

Abstract
Clinical studies indicate that sepsis-induced diaphragm dysfunction is a major contributor to respiratory failure in mechanically ventilated patients. Currently there is no drug to treat this form of diaphragm weakness. Sepsis-induced muscle dysfunction is thought to be triggered by excessive mitochondrial free radical generation; we therefore hypothesized that therapies that target mitochondrial free radical production may prevent sepsis-induced diaphragm weakness. The present study determined whether MitoTEMPOL, a mitochondrially targeted free radical scavenger, could reduce sepsis-induced diaphragm dysfunction. Using an animal model of sepsis, we compared four groups of mice: 1) sham-operated controls, 2) animals with sepsis induced by cecal ligation puncture (CLP), 3) sham controls given MitoTEMPOL (10 mg·kg−1·day−1 ip), and 4) CLP animals given MitoTEMPOL. At 48 h after surgery, we measured diaphragm force generation, mitochondrial function, proteolytic enzyme activities, and myosin heavy chain (MHC) content. We also examined the effects of delayed administration of MitoTEMPOL (by 6 h) on CLP-induced diaphragm weakness. The effects of MitoTEMPOL on cytokine-mediated alterations on muscle cell superoxide generation and cell size in vitro were also assessed. Sepsis markedly reduced diaphragm force generation. Both immediate and delayed MitoTEMPOL administration prevented sepsis-induced diaphragm weakness. MitoTEMPOL reversed sepsis-mediated reductions in mitochondrial function, activation of proteolytic pathways, and decreases in MHC content. Cytokines increased muscle cell superoxide generation and decreased cell size, effects that were ablated by MitoTEMPOL. MitoTEMPOL and other compounds that target mitochondrial free radical generation may be useful therapies for sepsis-induced diaphragm weakness.
Keywords: diaphragm weakness, mitochondrial dysfunction, MitoTEMPOL, proteolysis, sepsis
INTRODUCTION
Recent clinical studies suggest that sepsis-induced diaphragm dysfunction contributes to respiratory failure in mechanically ventilated, critically ill patients (11, 41, 52). Moreover, these studies also show that diaphragm weakness in this patient population has serious consequences (13, 41, 52). Specifically, patients with weaker diaphragms require longer durations of mechanical ventilation and have a much higher mortality than patients with stronger diaphragms. It has been suggested that diaphragm weakness contributes to mortality by making it far more difficult to wean patients from mechanical ventilation, influencing the decision to withdraw care because of the inability to be liberated from this form of life support (41, 52).
Currently there are no pharmacologic agents to treat this form of diaphragm dysfunction. However, animal studies indicate that sepsis-induced muscle weakness may be precipitated by excessive mitochondrial free radical generation (3, 36, 37). Previous studies have also suggested that sepsis-induced weakness may also be associated with activation of muscle proteolytic pathways (i.e., the proteasome, caspase, calpain) (24, 39, 40, 50, 51) and subsequent loss of critical skeletal muscle proteins (19, 50, 54). Theoretically, these two processes, infection-induced muscle mitochondrial free radical generation and activation of proteolytic pathways, may be linked. If so, administration of mitochondrially targeted antioxidants should be effective in suppressing mitochondrial free radical generation, reducing proteolytic enzyme activation, and preventing weakness.
The purpose of the present study, therefore, was to determine whether administration of MitoTEMPOL, a mitochondrially targeted free radical scavenger, would attenuate sepsis-induced diaphragm dysfunction (23, 53). We employed the cecal ligation puncture (CLP) model of sepsis and compared diaphragm force generation in control and CLP mice with and without concomitant administration of MitoTEMPOL. To further test the hypotheses presented in the previous paragraph, we also assessed diaphragm mitochondrial function, proteolytic enzyme pathway activation (caspase, calpain, the proteasome), levels of a key contractile protein (myosin heavy chain), and an index of muscle mitochondrial superoxide generation (aconitase) in these animals (19, 50, 54). In addition, since there is an inevitable delay between the time that a patient develops an infection and the point that same patient presents for medical care, we also determined whether delayed administration of MitoTEMPOL (i.e., by 6 h) (56) after the induction of sepsis was also effective in preventing diaphragm weakness.
Finally, to determine whether MitoTEMPOL acts directly on muscle cells to suppress mitochondrial superoxide generation, we also examined the effects of MitoTEMPOL on superoxide generation in an isolated muscle cell line model of sepsis, i.e., cytokine administration in vitro to C2C12 muscle cells.
MATERIALS AND METHODS
In vivo animal experimental protocols.
All animal protocols were approved by the University of Kentucky Institutional Animal Care and Use Committee. For our initial set of experiments, we studied four groups of mice (n = 5–7/group), including 1) sham-operated, vehicle-treated controls (0.3 mL of saline injected intraperitoneally after surgery); 2) cecal ligation puncture (CLP)-operated, vehicle-treated animals; 3) sham-operated animals treated with MitoTEMPOL (10 mg/kg in 0.3 mL of saline, injected intraperitoneally after surgery and at 24 h); and 4) CLP-operated animals given MitoTEMPOL (38, 50). MitoTEMPOL was purchased from Abcam (Cambridge, MA) (catalog no. ab144644). The ICR (CD-1) mouse strain was used for these experiments. Male mice (25–30 g) were purchased from Charles River Laboratories (Wilmington, MA). The dose of MitoTEMPOL used was chosen based on data from pilot studies and from previous literature supporting the effectiveness of this dosage in a previous animal model (33).
At 48 h after surgery, animals were euthanized and diaphragms were removed. Muscle strips with intact phrenic nerves were dissected from a portion of the diaphragm for assessments of force-frequency relationships, fatigue, and neuromuscular transmission failure. The remaining diaphragm was frozen, stored at −80°C, and subsequently used to determine diaphragm protein levels, proteolytic enzyme activities, and myosin heavy chain levels.
In a second group of experiments, diaphragms were excised from the four experimental groups (n = 3/group) at 48 h after surgery, diaphragm mitochondria were isolated (5, 43, 46), and mitochondrial function was assessed as described below.
In a third group of studies, to determine whether delayed administration of MitoTEMPOL would prevent CLP-induced reductions in diaphragm force generation, CLP animals were given MitoTEMPOL (10 mg/kg) initially at 6 h after surgery and then again at 24 h after surgery. Animals (n = 4–7/group) were euthanized at 48 h after surgery, diaphragms were dissected, and diaphragm force generation was assessed.
In a fourth group of experiments, we assessed the relationship between the evolution of early activation of diaphragm proteolytic pathways (i.e., caspase, calpain 20S proteasomal activity) and early increases in diaphragm superoxide generation, as assessed using the aconitase assay. For these studies, we examined seven groups of animals (n = 3/group) including 1) sham-operated controls; 2) CLP, vehicle treated, and euthanized at 4 h after surgery; 3) CLP, vehicle treated, and euthanized at 8 h after surgery; 4) CLP, vehicle treated, and euthanized at 24 h after surgery; 5) CLP, given MitoTEMPOL (10 mg/kg) and euthanized at 4 h; 6) CLP, given MitoTEMPOL (10 mg/kg), and euthanized at 8 h; and 7) CLP, given MitoTEMPOL (10 mg/kg), and euthanized at 24 h. Diaphragms were harvested and assayed for caspase, calpain, 20S proteasome activity, and aconitase activity.
Cecal ligation puncture-induced sepsis.
Cecal ligation puncture was performed as previously described (37, 38, 48–50). Briefly, male CD1 mice were anesthetized with isoflurane (2–4%) using a nose cone attached to an anesthetic gas vaporizer. After a steady plane of anesthesia was achieved, the abdomen was prepped and draped in a sterile fashion. Following an abdominal incision, the cecum was isolated, ligated (1.0 cm portion), punctured through and through with an 18-gauge sterile needle, and a small amount of feces expressed through the hole followed by reintroduction of the punctured cecum into the abdominal cavity. The abdominal musculature was sutured and the skin closed with surgical staples. The same procedure was used for sham surgeries, except that the abdomen was opened and closed without cecal ligation or puncture. Postoperatively, animals were resuscitated with 60 mL/kg of saline administered subcutaneously. For analgesia, buprenorphine (0.05 mg/kg sc) was given immediately and every 12 h following surgery. At 48 h following surgery, animals were euthanized with an intraperitoneal injection of pentobarbital (150 mg/kg).
Assessment of diaphragm strength, neuromuscular transmission, and fatigue.
To assess diaphragm maximal-specific force generation and the force-frequency relationship in animals from the four experimental groups, muscle strips with intact phrenic nerves were dissected from the left midcostal diaphragm and mounted in water-jacketed glass organ baths at 25°C containing physiologic Krebs-Henselheit solution, as previously described (37–39, 48, 50), except that curare was not added to the bath. One end of each strip was tied to the base of the organ bath and the other to a force transducer. The intact phrenic nerve was carefully placed in a cuff electrode. Stimulation of the nerve was accomplished using direct output from a Grass S48 stimulator (Grass, West Warwick, RI), and voltage was adjusted to achieve supramaximal stimulation. In addition, platinum mesh field stimulation electrodes were placed around the diaphragm muscle strip. Direct muscle stimulation was accomplished using a Grass S88 stimulator (Grass) interfaced with a constant current amplifier (iWorx, Dover, NH) connected to the field stimulation electrodes. With this dual stimulation setup, we were able to stimulate the phrenic nerve alone, the muscle, or a combination of nerve and muscle.
The diaphragm muscle strip was allowed to equilibrate for 15 min, and then muscle length was adjusted to Lo. To determine force-frequency relationships, diaphragm muscle strips were first stimulated with trains of 1-, 20-, and 50-Hz stimuli (train duration: 800 ms; 30 s between adjacent trains) delivered via the phrenic nerve electrode and subsequently via direct muscle field stimulation. Force was recorded with a BIOPAC transducer/computer system (BIOPAC Systems, Inc., Goleta, CA).
To assess neuromuscular transmission failure, we employed the technique described by Greising and colleagues (20, 21). For this assessment, the diaphragm was made to repetitively contract in response to phrenic nerve stimulation (1 train of phrenic stimuli per second) with intermittent superimposition of field muscle stimulation (1 train of field muscle stimulation every 10 s). We then calculated the rates of decline of force in response to nerve and field muscle stimulation using the formula transmission failure index = 100 × (NF − MF)/(1 − MF) (20). NF is defined as the percentage reduction in force generated in response to neural stimulation between the beginning and end of the fatigue protocol, while MF is the percentage reduction in force generated in response to field muscle stimulation. Greater levels of transmission failure are represented as increases in the transmission failure index.
After a 15-min rest period, we then assessed diaphragm force over time during a repetitive contraction trial in response to supramaximal direct muscle field stimulation (trains of 330 ms, 40-Hz stimulation, at a rate of 1 train per second over 2 min). For the purpose of reporting these data, we measured force at 30-s intervals to construct average force-time curves.
Muscle cross-sectional area was calculated as muscle strip weight divided by muscle density (1.06), and muscle length and muscle-specific force was calculated as force divided by cross-sectional area, as previously described (8). The total costal diaphragm weight was recorded. Diaphragm tissues not used for force determinations were frozen, stored at −80°C, and subsequently used for assays.
Mitochondrial activity assay.
To assess mitochondrial function, mitochondria were isolated from diaphragm muscle, as previously described (3, 7, 43, 46, 48). Mitochondria were then resuspended in mitochondrial assay buffer and placed in a fiberoptic fluorescence monitoring system (Instech Laboratories, Inc., Plymouth Meeting, PA) to measure rates of oxygen consumption (state 3, state 4), respiratory control ratio (RCR), ADP/O ratios, and ATP production rates. In our previous studies using this technique, we found that sepsis very reproducibly induced significant reductions in mitochondrial function by the 48-h time point (3, 7, 43, 46, 48). For this reason, we chose to study only a small number of animals/group (n = 3) for this particular group of experiments.
Calpain activity assay.
Diaphragm calpain activity was assessed using a commercially available kit (Abcam) according to the manufacturer’s instructions, as previously reported (32, 42). Briefly, samples (n = 4/group) were prepared in the proprietary extraction buffer, and protein levels were determined using the Bradford Assay (Bio-Rad, Hercules, CA). One-hundred micrograms of protein from diaphragm samples were used for the assay. Parallel samples containing a calpain inhibitor were also run to determine the specificity of the signal. Extraction buffer alone was used as a blank. Mixtures were placed in a 96-well plate, reaction buffer (10 microliters) and calpain substrate (5 microliters) were added, and an initial reading was recorded (excitation 400 nm, emission 505 nm). Samples were incubated in the dark at 37°C for 1 h, followed by a final fluorescent reading. Differences in fluorescent activity over time for a given sample in the presence and absence of the specific calpain inhibitor were used as an index of calpain enzyme activity.
Caspase activity assay.
Diaphragm caspase-3 activity was assessed as previously described (n = 4/group) using the caspase-3-specific fluorogenic substrate (30 μM N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin, Ac-DEVD-AMC) (42, 48, 49). For this assay, diaphragm samples were homogenized using a buffer consisting of 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 mM sodium chloride, 1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, 10 mM dithiothreitol, 1 mM ethylene diamine tetraacetic acid, and 10% glycerol. Duplicate measurements were performed in the presence of a specific caspase-3 inhibitor (DEVD-CHO, 20 nM). Immediately following substrate addition, baseline fluorescent measurements were recorded using a Molecular Devices spectrofluorophotometer (excitation 360 nm, emission 460 nm), followed by repeat measurements after 0.5 h of incubation at 30°C. Raw increases in fluorescence were calculated from the initial reading and the final reading; increases in fluorescence for the DEVD-CHO duplicate were subtracted from the raw reading to determine the caspase-3 activity.
20S proteasome subunit activity assay.
Proteasome activity of diaphragm samples (n = 4/group) was measured using a commercially available kit (20 S Proteasome Subunit Activity kit; Calbiochem, San Diego, CA), following the manufacturer’s protocol. AMC standards were employed to calibrate measurements of proteasomal activity (39, 47).
Aconitase activity assay.
We measured aconitase activity, an index of mitochondrial superoxide generation, for diaphragm homogenates obtained from the fourth set of experiments (i.e., assessment of the time course of muscle superoxide generation) using the Abcam Aconitase kit assay (Cambridge, MA), following the manufacturer’s protocol. For these measurements, diaphragm muscles were homogenized in the following buffer: 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 mM sodium chloride, 1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, 10 mM dithiothreitol, 1 mM ethylene diamine tetraacetic acid, and 10% glycerol.
Protein determination by Western blotting.
Western blotting was employed to measure MHC protein levels (n = 5/group). For these determinations, muscles were homogenized in buffer, and protein levels were determined using the Bradford assay (Bio-Rad, Hercules, CA). Buffer used for these homogenizations contained 10 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 20 mM HEPES, 2 mM ethylenediaminetetraacetic acid, 250 mM sodium chloride, 2 µg/mL leupeptin, 2 µg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, 0.5 µg/mL benzamidine, and 1 mM dithiothreitol.
Proteins from muscle samples were then diluted with an equal volume of Laemmli loading buffer and equal amounts of protein loaded onto Mini-Protean TGX Stain-Free gels (Bio-Rad). The V3 Western Workflowsystem and ChemiDoc Touch Imaging System were used to perform Western blot analyses (Bio-Rad). MHC levels were probed using the sc-20641 antibody from Santa Cruz Biotechnology (Dallas, TX). MHC densities were normalized to the total protein loaded for each lane using Image Lab 5.2.1 software (Bio-Rad). This analysis technique measures the density of the total protein loaded for each lane, measures the densitometry of the antibody-probed protein (i.e., MHC in this case), and normalizes the latter signal to the former. This approach avoids the artifacts associated with traditional approach of reprobing for “housekeeping” proteins, since many stresses can alter the levels of the usual proteins thought to represent housekeeping controls (14–16, 32).
In vitro myocyte cell protocols.
Experiments performed on intact animals have the inherent limitation that the direct effects of a therapy, such as MitoTEMPOL, on a given organ or cell type can be difficult to separate from indirect effects mediated by actions of the drug on systemic factors (e.g., immune cell function, alterations in blood flow, etc.). For that reason, we also examined the effects of MitoTEMPOL on isolated myocytes in culture media to demonstrate that this agent can block mitochondrial free radical generation. To simulate the effects of sepsis, we incubated cells with a mixture of cytokines. To assess myocyte superoxide generation, we used the MitoSOX assay, as previously described (38).
For these studies, C2C12 muscle myoblasts (ATCC, Manassas, VA) (n = 5/group) were grown to 70% confluency and differentiated for 5 days (37, 38, 44, 45). Mature myocytes (n = 5/condition) were then incubated with either control media (DMEM), cytomix (20 ng/mL TNFα, 50 U/mL IL-1β, 100 U/mL IFNγ, and 10 μg/mL LPS) (37, 38), MitoTEMPOL (10 mg/L), or cytomix plus MitoTEMPOL (10 mg/L) for 24 h. Following this, MitoSOX (5 µM diluted in DMSO; Invitrogen, San Diego, CA) was added to each plate, and cells were incubated for 20 min. Images were collected using a fluorescent microscope (an excitation frequency of 396 and an emission frequency of 560), stored on a hard drive, and analyzed using SigmaScan imaging software (57). The latter software was used to quantify the intensity of the MitoSOX fluorescent signal using the software ROI intensity (region of interest measurement of color intensity) utility.
In addition, we also assessed the effects of cytokines and MitoTEMPOL on in vitro myocyte cell size. For this assay, C2C12 skeletal muscle myoblasts were first transfected with a GFP-fluorescent marker (pmaxGFP vector; Lonza America, Inc., Alpharetta, GA) and then differentiated for 4 days (1, 28). Myocytes were incubated with either (n = 5/group) control media, cytomix, MitoTEMPOL (10 mg/L), or cytomix plus MitoTEMPOL (10 mg/L) for 24 h. To measure myotube width, six fluorescent images per well were captured. Two diagonal lines were drawn across each image, and cell width was measured where the diagonal lines transected the myotubes using ImageJ software (17, 28).
Statistical analysis.
ANOVA was used to compare variables (e.g., force) across groups of cells and animals treated with different agents, with post hoc testing (Tukey) to determine differences between groups. A P value of <0.05 was taken as indicating statistical significance for experiments.
RESULTS
Diaphragm force generation.
Diaphragm strength was assessed both by measuring the force generated by diaphragm strips in response to increasing frequencies (1–50 Hz) of electrical stimulation of the phrenic nerve and in response to increasing frequencies (1–50 Hz) of direct muscle electrical stimulation using field electrodes. CLP sepsis induced a downward shift in the diaphragm force-frequency relationship, assessed by either phrenic nerve or direct muscle stimulation (Fig. 1), and administration of MitoTEMPOL at the time of surgery blocked this sepsis-induced reduction in diaphragm strength. For example, using direct muscle stimulation at a frequency of 50 Hz, diaphragm force averaged 27.2 ± 1.5, 26.9 ± 1.8, 12.6 ± 1.3, and 27.0 ± 1.3 N/cm2, respectively, for sham-operated controls, sham + MitoTEMPOL, CLP, and CLP + MitoTEMPOL-treated groups (P < 0.001 for comparison of CLP with the other 3 conditions).
Fig. 1.
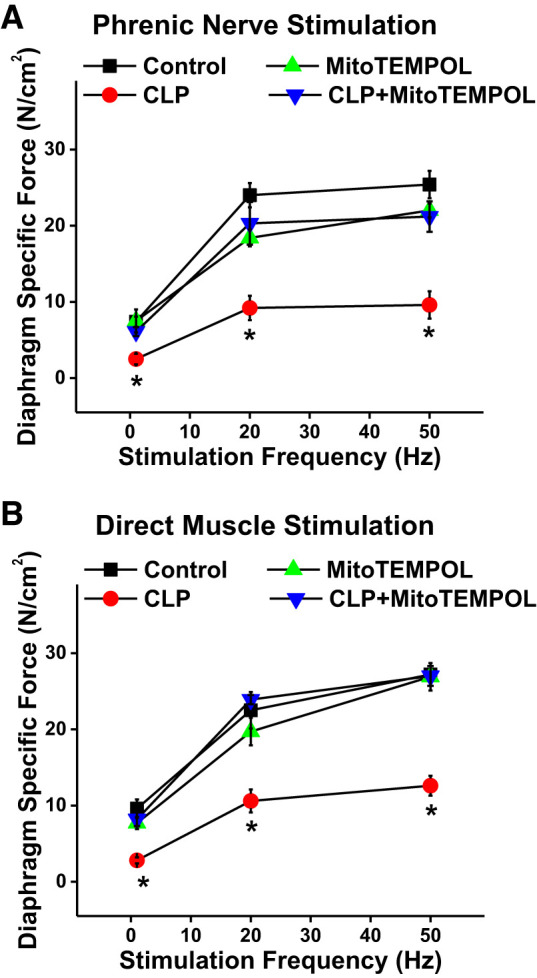
A: diaphragm-specific force generation (i.e., force per cross-sectional area) is shown as a function of phrenic nerve stimulation frequency. Data represent values for the following groups: sham control (black symbols; n = 6), cecal ligation puncture (CLP; red symbols; n = 7), sham control + MitoTEMPOL (green symbols; n = 3), and CLP + MitoTEMPOL (blue symbols; n = 3). B: diaphragm force is shown as a function of direct muscle electrical field stimulation frequency (n = 7 for controls, n = 7 for CLP, n = 5 for MitoTEMPOL, and n = 6 for CLP + MitoTEMPOL). Symbols represent group mean results and error bars represent 1 SE. CLP-induced sepsis produced a large reduction in diaphragm force generation, with either phrenic or direct muscle stimulation, at all excitation frequencies when compared with values for controls (P < 0.01 for comparison at each frequency). MitoTEMPOL administration prevented sepsis-induced reductions in diaphragm force, with diaphragm-specific force for the CLP + MitoTEMPOL group higher for CLP. Force generation for the control + MitoTEMPOL group was similar to that for the control group (not significant). *Statistical significance when compared with other groups.
We assessed diaphragm neuromuscular transmission using the technique of Greising et al. (Fig. 2A) (20). A transmission failure index (TFI) for diaphragms from the various groups of animals was generated by this technique; a higher index indicates greater transmission failure, and a lower index represents less transmission failure (16). We found that TFI was similar across the four experimental groups (Fig. 2B), indicating that CLP did not preferentially alter neuromuscular transmission in our animal model.
Fig. 2.
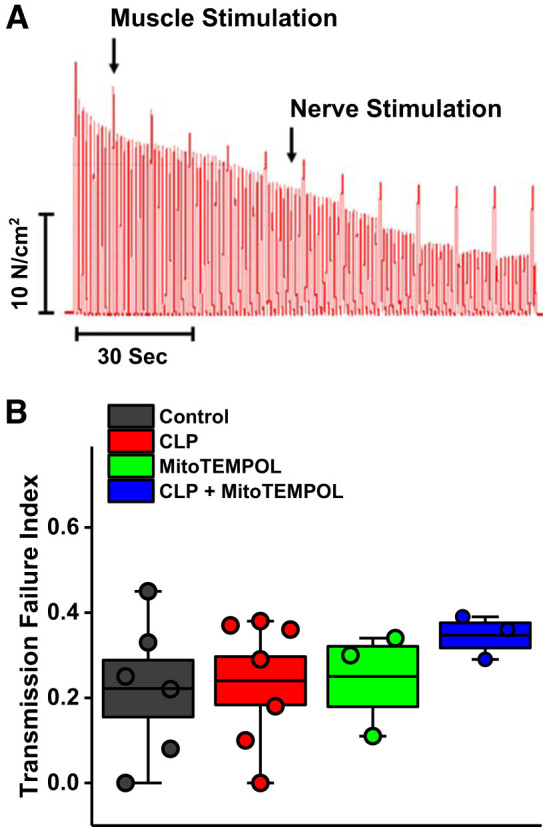
A: neuromuscular transmission failure during repetitive contraction trials was assessed as previously described (20, 48), with a representative experimental tracing shown (A). There was a proportionately greater fall, over time, of force generated in response to phrenic nerve stimulation as compared with direct muscle stimulation in this representative study. Similar results were observed in all experiments from all groups [n = 6 controls, n = 7 cecal ligation puncture (CLP), n = 3 MitoTEMPOL, and n = 3 CLP + MitoTEMPOL]. As shown in B, the calculated transmission failure index was similar for all groups (not significant). These data indicate that neuromuscular transmission was not altered in our animal model of sepsis. Data are plotted with boxes representing 1 SE; the dark line in the box indicates the mean, and whiskers indicate the minimum and maximum values.
We also measured force generation during a repetitive contraction trial induced by direct muscle field stimulation (Fig. 3). We found that the force generated at all points during these repetitive contraction trials was markedly reduced for diaphragms from septic CLP animals as compared with muscles from the other three experimental groups (P < 0.001 for all time points). At the end of these repetitive contraction trials, diaphragm force averaged 12.3 ± 1.8, 11.9 ± 1.7, 4.0 ± 0.7, and 12.6 ± 1.7 N/cm2 for sham controls, sham + MitoTEMPOL, CLP, and CLP + MitoTEMPOL treated groups, respectively (P < 0.01 for comparison of the control group with the other 3 conditions).
Fig. 3.
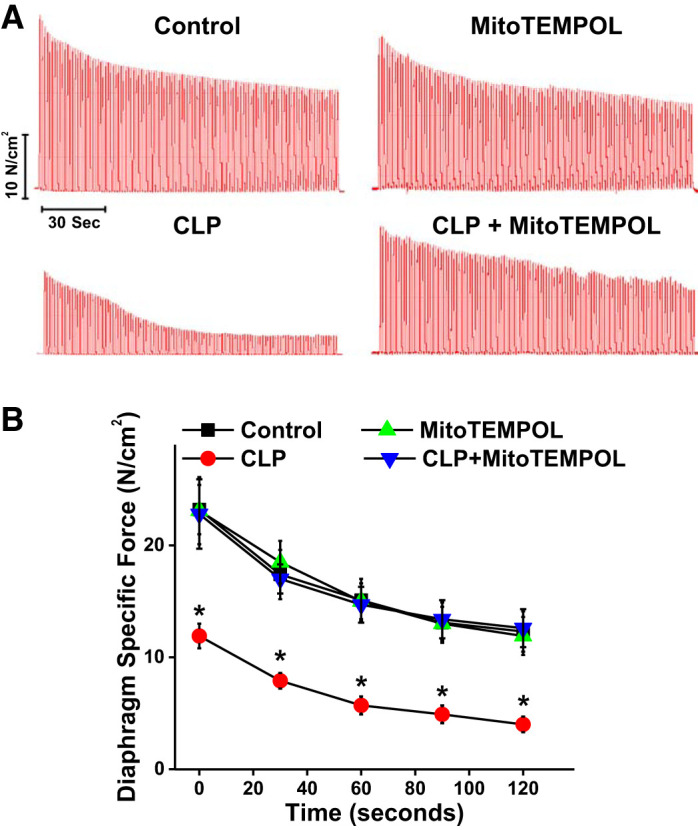
A: representative tracing of repetitive contraction trials from each experimental group [n = 7 for controls, n = 7 for cecal ligation puncture (CLP), n = 4 for MitoTEMPOL, and n = 4 for CLP+ MitoTEMPOL]. B: force generation was significantly lower at all times in diaphragms from septic animals compared with the other experimental groups. *Statistical significance.
While CLP sepsis markedly reduced diaphragm specific force, i.e., force normalized to muscle size, this stress did not significantly reduce diaphragm mass at the time point we examined. Specifically, diaphragm weight normalized to animal weight was not significantly different across the four experimental groups, averaging 1.88 ± 0.06, 1.84 ± 0.11, 1.91 ± 0.10, and 1.95 ± 0.15 mg/g for sham controls, sham + MitoTEMPOL, CLP, and CLP + MitoTEMPOL treated groups, respectively [not significant (NS) for comparison of the CLP group with the other three conditions].
Mitochondrial function.
Mitochondrial function was assessed in diaphragm mitochondria in the four experimental groups (Fig. 4) (3, 5, 7, 43, 46). CLP sepsis markedly reduced ADP-stimulated oxygen consumption (i.e., state 3 respiration, P < 0.001) but did not appreciably alter basal mitochondrial oxygen consumption (state 4 rates). The calculated respiratory control ratios (RCR) and maximal ATP generation rates were also significantly lower for mitochondrial samples from septic animals (P < 0.001 and P < 0.002, respectively). MitoTEMPOL administration prevented all of these CLP-induced reductions in mitochondrial function, increasing state 3 oxygen consumption and RCR and ATP production rates to levels significantly higher than for CLP septic animals (P < 0.01, P < 0.01, and P < 0.02, respectively).
Fig. 4.

A: mitochondrial respiration in response to ADP stimulation was reduced for diaphragms from septic animals and MitoTEMPOL administration prevented this reduction. B: basal, state 4 respiration rates were not, however, different between the 4 experimental groups (NS, not significant). C and D: both respiratory control ratios (C) and ATP generation rates (D) were lower for the cecal ligation puncture (CLP) group and MitoTEMPOL administration increased ATP production and respiratory control ratio (RCR) values compared with that found in the CLP group. We also found that the RCR for the CLP + MitoTEMPOL group was lower than the value for the control group. Data were obtained from n = 3 animals/group. Data are plotted with boxes representing 1 SE; the dark line in the box indicates the mean, and whiskers indicate the minimum and maximum values. *Statistical significance.
Proteolytic activation.
CLP sepsis elicited significant increases in the activity of three proteolytic enzyme systems (Fig. 5). Specifically, diaphragm calpain activity increased by 355% (P < 0.05), caspase 3 activity increased by 352% (P < 0.002), and 20S proteasome activity increased by 226% (P < 0.05; comparison of sham controls and septic groups). MitoTEMPOL administration at the time of surgery completely prevented each of these CLP-induced increases, maintaining proteolytic enzyme activity assays at basal control levels (NS for comparison of controls and CLP + MitoTEMPOL groups). We also found that diaphragm levels of myosin heavy chain were significantly reduced by CLP sepsis (Fig. 6), and MitoTEMPOL administration prevented this sepsis-induced reduction (P < 0.04 for comparison of the CLP group with the other 3 experimental groups).
Fig. 5.
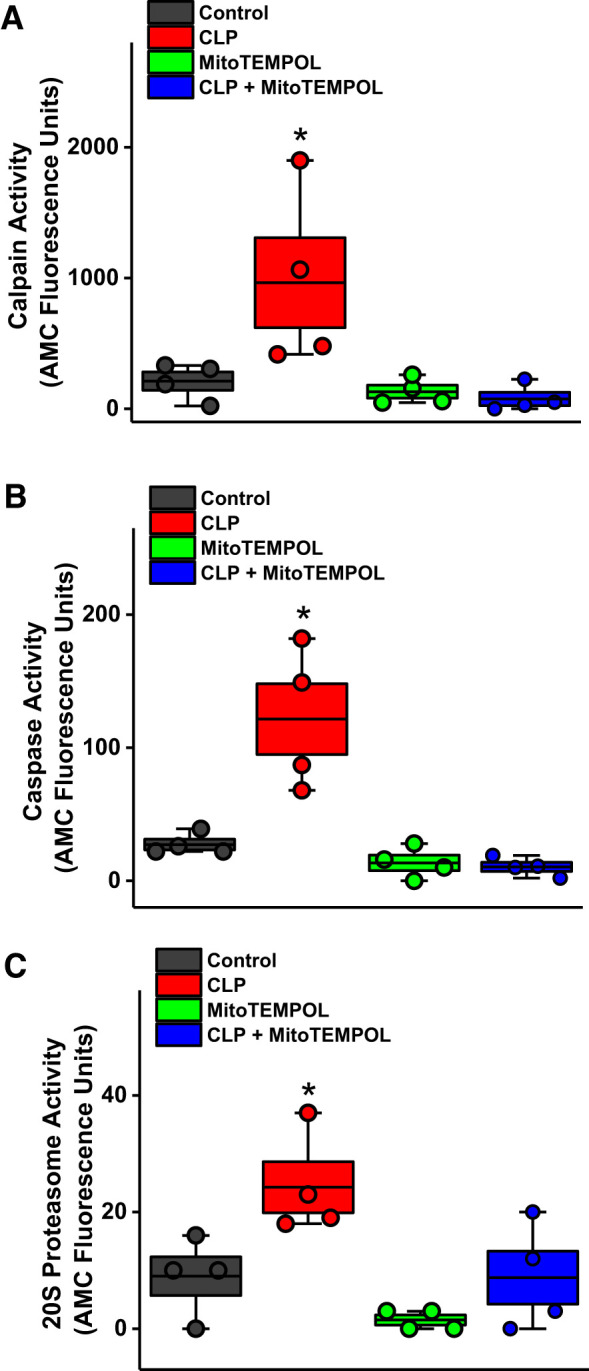
Sepsis-induced increases in calpain (A), caspase (B), and 20S proteasome proteolytic activity (C). MitoTEMPOL administration blocked each of these cecal ligation puncture (CLP)-mediated effects. Data were obtained from n = 4 animals/group. Data are plotted with boxes representing 1 SE; the dark line in the box indicates the mean, and whiskers indicate the minimum and maximum values. *Statistical significance.
Fig. 6.
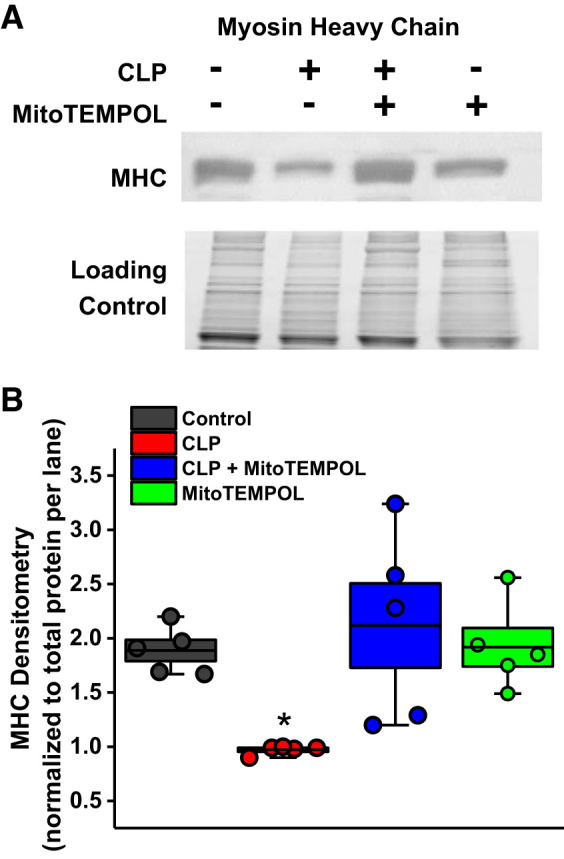
Sepsis-induced reductions in myosin heavy chain levels (MHC); assays were performed on samples from n = 5 animals/group. A: representative blot with the total protein gel which was used for normalization for densitometry. B: mean data for all experimental groups. As shown, administration of MitoTEMPOL prevented cecal ligation puncture (CLP)-induced reductions in MHC. Data are plotted with boxes representing 1 SE; the dark line in the box indicates the mean, and whiskers indicate the minimum and maximum values. *Statistical significance.
Effects of delayed MitoTEMPOL administration.
Patients with sepsis typically present to medical centers only after symptoms develop, so there is an obligatory delay between the onset of disease and the point in time that therapies can be administered. For this reason, we also examined the impact of delaying treatment with MitoTEMPOL for 6 h after CLP surgery. As shown in Fig. 7, delayed therapy with MitoTEMPOL was as effective as immediate therapy in preventing sepsis-induced loss of diaphragm strength, with diaphragm-specific force generation for both CLP + delayed MitoTEMPOL and CLP + immediate MitoTEMPOL groups significantly higher than levels for the CLP group (P < 0.001 for both comparisons). Similar findings were observed for force measurements during repetitive contraction trials (Fig. 7B), with force at every time point similar for CLP + delayed MitoTEMPOL and CLP + immediate MitoTEMPOL groups, with both MitoTEMPOL treatment groups significantly higher than CLP animals (P < 0.01 for both comparisons).
Fig. 7.
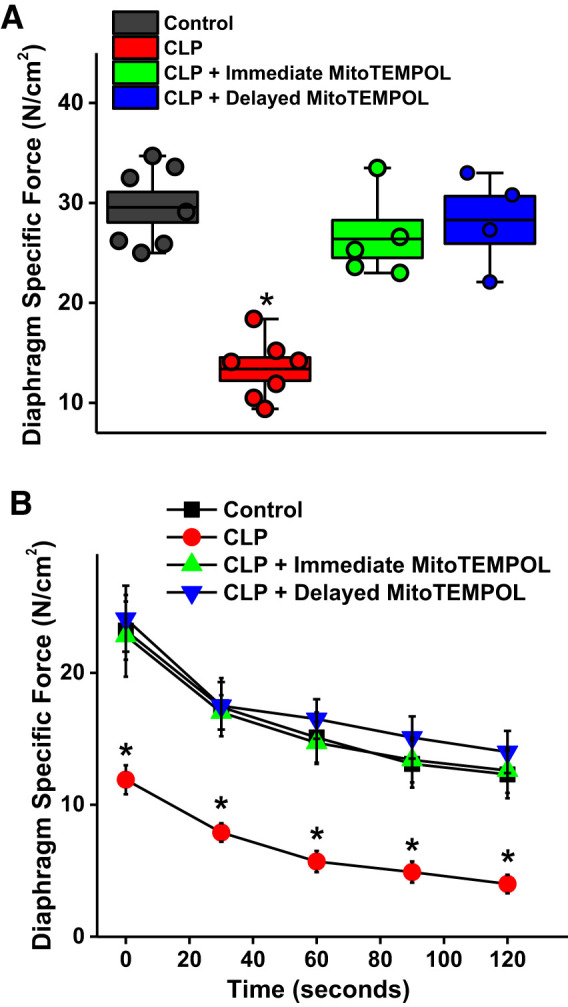
A: delayed administration of MitoTEMPOL (blue) was as effective as immediate postsurgical administration of MitoTEMPOL (green) in preventing sepsis-induced reductions in diaphragm force generation. [P < 0.001 for comparison of both cecal ligation puncture (CLP) + immediate MitoTEMPOL and CLP + delayed MitoTEMPOL groups to CLP alone]. Data were obtained for controls (n = 7), CLP (n = 7), immediate MitoTEMPOL (n = 5), and delayed MitoTEMPOL (n = 4) groups. Boxes represent 1 SE, center line in the box indicates the mean, and whiskers indicate the minimum and maximum values. B: demonstration that these effects of delayed MitoTEMPOL administration were maintained across all excitation frequencies and were significantly different when compared with values for septic animals (n = 7 control, n = 7 CLP, immediate n = 4 MitoTEMPOL, and n = 4 delayed MitoTEMPOL groups). *Statistical significance.
Time course of superoxide generation relative to proteolytic activity.
We also examined the time course of development of proteolytic pathway activation relative to increases in diaphragm superoxide generation by measuring diaphragm aconitase activity. Aconitase is present only in skeletal muscle mitochondria and is inactivated by mitochondrial superoxide generation, so reductions in aconitase activity can be used as an index of mitochondrial superoxide generation (27). As shown in Fig. 8, aconitase activity decreased at an early time point (4 h) after CLP, while proteolytic enzyme activity increased at later time points (8 and 24 h). Administration of MitoTEMPOL at the time of induction of CLP blocked both alterations in aconitase activity at all time points and prevented proteolytic enzyme activation at 24 h.
Fig. 8.

Diaphragm aconitase activity decreased by 4 h after cecal ligation puncture (CLP) (A) and remained low until 24 h after CLP (P < 0.05 for comparison of control levels to the other 3 time points). MitoTEMPOL-treated CLP animals had higher aconitase levels at all time points after CLP as compared with saline-treated CLP groups (P < 0.001, P < 0.02, and P < 0.01 for comparison of aconitase levels at 4 h, 8 h, and 24 h after CLP between CLP alone and CLP + MitoTEMPOL groups. Proteolytic enzyme activities (caspase in B, calpain in C, and 20S proteasome in D) were not significantly different from control animal values at 4 h but increased by 24 h. MitoTEMPOL administration reduced all 3 proteolytic enzyme activities at the 24-h time point (P = 0.009, P = 0.006, and P = 0.033 for caspase, calpain, and 20S proteasome activities, respectively, for comparison of CLP and CLP + MitoTEMPOL groups at the 24-h time point). Data were obtained from n = 3 animals/group/time point. *Statistical significance.
Effects of MitoTEMPOL on a muscle cell line.
We examined the effect of a mixture of cytokines (cytomix; 20 ng/mL TNFα, 50 U/mL IL-1β, 100 U/mL IFNγ, and 10 μg/mL LPS) (20) on superoxide generation and size in a muscle cell line (i.e., C2C12 cells) cultured in vitro. As shown in Fig. 9, cytomix induced a large increase in muscle cell superoxide generation, as assessed using the MitoSOX assay (P < 0.001), and also induced significant reductions in muscle cell width (P < 0.001; Fig. 10). Administration of MitoTEMPOL to cytomix-treated cells ablated cytomix-induced cell superoxide generation and prevented cytomix-induced reductions in cell width (P < 0.01 and P < 0.001, respectively, for comparison of superoxide production and cell width for cytomix and cytomix + MitoTEMPOL groups).
Fig. 9.
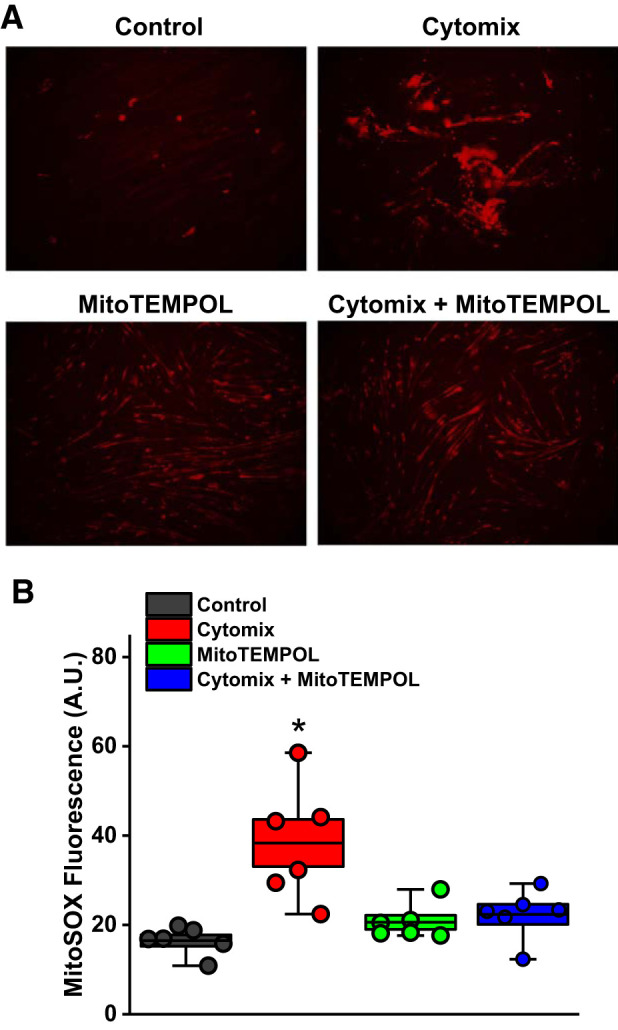
Addition of cytokines (i.e., cytomix, a combination of TNFα, IL1β, IFNγ, and LPS) elicited a large increase in superoxide generation of C2C12 myocytes (representative examples of MitoSOX activity in A, mean data in B; images obtained at ×20 magnification). Addition of MitoTEMPOL ablated cytomix-induced increases in MitoSOX activity. Data were obtained from n = 5 plates of cells/group. Boxes represent 1 SE; center line in the box indicates the mean, and whiskers indicate the minimum and maximum values. *Statistical significance.
Fig. 10.
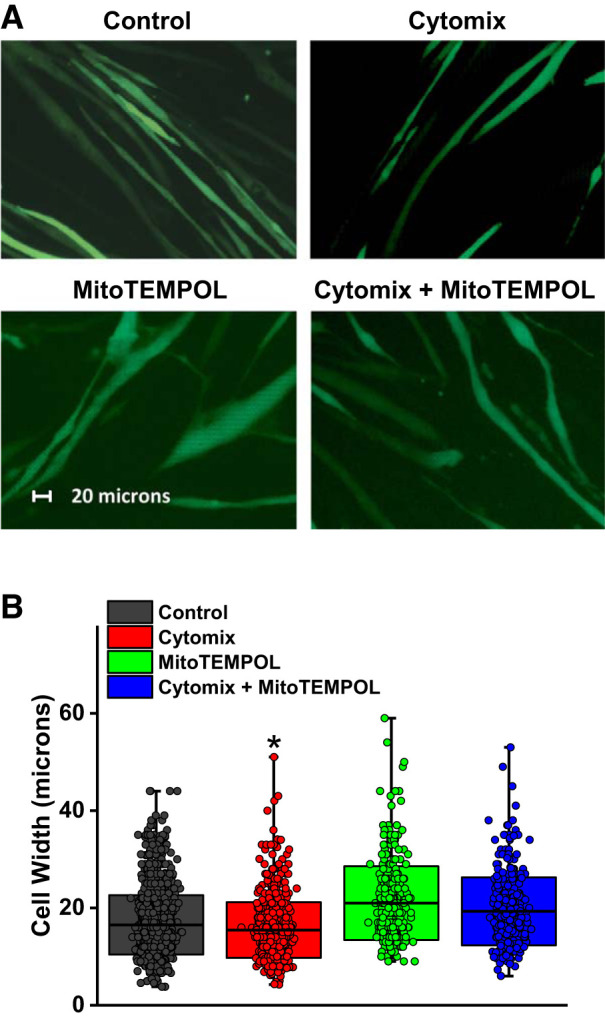
Cytomix administration reduced mean myocyte cell width (representative microphotographs in A, mean data in B). Concomitant administration of MitoTEMPOL prevented cytomix-mediated reductions in myocyte width. Data were obtained from n = 5 plates of cells/group with the following numbers of cells counted (n = 1,440 for controls, n = 720 for cytomix, n = 360 for MitoTEMPOL, and n = 360 for cytomix + MitoTEMPOL). In B, boxes represent 1 SD; center line in the box indicates the mean, and whiskers indicate the minimum and maximum values. *Statistical significance.
DISCUSSION
The major finding of the present study is that it is possible to prevent sepsis-induced reductions in diaphragm function by administration of MitoTEMPOL, a mitochondrially targeted superoxide scavenger. We found that sepsis dramatically reduced diaphragm strength, decreasing maximal force generation by 54%, with MitoTEMPOL preventing diaphragm weakness. Diaphragm force generation during repetitive contraction trials was also severely reduced throughout trials in septic muscles when compared with muscles from control animals, and MitoTEMPOL administration maintained force during these trials to that observed for diaphragms from control animals. We also found that MitoTEMPOL reversed sepsis-mediated reductions in diaphragm mitochondrial function, prevented diaphragm activation of three important proteolytic pathways (calpain, caspase, 20S proteasome), and prevented sepsis-induced reductions in diaphragm myosin heavy chain (MHC) content. Both immediate and delayed MitoTEMPOL administration prevented sepsis-induced diaphragm weakness, suggesting that this drug would be effective even when short delays prevent immediate administration. Finally, we assessed indices of mitochondrial superoxide generation in intact animals (i.e., the aconitase assay) and in isolated muscle cells (i.e., the MitoSOX assay). We found that mitochondrial superoxide generation increased in diaphragms in response to sepsis and in isolated cells in response to inflammatory cytokines and, moreover, that MitoTEMPOL prevented increases in mitochondrial superoxide in response to these stresses in both animals in vivo and in cells in vitro.
The rationale for the current experiment is linked to recent studies demonstrating that mechanically ventilated patients cared for in medical intensive care units (MICU) develop severe diaphragm weakness (11, 13, 22, 25, 41, 52, 55). Importantly, these prior studies also suggest that diaphragm weakness in this population has serious consequences, with weaker patients requiring longer durations of mechanical ventilation and manifesting a much higher mortality (41, 52). It has been suggested, moreover, that diaphragm weakness contributes to mortality by making it far more difficult to wean patients from mechanical ventilation, influencing the decision to withdraw care because of lack of progress (41, 52). One of the major clinical factors that appears to influence the development of diaphragm weakness in patients is sepsis (11, 41, 52). In support of such a possibility, numerous animal studies have shown that models of infection (cecal ligation perforation, endotoxin administration, pseudomonas pneumonia) result in severe diaphragm muscle weakness (12, 24, 39, 51).
Animal studies examining the relationship between infection and diaphragm function have shown that there are multiple mechanisms by which infections may alter muscle function, including impairment of excitation-contraction coupling and reductions in protein synthesis (2, 10, 26, 30, 58). Evidence suggests, however, that the major cellular processes by which infections affect muscle function may be induction of excessive muscle free radical generation, increased muscle proteolysis, and reductions in muscle mitochondrial function (3, 6, 19, 24, 36, 37, 39, 40, 50, 51, 54). It has also been hypothesized that these three phenomena may be pathophysiologically linked. According to this theory, infections induce excessive muscle mitochondrial free radical generation (3, 36, 37), which then activates proteolysis (24, 39, 40, 50, 51). Proteolytic pathway activation (e.g., the proteasome, caspase, calpain) is then thought to induce loss of critical skeletal muscle proteins (19, 50, 54), leading to weakness.
The current findings are consistent with this theory. Specifically, we found that sepsis led to increases in diaphragm caspase activity, calpain activity, and 20S proteasome activity, with MitoTEMPOL administration attenuating each of these sepsis-induced alterations. This finding is consistent with a link between sepsis-induced increases in muscle mitochondrial free radical generation and the activation of muscle proteolytic pathways. We also found that diaphragm myosin heavy chain (MHC) levels decreased in response to CLP-induced sepsis, and MitoTEMPOL blocked this depletion. Previous reports indicate that both caspase and calpain and the proteasome proteolytic pathways have the capacity to induce MHC cleavage, and as a result, the ability of MitoTEMPOL to prevent MHC loss would be consistent with an effect to reduce proteolytic enzyme activation (17, 34, 35).
A second process by which sepsis-induced mitochondrial superoxide generation may influence muscle function is by an effect of superoxide generation to induce mitochondrial dysfunction. Maintenance of muscle force development during repetitive muscle contractions is dependent upon the ability of the muscle to resynthesize ATP, thereby maintaining levels of high-energy phosphate compounds (ATP and CP) and low levels of inorganic phosphate (9, 18). Previous work has shown that sepsis leads to depletion of several key electron transport chain proteins, reducing mitochondrial state 3 respiration rates and ATP generation (7). In addition, sepsis also causes sarcomeric creatine kinase dysfunction, impairing transport of ATP from mitochondria to the cytosol, and reduces mRNA transcription of mitochondrial electron transport chain protein subunits, impairing the ability of muscle to replace damaged mitochondrial proteins (4). Importantly, infections are known to reduce muscle endurance, with muscles excised from infected animals demonstrating an inability to maintain force generation when compared with muscles taken from healthy controls (59). These phenomena may be pathophysiologically linked, with excessive free radical generation the cause of mitochondrial dysfunction and mitochondrial dysfunction, in turn, making it difficult for muscles to maintain force generation during repetitive contractions. The findings of the present study are consistent with this possibility. We found that sepsis reduced mitochondrial state 3 respiration rate and ATP generation rate, and MitoTEMPOL administration to septic animals blocked these sepsis-induced alterations. This ability of MitoTEMPOL to preserve mitochondrial respiration provides the likely explanation for the effect of this agent to improve diaphragm function during repetitive contraction trials.
Critically ill patients are known to develop myopathies, but critical care illnesses, and sepsis in particular, are also known to induce neuropathies and impaired neuromuscular transmission (29). To evaluate the possibility that CLP alters diaphragm function, at least in part, by effect on neural transmission, we assessed diaphragm force generation in response to electrical stimulation of the phrenic nerve and also examined relative fatigue rates with repetitive muscle and nerve stimulation using the technique described by Greising et al. (20). Under the conditions of our experimental model, however, we did not find that sepsis had detrimental effects on neuromuscular transmission during repetitive stimulation. In addition, while sepsis elicited a large downward shift in the diaphragm force-frequency curve assessed in response to phrenic nerve stimulation, the magnitude of this reduction was the same as that observed in response to direct muscle stimulation. The current data support the concept that sepsis-induced diaphragm dysfunction is primarily the result of a myopathic process rather than alterations in neuromuscular transmission.
MitoTEMPOL is a mitochondrially targeted agent that is a dismutase mimetic that directly catalyzes superoxide and also exerts other antioxidant actions through the effects of intracellular derivatives (53). In addition to reducing mitochondrial superoxide, it is also theoretically possible that this agent can reduce cellular levels of superoxide generated by nonmitochondrial cellular pathways of oxidant formation. To provide evidence that MitoTEMPOL did, in fact, influence mitochondrial superoxide formation in the present study, we measured indices of mitochondrial superoxide generation in muscles from septic animals and in isolated muscle cells exposed to a mixture of cytokines (38). In both systems, we found that superoxide generation increased in response to septic stimuli (CLP in intact animals, cytokines in isolated cells), and MitoTEMPOL administration blocked these increases. These findings are consistent with the interpretation that this agent primarily acts in muscle by suppressing mitochondrial sources of superoxide. Additional studies will be needed in the future to determine whether this agent also has effects on other potential sources of infection-induced oxidant generation (e.g., NADPH oxidase).
Finally, we found that delayed administration of MitoTEMPOL was as effective as immediate administration (i.e., at the time of induction of cecal ligation perforation) in maintaining diaphragm force generation. This is an important consideration since there is inevitably a delay between the time that a patient develops an infection and the point that the same patient presents for medical care. Some work suggests that examination of the effectiveness of a therapy after a delay of 5–8 h from the induction of sepsis is clinically relevant and represents a potentially achievable therapeutic application of a given treatment (56). If so, our data regarding the effectiveness of delayed MitoTEMPOL administration reinforces the potential translational relevance of this treatment.
There are, however, several limitations to this work. First, we chose to examine the effects of sepsis on the diaphragm primarily because dysfunction of this muscle may be a cause of poor outcomes for intensive care unit (ICU) patients. Sepsis also reduces limb muscle function, however, and dysfunction of these muscles is linked to long-term post-ICU exercise intolerance and reductions in activities of daily living. Future studies will be needed to determine whether the same mechanisms and responses observed in the diaphragm with MitoTEMPOL also occur in the limb musculature. Sepsis is also known to induce mitochondrial dysfunction in a wide variety of other organs during sepsis; additional work will also be needed to see whether this agent can prevent mitochondrial dysfunction and tissue injury in these other organs. We also looked at a relatively circumscribed duration of sepsis and treatment. Future studies will need to determine how long diaphragm dysfunction lasts in this and other models of sepsis (e.g., fecal slurry induced sepsis) and what treatment duration with MitoTEMPOL is effective.
In conclusion, our data indicate that MitoTEMPOL administration prevents sepsis-induced reductions in diaphragm force generation and diaphragm mitochondrial function. Our findings support the hypothesis that infection-induced muscle mitochondrial superoxide generation is linked to muscle proteolytic pathway activation, depletion of critical contractile proteins (i.e., myosin heavy chain), and muscle weakness. These data also support the hypothesis that infection-induced mitochondrial superoxide generation causes mitochondrial dysfunction, which further reduces the ability of muscles to maintain force generation during repetitive contractions. It is important to recognize that a variety of additional mitochondrially targeted antioxidants with mechanisms of action similar to those of MitoTEMPOL are currently in varying stages of testing in clinical trials in the United States and Europe (31). In general, these agents have been found to be safe in early clinical studies, and several are expected to be approved for clinical use in the near future. As a result, there is the potential for translation of MitoTEMPOL and/or agents with similar mechanisms of action into clinical trials of infected patients in the future, with the possibility that these treatments can preserve diaphragm function and clinical outcomes in critically ill patients.
GRANTS
G. S. Supinski is supported by R01HL113494 and R01HL141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health and by 5I01BX002132 from the Department of Veterans Affairs. L. A. P. Callahan is supported by R01HL112085 and R01HL141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health. E. A. Schroder is supported by R01HL141356 from the National Heart, Lung, and Blood Institute of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.S.S. conceived and designed research; G.S.S., L.W., and E.A.S. performed experiments; G.S.S., L.W., E.A.S., and L.A.P.C. analyzed data; G.S.S., L.W., E.A.S., and L.A.P.C. interpreted results of experiments; L.A.P.C. prepared figures; G.S.S. drafted manuscript; G.S.S., L.W., E.A.S., and L.A.P.C. edited and revised manuscript; G.S.S., L.W., E.A.S., and L.A.P.C. approved final version of manuscript.
REFERENCES
- 1.Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, Zimmers TA. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 6: e22538, 2011. doi: 10.1371/journal.pone.0022538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callahan LA, Nethery D, Stofan D, DiMarco A, Supinski G. Free radical-induced contractile protein dysfunction in endotoxin-induced sepsis. Am J Respir Cell Mol Biol 24: 210–217, 2001. doi: 10.1165/ajrcmb.24.2.4075. [DOI] [PubMed] [Google Scholar]
- 3.Callahan LA, Stofan DA, Szweda LI, Nethery DE, Supinski GS. Free radicals alter maximal diaphragmatic mitochondrial oxygen consumption in endotoxin-induced sepsis. Free Radic Biol Med 30: 129–138, 2001. doi: 10.1016/S0891-5849(00)00454-8. [DOI] [PubMed] [Google Scholar]
- 4.Callahan LA, Supinski GS. Diaphragm and cardiac mitochondrial creatine kinases are impaired in sepsis. J Appl Physiol (1985) 102: 44–53, 2007. doi: 10.1152/japplphysiol.01204.2005. [DOI] [PubMed] [Google Scholar]
- 5.Callahan LA, Supinski GS. Downregulation of diaphragm electron transport chain and glycolytic enzyme gene expression in sepsis. J Appl Physiol (1985) 99: 1120–1126, 2005. doi: 10.1152/japplphysiol.01157.2004. [DOI] [PubMed] [Google Scholar]
- 6.Callahan LA, Supinski GS. Sepsis-induced myopathy. Crit Care Med 37, Suppl: S354–S367, 2009. doi: 10.1097/CCM.0b013e3181b6e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callahan LA, Supinski GS. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am J Respir Crit Care Med 172: 861–868, 2005. doi: 10.1164/rccm.200410-1344OC. [DOI] [PubMed] [Google Scholar]
- 8.Close RI. Dynamic properties of mammalian skeletal muscles. Physiol Rev 52: 129–197, 1972. doi: 10.1152/physrev.1972.52.1.129. [DOI] [PubMed] [Google Scholar]
- 9.Cooke R, Franks K, Luciani GB, Pate E. The inhibition of rabbit skeletal muscle contraction by hydrogen ions and phosphate. J Physiol 395: 77–97, 1988. doi: 10.1113/jphysiol.1988.sp016909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowell KT, Soybel DI, Lang CH. Restorative mechanisms regulating protein balance in skeletal muscle during recovery from sepsis. Shock 47: 463–473, 2017. doi: 10.1097/SHK.0000000000000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demoule A, Jung B, Prodanovic H, Molinari N, Chanques G, Coirault C, Matecki S, Duguet A, Similowski T, Jaber S. Diaphragm dysfunction on admission to the intensive care unit. Prevalence, risk factors, and prognostic impact-a prospective study. Am J Respir Crit Care Med 188: 213–219, 2013. doi: 10.1164/rccm.201209-1668OC. [DOI] [PubMed] [Google Scholar]
- 12.Divangahi M, Matecki S, Dudley RW, Tuck SA, Bao W, Radzioch D, Comtois AS, Petrof BJ. Preferential diaphragmatic weakness during sustained Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med 169: 679–686, 2004. doi: 10.1164/rccm.200307-949OC. [DOI] [PubMed] [Google Scholar]
- 13.Dres M, Goligher EC, Heunks LMA, Brochard LJ. Critical illness-associated diaphragm weakness. Intensive Care Med 43: 1441–1452, 2017. doi: 10.1007/s00134-017-4928-4. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh R, Gilda JE, Gomes AV. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev Proteomics 11: 549–560, 2014. doi: 10.1586/14789450.2014.939635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilda JE, Gomes AV. Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal Biochem 440: 186–188, 2013. doi: 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilda JE, Gomes AV. Western blotting using in-gel protein labeling as a normalization control: stain-free technology. Methods Mol Biol 1295: 381–391, 2015. doi: 10.1007/978-1-4939-2550-6_27. [DOI] [PubMed] [Google Scholar]
- 17.Gilliam LA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 302: C195–C202, 2012. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godt RE, Nosek TM. Changes of intracellular milieu with fatigue or hypoxia depress contraction of skinned rabbit skeletal and cardiac muscle. J Physiol 412: 155–180, 1989. doi: 10.1113/jphysiol.1989.sp017609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gómez-SanMiguel AB, Villanúa MA, Martín AI, López-Calderón A. D-TRP(8)-gammaMSH prevents the effects of endotoxin in rat skeletal muscle cells through TNFα/NF-KB signalling pathway. PLoS One 11: e0155645, 2016. doi: 10.1371/journal.pone.0155645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greising SM, Ermilov LG, Sieck GC, Mantilla CB. Ageing and neurotrophic signalling effects on diaphragm neuromuscular function. J Physiol 593: 431–440, 2015. doi: 10.1113/jphysiol.2014.282244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greising SM, Vasdev AK, Zhan WZ, Sieck GC, Mantilla CB. Chronic TrkB agonist treatment in old age does not mitigate diaphragm neuromuscular dysfunction. Physiol Rep 5: e13103, 2017. doi: 10.14814/phy2.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermans G, Agten A, Testelmans D, Decramer M, Gayan-Ramirez G. Increased duration of mechanical ventilation is associated with decreased diaphragmatic force: a prospective observational study. Crit Care 14: R127, 2010. doi: 10.1186/cc9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu H, Li M. Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem Biophys Res Commun 478: 174–180, 2016. doi: 10.1016/j.bbrc.2016.07.071. [DOI] [PubMed] [Google Scholar]
- 24.Kovarik M, Muthny T, Sispera L, Holecek M. Effects of β-hydroxy-β-methylbutyrate treatment in different types of skeletal muscle of intact and septic rats. J Physiol Biochem 66: 311–319, 2010. doi: 10.1007/s13105-010-0037-3. [DOI] [PubMed] [Google Scholar]
- 25.Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120–127, 2003. doi: 10.1164/rccm.200210-1246OC. [DOI] [PubMed] [Google Scholar]
- 26.Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab 293: E453–E459, 2007. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- 27.Larsen FJ, Schiffer TA, Ørtenblad N, Zinner C, Morales-Alamo D, Willis SJ, Calbet JA, Holmberg HC, Boushel R. High-intensity sprint training inhibits mitochondrial respiration through aconitase inactivation. FASEB J 30: 417–427, 2016. doi: 10.1096/fj.15-276857. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Moylan JS, Chambers MA, Smith J, Reid MB. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 297: C706–C714, 2009. doi: 10.1152/ajpcell.00626.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nayci A, Atis S, Comelekoglu U, Ozge A, Ogenler O, Coskun B, Zorludemir S. Sepsis induces early phrenic nerve neuropathy in rats. Eur Respir J 26: 686–692, 2005. doi: 10.1183/09031936.05.0111004. [DOI] [PubMed] [Google Scholar]
- 30.Novak KR, Nardelli P, Cope TC, Filatov G, Glass JD, Khan J, Rich MM. Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J Clin Invest 119: 1150–1158, 2009. doi: 10.1172/JCI36570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oyewole AO, Birch-Machin MA. Mitochondria-targeted antioxidants. FASEB J 29: 4766–4771, 2015. doi: 10.1096/fj.15-275404. [DOI] [PubMed] [Google Scholar]
- 32.Posch A, Kohn J, Oh K, Hammond M, Liu N. V3 stain-free workflow for a practical, convenient, and reliable total protein loading control in western blotting. J Vis Exp: 50948, 2013. doi: 10.3791/50948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocha VC, França LS, de Araújo CF, Ng AM, de Andrade CM, Andrade AC, Santos ES, Borges-Silva MC, Macambira SG, Noronha-Dutra AA, Pontes-de-Carvalho LC. Protective effects of mito-TEMPO against doxorubicin cardiotoxicity in mice. Cancer Chemother Pharmacol 77: 659–662, 2016. doi: 10.1007/s00280-015-2949-7. [DOI] [PubMed] [Google Scholar]
- 34.Samengo G, Avik A, Fedor B, Whittaker D, Myung KH, Wehling-Henricks M, Tidball JG. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell 11: 1036–1045, 2012. doi: 10.1111/acel.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 271: 26690–26697, 1996. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 36.Supinski G, Nethery D, DiMarco A. Effect of free radical scavengers on endotoxin-induced respiratory muscle dysfunction. Am Rev Respir Dis 148: 1318–1324, 1993. doi: 10.1164/ajrccm/148.5.1318. [DOI] [PubMed] [Google Scholar]
- 37.Supinski GS, Alimov AP, Wang L, Song XH, Callahan LA. Calcium-dependent phospholipase A2 modulates infection-induced diaphragm dysfunction. Am J Physiol Lung Cell Mol Physiol 310: L975–L984, 2016. doi: 10.1152/ajplung.00312.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Supinski GS, Alimov AP, Wang L, Song XH, Callahan LA. Neutral sphingomyelinase 2 is required for cytokine-induced skeletal muscle calpain activation. Am J Physiol Lung Cell Mol Physiol 309: L614–L624, 2015. doi: 10.1152/ajplung.00141.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Supinski GS, Callahan LA. β-hydroxy-β-methylbutyrate (HMB) prevents sepsis-induced diaphragm dysfunction in mice. Respir Physiol Neurobiol 196: 63–68, 2014. doi: 10.1016/j.resp.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Supinski GS, Callahan LA. Calpain activation contributes to endotoxin-induced diaphragmatic dysfunction. Am J Respir Cell Mol Biol 42: 80–87, 2010. doi: 10.1165/rcmb.2008-0275OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Supinski GS, Callahan LA. Diaphragm weakness in mechanically ventilated critically ill patients. Crit Care 17: R120, 2013. doi: 10.1186/cc12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Supinski GS, Callahan LA. Double-stranded RNA-dependent protein kinase activation modulates endotoxin-induced diaphragm weakness. J Appl Physiol (1985) 110: 199–205, 2011. doi: 10.1152/japplphysiol.01203.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Supinski GS, Callahan LA. Hemin prevents cardiac and diaphragm mitochondrial dysfunction in sepsis. Free Radic Biol Med 40: 127–137, 2006. doi: 10.1016/j.freeradbiomed.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 44.Supinski GS, Ji X, Callahan LA. The JNK MAP kinase pathway contributes to the development of endotoxin-induced diaphragm caspase activation. Am J Physiol Regul Integr Comp Physiol 297: R825–R834, 2009. doi: 10.1152/ajpregu.90849.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Supinski GS, Ji X, Wang W, Callahan LA. The extrinsic caspase pathway modulates endotoxin-induced diaphragm contractile dysfunction. J Appl Physiol (1985) 102: 1649–1657, 2007. doi: 10.1152/japplphysiol.00377.2006. [DOI] [PubMed] [Google Scholar]
- 46.Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol 297: R1095–R1102, 2009. doi: 10.1152/ajpregu.90902.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Supinski GS, Vanags J, Callahan LA. Effect of proteasome inhibitors on endotoxin-induced diaphragm dysfunction. Am J Physiol Lung Cell Mol Physiol 296: L994–L1001, 2009. doi: 10.1152/ajplung.90404.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Supinski GS, Wang L, Schroder EA, Callahan LAP. SS31, a mitochondrially targeted antioxidant, prevents sepsis-induced reductions in diaphragm strength and endurance. J Appl Physiol (1985) 128: 463–472, 2020. doi: 10.1152/japplphysiol.00240.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Supinski GS, Wang L, Schroder EA, Callahan LAP. Taurine administration ablates sepsis induced diaphragm weakness. Respir Physiol Neurobiol 271: 103289, 2020. doi: 10.1016/j.resp.2019.103289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Supinski GS, Wang L, Song XH, Moylan JS, Callahan LA. Muscle-specific calpastatin overexpression prevents diaphragm weakness in cecal ligation puncture-induced sepsis. J Appl Physiol (1985) 117: 921–929, 2014. doi: 10.1152/japplphysiol.00975.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Supinski GS, Wang W, Callahan LA. Caspase and calpain activation both contribute to sepsis-induced diaphragmatic weakness. J Appl Physiol (1985) 107: 1389–1396, 2009. doi: 10.1152/japplphysiol.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Supinski GS, Westgate P, Callahan LA. Correlation of maximal inspiratory pressure to transdiaphragmatic twitch pressure in intensive care unit patients. Crit Care 20: 77, 2016. doi: 10.1186/s13054-016-1247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trnka J, Blaikie FH, Logan A, Smith RA, Murphy MP. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic Res 43: 4–12, 2009. doi: 10.1080/10715760802582183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Hees HW, Schellekens WJ, Linkels M, Leenders F, Zoll J, Donders R, Dekhuijzen PN, van der Hoeven JG, Heunks LM. Plasma from septic shock patients induces loss of muscle protein. Crit Care 15: R233, 2011. doi: 10.1186/cc10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watson AC, Hughes PD, Louise Harris M, Hart N, Ware RJ, Wendon J, Green M, Moxham J. Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 29: 1325–1331, 2001. doi: 10.1097/00003246-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 56.Wu R, Dong W, Qiang X, Wang H, Blau SA, Ravikumar TS, Wang P. Orexigenic hormone ghrelin ameliorates gut barrier dysfunction in sepsis in rats. Crit Care Med 37: 2421–2426, 2009. doi: 10.1097/CCM.0b013e3181a557a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao Y, Karam C, Yi J, Zhang L, Li X, Yoon D, Wang H, Dhakal K, Ramlow P, Yu T, Mo Z, Ma J, Zhou J. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol Res 138: 25–36, 2018. doi: 10.1016/j.phrs.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Liu H, Li S, Wu J, Sun J. SERCA1 attenuates diaphragm relaxation and uptake rate of SERCA in rats with acute sepsis. Mol Med Rep 16: 5015–5022, 2017. doi: 10.3892/mmr.2017.7134. [DOI] [PubMed] [Google Scholar]
- 59.Zolfaghari PS, Carré JE, Parker N, Curtin NA, Duchen MR, Singer M. Skeletal muscle dysfunction is associated with derangements in mitochondrial bioenergetics (but not UCP3) in a rodent model of sepsis. Am J Physiol Endocrinol Metab 308: E713–E725, 2015. doi: 10.1152/ajpendo.00562.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]