

Summary
DNA mismatch repair (MMR) corrects replication errors and is recruited by the histone mark H3K36me3, enriched in exons of transcriptionally active genes. To dissect in vivo the mutational landscape shaped by these processes, we employed single-cell exome sequencing on T cells of wild-type and MMR-deficient (Mlh1−/−) mice. Within active genes, we uncovered a spatial bias in MMR efficiency: 3′ exons, often H3K36me3-enriched, acquire significantly fewer MMR-dependent mutations compared with 5′ exons. Huwe1 and Mcm7 genes, both active during lymphocyte development, stood out as mutational hotspots in MMR-deficient cells, demonstrating their intrinsic vulnerability to replication error in this cell type. Both genes are H3K36me3-enriched, which can explain MMR-mediated elimination of replication errors in wild-type cells. Thus, H3K36me3 can boost MMR in transcriptionally active regions, both locally and globally. This offers an attractive concept of thrifty MMR targeting, where critical genes in each cell type enjoy preferential shielding against de novo mutations.
Subject Areas: Genetics, Genomics, Molecular Biology
Graphical Abstract
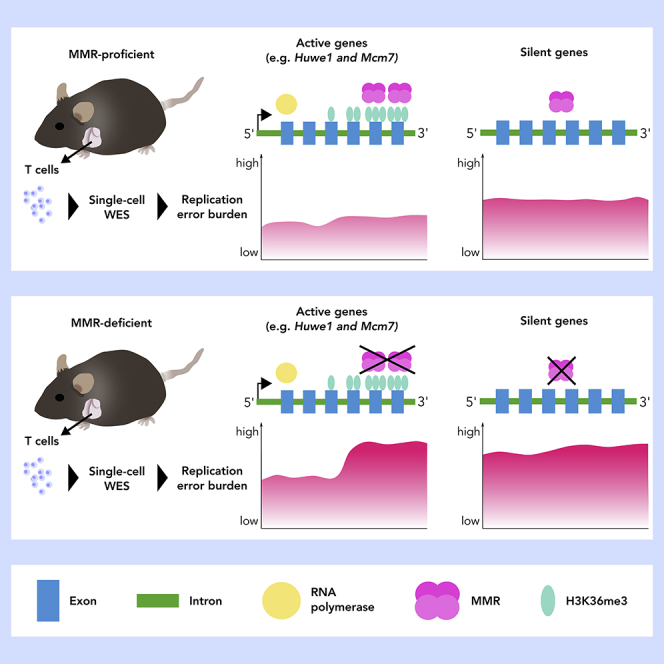
Highlights
-
•
Mutational hotspots can be identified using single-cell sequencing in Mlh1−/− mice
-
•
Mcm7 and Huwe1 genes represent mutational hotspots in non-malignant T cells
-
•
In vivo, 3′ exons of active genes enjoy MMR-mediated protection against mutations
Genetics; Genomics; Molecular Biology
Introduction
Maintaining genomic integrity during DNA replication is crucial for cellular homeostasis, especially in protein-coding regions. Occasionally, DNA replication errors occur, of which most, but not all, are corrected by the intrinsic proofreading activity of DNA polymerases (St Charles et al., 2015). DNA mismatch repair (MMR) corrects base-base mismatches and small insertion-deletion (indel) loops that have escaped proofreading and thereby protects the genome from replication-induced permanent mutations (Li, 2008). MMR initiates when the MSH2/MSH6 (MutSα) or MSH2/MSH3 (MutSβ) complex recognizes and binds DNA lesions, a step followed by recruitment of the MLH1/PMS2 (MutLα) complex that triggers the excision and repair of the mismatch (Lahue et al., 1989; Zhang et al., 2005).
MSH6 of MutSα can bind to trimethylated histone H3 lysine 36 (H3K36me3) and recruit the MMR machinery to chromatin (Li et al., 2013). H3K36me3 is found in exonic regions and enriched at the 3′ ends of transcribed genes (Kolasinska-Zwierz et al., 2009) and also in constitutive and facultative heterochromatin (Chantalat et al., 2011). Recently, H3K36me3 has been shown to also guide m6A deposition to mRNA (Huang et al., 2019), which is known to affect mRNA stability and translation (Huang et al., 2018a; Wang et al., 2014, 2015). Genome-wide mutational analyses of MMR-deficient cell lines and tumors have shown that presence of H3K36me3 reduces local mutation rate (Supek and Lehner, 2015, 2017). Moreover, in tumors and cell lines, MMR operates more efficiently in H3K36me3-enriched exons compared with introns (Frigola et al., 2017), and in actively transcribed genes compared with silent genes, and lowers the mutation frequency in the 3′ ends of the genes (Huang et al., 2018b). Mutation signature of the error-prone polymerase η, which is part of the somatic hypermutation specific MMR pathway, is targeted to 3′ ends of genes via H3K36me3 in solid tumors (Supek and Lehner, 2017).
MMR deficiency has been extensively modeled in Mlh1−/− mice, which display high microsatellite instability (MSI) and increased tumor mortality (Baker et al., 1996; Edelmann et al., 1996, 1999; Prolla et al., 1998). Female Mlh1−/− mice frequently develop lymphomas, mainly thymic, whereas males tend to develop gastrointestinal tumors (Gladbach et al., 2019). MSI occurs owing to the propensity of microsatellites (short tandem repeat sequences) to undergo strand slippage during DNA replication, which in MMR-deficient cells leads to deletion or insertion mutations within repeats. Recently, analysis of genome-wide mutations in Mlh1−/− lymphomas revealed several putative drivers of tumorigenesis (Daino et al., 2019; Gladbach et al., 2019).
To delineate how the mutational landscape in normal mammalian cells is shaped in vivo, on one hand, by replication errors, and on the other hand, by H3K36me3-mediated MMR correction, we performed single-cell whole-exome sequencing (scWES) on T cells isolated from MMR-proficient (Mlh1+/+) and MMR-deficient (Mlh1−/−) mice. Comparison of mutation distribution and frequency between MMR-proficient and -deficient mice revealed Huwe1 and Mcm7 genes as mutational hotspots exclusive to Mlh1−/− cells, implying that these regions present an inherent challenge to faithful DNA replication in T cells. Both hotspots are located in H3K36me3-enriched regions and expressed during T cell development. Analysis of MMR-dependent mutations indicate that H3K36me3-enriched 3′ exons are more protected against transcription-associated replication errors.
Results
Deletions Report on MMR-Dependent Mutations in Single-Cell Exome Sequencing
We isolated naive T cells from thymi of Mlh1+/+ and Mlh1−/− mice, followed by single-cell capture and whole-genome amplification on the Fluidigm C1 system, and then, by whole-exome enrichment and sequencing (Figure 1). Previous studies have utilized single-cell DNA sequencing to study clonality and mutation profiles of human cancers and normal cells (Leung et al., 2017; Wu et al., 2017; Zhang et al., 2019; Pellegrino et al., 2018). To check whether T cells were drawn from a similar cell population in both genotypes, we analyzed the proportions of distinct developmental thymic T cell populations (double negative, double positive, TCR αβ single positive [CD4 or CD8], TCR γδ) (Shah and Zuniga-Pflucker, 2014) by FACS. Cell frequencies of different thymic T cell populations between Mlh1−/− and Mlh1+/+ mice were similar to each other (Figure S1), indicating no defect in normal T cell developmental progression in Mlh1−/− mice, and that T cells analyzed by scWES from Mlh1+/+ and Mlh1−/− mice are drawn from similar thymic T cell populations. In both genotypes, the vast majority of cells were CD4+CD8+ double-positive T cells (67% for Mlh1+/+ and 65% for Mlh1−/− mice, respectively, Figure S1).
Figure 1.

Whole-Exome Sequencing of Single T Cells: Experimental Overview
Thymi of Mlh1−/− and Mlh1+/+ mice were dissected and used for enrichment of naive T cells, followed by single-cell capture, cell lysis, and whole-genome amplification in a Fluidigm C1. Amplified genomes were used for whole-exome sequencing (WES), and sequencing reads were analyzed for genetic variants. Shown is a read pileup and coverage of sample WT1-C26 in a ~5-kb-long region on chromosome 1 that contains three exons of Raph1. In addition to exons (green bar in exome panel), WES also partially covers non-coding regions adjacent to exons (blue bar in exome panel), enabling the comparison of mutation frequency between exonic and non-coding regions.
We sequenced 56 single-cell exomes in total, from 28 Mlh1−/− and 28 Mlh1+/+ T cells, to an average depth of 32X and coverage of 66% at depth ≥1X (Figures S2A and S2B). After excluding samples with low (<50%) coverage, 44 exomes (22 Mlh1+/+ and 22 Mlh1−/− exomes) were further analyzed for genetic variants. All detected variants with annotations (Related to Transparent Methods sections “Variant calling and filtering” and “Mutation annotation”) are listed in Table S2 titled “Annotated variants in single-cell exomes.” Overall, Mlh1−/− T cells had increased percentage (odds ratio [OR] = 1.56, 95% confidence interval [CI] = 1.44–1.69, p < 2.2 × 10−16) and frequencies (p = 5.487 × 10−6, Figures 2A and 2B and Table S1) of indels when compared with Mlh1+/+ T cells. Even though MMR deficiency increases also base substitutions (Meier et al., 2018), single nucleotide variant (SNV) frequencies between Mlh1−/− and Mlh1+/+ did not differ significantly in our dataset (p = 0.127, Figure 2B and Table S1). Analyzing insertions and deletions separately revealed that Mlh1−/− T cells had significantly higher deletion (p = 8.175 × 10−12) but not insertion frequencies (p = 0.1801) than Mlh1+/+ T cells (Figure 2C and Table S1). Taken together, deletions behaved in a genotype-dependent manner and thus represent MMR-dependent mutations.
Figure 2.

Global and Local Mutation Frequencies in Single T cells
(A–C) (A) Mlh1−/− T cells have an increased amount of indels out of total mutations in the whole exome compared with Mlh1+/+ Tcells (p << 0.0001, Fisher's exact test). Global (B) indel and SNV frequencies and (C) deletion and insertion frequencies, in Mlh1+/+ and Mlh1−/− T cells.Mlh1−/− T cells have significantly higher indel, and especially deletion, frequencies than Mlh1+/+ T cells (p << 0.001, two-tailed Mann-Whitney U test). Data in (B) is shown as boxplots together with kernel probability density and individual datapoints, and data in (C) is shown as median and interquartile range together with kernel probability density and individual datapoints. Outlier cells (see Transparent Methods) section ""Outlier cells in single-cell exomes) are marked with red color in (B).
(D) Mutation frequencies in 1-Mb windows across the mouse genome. Mlh1−/− T cells have multiple high local mutation peaks originating from only a single T cell.
(E) Mcm7 and Huwe1 are mutational hotspots in Mlh1−/− T cells. Columns are sorted by genotype and cell ID (outliers excluded), rows based on the average mutation frequency. Mlh1+/+ cells have label WT and Mlh1−/− cells have label KO, biological replicates are marked with 1 and 2. Each cell has a cell identifier that originates from the Fluidigm C1 plate capture site.
Bar plots on the right show proportions of mutation types, locations, and consequences in genes. Left-hand-side columns show positivity or negativity for RNApol2 and H3K36me3 peaks (See also Figure S3).
Huwe1 and Mcm7 Genes Are Mutational Hotspots in Mlh1−/− T Cells
Mlh1−/− cells provide a unique opportunity to reveal which chromosomal regions represent a particular challenge to the fidelity of the replication machinery, as any errors that are introduced will remain uncorrected by MMR. To identify such regions, we analyzed mutation frequencies in 1 Mb windows across single-cell exomes. On a megabase scale, local mutational frequencies were highly heterogeneous. The majority of the high-mutation-frequency peaks originated only from single T cells, and mutational hotspot windows shared between individual cells were sparse (Figure 2D). To establish whether any genes would emerge as MMR-dependent mutational hotspots, we scored all genes for mutations and asked which ones were mutated frequently in Mlh1−/− T cells (in more than 5 Mlh1−/− cells). Two genes, Huwe1 and Mcm7, stood out with their high mutational frequencies, exclusive to Mlh1−/− single-cell exomes (Figure 2E). Huwe1 encodes an E3 ubiquitin ligase, shown to regulate hematopoietic stem cell self-renewal and proliferation, and commitment to the lymphoid lineage (King et al., 2016). Mcm7 encodes a component of the MCM2-7 complex that forms the core of the replicative helicase, responsible for unwinding DNA ahead of the replication fork (Deegan and Diffley, 2016). Both genes are positive for RNA polymerase 2 and H3K36me3 in the mouse thymus and expressed from hematopoietic stem cells all the way to thymic T cells (Figures 2E, S3A, and S3B).
We then compared the mutational hotspots in Mlh1+/+ and Mlh1−/− normal T cells (this study) and with those in Mlh1−/− mouse lymphomas (Kakinuma et al., 2007; Daino et al., 2019; Gladbach et al., 2019). Only one shared mutational hotspot gene was found: Ttn, a massive gene with 324 exons, was mutated in both Mlh1−/− and Mlh1+/+ single-cell exomes (Figure 2E), in line with the findings of Daino et al. We did not identify any mutations in Ikzf1, previously reported as a mutational target gene in Mlh1-deficient T cell lymphomas (Daino et al., 2019; Kakinuma et al., 2007).
Other identified hotspot genes (Gm7361, Vps13c, Gm37013, Gm38667, Gm38666) were mutated in both Mlh1−/− and Mlh1+/+ T cells and thus were not specific for Mlh1 deficiency. All except Vps13c were negative or inconclusive for the presence of H3K36me3 and RNA polymerase 2, suggesting that these genes are not transcribed in mouse thymus (Figures 2E and S3A). Gm37013, Gm38667, and Gm38666 are predicted genes and they physically overlap with each other on chromosome 18 (Figure S3A), which explains their identical mutational pattern.
Insertions and Deletions Accumulate Differently within Repeats in Mlh1+/+ and Mlh1−/− T Cells
Next, we analyzed the size distribution of detected indels in single-cell exomes. Mlh1+/+ cells had more 1-nucleotide (nt) insertions than deletions, whereas this difference in Mlh1−/− T cells was evened out by increased 1-nt deletions (OR = 1.794, 95% CI = 1.531–2.101, p = 1.134 × 10−13, Figure 3A). The same trend for 1-nt insertions as the dominant indel type in Mlh1+/+ cells was observed in bulk T cell DNA samples from the same mice (Figure S4).
Figure 3.

Small Deletions Report on MMR-Dependent Mutations in Mouse T cells
(A) Indel length distribution as relative frequencies with Sison and Glaz 95% multinomial confidence intervals in Mlh1+/+ and Mlh1−/− T cells. Mlh1−/− and Mlh1+/+ cells have different ratios of 1-nt indels (p << 0.001, two-tailed Fisher's exact test). Indels of length ≥10 bp are binned together. See also Figure S4.
(B and C) (B) Relative and (C) normalized frequencies of deletions in microsatellites (MS) (mono-, di-, and trinucleotide repeats) and in non-microsatellite (random) sequence in single-cell samples.
(D and E) (D) Relative and (E) normalized frequencies of insertions in microsatellites (mono-, di-, and trinucleotide repeats) and in non-microsatellite (random) sequence in single-cell samples. Bar plots are ranked by descending mutation fraction within mononucleotide repeats. Mlh1−/− cells have a significantly higher deletion frequencies in microsatellites than Mlh1+/+ (p << 0.001, two-tailed Mann-Whitney U test). Mutation frequencies are shown as boxplots. Outliers (see Transparent Methods section "Outlier cells in single-cell samples") are labeled with red in (B) and (D).
We then analyzed the sequence context of the detected indels. As expected, most deletions in Mlh1−/− cells occurred at mononucleotide microsatellites, whereas in Mlh1+/+ cells, most deletions were found in non-microsatellite sequences (Figure 3B). When deletion counts were corrected for the number of base pairs of either microsatellite or non-microsatellite sequences, deletion frequencies were higher in microsatellites than in non-microsatellite sequences, regardless of MMR status (Figure 3C). This underscores the well-documented intrinsic propensity of microsatellites to slippage during replication. As expected, Mlh1−/− cells had significantly higher deletion frequencies in microsatellite sequences compared with Mlh1+/+ cells (p = 9.505 × 10−13, Figure 3C and Table S1). Insertion frequencies within repeats were more similar between Mlh1−/− and Mlh1+/+ T cells, occurring especially in mononucleotide repeats (Figure 3D). Mlh1−/− cells had somewhat higher insertion frequencies in the context of microsatellite sequences when compared with Mlh1+/+ cells (p = 0.039, Figure 3E and Table S1).
Exons Show a Decreased Burden of MMR-Dependent Mutations
Exome sequencing, despite its name, captures not only exons but also exon-adjacent, non-coding regions (3′ and 5′ UTR, promoter, or introns) (Figure 1) (Guo et al., 2012). This enabled us to ask whether de novo mutations accumulate differently in these two functionally distinct genic regions (exonic versus non-coding) in Mlh1−/− and Mlh1+/+ cells.
No significant difference in SNV frequencies or insertions was observed in either exonic or non-coding regions in Mlh1−/− cells compared with Mlh1+/+ cells (Figures 4A and 4B). In contrast, deletion frequencies increased in Mlh1−/− cells in non-coding regions compared with Mlh1+/+ cells (p = 9.94 × 10- 5, Figure 4C and Table S1). Exonic deletion frequencies in Mlh1−/− cells did not differ from those observed in Mlh1+/+ cells (Figure 4C), indicating that, in the absence of functional MMR, the integrity of coding regions is still maintained, likely by purifying selection, as suggested for MMR-deficient tumors by Kim et al., 2013. In conclusion, deletions, which we determined to be MMR-dependent mutations, increased more in non-coding regions adjacent to exons, as compared with exons themselves.
Figure 4.

Mlh1−/− Cells Accumulate Mutations to Non-coding Regions of Genome
(A) SNV, (B) insertion, and (C) deletion frequencies in exonic and non-coding (3′ and 5′ UTRs, promoters, splice sites, introns) regions of the exome in Mlh1+/+ and Mlh1−/− T cells. Mlh1−/− T cells have significantly higher frequencies of non-coding deletions (p<<0.001, two-tailed Mann-Whitney U test). Data is shown as boxplots together with kernel probability density and individual datapoints
H3K36me3-Enriched Regions Are Depleted of MMR-Dependent Mutations
Results from large tumor datasets strongly indicate that exons have a decreased mutation burden due to H3K36me3-mediated MMR (Frigola et al., 2017), but evidence of this in normal cells and tissues in vivo is still lacking. To assess whether replication errors in transcribed genes are buffered by MMR by virtue of their H3K36me3 enrichment, we first analyzed H3K36me3 abundance in RNA polymerase 2 (RNApol2)-positive (RNApol2+) and -negative (RNApol2-) genes in thymus using publicly available ChIP-seq data (ENCODE Project Consortium, 2012; Sloan et al., 2016). Presence of RNA polymerase 2 in the promoter region is a strong indicator of transcriptional activity (Barski et al., 2007), and we used it to score genes as either active (RNApol2+) or silent (RNApol2-). H3K36me3 levels in RNApol2+ genes were higher than in RNApol2- and peaked at the centers of the exons in these genes (Figure 5A), confirming that H3K36me3 is associated with transcriptional activity also in mouse thymus. However, not all RNApol2+ genes were positive for H3K36me3. Approximately 65% of RNApol2+ genes were also positive for H3K36me3, whereas 80% of H3K36me3-positive (H3K36me3+) genes were positive for RNApol2 (Figure 5B).
Figure 5.

H3K36me3 Reduces the Amount of MMR-Dependent Mutations in Exons
(A) H3K36me3 fold change (FC) (mean ± SD) in 1,000-bp window around exon centers in RNApol2-positive and -negative genes.
(B) Venn diagram of RNApol2-positive (+) and H3K36me3-positive (+) gene counts. Proportions of small deletions in genes positive or negative for H3K36me3 and RNApol2 in (C) Mlh1+/+ and (D) Mlh1−/− cells. Coding regions in genes positive for H3K36me3 have fewer deletions relative to H3K36me3-negative genes in Mlh1+/+ cells (p = 0.018, OR = 0.44, two-tailed Fisher's exact test), but not in Mlh1−/− cells. Deletion frequencies in the first to second exons (5′ exons) and the third to last exons (3′ exons) in RNApol2 (E) -positive and (F) -negative genes. In RNApol2-positive genes, Mlh1−/− cells have higher deletion frequency especially in the third to last exons (high H3K36me3) than Mlh1+/+ cells, and to lesser degree, in the first to second exons (low H3K36me3). The first panel shows the deletion frequencies in Mlh1+/+ and Mlh1−/− cells. data is shown as median and interquartile range together with kernel probability density and individual datapoints See also Figures S5 and S6. The second panel shows a schematic of H3K36me3 enrichment along a gene. The third panel shows a schematic of a gene structure. The fourth panel shows H3K36me3 signal as mean ± SD of FC in the first to second exons and third to last exons together with effect size as Cohen's d with Bessel's correction. Deletion frequencies were tested using two-tailed Mann-Whitney U test.
We analyzed how small deletions (that is, MMR-dependent mutations) were distributed to exons and non-coding regions based on either RNApol2 or H3K36me3 status of genes. The proportion of exonic deletions over non-coding deletions was decreased in H3K36me3+ genes compared with H3K36me3-negative (H3K36me3-) genes in Mlh1+/+ (p = 0.018, OR = 0.44, 95% CI = 0.198–0.906) but not in Mlh1−/− T cells (p = 1, OR = 0.972, 95% CI = 0.542–1.694, Figures 5C and 5D). Lower exonic deletion burden in RNApol2+ genes was also observed in Mlh1+/+ cells, similar to H3K36me3+ genes (p = 0.062, OR = 0.528, 95% C1 = 0.250–1.060, Figure 5C). The similar trends are not surprising, given the overlap between RNApol2+ and H3K36me3+ genes (Figure 5B). These results strongly support H3K36me3-guided, MMR-dependent protection of exons against genetic alterations.
The H3K36me3 mark is less abundant in 5′ exons, compared with 3′ exons of genes (Kolasinska-Zwierz et al., 2009; Frigola et al., 2017). To test whether local H3K36me3 levels affect the intra-genic distribution of mutations within genes in vivo, we compared deletion frequencies in the first and second exons (from here on referred to as 5′ exons) with those in the third to last exons (from here on referred to as 3′ exons), both in RNApol2+ and RNApol2- genes. In RNApol2+ genes, H3K36me3 signal increased in 3′ exons compared with 5′ exons (d = 0.335, Figures 5E and S6A), whereas in RNApol2- genes, there was no difference in H3K36me3 levels between 3′ and 5′ exons (d = 0.002, Figures 5F, S6A, and Table S1). In RNApol2+ genes, Mlh1−/− cells had higher deletion frequencies in 3′ exons (high in H3K36me3) compared with Mlh1+/+ cells (p = 4.57 × 10−5, Figures 5E, S6B, and Table S1). In 5′ exons (low in H3K36me3), the difference in deletion frequencies between Mlh1−/− and Mlh1+/+ was smaller, yet significant (p = 0.016, Figures 5E, S6B, and Table S1). Mlh1+/+ cells also had somewhat increased deletion frequencies in the 3′ exons compared with 5′ exons (p = 0.020, Figures 5E, S6B, and Table S1). Sequencing coverage was similar between samples with or without mutations in the analyzed exons, except in the 5′ exons in RNApol2+ regions in Mlh1+/+ cells (p = 0.04, Figure S5). Taken together, these results suggest that 3′ exons in transcriptionally active genes are more prone to acquiring replication-induced mutations compared with 5′ exons and that this effect is tempered by H3K36me3-guided MMR. No difference was observed in the deletion frequencies between Mlh1+/+ and Mlh1−/− cells in RNApol2- genes in 5′ exons (p = 0.539) or 3′ exons (p = 0.296, Figures 5F, S6B, and Table S1). Mlh1−/− cells, however, showed slightly higher deletion frequencies in 3′ exons compared with 5′ exons (p = 0.049, Figures 5F, S6B, and Table S1). H3K36me3- exons in RNApol2- genes accumulated mutations in similar frequencies in both Mlh1+/+ and Mlh1−/− cells. We interpret this to mean that the MMR machinery does not operate efficiently in these regions even in wild-type cells. RNApol2+, but not RNApol2-, genes showed genotype-dependent spatial variability in deletion frequencies; thus transcriptional activity appears to affect accumulation and/or repair of replication errors.
Discussion
Using single-cell sequencing of mouse thymic T cells, we uncovered how the exome-wide mutational landscape is shaped in vivo by replication errors, along with MMR-mediated error correction. We identify the Huwe1 and Mcm7 genes as novel mutational hotspots in normal Mlh1−/− thymic T cells. We further provide evidence for transcription-associated vulnerability to replication errors and for H3K36me3-guided MMR at 3′ exons of genes.
We show that scWES is a sensitive approach for unraveling signatures of replication errors and MMR activity. This is highlighted by the fact that we detected a substantial increase of deletions in Mlh1−/− T cells and found evidence of insertional bias in Mlh1+/+ T cells. DNA polymerases tend to create more deletions than insertions, especially in repeat sequences (Baptiste et al., 2015; Kunkel, 1986; Kim et al., 2013; Lujan et al., 2015; Woerner et al., 2015; Garcia-Diaz and Kunkel, 2006), and in the absence of MMR (which is the situation in Mlh1−/− cells), one would expect to directly detect replication errors. Indeed, we observed a significant increase of small deletions in Mlh1−/− cells compared with Mlh1+/+ cells. Taken together, we conclude that deletions reliably report on replication errors that would otherwise be repaired by MMR. In addition, we found that Mlh1+/+ cells had more insertions than deletions. Increase in 1-nt insertions rather than deletions in Mlh1+/+ cells has also been observed at unstable microsatellite loci in other MMR-proficient normal mouse tissues (Shrestha et al., 2019). Our findings are in line with the previously reported bias for MMR to correct deletion loops more efficiently than insertion loops, thereby creating an insertional bias at microsatellite sequences (Baptiste et al., 2013).
MMR-deficient cells (Mlh1−/−) accumulate replication-induced errors with every cell division. Developing lymphocytes are particularly susceptible to replication errors because they undergo multiple rounds of proliferative expansions during development and maturation. Comparison of mutational frequencies in Mlh1−/− versus Mlh1+/+ T cell exomes revealed two hotspots for replication errors, Huwe1 and Mcm7 genes. Mutations in Mcm7 affected both exons and introns, whereas mutations in Huwe1 were found exclusively in introns (Figure 2E). Exonic Mcm7 mutations comprised both synonymous and non-synonymous mutations. Synonymous exonic Mcm7 mutations, although they do not alter amino acid sequence, may still affect Mcm7 splicing regulatory sites or miRNA binding sites or cause changes in mRNA stability or translation efficiency. Intronic mutations may cause splicing defects, resulting in exon skipping or intron retention (Diederichs et al., 2016). A small fraction (1%–2%) of somatic mutations that alter amino acid sequence create neoepitopes that, when presented on the cell membrane, can provoke immune cell attack (Yamamoto et al., 2019). Mlh1-deficient mouse cancer cell lines have been shown to produce persistently neoantigens, both in vitro and in vivo (Germano et al., 2017). Neoantigenicity is unlikely, however, for MCM7 and HUWE1 that reside in the nucleus and/or cytosol, and thus, they lack the appropriate cellular localization to function as neoantigens. Because Huwe1 and Mcm7 are vulnerable to replication errors, we propose that over time, in Mlh1-deficient cells, damaging mutations will emerge in these genes, some with potentially tumorigenic effects. Indeed, deleterious mutations in Huwe1 and Mcm7 have been reported in Mlh1-deficient murine T cell lymphomas (Daino et al., 2019). The propensity of Mcm7, coding for an integral component of the replication machinery, to acquire deleterious mutations in MMR-deficient cells (Figure 2E) conceivably can further accelerate the accumulation of replication-associated errors, thereby adding insult to injury.
Both Huwe1 and Mcm7 are expressed in the T lymphocyte lineage and required for lymphocyte development. Shielding them from permanent mutations is likely important for cellular homeostasis and normal development, and Huwe1 and Mcm7 were in fact devoid of mutations in Mlh1+/+ T cells. In the face of frequent replication errors, how is efficient targeting of MMR to these regions ensured in wild-type cells? Both Huwe1 and Mcm7 were enriched for H3K36me3 in the mouse thymus, and H3K36me3-mediated MMR has been shown to protect actively transcribed genes (Huang et al., 2018b). Thus, H3K36me3-mediated recruitment of MMR to these genes provides an explanation for efficient error correction in wild-type cells; in the absence of MMR, H3K36me3 no longer has a protective effect.
Also, at single-cell resolution, the protective effect of H3K36me3-mediated MMR on active genes appears to hold true more globally. In wild-type cells, coding regions in H3K36me3-enriched genes exhibited lower mutation frequencies, compared with coding regions in H3K36me3-depleted genes. This effect was abolished in MMR-deficient cells. Our results indicate that H3K36me3-mediated MMR preserves the integrity of active genes in normal tissues in vivo, similarly as shown previously for tumors and cell lines (Supek and Lehner, 2015; Frigola et al., 2017; Huang et al., 2018b).
Moreover, we provide in vivo evidence that 3′ ends of actively transcribed genes are more prone to replication-associated errors and that more efficient recruitment of MMR via H3K36me3 protects these regions, ensuring that most of these errors do not become permanent mutations. Head-on collisions of the replication and transcription machineries can cause indels and base substitutions and especially increase the deletion burden within 3′ ends (and to a lesser degree 5′ ends) of genes under active transcription (Sankar et al., 2016). In HeLa cells, mutation frequency has been shown to decrease toward the 3′ end of the gene body, as H3K36me3 increases, implying more efficient MMR-mediated repair in these regions (Huang et al., 2018b). SNVs also accumulate more to 3′ UTRs than to 5′ UTRs in aging B lymphocytes (Zhang et al., 2019), in line with the notion that 3′ regions are in fact more prone to mutations. Efficient recruitment of the MMR machinery via H3K36me3 can shield against replication-induced errors specifically in transcribed genes, whose integrity is particularly important.
Here, we delineate the mutational landscape of T cells shaped by the status of DNA repair (functional versus impaired), dissected at the single-cell level in the context of H3K36me3. We provide evidence that, in normal thymocytes in vivo, MMR preferentially protects H3K36me3-positive genes and especially 3′ exons transcribed in T cell lineage, against accumulation of de novo mutations, providing an additional layer to the regional dynamics of H3K36me3-guided MMR. In addition, we identify Huwe1 and Mcm7 as novel mutational hotspots in (still phenotypically normal) Mlh1−/− T cells, both genes which are of importance during T cell development. Taken together, our results suggest an attractive concept of thrifty MMR targeting, where genes critical for the development of a given cell type and under mutational stress due to active transcription are preferentially shielded from acquiring deleterious mutations.
Limitations of the Study
-
-
The number of sequenced single T cells in our study is limited. For a comprehensive view of mutational hotspots and mutation frequencies, more single cells should be sequenced.
-
-
Owing to limited starting amount of DNA, single-cell genomes were amplified extensively in order to have enough material for sequencing. This amplification introduces in vitro artifacts, which affect the analysis of mutation frequencies and mutational features. Especially genuine de novo SNV mutations are expected to be masked by such artifacts.
-
-
Our exomic dataset represents less than 2% of the whole mouse genome, and specifically the coding portion where de novo mutations are under highest natural selection. In order to understand how mutation frequency plays out in intergenic regions and the factors contributing to this dynamic, whole-genome sequencing should be conducted.
Resource Availability
Lead Contact
Any further queries and requests should be addressed to corresponding author and lead contact Liisa Kauppi (liisa.kauppi@helsinki.fi) or to corresponding authors Elli-Mari Aska (elli.aska@helsinki.fi) and Denis Dermadi (ddermadi@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Single-cell exome sequencing data generated and analyzed during the current study are deposited as raw reads in FASTQ format to SRA: PRJNA575619. The variants observed in single T cells supporting the conclusions of this article are provided with the article as Table S2 titled “Annotated variants in single-cell exomes” in xlsx file format. Publicly available H3K36me3 (ENCODE: ENCFF853BYO, ENCFF287DIJ) and RNApol2 (ENCODE: ENCFF119XEH) ChIPSeq data can be found from ENCODE (https://www.encodeproject.org) database.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are grateful to Fran Supek, Esa Pitkänen, Niko Välimäki, and Julia Casado for discussions and advice. We wish to acknowledge CSC – IT Center for Science, Finland for computing resources, the Functional Genomics Unit (University of Helsinki) for sequencing services, Minna Nyström (University of Helsinki) for providing mice, Jussi Taipale and Anna Vähärautio for access to Fluidigm C1 system, and Kul Shanker Shrestha and Minna Tuominen for technical assistance. Assistance was also provided by the following core facilities: Laboratory Animal Center and Biomedicum Imaging Unit at University of Helsinki and Palo Alto Veterans Institute for Research (PAVIR) FACS Core.
E.A. is supported by a funded position in the Doctoral Program in Integrative Life Sciences, Doctoral School of Health, University of Helsinki, and ASLA-Fulbright Pre-Doctoral Fellowship 2018-2019. This work was supported by the Academy of Finland (grants 263870, 292789, 256996, 306026 to L.K.), the Sigrid Juséliuksen Säätiö (to L.K), and Emil Aaltonen Säätiö (to E.A.).
Author Contributions
Conceptualization, D.D. and L.K.; Methodology, E.A. and D.D.; Formal Analysis, E.A. and D.D.; Resources, D.D. and L.K.; Data Curation, E.A.; Writing – Original Draft, E.A., D.D., and L.K.; Writing – Review & Editing, E.A., D.D., and L.K.; Visualization, E.A.; Supervision, D.D. and L.K.; Project Administration, D.D. and L.K.; Funding Acquisition, L.K.
Declaration of Interests
The authors declare that they have no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101452.
Contributor Information
Elli-Mari Aska, Email: elli.aska@helsinki.fi.
Denis Dermadi, Email: ddermadi@stanford.edu.
Liisa Kauppi, Email: liisa.kauppi@helsinki.fi.
Supplemental Information
Annotated variants in single-cell exomes
References
- Baker S.M., Plug A.W., Prolla T.A., Bronner C.E., Harris A.C., Yao X., Christie D.M., Monell C., Arnheim N., Bradley A. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Baptiste B.A., Ananda G., Strubczewski N., Lutzkanin A., Khoo S.J., Srikanth A., Kim N., Makova K.D., Krasilnikova M.M., Eckert K.A. Mature microsatellites: mechanisms underlying dinucleotide microsatellite mutational biases in human cells. G3 (Bethesda) 2013;3:451–463. doi: 10.1534/g3.112.005173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptiste B.A., Jacob K.D., Eckert K.A. Genetic evidence that both dNTP-stabilized and strand slippage mechanisms may dictate DNA polymerase errors within mononucleotide microsatellites. DNA Repair (Amst) 2015;29:91–100. doi: 10.1016/j.dnarep.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., WEI G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Chantalat S., Depaux A., Hery P., Barral S., Thuret J.Y., Dimitrov S., Gerard M. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 2011;21:1426–1437. doi: 10.1101/gr.118091.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daino K., Ishikawa A., Suga T., Amasaki Y., Kodama Y., Shang Y., Hirano-Sakairi S., Nishimura M., Nakata A., Yoshida M. Mutational landscape of T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1: no synergism with ionizing radiation. Carcinogenesis. 2019;40:216–224. doi: 10.1093/carcin/bgz013. [DOI] [PubMed] [Google Scholar]
- Deegan T.D., Diffley J.F. MCM: one ring to rule them all. Curr. Opin. Struct. Biol. 2016;37:145–151. doi: 10.1016/j.sbi.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Diederichs S., Bartsch L., Berkmann J.C., Frose K., Heitmann J., Hoppe C., Iggena D., Jazmati D., Karschnia P., Linsenmeier M. The dark matter of the cancer genome: aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016;8:442–457. doi: 10.15252/emmm.201506055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann W., Cohen P.E., Kane M., Lau K., Morrow B., Bennett S., Umar A., Kunkel T., Cattoretti G., Chaganti R. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- Edelmann W., Yang K., Kuraguchi M., Heyer J., Lia M., Kneitz B., Fan K., Brown A.M., Lipkin M., Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1Apc1638N mutant mice. Cancer Res. 1999;59:1301–1307. [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J., Sabarinathan R., Mularoni L., Muinos F., Gonzalez-Perez A., Lopez-Bigas N. Reduced mutation rate in exons due to differential mismatch repair. Nat. Genet. 2017;49:1684–1692. doi: 10.1038/ng.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz M., Kunkel T.A. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem. Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Germano G., Lamba S., Rospo G., Barault L., Magri A., Maione F., Russo M., Crisafulli G., Bartolini A., Lerda G. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- Gladbach Y.S., Wiegele L., Hamed M., Merkenschlager A.M., Fuellen G., Junghanss C., Maletzki C. Unraveling the heterogeneous mutational signature of spontaneously developing tumors in MLH1(-/-) mice. Cancers (Basel) 2019;11:1485. doi: 10.3390/cancers11101485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Long J., He J., Li C.I., Cai Q., Shu X.O., Zheng W., Li C. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:194. doi: 10.1186/1471-2164-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Weng H., Sun W., Qin X., Shi H., Wu H., Zhao B.S., Mesquita A., Liu C., Yuan C.L. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Gu L., Li G.M. H3K36me3-mediated mismatch repair preferentially protects actively transcribed genes from mutation. J. Biol. Chem. 2018;293:7811–7823. doi: 10.1074/jbc.RA118.002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Weng H., Zhou K., Wu T., Zhao B.S., Sun M., Chen Z., Deng X., Xiao G., Auer F. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 2019;567:414–419. doi: 10.1038/s41586-019-1016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma S., Kodama Y., Amasaki Y., Yi S., Tokairin Y., Arai M., Nishimura M., Monobe M., Kojima S., Shimada Y. Ikaros is a mutational target for lymphomagenesis in Mlh1-deficient mice. Oncogene. 2007;26:2945–2949. doi: 10.1038/sj.onc.1210100. [DOI] [PubMed] [Google Scholar]
- Kim T.M., Laird P.W., Park P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013;155:858–868. doi: 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B., Boccalatte F., Moran-Crusio K., Wolf E., Wang J., Kayembe C., Lazaris C., Yu X., Aranda-Orgilles B., Lasorella A., Aifantis I. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat. Immunol. 2016;17:1312–1321. doi: 10.1038/ni.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X.S., Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T.A. Frameshift mutagenesis by eucaryotic DNA polymerases in vitro. J. Biol. Chem. 1986;261:13581–13587. [PubMed] [Google Scholar]
- Lahue R.S., Au K.G., Modrich P. DNA mismatch correction in a defined system. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- Leung M.L., Davis A., Gao R., Casasent A., Wang Y., Sei E., Vilar E., Maru D., Kopetz S., Navin N.E. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017;27:1287–1299. doi: 10.1101/gr.209973.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Li F., Mao G., Tong D., Huang J., Gu L., Yang W., Li G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan S.A., Clark A.B., Kunkel T.A. Differences in genome-wide repeat sequence instability conferred by proofreading and mismatch repair defects. Nucleic Acids Res. 2015;43:4067–4074. doi: 10.1093/nar/gkv271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B., Volkova N.V., Hong Y., Schofield P., Campbell P.J., Gerstung M., Gartner A. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018;28:666–675. doi: 10.1101/gr.226845.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino M., Sciambi A., Treusch S., Durruthy-Durruthy R., Gokhale K., Jacob J., Chen T.X., Geis J.A., Oldham W., Matthews J. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018;28:1345–1352. doi: 10.1101/gr.232272.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolla T.A., Baker S.M., Harris A.C., Tsao J.L., Yao X., Bronner C.E., Zheng B., Gordon M., Reneker J., Arnheim N. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat. Genet. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- Sankar T.S., Wastuwidyaningtyas B.D., Dong Y., Lewis S.A., Wang J.D. The nature of mutations induced by replication-transcription collisions. Nature. 2016;535:178–181. doi: 10.1038/nature18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D.K., Zuniga-Pflucker J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014;192:4017–4023. doi: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- Sloan C.A., Chan E.T., Davidson J.M., Malladi V.S., Strattan J.S., Hitz B.C., Gabdank I., Narayanan A.K., Ho M., Lee B.T. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44:D726–D732. doi: 10.1093/nar/gkv1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Charles J.A., Liberti S.E., Williams J.S., Lujan S.A., Kunkel T.A. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair (Amst) 2015;31:41–51. doi: 10.1016/j.dnarep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Lehner B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature. 2015;521:81–84. doi: 10.1038/nature14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Lehner B. Clustered mutation signatures reveal that error-prone DNA repair targets mutations to active genes. Cell. 2017;170:534–547.e23. doi: 10.1016/j.cell.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner S.M., Tosti E., Yuan Y.P., Kloor M., Bork P., Edelmann W., Gebert J. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol. Carcinog. 2015;54:1376–1386. doi: 10.1002/mc.22213. [DOI] [PubMed] [Google Scholar]
- Wu H., Zhang X.Y., Hu Z., Hou Q., Zhang H., Li Y., Li S., Yue J., Jiang Z., Weissman S.M. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene. 2017;36:2857–2867. doi: 10.1038/onc.2016.438. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.N., Kishton R.J., Restifo N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019;25:1488–1499. doi: 10.1038/s41591-019-0596-y. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yuan F., Presnell S.R., Tian K., Gao Y., Tomkinson A.E., GU L., LI G.M. Reconstitution of 5'-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Zhang L., Dong X., Lee M., Maslov A.Y., Wang T., Vijg J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. U S A. 2019;116:9014–9019. doi: 10.1073/pnas.1902510116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha, K., Aska, E., Tuominen, M. & Kauppi, L. (2019). Mlh1 haploinsufficiency induces microsatellite instability specifically in intestine, 10.1101/652198. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotated variants in single-cell exomes
Data Availability Statement
Single-cell exome sequencing data generated and analyzed during the current study are deposited as raw reads in FASTQ format to SRA: PRJNA575619. The variants observed in single T cells supporting the conclusions of this article are provided with the article as Table S2 titled “Annotated variants in single-cell exomes” in xlsx file format. Publicly available H3K36me3 (ENCODE: ENCFF853BYO, ENCFF287DIJ) and RNApol2 (ENCODE: ENCFF119XEH) ChIPSeq data can be found from ENCODE (https://www.encodeproject.org) database.