

Abstract
Trisomy 16 is the most common type of autosomal trisomy associated with spontaneous abortion and is incompatible with life. Upon examining previously reported cases of partial chromosome 16q duplication, it was noted that the majority of cases had complex chromosomal abnormalities due to parental balanced chromosomal translocation carriage. The clinical presentation of very rare pure partial trisomy 16q cases was associated with congenital anomalies, facial dysmorphic findings and intellectual disability. In this study, we evaluated the physical characteristics and genetic data of an 8-month-old girl with developmental delay and facial dysmorphic features. Dysmorphic features including prominent metopic suture, synophrys, asymmetric head shape, triangular and asymmetric face, telecanthus, epicanthal folds, down-slanting palpebral fissures, microphthalmia of the left eye, anteverted nares, smooth and tented philtrum, microretrognathia, low-set posteriorly rotated ears, auricular pits, high-arched palate, thin upper lip and hypotonia were recorded. Her karyotype was 46,XX,add(16)(q24). To identify the extension of the duplicated section, array comparative genomic hybridization (aCGH) analysis was performed, which showed a de novo 29.8 Mb duplication [arr[hgl9] 16q12.1q23.3(52459169-82285105) x 3], interpreted to be pathogenic. We present this case report to clarify the clinical findings of a rare chromosomal anomaly, discuss the genes that may be related to the phenotype and advance the literature in terms of knowledge regarding genotypephenotype correlation.
Keywords: Array comparative genomic hybridization (aCGH), Partial trisomy 16, 16q duplication
Introduction
Trisomy 16 is the most common type of autosomal trisomy associated with spontaneous abortion and is incompatible with life [1]. Among previously reported cases of partial chromosome 16q duplication, the majority presented complex chromosomal abnormalities due to parental balanced chromosomal translocation carriage [2]. The clinical presentation of very rare pure partial trisomy 16q cases was associated with congenital anomalies, facial dysmorphic findings and intellectual disability [3]. To the best of our knowledge, 22 pure partial trisomy 16q cases have been described in the literature so far (Table 1).
Table 1.
Clinical features of previously reported pure partial trisomy 16q cases in the literature and in our patient.
| Partial 16q trisomy region | Prx. D. 16q11.1-q13 | Prx. D. 16q11.2-q12.2 | Prx. D. 16q11.2-q12.1 | Prx. D. 16q11.2-q13 | Prx.D. 16q 12.1-q13 | Prx-Inter. D. 16q11.2-q22.1 | Prx-Inter. D. 16q11.2-q22.3 | Prx-Inter. D. 16q12.1-q21 | Prx-Inter. D. 16q13-q22.3 | Prx-Inter. D. 16q12.1-q22.1 | Inter-Dis. D. 16q22.1-q23.1 | Inter-Dis. D. 16q13-q24 | Prx-Dis. D. 16q12.1-q23.3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methodology for genetic analysis | Conv. Cyto./ FISH | Conv. Cyto./ FISH | Conv. Cyto./ FISH | Conv. Cyto./ FISH | Conv. Cyto./ FISH | SNP-aCHG (45,086,927- 71,261,259) 26.17 Mb | Oligonu-cleotide aCGH 22.5 Mb | Oligonucleo-tide aCGH (50,843,158-60,770,795) 9.92 Mb | Conv. Cyto. | aCGH 19.8 Mb | Conv. Cyto./ FISH | Conv. Cyto. | aCGH (52,459,169- 82,285,105) 29.8 Mb |
| References | [5] | [6] | [6] | [6] | [7] | [1] | [8] | [2] | [9] | [3] | [10] | [11] | this study |
| Gender | F | M | M;M;M;M | M;M;F | M | F | F | F | F | F | M | F | F |
| Age (years) | 3 | 5 | 5.3;3;32;32 | 9.5;5;39 | NR | 2 | 5 | 3.5 | 26 | 7 | 10 | 0 | 0 |
| Growth retardation | [–] | [–] | NR | 2/3 | NR | [–] | [–] | [–] | [+] | [+] | [+] | NR | [+] |
| Intellectual disability | [+] | [+] | 3/2 | 3/3 | RN | [+] | [+] | [+] | [+] | [+] | [+] | NR | [+] |
| Micro-cephaly | [–] | NR | NR | [–] | NR | [–] | [+] | [–] | [–] | [–] | [–] | NR | [+] |
| Behavioral problems | [+] | [+] | NR | 2/3 | NR | NR | [+] | [+] | [+] | NR | [+] | NR | NR |
| Epilepsy | [-] | NR | NR | NR | NR | [–] | [–] | [–] | NR | [+] | [+] | NR | [–] |
| Speech delay | [+] | [+] | 2/4 | 2/3 | NR | [+] | [–] | [+] | [+] | NR | [+] | NR | [+] |
| Obesity | [-] | [+] | NR | 3/3 | NR | [–] | [+] | [+] | NR | NR | NR | NR | [–] |
| Dysmorphic features | [+] | [+] | NR | [–] | NR | [+] | [+] | [+] | [+] | [+] | NR | [+] | [+] |
| Ear anomalies | [+] | NR | NR | NR | NR | [+] | NR | [+] | [–] | [–] | [+] | NR | [+] |
| Eye anomalies | [-] | [+] | NR | 2/3 | NR | [+] | [+] | [+] | [–] | [+] | [–] | NR | [+] |
| Skeletal anomalies | [+] | [+] | NR | 3/3 | NR | [+] | [–] | [+] | [+] | NR | [+] | [+] | [–] |
| Heart defects | [-] | [+] | [–] | [–] | NR | [+] | [–] | [–] | NR | [–] | [–] | [+] | [–] |
| Urogenital anomalies | [-] | NR | NR | NR | NR | [–] | [–] | [–] | NR | [–] | [–] | [+] | [–] |
| CNV anomalies | [-] | NR | NR | NR | NR | [–] | [–] | [–] | NR | [+] | [+] | NR | [–] |
Prx. D.: proximal duplication; Prx-Inter. D.: proximal intermediate duplication; Inter-Dis. D.: intermediate distal duplication;
Prx-Dis. D.: proximal-distal duplication; Conv. Cyto.: conventional cytogenetic; FISH: fluorescence in situ hybridization;
SNP: single nucleotide polymorphism; aCGH: array comparative genomic hybridization; F: female; M: male; NR: not reported;
CNV: central nervous system.
Case Report. In this study, we evaluated the physical characteristics and genetic data of an 8-month-old girl with developmental delay and facial dysmorphic features. She was bom as the fourth child by Cesarean section to healthy non consanguineous parents at 33 weeks’ gestation because of intrauterine growth retardation and fetal distress. At birth, her weight was 1000 g [-5.89 standard deviation score (SDS)], her length was 40 cm (-4.47 SDS) and her head circumference was 30 cm (-3.33 SDS). She was cared for in an incubator for 3 months due to respiratory distress and necrotising enterocolitis. At the age of 8 months, she weighed 4000 g (-5.55 SDS), was 60 cm in height (-3.6 SDS) and had a head circumference of 36.5 cm (-5.64 SDS). Dysmorphic features included prominent metopic suture, synophrys, asymmetric head shape, triangular and asymmetric face, telecanthus, epicanthal folds, down-slanting palpebral fissures, microphthalmia of the left eye, anteverted nares, smooth and tented philtrum, microretrognathia, low-set posteriorly rotated ears, auricular pits, high-arched palate, thin upper lip and hypotonia (Figure 1). She had a history of a single febrile seizure. At the age of 8 months, she was not able to babble or sit independently. Her echocardiography, urinary ultrasonography and brain magnetic resonance imaging (MRI) findings were normal. During her hearing examination, bilateral sensorineural hearing loss was detected.
Figure 1.

The patient’s pictures at the age of 8 months. (written informed consent was obtained from the patient’s parents for publication of proband’s)
Methods
Chromosome analysis was conducted from peripheral blood lymphocytes derived from the patient, and the analysis of GTG-banded chromosomes was performed according to standard procedures. To identify the extension of the duplicated section, array comparative genomic hybridisation (aCGH) analysis was performed. First, 400 μL of peripheral venous blood was drawn into EDNA vacutainers. Then, genomic DNA was extracted from the peripheral venous blood using the QIAamp® DNA Mini Kit (Qiagen GmbH, Hilden, Germany). The detection of genomic copy number variations (CNVs) was performed with Affymetrix CytoScan Optima 315 K arrays (Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Array results were visualized and evaluated using the Chromosome Analysis Suite version 3.2.0 software program (Thermo Fisher Scientific).
Results
The patient’s routine hematological, biochemical and blood gas analysis results were normal. Outcomes of metabolic tests including tandem mass spectrometry (MS/ MS) together with plasma amino acid, urine organic acid, lysosomal enzyme panel and very-long-chain fatty acid findings, were all normal. Her karyotype (450-band level) was 46,XX,add(16)(q24) (Figure 2). The aCGH analysis showed a29.8 Mb duplication [arr[hgl9]16ql2.1q23.3(52, 459, 169-82,285,105) x 3] (Figure 3), interpreted to be pathogenic.
Figure 2.
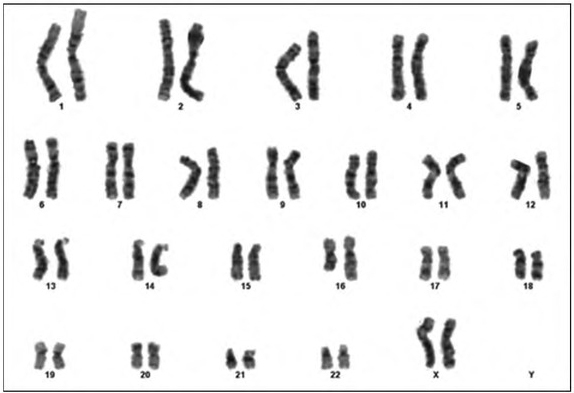
The patient’s karyotype: 46,XX,add(16)(q24).
Figure 3.
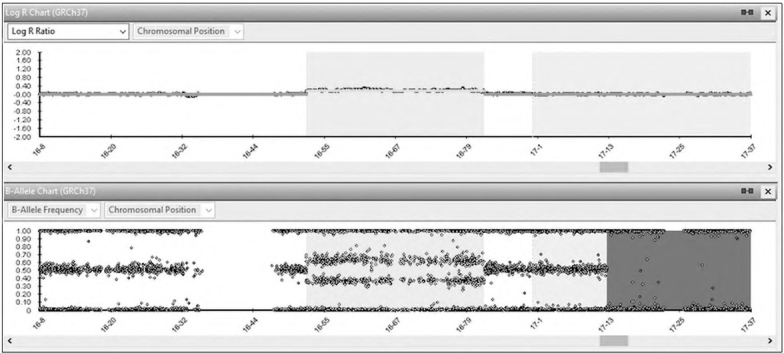
Result of 315k Affymetrix CyctoScan Optima array of chromosome 16. Enlargement of aCGH results on chromosome 16 show gain of 16ql2.1q23.3 corresponding to 29.8 Mb of DNA. X-axis corresponds to the genomic position in megabases of DNA and Y-axis show the log 2 ratio of signal intensity.
There were 343 genes in the duplication region, 211 of which were Online Mendelian Inheritance in Man (OMIM) genes and 62 (including 18 autosomal dominant genes and 44 autosomal recessive genes) of which were associated with the OMIM phenotype. Her parents’ karyotype (450-band level) and aCGH analysis findings were normal, suggesting a de novo origin for the duplication in the proband.
Discussion
The human chromosome 16 contains highly segmental duplication regions (i.e. low copy repeats). Approximately 10.0% of chromosome 16 consists of segmental duplication regions known to be prone to nonhomologous recombination (i.e. unequal crossover). In particular, the 16q22.2 and 16q23 regions can be considered hot-spots for non-homologous recombination. Microdeletions and duplications of different sizes seen in this chromosome are predicted to occur with this mechanism [4].
To investigate genotype-phenotype correlation, pure partial trisomy 16q cases can be stratified into three groups (proximal, proximal-intermediate, intermediate-distal) according to the location of chromosomal anomaly [2]. Most cases fall into the proximal 16q duplication group but many reports provide insufficient information about clinical findings and the sizes of duplicated segments characterized by aCGH analysis [5, 6, 7]. Almost all patients display intellectual disabilities, speech delays and behavioral problems (e.g. attention-deficit hyperactivity disorder, aggressiveness and autistic spectrum disorder). Half of the reported cases present with obesity, skeletal anomalies (e.g. short and thick fingers, foot deformities) and dysmorphic features (e.g. round face, flattened nasal bridge, dysplastic and low-set ears, long philtrum, thin upper Up, microretrognathia and short neck). In the literature, five cases have so far been identified as part of the proximal-intermediate 16q duplication group. Growth retardation and obesity were recorded in two of these cases, while intellectual disability, speech retardation, behavioral problems and dysmorphic features (e.g. round face, flattened nasal bridge, dysplastic and low-set ears, long philtrum, thin upper lip, microretrognathia and short neck) were present in almost all cases. Unlike the other patients in this group, one patient also showed epilepsy and lumbosacral meningomyelocele [1, 2, 3,8,9]. Finally, two cases have so far been reported in the intermediate-distal 16q duplication group. The clinical findings of one duplication 16q22.1q23.1 patient were similar to those in our case [10,11]. Compared to all pure 16q duplication cases, our case had more dysmorphic findings, but the neurodevelopmental retardation profile was similar. To the best of our knowledge, the dysmorphic findings of the present case such as an asymmetric face, microphthalmia, down-slanting palpebral fissures and synophrys have not previously been associated with this chromosomal anomaly.
In the Decipher database, nine partial 16q duplication cases (similar duplications as seen in our case), where the duplications were considered pathogenic, have been reported (Decipher identification #s. 362109, 368652, 294549, 389723, 400900, 401664, 331631, 398404 and 400834). In four of these cases, the duplications were de novo, whereas the inheritance was unknown in the others. Notable clinical features of the nine reported cases included intellectual disability in seven, short stature in two, asymmetric head shape in one, asymmetric face in one, epicanthus in one, anteverted nares in one, hypotonia in one and preauricular pits in one patient. Our case differs from these cases due to some clinical features such as microcephaly, intrauterine growth retardation, down-slanting palpebral fissures, microretrognathia, low-set posteriorly rotate ears, bilateral sensorineural hearing loss and microphtalmia.
The 29.8 Mb duplication in the present case involves 343 genes (including 62 genes associated with the OMIM phenotype). Although genotype-phenotype correlation is difficult, some of these genes are candidates for explaining our patient’s phenotype. The CTCF gene [OMIM #604167 Ch-Band: 16q22.1, genomic coordinates (GRCh3 8): 16:67, 562,525-67,639,184] pathogenic variants are known to bring on an autosomal dominant intellectual disability phenotype (OMIM #615502). Thus, the disruption of this gene in our patient may explain her intellectual disability. Separately, the MAF gene [OMIM #177075 Ch-Band: 16q23.2, genomic coordinates (GRCh38): 16:79,202,623-79, 601, 951 ] pathogenic variants are known to cause auto-somal dominant Ayme-Gripp syndrome (OMIM #601088). Clinical findings in the present case such as short stature, sensorineural hearing loss, low-set posteriorly rotated ears, thin upper lip and intellectual disability, are also found in this syndrome. Finally, the GNAO1 gene [OMIM #139311 Ch-Band: 16q13 genomic coordinates (GRCh38): 16:56, 191,488-56,357,443] de novo pathogenic variants are known to cause early infantile epileptic encephalopathy type 17 (OMIM #615473). A febrile seizure was detected once in our patient, and she was monitored for epilepsy thereafter. We present this case report to clarify the clinical findings of a rare chromosomal anomaly, discuss the genes that may be related to the phenotype and contribute new information to the literature concerning genotypephenotype correlation.
Footnotes
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Hansson K, Dauwerse H, Gijsbers A, van Diepen M, Ruivenkamp C, Kant S. Interstitial duplication in the proximal long armof chromosome 16. Am J Med Genet A. 2010;152A(7):1858–1761. doi: 10.1002/ajmg.a.33434. [DOI] [PubMed] [Google Scholar]
- 2.Odak L, Barisić I, Morozin Pohovski L, Riegel M, Schinzel A. Novel duplication on chromosome 16 (ql2.1-q21) associated with behavioral disorder, mild cognitive impairment speech delay, and dysmorphic features: case report. Croat Med J. 2011;52(3):415–422. doi: 10.3325/cmj.2011.52.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gustavsson P, Schoumans J, Staaf J, Borg A, Nordenskjöld M, Anneren G. Duplication 16q12. 1-q22.1 characterized by array CGh in a girl with spina bifida. Eur J Med Genet. 2007;50(3):237–241. doi: 10.1016/j.ejmg.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, Xu L, Li Y, Lin N, Su L, Cai M. Submicroscopic aberrations of chromosome 16 in prenatal diagnosis. Mol Cytogenet. 2019;12:36. doi: 10.1186/sl3039-019-0448-y. et al. doi. eCollection 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fryns JP, Kleczkowska A, Decock P, Van den Berghe H. Direct duplication 16q11.1-----16q13 is not associated with a typical dysmorphic syndrome. Ann Genet. 1990;33(1):46–48. [PubMed] [Google Scholar]
- 6.Barber JC, Zhang S, Friend N, Collins AL, Maloney VK, Hastings R. Duplications of proximal 16q flanked by heterochromatin are not euchromatic variants and show no evidence of heterochromatic position effect. Cytogenet Genome Res. 2006;114(3-4):351–358. doi: 10.1159/000094225. et al. [DOI] [PubMed] [Google Scholar]
- 7.Mascarello JT, Hubbard V. Routine use of methods for improved G-band resolution in a population of patients with malformations and developmental delay. Am J Med Genet. 1991;38:37–42. doi: 10.1002/ajmg.1320380110. [DOI] [PubMed] [Google Scholar]
- 8.Lonardo F, Perone L, Maioli M, Ciavarella M, Ciccone R, Monica MD. Clinical, cytogenetic and molecular-cytogenetic characterization of a patient with a de novo tandem proximal-intermediate duplication of 16q and review of the literature. Am J Med Genet A. 2011;155A(4):769–777. doi: 10.1002/ajmg.a.33852. et al. [DOI] [PubMed] [Google Scholar]
- 9.Mariner R, Jackson AW. Levitas A, Hagerman RJ, Braden M, McBogg PM. Autism, mental retardation, and chromosomal abnormalities. J Autism Dev Disord. 1986;16(4):425–440. doi: 10.1007/BF01531709. 3rd. et al. [DOI] [PubMed] [Google Scholar]
- 10.Tokutomi T, Wada T, Nakagawa E, Saitoh S, Sasaki M. A de novo direct duplication of 16q22.1➝q23.1 in a boy with midface hypoplasia and mental retardation. Am J Med Genet A. 2009;149A(11):2560–2563. doi: 10.1002/ajmg.a.33049. [DOI] [PubMed] [Google Scholar]
- 11.Hirai S, Ujue J, Suzuki J, Ishiama S, Tsukanishi A, Muramoto J. Duplication of the long arm of chromosome 16. Jpn J Pediatr. 1981;34(2):1963–1967. et al. [Google Scholar]