunoprecipitation (CLIP).13 These experimental methods laid important foundations for the detection of m6A modification sites. Accordingly, some bioinformatics tools that are able to detect m6A sites directly from the reads generated by the experiments were proposed.14,15 However, as the amount of sequencing data increases, we need to find an effective and efficient way to detect m6A in the transcriptome. Accordingly, sequence information-based computational methods were proposed to identify m6A sites. These methods can be queried in a recent review.16 With the research on the spatial specificity of gene expression, it has been found that the location of the m6A site is distinct in different tissues and species. Therefore, Dao et al.17 proposed a tool, called iRNA-m6A that can identify m6A modification sites in different tissues in human, mouse, and rat by using the algorithm of SVM, based on the data of Zhang et al.18. This method greatly improves the accuracy of predicting the m6A site. However, the performance for predicting the m6A site still has great potential to be improved.
In recent years, the deep learning algorithms made great contributions to bioinformatics. A large number of computational methods based on deep-learning algorithm, such as Gene2Vec,19 BERMP,20 DeepM6ASeq,21 and iPseU-CNN22 have been proposed. Inspired by these successful applications of the deep-learning algorithm in identifying RNA modifications, in the present work, we proposed a convolutional neural network (CNN)-based method, called im6A-TS-CNN, to identify m6A sites in different tissues from human, mouse, and rat. Results from a 5-fold cross-validation test and independent dataset test demonstrated that the performance of im6A-TS-CNN is better than or comparable with that of the existing method for the same aim. Moreover, the universality of im6A-TS-CNN was also demonstrated by a cross-species validation test. The framework of im6A-TS-CNN is illustrated in Figure 1.
Figure 1.

The Framework of the im6A-TS-CNN
The first step is to collect tissue-specific m6A data from the human, mouse, and rat. The second step is encoding the sequences by using the one-hot scheme. The third step is model construction.
Results and Discussion
Model Performance
In this article, the Keras in TensorFlow 2.0 under Python 3.6 was used to perform the predictions. The results from a 5-fold cross-validation test and independent dataset test of the proposed method for identifying the tissue-specific m6A modification sites in the human, mouse, and rat were shown in Table 1. With the comparison of results from a 5-fold cross-validation test and independent test, it was found that the proposed method is stable for identifying the m6A sites.
Table 1.
The Performance of im6A-TS-CNN for Identifying m6A Sites
| 5-Fold Cross Validation |
Independent Test |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sn (%) | Sp (%) | Acc (%) | MCC | AUC | Sn (%) | Sp (%) | Acc (%) | MCC | AUC | |
| h_b | 75.35 | 69.71 | 72.53 | 0.4523 | 0.8029 | 75.17 | 70.20 | 72.69 | 0.4543 | 0.8056 |
| h_k | 81.70 | 78.25 | 79.98 | 0.6006 | 0.8781 | 79.95 | 78.53 | 79.24 | 0.5848 | 0.8727 |
| h_l | 80.18 | 79.69 | 79.94 | 0.5992 | 0.8811 | 84.81 | 75.02 | 79.92 | 0.6012 | 0.8805 |
| m_b | 81.50 | 75.85 | 78.67 | 0.5749 | 0.8705 | 86.22 | 70.74 | 78.48 | 0.5765 | 0.8722 |
| m_h | 78.37 | 67.60 | 72.99 | 0.4633 | 0.8115 | 75.82 | 71.36 | 73.59 | 0.4723 | 0.8161 |
| m_k | 79.91 | 81.00 | 80.46 | 0.6094 | 0.8842 | 80.52 | 81.00 | 80.76 | 0.6151 | 0.8855 |
| m_l | 72.39 | 70.24 | 71.32 | 0.4288 | 0.7953 | 75.56 | 67.58 | 71.57 | 0.4328 | 0.7927 |
| m_t | 75.21 | 75.61 | 75.41 | 0.5090 | 0.8380 | 83.45 | 68.87 | 76.16 | 0.5288 | 0.8467 |
| r_b | 79.04 | 74.23 | 76.64 | 0.5379 | 0.8469 | 78.05 | 75.84 | 76.95 | 0.5391 | 0.8516 |
| r_k | 84.15 | 80.77 | 82.46 | 0.6500 | 0.9017 | 84.85 | 80.59 | 82.72 | 0.6550 | 0.9077 |
| r_l | 81.56 | 79.63 | 80.59 | 0.6126 | 0.8830 | 84.51 | 75.94 | 80.22 | 0.6067 | 0.8847 |
h, m and r before the hyphen stand for human, mouse, and rat, respectively; after the hyphen stand for brain, heart, kidney, liver, and testis, respectively.
To measure objectively the performance of the proposed method, the receiver operating characteristic (ROC) curves23,24 from a 5-fold cross-validation test and independent test were plotted in Figure 2 as well. It was found that most of the areas under the ROC curve (AUCs) are higher than 0.8 in both the 5-fold cross-validation test and independent test, demonstrating the reliability of the proposed method for identifying tissue-specific m6A sites.
Figure 2.

The ROC Curves for Identifying m6A in Different Tissues in the Three Species under the 5-Fold Cross-Validation Test and Independent Dataset Test
The value of AUC is given in the right corner of each graph.
Comparison with Existing Method
To further testify the superiority of im6A-TS-CNN, we compared its performance with that of Zhang et al.’s18 iRNA-m6A model, based on both the 5-fold cross-validation test and independent test. The comparative results in terms of AUC are shown in Table 2. Except for the identification of the m6A sites from the brain of mouse and rat, im6A-TS-CNN outperforms iRNA-m6A for the identification of m6A sites in the other tissues in the human, mouse, and rat. These results demonstrate that im6A-TS-CNN is a powerful tool for identifying tissue-specific m6A sites from different species.
Table 2.
Comparative Results between im6A-TS-CNN and iRNA-m6A under the 5-Fold Cross-Validation Test and Independent Test
| 5-Fold Cross Validation (AUC) |
Independent Test (AUC) |
|||||
|---|---|---|---|---|---|---|
| m6A-TS-CNN | iRNA-m6A | Difference | im6A-TS-CNN | iRNA-m6A | Difference | |
| h_b | 0.8029 | 0.7756 | 0.0273∗ | 0.8056 | 0.7845 | 0.0211∗ |
| h_k | 0.8781 | 0.8634 | 0.0147∗ | 0.8727 | 0.8565 | 0.0162∗ |
| h_l | 0.8811 | 0.8738 | 0.0073∗ | 0.8805 | 0.8681 | 0.0124∗ |
| m_b | 0.8705 | 0.8731 | −0.0026 | 0.8722 | 0.8613 | 0.0109∗ |
| m_h | 0.8115 | 0.7948 | 0.0167∗ | 0.8161 | 0.7878 | 0.0283∗ |
| m_k | 0.8842 | 0.8726 | 0.0116∗ | 0.8855 | 0.8697 | 0.0158∗ |
| m_l | 0.7953 | 0.7743 | 0.0210∗ | 0.7927 | 0.762 | 0.0307∗ |
| m_t | 0.8380 | 0.8156 | 0.0224∗ | 0.8467 | 0.8182 | 0.0285∗ |
| r_b | 0.8469 | 0.8282 | 0.0187∗ | 0.8516 | 0.8968 | −0.0452 |
| r_k | 0.9017 | 0.8877 | 0.0140∗ | 0.9077 | 0.8761 | 0.0316∗ |
| r_l | 0.8830 | 0.8766 | 0.0064∗ | 0.8847 | 0.8265 | 0.0582∗ |
h, m and r before the hyphen stand for human, mouse and rat; b, h, k, l, t after the hyphen stand for brain, liver, kidney, heart and testis, respectively.
inidcates the performance of im6A-TS-CNN is better than iRNA-m6A for identifying m6A sites.
Cross-Species and Cross-Tissue Validation
Since the datasets are from different species and tissues, it is interesting to test whether the model, trained based on the data from a specific tissue in a species, is able to identify m6A from other tissues and species. Accordingly, the cross-species and cross-tissue validation was performed. The AUCs of im6A-TS-CNN for identifying m6A sites from other species and tissues are shown in Figure 3. As shown in Figure 3, it can be concluded that im6A-TS-CNN is also effective for the cross-species and cross-tissue identification of m6A sites, demonstrating the universality of the proposed method.
Figure 3.

Heatmap Showing the AUC Values of Cross-Species and Cross-Tissue Validation
The abscissa represents the independent dataset, and the ordinate represents the model.
Conclusions
In this article, we proposed a CNN-based method, called i6mA-TS-CNN, for identifying m6A in the brain, liver, kidney, heart, and testis from the human, mouse, and rat. The results from a 5-fold cross-validation test and independent test demonstrate that i6mA-TS-CNN is better than the existing method for identifying tissue-specific m6A. For the convenience of the scientific community, the command-line version of i6mA-TS-CNN, together with its source code and user manual, is provided at https://github.com/liukeweiaway/DeepM6A_cnn. In addition, the high-, normal-, and low-threshold options were provided to control the false-positive rate. The corresponding performance with different options was listed in Table S1. Taken together, we hope that the i6mA-TS-CNN will become a useful tool for identifying m6A sites.
Materials and Methods
Datasets
A high-quality dataset is very important for the construction of a computational model. In 2019, Zhang et al.18 developed a high-throughput, antibody-independent m6A detection method based on the m6A-sensitive RNA endoribonuclease to identify the m6A site in different tissues, namely the brain, liver, kidney, heart, and testis from the human, mouse, and rat. Based on these data, Dao et al.17 built a high-quality benchmark dataset that can be used to train a computational method for identifying m6A sites, which contains both m6A site- and non-m6A site-containing sequences with the length of 41 nt. The CD-HIT program25 was used to make sure that the sequence similarity of the dataset was less than 80%. The detailed information of this dataset is provided in Table 3.
Table 3.
The Information of Benchmark Datasets for Predicting RNA m6A Sites
| Name | Training |
Testing |
||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| h_b | 4,605 | 4,605 | 4,604 | 4,604 |
| h_k | 2,634 | 2,634 | 2,634 | 2,634 |
| h_l | 4,574 | 4,574 | 4,573 | 4,573 |
| m_b | 8,025 | 8,025 | 8,025 | 8,025 |
| m_h | 4,133 | 4,133 | 4,133 | 4,133 |
| m_k | 3,953 | 3,953 | 3,952 | 3,952 |
| m_l | 2,201 | 2,201 | 2,200 | 2,200 |
| m_t | 4,707 | 4,707 | 4,706 | 4,706 |
| r_b | 2,352 | 2,352 | 2,351 | 2,351 |
| r_k | 1,762 | 1,762 | 1,762 | 1,762 |
| r_l | 3,433 | 3,433 | 3,432 | 3,432 |
h, m and r before the hyphen stand for human, mouse and rat; b, h, k, l, t after the hyphen stand for brain, liver, kidney, heart and testis, respectively.
One-Hot Encoding
One-hot encoding is a common and effective method.26 According to such a scheme, in an RNA segment, A is represented as (1,0,0,0), U as (0,1,0,0), C as (0,0,1,0), and G as (0,0,0,1). Therefore, an RNA sequence of length l can be converted into a 4-l dimensional vector.
Convolutional Neural Network
In recent years, Convolutional Neural Network (CNN) has been widely used to solve biological problems.22,27,28 The structure of the CNN is shown in Figure 1. It contains a convolutional layer with 200 filters in which the kernel size is 6. After convolution operation, a max-pooling layer with the size of 4 was added. The convolution layer is mathematically represented and computed as the following:
| (Equation 1) |
where R represents the RNA segment, f denotes the index of the kernel, and j denotes the index of the output position. In Equation 1, each filter Wf is an S × N weight matrix, where S is the filter size, and N is the input channels. The rectified linear function (ReLU) is expressed as the following:
| (Equation 2) |
In order to prevent overfitting, we choose to lose some parameters and set the dropout rate of 0.16. The results were output to a fully connected layer containing 164 neural units and then compressed to 32 neural units. Finally, the softmax function was used to predict whether the RNA segment contains m6A sites or not and is expressed as the following:
| (Equation 3) |
When building the model, the stochastic gradient descent (SGD) was used as the optimizer with a learning rate of 0.001, and the categorical cross entropy was used as the loss function. In the training process, a total of 2,000 epochs were carried out by using the early stopping method with the patience of 50 and min_delta of 0.001.
Evaluation Metrics
In order to evaluate the model, we use the sensitivity (Sn), specificity (Sp), accuracy (Acc), and Matthews correlation coefficient (MCC), which are defined as the following,26,29,30 to evaluate the performance of the following model:
| (Equation 4) |
where N+ is the total number of the RNA sequence containing modification site, is the number of false-negative samples, N− is the total number of the RNA sequence that did not contain any modification site, and is teh number of false-positive samples.
In addition, we also used the ROC curve31 and the area under the ROC curve (AUC) to evaluate the proposed model.
Author Contributions
W.C. conceived and designed the study. K.L. conducted the experiments and implemented the algorithms. W.C., L.C., P.D., and K.L. performed the analysis and wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors would like to express their gratitude to Prof. Quan Zou for his help in optimizing the parameters of CNN. This work was supported by the National Nature Scientific Foundation of China (31771471) and Natural Science Foundation for Distinguished Young Scholar of Hebei Province (number C2017209244).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.07.034.
Supplemental information
References
- 1.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bokar J.A. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. In: Grosjean H., editor. Fine-Tuning of RNA Functions by Modification and Editing. Topics in Current Genetics. Springer; 2005. pp. 141–177. [Google Scholar]
- 4.Wu R., Jiang D., Wang Y., Wang X. N (6)-Methyladenosine (m(6)A) Methylation in mRNA with A Dynamic and Reversible Epigenetic Modification. Mol. Biotechnol. 2016;58:450–459. doi: 10.1007/s12033-016-9947-9. [DOI] [PubMed] [Google Scholar]
- 5.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiliger K.J., Hess J., Vitagliano D., Salerno P., Braselmann H., Salvatore G., Ugolini C., Summerer I., Bogdanova T., Unger K. Novel candidate genes of thyroid tumourigenesis identified in Trk-T1 transgenic mice. Endocr. Relat. Cancer. 2012;19:409–421. doi: 10.1530/ERC-11-0387. [DOI] [PubMed] [Google Scholar]
- 7.Machiela M.J., Lindström S., Allen N.E., Haiman C.A., Albanes D., Barricarte A., Berndt S.I., Bueno-de-Mesquita H.B., Chanock S., Gaziano J.M. Association of type 2 diabetes susceptibility variants with advanced prostate cancer risk in the Breast and Prostate Cancer Cohort Consortium. Am. J. Epidemiol. 2012;176:1121–1129. doi: 10.1093/aje/kws191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lichinchi G., Zhao B.S., Wu Y., Lu Z., Qin Y., He C., Rana T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe. 2016;20:666–673. doi: 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bansal H., Yihua Q., Iyer S.P., Ganapathy S., Proia D.A., Penalva L.O., Uren P.J., Suresh U., Carew J.S., Karnad A.B. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171–1174. doi: 10.1038/leu.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 12.Chen K., Lu Z., Wang X., Fu Y., Luo G.-Z., Liu N., Han D., Dominissini D., Dai Q., Pan T., He C. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 2015;54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A., Haripal B., Zucker-Scharff I., Moore M.J., Park C.Y. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29:2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen K., Wei Z., Zhang Q., Wu X., Rong R., Lu Z., Su J., de Magalhães J.P., Rigden D.J., Meng J. WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019;47:e41. doi: 10.1093/nar/gkz074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng J., Lu Z., Liu H., Zhang L., Zhang S., Chen Y., Rao M.K., Huang Y. A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods. 2014;69:274–281. doi: 10.1016/j.ymeth.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Zhao P., Li F., Wang Y., Smith A.I., Webb G.I., Akutsu T., Baggag A., Bensmail H., Song J. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief. Bioinform. 2019 doi: 10.1093/bib/bbz112. Published online November 11, 2019. [DOI] [PubMed] [Google Scholar]
- 17.Dao F.-Y., Lv H., Yang Y.H., Zulfiqar H., Gao H., Lin H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020;18:1084–1091. doi: 10.1016/j.csbj.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z., Chen L.-Q., Zhao Y.-L., Yang C.-G., Roundtree I.A., Zhang Z., Ren J., Xie W., He C., Luo G.-Z. Single-base mapping of m(6)A by an antibody-independent method. Sci. Adv. 2019;5:eaax0250. doi: 10.1126/sciadv.aax0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou Q., Xing P., Wei L., Liu B. Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA. 2019;25:205–218. doi: 10.1261/rna.069112.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y., He N., Chen Y., Chen Z., Li L. BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018;14:1669–1677. doi: 10.7150/ijbs.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Hamada M. DeepM6ASeq: prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinformatics. 2018;19(Suppl 19):524. doi: 10.1186/s12859-018-2516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tahir M., Tayara H., Chong K.T. iPseU-CNN: Identifying RNA Pseudouridine Sites Using Convolutional Neural Networks. Mol. Ther. Nucleic Acids. 2019;16:463–470. doi: 10.1016/j.omtn.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasan M.M., Manavalan B., Shoombuatong W., Khatun M.S., Kurata H. i4mC-Mouse: Improved identification of DNA N4-methylcytosine sites in the mouse genome using multiple encoding schemes. Comput. Struct. Biotechnol. J. 2020;18:906–912. doi: 10.1016/j.csbj.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasan M.M., Manavalan B., Shoombuatong W., Khatun M.S., Kurata H. i6mA-Fuse: improved and robust prediction of DNA 6 mA sites in the Rosaceae genome by fusing multiple feature representation. Plant Mol. Biol. 2020;103:225–234. doi: 10.1007/s11103-020-00988-y. [DOI] [PubMed] [Google Scholar]
- 25.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 26.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. Nucleic Acids. 2019;16:733–744. doi: 10.1016/j.omtn.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aoki G., Sakakibara Y. Convolutional neural networks for classification of alignments of non-coding RNA sequences. Bioinformatics. 2018;34:i237–i244. doi: 10.1093/bioinformatics/bty228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren F., Yang C., Qiu Q., Zeng N., Cai C., Hou C., Zou Q. Exploiting Discriminative Regions of Brain Slices Based on 2D CNNs for Alzheimer’s Disease Classification. IEEE Access. 2019;7:181423–181433. [Google Scholar]
- 29.Liu K., Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinformatics. 2020;36:3336–3342. doi: 10.1093/bioinformatics/btaa155. [DOI] [PubMed] [Google Scholar]
- 30.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. AtbPpred: A Robust Sequence-Based Prediction of Anti-Tubercular Peptides Using Extremely Randomized Trees. Comput. Struct. Biotechnol. J. 2019;17:972–981. doi: 10.1016/j.csbj.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh F., Turnbull B.W. Non- & semi-parametric estimation of the receiver operating characteristics (ROC) curve. Ann. Stat. 1996;24:25–40. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.