

Abstract
Mechanisms that contribute to myocardial fibrosis, particularly in response to left ventricular pressure overload (LVPO), remain poorly defined. To test the hypothesis that a myocardial-specific profile of secreted factors is produced in response to PO, levels of 44 factors implicated in immune cell recruitment and function were assessed in a murine model of cardiac hypertrophy and compared with levels produced in a model of pulmonary fibrosis (PF). Mice subjected to PO were assessed at 1 and 4 wk. Protein from plasma, LV, lungs, and kidneys were analyzed by specific protein array analysis in parallel with protein from mice subjected to silica-instilled PF. Of the 44 factors assessed, 13 proteins were elevated in 1-wk PO myocardium, whereas 18 proteins were found increased in fibrotic lung. Eight of those increased in 1-wk LVPO were not found to be increased in fibrotic lungs (CCL-11, CCL-12, CCL-17, CCL-19, CCL-21, CCL-22, IL-16, and VEGF). Additionally, six factors were increased in plasma of 1-wk LVPO in the absence of increases in myocardial levels. In contrast, in mice with PF, no factors were found increased in plasma that were not elevated in lung tissue. Of those factors increased at 1 wk, only TIMP-1 remained elevated at 4 wk of LVPO. Immunohistochemistry of myocardial vasculature at 1 and 4 wk revealed similar amounts of total vasculature; however, evidence of activated endothelium was observed at 1 wk and, to a lesser extent, at 4 wk LVPO. In conclusion, PO myocardium generated a unique signature of cytokine expression versus that of fibrotic lung.
NEW & NOTEWORTHY Myocardial fibrosis and the resultant increases in myocardial stiffness represent pivotal consequences of chronic pressure overload (PO). In this study, cytokine profiles produced in a murine model of cardiac fibrosis induced by PO were compared with those produced in response to silica-induced lung fibrosis. A unique profile of cardiac tissue-specific and plasma-derived factors generated in response to PO are reported.
Keywords: cytokine, fibrosis, heart, inflammation, pressure overload, pulmonary
INTRODUCTION
Although left ventricular (LV) pressure overload (PO) induced by transverse aortic constriction (TAC) causes myocardial fibrosis, the underlying cellular mechanisms driving increases in collagen are poorly defined. Studies by Moore-Morris et al. (19) demonstrated a primary role for resident cardiac fibroblasts in the expression of type I collagen mRNA in response to left ventricular pressure overload (LVPO). However, recruitment of bone marrow-derived monocyte/macrophage populations to the myocardium has also been shown to contribute to extracellular matrix (ECM) accumulation in PO hearts of mice and man. For example, in mice with LVPO, antibody depletion or inhibition of the monocyte receptor critical for myocardial recruitment, CCR2, significantly reduced ECM remodeling (22). Using other murine models of diastolic dysfunction, Hulsmans et al. (11) reported that macrophage production of interleukin (IL)-10 is required for fibrotic deposition of collagen. This study also described myocardial macrophage expansion in human subjects with heart failure. Hence, knowledge of the factors that drive inflammatory cell recruitment, including monocytes, to the PO myocardium is of critical importance.
McDonald et al. (17) recently presented a time-dependent analysis of collagen deposition that coincided with expansion of the myocardial macrophage population in response to LVPO. In these studies, insoluble collagen deposition in LVPO myocardium was found to be significant at 2 wk, although increases in soluble collagen were initially detected at 1 wk of PO. Progressive increases in insoluble collagen were apparent up to 4 wk after LVPO and notably associated with elevated levels of myocardial macrophages. The increase in myocardial macrophages at 1 wk suggested an active organ-specific recruitment of circulating cells to LVPO myocardium. Furthermore, whether this profile for fibrotic LVPO is distinct from that of other fibrotic organs, such as fibrotic lung, is not yet characterized.
Increases in load placed on the heart, from conditions such as hypertension, are sensed by resident myocardial cells, including myocytes, fibroblasts, endothelial cells, and resident inflammatory cells. In PO, myocytes respond by undergoing addition of sarcomeres, leading to myocyte hypertrophy (10). Resident fibroblasts increase in number, as does ECM deposition. Both myocytes and fibroblasts, as well as resident immune cells, release soluble factors, such as cytokines and chemokines, that lead to recruitment of circulating cells to the myocardium (6). Soluble factors also induce endothelial cells to undergo changes in gene expression to generate activated endothelium, represented by increased levels of leukocyte adhesion receptors that facilitate recruitment of circulating inflammatory cells (12). Examples of activated endothelial adhesion receptors include E-selectin, intracellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) (1).
The myocardium is known to demonstrate several unique properties in terms of inflammatory responses (6, 15, 27). Therefore, in the present study, we sought to determine whether LVPO myocardium produced a unique cytokine profile that was organ specific at different times post-PO. To this end, plasma and tissue samples from two murine models of fibrosis were analyzed for protein expressin of 44 cytokine/chemokines to determine whether cardiac PO generated a distinct expression pattern versus a murine model of lung fibrosis induced by silica. A murine model of pulmonary fibrosis was selected for comparison, as lung is the organ predicted to be most affected by LVPO. Lungs are in closest proximity to and with immediate physiological ties to heart function. In addition to cytokine analysis, alterations in myocardial vasculature and the expression of activated endothelial adhesion molecules were also assessed at distinct times following induction of PO.
MATERIALS AND METHODS
Murine models of fibrosis: pressure overload and silica-instilled pulmonary fibrosis.
C57Bl/6 mice (bred in house, stock no. 002014, n = 58; Jackson Laboratories, Bar Harbor, ME), ∼3 mo of age, males and females, were randomized into groups as outlined in Fig. 1. Control mice (no intervention), sham operated, and mice subjected to transaortic constriction (TAC) were harvested at 1-wk and 4-wk time points. Mice underwent TAC surgery to induce LVPO, as previously described in Bradshaw et al. (5).
Fig. 1.

Consort diagram describing the animal groups used in the present study. Forty mice were used in the cardiac pressure-overload (PO) arm of the experiment, whereas 18 animals were used for the silica-induced lung fibrosis studies. Both male (M) and female (F) mice were used in each arm.
Silica-instilled pulmonary fibrosis (PF) was initiated as previously described (16). Briefly, mice were were intratracheally instilled with 6 mg of silica (Sigma-Aldrich, St. Louis, MO) suspended in 60 μl of sterile 0.9% saline solution. Eight weeks postinstillation, mice were anesthetized by isoflurane anesthesia and euthanized via bilateral thoracotomy, followed by exsanguination. The 8-wk time point was chosen based on previous experience with this model that indicated robust fibrosis at 8 wk. Samples from animals exhibiting a robust fibrotic response by gross inspection of lung tissue were analyzed. All animal procedures were reviewed and approved by the Ralph H. Johnson Veterans Affairs Institutional Animal Care and Use Committee.
Tissue processing.
Following euthanasia, individual left ventricles of heart, kidney, and lungs from each mouse were homogenized in 20 mM Tris·HCl, pH 7.5, and 150 mM NaCl with protease inhibitors (complete mini inhibitor cocktail; Roche) on ice for 1 h, and insoluble material was removed by centrifugation. Total protein per samples was assessed by BCA assay according to the manufacturer’s instructions (Thermo Fisher Scientific) (Supplemental Fig. S1; Supplemental Material for this article is available online at https://doi.org/10.6084/m9.figshare.12167760.v1). Blood from each mouse was also collected at termination and placed in a bullet tube with 2 mM EDTA and centrifuged for 10 min at 1,400 rpm. All biological samples were flash-frozen and stored at −80°C before array analysis.
Cytokine array analysis.
Plasma and tissue samples were subjected to array analysis using the murine cytokine/chemokine array 44-plex (MD-44; Eve Technologies Corp., Calgary, AB, Canada). A complete list of chemokine proteins assessed is provided in the Supplemental Material (Supplemental Table S1). Undiluted samples were assessed for cytokine/protein concentration and reported as picograms per milliliter.
Immunohistochemistry.
Myocardial sections were fixed in formalin and embedded in paraffin before sectioning. Picrosirius red staining was performed as previously described (5). For immunohistochemistry, following antigen retrieval, primary antibodies against CD31/platelet endothelial cell adhesion molecule-1 (PECAM-1) (clone: SZ31; SKU: DIA-310) or ICAM1 (LS-313412, lot. no. 62019) were incubated on tissue sections followed by horseradish peroxidase (HRP) anti-rat (Novus Biologicals)or HRP anti-mouse secondary (Vector Labortories, Inc.) antibodies. Sections were treated with a Tyramide Amplification Buffer (cat. no. 22027, lot. no. 19R1017), followed by TrueBlack Lipofuscin Autofluorescence Quencher (cat. no. 23007, lot. no.18T0712-1078131), and mounted with ProLong Gold antifade reagent with DAPI (REF: P36931; lot. no. 2097915). Stained sections were viewed on an Olympus DP80 microscope using fluorescent microscopy and images captured on an Olympus BX53 camera. Quantification of total blood vessels (PECAM+) was performed by adjusting the color threshold in ImageJ to indicate PECAM-positive vessel staining only for each region of interest (ROI), followed by conversion to 8-bit, binary images. ImageJ was then used to calculate PECAM+ vessel staining by total area per ROI and average size of stained vessel per ROI. PECAM staining was normalized to total nuclei (by DAPI) for each ROI. A total of five ROIs per heart sections from five separate mice were analyzed and quantified.
Statistics.
Data are reported as means ± SE (Table 1) and as medians ± interquartile ranges (IQR) (KaleidaGraph 4.5.3; Synergy Software, Reading, PA). Outliers are the values lower than Q1 − 1.5 × IQR and values larger than Q3 + 1.5 × IQR. To detect significant differences between all measured variables, we used a one-way ANOVA followed by Bonferroni t test for multiple comparisons. For those measured variables that had a nonnormal distribution, statistical differences were examined using Kruskall-Wallis H test, followed by the Dunn’s method for multiple comparisons (SigmaStat 4.0, Systat Software, Inc., San Jose, CA). A P value of <0.05 was considered significant.
Table 1.
Gravimetric measurements (mass)
| n | Body, g | Heart, mg | Left Ventricle, mg | Lung, mg | Kidney, mg | LV/Body, mg/g | Lung/Body, mg/g | Kidney/Body, mg/g | |
|---|---|---|---|---|---|---|---|---|---|
| Control | 16 | 26 ± 1 | 129 ± 6 | 87 ± 4 | 141 ± 4 | 157 ± 8 | 3.3 ± 0.1 | 5.5 ± 0.1 | 6.0 ± 0.1 |
| Sham | 16 | 24 ± 1 | 128 ± 6 | 87 ± 4 | 134 ± 4 | 153 ± 7 | 3.6 ± 0.1 | 5.7 ± 0.1 | 6.3 ± 0.2 |
| LVPO, 1 wk | 9 | 26 ± 1 | 184 ± 16*# | 137 ± 12*,# | 229 ± 35*# | 137 ± 6 | 6.0 ± 0.6*,# | 10.2 ± 1.8*,# | 5.3 ± 0.2*,# |
| LVPO, 4 wk | 8 | 24 ± 1 | 248 ± 15*# | 175 ± 9*,# | 372 ± 52*# | 138 ± 3 | 7.4 ± 0.5*,# | 15.7 ± 2.3*,# | 5.8 ± 0.2 |
| Silica | 10 | 33 ± 1*# | 152 ± 6 | 105 ± 4*,# | 248 ± 22*# | 183 ± 7*# | 3.1 ± 01. | 7.3 ± 0.5*,# | 5.5 ± 0.1# |
Values are means ± SE. LV, left ventricle; LVPO, left ventricular pressure overload.
P < 0.05 vs. control;
P < 0.05 vs. sham.
RESULTS
To address the hypothesis, study groups included mice with LVPO at 1 and 4 wk post-transverse aortic constriction (TAC) and mice with silica-induced pulmonary fibrosis (PF) (Fig. 1). The left ventricle (LV) and kidneys from mice with silica-induced PF were used to represent noninvolved tissue controls for LVPO myocardium. Gravimetric data (Table 1) from myocardial tissue of LVPO mice at both 1 and 4 wk indicated significantly increased LV and LV/body mass, demonstrating hypertrophic response to banding consistent with previous results from our and other laboratories (5, 17). LV/body mass was not significantly increased in mice with silica-induced fibrosis. The lungs of mice with silica-induced fibrosis demonstrated significant increases in mass versus sham and control mice, consistent with increased fibrosis and lung pathology. Lungs in mice with LVPO also demonstrated increases over control and sham values indicative of LVPO-induced increases in diastolic pressure.
As evidence of a significant fibrotic response, picrosirius red-stained images of 1-wk and 4-wk LVPO myocardium demonstrate a robust increase in insoluble collagen deposition versus sham (Fig. 2, A–C), as previously reported by our group and others (5, 9, 17). In addition, lungs from silica-treated mice also showed a notable increase in insoluble collagen versus control lungs 8 wk following induction of fibrosis, as previously reported (Fig. 2, D and E) (16). Hence, fibrotic deposition of collagen was present in the involved tissues: LV in LVPO and lungs in the silica model.
Fig. 2.

Picrosirius red (PSR)-stained images of tissue sections taken from left ventricle of sham (A), 1-wk left ventricular pressure overload (LVPO; B), and 4-wk LVPO (C), demonstrating increases in histochemical staining of collagen with LVPO. In the silica-induced lung fibrosis model, sections of lung from 8-wk silica mice (E) had robust staining of fibrillar collagen vs. control (D). Images are representative of n = 5 mice (n = 3 sections/mouse). Size bar, 50 μm.
The expression of 44 separate cytokines (a complete list of cytokines is provided in Supplemental Table S1) known to be involved in inflammatory cell activity and recruitment was interrogated in tissues from mice subjected to no intervention (control), sham operation, 1-wk LVPO, 4-wk LVPO, and 8 wk of silica-induced PF. Proteins were extracted from the LV of heart, lung, and kidney from 1- and 4-wk LVPO and from silica-induced PF and subjected to array analysis. Extracted protein concentrations (total protein) were found to be equivalent in each tissue (cardiac protein concentrations are shown in Supplemental Fig. S1).
Thirteen of the 44 inflammatory factors assayed were found to be elevated in 1-wk PO myocardium. Of these, eight cytokines were uniquely increased in 1-wk LVPO, and the remaining five were also found to be increased in fibrotic lung tissues (Fig. 3). Factors increased in myocardium at 1 wk were eotaxin [CC ligand (CCL)-11], CCL 2, CCL12, CCL17, CCL20, CCL21, CCL22, CXC ligand (CXCL)-9, CXCL-10, leukemia inhibitory factor [LIF; and interleukin (IL)-6 family member], IL-16, vascular endothelial growth factor (VEGF), and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1). TIMP-1 was the only factor found to be significantly expressed in myocardial tissues in 4 wk LVPO. Representative profiles of protein expression in tissues from each fibrotic model are shown in Fig. 4. CC ligand (CCL)-11 or eotaxin is known to play a role in eosoniphil recruitment, among other activities, and was also increased in the plasma of 1-wk LVPO mice (8). Increases in CCL-11 were noted in the lungs of 1-wk LVPO mice but not in fibrotic lung tissue. No increases in CCL-11 were detected in tissue or plasma from 4-wk LVPO or with silica-induced PF.
Fig. 3.
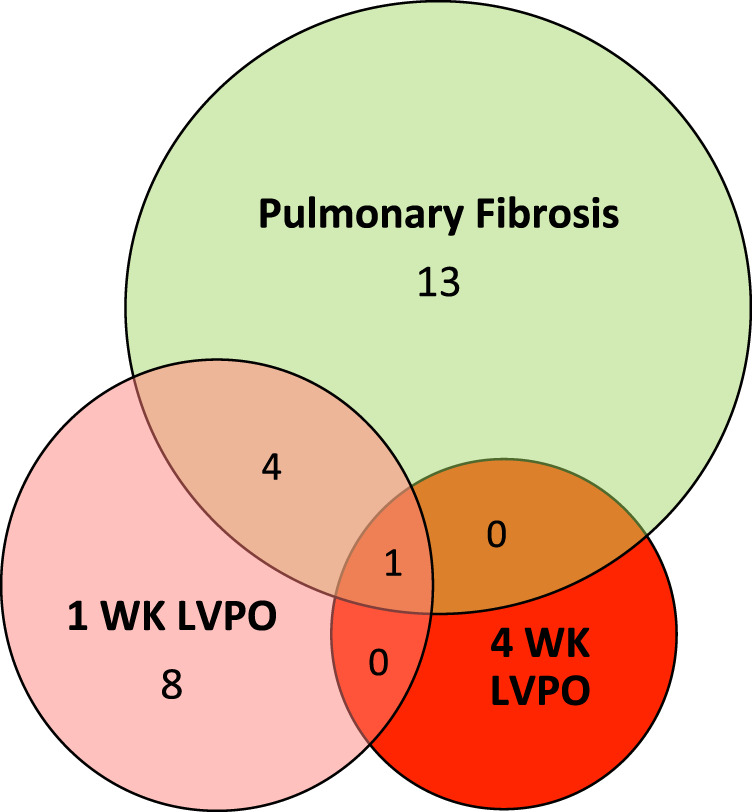
Venn diagram illustrating differences in cytokine/factor expression found in involved tissue in 1-wk left ventricular pressure overload (LVPO) myocardium (pink circle), silica-induced fibrotic lung (green circle), and 4-wk LVPO myocardium (red circle). For a complete list of cytokine/factors assessed, see Supplemental Table S1.
Fig. 4.

Representative factors elevated in left ventricular pressure overload (LVPO) myocardium. CC ligand (CCL)-11 (A–D) and CCL-12 (E–H) were increased in hearts and in lungs of 1 wk LVPO mice. No increases in CCL-11 or CCL-12 were found in fibrotic lung or in hearts or kidneys from silica-treated mice. Tissue inhibitor of matrix metalloproteinase-1 (TIMP-1; I–L) was found to be increased in both LVPO myocardium and in lung as well as in silica-treated lung. TIMP-1 was the only factor assayed found to be significantly elevated in 4 wk LVPO myocardium, albeit in lower amounts than that of 1-wk LVPO. Plasma concentrations of of TIMP-1 were found to be elevated at both 1 and 4 wk of LVPO. Levels of CCL-11 were also found to be elevated in plasma from LVPO mice at 1 wk only, whereas plasma levels of CCL-12 were not increased in any of the tested mice. *P < 0.05 LVPO vs. control; #P < 0.05 LVPO vs. corresponding sham; &P < 0.05 silica vs. control. MCP-5, monocyte chemoattractant protein-5; PO, pressure overload.
Levels of CCL-12 (or MCP-5), a cytokine produced by macrophages in response to cardiac ischemic injury, were increased in 1-wk LVPO myocardium (Fig. 4) (7). Similarly to CCL-11, elevated levels of CCL-12 were also detected in the lungs of 1-wk LVPO mice in addition to LV tissue but were not elevated in fibrotic lung tissue.
Plasma levels of CCL-11 were also increased in 1-wk LVPO mice, whereas CCL-12 was not elevated in plasma at this time point (Fig. 4). Levels of both CCL-11 and CCL-12 returned to baseline levels in 4-wk LVPO myocardium, as did all 13 of the factors increased at 1-wk PO, with the exception of TIMP-1 (Fig. 4). As stated, TIMP-1 was the only factor assayed with statistically significant elevations in 4-wk LVPO versus sham control. TIMP-1 levels were also increased in lungs from 1- and 4-wk LVPO mice as well as in fibrotic lung, albeit at reduced levels compared with the 1-wk time point. Plasma levels of TIMP-1 were increased in 1-wk LVPO and remained elevated, albeit reduced, in 4 wk LVPO. No increases in plasma levels of TIMP-1 were observed in silica-instilled mice.
Eighteen cytokines were detected with increased expression in the lungs with silica-induced PF. Five cytokines were elevated in fibrotic tissues from both models, whereas 13 were not elevated in LV of LVPO mice (Fig. 3). Several notable factors that were increased in fibrotic lung but were not increased in LVPO myocardium included CCL-3, TNFα, and CXCL-2 (Fig. 5). Interestingly, of these factors, only CCL-3 was found to be elevated in both lung and plasma from mice with silica-induced PF. CCL-3 is secreted by activated macrophages, lymphocytes, and epithelial and endothelial cells in the lung and has been implicated previously in PF (2). TNFα has also been implicated in PF and is primarily expressed by activated macrophages, lymphocytes, endothelial cells and fibroblasts (2). CXCL-2, implicated in angiogenesis in other fibrotic lung models, was the most highly represented of the three cytokines that were not elevated in the LV of LVPO myocardium (Fig. 5) (2).
Fig. 5.

Representative factors increased in lungs from silica-instilled mice that were not significantly elevated in fibrotic hearts from 1-wk or 4-wk left ventricular pressure overload (LVPO). CC ligand-3 (CCL-3; A and B), CXC ligand-2 (CXCL-2; C and D), and TNFα (E and F) were increased in fibrotic lungs but not induced in left ventricle (LV) at 1 wk or 4 wk of LVPO. Levels of CXCL-2 were also slightly elevated in the LV of silica-instilled mice. TNFα was found to slightly increase in the lungs of 1-wk LVPO, but not to the extent seen in fibrotic lung. CCL-3, but not CXCL-2 or TNFα, was significantly elevated in plasma from silica-instilled mice (data not shown). &P < 0.05 silica vs. control. MIP-1a and -2, macrophage inflammatory protein-1a and -2, respectively; PO, pressure overload.
Five factors were found to be elevated in both 1-wk LVPO and fibrotic lung: CCL-2, CXCL-9, CXCL-10, LIF, and TIMP-1. Levels of CCL-2 (MCP-1) and CXCL-10 in tissues and plasma are shown in Fig. 6. CCL-2, expressed by macrophages, lymphocytes, endothelial cells, and fibroblasts, is highly implicated in monocyte recruitment to tissues (15). Heightened expression of CXCL-10 and CXCL-9 is implicated in T cell recruitment in both heart and lung models of fibrosis (2, 27). Plasma levels of CXCL-10 were also increased in 1-wk LVPO mice but not in plasma with silica-induced PF. Increases in plasma levels of CCL-2 were not noted in either cardiac or lung fibrotic models.
Fig. 6.

Representative factors significantly elevated in fibrotic left ventricle (LV) from 1-wk left ventricular pressure overload (LVPO) myocardium and also increased in fibrotic lung. A–C: levels of CC ligand-2 (CCL-2) were found to be elevated in lungs of 1-wk LVPO and plasma, whereas levels were not increased in hearts from silica-instilled mice nor in plasma. D–F: levels of CXC ligand-10 (CXCL-10) were increased in LV, lungs, and plasma in 1-wk LVPO mice and in hearts from silica-instilled mice but were not found elevated in plasma from silica-instilled mice. *P < 0.05 LVPO vs. control; #P < 0.05 LVPO vs. corresponding sham; &P < 0.05 silica vs. control.
Additionally, six factors were found to be increased in plasma of 1-wk LVPO mice, although these specific cytokines were not in increased in myocardium. In contrast, in mice with silica-induced PF, no factors were found to be increased in plasma only that were not also found to be elevated in lung tissue. Two representative examples of cytokines increased in plasma from LVPO mice are shown in Fig. 7: granulocyte macrophage-colony stimulating factor (GM-CSF) and CCL-21. Whereas CCL-21 was also increased in LV at 1 wk following LVPO, GM-CSF was not found to be significantly increased in heart tissue. On the other hand, levels of GM-CSF, secreted by cells, including fibroblasts, were significantly elevated in lungs from silica-instilled mice, although plasma levels did not reach statistical significance in mice from this model. Levels of CCL-21, a factor implicated in the stimulation of naive T cells, remained elevated in plasma from mice at the 4-wk LVPO time point (4).
Fig. 7.
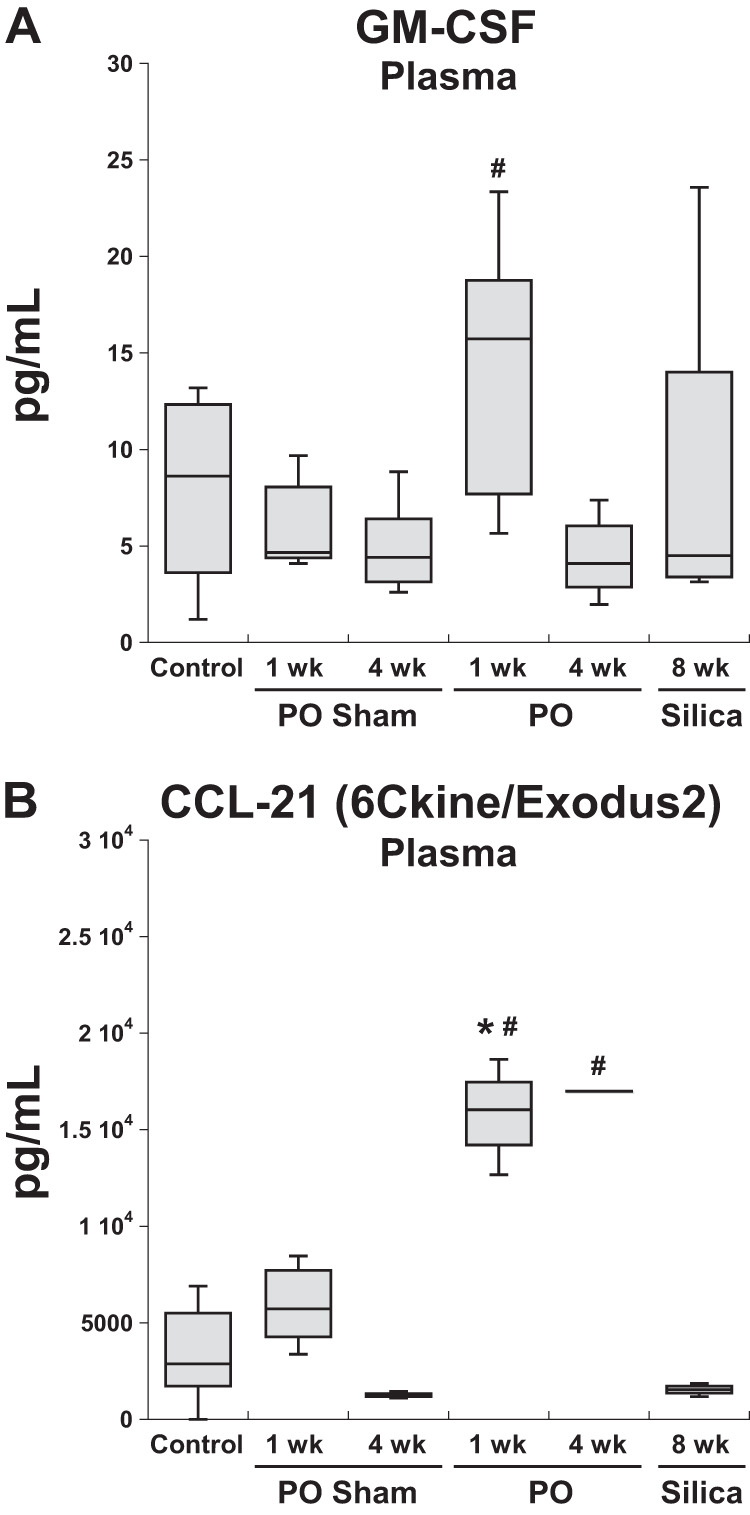
Representative factors found to be significantly elevated in plasma of 1-wk wk left ventricular pressure overload (LVPO) mice that were not elevated in plasma of mice with silica-instilled pulmonary fibrosis (PF). CC ligand-21 (CCL-21) remained elevated in plasma of mice with 4 wk of pressure overlaod (PO). Although CCL-21 was also elevated in 1 wk LVPO myocardium (B), granulocyte macrophage-colony stimulating factor (GM-CSF) was not increased in hearts at 1 wk of LVPO (data not shown; A). *P < 0.05, LVPO vs. control; #P < 0.05, LVPO vs. corresponding sham.
To determine whether changes in myocardial vasculature were associated with increases in expression of factors implicated in the recruitment of inflammatory cells to the heart, immunohistochemistry was performed on 1-wk and 4-wk LVPO myocardium. As shown in Fig. 8, overall changes in vasculature, detected with PECAM staining, were not apparent in 1-wk (Fig. 8B) and 4-wk (Fig. 8C) LVPO versus control (Fig. 8A). Quantification of stained images confirmed no significant alterations in the vasculature of PO myocardium versus control or sham (Supplemental Fig. S2). However, a marker of activated endothelium, intercellular adhesion molecule (ICAM), demonstrated stronger staining in 1-wk LVPO over that of control (23). Evidence of persistent ICAM staining was also detectable in 4-wk LVPO, although levels appeared to be lessened over that identified in 1 wk (Fig. 8, D, E, and F; for no primary controls, see Supplemental Fig. S3). Hence, during times of active inflammatory cell recruitment following PO at 1 wk, robust endothelial cell expression of ICAM was consistent with activation of the myocardial vasculature and persisted for ≤4 wk of LVPO.
Fig. 8.

Immunohistochemical staining of vasculature in control (A), 1-wk (B), and 4-wk (C) left ventricular pressure overload (LVPO) using anti-platelet endothelial cell adhesion molecule (PECAM) PECAM antibodies revealed no overt differences in total vessel density or size after LVPO (quantification in Supplemental Material). Intercellular adhesion molecule (ICAM) immunoreactivity, indicative of activated endothelium, was visibly increased at 1 wk of LVPO (E) over control (D) with diminished but persistent staining at 4 wk (F). Scale bar, 20 μm.
DISCUSSION
The present studies were designed to address the hypothesis that an organ-specific panel of inflammatory factors derived from PO myocardium and present in the circulation directs a specific myocardial inflammatory cell recruitment that, in turn, contributes to LVPO-induced fibrosis. Analysis of 44 cytokines revealed a novel signature of expression in PO myocardium and in plasma from PO mice that was distinct from those in a fibrotic lung model. We report a panel of factors that represent a LVPO organ-specific profile, as these factors were increased in plasma and tissue of LVPO mice but were not increased in lungs from a silica-induced model of PF. Thus, PO induces myocardial cells to express a panel of cytokines and chemokines that 1) increase circulating levels of activators of inflammatory cells, 2) activate endothelial cells to facilitate myocardial recruitment of inflammatory cells, and 3) influence activation of recruited inflammatory cells to the myocardium (Fig. 9).
Fig. 9.

Schematic representation of increased fibrotic deposition of collagen in left ventricular pressure overload (LVPO)-inducing increases in organ-specific factors such as CC ligand (CCL)-11 and -12 in the myocardium as well as increases in circulating cytokines and chemokines, including CCL-21 and granulocyte macrophage-colony stimulating factor (GM-CSF). The majority of factors increased in 1-wk LVPO returned to baseline levels at 4 wk. Evidence for activated endothelium [intercellular adhesion molecule (ICAM) + staining] was strongest at 1 wk, but persistent increases over sham were observed at 4-wk LVPO.
Overall, the LVPO cytokine signature identified here was consistent with established profiles associated with recruitment of monocyte/macrophage and T cell populations to PO myocardium (17). Specifically, evidence for the presence of alternatively activated macrophages (M2) and Th1 T cells in 1-wk LVPO tissue was found. For example, increased levels of CXCL9 and CXCL10 were consistent with Th1 cell recruitment, whereas increases in VEGF and the TGFβ-inducing cytokine IL-16 were consistent with M2 macrophage polarization (4, 15, 26). In contrast, cytokine expression in fibrotic lung reflected a more proinflammatory milieu, including cytokine expression consistent with classically activated macrophages (M1), notably IL-1β and TNFα (2). Unlike PO mice, no increases in plasma levels of cytokines increased in fibrotic lung were found, with the exception of IL-2 and CCL-3. Activation of myocardial endothelium, marked by increases in ICAM expression in 1- and 4-wk LVPO, was consistent with active recruitment of inflammatory cells to the myocardium. Given that previous studies have implicated recruitment of inflammatory cells in the development of fibrosis and progression of heart failure, the profile of cytokines that participate in a LVPO-specific signature has the potential to yield valuable insight into strategies to target these signaling molecules for future therapies (9, 11).
CCL11 and CCL12 may be of specific note for potential future targeting, as these factors were elevated early (1-wk LVPO) but not at the later time point (4-wk LVPO) and were not associated with fibrotic lung. Previously, others have reported that CCL-11 (a.k.a. eotaxin) is expressed by cardiac macrophages and by mast cells in acute transplant heart studies where elevated levels of this cytokine were correlated with increases in myocardial fibrosis (28). CCL-11 is a member of the eotaxin family with potent eosinophil chemoattractant activity. A role for eosinophils in cardiac PO is currently undefined; however, as functions of CCL-11 in modulating macrophage and mast cell activity are also proposed, CCL-11 production in PO myocardium might reflect activity for these cell types (8). The return of expression of CCL-11 at the 4-wk time point following LVPO to that of control/sham LV suggested that CCL-11 serves primarily as an early modulator in PO hearts. CCL-11 activity is also associated with angiogenesis and coincided with elevated levels of another factor not found to be increased in fibrotic lung: vascular endothelial growth factor (VEGF) (14). Increased levels of CCL-11 and VEGF at 1 wk could potentially result in increased angiogenic activity. However, PECAM staining of myocardial vasculature showed no differences in blood vessel density in 1- or 4-wk LVPO versus control hearts, which argued against a potent angiogenic activity of CCL-11 and VEGF that would result in an increase in blood vessel density. However, increases in CCL-11 and VEGF might influence endothelial activation and permeability in PO myocardium. Increases in ICAM staining at 1 wk demonstrated endothelial activation in LVPO myocardium that persisted at 4 wk (Fig. 8).
CCL-12, also known as monocyte chemoattractant protein (MCP)-5, exhibited elevated levels in LVPO myocardium and in the lungs of mice with 1 wk of PO but not in the lungs from silica-instilled mice. Several factors increased in LVPO myocardium were also found to be elevated in the lungs of mice, with LVPO likely representing secondary inflammatory triggers in lungs of mice with LVPO. Recently, elevated levels of CCL-12 were also reported in infarcted myocardium during periods of chronic inflammation following injury, consistent with CCL-12/MCP-5 being a key monocyte recruiting factor for the heart (7). CCL-2 (a.k.a. MCP-1) also demonstrated increased levels in 1-wk LVPO and plasma from mice with LVPO while also being significantly increased in fibrotic lung from silica-instilled mice. Hence, increases in CCL-12 were seen in LVPO myocardium, whereas increases in CCL-2 were also detected in fibrotic lungs (Fig. 6). CCL-2 and CCL-12 both bind CC receptor 2 (CCR2) expressed on monocytes and promote monocyte recruitment, increasing the number of macrophages in a given tissue. Pharmacological or antibody-mediated inhibition of CCR2 activity significantly reduced fibrosis in mice with LVPO, confirming the pivotal function of CCR2-expressing cells in PO-induced collagen accumulation (22). CCL-12 (MCP-5) is not expressed in humans, although murine CCL-12 demonstrates higher structural homology to human CCL-2 (MCP-1) than to murine CCL-2 (18). Future studies to address precise signatures of monocyte-recruiting factors specific to myocardium in both murine and human conditions will benefit the design of heart-specific regulation of inflammatory cell recruitment. Notably, IL-16 was also specifically increased in 1-wk LVPO and previously implicated in the development of myocardial fibrosis (26).
In addition to factors that were consistent with monocyte recruitment, factors implicated in T cell recruitment were also represented in PO myocardium. Levels of CXCL-9 and CXCL-10, ligands for CXCR3 that are expressed primarily on CD4+ Th1 T cells, were elevated in LVPO myocardium. CXCR3 expression has been identified in transplant studies as an important mediator of cardiotropism for primed T cells (13, 21). Elevated levels of CCL21 were detected in plasma of 1-wk LVPO mice, another cytokine implicated in T cell migration (4, 21). Factors associated with CD4+ Th2 T cells, IL-4 and IL-13, were not found to be elevated in myocardium or plasma, consistent with previous observations that Th1 T cells are more highly represented in PO myocardium than Th2 T cells (20). Likewise, increased levels of IL-17, a secreted product of Th17 cells, were not detected in PO myocardium. ICAM expression is a known binding factor for T cell recruitment mediated through T cell expression of LFA-1, a binding partner for ICAM (23). Hence, the cytokine profile in 1-wk LVPO myocardium was consistent with active recruitment of CD4+ Th1 T cells facilitated by increases in ICAM expression in myocardial vasculature.
Expression of factors induced by LVPO was also dependent upon duration of PO. At 4 wk of LVPO, factors found to be elevated at 1 wk had returned to those of baseline/sham control, with the exception of myocardial TIMP-1 and plasma levels of CCL21. We conclude that peak recruitment of inflammatory cells occurs in the first weeks following induction of PO. Persistent increases in ICAM staining in 4-wk myocardium might indicate continued inflammatory cell recruitment, although levels of chemokine/cytokines profile assessed herein were notably absent in 4-wk LVPO myocardium. In congruence with previous reports, TIMP-1 was significantly elevated in 1-wk LVPO mice in LV, lung, and plasma (25). Smaller increases in TIMP-1 were also observed in fibrotic lungs. Interestingly, plasma levels of TIMP-1 were found to be predictive of cardiac heart failure in patients (3). TIMP-1, as an inhibitor of matrix metalloproteinases (MMPs), is predicted to contribute to increases in ECM content by reducing MMP-mediated ECM degradation. Although TIMP-1 remained significantly increased in 4-wk LVPO myocardium versus levels in control and sham, levels were reduced from that of 1 wk LVPO. Nonetheless, TIMP-1 represented the only factor interrogated in these studies that remained elevated in 4-wk LVPO myocardium. Hence, these studies confirmed TIMP-1 as a valid marker of PO myocardium and likely to function as an active participant in cardiac fibrosis.
Interestingly, tumor necrosis factor-α (TNFα), which has previously been reported to influence inflammation and the development of cardiac hypertrophy, exhibited no significant differences in levels of expression in LVPO myocardium versus control or sham tissue at the time points assayed (24). However, TNFα was significantly increased in lungs from silica-instilled mice (Fig. 5). Similarly, CCL-3 and CXCL-2 were also representative of factors increased in fibrotic lung but not in LVPO. Generally, the baseline levels of many cytokines examined in this study were higher in lung than in LV, which was indicative of the heightened immune response necessitated for lung that is continually exposed to extrinsic environmental factors versus the more protected milieu of the myocardium. The studies presented herein identify a selective increase in cytokines and factors produced by PO myocardium that was distinct from that produced by fibrotic lung (Fig. 9).
Limitations of the present study include the finite list of immune factors that were interrogated, the detection limitations of the analysis for each analyte, the inclusion of only two time points following LVPO, and the one time point assessed for the silica model of lung fibrosis. Future studies are planned to broaden both the cytokine profile and the time course of both fibrotic models to elaborate the PO myocardial signature of cytokine/chemokine factors that control inflammatory cell cardiac recruitment as well as the inclusion of patient samples. Of further interest would be comparisons to other organ-specific fibrotic models such as liver or kidney. In addition, future studies to examine myocardial inflammatory cell production of proteins that specifically affect ECM assembly, deposition, and remodeling will yield valuable insight into mechanisms of cardiac fibrosis in response to PO.
GRANTS
This work was partially supported by National Heart, Lung, and Blood Institute Grant 5R01-HL-123478-01 (to M.R.Z.) and Department of Veterans Affairs Grants CX001608 (to A.D.B. and M.R.Z.), BX000333 (to A.C.L.), and BX004072 (to L.T.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.O., C.F.B., A.O.V.L., Y.Z., L.T.M., A.C.L., M.R.Z., and A.D.B. conceived and designed research; M.O., C.F.B., A.O.V.L., Y.Z., L.T.M., A.C.L., M.R.Z., and A.D.B. performed experiments; M.O., C.F.B., A.O.V.L., Y.Z., A.C.L., M.R.Z., and A.D.B. analyzed data; M.O., C.F.B., A.O.V.L., Y.Z., L.T.M., A.C.L., M.R.Z., and A.D.B. interpreted results of experiments; M.O., C.F.B., A.O.V.L., Y.Z., A.C.L., M.R.Z., and A.D.B. prepared figures; M.O., C.F.B., M.R.Z., and A.D.B. drafted manuscript; M.O., C.F.B., A.O.V.L., Y.Z., L.T.M., A.C.L., M.R.Z., and A.D.B. edited and revised manuscript; M.O., C.F.B., A.O.V.L., Y.Z., L.T.M., A.C.L., M.R.Z., and A.D.B. approved final version of manuscript.
REFERENCES
- 1.Adams DH, Shaw S. Leucocyte-endothelial interactions and regulation of leucocyte migration. Lancet 343: 831–836, 1994. doi: 10.1016/S0140-6736(94)92029-X. [DOI] [PubMed] [Google Scholar]
- 2.Agostini C, Gurrieri C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 3: 357–363, 2006. doi: 10.1513/pats.200601-010TK. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation 113: 2089–2096, 2006. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 4.Blanton RM, Carrillo-Salinas FJ, Alcaide P. T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol 317: H124–H140, 2019. doi: 10.1152/ajpheart.00028.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Boggs J, Lacy JM, Zile MR. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 119: 269–280, 2009. doi: 10.1161/CIRCULATIONAHA.108.773424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev 15: 415–422, 2010. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML. Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing. JCI Insight 2: e94207, 2017. doi: 10.1172/jci.insight.94207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diny NL, Hou X, Barin JG, Chen G, Talor MV, Schaub J, Russell SD, Klingel K, Rose NR, Čiháková D. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur J Immunol 46: 2749–2760, 2016. doi: 10.1002/eji.201646557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med, 65: 70–99, 2019. doi: 10.1016/j.mam.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Gaasch WH, Zile MR. Left ventricular structural remodeling in health and disease: with special emphasis on volume, mass, and geometry. J Am Coll Cardiol 58: 1733–1740, 2011. doi: 10.1016/j.jacc.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M. Cardiac macrophages promote diastolic dysfunction. J Exp Med 215: 423–440, 2018. doi: 10.1084/jem.20171274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt BJ, Jurd KM. Endothelial cell activation. A central pathophysiological process. BMJ 316: 1328–1329, 1998. doi: 10.1136/bmj.316.7141.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komarowska I, Coe D, Wang G, Haas R, Mauro C, Kishore M, Cooper D, Nadkarni S, Fu H, Steinbruchel DA, Pitzalis C, Anderson G, Bucy P, Lombardi G, Breckenridge R, Marelli-Berg FM. Hepatocyte growth factor receptor c-Met instructs T cell cardiotropism and promotes T cell migration to the heart via autocrine chemokine release. Immunity 42: 1087–1099, 2015. doi: 10.1016/j.immuni.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laakkonen JP, Lähteenvuo J, Jauhiainen S, Heikura T, Ylä-Herttuala S. Beyond endothelial cells: vascular endothelial growth factors in heart, vascular anomalies and placenta. Vascul Pharmacol 112: 91–101, 2019. doi: 10.1016/j.vph.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res 191: 15–28, 2018. doi: 10.1016/j.trsl.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald LT, Johnson SD, Russell DL, Young MRI, LaRue AC. Role of a novel immune modulating DDR2-expressing population in silica-induced pulmonary fibrosis. PLoS One 12: e0180724, 2017. doi: 10.1371/journal.pone.0180724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald LT, Zile MR, Zhang Y, Van Laer AO, Baicu CF, Stroud RE, Jones JA, LaRue AC, Bradshaw AD. Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis. Am J Physiol Heart Circ Physiol 315: H92–H100, 2018. doi: 10.1152/ajpheart.00719.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 35: 175–181, 2006. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124: 2921–2934, 2014. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med 214: 3311–3329, 2017. doi: 10.1084/jem.20161791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ngwenyama N, Salvador AM, Velázquez F, Nevers T, Levy A, Aronovitz M, Luster AD, Huggins GS, Alcaide P. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight 4: e125527, 2019. doi: 10.1172/jci.insight.125527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 3: 230–244, 2018. doi: 10.1016/j.jacbts.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salvador AM, Nevers T, Velázquez F, Aronovitz M, Wang B, Abadía Molina A, Jaffe IZ, Karas RH, Blanton RM, Alcaide P. Intercellular adhesion molecule 1 regulates left ventricular leukocyte infiltration, cardiac remodeling, and function in pressure overload-induced heart failure. J Am Heart Assoc 5: e003126, 2016. doi: 10.1161/JAHA.115.003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 115: 1398–1407, 2007. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 25.Takawale A, Zhang P, Patel VB, Wang X, Oudit G, Kassiri Z. Tissue inhibitor of matrix metalloproteinase-1 promotes myocardial fibrosis by mediating CD63-integrin β1 interaction. Hypertension 69: 1092–1103, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09045. [DOI] [PubMed] [Google Scholar]
- 26.Tamaki S, Mano T, Sakata Y, Ohtani T, Takeda Y, Kamimura D, Omori Y, Tsukamoto Y, Ikeya Y, Kawai M, Kumanogoh A, Hagihara K, Ishii R, Higashimori M, Kaneko M, Hasuwa H, Miwa T, Yamamoto K, Komuro I. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS One 8: e68893, 2013. doi: 10.1371/journal.pone.0068893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Linthout S, Tschöpe C. The quest for antiinflammatory and immunomodulatory strategies in heart failure. Clin Pharmacol Ther 106: 1198–1208, 2019. doi: 10.1002/cpt.1637. [DOI] [PubMed] [Google Scholar]
- 28.Zweifel M, Mueller C, Schaffner T, Dahinden C, Matozan K, Driscoll R, Mohacsi P. Eotaxin/CCL11 expression by infiltrating macrophages in rat heart transplants during ongoing acute rejection. Exp Mol Pathol 87: 127–132, 2009. doi: 10.1016/j.yexmp.2009.07.006. [DOI] [PubMed] [Google Scholar]