

Abstract
Myocardial ischemia leads to conduction slowing, cell-to-cell uncoupling, and arrhythmias. We previously demonstrated that varying perfusate sodium (Na+) and calcium (Ca2+) attenuates conduction slowing and arrhythmias during simulated ischemia with continuous perfusion. Cardioprotection was selectively associated with widening of the perinexus, a gap junction adjacent nanodomain important to ephaptic coupling. It is unknown whether perfusate composition affects the perinexus or ischemic conduction during nonsimulated ischemia, when coronary flow is reduced or halted. We hypothesized that altering preischemic perfusate composition could facilitate perinexal expansion and attenuate conduction slowing during global ischemia. To test this hypothesis, ex vivo guinea pig hearts (n = 49) were Langendorff perfused with 145 or 153 mM Na+ and 1.25 or 2.0 mM Ca2+ and optically mapped during 30 min of no-flow ischemia. Altering Na+ and Ca2+ did not substantially affect baseline conduction. Increasing Na+ and decreasing Ca2+ both lowered pacing thresholds, whereas increasing Ca2+ narrowed perinexal width (Wp). A least squares mean estimate revealed that reduced perfusate Na+ and Ca2+ resulted in the most severe conduction slowing during ischemia. Increasing Na+ alone modestly attenuated conduction slowing, yet significantly delayed the median time to conduction block (10 to 16 min). Increasing both Na+ and Ca2+ selectively widened Wp during ischemia (22.7 vs. 15.7 nm) and attenuated conduction slowing to the greatest extent. Neither repolarization nor levels of total or phosphorylated connexin43 correlated with conduction slowing or block. Thus, perfusate-dependent widening of the perinexus preserved ischemic conduction and may be an adaptive response to ischemic stress.
NEW & NOTEWORTHY Conduction slowing during acute ischemia creates an arrhythmogenic substrate. We have shown that extracellular ionic concentrations can alter conduction by modulating ephaptic coupling. Here, we demonstrate increased extracellular sodium and calcium significantly attenuate conduction slowing during no-flow ischemia. This effect was associated with selective widening of the perinexus, an intercalated disc nanodomain and putative cardiac ephapse. These findings suggest that acute changes in ephaptic coupling may serve as an adaptive response to ischemic stress.
Keywords: arrhythmia, calcium, conduction, ischemia, sodium
INTRODUCTION
Myocardial ischemia, a condition in which coronary blood flow is inadequate to maintain steady-state myocardial function, is a leading cause of cardiovascular death and, accordingly, remains a key topic of biomedical research (7, 32). A lack of oxygen and substrate exchange between blood and tissue produces metabolic, ionic, and electrical instabilities within the heart. In turn, these changes alter cardiomyocyte membrane properties, and within minutes, ischemia can lead to changes in cardiac excitability, conduction, and repolarization (11, 42, 79). If blood flow is not restored, ischemia eventually leads to gap junction (GJ) remodeling and a decrease in cell-to-cell coupling. Altogether, these changes create a multilayered substrate for lethal arrhythmias.
To date, significant focus has been placed on developing therapies to prevent arrhythmias during ischemia by targeting GJ coupling (GJC) to preserve cardiac conduction (13, 14, 48, 53). It is important to note that multiple factors beyond GJC modulate cardiac conduction (63). For example, during an acute ischemic event, significant changes in conduction (within 2 min) (11, 37, 49, 79) and an initial peak in arrhythmia risk (5–10 min) (30, 31, 34, 43) can precede the onset of GJ remodeling and subsequent changes in GJC (15–30 min) (4, 6, 60, 71). Thus, it is clear that preventing conduction slowing and arrhythmias during this acute, initial phase of ischemia may not rely solely on mechanisms related to GJC.
It is well established that interventions designed to reduce intracellular calcium accumulation during ischemia attenuate conduction slowing (11, 12, 19, 35, 41, 47, 59), and increasing intracellular calcium (11, 41, 59) exacerbates conduction slowing. The proposed mechanisms include calcium modulation of resting membrane potential (RMP), action potential (AP) maximum upstroke velocity, and tissue resistance (extracellular, membrane, and intracellular). Yet, mechanistic interpretation is often difficult to reconcile between studies. For example, the L-type calcium channel blocker verapamil, which presumably reduces intracellular calcium accumulation during ischemia, has been reported to attenuate (11, 41), not change (47, 59), and exacerbate (35) the rise in RMP observed during ischemia. Despite this, verapamil consistently attenuated cardiac conduction slowing in these studies, suggesting that other factors contribute to conduction slowing during the acute phases of ischemia before GJ uncoupling and irreversible myocardial damage.
Previous work by our group and others suggests that an additional form of cell-to-cell electrical coupling, referred to as ephaptic coupling (EpC), plays a role in conduction, and in particular can act in parallel with GJC to regulate conduction velocity (CV) (20, 25, 26, 39, 56, 58, 81, 83). We have proposed that the perinexus, an intercalated disc (ID) nanodomain adjacent to GJs, serves as a candidate structure for the cardiac ephapse (68, 69, 74, 76). Both in silico and ex vivo findings have demonstrated that perfusate composition and restricted extracellular nanodomains are critical determinants of EpC and its effects on conduction. (20, 22–24, 26, 29, 33, 39, 56–58, 73–75, 83).
Given that hallmark features of ischemia (e.g., ionic imbalances and changes in extracellular volume) also serve as underpinnings of EpC, it provokes the question of whether EpC plays a role in acute conduction changes during the onset of cardiac ischemia. Recently, in an ex vivo model of simulated metabolic ischemia (metabolic stress), we demonstrated that different combinations of sodium and calcium concentrations ([Na+] and [Ca2+], respectively) perfused into the heart during ischemia either attenuate or exacerbate conduction slowing (24). Contrary to the original hypothesis, conduction was selectively preserved with specific perfusate compositions that were associated with perinexal expansion, which in theory would reduce EpC (24).
While the simulated ischemia model mimics several key features of metabolic stress/ischemia (e.g., hypoxia, aglycemia, hyperkalemia, acidosis), the use of continuous flow results in more controlled extracellular ionic concentrations (and pH), and continuously washes out cellular waste products (46, 51). In particular, at the onset of ischemia, the dynamic change in extracellular potassium accumulation is a critical factor shaping the acute changes in RMP, excitability, and conduction (11, 42, 79). Additionally, as the simulated ischemia model maintains intravascular pressure, the continuous flow likely affects fluid balance within the ID and influences the remodeling of perinexal structure during ischemia. Therefore, we thought that employing a more physiological model of ischemia was necessary to better understand the broader relevance of perinexal adaptation during true ischemia. In contrast, a no-flow ischemia model allows for the dynamic accumulation/depletion of ionic gradients and metabolites that are key determinants of excitability and conduction at the onset of ischemia. At present, it is unknown whether structural changes within the perinexus mediated by preischemic perfusate composition would still mitigate conduction slowing or arrhythmias during no-flow ischemia. To test this, we preperfused isolated guinea pig hearts with buffer solutions containing varying [Na+] and [Ca2+] before halting flow and inducing global ischemia for 30 min. The [Na+] and [Ca2+] values tested were based off of guinea pig blood chemistries and concentrations previously tested (20, 23, 24).
Here, we confirm that altered ionic fluid composition modulates electrophysiology in emergent ways. This, in of itself, has clear implications for studies involving perfused cardiac tissue preparations, as overlooked or unreported differences in perfusate composition could underlie seemingly inconsistent experimental results within the literature. Preceding ischemia, differences in perfusate [Na+] and [Ca2+] significantly affected tissue excitability and perinexal width (Wp); however, conduction was not substantially altered, which raises the important issue that in cardiac tissue, excitability and conduction are codependent but distinct electrophysiological phenomena. Accordingly, a perfusate-dependent increase in baseline tissue excitability (but not CV) was correlated with a delay in ischemia-induced conduction block. Overall, ischemia was associated with perinexal widening, but this effect was only significant in hearts perfused with elevated [Na+] and [Ca2+]. Importantly, perfusate-dependent widening of the perinexus selectively attenuated conduction slowing during metabolic and now no-flow ischemia, suggesting that perinexal expansion during cardiac stress could be an important adaptive response.
METHODS
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at Virginia Polytechnic Institute and State University and conducted in compliance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Langedorff-Perfused Hearts
Male, retired breeder, albino, Hartley guinea pigs (Hilltop Laboratory Animals, Scottdate, PA; n = 73, ~800–1,200 g, 13–20 mo old) were anesthetized with isoflurane as previously described (36). Once in a surgical plane, a thoracotomy was performed and the heart was excised and rinsed in perfusate bubbled with 100% oxygen. The ascending aorta was cannulated (3-mm tip, glass aortic cannula, Radnoti No. 140153-30) within 2–4 min and Langendorff perfused at 37.0 ± 0.5°C in nonrecirculating fashion with one of four HEPES-based buffer solutions that only varied in the NaCl and/or CaCl2 concentration(s) (see Table 1 for perfusate compositions). We chose to omit proteins and colloids from our crystalline buffers for continuity with our previous studies and to allow for optical mapping throughout the duration of the 30+-min protocol, as we have previously shown that albumin can rapidly decrease the signal-to-noise ratio of 4-(2-(6-(dibutylamino)-2-naphthalenyl)ethenyl)-1-(3-sulfopropyl)pyridinium hydroxide inner salt (di-4-ANEPPS) fluorescence signals (24). Perfusate compositions were not corrected for differences in osmolarity; the maximal difference in osmolarity was ~17 mosmol/L (5%). Once cannulated, the atria were excised to reduce competitive ventricular stimulation. The heart was positioned in front of an imaging window in a custom-made, three-dimensional (3D) printed tissue bath as previously described (21). Hearts were perfused with constant flow (typically 20 ml/min) via a peristaltic pump to maintain a perfusion pressure of 40–60 mmHg and monitored by an inline pressure transducer (Millar SPR-524). The heart was immersed in the effluent, which was subsequently recirculated and warmed in a separate circuit from the primary perfusion system to control bath temperature.
Table 1.
Perfusate compositions
| Name | CaCl2 | KCl | MgCl2 | Dextrose | HEPES | NaCl | NaOH | Total Na+ | Osmolarity, mosmol/L |
|---|---|---|---|---|---|---|---|---|---|
| 145Na+/1.25Ca2+ | 1.25 | 4.56 | 0.7 | 5.5 | 10.0 | 140.0 | 5.5 | 145.5 | 328.9 |
| 145Na+/2Ca2+ | 2.0 | 4.56 | 0.7 | 5.5 | 10.0 | 140.0 | 5.5 | 145.5 | 331.2 |
| 153Na+/1.25Ca2+ | 1.25 | 4.56 | 0.7 | 5.5 | 10.0 | 147.5 | 5.5 | 153.0 | 344.0 |
| 153Na+/2Ca2+ | 2.0 | 4.56 | 0.7 | 5.5 | 10.0 | 147.5 | 5.5 | 153.0 | 346.2 |
Composition (in mM) of the 4 experimental perfusates indicating the changes in CaCl2 and NaCl to produce total [Na+] (=NaCl + NaOH) of either 145 or 153 mM and total [Ca2+] of either 1.25 or 2 mM. Calculated osmolarity (in mosmol/L) is shown for each perfusate.
For external pacing, a unipolar silver-chloride electrode was placed on the midapicobasal region of the anterior left ventricular epicardium. A second ground electrode was placed in the bath. The tissue was electrically stimulated with a 5-ms square pulse, and the diastolic threshold (current, mA) was obtained at a cycle length (CL) of 300 ms. During the experimental protocol, the heart was paced at 1.5× diastolic threshold. Hearts were only paced immediately preceding and during the indicated time points of data acquisition; otherwise, the heart was allowed to beat according to the intrinsic rhythm. A volume-conducted electrocardiogram (ECG) was obtained from three silver-chloride needle electrodes (2 on either side of the ventricles and 1 in the rear of the bath) (21). ECG signals were notch filtered (0.5–100 Hz), and ECG, inline perfusion pressure, and bath temperature were sampled at 1 kHz and continuously recorded throughout the entire experiment using LabChart.
Optical Mapping
After a 30-min stabilization period, hearts (n = 25) were perfused with the voltage-sensitive fluorophore, di-4-ANEPPS (100 mL, 7.5 µM), followed by a 10-min washout period. The fluorophore was excited by a halogen light source (MHAB-150W, Moritex), equipped with a fiber light guide and 520/35-nm bandpass filter (Brightline). The filtered excitation light was directed onto a dichroic mirror (565 nm, Chroma Technology) and reflected onto the heart via epi-illumination. Emitted light was collected via a tandem lens system and transmitted through a 610-nm long-pass filter (Andover Corp.) before detection by a MiCam Ultima L-type CMOS camera (SciMedia: 100 × 100 pixels, field of view = 15.9 × 15.9 mm). Baseline optical APs were recorded at a 1-kHz sampling rate for a duration of ~2 s during intrinsic activity and steady-state pacing at a CL of 300 ms.
No-flow ischemia protocol.
After the baseline recordings, the perfusion pump was turned off to halt the flow of perfusate, leading to the onset of global ischemia. Optical APs were recorded during steady-state pacing (300 ms) and intrinsic activity every 2 min, up to 30 min of no-flow ischemia. If loss of pacing capture occurred during ischemia, the pacing stimulation amplitude was increased until 1:1 capture was reestablished, with an upper limit of 5× diastolic threshold. If 1:1 capture was not achieved at this maximum level, the protocol was halted.
Data analysis.
Fluorescent signals were spatially binned 2 × 2 (effective pixel resolution = 0.318 mm) and subsequently analyzed using custom MatLab programs to determine AP activation time and calculate CV (transverse, CVT, and longitudinal, CVL), anisotropy ratio (AR = CVL/CVT), and AP duration at 90% repolarization (APD90) as previously described (5, 27, 28, 36). Conduction block was identified from movies of wave-front propagation generated from the fluorescent signals and isochrone maps of AP activation time. Conduction block was never detected at baseline. The earliest occurrence of conduction block observed during ischemia was identified for each heart, and a survival analysis was conducted to test for differences in time to conduction block.
Western blot analysis.
In a separate set of studies, left ventricular tissue was collected to assess the ventricular GJ protein, connexin43 (Cx43), at 10 min of ischemia or equivalent nonischemic time control in hearts perfused with each of the perfusate compositions (n = 6 hearts per perfusate, per condition; total n = 48 hearts). The time point of tissue collection was chosen to assess acute ischemia-induced conduction changes, preceding the significant rise in tissue resistance associated with GJ remodeling and the onset of irreversible myocardial damage (2, 6, 10, 11, 60, 61, 67, 71). Tissue samples were snap frozen in liquid nitrogen, and Western blot analysis for total Cx43 and phospho-Cx43-Ser368 was performed. Samples were weighed and homogenized in RIPA lysis buffer at a final volume of 100 mg/ml, consisting of 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 2 mM NaF, and 200 μM Na3VO4, supplemented with HALT protease and phosphatase inhibitors (Thermo Scientific). After sonication, lysates were clarified by centrifugation at 10,000 g for 20 min at 4°C. Sample protein concentration was measured using the Bio-Rad DC Protein Assay and normalized. Then, 4X Bolt LDS Sample Buffer (Thermo Scientific) supplemented with DTT (100 mM; Fisher) was added, and samples denatured for 10 min at 70°C. Samples were subjected to SDS-PAGE and transferred to PVDF membranes, which underwent methanol fixation before rewetting and blocking for 1 h at room temperature in 5% BSA (Fisher). Membranes were incubated with rabbit anti-pCx43-Ser368 (1:1,000, Cell Signaling Technologies) overnight at 4°C and washed four times before incubation with anti-rabbit HRP (1:5,000, Abcam) for 1 h at room temperature. After development with Clarity ECL Substrate (Bio-Rad), membranes were imaged and stripped with ReBlot Plus Strong Solution (MilliporeSigma). Membranes were blocked and reprobed overnight at 4°C with rabbit anti-Cx43 (1:3,000; MilliporeSigma) and mouse anti-α-tubulin (1: 3,000; MilliporeSigma), followed by secondary antibodies for 1 h at room temperature (both 1:1,000, goat anti-mouse AlexaFluor555, goat anti-rabbit AlexaFluor647). Membranes were imaged, and total Cx43, phospho-Cx43-Ser368, and tubulin were quantified using a Bio-Rad Chemidoc MP. Total Cx43 was normalized to α-tubulin, and phospho-Cx43-Ser368 was normalized to total Cx43.
Transmission Electron Microscopy
In a subset of hearts used for immunoblotting, left ventricular tissue was also collected for transmission electron microscopy (TEM) to assess the intermembrane separation in the GJ adjacent nanodomain termed the perinexus. Tissue was collected at 10 min of ischemia or equivalent nonischemic time control in hearts perfused with each of the perfusate compositions (n = 3 hearts per perfusate, per condition; total n = 24 hearts). Tissue sections were prepared, fixed in 2% glutaraldehyde and processed for TEM as previously described (26). Images (average of 16 per heart) of perinexi and their associated GJs within the ID were obtained using a JEOL JEM1400 electron microscope at 150,000× magnification. Perinexal width (Wp) was measured within the range of 30–105 nm from the edge of the GJ plaque using ImageJ and a MatLab-based algorithm recently developed by our group (65).
Statistical Analysis
Statistical analyses were performed using R Statistical Software v3.5.2 and GraphPad Prism 8. Summary data are presented as means ± SE unless otherwise noted. An ordinary one-way analysis of variance (ANOVA) was used to test for baseline differences in CV, pacing threshold, APD90, and ∆APD90 (ischemia vs. baseline) across all four perfusate groups; each perfusate was tested against 145Na+/1.25Ca2+ with post hoc correction using Dunnett’s multiple comparisons test. Additionally, a two-way ANOVA was performed to investigate an additive relationship between [Na+] and [Ca2+] on pacing threshold. Time to onset of conduction block for each perfusate was compared using a Cox proportional hazard model where all samples with an onset of conduction block greater than 30 min were censored data points. CV was measured up to the time point of conduction block, i.e., CV was not evaluated at time points post-block. To evaluate differences in the rate of change for CV among the perfusates, we used a mixed-effects linear (LS Means) model to test for an interaction among ischemia time, [Ca2+], and [Na+], adjusting for baseline CV, and using each sample as a random effect (to account for repeated measures). The model also accounted for CV data drop out due to the onset of conduction block. Contrasts were employed to evaluate differences in the rate of change in CV among each of the perfusate groups. For TEM image analysis, quantification revealed skewed distributions of Wp. To address this, all analyses of Wp used the natural log transform of Wp to normalize the distributions. A mixed-effects linear model was used to evaluate differences in log(Wp) for perfusates and intervention (time control vs. ischemia). A post hoc Tukey’s honestly significant difference test was performed to compare pairwise differences. Lastly, we used a Jonckheere-Terpstra test from the clinfun package (78) to test for the predetermined rank order of means for Wp. An α value of 0.05 was used to determine significance.
RESULTS
Baseline Conduction Response to [Na+] and [Ca2+]
To assess whether small changes in perfusate ionic composition affect conduction at baseline (preischemia), we obtained optical APs from the anterior epicardial surface of the left and right ventricle (LV and RV, respectively). Previously, we have found that under otherwise normal conditions, modest changes in ionic concentration within near physiological ranges (e.g., 145–155 mM [Na+] and 1.25–2.5 mM [Ca2+]) do not significantly affect CV (20, 24, 26). Figure 1A shows an image of a cannulated heart with the corresponding field of view (FOV, black box) and a single frame from the mapping window showing the placement of the pacing wire adjacent to the left anterior descending artery (LAD) in the midapicobasal LV. In Fig. 1B, representative maps of AP activation isochrones demonstrate that preceding the onset of ischemia, excitation spreads elliptically from the site of pacing (denoted by *), consistent with normal anisotropic conduction, without any measurable differences in conduction pattern or total activation time due to alterations in [Na+] or [Ca2+]. CV (transverse, CVT, and longitudinal, CVL) was quantified from the isochrone maps during steady-state perfusion with each of the four perfusate compositions tested (145 or 153 mM [Na+] and 1.25 or 2 mM [Ca2+]). Summary data (Fig. 1C) revealed no significant differences in baseline CVT (P = 0.33) and only hearts perfused with 153Na+/2Ca2+ demonstrated a significant change in baseline CVL (P < 0.05). No perfusate differences in baseline AR were observed (P = 0.08).
Fig. 1.

At baseline, changes in perfusate [Na+] and [Ca2+] significantly modulate excitability but have a minimal effect on conduction. A, left: image of a Langendorff-perfused guinea pig heart with the black box representing the field of view (FOV). A, right: representative imaging frame of the anterior ventricular epicardium (LAD, left anterior descending coronary artery), with pacing electrode in the center of the FOV. B: representative isochrone maps of action potential activation times with pacing (*site of pacing) during baseline conditions for each perfusate composition. Isochrones represent ∆3 ms, scale bar = 5 mm. C: summary data for baseline transverse conduction velocity (CVT; ANOVA, P = 0.33; left), longitudinal conduction velocity (CVL; *P < 0.05 vs. 145Na+/1.25Ca2+; middle), and anisotropy ratio (AR; ANOVA, P = 0.08; right); data present as individual values with means ± SE. D: summary data for baseline pacing threshold (#P < 0.05 by 2-way ANOVA; means ± SE); n = 6, 6, 6, and 7 for 145Na+/1.25Ca2+, 145Na+/2Ca2+, 153Na+/1.25Ca2+, and 153Na+/2Ca2+, respectively.
Despite the minimal effect on baseline CV, the perfusate compositions tested significantly influenced tissue excitability. Specifically, increased [Na+] and decreased [Ca2+] were both associated with a lower diastolic pacing threshold (Fig. 1D). With 145 mM [Na+] (combined [Ca2+] groups) the mean threshold for pacing was 0.88 mA, whereas increasing [Na+] to 153 mM significantly decreased mean threshold to 0.61 mA (P < 0.05 vs. 145 mM [Na+]). Interestingly, the relationship between [Ca2+] and pacing threshold was even stronger. Specifically, with 1.25 mM [Ca2+] (combined [Na+] groups), the threshold for pacing was 0.56 mA, whereas increasing [Ca2+] to 2 mM significantly increased the threshold to 0.90 mA (P < 0.01).
Sensitivity of Ischemic Conduction Block to [Na+] and [Ca2+]
After the baseline period, global ischemia was induced by turning off the perfusion pump and halting perfusate flow through the cannula. During ischemia, the hearts were paced at CL = 300 ms, and optical APs were recorded every 2 min to assess the effects of perfusate composition on ischemia-induced CV changes and development of conduction block. If the ventricles failed to capture 1:1, the stimulus current amplitude was increased until capture was achieved or the stimulus reached the upper limit of 5× the diastolic threshold. The end points of the study were 30 min of ischemia or failure to capture the heart 1:1 at the upper limit of pacing stimulation.
Figure 2A, left, shows an activation map at 10 min of ischemia with crowding of isochrones indicating slowed (total activation time = 69 ms) but still relatively normal elliptical patterns of activation typical of anisotropic conduction (as shown in Fig. 1B). By 14 min of ischemia (Fig. 2A, right), the observed conduction slowing became more severe (total activation time = 97 ms), and unidirectional conduction block developed proximal to the site of pacing (between sites B′ and C′ on the map). Optical APs taken from the map at 14 min of ischemia (Fig. 2B, right) show the initiation of the wave front at the site of pacing (trace A) and reveal normal wave-front propagation from site A to E (blue arrow) but failed propagation from site A to E′ (into the RV, toward the left of the mapping field, red line in traces). An initial voltage deflection was observed in the trace from site B′. This activity failed to conduct to site C′ and instead the wave front wrapped around the line of conduction block and activated sites C′, D′, and E′ after a significant delay. This is in contrast to the traces recorded at 10 min of ischemia (Fig. 2B, left), where activation spreads out from the site of pacing (trace A) continuously and uninterrupted in both directions (blue arrows).
Fig. 2.
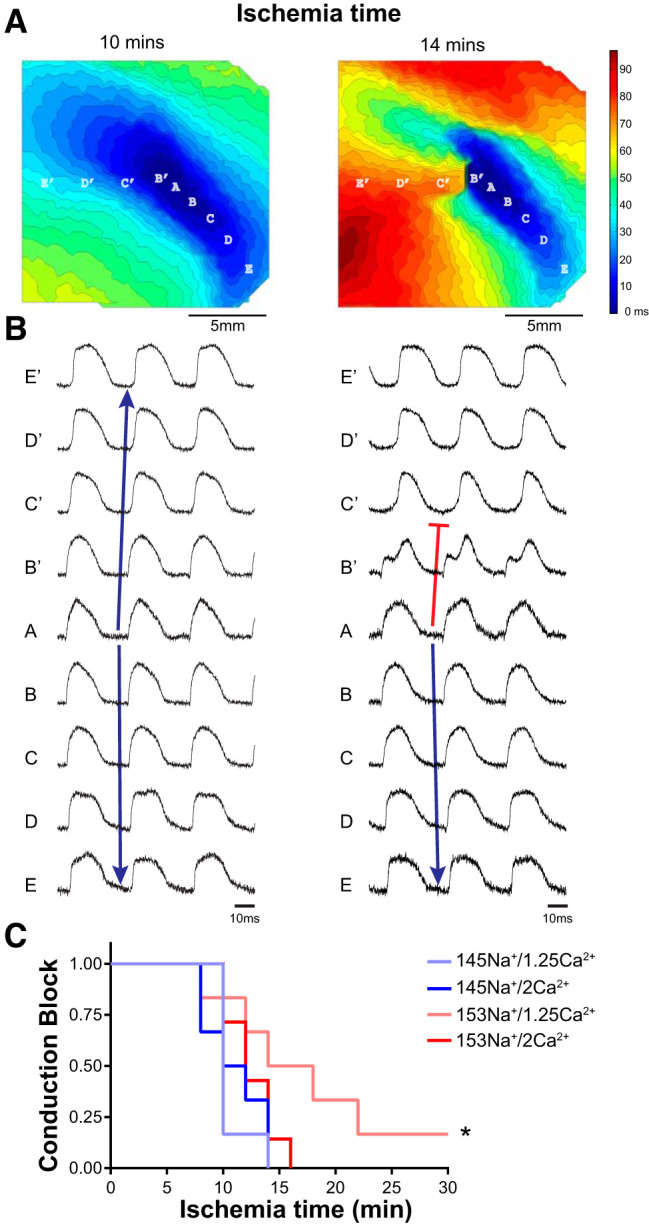
Increased perfusate [Na+] delays ischemia-induced conduction block. A: representative isochrone maps of action potential (AP) activation times at 10 (left) and 14 (right) min of ischemia with resulting conduction block. Isochrones represent ∆3 ms; scale bar = 5 mm. B: individual traces of 3 consecutive optical APs recorded from sites indicated in A; time scale = 10 ms. Blue arrows represent continuous conduction from the site of pacing (A). Red line indicates sites of unidirectional conduction block. C: Kaplan-Meier survival curves for onset of conduction block as a function of ischemia time. *P < 0.05 median time to block vs. 145Na+/1.25Ca2+ by Cox proportional hazard model; n = 6, 6, 6, and 7 for 145Na+/1.25Ca2+, 145Na+/2Ca2+, 153Na+/1.25Ca2+, and 153Na+/2Ca2+, respectively.
For each heart, the earliest instance of unidirectional block was determined from the activation maps, and Kaplan-Meier curves were generated for each perfusate (Fig. 2C). Overall, a Cox proportional hazard model found a significant effect of perfusate composition on the time to ischemia-induced conduction block (P < 0.05). Summary data reveal that increasing [Na+] with 1.25 mM [Ca2+] delayed the onset of conduction block from a median of 10 to 16 min of ischemia (60% delay for 153Na+/1.25Ca2+ vs. 145Na+/1.25Ca2+, hazard ratio = 1/6.94, P < 0.01). However, with elevated [Ca2+] (2 mM), increasing [Na+] did not significantly affect the time to conduction block.
From the traces in Fig. 2B, it is clear that pacing (CL = 300 ms) did not impinge on repolarization as there is a visualizable phase 4, with a period of full repolarization in the traces of transmembrane potential fluorescence. Instead, the development of unidirectional block appears to be associated with refractoriness due to reduced excitability. To investigate this more thoroughly, we next quantified repolarization at baseline and the time point at which unidirectional block first occurred. Figure 3A shows representative optical APs recorded at baseline and during ischemia with each of the perfusates. Paired APs recorded at the time of ischemia-induced conduction block are superimposed with those recorded from the same channel in the same heart during baseline (preischemia) conditions. At baseline, there were no apparent differences in APs, with no significant difference in APD90 among the perfusates tested (Fig. 3B, P = 0.95). The superimposed APs demonstrate that acute no-flow ischemia modestly shortened APD, independent of perfusate composition. Over all experiments, APD90 measured at the time of conduction block was significantly shorter than baseline APD90 for each perfusate (Fig. 3C, paired t test, #P < 0.05), with no significant differences among perfusates (ANOVA, P = 0.38). As a result, the change in APD90 (∆APD90, ischemia vs. baseline, Fig. 3D) was relatively uniform (7–16% change), with no significant differences among perfusates (P = 0.72). Furthermore, APD dispersion (standard deviation of APD90 within the field of view) was unaffected by perfusate composition at baseline or the time of conduction block (ANOVA, P = 0.28 and P = 0.23, respectively). In short, while acute ischemia was associated with APD shortening, the data do not present a clear relationship between perfusate composition and APD changes that would explain the onset of conduction block.
Fig. 3.
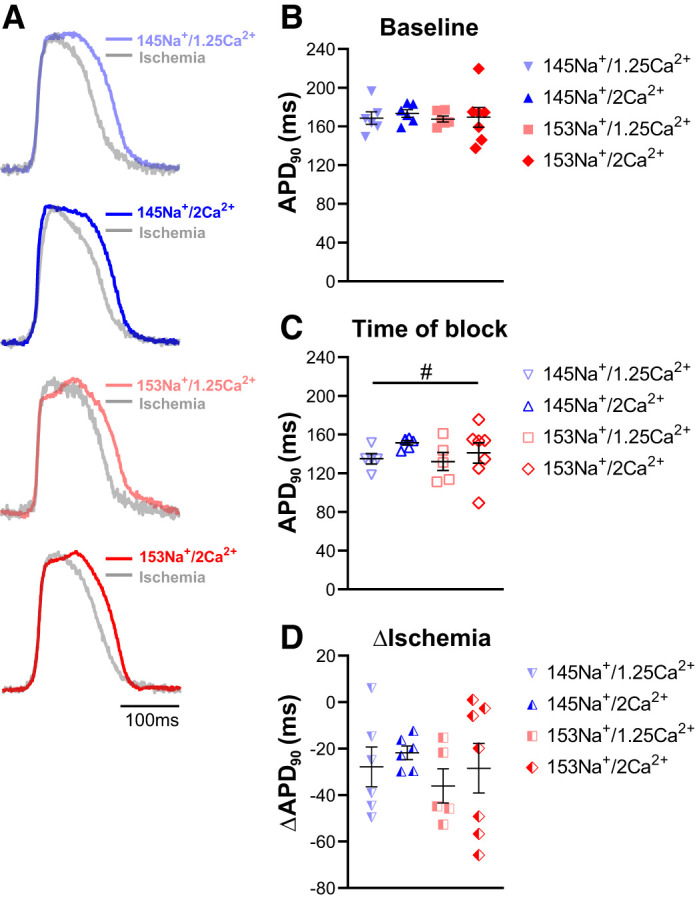
Perfusate differences in conduction block are not associated with differences in repolarization. A: representative optical action potentials (APs) recorded at baseline (bold color traces) and at the time of conduction block (light gray traces) for each perfusate, superimposed to show the effect of ischemia on repolarization. Paired traces are from the same recording site before and during ischemia; time scale = 100 ms. B: summary data demonstrating no significant differences in baseline AP duration at 90% repolarization (APD90) among the perfusate groups (ANOVA, P = 0.95). C: at the onset of conduction block, no significant differences in APD90 were observed among the perfusates (ANOVA, P = 0.38). Ischemic APD90 was significantly shorter than baseline for each perfusate (paired t test, #P < 0.05). D: summary data for ∆APD90 (ischemia – baseline APD90) show no perfusate differences in APD shortening at the time of conduction block (ANOVA, P = 0.72); n = 6, 6, 6, and 7 for 145Na+/1.25Ca2+, 145Na+/2Ca2+, 153Na+/1.25Ca2+, and 153Na+/2Ca2+, respectively. Summary data presented as individual values with means ± SE.
Sensitivity of Ischemic Conduction Velocity to [Na+] and [Ca2+]
We next sought to assess the effect of perfusate composition on ischemia-induced changes in CV. Representative activation isochrone maps (Fig. 4A) generated from APs recorded during acute ischemia (8 min), before the onset of conduction block, show dramatic conduction slowing relative to baseline (preischemia, Fig. 1B) in hearts perfused with 145Na+/1.25Ca2+. In contrast to 145Na+/1.25Ca2+, at 8 min of ischemia, conduction slowing was subtler in hearts perfused with 145Na+/2Ca2+ and 153Na+/1.25Ca2+, while CV slowed the least with 153Na+/2Ca2+. CV (CVT, CVL, and AR) was measured up to the time point of conduction block, and summary data are plotted in Fig. 4B; note that the data have been truncated at 14 min of ischemia as a result of data dropout due to the onset of conduction block. In general, ischemia was associated with CV slowing, but relative to 145Na+/1.25Ca2+, there were no significant differences in CVT, CVL, or AR among the perfusates at any time during ischemia (Fig. 4B, left; multiple t tests, P > 0.05). However, when CV during ischemia was normalized to the respective baseline value (Fig. 4B, right), hearts perfused with 145Na+/1.25Ca2+, 145Na+/2Ca2+, and 153Na+/1.25Ca2+ each demonstrated a time-dependent decrease in CVT and CVL (simple linear regression, P < 0.01). In contrast, hearts perfused with 153Na+/2Ca2+ failed to demonstrate a significant decrease in CVT or CVL during acute ischemia (P = 0.12 and P = 0.89, respectively). There were no significant time-dependent changes in AR in any of the perfusates tested.
Fig. 4.

Increased [Na+] and [Ca2+] selectively preserves conduction during acute no-flow ischemia. A: representative isochrone maps of action potential activation time at 8 min of ischemia (before the onset of conduction block) demonstrating conduction differences between the perfusates tested. Isochrones represent ∆3 ms; scale bar = 5 mm. B: summary data (n = 6, 6, 6, and 7 for 145Na+/1.25Ca2+, 145Na+/2Ca2+, 153Na+/1.25Ca2+, and 153Na+/2Ca2+, respectively) for transverse CV (CVT, top), longitudinal CV (CVL, middle), and anisotropy ratio (AR, bottom); summary data are presented as means ± SE (left, #P < 0.05 vs. 145Na+/1.25Ca2+ at the indicated time point), and as a percent change of baseline (right; simple linear regression testing for a time-dependent CV change, *P < 0.01). C: least squares means (LS Means) model of CVT (top) and CVL (bottom) slowing during ischemia (presented as estimated value with 95% confidence interval); *adjusted P < 0.05 for slope vs. 145Na+/1.25Ca2+.
To further characterize the conduction slowing during ischemia and account for data drop out at later time points due to the onset of conduction block, we used a least squares means model (Fig. 4C) to test for perfusate-dependent effects on ischemic conduction. The model revealed a significantly nonzero slope for CVT and CVL with each of the four perfusates (see Table 2 for slope values). Relative to 145Na+/1.25Ca2+, hearts perfused with 145Na+/2Ca2+ had a significantly less negative slope for CVT (P < 0.01), but not for CVL (P = 0.31). For both CVT and CVL, the slopes for 153Na+/1.25Ca2+ and 153Na+/2Ca2+ were all statistically less negative than the slope for 145Na+/1.25Ca2+ (P < 0.02), with the least negative slope (i.e., the least degree of CV slowing) reported in 153Na+/2Ca2+ hearts (P < 0.05). To assess the relative impact of changes in [Na+] and [Ca2+] on CV slowing during ischemia, we used the least squares model to determine the overall [Na+] and [Ca2+] effects, with combined [Ca2+] and [Na+], respectively. The model found both a significant [Na+] and [Ca2+] effect on CVT (P < 0.02), only an effect of [Na+] on CVL (P < 0.01), and no [Na+]-[Ca2+] interaction for either. Hence, the CV slope for 153 mM [Na+] was less negative than 145 mM [Na+], and the CVT slope for 2 mM [Ca2+] was less negative than 1.25 mM [Ca2+], indicating that both elevating [Na+] and [Ca2+] were associated with attenuated CV slowing during ischemia. Thus, the data and model both show that preischemic perfusion with 153Na+/2Ca2+ selectively preserves CV during acute, no-flow ischemia.
Table 2.
Estimated CV slope values from LS Means model
| Name | CVT | P Value | CVL | P Value |
|---|---|---|---|---|
| 145Na+/1.25Ca2+ | −1.12 | — | −2.46 | — |
| 145Na+/2Ca2+ | −0.76* | 0.01 | −1.93 | 0.31 |
| 153Na+/1.25Ca2+ | −0.79* | 0.01 | −1.37* | <0.001 |
| 153Na+/2Ca2+ | −0.66* | <0.001 | −0.96* | <0.001 |
Conduction velocity (CV) slope values [in (cm/s)/min of ischemia] estimated from the lease-squares means model; differences in slope compared with 145Na+/1.25Ca2+ with Tukey’s correction. CVT and CVL, transverse and longitudinal CV, respectively.
P < 0.05.
Connexin43 Protein Expression During Acute No-Flow Ischemia
To assess whether effects of perfusate changes on GJC could account for the observed changes in conduction during acute no-flow ischemia, we investigated whether there were perfusate-dependent differences in expression and phosphorylation of the ventricular GJ protein connexin43 (Cx43). In a separate set of studies, tissue samples were obtained at either 10 min of no-flow ischemia (n = 6 hearts/perfusate) or 10 min of nonischemic time control (baseline flow rate, n = 6 hearts/perfusate). We did not expect to observe significant changes in Cx43 at this time point, as it proceeds most ischemia-induced GJ remodeling and the onset of irreversible cell damage (2, 6, 10, 60, 61, 67, 71). Furthermore, by 10 min of ischemia, we observed significant CV slowing (Fig. 4) without the significant drop out of CV data due to conduction block (median time to conduction block was ≥10 min, Fig. 2B) at later time points. To rule out the effect of ischemia-induced changes in Cx43 on acute conduction slowing, samples were probed for total Cx43 expression and phosphorylation at Cx43-Ser368 (pCx43) (Fig. 5A). Total Cx43 expression was normalized to α-tubulin expression and pCx43 expression was normalized to total Cx43 expression; the results are presented as fold change relative to 145Na+/1.25Ca2+ time control (Fig. 5, B and C). The representative Western blot (Fig. 5A) and quantification of Cx43 and pCx43 (Fig. 5, B and C) indicate that Cx43 expression and phosphorylation at Ser368 were not significantly altered by perfusate composition (P = 0.63 and P = 0.50, respectively), nor did they significantly change during acute (10 min) ischemia (ischemia vs. time control; P = 0.47 and P = 0.25, respectively). These data suggest that the observed perfusate differences in conduction slowing precede measurable changes in Cx43 expression or phosphorylation status of Cx43-Ser368.
Fig. 5.

Expression of total and phosphorylated Cx43 is unaffected by perfusate composition during acute no-flow ischemia. A: Western blots of phosphorylated Cx43-Ser368 (pCx43), total Cx43, and α-tubulin (loading control) at 10 min of ischemia (+) or nonischemic time control (−) for each of the four perfusate groups with molecular mass markers (arrows) indicated to the right of the blots. Summary data for expression of total Cx43 protein normalized to loading control (B) and pCx43 normalized to total Cx43 (C). Summary data presented as individual values and means ± SE with fold change relative to 145Na+/1.25Ca2+ time control. No significant differences among perfusates or treatment groups detected by ANOVA; n = 6 for each perfusate and condition.
Change of Perinexal Structure During Acute No-Flow Ischemia
Previously, we reported that increasing perfusate [Ca2+] can decrease Wp and that structural changes in the perinexus are associated with changes in conduction via effects on EpC (22, 24, 26, 74). To determine whether changes in EpC contributed to the observed changes in conduction during ischemia, intermembrane separation was quantified from transmission electron micrographs of the GJ-adjacent perinexal nanodomain. The purpose of these measurements was to determine if there were baseline (preischemia) differences in Wp among the perfusates tested or if they affected changes in Wp at 10 min of ischemia, a time point at which perfusate-dependent differences in CV were observed. Representative TEM images of a GJ and the adjacent perinexus nanodomain for each perfusate under nonischemic (time control, top) and ischemic (bottom) conditions are shown in Fig. 6A. The differences in perinexal structure among the time control images were subtle. Strikingly, the perinexus within the 153Na+/2Ca2+ group significantly expanded during ischemia. Summary data for Wp are shown in Fig. 6, B–D. A mixed model determined the interactions between perfusate composition and intervention (time control or ischemia). Among time controls, there was a significant main effect of [Ca2+] (Fig. 6B, P < 0.01), such that elevated [Ca2+] (i.e., 145Na+/2Ca2+ and 153Na+/2Ca2+) had narrower Wp than 1.25 mM [Ca2+], which is consistent with our previous observations (22, 24). More specifically, the rank order of means for nonischemic Wp was (from narrowest to widest): 153Na+/2Ca2+, 145Na+/2Ca2+, 153Na+/1.25Ca2+, and 145Na+/1.25Ca2+ (P < 0.01). For all perfusates combined, mean Wp from ischemic samples was wider than mean Wp from nonischemic time controls (Fig. 6C, P < 0.02). However, when evaluating individual perfusate compositions, ischemia-associated widening of Wp only reached significance for 153Na+/2Ca2+ (Fig. 6D, P < 0.05). Thus, similar to our previous findings during simulated metabolic ischemia (24), selective widening of the perinexus during acute ischemia was associated with attenuated conduction slowing.
Fig. 6.

Increased [Na+] and [Ca2+] is associated with perinexal expansion during acute no-flow ischemia. A: representative transmission electron micrographs of the gap junction-adjacent perinexus nanodomain (shaded yellow) for each perfusate under nonischemic (time control) and ischemic conditions. The images demonstrate perinexal expansion [increased perinexal width (Wp)] induced by 10 min of no-flow ischemia in hearts perfused with 153Na+/2Ca2+; scale bar = 100 nm. Differences in Wp were evaluated using a mixed-effects linear model (B–D); note: y-axes plotted on natural log scale for transformed data (see methods). B: violin plots of individual data points and summary data (means ± SE) for Wp in nonischemic time controls as a function of [Ca2+] (n = 6 hearts each, ≥15 perinexal images per heart). Main effect for [Ca2+] demonstrating that mean Wp decreased in perfusates containing 2 vs. 1.25 mM [Ca2+] (black data points with gray bars vs. gray data points with black bars, respectively; *P < 0.01). C: violin plots of individual data points with summary data (means ± SE) for Wp at 10 min of ischemia (Isch, gray data points with black bars) or nonischemic time control (TC, black data points with gray bars) (n = 6 hearts each, ≥15 perinexal images per heart). For all perfusate groups combined, mean Wp was greater in Isch vs. TC, *P < 0.05. D: individual data points and summary data (means ± SE) for WP by individual perfusate composition (n = 3 hearts for each perfusate and condition, ≥15 perinexal images per heart). For individual perfusates, perinexal expansion was only significant for 153Na+/2Ca2+ (*P < 0.05 vs. nonischemic time control by Tukey’s honestly significant difference test).
DISCUSSION
In the present study, we describe the effects of modulating perfusate sodium ([Na+]) and calcium ([Ca2+]) concentrations on electrophysiological behavior before and during no-flow ischemia in Langendorff-perfused guinea pig hearts. Consistent with our previous studies, modest differences in perfusate ionic composition had minimal effects on baseline conduction. We speculate that in isolation, these small perfusate changes result in mild perturbations of EpC with corresponding subtle or concealed effects on conduction that could possibly fall below our resolution to detect. The effects of perfusate composition on conduction are unmasked in the presence of additional stressors, e.g., loss of Cx43, GJ uncoupling, inflammation, or ischemia (20, 23–26). In this study, we hypothesized that perfusate composition(s) facilitating perinexal expansion during no-flow ischemia would preserve conduction. The data support this hypothesis but also suggest a surprisingly more complex relationship between excitability, CV, [Na+], and perinexal expansion modulated by [Ca2+]. Specifically, elevating [Na+] from 145 to 153 mM increased preischemic excitability (reduced pacing threshold) and delayed ischemia-induced conduction block. Elevating [Ca2+] from 1.25 to 2 mM reduced preischemic excitability (increased pacing threshold), decreased perinexal membrane separation (perinexal width, Wp), and attenuated ischemia-induced conduction slowing in the presence of 153 mM, but not 145 mM, [Na+]. Importantly, the data suggest that ischemia-induced perinexal expansion may be an acute, adaptive intercellular response that preserves conduction during the onset of global ischemia.
Recently, we provided evidence that small changes in perfusate ionic composition can significantly affect conduction in isolated guinea pig hearts subjected to simulated metabolic ischemia (24). In earlier studies we found that narrow perinexi enhanced CV, whereas wider perinexi were associated with conduction slowing and/or arrhythmias in animal models of compromised GJC, ID dehiscence, or inflammation (23, 26, 73). In contrast, we found that perinexal expansion was antiarrhythmic during simulated metabolic ischemia with continuous flow. However, it remained unknown whether these findings were broadly applicable or instead specific to the simulated metabolic ischemia model with continuous flow maintaining a more controlled extracellular milieu and normal intravascular tone. The present study investigated how changes in [Na+] and [Ca2+] induce perinexal modulation during no-flow ischemia, a model which more faithfully incorporates key features of the acute onset of ischemia, including the accumulation/depletion of extra- and intracellular ionic concentrations and metabolites and the loss of intravascular pressure with concomitant effects on tissue hydration levels.
Excitability vs. Conduction
Cell excitability has been defined as the “ease with which a response may be triggered” (8). Functionally speaking, it is often expressed as the minimum stimulus required to depolarize the membrane to the threshold voltage and elicit an action potential (AP). Membrane excitability is a critical determinant of impulse propagation and CV in cardiac tissue (8, 63, 82), and reductions in either are well-established arrhythmia mechanisms. Though they share common molecular and structural determinants (e.g., sodium channel availability), excitability and conduction are distinct phenomena, as epitomized by the concept of slow but safe conduction, i.e., safety factor (70).
In the present study, tissue excitability was quantified at baseline as the stimulation threshold (in mA of applied current) for external pacing with a fixed pulse width, acknowledging that the parameter could be quantified more robustly with strength-duration curves. Of the electrophysiologically relevant cations, the effects of potassium concentration ([K+]) on excitability are perhaps the most widely studied. For example, elevating extracellular [K+] increases resting membrane potential (RMP), which initially increases excitability by reducing the potential difference between RMP and the threshold voltage, but subsequently decreases excitability via progressive inactivation of sodium channels (42, 63). The biphasic response of excitability to [K+] is paralleled by the biphasic response of conduction to [K+] (15, 42).
Despite the dependence of excitability on Na+ current (INa) availability and voltage-dependent activation of INa, the relationship between physiological levels of [Na+] and tissue excitability and conduction is not well understood. A significant electrophysiological effect of increasing [Na+] is to increase the reversal potential for Na+ (ENa) and the driving force of INa, which is most relevant to active depolarization. While we report [Na+] affected baseline stimulus threshold over the investigated concentrations, the effect of changing [Ca2+] from 1.25 to 2.0 mM was greater.
Previous reports demonstrated that increasing extracellular [Ca2+] to either 5 or 10 mM reduces excitability (63, 82). According to Brown and Noble, a 400% increase in extracellular [Ca2+] was required to increase the sodium channel activation threshold by 5 to 6 mV (9). Pressler and Baily found a similar result of a 14-mV shift in the all-or-nothing activation threshold with a 400% increase in extracellular [Ca2+] (64). The Peon, Ferrier, and Moe study, while not adequately powered (n = 3 per group), might suggest that threshold current is modestly increased by 10% or less when increasing extracellular [Ca2+] by 100% (1.25 to 2.5 mM) (63). To our knowledge, this is the first report demonstrating that increasing [Ca2+] from 1.25 to 2.0 mM (60% increase) can affect tissue excitability. However, the current results would be consistent with reduced excitability due to increased intracellular [Ca2+] subsequent to elevated extracellular [Ca2+].
Another possible mechanism of hypercalcemia reducing excitability may be linked to the pathway of calcium-induced modulation of INa; however, sodium channel inhibition with flecainide does not alter extracellularly mediated measures of excitability. Thus, this mechanism may be incompletely understood (3). Alternatively, the ID densely expresses sodium channels and is two orders of magnitude narrower than the axial separation of myocytes. Thus, it is possible that tissue excitability is modulated by postulated differences between bulk extracellular potentials and those within narrower ID nanodomains via a voltage divider effect.
Ischemic Excitability, Conduction, and Block
Ischemia is associated with increased extracellular [K+] accumulation, acidosis, a rise in RMP, and eventual GJ uncoupling (6, 11, 40, 50, 62). Interestingly, CV has been reported to initially increase in the first 2 min of ischemia and then decrease thereafter (37, 40). This behavior has been attributed to the temporal rise in extracellular [K+] and the associated effects on RMP and diastolic threshold (RMP approaching the threshold potential) as discussed above. The subsequent early depression of CV during ischemia is thought to be related to further [K+]-induced depolarization of RMP, thereby inactivating INa, combined with reduced pH leading to reduced sodium channel availability (decreased excitability) (40, 42, 50, 85).
This highlights a critical difference between our previous and current ischemia models, as the switch in perfusate [K+] used in the simulated ischemia model resulted in an abrupt and fixed elevation of extracellular [K+] (24). In the present study, we wanted to investigate whether our previous observations on the relationship between perinexal expansion and conduction were dependent on a constant extracellular milieu during ischemia. Our results demonstrate that 153Na+/2Ca2+ attenuated conduction slowing more than other ionic concentrations, and further studies are necessary to determine if pH is significantly affected by preischemia perfusates. As such, the consistency of the present results with our previous results from the simulated ischemia model suggests that the association between perinexal expansion and attenuated conduction slowing during acute ischemia are not solely due to disparate temporal changes in extracellular [K+] or the associated changes in RMP (24). However, we did not measure extracellular [K+] or RMP in this study; therefore, we cannot rule out the involvement of these mechanisms.
Exacerbating intracellular [Ca2+] overload enhances conduction slowing during ischemia, and alleviating [Ca2+] overload by inhibiting the L-type calcium channel with verapamil attenuates this conduction loss (35, 41, 59). Given that increased extracellular [Ca2+] will lead to increased intracellular [Ca2+], our finding that increased extracellular [Ca2+] (under conditions of increased extracellular [Na+]) attenuates conduction slowing during ischemia is seemingly inconsistent with these previous studies. Likewise, reduced extracellular [Na+] should also lead to increased intracellular [Ca2+] (1, 52, 84). Accordingly, one might expect that the perfusate with the lowest [Na+] and highest [Ca2+], i.e., 145Na+/2Ca2+, should produce the most severe conduction slowing during ischemia. Since this is not what we observed, factors beyond intracellular [Ca2+] and extracellular [K+] accumulation may be important for modulating conduction during ischemia.
Interestingly, preservation of CV and conduction block were not consistent among perfusates. Conduction block during ischemia correlated with preischemic measures of excitability more so than CV. The data support the concept that excitability and CV rely on similar proteins and electrophysiological pathways, but nevertheless are distinct mechanisms that can be regulated independently from one another. One potential explanation is that excitability was determined by tissue response to an extracellularly induced electric field that may predominantly affect sarcolemmal ion channels as suggested above. Conduction, on the other hand, is a myocyte generated phenomenon that will interact differently with discrete cellular domains according to protein distributions, restricted extracellular volumes (80), and local molecular control.
Conduction Block and Repolarization
Electrophysiological refractoriness is a well-established mechanism of conduction block that can be independent of steady-state conduction (16–18). If increased refractory periods were a mechanism of conduction block in this model, one might expect APD prolongation, as an index of post-repolarization refractoriness, with specific interventions. Interestingly, while APD shortening is considered a hallmark of ischemia, APD does not always shorten acutely (11, 44, 51, 72), and our present and previous results suggest that ionic composition and model of ischemia may affect the time course and extent of APD shortening due to ischemia (24). Regardless, at the time of ischemic conduction block, we generally observed modest APD shortening (16% or less) with all perfusates, which did not appear to be associated with the development of conduction block. Therefore, the increased risk of post-refractory associated conduction block observed in the ischemic guinea pig heart is likely independent of APD and requires further investigation.
Ischemic Conduction and Electrical Coupling
GJs are generally accepted to mediate cell-to-cell communication and cardiac conduction, although the majority of studies that find a correlation conclude that reduced functional GJC can sustain very slow but safe conduction. Regardless, we did not see significant changes in total or Ser368 phosphorylated Cx43 at 10 min of ischemia, consistent with previous work (4, 6, 60, 71). However, Cx43 can be posttranslationally modified in a number of ways, and the experimental design did not address these possibilities.
EpC, the cell-to-cell communication pathway parallel to GJC, suggests intercellular separation within the ID may be equally important to conduction. This and previous studies from our group suggest that ID volume modulation is a dynamic process, and like other biological processes, the conditions separating an adaptive from a pathophysiological response is nuanced. While interstitial volume collapse is a recognized feature of ischemia and is thought to play a key role in conduction slowing and contractile failure at the onset of ischemia (6, 51), the corresponding changes in ID volume are not yet as clear. Based on this no-flow ischemia study and our previous simulated metabolic ischemia study (24), perinexal expansion appears to be an adaptive response that supports electrical propagation during cardiac stress. Specific and unique to this study, hearts perfused with 153Na+/2Ca2+ preischemia experienced perinexal expansion and attenuated conduction slowing during the ischemic period.
The adaptive mechanism of preserved conduction during ischemia could be related to reduced ephaptic self-attenuation. Specifically, EpC occurs by the depletion of extracellular [Na+] during voltage-gated sodium channel activation, which raises intracellular potential, reduces extracellular potential, and reduces the Na+ reversal potential, all of which reduce the driving force of Na+. Under normal conditions, this mechanism may adaptively permit stable CV and uniform myocardial activation beat to beat. As sodium channel availability or excitability decreases, we hypothesize that reducing ephaptic self-attenuation adaptively preserves conduction, but this requires significantly more experimental evidence.
Limitations
The isolated heart preparation is experimentally tractable, but findings cannot be directly applied to in vivo conditions. Limitations of optical mapping in the Langendorff-perfused heart to study cardiac ischemia were previously discussed (24). Briefly, hearts were optically mapped with an electromechanical uncoupler that could modify calcium handling (45) and metabolic demand. Also, the isolated heart preparation lacks neurohumoral control, and perfusates are oversimplifications of blood, particularly if proteins or colloids are omitted. Isoflurane anesthesia is a known sodium channel antagonist and has been shown to be cardioprotective (38), and the effects may not completely wash out after isolation of the heart. Finally, di-4-ANEPPS can slow ventricular conduction (54).
Furthermore, in this study we did not correct the test perfusates for differences in osmolarity. Previously, we have shown that increased osmolarity can increase interstitial volume and slow CV (77), but these changes in osmolarity (116 mosmol/L, approximately a 40% change) were significantly greater than the maximal differences in osmolarity in the current study (17 mosmol/L, approximately a 5% change). With relatively small changes in osmolarity (<10%), the effects of perfusate composition on conduction did not correlate with perfusate osmolarity in this study or previous studies (24, 26). So, while the test perfusates do have small differences in osmolarity, these differences do not appear to be able to explain the differences in conduction slowing in response to ischemia. Finally, cardiac ischemia is a complex and multifactorial process. In addition to the factors discussed above, ischemia has also been associated with other significant changes including levels of inorganic phosphates, phosphocreatine, ATP, intracellular pH, mitochondrial and sarcoplasmic reticulum function, and intracellular resistance (51). It is not clear how these factors affect or are affected by differences in perfusate composition, but regardless, this study suggests that perfusate ion concentration can have a significant effect on conduction slowing during acute ischemia. The implication is that in ischemia studies using perfused cardiac tissue preparations, the choice of perfusate could affect results, interpretation, and data reproducibility if not properly reported or accounted for.
Perinexal measurements with TEM estimate Wp from a two-dimensional (2D) tissue section and do not fully describe the 3D structure of the perinexus. However, the spatial resolution of existing 3D electron microscopy methods [7–20 nm/pixel (55)] are insufficient to assess intercellular membrane separation on the scale of 10–20 nm. Additionally, the measure of Wp from Langendorff perfused and fixed tissue should not be interpreted to represent physiological values, but rather the relative and directional changes (wider, narrower) in Wp are the relevant parameters in our studies. Importantly, the 2D Wp measurement has proven to be consistently useful for structure-function investigations in cardiac myocardium (20, 22–26, 29, 66, 73, 74).
The major and significant difference between this and our previous study (24) is that flow was discontinued in this model. Distinct limitations when comparing the simulated metabolic ischemia and no-flow ischemia models may be that heart temperature decreases during no-flow ischemia, and importantly, the extracellular ionic milieu will likely respond differently without continuous perfusion.
Conclusions
Extracellular ionic composition and perinexal structure are critical determinants of ventricular conduction in response to cardiac ischemia. Accordingly, differences in perfusate composition could have significant implications for scientific reproducibility if not considered when interpreting experimental results. It could also contribute to the difficulty in translating promising preclinical results for antiarrhythmic therapies into clinical practice.
The data show that perinexal expansion can serve as an adaptive response during cardiac ischemia and mitigate conduction slowing. By modulating the effects of ischemia, acute adaptation of perinexal structure is a critical factor that should be considered in studies evaluating antiarrhythmic agents. Finally, it raises the question of whether optimized intravenous fluid ionic composition could be combined with drug therapies directed at GJC to further enhance cardioprotection during ischemia-reperfusion.
GRANTS
This work was supported by Heart Rhythm Society Fellowship, Mark Josephson and Hein Wellens Clinical 985 Research Award (to G.S.H.); and National Heart, Lung, and Blood Institute Grants F31 HL140909 (to C.C.J.), R01 HL132236 (to J.W.S.), R01 H156728 and R01 HL141855 to (R.G.G.), and R01 HL141855, R01 HL102298, and R01 HL138003 (to S.P.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.S.H. and S.P. conceived and designed research; G.S.H. and C.C.J. performed experiments; G.S.H., C.C.J., A.N.T., and J.W.S. analyzed data; G.S.H., C.C.J., A.N.T., J.W.S., and S.P. interpreted results of experiments; G.S.H. and C.C.J. prepared figures; G.S.H. drafted manuscript; G.S.H., C.C.J., A.N.T., R.G.G., J.W.S., and S.P. edited and revised manuscript; G.S.H., C.C.J., A.N.T., R.G.G., J.W.S., and S.P. approved final version of manuscript.
REFERENCES
- 1.Allen DG, Eisner DA, Lab MJ, Orchard CH. The effects of low sodium solutions on intracellular calcium concentration and tension in ferret ventricular muscle. J Physiol 345: 391–407, 1983. doi: 10.1113/jphysiol.1983.sp014984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ames A, Nesbett FB. Pathophysiology of ischemic cell death: I. Time of onset of irreversible damage; importance of the different components of the ischemic insult. Stroke 14: 219–226, 1983. doi: 10.1161/01.str.14.2.219. [DOI] [PubMed] [Google Scholar]
- 3.Arnsdorf MF, Sawicki GJ. Flecainide and the electrophysiologic matrix: the effects of flecainide acetate on the determinants of cardiac excitability in sheep Purkinje fibers. J Cardiovasc Electrophysiol 7: 1172–1182, 1996. doi: 10.1111/j.1540-8167.1996.tb00496.x. [DOI] [PubMed] [Google Scholar]
- 4.Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein-Rathlou NH, Andersen S, Jensen ON, Hennan JK, Kjølbye AL. Identification of ischemia-regulated phosphorylation sites in connexin43: a possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123). J Mol Cell Cardiol 40: 790–798, 2006. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng 45: 563–571, 1998. doi: 10.1109/10.668746. [DOI] [PubMed] [Google Scholar]
- 6.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kléber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 87: 656–662, 2000. doi: 10.1161/01.RES.87.8.656. [DOI] [PubMed] [Google Scholar]
- 7.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UK, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 137: e67–e492, 2018. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 8.Boyett MR, Jewell BR. Analysis of the effects of changes in rate and rhythm upon electrical activity in the heart. Prog Biophys Mol Biol 36: 1–52, 1980. doi: 10.1016/0079-6107(81)90003-1. [DOI] [PubMed] [Google Scholar]
- 9.Brown RH Jr, Noble D. Displacement of activator thresholds in cardiac muscle by protons and calcium ions. J Physiol 282: 333–343, 1978. doi: 10.1113/jphysiol.1978.sp012466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmeliet E. Myocardial ischemia: reversible and irreversible changes. Circulation 70: 149–151, 1984. doi: 10.1161/01.CIR.70.1.149. [DOI] [PubMed] [Google Scholar]
- 11.Cascio WE, Yan GX, Kléber AG. Passive electrical properties, mechanical activity, and extracellular potassium in arterially perfused and ischemic rabbit ventricular muscle. Effects of calcium entry blockade or hypocalcemia. Circ Res 66: 1461–1473, 1990. doi: 10.1161/01.RES.66.6.1461. [DOI] [PubMed] [Google Scholar]
- 12.Clusin WT, Buchbinder M, Ellis AK, Kernoff RS, Giacomini JC, Harrison DC. Reduction of ischemic depolarization by the calcium channel blocker diltiazem. Correlation with improvement of ventricular conduction and early arrhythmias in the dog. Circ Res 54: 10–20, 1984. doi: 10.1161/01.RES.54.1.10. [DOI] [PubMed] [Google Scholar]
- 13.De Vuyst E, Boengler K, Antoons G, Sipido KR, Schulz R, Leybaert L. Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br J Pharmacol 163: 469–483, 2011. doi: 10.1111/j.1476-5381.2011.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhein S, Hagen A, Jozwiak J, Dietze A, Garbade J, Barten M, Kostelka M, Mohr FW. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: the action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch Pharmacol 381: 221–234, 2010. doi: 10.1007/s00210-009-0473-1. [DOI] [PubMed] [Google Scholar]
- 15.Dominguez G, Fozzard HA. Influence of extracellular K+ concentration on cable properties and excitability of sheep cardiac Purkinje fibers. Circ Res 26: 565–574, 1970. doi: 10.1161/01.RES.26.5.565. [DOI] [PubMed] [Google Scholar]
- 16.Downar E, Janse MJ, Durrer D. The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation 56: 217–224, 1977. doi: 10.1161/01.CIR.56.2.217. [DOI] [PubMed] [Google Scholar]
- 17.El-Sherif N, Scherlag BJ, Lazarra R, Samet P. Pathophysiology of tachycardia- and bradycardia-dependent block in the canine proximal His-Purkinje system after acute myocardial ischemia. Am J Cardiol 33: 529–540, 1974. doi: 10.1016/0002-9149(74)90613-4. [DOI] [PubMed] [Google Scholar]
- 18.El Sherif N, Scherlag BJ, Lazzara R. Electroide cather recording during malignant ventricular arrythmia following experimental acute myocardial ischemia. Evidence for re-entry due to conduction delay and block in ischemic myocardium. Circulation 51: 1003–1014, 1975. doi: 10.1161/01.CIR.51.6.1003. [DOI] [PubMed] [Google Scholar]
- 19.Elharrar V, Gaum WE, Zipes DP. Effect of drugs on conduction delay and incidence of ventricular arrhythmias induced by acute coronary occlusion in dogs. Am J Cardiol 39: 544–549, 1977. doi: 10.1016/S0002-9149(77)80164-1. [DOI] [PubMed] [Google Scholar]
- 20.Entz M 2nd, George SA, Zeitz MJ, Raisch T, Smyth JW, Poelzing S. Heart rate and extracellular sodium and potassium modulation of gap junction mediated conduction in guinea pigs. Front Physiol 7: 16, 2016. doi: 10.3389/fphys.2016.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Entz M 2nd, King DR, Poelzing S. Design and validation of a tissue bath 3-D printed with PLA for optically mapping suspended whole heart preparations. Am J Physiol Heart Circ Physiol 313: H1190–H1198, 2017. doi: 10.1152/ajpheart.00150.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.George SA, Bonakdar M, Zeitz M, Davalos RV, Smyth JW, Poelzing S. Extracellular sodium dependence of the conduction velocity-calcium relationship: evidence of ephaptic self-attenuation. Am J Physiol Heart Circ Physiol 310: H1129–H1139, 2016. doi: 10.1152/ajpheart.00857.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.George SA, Calhoun PJ, Gourdie RG, Smyth JW, Poelzing S. TNFα modulates cardiac conduction by altering electrical coupling between myocytes. Front Physiol 8: 334, 2017. doi: 10.3389/fphys.2017.00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George SA, Hoeker G, Calhoun PJ, Entz M 2nd, Raisch TB, King DR, Khan M, Baker C, Gourdie RG, Smyth JW, Nielsen MS, Poelzing S. Modulating cardiac conduction during metabolic ischemia with perfusate sodium and calcium in guinea pig hearts. Am J Physiol Heart Circ Physiol 316: H849–H861, 2019. doi: 10.1152/ajpheart.00083.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.George SA, Poelzing S. Cardiac conduction in isolated hearts of genetically modified mice–Connexin43 and salts. Prog Biophys Mol Biol 120: 189–198, 2016. doi: 10.1016/j.pbiomolbio.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George SA, Sciuto KJ, Lin J, Salama ME, Keener JP, Gourdie RG, Poelzing S. Extracellular sodium and potassium levels modulate cardiac conduction in mice heterozygous null for the Connexin43 gene. Pflugers Arch 467: 2287–2297, 2015. doi: 10.1007/s00424-015-1698-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girouard SD, Laurita KR, Rosenbaum DS. Unique properties of cardiac action potentials recorded with voltage-sensitive dyes. J Cardiovasc Electrophysiol 7: 1024–1038, 1996. doi: 10.1111/j.1540-8167.1996.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 28.Girouard SD, Pastore JM, Laurita KR, Gregory KW, Rosenbaum DS. Optical mapping in a new guinea pig model of ventricular tachycardia reveals mechanisms for multiple wavelengths in a single reentrant circuit. Circulation 93: 603–613, 1996. doi: 10.1161/01.CIR.93.3.603. [DOI] [PubMed] [Google Scholar]
- 29.Greer-Short A, George SA, Poelzing S, Weinberg SH. Revealing the concealed nature of long-QT type 3 syndrome. Circ Arrhythm Electrophysiol 10: e004400, 2017. doi: 10.1161/CIRCEP.116.004400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris AS, Bisteni A, Russell RA, Brigham JC, Firestone JE. Excitatory factors in ventricular tachycardia resulting from myocardial ischemia; potassium a major excitant. Science 119: 200–203, 1954. doi: 10.1126/science.119.3085.200. [DOI] [PubMed] [Google Scholar]
- 31.Harris AS, Estandia A, Tillotson RF. Ventricular ectopic rhythms and ventricular fibrillation following cardiac sympathectomy and coronary occlusion. Am J Physiol 165: 505–512, 1951. doi: 10.1152/ajplegacy.1951.165.3.505. [DOI] [PubMed] [Google Scholar]
- 32.Hearse DJ. Myocardial ischaemia: can we agree on a definition for the 21st century? Cardiovasc Res 28: 1737–1744, 1994. doi: 10.1093/cvr/28.12.1737. [DOI] [PubMed] [Google Scholar]
- 33.Hichri E, Abriel H, Kucera JP. Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J Physiol 596: 563–589, 2018. doi: 10.1113/JP275351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation 61: 768–778, 1980. doi: 10.1161/01.CIR.61.4.768. [DOI] [PubMed] [Google Scholar]
- 35.Hiramatsu Y, Buchanan JW Jr, Knisley SB, Koch GG, Kropp S, Gettes LS. Influence of rate-dependent cellular uncoupling on conduction change during simulated ischemia in guinea pig papillary muscles: effect of verapamil. Circ Res 65: 95–102, 1989. doi: 10.1161/01.RES.65.1.95. [DOI] [PubMed] [Google Scholar]
- 36.Hoeker GS, Skarsfeldt MA, Jespersen T, Poelzing S. Electrophysiologic effects of the IK1 inhibitor PA-6 are modulated by extracellular potassium in isolated guinea pig hearts. Physiol Rep 5: e13120, 2017. doi: 10.14814/phy2.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holland RP, Brooks H. The QRS complex during myocardial ischemia. An experimental analysis in the porcine heart. J Clin Invest 57: 541–550, 1976. doi: 10.1172/JCI108309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ismaeil MS, Tkachenko I, Gamperl AK, Hickey RF, Cason BA. Mechanisms of isoflurane-induced myocardial preconditioning in rabbits. Anesthesiology 90: 812–821, 1999. doi: 10.1097/00000542-199903000-00024. [DOI] [PubMed] [Google Scholar]
- 39.Jæger KH, Edwards AG, McCulloch A, Tveito A. Properties of cardiac conduction in a cell-based computational model. PLOS Comput Biol 15: e1007042, 2019. doi: 10.1371/journal.pcbi.1007042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janse MJ, Kléber AG. Electrophysiological changes and ventricular arrhythmias in the early phase of regional myocardial ischemia. Circ Res 49: 1069–1081, 1981. doi: 10.1161/01.RES.49.5.1069. [DOI] [PubMed] [Google Scholar]
- 41.Kabell G. Modulation of conduction slowing in ischemic rabbit myocardium by calcium-channel activation and blockade. Circulation 77: 1385–1394, 1988. doi: 10.1161/01.CIR.77.6.1385. [DOI] [PubMed] [Google Scholar]
- 42.Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res 51: 614–623, 1982. doi: 10.1161/01.RES.51.5.614. [DOI] [PubMed] [Google Scholar]
- 43.Kaplinsky E, Ogawa S, Balke CW, Dreifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation 60: 397–403, 1979. doi: 10.1161/01.CIR.60.2.397. [DOI] [PubMed] [Google Scholar]
- 44.Kenigsberg DN, Khanal S, Kowalski M, Krishnan SC. Prolongation of the QTc interval is seen uniformly during early transmural ischemia. J Am Coll Cardiol 49: 1299–1305, 2007. doi: 10.1016/j.jacc.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 45.Kettlewell S, Walker NL, Cobbe SM, Burton FL, Smith GL. The electrophysiological and mechanical effects of 2,3-butane-dione monoxime and cytochalasin-D in the Langendorff perfused rabbit heart. Exp Physiol 89: 163–172, 2004. doi: 10.1113/expphysiol.2003.026732. [DOI] [PubMed] [Google Scholar]
- 46.Kihara Y, Grossman W, Morgan JP. Direct measurement of changes in intracellular calcium transients during hypoxia, ischemia, and reperfusion of the intact mammalian heart. Circ Res 65: 1029–1044, 1989. doi: 10.1161/01.RES.65.4.1029. [DOI] [PubMed] [Google Scholar]
- 47.Kimura S, Bassett AL, Kohya T, Kozlovskis PL, Myerburg RJ. Regional effects of verapamil on recovery of excitability and conduction time in experimental ischemia. Circulation 76: 1146–1154, 1987. doi: 10.1161/01.CIR.76.5.1146. [DOI] [PubMed] [Google Scholar]
- 48.Kjølbye AL, Haugan K, Hennan JK, Petersen JS. Pharmacological modulation of gap junction function with the novel compound rotigaptide: a promising new principle for prevention of arrhythmias. Basic Clin Pharmacol Toxicol 101: 215–230, 2007. doi: 10.1111/j.1742-7843.2007.00123.x. [DOI] [PubMed] [Google Scholar]
- 49.Kléber AG, Janse MJ, Wilms-Schopmann FJ, Wilde AA, Coronel R. Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation 73: 189–198, 1986. doi: 10.1161/01.CIR.73.1.189. [DOI] [PubMed] [Google Scholar]
- 50.Kléber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res 61: 271–279, 1987. doi: 10.1161/01.RES.61.2.271. [DOI] [PubMed] [Google Scholar]
- 51.Koretsune Y, Corretti MC, Kusuoka H, Marban E. Mechanism of early ischemic contractile failure. Inexcitability, metabolite accumulation, or vascular collapse? Circ Res 68: 255–262, 1991. doi: 10.1161/01.RES.68.1.255. [DOI] [PubMed] [Google Scholar]
- 52.Kozeny GA, Murdock DK, Euler DE, Hano JE, Scanlon PJ, Bansal VK, Vertuno LL. In vivo effects of acute changes in osmolality and sodium concentration on myocardial contractility. Am Heart J 109: 290–296, 1985. doi: 10.1016/0002-8703(85)90596-4. [DOI] [PubMed] [Google Scholar]
- 53.Laird DW, Lampe PD. Therapeutic strategies targeting connexins. Nat Rev Drug Discov 17: 905–921, 2018. doi: 10.1038/nrd.2018.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larsen AP, Sciuto KJ, Moreno AP, Poelzing S. The voltage-sensitive dye di-4-ANEPPS slows conduction velocity in isolated guinea pig hearts. Heart Rhythm 9: 1493–1500, 2012. doi: 10.1016/j.hrthm.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, Keegan S, Lin X, Arcos T, Feng-Xia-Liang, Korchev YE, Gorelik J, Fenyö D, Rothenberg E, Rothenberg E, Delmar M. Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun 7: 10342, 2016. doi: 10.1038/ncomms10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin J, Keener JP. Modeling electrical activity of myocardial cells incorporating the effects of ephaptic coupling. Proc Natl Acad Sci USA 107: 20935–20940, 2010. doi: 10.1073/pnas.1010154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin J, Keener JP. Ephaptic coupling in cardiac myocytes. IEEE Trans Biomed Eng 60: 576–582, 2013. doi: 10.1109/TBME.2012.2226720. [DOI] [PubMed] [Google Scholar]
- 58.Lin J, Keener JP. Microdomain effects on transverse cardiac propagation. Biophys J 106: 925–931, 2014. doi: 10.1016/j.bpj.2013.11.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maruyama T, Cascio WE, Knisley SB, Buchanan J, Gettes LS. Effects of ryanodine and BAY K 8644 on membrane properties and conduction during simulated ischemia. Am J Physiol 261: H2008–H2015, 1991. doi: 10.1152/ajpheart.1991.261.6.H2008. [DOI] [PubMed] [Google Scholar]
- 60.Miura T, Ohnuma Y, Kuno A, Tanno M, Ichikawa Y, Nakamura Y, Yano T, Miki T, Sakamoto J, Shimamoto K. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol 286: H214–H221, 2004. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- 61.Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res 66: 913–931, 1990. doi: 10.1161/01.RES.66.4.913. [DOI] [PubMed] [Google Scholar]
- 62.Owens LM, Fralix TA, Murphy E, Cascio WE, Gettes LS; Experimental Working Group . Correlation of ischemia-induced extracellular and intracellular ion changes to cell-to-cell electrical uncoupling in isolated blood-perfused rabbit hearts. Circulation 94: 10–13, 1996. doi: 10.1161/01.CIR.94.1.10. [DOI] [PubMed] [Google Scholar]
- 63.Peon J, Ferrier GR, Moe GK. The relationship of excitability to conduction velocity in canine Purkinje tissue. Circ Res 43: 125–135, 1978. doi: 10.1161/01.RES.43.1.125. [DOI] [PubMed] [Google Scholar]
- 64.Pressler ML, Elharrar V, Bailey JC. Effects of extracellular calcium ions, verapamil, and lanthanum on active and passive properties of canine cardiac purkinje fibers. Circ Res 51: 637–651, 1982. doi: 10.1161/01.RES.51.5.637. [DOI] [PubMed] [Google Scholar]
- 65.Raisch T, Khan M, Poelzing S. Quantifying intermembrane distances with serial image dilations. J Vis Exp 139: 2018. doi: 10.3791/58311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raisch TB, Yanoff MS, Larsen TR, Farooqui MA, King DR, Veeraraghavan R, Gourdie RG, Baker JW, Arnold WS, AlMahameed ST, Poelzing S. Intercalated disk extracellular nanodomain expansion in patients with atrial fibrillation. Front Physiol 9: 398, 2018. doi: 10.3389/fphys.2018.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reimer KA, Jennings RB, Hill ML. Total ischemia in dog hearts, in vitro 2. High energy phosphate depletion and associated defects in energy metabolism, cell volume regulation, and sarcolemmal integrity. Circ Res 49: 901–911, 1981. doi: 10.1161/01.RES.49.4.901. [DOI] [PubMed] [Google Scholar]
- 68.Rhett JM, Jourdan J, Gourdie RG. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol Biol Cell 22: 1516–1528, 2011. doi: 10.1091/mbc.e10-06-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rhett JM, Veeraraghavan R, Poelzing S, Gourdie RG. The perinexus: sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc Med 23: 222–228, 2013. doi: 10.1016/j.tcm.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rudy Y. Lessons learned about slow discontinuous conduction from models of impulse propagation. J Electrocardiol 38, Suppl: 52–54, 2005. doi: 10.1016/j.jelectrocard.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 71.Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J 17: 1355–1357, 2003. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- 72.Spear JF, Michelson EL, Moore EN. Reduced space constant in slowly conducting regions of chronically infarcted canine myocardium. Circ Res 53: 176–185, 1983. doi: 10.1161/01.RES.53.2.176. [DOI] [PubMed] [Google Scholar]
- 73.Veeraraghavan R, Hoeker GS, Alvarez-Laviada A, Hoagland D, Wan X, King DR, Sanchez-Alonso J, Chen C, Jourdan J, Isom LL, Deschenes I, Smyth JW, Gorelik J, Poelzing S, Gourdie RG. The adhesion function of the sodium channel beta subunit (β1) contributes to cardiac action potential propagation. eLife 7: e37610, 2018. doi: 10.7554/eLife.37610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch 467: 2093–2105, 2015. doi: 10.1007/s00424-014-1675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veeraraghavan R, Lin J, Keener JP, Gourdie R, Poelzing S. Potassium channels in the Cx43 gap junction perinexus modulate ephaptic coupling: an experimental and modeling study. Pflugers Arch 468: 1651–1661, 2016. doi: 10.1007/s00424-016-1861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Veeraraghavan R, Poelzing S, Gourdie RG. Intercellular electrical communication in the heart: a new, active role for the intercalated disk. Cell Commun Adhes 21: 161–167, 2014. doi: 10.3109/15419061.2014.905932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veeraraghavan R, Salama ME, Poelzing S. Interstitial volume modulates the conduction velocity-gap junction relationship. Am J Physiol Heart Circ Physiol 302: H278–H286, 2012. doi: 10.1152/ajpheart.00868.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Venkatraman ES. clinfun: Clinical Trial Design and Data Analysis Functions. R package version 1.0.14. [Online]. 2017. https://cran.r-project.org/package=clinfun.
- 79.Vermeulen JT, Tan HL, Rademaker H, Schumacher CA, Loh P, Opthof T, Coronel R, Janse MJ. Electrophysiologic and extracellular ionic changes during acute ischemia in failing and normal rabbit myocardium. J Mol Cell Cardiol 28: 123–131, 1996. doi: 10.1006/jmcc.1996.0012. [DOI] [PubMed] [Google Scholar]
- 80.Vermij SH, Abriel H, Kucera JP. Modeling depolarization delay, sodium currents, and electrical potentials in cardiac transverse tubules. Front Physiol 10: 1487, 2019. doi: 10.3389/fphys.2019.01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wei N, Mori Y, Tolkacheva EG. The dual effect of ephaptic coupling on cardiac conduction with heterogeneous expression of connexin 43. J Theor Biol 397: 103–114, 2016. doi: 10.1016/j.jtbi.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 82.Weidmann S. Effects of calcium ions and local anesthetics on electrical properties of Purkinje fibres. J Physiol 129: 568–582, 1955. doi: 10.1113/jphysiol.1955.sp005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinberg SH. Ephaptic coupling rescues conduction failure in weakly coupled cardiac tissue with voltage-gated gap junctions. Chaos 27: 093908, 2017. doi: 10.1063/1.4999602. [DOI] [PubMed] [Google Scholar]
- 84.Wilbrandt W, Koller H. Die Calciumwirkung Am Froschherzen Als Funktion Des Ionengleichgewichts Zwischen Zellmembran Und Umgebung Helv Physiol Pharmacol Acta 6: 208–221, 1948. http://www.ncbi.nlm.nih.gov/pubmed/18874558. [PubMed] [Google Scholar]
- 85.Yatani A, Brown AM, Akaike N. Effect of extracellular pH on sodium current in isolated, single rat ventricular cells. J Membr Biol 78: 163–168, 1984. doi: 10.1007/BF01869203. [DOI] [PubMed] [Google Scholar]