

Abstract
N-methyl-d-aspartate receptors (NMDARs) are fundamental coincidence detectors of synaptic activity necessary for the induction of synaptic plasticity and synapse stability. Adjusting NMDAR synaptic content, whether by receptor insertion or lateral diffusion between extrasynaptic and synaptic compartments, could play a substantial role defining the characteristics of the NMDAR-mediated excitatory postsynaptic current (EPSC), which in turn would mediate the ability of the synapse to undergo plasticity. Lateral NMDAR movement has been observed in dissociated neurons; however, it is currently unclear whether NMDARs are capable of lateral surface diffusion in hippocampal slices, a more physiologically relevant environment. To test for lateral mobility in rat hippocampal slices, we rapidly blocked synaptic NMDARs using MK-801, a use-dependent and irreversible NMDAR blocker. Following a 5-min washout period, we observed a strong recovery of NMDAR-mediated responses. The degree of the observed recovery was proportional to the amount of induced blockade, independent of levels of intracellular calcium, and mediated primarily by GluN2B-containing NMDA receptors. These results indicate that lateral diffusion of NMDARs could be a mechanism by which synapses rapidly adjust parameters to fine-tune synaptic plasticity.
NEW & NOTEWORTHY N-methyl-d-aspartate-type glutamate receptors (NMDARs) have always been considered stable components of synapses. We show that in rat hippocampal slices synaptic NMDARs are in constant exchange with extrasynaptic receptors. This exchange of receptors is mediated primarily by NMDA receptors containing GluN2B, a subunit necessary to undergo synaptic plasticity. Thus this lateral movement of synaptic receptors allows synapses to rapidly regulate the total number of synaptic NMDARs with potential consequences for synaptic plasticity.
Keywords: extrasynaptic receptors, NMDA receptors, synaptic receptors, synaptic transmission
INTRODUCTION
The N-methyl-d-aspartate receptor (NMDAR) is an ionotropic glutamate receptor expressed throughout neocortex, is highly permeable to calcium, and is fundamental for both synaptogenesis and experience-driven synaptic plasticity (Cline and Haas 2008; Gambrill and Barria 2011; Lau and Zukin 2007). Although historically NMDARs have been thought to be relatively stable components of synapses based on their tight association with scaffolding proteins (Malenka and Nicoll 1999; Malinow and Malenka 2002), evidence shows that NMDARs can be dynamic and regulated by synaptic activity within hours or days. Sensory deprivation in rodents alters NMDAR-subunit composition at synapses consequently affecting the threshold for synaptic plasticity (Philpot et al. 2001, 2003; Quinlan et al. 1999). In cultured hippocampal neurons, prolonged blockade of synaptic activity with TTX increases NMDARs surface expression (Lissin et al. 1998). Similarly, blockade of receptor activation with the competitive antagonist 2-amino-5-phosphonopentanoic acid (APV) increases NMDAR phosphorylation in a manner to promote surface expression (Chung et al. 2004), increases receptor localization to synapses (Rao and Craig 1997), and prevents the normal switch in subunit composition of NMDARs (Barria and Malinow 2002), once again altering synaptic plasticity (Barria and Malinow 2005).
Synaptic NMDAR content in hippocampal slices can also be regulated in short time scales of minutes and seconds in response to changes in synaptic activity. This allows synapses to dynamically adjust NMDAR-mediated transmission and the threshold for synaptic plasticity (Gambrill et al. 2011). Because this change in synaptic NMDAR content is rapid, it was suggested that lateral diffusion of NMDARs, rather than membrane insertion from intracellular compartments, was responsible. This idea is supported by studies in cultured dissociated neurons, where single-receptor tracking using quantum dots shows that, in this system, NMDARs can diffuse quite readily on the plasma membrane surface at a rate determined by their subunit composition (Groc et al. 2004, 2006). Additionally, by using the activity-dependent inhibitor MK-801, it has been shown in cultured autaptic hippocampal neurons that NMDARs are capable of rapidly moving laterally between extrasynaptic and synaptic compartments (Tovar and Westbrook 2002). However, with the use of a similar approach in hippocampal slices, a more physiologically relevant system, this lateral motility has been called into question. It was concluded that extrasynaptic and synaptic NMDARs form stable pools (Harris and Pettit 2007). In addition, questions regarding whether quantum dots used for single-molecule tracking can reliably access the synaptic space makes previous observations of rapid lateral diffusion of synaptic receptors using this method of tracking unclear (Delgado and Selvin 2018).
On postsynaptic sites, synaptic and extrasynaptic NMDARs are proposed to have different subunit compositions and functions via association with different signaling platforms (Hardingham and Bading 2010; Papouin and Oliet 2014). Whether there is exchange between synaptic and extrasynaptic NMDARs pools remains unclear. Considering the fundamental role of NMDARs in learning and their implication in neuropathologies, it is of crucial importance to clarify if and/or how these receptors traffic laterally between compartments as a means of fine-tuning synaptic characteristics on a minute-by-minute basis. In this report we investigate whether NMDARs can rapidly move between synaptic and extrasynaptic compartments in hippocampal slices. We have found evidence supportive of NMDAR exchange between synaptic and extrasynaptic compartments. This exchange might represent a mechanism by which synapses can rapidly change their susceptibility to plasticity.
MATERIALS AND METHODS
Hippocampal slices.
Organotypic hippocampal slices (400 μm thick) were prepared according to standard procedures (Opitz-Araya and Barria 2011) from postnatal day (P)6–9 male and female Sprague-Dawley rats and maintained in culture for 3–8 days at 35°C. Animals were handled according to a protocol approved by the University of Washington (Seattle, WA) institutional animal care and use committee.
Electrophysiology.
For each experiment, the CA1 area of a hippocampal slice was isolated by making two cuts flanking the CA1 region, and then the slice was placed into the recording chamber containing modified ACSF (in mM): 10 glucose, 2.5 KCl, 118 NaCl, 1 NaH2PO4, 2 CaCl2, 2 MgCl2, and 26 NaHCO3, pH 7.4, constantly circulating and bubbled with 95% O2-5% CO2. NMDAR-mediated currents were recorded from CA1 neurons patched under visual guidance with glass pipettes (~2–4 MΩ) filled with a cesium-based internal solution (in mM): 115 CsMeSO4, 20 CsCl2, 10 HEPES, 2.5 MgCl2, 4 MgATP, 0.4 Na3GTP, 10 Na- phosphocreatine, and 0.6 EGTA, pH 7.25. Currents were evoked by holding the cells at depolarized potentials below the NMDAR reversal potential (between −40 and −15 mV, average −30 mV) and stimulating the Schaffer collaterals with a bipolar cluster electrode (CE2C55; FHC) placed ~100 μm away from the cell of interest. To isolate NMDAR currents, 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX; 2 μM) and picrotoxin (100 μM) were also included in the bath. Temperature was kept between 22.8 and 23.0°C. Recordings were obtained with a MultiClamp 700B amplifier (Axon Instruments) and pClamp 10.1 software.
Statistics.
The amplitude of EPSCs for individual experiments was normalized to the average of the baseline period to aggregate data from different cells. Where appropriate, statistical significance was determined using a nonparametric test, Mann-Whitney U test, or repeated measures ANOVA with post hoc Bonferroni correction for multiple comparisons. A value of P < 0.05 was considered statistically significant. Data are presented as average (±SE) values of recorded cells.
RESULTS
Activity bidirectionally regulates synaptic NMDARs.
It has been proposed that NMDARs can diffuse laterally in the plane of the membrane (Groc et al. 2006; Tovar and Westbrook 2002) and regulate synaptic responses in a dynamic manner (Gambrill et al. 2011). To test whether synapses can rapidly adjust their synaptic content of NMDARs in response to varying frequencies of synaptic stimulation, we recorded isolated NMDAR-mediated postsynaptic currents (EPSCs) in rat hippocampal CA3–CA1 synapses in the presence of the AMPA receptor blocker NBQX and 2 mM Mg2+. NMDAR-mediated EPSCs were obtained by stimulation of Schaffer collaterals at 0.1 Hz while relieving the Mg2+ block by holding the cell at a negative potential around −30 mV, where inward NMDAR currents were maximal. Stimulation intensity was adjusted for each cell to produce responses between 25 and 50 pA. The frequency of stimulation was then decreased to 0.05 Hz. This decrease in stimulation frequency caused a progressive increase in the amplitude of NMDAR-mediated EPSCs that stabilized after 10 min (Fig. 1, A and B; P < 0.05, Mann-Whitney test) as has been observed before (Gambrill et al. 2011).
Fig. 1.

Synaptic activity rapidly regulates NMDA receptor (NMDAR)-mediated excitatory postsynaptic currents (EPSCs). A, top: sample traces of isolated NMDAR currents from CA1 cells in rat hippocampal slices recorded in whole cell configuration at approximately −30 mV during baseline (a1) or after 15 min of a decreased sampling frequency (a2). Bottom, normalized peak amplitude of evoked NMDAR responses obtained by stimulation of Schaffer collaterals at 0.1 Hz. After 5 min of baseline, sampling frequency was reduced in half (n = 4). B: average peak amplitude of NMDAR EPSCs normalized to baseline during a 5-min window as indicated. *P < 0.05 (n = 4). C, top: sample traces of isolated NMDAR currents from CA1 cells in rat hippocampal slices recorded in whole cell configuration at approximately −30 mV during baseline (c1), after 5 min of an increase in sampling frequency (c2), and after sampling frequency was returned to the same frequency as baseline (c3). Bottom, normalized peak amplitude of evoked NMDAR responses obtained by stimulation of Schaffer collaterals at 0.1 Hz. After 5 min of baseline, sampling frequency was increased to 0.5 Hz and then returned to same frequency as baseline (n = 10). D: average peak amplitude of NMDAR EPSCs normalized to baseline at indicated time. *P < 0.05 (n = 4).
Increasing the stimulation frequency from 0.1 to 0.5 Hz produced the opposite effect, i.e., a gradual decrease in the amplitude of evoked NMDAR EPSCs (P < 0.05, Mann-Whitney test). When the frequency of stimulation was returned to 0.1 Hz, the amplitude of the responses gradually returned to baseline levels (Fig. 1C). This rapid adjustment in the amplitude of evoked NMDARs to varying input activity suggests that synaptic and extrasynaptic NMDARs, at least a fraction of them, are mobile and can respond to changes in the frequency of stimulation.
Incomplete blockade by MK-801 of synaptic NMDARs.
To further investigate whether synaptic NMDARs can drift in and out of synapses, we used MK-801, a noncompetitive and activity-dependent NMDAR antagonist (Halliwell et al. 1989). MK-801 blocks only activated receptors within stimulated synapses. Since MK-801 blockade is irreversible at negative holding potentials (Huettner and Bean 1988; Rosenmund et al. 1993), should receptors be fixed within or outside of synapses, each stimulus iteration should decrease the overall NMDAR EPSC amplitude until all receptors are blocked and evoked responses are indistinguishable from noise.
Evoked NMDAR-mediated responses were obtained in CA1 neurons by stimulation of Schaffer collaterals at 0.1 Hz. After a stable baseline was obtained, MK-801 was added to the perfusion bath and stimulation stopped to allow for an even distribution of the drug in the tissue. Three concentrations, 5, 10, and 40 μM, all well above the described MK-801 IC50, were used (Wamil and McLean 1992). When stimulation was resumed, NMDAR EPSC amplitudes rapidly declined as expected due to blockade of the activated synaptic NMDARs. Within the next 10 min, responses became progressively smaller but were never eliminated at any of the concentrations of MK-801 used (Fig. 2, A–C). Even 40 μM MK-801 after 10 min did not eliminate NMDAR-mediated EPSCs (Fig. 2, C and D). Only the addition of 100 μM dl-APV, a competitive antagonist for the glutamate binding site, rapidly and completely eliminated NMDAR-mediated responses (Fig. 2, A–C). The blockade of synaptic NMDARs by MK-801 can be fit with a single-order exponential decay function (Fig. 2E). Fits for all three concentrations reveal that the time constant of the blockade was dependent on MK-801 concentration in a nonlinear manner, with 40 μM being close to saturation (Fig. 2F). Importantly, the percentage of MK-801 blockade after 10 min seems to reach a plateau at a maximum of 90% blockade (Fig. 2G). Similar incomplete blockade of NMDAR-mediated synaptic currents after 15–20 min of MK-801 treatment have been observed before in hippocampal slices (Harris and Pettit 2007; Hessler et al. 1993) or single cultured neurons that form autapses (Rosenmund et al. 1993). Given the saturating concentration of MK-801, the preincubation period, and the long stimulation period in the presence of the drug, it is unlikely the remaining current is due to incomplete pharmacological blockade.
Fig. 2.

Incomplete blockade of NMDA receptor (NMDAR) responses by MK-801. A–C, top: examples of NMDAR excitatory postsynaptic currents (EPSCs) obtained at baseline (a1, b1, c1), after 10 min of 0.1-Hz stimulation in the presence of varying concentration of MK-801 (a2, b2, c2), and in the presence of 100 μM dl-2-amino-5-phosphonopentanoic acid (APV; a3, b3, c3). Bottom, NMDAR EPSC peak amplitude normalized to baseline (n = 5 for each MK-801 concentration used). Stimulation was suspended for 5 min when MK-801 was added to the bath (gray shading). After 5 min, stimulation was resumed at same frequency as baseline. After 10 min, APV was added to the bath. D: enlarged example NMDAR EPSCs from C (c1 and c2) evoked at baseline and after 10 min of 0.1-Hz stimulation in the presence of 40 μM MK-801, normalized to the peak. Notice different amplitude scales. E: the decay in NMDAR-mediated transmission due to MK-801 blockade was fitted by a single exponential curve. NMDAR EPSC blockade is shown with 5, 10, or 40 μM MK-801. F: time constant (τ) of the MK-801 blockade of NMDAR EPSCs as a function of MK-801 concentration. G: percentage of baseline blocked by 60 stimuli (10 min at 0.1 Hz) as a function of MK-801 concentration.
Incomplete blockade suggests that either a fraction of synaptic NMDARs are not fixed within the synapse and can constantly be replaced with nonblocked extrasynaptic receptors or that nonblocked extrasynaptic receptors are added to the synapse on top of receptors that remain synaptically fixed. Either mechanisms could prevent a complete blockade on this timescale and at this stimulation frequency.
Recovery of synaptic NMDARs after MK-801 blockade.
If synaptic NMDARs are in a constant exchange with extrasynaptic receptors, this could explain 1) the adjustment of NMDAR synaptic content depending on the stimulation frequency (Fig. 1) and 2) the incomplete blockade of synaptic NMDARs by MK-801 (Fig. 2). To test whether a rapid exchange of synaptic and extrasynaptic NMDA receptors occurs, we hypothesized that it should be possible to recover NMDARs responses after MK-801 has blocked a fraction of synaptic NMDARs and the drug washed away. A partial recovery after MK-801 blockade has been observed in autaptic synapses (Tovar and Westbrook 2002), where the exchange of solutions is almost instantaneous, but it has failed to be observed in hippocampal slices (Harris and Pettit 2007).
To test whether extrasynaptic receptors can drift into synapses and replace blocked receptors, we first blocked a fraction of synaptic receptors by 10 μM MK-801 with 30 pulses delivered at 0.1 Hz (5 min). This rapidly reduced the amplitude of evoked NMDAR EPSCs to 31 ± 5% of baseline levels (Fig. 3, A and B; P < 0.01, repeated measures ANOVA with post hoc Bonferroni correction test). The drug was then washed out for 5 min with the stimulation turned off to prevent further blockade of synaptic NMDARs during this period. On resuming stimulation at 0.1 Hz, we observed NMDAR-mediated EPSCs had recovered to, on average, 61 ± 9% of baseline (Fig. 3, A and B). This increase in the amplitude of NMDAR EPSCs was significantly larger compared with that at the end of the blockade period (Fig. 3B; P < 0.01, repeated measures ANOVA with post hoc Bonferroni correction test). Over the course of the next 10 min, response amplitudes decreased back to values slightly above the values observed at the end of the MK-801 block period (33 ± 6% of baseline) but did not decrease further.
Fig. 3.

NMDA receptor (NMDAR)-mediated excitatory postsynaptic current (EPSC) recovery post-MK-801 blockade. A, top: example NMDAR EPSCs obtained at baseline (a1), after 5 min of stimulation in the presence of 10 μM MK-801 (a2), and immediately following washout (a3). Bottom, NMDAR EPSC peak amplitude normalized to baseline (n = 11). Stimulation was suspended for 5 min when MK-801 was added to the bath and during washout period (gray shading). B: average peak amplitude of NMDAR EPSCs normalized to baseline. Amplitude of last 3 traces at the end of MK-801 blockade period (a2; MK-801) and the first 3 traces immediately after stimulation resumed following washout of the drug (a3; recovery) were averaged. The last 3 min after stimulation resumed were also averaged (a4; recovery). *P < 0.01 (n = 11). C: percent blockade by 10 μM MK-801 as a function of the initial amplitude of NMDAR EPSCs. D: percent recovery after MK-801 washout as a function of the initial amplitude of NMDAR EPSCs. E: relationship between the percent recovery after MK-801 washout and percent blockade on each experiment. Dotted line is best fit for a linear regression analysis. Coefficient of determination (R2) and P values from linear regression analysis are indicated in C–E. F: peak amplitude of isolated NMDAR EPSCs (n = 4). Stimulation of Schaffer collaterals was suspended (gray shading) during coapplication of MK-801 and NMDA to block all surface NMDARs. After a 5-min washout period, stimulation was resumed. G: membrane resistance (top) and series resistance (bottom) of an experiment from F to show stability of the whole cell recording. Stim., stimulation.
Early control experiments with the same sequence of events as in this block and recovery paradigm were performed, but without MK-801. This control showed that after a period of no stimulation, a temporary increase in NMDAR transmission is observed that subsequently decays (also see Gambrill et al. 2011), indicating that the decay postrecovery is not entirely due to residual MK-801. A similar control was performed where MK-801 was not removed during the washout period. In this case, NMDAR decayed much faster than when MK-801 had been removed from the bath (not shown). Together, these results indicate that while residual MK-801 may remain in the bath, it alone is not responsible for the decay in response amplitude when stimulation is resumed.
Neither the percent blockade, measured with the last three stimuli compared with baseline, nor the percent recovery, measured with the first three stimuli after the washout period, were correlated with the baseline average amplitude of NMDARs EPSCs (Fig. 3, C and D). A small, nonsignificant negative correlation between the degree of MK-801 blockade and the percent of recovery relative to baseline was observed (Fig. 3E). The P value shown in Fig. 3, C–E, was calculated from an F test, and the R2 value represents the goodness of the linear regression fit. This partial and temporary recovery of NMDAR responses suggests that after MK-801 blockade and during the washout period, unblocked extrasynaptic NMDARs move into the synapses.
This increase in synaptic NMDAR-mediated transmission is consistent with the adjustment to synaptic activity observed in Fig. 1A (see also Gambrill et al. 2011). Receptors that move into the synapses could either replace existing blocked receptors or be added to the synapse. The fact that response amplitudes decay again, when stimulation is resumed, to values observed at the end of the MK-801 block period suggests that NMDARs move out of the synapse to adjust for the new stimulation frequency, but that previously blocked NMDARs do not leave the synapses. It is plausible that synaptic receptors, to adjust to stimulation frequency and leave the synapse, need channel opening to break free from their synaptic anchorage, something that could be prevented by MK-801.
To test whether the temporary recovery of NMDAR-mediated currents observed after MK-801 blockade results from the trafficking of intracellular receptors to the surface, we suspended stimulation of the Schaffer collaterals and coapplied the agonist NMDA (25 μM) with MK-801 to block all surface receptors. This was followed by a period of washout still in the absence of synaptic stimulation. When stimulation was resumed, we observed no recovery of NMDAR-mediated currents (Fig. 3F). These results support that NMDAR recovery from MK-801 block is not mediated by receptors trafficking into synapses from intracellular compartments, but by receptors located on the surface that can move laterally into synapses.
The small negative correlation between the degree of MK-801 blockade (how much of baseline was blocked) and the percentage of baseline recovered after washout of the drug suggests that the receptors contributing to the recovery are influenced by the amount of induced blockade (Fig. 3E). To further examine this observation, we repeated the experiment while increasing the percentage of blockade by stimulating synapses at a slightly higher rate.
We hypothesized that because the constant exchange between synaptic and extrasynaptic receptors, a larger number of synaptically blocked receptors will concomitantly decrease the number of receptors available for recovery. Stimulation of synapses at 0.2 Hz in the presence of 10 μM MK-801 blocked NMDAR-mediated EPSCs to 27 ± 7% of baseline values. After washout of the drug, NMDAR responses still recovered significantly to 53 ± 7% of baseline values (Fig. 4, A and B; P < 0.05, repeated measures ANOVA with post hoc Bonferroni correction test). This recovery, however, is less than that observed when NMDAR-mediated currents were blocked at 0.1 Hz (Fig. 3B). When NMDAR-mediated synaptic currents were stimulated at 0.3 Hz, MK-801 blocked NMDAR currents to 20 ± 7% of baseline values. After washout of the drug, NMDAR-mediated EPSCs recovered to 33 ± 6% of baseline; in this case, the recovery was not statistically significant (Fig. 4, C and D; repeated measures ANOVA with post hoc Bonferroni correction test). In both cases, as before, after the initial recovery, the amplitude of NMDAR-mediated EPSCs decayed back to the level observed at the end of the blockade period. As expected, increasing the stimulation frequency sped the rate of block (Fig. 4E) and increased the percentage of blockade in the 5 min before washout of the drug (Fig. 4F). The increased blockade observed with stimulation at either 0.2 or 0.3 Hz correlated with decreased recovery of NMDAR currents (Fig. 4G).
Fig. 4.

Increased level of MK-801 blockade limits recovery of NMDA receptor (NMDAR)-mediated excitatory postsynaptic currents (EPSCs). A–D: normalized peak amplitude of evoked NMDAR responses obtained by stimulation of Schaffer collaterals at 0.2 (A; n = 5) or 0.3 Hz (C; n = 6). Stimulation was suspended for 5 min when MK-801 was added to the bath and during washout period (gray shading). Bar graphs are averages of the peak amplitude of last 3 traces at the end of MK-801 blockade period (MK-801) and the first 3 traces immediately after stimulation resumed following washout of the drug (recovery) from cells stimulated at 0.2 (B; n = 5) or 0.3 Hz (D; n = 6). All values are normalized to baseline. *P < 0.05; n.s., not significant. E: single-order exponentials fitted to the average decay in the presence of MK-801 for each of the stimulation frequencies indicated. F: percentage of baseline blocked as a function of stimulation frequency. G: percentage of baseline recovered as a function of stimulation frequency. Stim., stimulation.
These data support a rapid and activity-dependent exchange of receptors between synaptic and extrasynaptic compartments such that synaptic receptors blocked during MK-801 exposure get functionally replaced with extrasynaptic receptors, albeit temporarily until stimulation is resumed. An increase in the amount of blockade concomitantly decreases the number of functional receptors available for replacement once they reach a new equilibrium.
NMDAR diffusion is not mediated by intracellular calcium.
Intracellular calcium has been shown to regulate the trafficking of NMDARs from intracellular compartments to the surface (Hunt et al. 2013; Lau and Zukin 2007) as well as NMDAR current rundown and channel open time (Krupp et al. 1999; Legendre et al. 1993). We tested next whether intracellular calcium might play a role in the observed recovery of synaptic NMDAR currents following blockade with MK-801. To this end, we used BAPTA in the patch pipette while blocking synaptic receptors with MK-801 and 0.1-Hz stimulation of Schaffer collaterals. Neither 10 nor 15 mM BAPTA had any effect on the degree of MK-801 blockade or recovery after washout of the drug compared with the amount of blockade and recovery in control conditions (Fig. 5, A–C, two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test). These results suggest a calcium-independent mechanism governs receptor lateral diffusion responsible of the recovery observed after MK-801 blockade.
Fig. 5.
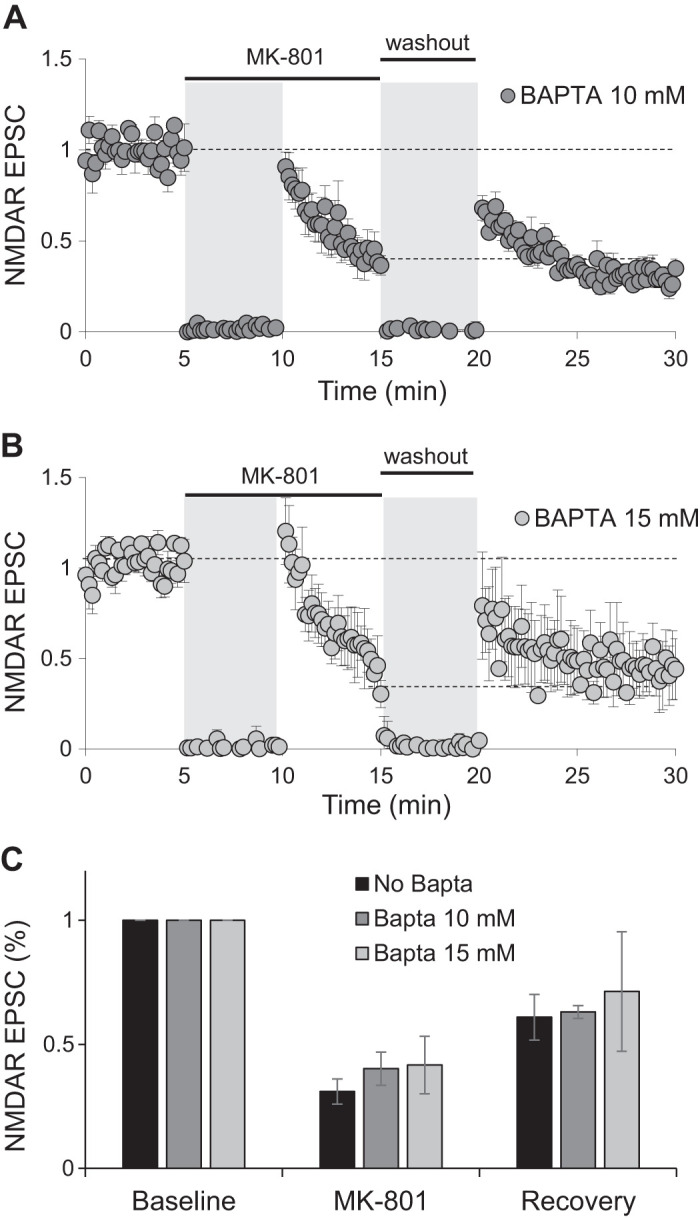
Recovery of NMDA receptor (NMDAR) currents does not require intracellular calcium. A and B: normalized peak amplitude of evoked NMDAR responses obtained by stimulation of Schaffer collaterals at 0.1 Hz with 10 (A; n = 5) or 15 mM BAPTA (B; n = 4) in the patch pipette. Stimulation was suspended for 5 min when MK-801 was added to the bath and during washout period (gray shading). C: average of peak amplitude of NMDAR excitatory postsynaptic currents (EPSCs) normalized to baseline from experiments shown in A and B. For easy comparison, data from Fig. 3B are also presented (no BAPTA). Amplitude of last 3 traces at the end of MK-801 blockade period (MK-801) and the first 3 traces immediately after stimulation resumed following washout of the drug (recovery) were averaged.
NMDAR diffusion is regulated by subunit composition.
NMDARs are heterotetramers containing two obligatory GluN1 subunits and two GluN2 subunits that confer the receptor its kinetic properties. GluN2A-containing receptors demonstrate faster decay kinetics (Traynelis et al. 2010) and weak, if any, association with CaMKII, while GluN2B-containing NMDA receptors demonstrate slower decay kinetics and heightened association with CaMKII (Barria and Malinow 2005; Mayadevi et al. 2002), an association necessary for synaptic plasticity (Barria and Malinow 2005; Halt et al. 2012; Zhou et al. 2007). The subunit composition of NMDARs located within synaptic versus extrasynaptic zones has been a source of contention over the last decade, with a prevailing view that extrasynaptic receptors contain primarily GluN2B, and synaptic receptors GluN2A, although drawing definitive lines between the two has been complicated by the limitations of various experimental paradigms (Papouin and Oliet 2014).
We have reported previously that potentiation of NMDAR currents induced by a reduction in the sampling rate is due to incorporation of GluN2B-containing NMDA receptors, as there is an increase in the decay time of NMDAR-mediated EPSCs and an increased sensitivity to ifenprodil, a specific GluN2B antagonist (Gambrill et al. 2011). Studies in cultured dissociated neurons also distinguished GluN2B-containing NMDA receptors as more mobile than their GluN2A counterparts (Groc et al. 2006).
To test whether subunit composition of NMDARs plays a role in either the blockade by MK-801 or the recovery observed after the drug has been washed out, we analyzed the time to half-decay of NMDAR-mediated EPSCs from data shown in Fig. 3A. NMDAR EPSCs were significantly faster at the end of MK-801 blockade compared with baseline or the beginning of blockade (Fig. 6A; repeated measures ANOVA with post hoc Bonferroni correction test). This suggests that synaptic GluN2B-containing receptors are more sensitive to MK-801 blockade in slices, perhaps because of their longer opening times (Erreger et al. 2005). After the drug is washed out and a recovery in the NMDAR-responses is observed as described in Fig. 3A, a significant increase in the time to half-decay is observed, suggesting that NMDAR diffusion is primarily mediated by GluN2B-containing receptors.
Fig. 6.

Overexpression of GluN2A does not prevent recovery. A: time to half-decay of isolated NMDA receptor (NMDAR) excitatory postsynaptic currents (EPSCs) from control cells during baseline, MK-801 blockade, or recovery periods. *P < 0.05. B: sample traces of synaptically evoked NMDAR responses recorded at +40 mV in a control CA1 neuron (black trace) and an adjacent neuron expressing green fluorescent protein (GFP)-tagged GluN2A and GluN1 (gray trace). C: normalized peak amplitude of evoked NMDAR responses obtained by stimulation of Schaffer collaterals at 0.1 Hz from CA1 neurons overexpressing GFP-tagged GluN2A and GluN1 (n = 6). Stimulation was suspended for 5 min when MK-801 was added to the bath and during washout period (gray shading). D: average of peak amplitude of NMDAR EPSCs normalized to baseline from neurons expressing GluN2A (NR2A; open bars). For easy comparison, data from Fig. 3B are also presented (control; closed bars). Amplitude of last 3 traces at the end of MK-801 blockade period (MK-801) and the first 3 traces immediately after stimulation resumed following washout of the drug (recovery) were averaged. *P < 0.05.
Next, we manipulated the synaptic content of GluN2A-containing NMDA receptors by overexpressing equimolar amounts of optically tagged GluN2A and GluN1 subunits (Barria and Malinow 2005) in organotypic hippocampal slices via biolistic transfection (Opitz-Araya and Barria 2011; Woods and Zito 2008). Synaptic NMDAR-mediated currents evoked in cells overexpressing GluN2A are insensitive to ifenprodil (Barria and Malinow 2005) and exhibit a faster decay time (Fig. 6B), indicating a dominance of GluN2A-containing NMDA receptors at synapses, while endogenous levels of GluN2B are not affected (Barria and Malinow 2005).
Similar to our previous results, these cells also demonstrated significant blockade and recovery following MK-801 washout (P < 0.05, repeated measures ANOVA with post hoc Bonferroni correction test), almost achieving baseline levels (88 ± 17%; Fig. 6C and Fig. 6D, open bars). However, in these cells MK-801 blocked NMDAR EPSCs to only 58 ± 13% of baseline levels (Fig. 6C), significantly less than the amount of blockade in control conditions (Fig. 6D; two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test). The lesser extent of the MK-801 blockade compared with control experiments may be related to a decreased efficacy of MK-801 for blocking GluN2A than GluN2B given the relatively faster kinetics of the GluN2A subunits. Importantly, responses were able to recover once MK-801 had been washed out, consistent with an endogenous GluN2B extracellular pool available.
Next, we tested directly whether GluN2B is the subunit necessary for the recovery of NMDAR responses observed after MK-801 blockade. We treated slices for 15 min with 1 μM Ro 25-6981, a specific GluN2B blocker (Fischer et al. 1997) while monitoring NMDAR-mediated synaptic responses. This treatment decreases the decay time of NMDAR-mediated EPSCs (Barria and Malinow 2005) and reduced NMDAR EPSCs to 57 ± 9% of baseline values. Once EPSCs had mostly stabilized, we repeated the MK-801 blockade and recovery paradigm as before.
A 5-min MK-801 blockade decreased EPSCs to 33 ± 6% of baseline amplitude (Fig. 7A and Fig. 7B, shaded bars), a similar blockade as in control neurons (Fig. 3), shown again in Fig. 7B for easy comparison (two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test). Following washout, in cells pretreated with Ro 25-6981, there was a small recovery of NMDAR responses that is not statistically different compared with the amount of recovery in control cells (Fig. 7B; two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test).
Fig. 7.
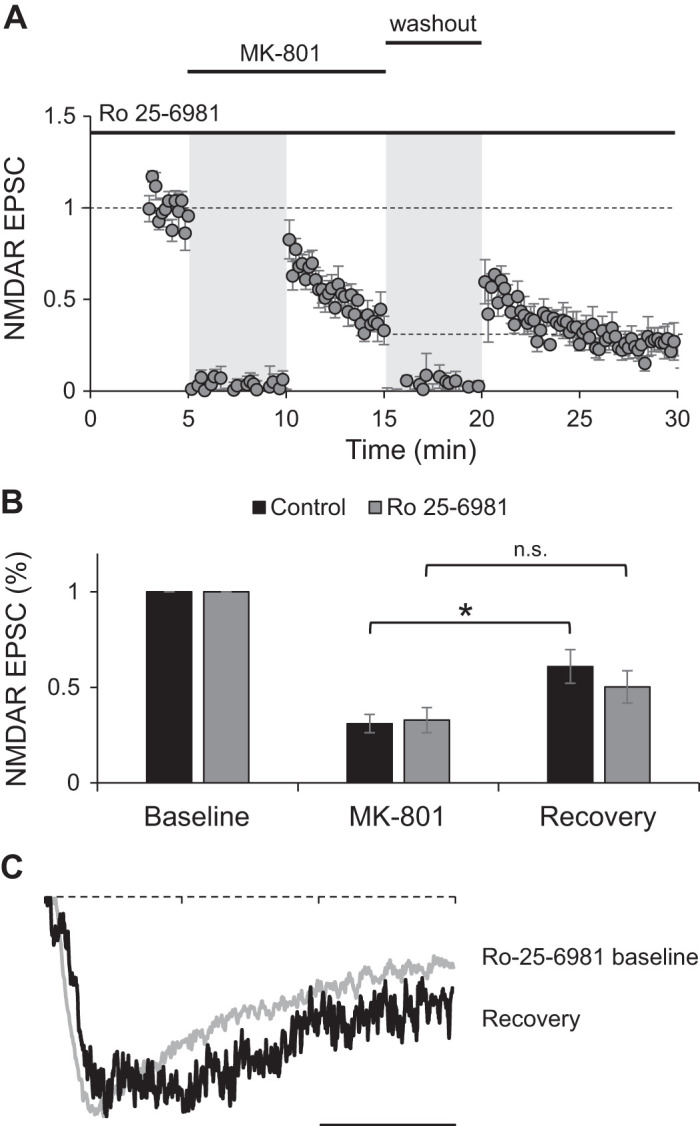
Blockade of endogenous GluN2B prevents recovery from MK-801 blockade. A: normalized peak amplitude of evoked NMDA receptor (NMDAR) responses obtained by stimulation of Schaffer collaterals at 0.1 Hz from CA1 neurons pretreated with Ro 25-6981 for 15 min (n = 7). Stimulation was suspended for 5 min when MK-801 was added to the bath and during washout period (gray shading). B: average of peak amplitude of NMDAR excitatory postsynaptic currents (EPSCs) normalized to baseline from neurons pretreated with Ro 25-6981 (shaded bars). For easy comparison, data from Fig. 3B are also presented (control; closed bars). Amplitude of last 3 traces at the end of MK-801 blockade period (MK-801) and the first 3 traces immediately after stimulation resumed following washout of the drug (recovery) were averaged. Student’s t test indicates no significant recovery after MK-801 washout (P = 0.132; n.s., not significant). C: example normalized traces of isolated NMDAR EPSCs from baseline period in the presence of Ro 25-6981 and from the recovery period after drug washout. Scale bar, 50 ms.
However, the small recovery observed in cells pretreated with Ro 25-6991 is not statistically different compared with recovery at the end of the MK-801 blockade period (Fig. 7B, shaded bars; two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test). This is different from control neurons, where responses during the recovery period are statistically different compared with those at the end of the blockade (Fig. 7B, closed bars; two-factor ANOVA with repeated measures on one factor with post hoc Bonferroni correction test).
Interestingly, the small recovery observed in neurons treated with Ro 25-6981 exhibited slower EPSCs, suggesting that it is mediated by GluN2B-containing NMDARs (Fig. 7C), presumably because of incomplete blockade with Ro 25-6981. All together, our data support that GluN2B-containing NMDA receptors are primarily responsible for the recovery observed after MK-801 block.
DISCUSSION
Lateral motility of NMDARs, in particular their capability to go in and out of synapses, has been observed in dissociated neurons using single-molecule labeling (Groc et al. 2004, 2006) and indirectly inferred by recovery of NMDAR function after MK-801 blockade (Tovar and Westbrook 2002). However, recovery from MK-801 blockade has not been observed in hippocampal slices (Harris and Pettit 2007), a more physiologically relevant system. In addition, the use of quantum dots to label synaptic single molecules has been called into question because of their size and accessibility to synaptic space (Delgado and Selvin 2018). In this study we used MK-801 blockade of synaptic receptors in organotypic hippocampal slices to study whether NMDARs are able to diffuse in the plane of the membrane entering and leaving synaptic areas. Organotypic slices, as opposed to acute slices, provide a more stable preparation that has recovered from cell death and excitotoxicity caused by sample preparation that could affect lateral motility.
Our results show that blockade of NMDAR currents by MK-801 is reversed partially once the drug is removed. This recovery in the NMDAR function is transitory, and once stimulation is resumed, amplitude of NMDAR EPSCs decays to a new equilibrium. This initial recovery indicates that unblocked extrasynaptic receptors are mobile and able to incorporate into synapses during the washout period, consistent with the rapid adjustment in the amplitude of evoked NMDARs to varying input activity (Fig. 1) or periods without stimulation (Gambrill et al. 2011).
While presynaptic changes affecting probability of release have not been ruled out in these experiments, there is no reason to think that could be the case: the frequency and intensity of stimulation is the same before and after addition of MK-801; i.e., there was no particular plasticity-inducing protocol used that could alter presynaptic probability of release, and, more importantly, previous results from our laboratory have shown no changes in probability of release following synaptic potentiation of NMDAR responses by varying stimulation (Gambrill et al. 2011).
Another possibility is that changes in tonic Ca2+-dependent inactivation (Krupp et al. 1999) are responsible for the recovery observed. Several considerations make us favor the hypothesis that a mobile fraction of synaptic NMDARs accounts for changes in the amplitude of EPSCs when stimulation frequency changes, and not changes in the properties of a stable synaptic pool. First, if synaptic NMDARs were not mobile, they should have been fully blocked by MK-801 independently of the Ca2+-dependent tonic inactivation (Fig. 2). Second, our experiment in Fig. 5 shows that BAPTA in the pipette does not prevent an increase in the EPSC amplitude after a period of no stimulation (washout), suggesting that the increase in EPSC amplitude is not due to changes in the tonic Ca2+-dependent inactivation. Third, the Ca2+-dependent inactivation phenomenon has been shown to affect GluN2A-containing receptors and is not observed in GluN2B-containing receptors (Krupp et al. 1996). Changes in the amplitude of EPSCs as a function of stimulation frequency involve an increase or decrease in GluN2B-containing receptors, as shown in Figs. 6 and 7 (also see Gambrill et al. 2011), making it unlikely that the mechanism is a change in the tonic Ca2+-dependent inactivation. Consistent with this, mobility of receptors has been shown in dissociated autaptic synapses (Tovar and Westbrook 2002), and GluN2B, but not GluN2A, receptors have been shown to be highly dynamic in the membrane plane (Groc et al. 2006).
The amount of recovery was inversely proportional to the amount of blockade, suggesting that a constant exchange of synaptic and extrasynaptic receptors allows for MK-801 to block not only synaptic receptors but also receptors from the extrasynaptic pool, rendering them unavailable for the recovery of NMDAR function. This could explain why recovery from MK-801 blockade was not observed in experiments where exposure to the drug was long and almost complete blockade of NMDAR EPSCs was first obtained (Harris and Pettit 2007). It is interesting to notice that not even a high concentration of MK-801 (40 μM) fully blocked NMDAR EPSCs after 10 min of stimulation, despite the fact that the tissue was preincubated in the drug. This incomplete blockade can also be observed in other experiments where longer times of MK-801 plus stimulation have been used (Harris and Pettit 2007; Hessler et al. 1993). It is possible that a constant delivery of receptors to the extrasynaptic pool from intracellular stores keeps replenishing NMDARs present in the membrane, making it difficult to completely eliminate synaptic NMDAR responses in this timescale.
Our data support a model where GluN2B receptors can move in and out of synapses to adjust the amplitude of NMDAR-mediated EPSCs, whereas GluN2A seems to be more stationary at synapses. This is consistent with previous observations showing that adaptation of NMDAR EPSCs to frequency stimulation is done by movement of GluN2B-containing NMDA receptors (Gambrill et al. 2011) and with single-molecule tracking experiments in dissociated neurons that show GluN2B present in abundance in the dendritic membrane, where they diffuse at a higher rate than GluN2A (Groc et al. 2006). NMDAR synaptic content is also regulated by Wnt signaling (Cerpa et al. 2011). In this case, noncanonical Wnt signaling regulates NMDAR-mediated EPSCs via rapid regulation of the synaptic content of GluN2B-containing NMDARs (Cerpa et al. 2011; McQuate et al. 2017). Thus evidence points to a model where NMDARs, primarily those containing the GluN2B subunit, can diffuse readily on the surface of neurons to fine-tune synaptic content in response to patterns of activity or be regulated by other signaling pathways. In the absence of activity, GluN2B are more likely to stop in a synaptic domain and associate with scaffolding proteins. Glutamate binding to NMDARs could release them from scaffolding and promote return to the diffusible pool. Under constant stimulation, receptors reach an equilibrium of movement between synaptic and extrasynaptic spaces that results in a constant number of receptors within synapses.
The mechanism by which glutamate binding results in movement of receptors away from synapses also warrants further study. A calcium-independent metabotropic function of NMDARs has only recently come to light, whereby glutamate binding stimulates movement of the NMDAR COOH terminus and changes receptor signaling properties (Dore et al. 2015; Nabavi et al. 2013). It is possible glutamate binding could also trigger a conformational change in receptors to promote diffusion away from synapses. However, our results suggest that channel opening is necessary, because MK-801 blockade seemed to prevent their diffusion away from the synapse. The potential effect of MK-801 in stabilizing receptors at synapses should be further addressed in the near future.
Our results support a body of literature indicating the ability of NMDARs to diffuse between the synaptic and extrasynaptic space on a short timescale. This ability of NMDARs might control the GluN2B content of synapses on a fast timescale, modifying the ability of synapses to undergo potentiation or depression on a minute-by-minute basis. Full understanding of processes that regulate NMDARs will help develop novel therapeutics for neuropathologies where the ability of these receptors to coordinate plasticity has gone awry.
GRANTS
This work was funded in part by National Science Foundation (NSF) Grant IOS-1755004 (to A. Barria), National Institutes of Health Training Grant 5T32GM007108 (to A. McQuate), and an NSF Graduate Research Fellowship (to A. McQuate).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.B. conceived and designed research; A.M. performed experiments; A.M. analyzed data; A.M. and A.B. interpreted results of experiments; A.M. prepared figures; A.B. drafted manuscript; A.M. and A.B. edited and revised manuscript; A.B. approved final version of manuscript.
REFERENCES
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345–353, 2002. doi: 10.1016/S0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48: 289–301, 2005. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Cerpa W, Gambrill A, Inestrosa NC, Barria A. Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J Neurosci 31: 9466–9471, 2011. doi: 10.1523/JNEUROSCI.6311-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci 24: 10248–10259, 2004. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol 586: 1509–1517, 2008. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado JY, Selvin PR. A revised view on the role of surface AMPAR mobility in tuning synaptic transmission: limitations, tools, and alternative views. Front Synaptic Neurosci 10: 21, 2018. doi: 10.3389/fnsyn.2018.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore K, Aow J, Malinow R. Agonist binding to the NMDA receptor drives movement of its cytoplasmic domain without ion flow. Proc Natl Acad Sci USA 112: 14705–14710, 2015. doi: 10.1073/pnas.1520023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563: 345–358, 2005. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther 283: 1285–92, 1997. [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci USA 108: 5855–5860, 2011. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, Storey GP, Barria A. Dynamic regulation of NMDA receptor transmission. J Neurophysiol 105: 162–171, 2011. doi: 10.1152/jn.00457.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci 7: 695–696, 2004. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc Natl Acad Sci USA 103: 18769–18774, 2006. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell RF, Peters JA, Lambert JJ. The mechanism of action and pharmacological specificity of the anticonvulsant NMDA antagonist MK-801: a voltage clamp study on neuronal cells in culture. Br J Pharmacol 96: 480–494, 1989. doi: 10.1111/j.1476-5381.1989.tb11841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31: 1203–1216, 2012. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11: 682–696, 2010. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol 584: 509–519, 2007. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature 366: 569–572, 1993. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-d-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA 85: 1307–1311, 1988. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P, Castillo PE. Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat Neurosci 16: 1049–1059, 2013. doi: 10.1038/nn.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. Calcium-dependent inactivation of recombinant N-methyl-d-aspartate receptors is NR2 subunit specific. Mol Pharmacol 50: 1680–1688, 1996. [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci 19: 1165–1178, 1999. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8: 413–426, 2007. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook GL. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci 13: 674–684, 1993. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M. Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc Natl Acad Sci USA 95: 7097–7102, 1998. doi: 10.1073/pnas.95.12.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science 285: 1870–1874, 1999. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126, 2002. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mayadevi M, Praseeda M, Kumar KS, Omkumar RV. Sequence determinants on the NR2A and NR2B subunits of NMDA receptor responsible for specificity of phosphorylation by CaMKII. Biochim Biophys Acta 1598: 40–45, 2002. doi: 10.1016/S0167-4838(02)00315-1. [DOI] [PubMed] [Google Scholar]
- McQuate A, Latorre-Esteves E, Barria A. A Wnt/calcium signaling cascade regulates neuronal excitability and trafficking of NMDARs. Cell Reports 21: 60–69, 2017. doi: 10.1016/j.celrep.2017.09.023. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci USA 110: 4027–4032, 2013. doi: 10.1073/pnas.1219454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz-Araya X, Barria A. Organotypic hippocampal slice cultures. J Vis Exp 48: e2462, 2011. doi: 10.3791/2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T, Oliet SH. Organization, control and function of extrasynaptic NMDA receptors. Philos Trans R Soc Lond B Biol Sci 369: 20130601, 2014. doi: 10.1098/rstb.2013.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci 23: 5583–5588, 2003. doi: 10.1523/JNEUROSCI.23-13-05583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29: 157–169, 2001. doi: 10.1016/S0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2: 352–357, 1999. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19: 801–812, 1997. doi: 10.1016/S0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science 262: 754–757, 1993. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron 34: 255–264, 2002. doi: 10.1016/S0896-6273(02)00658-X. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496, 2010. [Erratum in Pharmacol Rev 66: 1141, 2014.] doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamil AW, McLean MJ. Use-, concentration- and voltage-dependent limitation by MK-801 of action potential firing frequency in mouse central neurons in cell culture. J Pharmacol Exp Ther 260: 376–383, 1992. [PubMed] [Google Scholar]
- Woods G, Zito K. Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J Vis Exp 12: e675, 2008. doi: 10.3791/675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci 27: 13843–13853, 2007. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]