

Summary
During an immune response, natural killer (NK) cells activate specific metabolic pathways to meet the increased energetic and biosynthetic demands associated with effector functions. Here, we found in vivo activation of NK cells during Listeria monocytogenes infection-augmented transcription of genes encoding mitochondria-associated proteins in a manner dependent on the transcriptional coactivator PGC-1α. Using an Ncr1Cre-based conditional knockout mouse, we found that PGC-1α was crucial for optimal NK cell effector functions and bioenergetics, as the deletion of PGC-1α was associated with decreased cytotoxic potential and cytokine production along with altered ADP/ATP ratios. Lack of PGC-1α also significantly impaired the ability of NK cells to control B16F10 tumor growth in vivo, and subsequent gene expression analysis showed that PGC-1α mediates transcription required to maintain mitochondrial activity within the tumor microenvironment. Together, these data suggest that PGC-1α-dependent transcription of specific target genes is required for optimal NK cell function during the response to infection or tumor growth.
Subject Areas: Biological Sciences, Cellular Physiology, Immunology, Cancer
Graphical Abstract
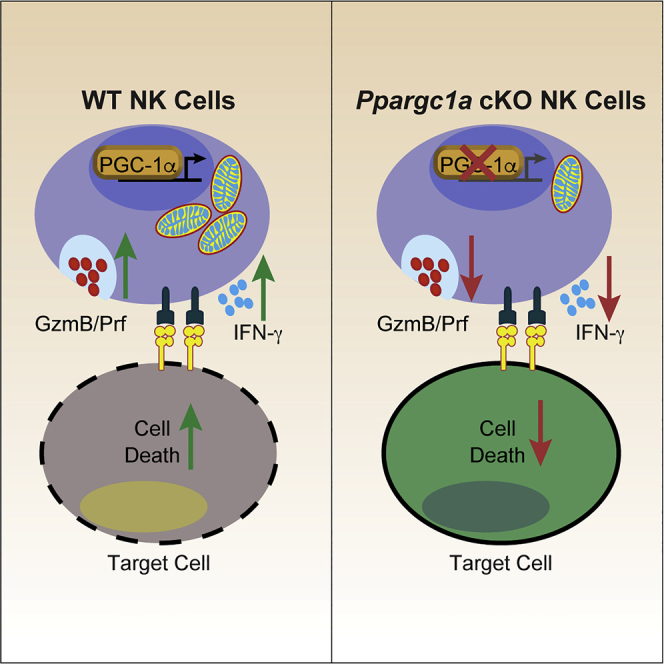
Highlights
-
•
NK cells activate mitochondria-associated gene transcription during in vivo challenge
-
•
NK cell-specific deletion of PGC-1α results in energetic and functional defects
-
•
PGC-1α-deficient NK cells fail to clear B16F10 tumors to the level of WT cells
-
•
Reduced tumor clearance is associated with mitochondrial dysfunction
Biological Sciences; Cellular Physiology; Immunology; Cancer
Introduction
Natural killer (NK) cells are innate lymphocytes that constitute a critical part of the immune response to infection and tumor growth (Cerwenka and Lanier, 2001; Morvan and Lanier, 2015; Vivier et al., 2008). Following infection or transformation, diseased cells upregulate the expression of ligands for NK cell activation receptors (NKRs) (Cerwenka and Lanier, 2001; Diefenbach et al., 2001; Pende et al., 2002), which renders them susceptible to NK cell-mediated cytotoxicity via perforin and granzyme B secretion and by ligation of death receptors on target cells such as Fas and Trail (Topham and Hewitt, 2009). NK cells are also capable of regulating both innate and adaptive immune cells by secretion of proinflammatory cytokines such as interferon (IFN)-γ and granulocyte/monocyte-colony-stimulating factor (GM-CSF) (Fauriat et al., 2010). The fact that these effector functions occur independently of interactions with major histocompatibility complex (MHC)-I gives NK cells a critical advantage in controlling infection and tumor growth, as their functional capacity is unaffected but rather augmented by the downregulation of MHC-I, which can occur in infected or transformed cells (Garrido et al., 2016; Seliger et al., 2006). The self-independent killing mechanism of NK cells also creates an expanded donor pool for NK cell-based immunotherapy, and the adoptive transfer of NK cells has demonstrated efficacy against multiple cancer types (Cheng et al., 2013; Iliopoulou et al., 2010; Miller et al., 2005; Ruggeri et al., 2002a). Therefore, it is essential to elucidate molecular mechanisms that regulate NK cell effector functions to optimize cell-based immunotherapy and disease treatment.
In NK cells, alterations in cellular metabolism enable cells to meet increased energetic and biosynthetic demands associated with control of infection and tumor growth (Donnelly et al., 2014; Keating et al., 2016; Keppel et al., 2015; Mah et al., 2017; Marcais et al., 2014). This connection between metabolism and function is critical to understand from a clinical perspective, as disruption of energy homeostasis can lead to immune suppression. For example, NK cells isolated from obese patients or patients with type 2 diabetes exhibit reduced cytokine production and cytotoxic potential compared with those isolated from metabolically healthy controls (Michelet et al., 2018; Piatkiewicz et al., 2013). Similarly, tumor progression and the associated changes in nutrient availability in the tumor microenvironment (TME) can also impact glycolysis in a way that leads to reduced antitumor functions (Chang et al., 2015; Siska et al., 2017). In NK cells, specifically, TME-induced activation of fructose bisphosphatase 1 (FBP1) leads to reduced glycolysis and reduced ability to mediate effector functions during tumor progression (Cong et al., 2018). Importantly, inhibition of FBP1 augments the ability of NK cells to produce cytokines and mediate cytotoxicity against target cells, demonstrating a clear link between glycolysis and NK cell effector functions (Cong et al., 2018). Given the prominent role of metabolism in NK cell activity, it is critical to identify proteins that initiate metabolic changes required for effector functions.
Several regulatory mechanisms control glycolytic increases required for effector functions. For example, in cytokine-stimulated NK cells, the mechanistic target of rapamycin (mTOR) mediates increases in glycolysis and nutrient uptake that are essential for cytokine production and control of viral infection (Donnelly et al., 2014; Mah et al., 2017; Marcais et al., 2014). Similar results were seen in human NK cells following cytokine-mediated activation (Keating et al., 2016). In addition, the mTOR complexes (mTORC1 and mTORC2) are also required for maturation and maintaining NK cell homeostasis in peripheral tissues (Yang et al., 2018). Transcriptionally, increases in HIF-1α activation have been shown to drive glycolytic gene transcription and augment cytokine production (Velasquez et al., 2016). Recent work also indicates that activation of c-Myc leads to increased amino acid uptake required to sustain increased metabolic activity in cytokine-stimulated NK cells (Loftus et al., 2018). Studies using human NK cells also demonstrate that increased pyruvate kinase M1/2 (PKM2) expression is associated with increased glycolysis and greater metabolic flexibility during cell-mediated cytotoxicity (Schafer et al., 2019). Together, these studies have been integral in defining how NK cells regulate glycolysis and overall metabolic activity to sustain effector functions.
The importance of proteins that control glycolysis is well-established, whereas the role of mitochondrial regulators remains controversial. Recent work shows the activation of lipid metabolism inhibits NK cell function (Michelet et al., 2018). Treatment of NK cells with agonists for PPARα and PPARδ, fatty acid-regulated transcription factors that control lipid metabolism, results in increased lipid uptake and reduces the expression of IFN-γ and GzmB. Supporting this notion, mice fed exogenous lipids or high-fat diet exhibit reduced IFN-γ and GzmB levels and are unable to control tumor growth to the level of control mice (Michelet et al., 2018). Conversely, the function of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a transcriptional coactivator that augments both PPARα and PPARδ activities, is required for optimal NK cell-mediated cytotoxicity in human NK cells following high-dose interleukin (IL)-2 culture (Miranda et al., 2016, 2018). Blockade of fatty acid oxidation (FAO) by treatment with the carnitine palmitoyltransferase 1 inhibitor etomoxir also demonstrates varying effects on NK cell function, as previous work shows that etomoxir treatment reduces cytokine production but augments cytotoxicity in NK cells exposed to exogenous lipids (Keppel et al., 2015; Michelet et al., 2018). Outside of fatty acid metabolism, the importance of mitochondria-derived ATP also exhibits varying regulation, as oligomycin-mediated inhibition of ATP synthase reduces NK cell-mediated cytokine production in NK cells cultured in IL-15 for 24 h, but extending the culture period to 72 h renders cells insensitive to oligomycin treatment (Keppel et al., 2015). These conflicting results and the varying activation conditions used in previous work highlight the need for direct analysis of mitochondria and associated regulatory proteins in vivo to determine the role of mitochondrial metabolism during an immune response.
PGC-1α is a transcriptional coactivator that drives the transcription of genes critical for mitochondrial function (Puigserver et al., 1998; Vega et al., 2000; Wu et al., 1999). Through interactions with different transcription factors, PGC-1α mediates a variety of tissue-specific functions associated with increased oxidative phosphorylation, including uncoupled thermogenesis in brown adipose tissue, mitochondrial biogenesis in skeletal muscle, and FAO in the liver (Michael et al., 2001; Puigserver et al., 1998; Wu et al., 2002; Yoon et al., 2001). As PGC-1α functions explicitly in response to energetic stress, we hypothesized that PGC-1α allows NK cells to adapt to changing nutrient and inflammatory settings during an immune response. Indeed, recent evidence demonstrates PGC-1α sustains mitochondrial activity in CD8 T cells, as loss of PGC-1α function during chronic infection or exposure to a TME induces a hyporesponsive phenotype (Bengsch et al., 2016; Scharping et al., 2016). In NK cells, knockdown of PGC-1α following culture in high doses of IL-2 results in reduced cytotoxic potential, and NK cells isolated from older individuals and cultured in high doses of IL-2 exhibit reduced cytotoxicity and decreased PGC-1α levels compared with cells isolated from younger donors (Miranda et al., 2016, 2018). This work demonstrated the importance of PGC-1α in maintaining the cytotoxic potential of cultured NK cells; however, whether PGC-1α functions in vivo has yet to be determined. The fact that activation of proteins that function in concert with PGC-1α is immunosuppressive in other models and during the control of tumor growth (Michelet et al., 2018) demonstrates the need for directly measuring the function of PGC-1α and for determining its importance in NK cell function in multiple models including in vivo immune challenge.
It is also critical to understand mitochondrial regulation from a clinical perspective, as the mitochondria may play a central role in the efficacy of cell-based immunotherapies. Persistence of engrafted cells, for example, is associated with improved anti-tumor immunity and patient survival in both pre-clinical models and actual clinical trials (Klebanoff et al., 2005; Louis et al., 2011; Rosenberg et al., 2011; Zhou et al., 2005). Subsequent studies have demonstrated a link between persistence and mitochondrial function, and sorting cells based on mitochondrial parameters is useful in separating short- and long-lived cells (Kishton et al., 2017; Nayar et al., 2015; Sukumar et al., 2016). In addition, T cell differentiation and memory formation are essential factors in determining graft persistence, and direct alteration of mitochondrial function in vitro can be used to promote memory formation that is retained in vivo following engraftment (Sukumar et al., 2013; van der Windt et al., 2012; Vannini et al., 2016; Wahl et al., 2012). Along with longevity and persistence, anti-tumor functions are improved by augmenting the mitochondrial function of T cells before engraftment that synergized with anti-PD-1 antibody therapy (Chamoto et al., 2017; Menk et al., 2018). These studies highlight the translational potential of utilizing metabolism to optimize lymphocyte functions during cell-based immunotherapies.
In this study, we sought to determine the importance of PGC-1α-mediated changes in mitochondrial function for the NK cell immune response. In vivo activation of NK cells augmented transcription of genes associated with mitochondrial function and significantly upregulated a set of known PGC-1α target genes. We then created a conditional knockout model of PGC-1α using the Ncr1Cre system (Ppargc1a cKO) and found the loss of PGC-1α reduced expression of these target genes following immune challenge and disrupted NK cell function in multiple settings. We observed in vitro defects in cytokine production and cell-mediated cytotoxicity that were associated with disrupted ATP homeostasis and OxPhos activity, and the inhibition of PGC-1α recapitulated these functional and energetic defects in human NK cells. Lack of PGC-1α resulted in reduced mitochondrial mass and membrane potential following inoculation with B16F10 melanoma, whereas NK cells from unchallenged mice demonstrated little difference in mitochondrial phenotype. These defects were associated with decreased tumor clearance as well as reduced expression of PGC-1α target genes, including genes that regulate mitochondrial nutrient utilization such as Lcad, Pdk1, Got1, and Got2. Together, our data identify a set of genes regulated by PGC-1α in NK cells and demonstrate the importance of mitochondrial regulation during the response to infection and tumor growth.
Results
PGC-1α Regulates Metabolic Gene Transcription in NK Cells during Bacterial Infection
We hypothesized that, in addition to glycolytic gene transcription, activation of NK cells in vivo would increase mitochondrial gene transcription during an immune response. To identify transcriptional changes in metabolic genes that occurred in activated NK cells, we analyzed our recently published RNA sequencing dataset in which wild-type (WT) mice were infected with Listeria monocytogenes (Dunn and North, 1991; Humann and Lenz, 2010; Shegarfi et al., 2009; Thale and Kiderlen, 2005) to induce an immune response in splenic NK cells (Nanbakhsh et al., 2019). In these studies, NK cells were isolated via negative selection, with a purity of 95% for all samples (Nanbakhsh et al., 2019). To determine if mitochondrial gene transcription was upregulated during infection, we used competitive gene set enrichment analysis (GSEA) to test for enrichment of gene sets comprising the Hallmark, MSigDB c2, and Gene Ontology databases to identify transcriptional changes induced by in vivo activation of NK cells. Utilizing competitive GSEA to determine enrichment score and the FRY variation of rotational gene set testing (Wu et al., 2010) (Table 1), we found NK cells from infected mice showed significant enrichment of Hallmark gene sets associated with inflammation and glycolysis compared with uninfected controls (Figures 1A and S1A, extended results in Table S1). In addition to these control gene sets, NK cells from infected mice also demonstrated significant enrichment of genes belonging to the Oxidative Phosphorylation Hallmark gene set (Figure 1A, Table 1). Analysis of the Oxidative Phosphorylation KEGG pathway revealed similar transcriptional regulation including upregulation of many of the protein subunits associated with the Electron Transport Chain (ETC) protein complexes (Figures S1B and S1C, Table 1). In addition to gene sets related to global OxPhos, these analyses show significant enrichment of multiple gene sets corresponding to specific mitochondrial functions, including Mitochondrial Membrane Organization, TCA Cycle, Respiratory Electron Transport, and Mitochondrial Translation (Figure 1B, Table 1). To determine the level of enrichment of individual gene sets during bacterial infection, we used the FRY variation of rotational gene set testing for differential expression analysis (Wu et al., 2010) and found that multiple mitochondria-associated gene sets were indeed significantly upregulated in NK cells from infected mice compared with uninfected controls (Table 1). These data demonstrate a coordinated increase in mitochondrial gene transcription in NK cells during bacterial infection.
Table 1.
Enrichment of Mitochondrial Gene Sets
| Gene List | NGenes | E.S. | Direction | p Value | FDR |
|---|---|---|---|---|---|
| HALLMARK_GLYCOLYSIS | 239 | 2.1 | Up | 5.56 × 10−6 | 1.90 × 10−5 |
| HALLMARK_OXIDATIVE_PHOSPHORYLATION | 227 | 2.3 | Up | 0.00048225 | 0.000618 |
| HALLMARK_INFLAMMATORY_RESPONSE | 244 | 3.5 | Up | 2.87 × 10−6 | 1.26 × 10−5 |
| GO_POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION | 448 | 2.5 | Up | 8.08 × 10−7 | 6.63 × 10−6 |
| KEGG_OXIDATIVE_PHOSPHORYLATION | 120 | 2.4 | Up | 0.00100761 | 0.001087 |
| GO_MITOCHONDRIAL_MEMBRANE_ORGANIZATION | 119 | 1.6 | Up | 1.08 × 10−5 | 2.77 × 10−5 |
| GO_MITOCHONDRIAL_TRANSLATION | 111 | 2.6 | Up | 0.00020051 | 0.00027403 |
| GO_MITOCHONDRIAL_TRANSMEMBRANE_TRANSPORT | 69 | 2.1 | Up | 0.0001381 | 0.00020221 |
| GO_MITOCHONDRIAL_TRANSPORT | 212 | 1.5 | Up | 6.87 × 10−5 | 0.00011741 |
| GO_PROTEIN_LOCALIZATION_TO_MITOCHONDRION | 82 | 1.6 | Up | 3.96 × 10−5 | 7.37 × 10−5 |
| GO_MITOCHONDRIAL_PROTEIN_COMPLEX | 133 | 2.6 | Up | 0.00237101 | 0.00243029 |
| GO_POSITIVE_REGULATION_OF_MITOCHONDRION_ORGANIZATION | 182 | 1.3 | Up | 4.92 × 10−6 | 1.83 × 10−5 |
| GO_PROTEIN_TARGETING_TO_MITOCHONDRION | 60 | 1.8 | Up | 9.41 × 10−5 | 0.00015427 |
| PPARGC1A_TARGET_GENE_LIST | 24 | 1.7 | Up | 0.00013528 | 0.00020221 |
| GO_GLUTAMINE_FAMILY_AMINO_ACID_METABOLIC_PROCESS | 55 | 1.9 | Up | 0.00011041 | 0.000174 |
| GO_CELLULAR_RESPONSE_TO_FATTY_ACID | 70 | 2.7 | Up | 5.46 × 10−7 | 6.63 × 10−6 |
| GO_PYRUVATE_METABOLIC_PROCESS | 53 | 2.0 | Up | 4.99 × 10−5 | 8.90 × 10−5 |
NGenes, number of genes; E.S, enrichment score; FDR, false discovery rate.
FRY statistical test results to determine the level of enrichment of various gene sets in NK cells from infected mice versus uninfected controls. Enrichment of different gene sets was determined in NK cells from mice following L. monocytogenes infection compared with uninfected controls, and table shows the number of genes in each gene set, the direction of the enrichment in terms of genes demonstrating upregulation or downregulation after infection, as well as the p value and false discovery rate obtained from applying the FRY variant of the rotational gene set test for differential expression analysis (Wu et al., 2010).
Figure 1.

PGC-1α Regulates Metabolic Gene Transcription in NK Cells during Bacterial Infection
GSEA was performed on RNA sequencing data collected previously by the laboratory and used for different analyses for a previously published article (Nanbakhsh et al., 2019). Briefly, NK cells were isolated from the spleens of WT mice 48 h after infection with L. monocytogenes or from uninfected controls to identify changes in gene expression following infection (n = 3 individual mice). Bar code plots depict the genes comprising individual gene sets positioned along the horizontal axis based on expression in infected versus control mice with the trace indicating gene clustering in the up or down direction along the horizontal axis.
(A and B) Enrichment of (A) Hallmark gene associated with glycolysis and OxPhos along with enrichment of (B) gene ontology datasets related to specific mitochondrial functions was measured by competitive GSEA to determine an enrichment score (y axis), and the FRY variation of rotational gene set testing was used to calculate the significance of enrichment (Table 1).
(C) PGC-1α target genes were identified from previous work (Table 2), and enrichment in the up or down direction of the curated gene set was determined in NK cells following L. monocytogenes infection.
(D) Analysis of individual genes shows the change in expression values of specific genes following infection.
(E and F) WT and Ppargc1a cKO littermate controls were infected with 2 × 104 CFU of L. monocytogenes. After 24 h, livers were resected and CFU assays were performed by making serial dilutions of liver homogenates, spotting the dilutions in triplicate, and quantifying the data across all dilutions tested (n = 3 individual mice for each genotype from 2 independent experiments).
(G) NK cells were isolated from the spleens of infected mice via negative selection, RT-qPCR was performed, and expression values for detectable PGC-1α target genes were quantified (n = 3 individual mice for each genotype from 2 independent experiments). Bar graphs present the mean ± SD for individual experiments or mean ± SEM, where multiple experiments are indicated. Statistical significance was calculated using Student's t test (E) and Student's t test corrected for multiple comparisons using the Holm-Sidak method (G, individual genes). Two-way ANOVA was used to determine an overall statistical difference of PGC-1α-target gene expression between WT and Ppargc1a cKO NK cells following L. monocytogenes infection (G).
The significance of GSEA results was determined using the FRY variation of rotational gene set testing for differential expression analysis (Table 1). ∗p < 0.05.
In response to changing nutrient and energetic environments, PGC-1α serves as the principal regulator of nuclear-encoded mitochondrial gene transcription in multiple cell types. We thus hypothesized that L. monocytogenes infection would increase transcription of PGC-1α target genes in NK cells. To test this hypothesis, we compiled a list of known PGC-1α target genes (Table 2) and tested for their enrichment via competitive GSEA. PGC-1α regulates several genes in various tissues; however, its specific target genes in lymphocytes are not known. Therefore, we included genes capable of regulating varying mitochondrial functions including biogenesis, ETC activity, and FAO (Table 2). We then added our compiled list to our competitive GSEA and saw a significant enrichment of the PGC-1α target genes in NK cells isolated from infected mice compared with uninfected controls (Figure 1C). Analyses of individual genes showed that in addition to transcripts responsible for ETC activity, genes associated with mitochondrial nutrient acquisition pathways, including glutaminolysis, FAO, and pyruvate metabolism, were upregulated in NK cells in mice infected with L. monocytogenes (Figure 1D). Results from competitive GSEA also revealed enrichment of gene sets associated with these pathways following in vivo activation, further demonstrating transcriptional regulation of mitochondrial function (Figure S1D).
Table 2.
Identification and Function of Previously Established PGC-1α Target Genes
| Gene | GC ID | Tissue | Reference | Function |
|---|---|---|---|---|
| Ppargc1a | GC04M023755 | Muscle | (Handschin et al., 2003) | Transcription |
| Ppara | GC22P046150 | Adipose | (Mazzucotelli et al., 2007) | Transcription |
| Pparg | GC03P012328 | Muscle | (Zhang et al., 2007) | Transcription |
| Nrf1 | GC07P129611 | Muscle | (Wu et al., 1999) | Transcription |
| Esrra | GC11P064305 | Muscle | (Lin et al., 2004) | Transcription |
| Srebf1 | GC17M017810 | Liver | (Oberkofler et al., 2004) | Transcription |
| Pck1 | GC20P057561 | Liver | (Yoon et al., 2001) | Gluconeogenesis |
| G6pc | GC17P042900 | Liver | (Yoon et al., 2001) | Gluconeogenesis |
| Cpt1a | GC11M068772 | Liver | (Zhang et al., 2004) | FA oxidation |
| Cpt1b | GC22M050569 | Muscle | (Kleiner et al., 2009) | FA oxidation |
| Acadm | GC01P075724 | Liver | (Rhee et al., 2003) | FA oxidation |
| Tfam | GC10P058385 | Muscle | (Wu et al., 1999) | Biogenesis/ETC |
| Atp5b | GC12M056639 | Muscle | (Puigserver et al., 2001) | Biogenesis/ETC |
| ATP5g1 | GC17P048893 | Muscle | (Gerhart-Hines et al., 2007) | Biogenesis/ETC |
| NdufA2 | GC05M140607 | Adipose | (Bai et al., 2011) | Biogenesis/ETC |
| Cox4i1 | GC16P085833 | Muscle | (Puigserver et al., 2001) | Biogenesis/ETC |
| Cycs | GC07M025158 | Muscle | (Puigserver et al., 2001) | Biogenesis/ETC |
| Pdk1 | GC02P172555 | Muscle | (Gudiksen and Pilegaard, 2017) | Pyr. metabolism |
| Pdk2 | GC17P050095 | Muscle | (Gudiksen and Pilegaard, 2017) | Pyr. metabolism |
| Pdk3 | GC0XP024393 | Kidney | (Degenhardt et al., 2007) | Pyr. metabolism |
| Pdk4 | GC07M095583 | Muscle | (Kleiner et al., 2009) | Pyr. metabolism |
| Mfn2 | GC01P011980 | Muscle | (Cartoni et al., 2005) | Morphology |
| Ucp1 | GC04M140559 | Adipose | (Puigserver et al., 1998) | Thermogenesis |
| Slc1a5 | GC19M046760 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Gpt2 | GC16P046885 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Gls | GC02P190880 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Gls2 | GC12M056470 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Glul | GC01M182350 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Got1 | GC10M099396 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Got2 | GC16M058707 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
| Glud1 | GC10M087050 | Breast cancer | (McGuirk et al., 2013) | AA metabolism |
GC ID, gene card ID; FA, fatty acid; ETC, electron transport chain; Pyr., pyruvate; AA, amino acid.
Previous studies were utilized to compile a list of target genes potentially regulated by PGC-1α in NK cells. Genes from studies using different cell types and metabolic stresses were considered. The list contains genes responsible for varying aspects of mitochondrial function to account for various potential outcomes of PGC-1α-mediated regulation in NK cells. Genes are listed by italicized gene symbol in the table.
Next, we tested for the presence of PGC-1α in NK cells during L. monocytogenes infection. As low NK cell numbers make analyzing protein levels in NK cells ex vivo difficult, we tested for PGC-1α gene expression through RT-qPCR. We isolated NK cells from the spleens of mice infected with L. monocytogenes at various time points, extracted RNA, and performed RT-qPCR. RNA from macrophages was used as a positive control (McCarthy et al., 2013). Although present at lower amounts compared with macrophage RNA, we were able to detect the expression of the PGC-1α gene (Ppargc1a) in NK cells at all time points (Figure S2A). The presence of PGC-1α has been demonstrated in human NK cells, whereas its presence in murine NK cells is not well established. We therefore sought to confirm its presence and subsequently functional relevance in NK cells using a genetic deletion model. We generated an NK cell-specific conditional knockout mouse model (Ppargc1a cKO) by crossing an Ncr1Cre mouse with a Ppargc1a-floxed mouse containing loxP sites outside of exons 3–5 of the Ppargc1a gene (Handschin et al., 2007; Lin et al., 2004; Narni-Mancinelli et al., 2011). We verified this deletion using RT-PCR (Figure S2B) and observed similar developmental trajectories between WT and Ppargc1a cKO NK cells, suggesting that the loss of PGC-1α did not disrupt NK cell homeostasis (Figures S2C–S2F). Based on our RNA sequencing data, we hypothesized that a loss of PGC-1α would result in reduced clearance of L. monocytogenes. To test this hypothesis, we inoculated WT and Ppargc1a cKO mice with L. monocytogenes and performed colony-forming unit (CFU) assays 24 h post infection. CFU counts revealed that Ppargc1a cKO mice were indeed unable to control L. monocytogenes infection to the level of WT mice (Figures 1E and 1F). We then sought to determine if PGC-1α was regulating gene expression in NK cells during bacterial infection. We did this by measuring transcript levels of potential PGC-1α target genes we found to be upregulated following bacterial infection (Figure 1D). Because RT-qPCR has different levels-of-detection requirements than RNA sequencing, we also tested the expression of different isoforms of upregulated genes and genes involved in the same pathway as those upregulated. We found that NK cells isolated by negative selection (Figure S2G) from the spleens of Ppargc1a cKO mice following L. monocytogenes infection exhibited reduced expression of PGC-1α target genes compared with infected WT controls (Figure 1G). We also used these cells to confirm the presence of PGC-1α in NK cells and were indeed able to detect Ppargc1a transcripts in WT NK cells but not those isolated from Ppargc1a cKO mice (Figure S2H), suggesting that PGC-1α is present and functional in murine NK cells.
PGC-1α Is Required for Optimal NK Cell Effector Functions In Vitro
We next tested the role of PGC-1α in specific NK cell effector functions, i.e., cell-mediated cytokine production and cytotoxicity. To do this, we first verified that the presence of various activation and inhibitory receptors was unchanged on Ppargc1a cKO NK cells (Figure S3) and then utilized IL-2-cultured splenic NK cells from WT and Ppargc1a cKO to determine the importance of PGC-1α downstream of specific mechanisms of activation. Using cells cultured from WT and Ppargc1a cKO mice, which show a complete absence of detectable Ppargc1a gene expression (Figure S2B), we found that NK cells from Ppargc1a cKO mice produced significantly lower amounts of inflammatory molecules compared with WT NK cells following activation through NKG2D (Figure 2A). Ppargc1a cKO NK cells exhibited similar decreases in IFN-γ levels downstream of several other activation receptors (Figure 2B), suggesting that PGC-1α-mediated mitochondrial regulation plays a role in cytokine production downstream of receptor-mediated activation. Under these same stimulatory conditions, we also observed that activation of the upstream regulator of PGC-1α, AMPK, and inhibition of AMPK via compound C resulted in similar defects in cytokine production as those seen in PGC-1α-deficient NK cells (Figures S4A and S4B), further demonstrating the importance of this pathway in NK cell effector functions. Interestingly, following IL-12- and IL-18-mediated stimulation of NK cells, the production of both IFN-γ and GM-CSF remained intact in Ppargc1a cKO NK cells compared with WT, suggesting that in cultured cells, PGC-1α functions primarily downstream of activation receptor stimulation (Figure S4C).
Figure 2.

PGC-1α Is Required for Optimal NK Cell Effector Functions In Vitro
(A) IL-2-cultured NK cells from WT and Ppargc1a cKO mice were activated with plate-bound anti-NKG2D activating antibody, and supernatants were collected after 6 h for analysis of cytokine and chemokine secretion via multiplex (n = 6 individual mice for each genotype from 2 independent experiments).
(B) IL-2-cultured NK cells from WT and Ppargc1a cKO mice were activated with plate-bound anti-NK1.1, anti-2B4, or anti-Ly49H activating antibodies, and IFN-γ secretion was quantified via ELISA (n = 3 individual mice for each genotype).
(C) Cytokine and chemokine secretion was measured after 6 days in normoxic culture followed by an additional 24 h in normoxic or hypoxic conditions (1% O2) and subsequent stimulation via plate-bound anti-NKG2D activating antibody (n = 3 individual mice for each genotype). Data are presented as production of inflammatory molecules where production of individual molecules was normalized to the levels seen from WT mice. The fold change in production of inflammatory molecules from Ppargc1a cKO NK cells under normoxic conditions was compared with production from WT NK cells under normoxic conditions, and fold change in production of inflammatory molecules from Ppargc1a cKO NK cells under hypoxic conditions was compared with production from WT NK cells under hypoxic conditions.
(D) IL-2-cultured NK cells were incubated with 51Cr-labeled EL4 control or B16F10 target cells, and cell-mediated cytotoxicity was quantified via 51Cr release (n = 3 individual mice for each genotype).
(E) NK cells were isolated from the spleens of WT and Ppargc1a cKO mice via negative selection and activated ex vivo in RPMI with 10% FBS via plate-bound anti-NKG2D activating antibody (0, 1, 5, and 10 μg/mL). After 6 h, NK cells were harvested and IFN-γ positivity was assessed by flow cytometry.
(F) Control splenocytes from C57BL/6 (Self) mice, and target splenocytes from B6.b2mtm1Unc/J (Missing-Self) and BALB/cJ (Non-Self) mice were labeled with CFSE alone or CFSE and CTV or CFSE and CTR and injected at a 1:1:1 ratio into C57BL/6 control mice, C57BL/6 mice depleted of NK cells (PK136), WT, or Ppargc1a cKO mice. After 18 h, labeled splenocytes were collected, and flow cytometry was used to determine changes in cell ratio as indicating elimination of target cells by NK cells. Flow plots are shown depicting each cell type as a percentage of total, labeled splenocytes.
Bar graphs present the mean ± SD or mean ± SEM where multiple, independent experiments are indicated. Statistical significance was calculated using two-way ANOVA corrected for multiple comparisons using the Holm-Sidak method (A–D). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We next sought to determine if the reduced cytokine production capacity seen in NK cells from Ppargc1a cKO mice was a result of these cells' inability to mediate metabolic changes required for inflammatory cytokine production. As it would be difficult to restore the myriad aspects of mitochondrial function regulated by PGC-1α, we instead attempted to activate the cells in conditions where metabolic reprogramming would be skewed toward glycolytic metabolism to reduce the potential reliance upon PGC-1α. Previous work showed that culturing NK cells in hypoxic conditions augments glycolytic gene expression along with rates of glycolysis (Velasquez et al., 2016). We therefore cultured WT and Ppargc1a cKO NK cells in 1% O2 before NKG2D-mediated activation. NK cells cultured in normoxic conditions revealed similar defects in cytokine and chemokine production seen in previous experiments, whereas culturing cells in hypoxic conditions was sufficient to rescue secretion of proinflammatory molecules in Ppargc1a cKO NK cells (Figure 2C), suggesting that PGC-1α contributes to cytokine production through bioenergetic regulation. We then evaluated the ability of PGC-1α-deficient NK cells to mediate cytotoxicity in 51Cr-release assays. When cultured with B16F10 target cells, NK cells isolated from Ppargc1a cKO mice were unable to mediate cytotoxicity to the level of WT NK cells (Figure 2D). Although this defect was significant, PGC-1α-deficient NK cells retained moderate degrees of cytotoxicity with B16F10 and other target cells (Figure S4D). These data suggest that in cultured NK cells, PGC-1α may play a preferential role in cytokine production compared with cell-mediated cytotoxicity, although a further investigation into how mitochondrial metabolism contributes to these processes and determining how the loss of PGC-1α affects these functions in vivo is required to sufficiently explore this phenomenon.
Our data show that changing oxygen concentrations alters the requirement of PGC-1α for optimal NK cell effector functions, which suggests that PGC-1α is dependent on the nutrient environment. This context dependence has been described in other work as well (Donnelly et al., 2014; Keating et al., 2016; Keppel et al., 2015; Mah et al., 2017; Marcais et al., 2014) and demonstrates the importance of evaluating metabolic proteins independent of cell culture systems. We, therefore, sought to determine the significance of PGC-1α in NK cell effector functions independent of in vitro cell culture. To do this, we first utilized NK cells isolated from the spleens of WT and Ppargc1a cKO mice and activated through NKG2D. We found that PGC-1α-deficient NK cells exhibited only moderate decreases in intracellular IFN-γ production compared with WT when activated through NKG2D (Figure 2E). To test the cytotoxic capacity of naive Ppargc1a cKO NK cells, we utilized a splenocyte rejection model in which WT and Ppargc1a cKO mice receive labeled target cells. The cells were recovered, and flow cytometry is used to measure the ability of NK cells to eliminate the target cells in vivo. When labeled cells were injected into C57BL/6 control mice, target cells that were not susceptible to NK cell-mediated killing (Self) were recovered at 4–6 times the rate of those sensitive to NK cell-mediated cytotoxicity (non-self, missing self), demonstrating robust elimination of target cells (Figure 2F). This killing was lost when NK cells were depleted through injection of the NK cell-depleting antibody NK1.1 (PK136). When Ppargc1a cKO mice and littermate controls were injected, control cells were recovered at roughly four times the rate of target cells in both WT and PGC-1α-deficient mice (Figure 2F). This suggests that, in the absence of PGC-1α, cytotoxicity is maintained in an acute model of NK cell-mediated cytotoxicity. These data demonstrate that in acute models of naive NK cell activation, PGC-1α is not required for cytotoxicity or inflammatory cytokine production. To determine if differences in functional capacity seen following IL-2 culture were due to differences in NK cell maturation after IL-2 treatment, we measured CD27 and CD11b expression and found that NK cell maturation was similar between WT and Ppargc1a cKO NK cells, suggesting that functional differences were not a result of developmental defects that arose during cell culture (Figure S4E).
We also evaluated IL-2-cultured NK cells for differences in proliferative capacity and viability. Ki67 staining and overall culture yields revealed little differences in proliferation between WT and Ppargc1a cKO NK cells after culture (Figure S4F). Annexin V and propidium iodide (PI) staining revealed inconsistent increases in Annexin V and PI double-positive populations in PGC-1α-deficient NK cells, whereas WT NK cells exhibited inconsistent increases in PI single-positive populations (Figure S4F). These data suggest that PGC-1α may be partially responsible for enabling NK cells to maintain viability during extended cytokine exposure, although the trend is not consistent enough to make a definitive conclusion at this time. These data also suggest that defects in effector functions are not due to overall cellular health, as we would expect viability to mirror differences in cytokine production, and for NK cells activated through IL-12/18 to exhibit defects similar to those seen downstream of activation receptors.
PGC-1α Is Required for Optimal NK Cell Bioenergetics In Vitro
We next tested if defects seen in NK cell effector functions were associated with altered bioenergetics in PGC-1α-deficient NK cells. As PGC-1α regulates mitochondrial function, we initially evaluated OxPhos in NK cells isolated from WT and Ppargc1a cKO mice using Seahorse analyses. Mitochondrial stress tests revealed subtle but consistent and significant reductions in various mitochondrial parameters in PGC-1α-deficient NK cells (Figure 3A). We also measured basal and maximal glycolysis on the same cells and found slight inconsistent reductions in both glycolytic parameters in PGC-1α-deficient NK cells compared with WT cells (Figure 3B), suggesting PGC-1α may regulate overall cellular energetics, although it preferentially regulates mitochondrial function as expected. To determine if these differences were sufficient to affect cell metabolism, we directly compared the levels of key metabolites (ADP/ATP ratios and NAD/NADH ratios) between WT and Ppargc1a cKO NK cells to determine if there were differences in global energetics. These analyses revealed that PGC-1α-deficient NK cells had significantly augmented ADP/ATP ratios and tended to have decreased NAD/NADH ratios (Figure 3C). Interestingly, we compared mitochondrial mass between WT and PGC-1α-deficient NK cells under these same conditions and found minimal differences (Figure 3D), suggesting the bioenergetic defects seen in IL-2-cultured NK cells are independent of overall mitochondrial biogenesis. Previous studies have indicated that increased levels of ADP are indicative of cells primed to make ATP (Menk et al., 2018). Thus it is possible that PGC-1α-deficient NK cells are not turning over ADP to ATP as efficiently as WT cells and subsequently slowing the conversion of NADH to oxidized NAD. Although a detailed examination of cell metabolism and metabolomics would be necessary to test this hypothesis, these data do demonstrate that differences in ATP homeostasis results from PGC-1α deletion.
Figure 3.

PGC-1α Is Required for Optimal NK Cell Bioenergetics In Vitro
(A and B) IL-2-cultured NK cells from WT and Ppargc1a cKO mice were washed of IL-2 and seeded onto V3 PET Cell Culture Microplates in unbuffered RPMI, and oxygen consumption rate (OCR) was determined using a Seahorse XF-96 Extracellular Flux Analyzer. Basal OCR was determined, and oligomycin injection was used to quantify ATP-linked OCR followed by FCCP injection to quantify maximal OCR. Basal and maximal glycolytic rates were also obtained from the same set of cells by measuring extracellular acidification rate before and after oligomycin injection. A representative OCR plot is shown followed by quantification over multiple experiments (n = 7 individual mice for each genotype from 4 independent experiments).
(C) IL-2-cultured NK cells from WT and Ppargc1a cKO mice were seeded in control, or anti-NKG2D-coated wells for 6 h. Cells were removed from plates and lysed in potassium phosphate (oxidized metabolites) or potassium hydroxide (reduced metabolites) solutions to collect metabolites. High-performance liquid chromatography was used to quantify levels of ADP, ATP, NAD, and NADH (n = 3 individual mice for each genotype).
(D) IL-2-cultured NK cells from WT and Ppargc1a cKO mice were washed of IL-2 and stained with NK cell markers (NK1.1/CD3ϵ) along with the MitoTracker Green FM dye to measure mitochondrial mass. Data representative of individual mice are shown as histograms with mitochondrial mass on the x axis. Compiled data from multiple animals are presented as well (n = 3 individual mice for each genotype).
Bar graphs present the mean ± SD or mean ± SEM where multiple experiments are indicated. Statistical significance was calculated using two-way ANOVA corrected for multiple comparisons using the Holm-Sidak method (A–C), whereas Student's t test corrected for multiple comparisons was used to determine significance of individual metabolic parameters (A,B). ∗p < 0.05; ∗∗∗p < 0.001.
Human NK Cells Require PGC-1α for Optimal Effector Functions and OxPhos
Considering the importance of PGC-1α in other human tissues and the functional defects observed in the Ppargc1a cKO model, we hypothesized that PGC-1α signaling is critical for NK cell effector functions and bioenergetics in human primary NK cells as well. Therefore, to investigate the role of PGC-1α in human primary NK cells, we utilized the PGC-1α inhibitor SR-18292, which augments inhibitory acetylation marks on PGC-1α by promoting the interaction between PGC-1α and the acetyltransferase GCN5 (Sharabi et al., 2017). We first verified the ability of SR-18292 to inhibit PGC-1α-mediated gene transcription in NK cells. To do this, we activated murine NK cells through NKG2D in the presence or absence of SR-18292 and evaluated the expression of genes shown to be upregulated following in vivo activation (Figure 1D). RT-qPCR analysis showed an induction of several PGC-1α target genes following antibody-mediated stimulation (Figure S5A), and that this induction was significantly reduced for a select set of genes following treatment with SR-18292 (Figure S5A). This reduction occurred without global suppression of mitochondrial genes or loss of cell viability (Figures S5A–S5C).
We also tested whether SR-18292 reduced the functional capacity of Ppargc1a cKO NK cells by measuring IFN-γ production in IL-2-cultured NK cells from WT and Ppargc1a cKO mice to determine if treatment with the inhibitor elicited any off-target effects. As expected, we found that treatment with SR-18292 reduced the ability of WT NK cells to produce IFN-γ following stimulation through NKG2D (Figure S5D). Treatment with SR-18292 also reduced the ability of Ppargc1a cKO NK cells to produce IFN-γ downstream of NKG2D (Figure S5D), suggesting some off-target effects do occur. Previous studies in IL-2-cultured human NK cells show that targeted disruption of PGC-1α function via Ppargca1-specific small interfering RNA (siRNA) is associated with reduced mitochondrial activity (Miranda et al., 2016, 2018). Our data in Figure S5A do demonstrate on-target effects on mitochondrial gene expression, so we then tested whether inhibitor treatment was associated with altered mitochondrial function. To determine if SR-18292 treatment had similar energetic effects in naive human NK cells as those seen in siRNA-treated, cultured NK cells (Miranda et al., 2016, 2018), we performed Seahorse analyses. Treatment of NK cells from healthy donors with SR-18292 caused a decrease in multiple mitochondrial parameters and significant reductions in maximal respiration and spare respiratory capacity (Figures 4A and 4B) similar to results seen in previous work with PGC-1α-targeted siRNA (Miranda et al., 2016, 2018). Importantly, these energetic defects were not associated with a loss of cell viability (Figures S5E and S5F). Along with these energetic defects, functional analyses of IL-2-cultured human NK cells treated with Ppargc1a-specific siRNA also revealed defects in cytotoxicity and cytokine production (Miranda et al., 2016, 2018), results which were recapitulated in naive NK cells following treatment with SR-18292 (Figures 4C and 4D). These data recapitulate results seen with PGC-1α-targeted siRNA in cultured human NK cells and suggest an association between mitochondrial activity and effector functions in naive human NK cells as well.
Figure 4.

Human NK Cells Require PGC-1α for Optimal Effector Functions and OxPhos
Human NK cells were isolated from healthy donors using an NK cell-negative selection kit and maintained in 100 units IL-2.
(A and B) NK cells were isolated via negative selection and immediately activated for 18 h with plate-bound anti-NKG2D ± SR-18292 (30 μM). Overall mitochondrial activity and various parameters were quantified using a Seahorse XF-96 flux analyzer. (A) OCR plots and (B) quantified values are shown as the mean ± SEM of 4 separate donors (2 independent experiments).
(C) After selection, NK cells were maintained overnight and subsequently co-cultured with 51Cr-labeled K562 target cells ± SR-18292 at various effector to target (E:T) ratios. After 4 h, supernatants were collected to determine cell-mediated cytotoxicity (n = 4 individual donors, two independent experiments).
(D) After selection, NK cells were maintained overnight and subsequently stimulated with plate-bound anti-NKG2D for 6 or 18 h ± SR-18292 (30 μM). Supernatants were collected for measurement of cytokine/chemokine release via multiplex (n = 4 individual donors, two independent experiments).
Bar graphs present the mean ± SEM. Statistical significance was calculated using paired two-way ANOVA corrected for multiple comparisons using the Holm-Sidak method. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Ppargc1a cKO Mice Fail to Control Tumor Growth to the Level of WT Mice
Although previous work and our data here demonstrate the importance of PGC-1α in IL-2-cultured NK cells in vitro, the role of PGC-1α in maintaining mitochondrial metabolism in a TME has yet to be determined. In addition, studies investigating the role of lipid metabolism pathways controlled by PPARα and PPARδ, transcription factors whose function is augmented by PGC-1α, show activation of these pathways suppresses NK cell function. Thus, we sought to clarify the role of PGC-1α in maintaining mitochondrial metabolism in a TME by directly testing the importance of PGC-1α in NK cells during tumor challenge. Given the PGC-1α-dependent gene expression seen following in vivo activation of NK cells and the functional defects seen in PGC-1α-deficient NK cells in vitro, we hypothesized that PGC-1α maintains cellular metabolism required for NK cell-mediated control of tumor growth. To test this hypothesis, we utilized the B16F10 lung metastasis model, as NK cells are the primary immune cells responsible for controlling tumor growth in this model. WT and Ppargc1a cKO mice were injected with 2 × 105 B16F10 cells, and lungs were isolated 10–14 days postinjection. Lung resection revealed significant impairment in the ability of PGC-1α-deficient NK cells to control B16F10 tumor growth, as lungs from Ppargc1a cKO mice contained, on average, over twice the number of nodules as their corresponding littermate controls (Figures 5A–5C). We next determined if the inability of PGC-1α-deficient NK cells to control tumor growth was associated with altered cellular metabolism in the TME. To test this, we isolated lymphocytes from the lungs of tumor-bearing mice and measured mitochondrial mass and membrane potential in CD3ϵ−NK1.1+ NK cells using flow cytometry. When these parameters were analyzed individually, we found that both mitochondrial mass and mitochondrial membrane potential were reduced in PGC-1α-deficient NK cells isolated from the lungs of tumor-bearing mice (Figures 5D and 5F). We also analyzed both parameters on the same flow plot to identify the percentage of cells that had high amounts of mitochondria as well as high levels of mitochondrial transmembrane potential. This staining profile identifies cells with high numbers of active mitochondria, which we and others term polarized cells (Bengsch et al., 2016; Scharping et al., 2016). In NK cells from WT mice, we found polarized cells at nearly double the rate as were present in NK cells from Ppargc1a cKO mice (Figures 5D and 5F), suggesting PGC-1α is required to maintain mitochondrial function in the TME. We also performed identical analyses on naive NK cells from the lungs of WT and Ppargc1a cKO mice and found that both mitochondrial mass and membrane potential were similar (Figure 5E), suggesting mitochondrial regulation downstream of PGC-1α occurs during tumor challenge specifically.
Figure 5.

Ppargc1a cKO Mice Fail to Control Tumor Growth to the Level of WT Mice
WT and Ppargc1a cKO mice were injected with 2 × 105 B16F10 melanoma cells intravenously, and lungs were collected 10–14 days later.
(A) Images of whole lungs were taken using a stereoscope.
(B) Lung sections were also obtained and fixed in zinc formalin followed by freezing and slicing for H&E staining. H&E images are representative and were not used for quantification. Scale bars on whole sections (left), 1 mm and on individual nodules (right), 100 μm.
(C) The total number of nodules was quantified on the resected lungs through blinded counting and averaging from multiple mice (n = 6 individual mice for each genotype from 2 independent experiments).
(D and E) WT and Ppargc1a cKO mice were injected with 2 × 105 B16F10 melanoma cells, and 10 days later, lungs were removed from tumor-bearing mice and lymphocytes were isolated. Lymphocyte suspensions were stained with NK cell surface markers (CD3ϵ−NK1.1+) and a live/dead stain (PI), along with dyes for mitochondrial mass (Mitotracker Green FM) and mitochondrial membrane potential (TMRE). Representative data of individual parameters are shown after gating on NK cells (histograms), along with both parameters in the same dot plot after gating on NK cells to quantify cells with high amounts of active mitochondria (polarized). Identical analyses were performed on NK cells from control mice that did not receive any tumor challenge.
(F) Data for mitochondrial mass and membrane potentials from (D and E) were quantified over multiple experiments (n = 4 individual mice for each genotype, 3 independent experiments).
(G) WT and Ppargc1a cKO mice were injected with 2 × 105 B16F10 melanoma cells, and 10 days later lungs were removed and NK cells were isolated via fluorescence-activated cell sorting (FACS). RT-qPCR was then used to evaluate gene expression in FACS-sorted NK cells to determine expression levels of PGC-1α-target genes seen upregulated in RNA sequencing data (n = 3 individual mice for each genotype from 2 independent experiments).
Bar graphs present the mean ± SD or mean ± SEM, where multiple experiments are indicated. Statistical significance was calculated using Student's t test (C) or Student's t test corrected for multiple comparisons using the Holm-Sidak method (F and G, individual genes). Two-way ANOVA was used to determine overall statistical difference of PGC-1α-target gene expression between WT and Ppargc1a cKO NK cells following tumor inoculation (G). ∗p < 0.05; ∗∗p < 0.01.
PGC-1α Regulates Metabolic Gene Transcription during Tumor Growth
Although these data show significant differences in mitochondrial phenotype between WT and PGC-1α-deficient mice, these may be simply due to the increased tumor burden seen in the Ppargc1a cKO mice, thus resulting in less metabolically fit cells. Therefore, we evaluated glucose uptake in NK cells isolated from the TME as a readout of non-mitochondrial metabolic function. We found that PGC-1α-deficient NK cells mediated glucose uptake to similar levels seen in WT NK cells (Figure S6), suggesting that the altered mitochondrial phenotype observed in Ppargc1a cKO NK cells is not due to global metabolic suppression resulting from an increased tumor burden. We then tested the function of PGC-1α in the TME directly by comparing the transcription of PGC-1α target genes between WT and PGC-1α-deficient NK cells isolated from the TME. To do this, we sorted NK cells by fluorescence-activated cell sorting FACS from the lungs of tumor-bearing mice and isolated RNA to test for differential expression of PGC-1α target genes. The analysis revealed significant decreases in expression of multiple PGC-1α target genes in NK cells isolated from Ppargc1a cKO mice compared with WT controls (Figure 5G). Similar to results from L. monocytogenes infection, we observed a reduction in the transcription of several genes that function in mitochondrial nutrient utilization pathways such as Lcad, Pdk1, Got1, and Got2. Further analysis beyond the scope of this study is needed to directly test the importance of specific genes in maintaining mitochondrial activity in this setting, although these data do suggest PGC-1α-mediated transcriptional regulation of mitochondria occurs as part of NK cell anti-tumor immunity.
Discussion
The data presented here demonstrate the critical role of PGC-1α in maintaining mitochondrial function in NK cells during an immune response. Using RNA sequencing analyses, we identified significant enrichment of a gene set composed of established target genes of PGC-1α following in vivo activation of NK cells via L. monocytogenes infection. The deletion of PGC-1α in NK cells resulted in augmented bacterial growth associated with reduced PGC-1α-target gene transcription in cells isolated from Ppargc1a cKO mice compared with WT controls. In terms of specific effector functions, we found that PGC-1α was required for both cytotoxicity and cytokine production in NK cells, and that these defects were associated with bioenergetic deficiencies induced by disruption of PGC-1α function. Using the B16F10 tumor model, we found that defects in mitochondrial function in the TME were associated with the reduced ability of Ppargc1a cKO NK cells to control the growth of B16F10 lung metastases. Analysis of mitochondria from control experiments demonstrated reductions in mitochondrial parameters were a result of tumor challenge, and transcriptional analyses suggest PGC-1α regulates mitochondria-associated gene expression during the NK cell response to tumorigenesis. Our data bring to light the critical role of PGC-1α-mediated mitochondrial regulation for the NK cell immune response.
Based on previous work demonstrating transcription-based control of metabolism in NK cells (Donnelly et al., 2014; Velasquez et al., 2016), we utilized whole-transcriptome data analysis to test for changes in metabolic regulation following in vivo activation of NK cells. As expected, GSEA of transcriptional changes in NK cells following infection with L. monocytogenes demonstrated robust activation of inflammatory gene signatures as well as increased expression of genes responsible for driving glycolysis. This is consistent with previous work in murine cytomegalovirus infection models where administration of 2-deoxyglucose or mTOR deletion inhibits the function of NK cells (Mah et al., 2017; Marcais et al., 2014) and provides further evidence supporting earlier studies suggesting glycolysis is a necessary component of NK cell effector functions. In addition to these expected changes, analyses of cells from infected mice show significant enrichment of gene sets critical for mitochondrial function, including genes associated with mitochondrial nutrient uptake, ETC subunits, as well as known target genes of PGC-1α. Our data show only a minimal transcriptional regulation of Ppargc1a in NK cells following infection, and our RNA sequencing results also suggest transcription of PGC-1α itself is not co-regulated along with its target genes. PGC-1α is subject to multiple post-translational modifications that control both protein activity and stability (Jager et al., 2007; Rodgers et al., 2005), suggesting a potential role for post-transcriptional mechanisms, although further biochemical evaluation is necessary to determine the mechanism that regulates PGC-1α in vivo.
Results from differential expression analysis of our RNA sequencing data show that whereas a set of PGC-1α-regulated genes was upregulated following infection, multiple genes were unchanged or downregulated. In addition, experiments with Ppargc1a cKO NK cells suggest that, in the case of Gpt2 specifically, PGC-1α-independent regulation may occur as well. Previous work shows the specific PGC-1α-dependent gene expression profile can vary between tissues and cell types, with the outcome being controlled both by the activation condition and the transcription factor to which PGC-1α binds. In hepatocytes, for example, PGC-1α activation from fasting increases transcription of the gluconeogenic genes Pck1 and G6pc through activation of HNF4 (Herzig et al., 2001; Rhee et al., 2003; Yoon et al., 2001). In addition to these gluconeogenesis genes, activation of PGC-1α in hepatocytes also induces expression of genes required for FAO, such as Cpt1 and Acadm (Rhee et al., 2003). Although still being PGC-1α-dependent, activation of these genes is regulated independently of the gluconeogenic targets Pck1 and G6pc, and loss of HNF4 does not disrupt the ability of PGC-1α to induce transcription of Cpt1 and Acadm (Rhee et al., 2003). In addition to specific metabolic pathways, PGC-1α-mediated mitochondrial biogenesis has also been shown to be context dependent, as activation of PPARδ in skeletal muscle augments PGC-1α-mediated FAO independent of changes in mitochondrial gene expression (Kleiner et al., 2009). Our studies utilizing Ppargc1a cKO NK cells reveal similar variations in the role of PGC-1α in mitochondrial biogenesis between in vitro and in vivo activation conditions and demonstrate that the function of PGC-1α in NK cells may change depending on activation and culture conditions as well. These findings are consistent with regulation of PGC-1α function seen in other cell types (Kleiner et al., 2009) and suggest that NK cells have an activation-specific PGC-1α target gene expression profile that warrants further, mechanistic investigation.
Functionally we were able to show PGC-1α is required for NK cell-mediated control of bacterial infection in vivo, as well as cytokine production and cytotoxicity in vitro. The defect in cytokine production seen in vitro was rescued upon short-term culture in hypoxic conditions, and analysis of cytotoxicity and cytokine production in naive NK cells also revealed little difference between WT and PGC-1α-deficient NK cells. We also observed differences in the requirement of PGC-1α for cytokine production downstream of NKG2D-mediated activation compared with cytokine receptor stimulation. Our observations are in line with several recent studies demonstrating that metabolic requirements vary with the experimental conditions in which the cells are activated. For example, cytokine production downstream of NK1.1 activation is sensitive to oligomycin treatment even after culture in high doses of IL-15; however, extending the culture period from 24 to 72 h rendered NK cell-mediated IFN-γ production insensitive to oligomycin treatment (Keppel et al., 2015). Oppositely, naive NK cells are insensitive to 2-DG treatment following cytokine-mediated activation, whereas IL-15-cultured NK cells show significant decreases in IFN-γ production downstream of cytokine receptors following activation in the presence of 2-DG (Donnelly et al., 2014; Keppel et al., 2015). Similarly, NK cell-mediated IFN-γ production is reduced by oligomycin treatment downstream of NK1.1 stimulation, whereas it is only minimally affected downstream of the IL-12 and IL-18 receptors (Keppel et al., 2015). These changes in sensitivity to metabolic inhibitors depending on different culture conditions or activation mechanisms demonstrate the context-dependent metabolic requirements of NK cells and highlight the need for further analysis of NK cell metabolism using in vivo models combined with in vitro mechanistic investigations.
PGC-1α is typically activated as part of the response to energetic stress when changing nutrient environments disrupt cell homeostasis Puigserver et al., 1998. Analyses of NK cell development and effector functions during acute models of activation suggest that the role of PGC-1α in NK cells is similar to what is observed in other cell types in that it does not contribute to mitochondrial regulation under all conditions. These data suggest that acute models of activation do not disrupt NK cell homeostasis to the degree that PGC-1α-mediated mitochondrial regulation is required, although it is also possible that loss of PGC-1α in these contexts is being compensated for by the other PGC-1 family members, PGC-1β or PGC-1α-related coactivator (PRC). In the case of PGC-1β specifically, overexpression studies of glucose metabolism in hepatocytes reveal several overlapping targets between the two proteins (Lin et al., 2003). Previous work shows the proteins do not completely compensate for one another, however, and single deletion of either coactivator causes significant mitochondrial defects in multiple tissues (Karkkainen et al., 2019; Shao et al., 2010; Chambers et al., 2012; Gurung et al., 2011; Lin et al., 2004), similar to what our data show here in NK cells following deletion of PGC-1α. These data do not rule out the possibility of PGC-1β regulating mitochondrial function in specific contexts, and in fact, the variation seen in mitochondrial defects across activation conditions suggest an additional investigation into the role that other PGC-1 family members play in NK cell metabolism is warranted.
The context-dependent nature of metabolic requirements is also demonstrated in comparing our in vitro cytokine production data downstream of IL-12 and IL-18 and our in vivo clearance of L. monocytogenes, which is partially dependent on IL-12- and IL-18-mediated NK cell activation (Nomura et al., 2002). The differences in bacterial clearance not being mirrored in in vitro cytokine production assays may be due to culturing cells in IL-2, which could have altered the metabolic requirements of NK cell activation. The other possibility, which is also likely based on the complex processes involved in an in vivo immune response versus in vitro activation is that the two mechanisms of stimulation are not the same. Although IL-12 and IL-18 have been shown to play a role in NK cell activation during L. monocytogenes infection (Humann and Lenz, 2010; Tripp et al., 1994), this same work also shows that induction of maximal NK cell function requires cell-cell contact, and that eliminating the interaction between NK cells and infected DCs abrogates NK cell-mediated IFN-γ production (Humann and Lenz, 2010). In addition, the L1S fragment of the L. monocytogenes protein p60 can enhance NK cell activation and stimulate IFN-γ production (Humann et al., 2007; Ortiz and Lenz, 2017). These previous studies and our data here strongly suggest that stimulation of NK cells during L. monocytogenes infection involves additional factors beyond IL-12- and IL-18-mediated activation.
In addition to PGC-1α, our in vitro analyses demonstrate that NKG2D stimulation activates AMPK, and that AMPK inhibition disrupts NK cell-mediated cytokine production. Interestingly, previous work in T cells shows that although AMPK is required to sustain T cell numbers in vivo, AMPK-deficient T cells exhibit a hyper-responsive phenotype following in vitro stimulation similar to the NKG2D-mediated activation used in this study (Blagih et al., 2015). These data suggest that AMPK activation has a varied effect in T cells compared with NK cells in terms of IFN-γ production. Because AMPK is required to sustain T cell numbers during infection, we suspect that the different role of AMPK is due to differences in proliferative demand in T cells versus NK cells following stimulation through cell surface receptors. Specifically, stimulation through the T cell receptor induces clonal expansion, whereas activation of NK cells through NKG2D occurs without any increase in proliferation. This likely results in different energetic demands in NK cells versus T cells following stimulation through these receptors, which could lead to different functions for proteins responsible for dealing with energetic stress like AMPK. The role of AMPK could be tested in NK cells by stimulating through pathways known to induce either proliferation or cytokine production, for example, IL-15- versus NKG2D-mediated activations.
Previous reports have established the importance of PGC-1α in human NK cells for cytotoxicity following culture in high doses of IL-2 (Miranda et al., 2016, 2018). Our results here complement these findings by demonstrating a similar association between disrupted mitochondrial function and reduced NK cell effector functions in naive human NK cells as well. We utilized primary human NK cells and a small molecule inhibitor previously shown to reduce PGC-1α target gene expression in vitro and in vivo, and although treatment of murine NK cells revealed off-target effects of the inhibitor, we were able to recapitulate the metabolic defects previously seen in cultured human NK cells (Miranda et al., 2016, 2018). Myriad studies of lymphocyte metabolism demonstrate the translational potential of utilizing metabolic regulation to optimize cell-based immunotherapy (Kishton et al., 2017). Previous work in T cells demonstrates that mitochondrial function can be utilized as a predictor of cellular persistence, and that in vitro metabolic reprogramming can induce prolonged memory formation following engraftment (Lugli et al., 2013; Gattinoni et al., 2011). In addition, previous work in T cells and NK cells shows metabolic alterations can be used to optimize anti-tumor functions during tumor progression (Menk et al., 2018; Cong et al., 2018). Our data here demonstrate an association between mitochondrial function and optimal NK cell cytotoxicity and cytokine production in primary human NK cells, thus corroborating previous studies in cultured NK cells and demonstrating the importance of optimizing mitochondrial function during NK cell-based immunotherapy.
In addition to the in vitro evaluation of specific effector functions, our data from the B16F10 lung melanoma model suggest PGC-1α regulates mitochondrial function during the NK cell response to tumor growth. Our data also identify multiple PGC-1α target genes whose reduced expression was associated with an inability to optimally control tumor growth, including Lcad, Pdk1, Got1, and Got2. Because PGC-1α regulates multiple pathways capable of augmenting mitochondrial function, it is necessary to determine the specific PGC-1α target genes responsible for maintaining mitochondrial function in NK cells to identify potential mechanisms by which metabolism can be controlled. Our data measuring gene expression in cells isolated directly from the TME provide a set of candidates for augmenting cell function through mitochondrial regulation. Previous work has demonstrated the ability of metabolic control to increase lymphocyte function. For example, over-expression of Pck1, the enzyme responsible for the synthesis of phosphoenolpyruvate, prolongs calcium signaling in activated T cells and enhances anti-tumor efficacy following adoptive transfer (Ho et al., 2015). Similarly, chimeric antigen receptors engineered to promote mitochondrial metabolism provide T cells with increased persistence and a more memory-like phenotype following transduction (Kawalekar et al., 2016). These studies exemplify the translational potential of immune cell metabolism and demonstrate the importance of elucidating mechanisms of metabolic regulation in lymphocytes.
During tumor growth, transformed cells are capable of sequestering glucose away from lymphocytes, thus creating a metabolically challenging environment that can limit the effectiveness of infiltrating immune cells (Andrejeva and Rathmell, 2017; Chang et al., 2015; Siska et al., 2017; Vander Heiden et al., 2009). Our data show that insufficient mitochondrial function contributes to immune cell dysfunction and that PGC-1α represents a potential pathway through which mitochondrial metabolism could be augmented during an immune response. Given that adoptive transfers of NK cells into patients have demonstrated efficacy against multiple cancer types (Cheng et al., 2013; Iliopoulou et al., 2010; Miller et al., 2005; Ruggeri et al., 2002b), these findings have the potential to optimize cell-based immunotherapy through metabolic regulation.
Limitations of the Study
The data obtained using the inhibitor SR-18292 on human NK cells are limited in that using only the inhibitor makes phenotypic changes difficult to interpret without considering previous studies. Our data serve as a validation of previous work in which siRNA was used to show that the functional loss of PGC-1α elicits mitochondrial defects similar to our results (Miranda et al., 2016, 2018). Our data are unique in that we used naive human NK cells as opposed to IL-2-cultured cells, but they are limited in that off-target effects of the inhibitor make it difficult to draw definitive conclusions. Irrespective of this, the previous work utilizing siRNA shows similar defects in the absence of off-target effects, thus supporting our findings from this study (Miranda et al., 2016, 2018). In addition, because the data in this study are from several models of an immune response, it is not possible to determine mechanistically how PGC-1α-mediated mitochondrial function may contribute to NK cell effector functions as its role may vary between the different models. Our data suggest PGC-1α is required for optimal NK cell effector functions under specific activation conditions, including in response to bacterial infection and tumor progression. Although the data thus present a broader scope of the importance of PGC-1α, they fail to elucidate one specific mechanistic pathway. Defining how PGC-1α-mediated mitochondrial function contributes to NK cell effector functions is important both from a basic science and clinical perspective, and the data presented in this study potentially serve as a starting point for this future work.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Subramaniam Malarkannan (SMalarkannan@Versiti.org).
Materials Availability
This study generated a conditional knockout mouse model of PGC-1α. Inquiries and material requests should be directed to and will be fulfilled by the Lead Contact, Subramaniam Malarkannan (SMalarkannan@Versiti.org).
Data and Code Availability
This study did not generate any new RNA seq datasets, and those data analyzed from previous studies (Nanbakhsh et al., 2019) can be found on the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI) under the accession number GEO: GSE136015 and are publicly accessible at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136015.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Ncr1iCre mice were a kind gift from Eric Vivier, Center d'Immunologie de Marseille-Luminy, France. This research was completed in part with computational resources and technical support provided by the Research Computing Center at the Medical College of Wisconsin. We thank the colleagues from Children's Research Institute Histology Core for help with the lung sections and H&E staining. We are grateful to Jason Siebert, Nathan Schloemer, and Alex Abel for inputs in the preparation of this manuscript. This work was supported in part by Ann’s Hope Melanoma Foundation (S.M. and M.S.T.); NIH R01 AI102893 (S.M.) and NCI R01 CA179363 (S.M. and M.S.T.); Alex's Lemonade Stand Foundation for Childhood Cancer (S.M.); HRHM Program of MACC Fund/Children's Hospital of Wisconsin (S.M.); Nicholas Family Foundation (S.M.); Gardetto Family (S.M.); MCW-Cancer Center-Large Seed Grant (S.M. & M.S.T.); MACC Fund/Children's Hospital of Wisconsin (M.S.T. and S.M.); and Advancing Healthier Wisconsin (S.M. & M.R.).
Author Contributions
Z.J.G. and S.M. were primarily responsible for designing and building the concept of the study. E.H., A.N., S.H., C.Y., and A.M. contributed to designing, developing, and carrying out the methodology and techniques used as well as interpretation of the data. A.N., S.-W.T., A.L., and M.J.F. helped collect and analyze the RNA sequencing data, and M.J.R. and M.S.T. played a critical role in guiding the direction of the study and supervising the work. Z.J.G. performed the bulk of the experiments, crafted the figures, and wrote the manuscript with guidance from S.M.
Declaration of Interests
The authors declare that they have no competing interests.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101454.
Supplemental Information
References
- Andrejeva G., Rathmell J.C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017;26:49–70. doi: 10.1016/j.cmet.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P., Canto C., Oudart H., Brunyanszki A., Cen Y., Thomas C., Yamamoto H., Huber A., Kiss B., Houtkooper R.H. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B., Johnson A.L., Kurachi M., Odorizzi P.M., Pauken K.E., Attanasio J., Stelekati E., McLane L.M., Paley M.A., Delgoffe G.M. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagih J., Coulombe F., Vincent E.E., Dupuy F., Galicia-Vazquez G., Yurchenko E., Raissi T.C., van der Windt G.J., Viollet B., Pearce E.L. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- Cartoni R., Leger B., Hock M.B., Praz M., Crettenand A., Pich S., Ziltener J.L., Luthi F., Deriaz O., Zorzano A. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005;567:349–358. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerwenka A., Lanier L.L. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- Chambers K.T., Chen Z., Crawford P.A., Fu X., Burgess S.C., Lai L., Leone T.C., Kelly D.P., Finck B.N. Liver-specific PGC-1beta deficiency leads to impaired mitochondrial function and lipogenic response to fasting-refeeding. PLoS One. 2012;7:e52645. doi: 10.1371/journal.pone.0052645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoto K., Chowdhury P.S., Kumar A., Sonomura K., Matsuda F., Fagarasan S., Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. U S A. 2017;114:E761–E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.H., Qiu J., O'Sullivan D., Buck M.D., Noguchi T., Curtis J.D., Chen Q., Gindin M., Gubin M.M., van der Windt G.J. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Chen Y., Xiao W., Sun R., Tian Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013;10:230–252. doi: 10.1038/cmi.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong J., Wang X., Zheng X., Wang D., Fu B., Sun R., Tian Z., Wei H. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. 2018;28:243–255. doi: 10.1016/j.cmet.2018.06.021. [DOI] [PubMed] [Google Scholar]
- Degenhardt T., Saramaki A., Malinen M., Rieck M., Vaisanen S., Huotari A., Herzig K.H., Muller R., Carlberg C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 2007;372:341–355. doi: 10.1016/j.jmb.2007.06.091. [DOI] [PubMed] [Google Scholar]
- Diefenbach A., Jensen E.R., Jamieson A.M., Raulet D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly R.P., Loftus R.M., Keating S.E., Liou K.T., Biron C.A., Gardiner C.M., Finlay D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014;193:4477–4484. doi: 10.4049/jimmunol.1401558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn P.L., North R.J. Early gamma interferon production by natural killer cells is important in defense against murine listeriosis. Infect. Immun. 1991;59:2892–2900. doi: 10.1128/iai.59.9.2892-2900.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauriat C., Long E.O., Ljunggren H.G., Bryceson Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115:2167–2176. doi: 10.1182/blood-2009-08-238469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido F., Aptsiauri N., Doorduijn E.M., Garcia Lora A.M., van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016;39:44–51. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z., Rodgers J.T., Bare O., Lerin C., Kim S.H., Mostoslavsky R., Alt F.W., Wu Z., Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudiksen A., Pilegaard H. PGC-1alpha and fasting-induced PDH regulation in mouse skeletal muscle. Physiol. Rep. 2017;5:e13222. doi: 10.14814/phy2.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung I.S., Medina-Gomez G., Kis A., Baker M., Velagapudi V., Neogi S.G., Campbell M., Rodriguez-Cuenca S., Lelliott C., McFarlane I. Deletion of the metabolic transcriptional coactivator PGC1beta induces cardiac arrhythmia. Cardiovasc. Res. 2011;92:29–38. doi: 10.1093/cvr/cvr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C., Choi C.S., Chin S., Kim S., Kawamori D., Kurpad A.J., Neubauer N., Hu J., Mootha V.K., Kim Y.B. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C., Rhee J., Lin J., Tarr P.T., Spiegelman B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc. Natl. Acad. Sci. U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Long F., Jhala U.S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Ho P.C., Bihuniak J.D., Macintyre A.N., Staron M., Liu X., Amezquita R., Tsui Y.C., Cui G., Micevic G., Perales J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humann J., Bjordahl R., Andreasen K., Lenz L.L. Expression of the p60 autolysin enhances NK cell activation and is required for listeria monocytogenes expansion in IFN-gamma-responsive mice. J. Immunol. 2007;178:2407–2414. doi: 10.4049/jimmunol.178.4.2407. [DOI] [PubMed] [Google Scholar]
- Humann J., Lenz L.L. Activation of naive NK cells in response to Listeria monocytogenes requires IL-18 and contact with infected dendritic cells. J. Immunol. 2010;184:5172–5178. doi: 10.4049/jimmunol.0903759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulou E.G., Kountourakis P., Karamouzis M.V., Doufexis D., Ardavanis A., Baxevanis C.N., Rigatos G., Papamichail M., Perez S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010;59:1781–1789. doi: 10.1007/s00262-010-0904-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen O., Tuomainen T., Mutikainen M., Lehtonen M., Ruas J.L., Hanhineva K., Tavi P. Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019;115:107–118. doi: 10.1093/cvr/cvy155. [DOI] [PubMed] [Google Scholar]
- Kawalekar O.U., O'Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Keating S.E., Zaiatz-Bittencourt V., Loftus R.M., Keane C., Brennan K., Finlay D.K., Gardiner C.M. Metabolic reprogramming supports IFN-gamma production by CD56bright NK cells. J. Immunol. 2016;196:2552–2560. doi: 10.4049/jimmunol.1501783. [DOI] [PubMed] [Google Scholar]
- Keppel M.P., Saucier N., Mah A.Y., Vogel T.P., Cooper M.A. Activation-specific metabolic requirements for NK Cell IFN-gamma production. J. Immunol. 2015;194:1954–1962. doi: 10.4049/jimmunol.1402099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton R.J., Sukumar M., Restifo N.P. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017;26:94–109. doi: 10.1016/j.cmet.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff C.A., Gattinoni L., Torabi-Parizi P., Kerstann K., Cardones A.R., Finkelstein S.E., Palmer D.C., Antony P.A., Hwang S.T., Rosenberg S.A. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner S., Nguyen-Tran V., Bare O., Huang X., Spiegelman B., Wu Z. PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. J. Biol. Chem. 2009;284:18624–18633. doi: 10.1074/jbc.M109.008797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Tarr P.T., Yang R., Rhee J., Puigserver P., Newgard C.B., Spiegelman B.M. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Loftus R.M., Assmann N., Kedia-Mehta N., O'Brien K.L., Garcia A., Gillespie C., Hukelmann J.L., Oefner P.J., Lamond A.I., Gardiner C.M. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018;9:2341. doi: 10.1038/s41467-018-04719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis C.U., Savoldo B., Dotti G., Pule M., Yvon E., Myers G.D., Rossig C., Russell H.V., Diouf O., Liu E. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli E., Dominguez M.H., Gattinoni L., Chattopadhyay P.K., Bolton D.L., Song K., Klatt N.R., Brenchley J.M., Vaccari M., Gostick E. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Invest. 2013;123:594–599. doi: 10.1172/JCI66327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah A.Y., Rashidi A., Keppel M.P., Saucier N., Moore E.K., Alinger J.B., Tripathy S.K., Agarwal S.K., Jeng E.K., Wong H.C. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight. 2017;2:e95128. doi: 10.1172/jci.insight.95128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais A., Cherfils-Vicini J., Viant C., Degouve S., Viel S., Fenis A., Rabilloud J., Mayol K., Tavares A., Bienvenu J. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014;15:749–757. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucotelli A., Viguerie N., Tiraby C., Annicotte J.S., Mairal A., Klimcakova E., Lepin E., Delmar P., Dejean S., Tavernier G. The transcriptional coactivator peroxisome proliferator activated receptor (PPAR)gamma coactivator-1 alpha and the nuclear receptor PPAR alpha control the expression of glycerol kinase and metabolism genes independently of PPAR gamma activation in human white adipocytes. Diabetes. 2007;56:2467–2475. doi: 10.2337/db06-1465. [DOI] [PubMed] [Google Scholar]
- McCarthy C., Lieggi N.T., Barry D., Mooney D., de Gaetano M., James W.G., McClelland S., Barry M.C., Escoubet-Lozach L., Li A.C. Macrophage PPAR gamma Co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol. Med. 2013;5:1443–1457. doi: 10.1002/emmm.201302587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirk S., Gravel S.P., Deblois G., Papadopoli D.J., Faubert B., Wegner A., Hiller K., Avizonis D., Akavia U.D., Jones R.G. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013;1:22. doi: 10.1186/2049-3002-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menk A.V., Scharping N.E., Rivadeneira D.B., Calderon M.J., Watson M.J., Dunstane D., Watkins S.C., Delgoffe G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018;215:1091–1100. doi: 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael L.F., Wu Z., Cheatham R.B., Puigserver P., Adelmant G., Lehman J.J., Kelly D.P., Spiegelman B.M. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. U S A. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelet X., Dyck L., Hogan A., Loftus R.M., Duquette D., Wei K., Beyaz S., Tavakkoli A., Foley C., Donnelly R. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018;19:1330–1340. doi: 10.1038/s41590-018-0251-7. [DOI] [PubMed] [Google Scholar]
- Miller J.S., Soignier Y., Panoskaltsis-Mortari A., McNearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- Miranda D., Jara C., Ibanez J., Ahumada V., Acuna-Castillo C., Martin A., Cordova A., Montoya M. PGC-1alpha-dependent mitochondrial adaptation is necessary to sustain IL-2-induced activities in human NK cells. Mediators Inflamm. 2016;2016:9605253. doi: 10.1155/2016/9605253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda D., Jara C., Mejias S., Ahumada V., Cortez-San M.M., Ibanez J., Hirsch S., Montoya M. Deficient mitochondrial biogenesis in IL-2 activated NK cells correlates with impaired PGC1-alpha upregulation in elderly humans. Exp. Gerontol. 2018;110:73–78. doi: 10.1016/j.exger.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Morvan M.G., Lanier L.L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer. 2015;16:7. doi: 10.1038/nrc.2015.5. [DOI] [PubMed] [Google Scholar]
- Nanbakhsh A., Srinivasamani A., Holzhauer S., Riese M.J., Zheng Y., Wang D., Burns R., Reimer M.H., Rao S., Lemke A.J.C.i.r. Mirc11 disrupts inflammatory but not cytotoxic responses of NK cells. Cancer Immunol. Res. 2019;7:1647–1662. doi: 10.1158/2326-6066.CIR-18-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narni-Mancinelli E., Chaix J., Fenis A., Kerdiles Y.M., Yessaad N., Reynders A., Gregoire C., Luche H., Ugolini S., Tomasello E. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. U S A. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar S., Dasgupta P., Galustian C. Extending the lifespan and efficacies of immune cells used in adoptive transfer for cancer immunotherapies-A review. Oncoimmunology. 2015;4:e1002720. doi: 10.1080/2162402X.2014.1002720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T., Kawamura I., Tsuchiya K., Kohda C., Baba H., Ito Y., Kimoto T., Watanabe I., Mitsuyama M. Essential role of interleukin-12 (IL-12) and IL-18 for gamma interferon production induced by listeriolysin O in mouse spleen cells. Infect. Immun. 2002;70:1049–1055. doi: 10.1128/IAI.70.3.1049-1055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberkofler H., Schraml E., Krempler F., Patsch W. Restoration of sterol-regulatory-element-binding protein-1c gene expression in HepG2 cells by peroxisome-proliferator-activated receptor-gamma co-activator-1alpha. Biochem. J. 2004;381:357–363. doi: 10.1042/BJ20040173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz A.L., Lenz L.L. A Listeria-derived polypeptide promotes in vivo activation of NK cells for antitumor therapy. Immunohorizons. 2017;1:53–62. doi: 10.4049/immunohorizons.1700013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende D., Rivera P., Marcenaro S., Chang C.C., Biassoni R., Conte R., Kubin M., Cosman D., Ferrone S., Moretta L. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178–6186. [PubMed] [Google Scholar]
- Piatkiewicz P., Milek T., Bernat-Karpinska M., Ohams M., Czech A., Ciostek P. The dysfunction of NK cells in patients with type 2 diabetes and colon cancer. Arch. Immunol. Ther. Exp. (Warsz) 2013;61:245–253. doi: 10.1007/s00005-013-0222-5. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Rhee J., Lin J., Wu Z., Yoon J.C., Zhang C.Y., Krauss S., Mootha V.K., Lowell B.B., Spiegelman B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rhee J., Inoue Y., Yoon J.C., Puigserver P., Fan M., Gonzalez F.J., Spiegelman B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. U S A. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri L., Capanni M., Tosti A., Urbani E., Posati S., Aversa F., Martelli M.F., Velardi A. Innate immunity against hematological malignancies. Cytotherapy. 2002;4:343–346. doi: 10.1080/146532402760271127. [DOI] [PubMed] [Google Scholar]
- Ruggeri L., Capanni M., Urbani E., Perruccio K., Shlomchik W.D., Tosti A., Posati S., Rogaia D., Frassoni F., Aversa F. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- Schafer J.R., Salzillo T.C., Chakravarti N., Kararoudi M.N., Trikha P., Foltz J.A., Wang R., Li S., Lee D.A. Education-dependent activation of glycolysis promotes the cytolytic potency of licensed human natural killer cells. J. Allergy Clin. Immunol. 2019;143:346–358 e346. doi: 10.1016/j.jaci.2018.06.047. [DOI] [PubMed] [Google Scholar]
- Scharping N.E., Menk A.V., Moreci R.S., Whetstone R.D., Dadey R.E., Watkins S.C., Ferris R.L., Delgoffe G.M. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:701–703. doi: 10.1016/j.immuni.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Seliger B., Ritz U., Ferrone S. Molecular mechanisms of HLA class I antigen abnormalities following viral infection and transformation. Int. J. Cancer. 2006;118:129–138. doi: 10.1002/ijc.21312. [DOI] [PubMed] [Google Scholar]
- Shao D., Liu Y., Liu X., Zhu L., Cui Y., Cui A., Qiao A., Kong X., Liu Y., Chen Q. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion. 2010;10:516–527. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Sharabi K., Lin H., Tavares C.D.J., Dominy J.E., Camporez J.P., Perry R.J., Schilling R., Rines A.K., Lee J., Hickey M. Selective chemical inhibition of PGC-1alpha gluconeogenic activity ameliorates type 2 diabetes. Cell. 2017;169:148–160. doi: 10.1016/j.cell.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shegarfi H., Sydnes K., Lovik M., Inngjerdingen M., Rolstad B., Naper C. The role of natural killer cells in resistance to the intracellular bacterium Listeria monocytogenes in rats. Scand. J. Immunol. 2009;70:238–244. doi: 10.1111/j.1365-3083.2009.02292.x. [DOI] [PubMed] [Google Scholar]
- Siska P.J., Beckermann K.E., Mason F.M., Andrejeva G., Greenplate A.R., Sendor A.B., Chiang Y.J., Corona A.L., Gemta L.F., Vincent B.G. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2:e93411. doi: 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M., Liu J., Ji Y., Subramanian M., Crompton J.G., Yu Z., Roychoudhuri R., Palmer D.C., Muranski P., Karoly E.D. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M., Liu J., Mehta G.U., Patel S.J., Roychoudhuri R., Crompton J.G., Klebanoff C.A., Ji Y., Li P., Yu Z. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thale C., Kiderlen A.F. Sources of interferon-gamma (IFN-gamma) in early immune response to Listeria monocytogenes. Immunobiology. 2005;210:673–683. doi: 10.1016/j.imbio.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Topham N.J., Hewitt E.W. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128:7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp C.S., Gately M.K., Hakimi J., Ling P., Unanue E.R. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. Reversal by IFN-gamma. J. Immunol. 1994;152:1883–1887. [PubMed] [Google Scholar]
- van der Windt G.J., Everts B., Chang C.H., Curtis J.D., Freitas T.C., Amiel E., Pearce E.J., Pearce E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N., Girotra M., Naveiras O., Nikitin G., Campos V., Giger S., Roch A., Auwerx J., Lutolf M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016;7:13125. doi: 10.1038/ncomms13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez S.Y., Killian D., Schulte J., Sticht C., Thiel M., Lindner H.A. Short term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J. Biol. Chem. 2016;291:12960–12977. doi: 10.1074/jbc.M116.721753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- Wahl D.R., Byersdorfer C.A., Ferrara J.L., Opipari A.W., Jr., Glick G.D. Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol. Rev. 2012;249:104–115. doi: 10.1111/j.1600-065X.2012.01148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Lim E., Vaillant F., Asselin-Labat M.L., Visvader J.E., Smyth G.K. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26:2176–2182. doi: 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Kanatous S.B., Thurmond F.A., Gallardo T., Isotani E., Bassel-Duby R., Williams R.S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R.C. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yang C., Tsaih S.W., Lemke A., Flister M.J., Thakar M.S., Malarkannan S. mTORC1 and mTORC2 differentially promote natural killer cell development. Elife. 2018;7:e35619. doi: 10.7554/eLife.35619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C.R., Granner D.K. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu C., Zhu L., Jiang X., Chen X., Qi X., Liang X., Jin S., Zhang P., Li Q. PGC-1alpha inhibits oleic acid induced proliferation and migration of rat vascular smooth muscle cells. PLoS One. 2007;2:e1137. doi: 10.1371/journal.pone.0001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Ma K., Song S., Elam M.B., Cook G.A., Park E.A. Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha) J. Biol. Chem. 2004;279:53963–53971. doi: 10.1074/jbc.M406028200. [DOI] [PubMed] [Google Scholar]
- Zhou J., Shen X., Huang J., Hodes R.J., Rosenberg S.A., Robbins P.F. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J. Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any new RNA seq datasets, and those data analyzed from previous studies (Nanbakhsh et al., 2019) can be found on the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI) under the accession number GEO: GSE136015 and are publicly accessible at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136015.