

Keywords: Slc26a4, pendrin, Cl−/HCO3− exchange, ENaC, intercalated cells, blood pressure
Abstract
Intercalated cells (ICs) are found in the connecting tubule and the collecting duct. Of the three IC subtypes identified, type B intercalated cells are one of the best characterized and known to mediate Cl− absorption and HCO3− secretion, largely through the anion exchanger pendrin. This exchanger is thought to act in tandem with the Na+-dependent Cl−/HCO3− exchanger, NDCBE, to mediate net NaCl absorption. Pendrin is stimulated by angiotensin II and aldosterone administration via the angiotensin type 1a and the mineralocorticoid receptors, respectively. It is also stimulated in models of metabolic alkalosis, such as with NaHCO3 administration. In some rodent models, pendrin-mediated HCO3− secretion modulates acid-base balance. However, of probably more physiological or clinical significance is the role of these pendrin-positive ICs in blood pressure regulation, which occurs, at least in part, through pendrin-mediated renal Cl− absorption, as well as their effect on the epithelial Na+ channel, ENaC. Aldosterone stimulates ENaC directly through principal cell mineralocorticoid hormone receptor (ligand) binding and also indirectly through its effect on pendrin expression and function. In so doing, pendrin contributes to the aldosterone pressor response. Pendrin may also modulate blood pressure in part through its action in the adrenal medulla, where it modulates the release of catecholamines, or through an indirect effect on vascular contractile force. In addition to its role in Na+ and Cl− balance, pendrin affects the balance of other ions, such as K+ and I−. This review describes how aldosterone and angiotensin II-induced signaling regulate pendrin and the contribution of pendrin-positive ICs in the kidney to distal nephron function and blood pressure.
Pendrin is a Cl−/HCO3− exchanger expressed in the apical region of a kidney minority cell type as well as in thyroid, inner ear, and the adrenal medulla.
In people and in mice, pendrin gene ablation can lead to hypothyroidism, deafness, volume contraction, and reduced blood pressure.
Angiotensin II and aldosterone stimulate pendrin, which increases renal Cl− absorption, thereby contributing to the pressor response seen in response to these hormones.
In kidney, pendrin increases renal ENaC activity and abundance, thereby stimulating the renal absorption of both Na+ and Cl−.
Metabolic alkalosis stimulates pendrin, thereby increasing the secretion of HCO3− into the pro-urine, which mitigates the alkalosis.
Some diuretics and antihypertensives, such as mineralocorticoid receptor antagonists, may target pendrin to achieve their effects.
I. INTRODUCTION
The epithelial cells of the connecting tubule (CNT) and cortical collecting duct (CCD) are referred to as intercalated cells, principal cells, and connecting tubule cells. The transporters expressed in the various cell types of these segments are displayed in FIGURE 1. Intercalated cells make up only ~1% of kidney volume (93, 150) and ~2–2.5% of kidney mass (25). Nonetheless, these cells play an important role in renal physiology. This minority cell type expresses the H+-ATPase on the apical or the basolateral plasma membrane, which coincides with whether the cell mediates H+ or secretion (38, 174). Intercalated cells (ICs) that express the H+-ATPase on the basolateral plasma membrane secrete and absorb Cl− by way of an apical plasma membrane Cl−/ exchanger, known as pendrin, which is encoded by Slc26a4 (95, 165, 230, 231), and are referred to as type B or β ICs. In treatment models associated with metabolic alkalosis, total and apical plasma membrane pendrin abundance increases, which augments net secretion, thereby helping to correct the alkalosis (48, 165, 217). Conversely, type A intercalated cells express the H+-ATPase on the apical plasma membrane in series with basolateral plasma membrane Cl−/ exchange, mediated by anion exchanger 1 (AE1), which is encoded by Slc4a1 (17–19, 30, 52). In this IC subtype, apical plasma membrane H+-ATPase abundance as well as apical H+ secretion increase in rodent models of metabolic acidosis (167, 220). A third IC subtype, referred to as non-A, non-B ICs, expresses both pendrin (95, 230) and the H+-ATPase (95) on the apical plasma membrane. However, much less is known about this third IC subtype than the others. Because ICs express the H+-ATPase on the apical or basolateral plasma membrane and because they mediate net secretion of H+ or , respectively, early studies focused on their role in acid-base balance (see sects. II and III).
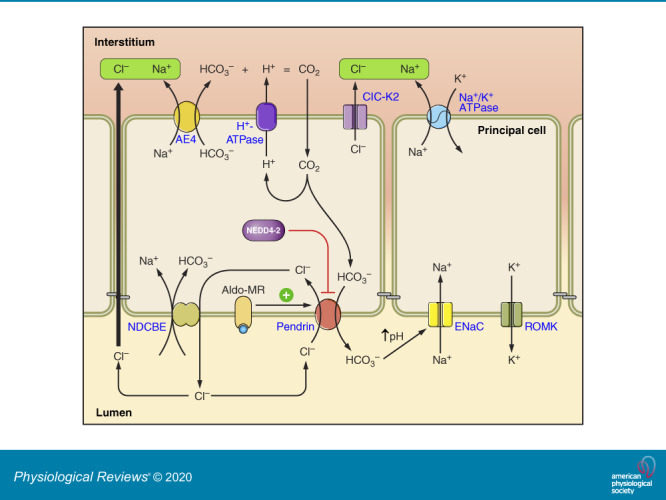
FIGURE 1.

Cell types and transporters in the cortical collecting duct (CCD). Intercalated and principal cell ion transporter distribution within of the CCD is shown. In type B intercalated cells (ICs), the Na+-dependent Cl−/HCO3− exchanger, NDCBE, mediates Na+ and HCO3− absorption, whereas pendrin mediates HCO3− secretion and Cl− absorption. Through the action of these 2 transporters, Cl− and HCO3− are recycled across the apical membrane. Net H+ and Cl− exit occur across the basolateral plasma membrane through the Cl− channel, ClC-K2, and the H+-ATPase. Na+ exits the cell through the Na+-HCO3− cotransporter, AE4. Type A ICs secrete H+ through the apical plasma membrane H+-ATPase with net HCO3− efflux across the basolateral plasma membrane through the Cl−/HCO3− exchanger, AE1. Type A ICs also express the ammonia channel, Rhbg, in the basolateral regions of the cell. Principal cells absorb Na+ through the epithelial Na+ channel, ENaC, which exits across the basolateral plasma membrane through the Na+-K+-ATPase. In so doing, it generates a lumen-negative transepithelial voltage, which provides the driving force for net secretion of K+. Most likely, Cl− is also absorbed through paracellular transport driven by the lumen-negative transepithelial voltage generated by the epithelial Na+ channel, ENaC. Non-A, non-B ICs are rare in mouse CCD. However, these cells express pendrin and the H+-ATPase on the apical plasma membrane and express ClC-K2, AE4, and Rhbg in the basolateral regions of the cell. Whether these cells express NDCBE is unclear.
Studies that identified the genes encoding these transporters led to important observations as to the role ICs play in Na+ (96, 104) and Cl− (219, 231) balance, as well as in blood pressure regulation (217, 231). In particular, pendrin regulates blood pressure and NaCl balance by mediating Cl− absorption, and by regulating epithelial Na+ channel (ENaC)-dependent Na+ absorption (96, 141, 144). Moreover, both rodent and human studies have shown that inactivation mutations in the gene encoding pendrin (Slc26a4) lead to a natriuresis, a chloriuresis, and lower blood pressure (89, 96, 209, 217, 219). As such, there has been interest in the development of compounds that inhibit pendrin for potential use in clinical practice as diuretics or antihypertensives (24, 63). Other transporters within these pendrin-positive ICs also modulate blood pressure. For example, in pendrin-positive ICs, a Na+-dependent Cl−/ exchanger (NDCBE), encoded by Slc4a8, is thought to operate in tandem with pendrin-mediated Cl−/ exchange to mediate net NaCl absorption (FIGURE 1) (104). Thus pendrin and NDCBE form a metabolon that mediates net NaCl absorption. If either is eliminated, this IC NaCl absorption mechanism is greatly reduced.
Pendrin abundance and activity are greatly stimulated by angiotensin II and by aldosterone (140, 217, 218, 231). Moreover, pendrin-positive ICs modulate blood pressure, at least in part, through their interaction with the renin-angiotensin-aldosterone system (see sect. XIII). Whether angiotensin II stimulates pendrin in vivo, independently of its impact on aldosterone release, however, is debated (73). Aldosterone stimulates pendrin, at least in part, through its unique interaction with the mineralocorticoid receptor (MR) within ICs. The MR has a phosphorylation site that is unique to ICs (187). When dephosphorylated at S843, aldosterone binding to the IC MR is enhanced. This MR S843 dephosphorylation increases in response to angiotensin II or aldosterone-induced hypokalemia.
A subset of ICs (type B) mediates electroneutral NaCl absorption across the plasma membrane through the action of pendrin-mediated Cl−/ exchange and NDCBE-mediated Na+-dependent Cl−/ exchange (NDCBE) across the apical plasma membrane. NaCl absorption that occurs through this mechanism is thought to be important during vascular volume contraction when renal K+ conservation is needed. In the absence of pendrin, i.e., in pendrin-null mice, hypokalemia is observed under some treatment conditions (248). How pendrin contributes to K+ homeostasis is not fully understood, but may occur through an interaction with “With No Lysine” (WNK) kinases (107).
Pendrin-positive cells communicate with other tissues as well as with other renal segments, through paracrine signaling, and probably through the sympathetic nervous system (8, 56, 102, 141). This interaction likely contributes to the effect pendrin-positive ICs have on blood pressure and NaCl homeostasis.
This review summarizes what is known about pendrin-positive ICs and their role in renal physiology. It also describes early studies that led to the discovery of ICs and their role in acid-base homeostasis and describes how our thinking about these cells was transformed following work made possible by the human genome project.
II. INITIAL OBSERVATIONS OF INTERCALATED CELLS AND THEIR ROLE IN H+ AND HCO3− SECRETION WITHIN THE CCD AND CNT
Over the better part of the past century, two different collecting duct cell populations have been appreciated when viewed by light microscopy. The first, which in the mammalian kidney are referred to as “principal cells,” are more abundant and appear “light” with relatively few organelles, such as mitochondria (158). The second, now known as “intercalated cells” in the mammalian kidney appear darker in some preparations, with abundant organelles, including mitochondria (158). In the early 1970s, robust carbonic anhydrase activity was observed in these dark, mitochondria-rich, minority cells of the turtle and toad bladder (180, 182) as well as in rat kidney (106, 161). Moreover, since either CO2 removal or carbonic anhydrase inhibitors eliminate H+ secretion in the turtle bladder (180), it was recognized that H+ secretion by these cells requires CO2 as a H+ source.
Early reports showed that these mitochondria-rich cells of the turtle and toad bladder as well as the mammalian kidney could be subdivided into at least two distinct groups based on the cellular ultrastructure observed by freeze-fracture as well as by scanning and transmission electron microscopy (198, 221, 226). The first cell subtype, termed type α (in amphibians) or A (in mammals), has apical microplicae and microvilli, whereas the type β or B intercalated cell, in the amphibian and mammalian kidneys, respectively, has apical plasma membrane microvilli without microplicae (158, 198). These scanning electron microscopy studies also showed that the apical microplicae and microvilli observed in the “dark cells” are more prominent following either NaHCO3 administration or respiratory acidosis (61).
Cytoplasmic studs and rod-shaped intramembrane particles are observed on the apical plasma membrane and in subapical vesicles of the α-type ICs, but in the basolateral regions of the β-type IC in toad and turtle bladder epithelium (17, 198, 226). In mammalian collecting duct, apical plasma membrane and cytoplasmic membrane vesicles contain electron-dense studs and intramembranous rod-shaped particles in the type A ICs of the outer medullary collecting duct (OMCD) and CCD (111, 159). Studs are also occasionally observed along the cytoplasmic surface of the type B IC basolateral plasma membrane (205). Thus, within ICs, the distribution of rods and studs is similar to the distribution of the H+-ATPase (18, 19). Using monoclonal antibodies raised against affinity-purified H+-ATPase protein taken from bovine kidney medullary extracts, Brown et al. (17) observed that these studlike structures represent multiple H+-ATPase subunits arranged around a central depression. In both the turtle bladder and in rabbit CCD, these H+-ATPase-containing subapical vesicles rapidly insert into the plasma membrane when stimulated with CO2 in vitro (51, 176, 199). Similarly, in the rat type A IC, apical plasma membrane H+ abundance increases markedly in rodent models of respiratory acidosis (110, 224) or in rodent models of metabolic acidosis, such as with NH4Cl administration (10, 167, 220). Thus H+-ATPase-containing cytoplasmic vesicles fuse with the apical plasma membrane during both in vivo and in vitro models of acidosis, thereby increasing net H+ secretion.
The first report of net H+ secretion by the collecting duct was made by Gottschalk, Lassiter, and Mylle in 1960 using micropuncture (54). However, measurement of ion transport in specific, native renal tubule segments became possible when Maurice Burg and colleagues developed the technique of perfusing renal tubules in vitro (20, 64). This advancement led to many reports in the 1970s and 1980s characterizing transport and H+ secretion in the rabbit and rat CCD and OMCD as well in rabbit CNT. Since virtually all intercalated cells in the OMCD express the H+-ATPase on the apical, but not the basolateral plasma membrane, studies of OMCDs perfused in vitro reflect transport of the type A, but not the B type IC. Proton secretion (or absorption) was observed in the OMCD and noted to be markedly stimulated in models of metabolic acidosis (212). These data were consistent with a large body of work done at the about the same time showing increased apical plasma membrane H+-ATPase abundance during metabolic acidosis (10, 220). Unlike the OMCD, the isolated perfused CCD is capable of either secreting or absorbing (7, 117). These observations are consistent with the presence of two IC subtypes in the CCD, one that can secrete (type B) and one that can absorb , reflecting H+ secretion by type A ICs. However, the OMCD only secretes H+ and contains only the H+-secreting IC subtype (7).
The type A and type B subclassification of ICs was originally based on the distinct cellular morphology of these two cell subtypes. A later subclassification of these cells was based on the subcellular distribution of the H+-ATPase and on whether or not they express the basolateral Cl−/ exchanger AE1 (FIGURE 1) (4, 19, 93, 205) or the apical Cl−/ exchanger pendrin (165). This classification has been useful in that it predicts whether the cell secretes H+ or (38, 174). Type A ICs have an apical H+-ATPase, and basolateral AE1 immunoreactivity, while type B ICs have both basolateral and cytoplasmic H+-ATPase, apical pendrin, but no AE1 immunoreactivity (95, 230). In addition, expression of the ammonia transporter Rhbg distinguishes pendrin-positive IC subtypes, such as non-A, non-B ICs from type B ICs, since this protein is found in the former, but not in the latter, cell type (225). Labeling of these transporters in the various cell types found in the mouse CCD and CNT are shown in FIGURE 2.
FIGURE 2.

Intercalated cell marker labeling in mouse cortical collecting duct (CCD) and connecting tubule (CNT). Characteristic immunolabeling of the three distinct intercalated cell subtypes in the CNT (top panels) and CCD (bottom panels) are shown by differential interference contrast microscopy (DIC). Type A intercalated cells express the basolateral anion exchanger AE1, apical H+-ATPase, and the basolateral ammonia transporter Rhbg. Type B intercalated cells express the apical anion exchanger, pendrin, and basolateral H+-ATPase, but not AE1 or Rhbg. Non-A, non-B intercalated cells express apical pendrin, apical H+-ATPase, and basolateral Rhbg, but not AE1. Left column: double labeling for pendrin (blue) and AE1 (brown), the latter of which is definitive for type A intercalated cells; apical pendrin (blue) is present in type B and non-A, non-B intercalated cells. Pendrin labeling is exclusively in AE1-negative cells. Middle column: double labeling for AE1 (brown) and the a4 subunit of H+-ATPase (blue). Type A intercalated cells (AE1-positive) have apical H+-ATPase label. Type B intercalated cells have basolateral H+-ATPase label (arrows), as well as diffuse apical label, which correlates with cytoplasmic vesicle labeling shown by immunogold electron microscopy. Type B intercalated cells are uncommon in the CNT, but represent virtually all of the non-A intercalated cells in the CCD. In the CNT, the majority of non-A intercalated cells have apical H+-ATPase label, but no basolateral label. These are non-A, non-B intercalated cells. Right column: double labeling for pendrin (blue) and Rhbg (brown). Type B intercalated cells and non-A, non-B intercalated cells, both pendrin-positive, can be discriminated by basolateral Rhbg expression. Non-A, non-B intercalated cells, which express basolateral Rhbg (arrowheads), are the predominant pendrin-positive cell type in the CNT. Type B intercalated cells do not express detectable Rhbg (arrows) and comprise virtually all of the pendrin-positive cells in the CCD. Rhbg immunolabel is also present in type A intercalated cells (open arrows), CNT cells, and CCD principal cells. [From Verlander and Clapp (216), with permission from Elsevier.]
Mammalian ICs are also classified as α or β based on functional measurements. The α-type IC shows apical H+ secretion and basolateral, stilbene-sensitive Cl−/ exchange (15, 67, 68, 105, 118, 119, 200), whereas the β-type IC showed apical, stilbene-insensitive Cl−/ exchange (39, 40, 195, 237, 240). Our ability to measure H+ secretion and apical anion exchange in individual intercalated cells became possible with the development of the pH-sensitive fluorophore 2′,7′-bis(2-carboxyethyl)-5,6-carboxyfluorescein (BCECF). When applied to the luminal fluid, this dye is preferentially taken up by ICs, because of the greater esterase content of ICs relative to principal cells (238). Because changes in intracellular pH (pHi) in individual cells of isolated, perfused CCDs and OMCDs reflect IC transport almost exclusively, anion exchange, such as Cl−/ exchange, across the apical and/or basolateral IC plasma membrane can be quantified and its inhibitor sensitivity tested in individual cells of CCDs or OMCDs perfused in vitro. This rate of anion exchange is determined by measuring the rate of change in IC pHi that follows the addition or removal of Cl− from the perfusate or the bath in the presence or absence of inhibitors. Moreover, apical H+ secretion by ICs can be determined by quantifying the effect of inhibitors of the H+-ATPase applied to the luminal fluid after the cell was acidified. In practice, this is done by measuring the change in IC pHi over time after the cell was loaded with H+, which is achieved by the addition and then removal of NH4+. The difference in dpHi/dt when measured in the presence or absence of an H+-ATPase inhibitor in the luminal fluid, e.g., bafilomycin, reflects the rate of apical H+ secretion. These studies showed that ICs of the OMCD, i.e., the α type, have an apical, bafilomycin-sensitive H+ pump and a stilbene-sensitive basolateral Cl−/ exchanger (15, 67, 68, 105, 118, 119, 200), which is thought to be band 3 (AE1)-mediated. Net H+ secretion by the α-type IC occurs in parallel with Cl− secretion, although the mechanism for the latter is still not well understood (126, 127, 228, 229).
With the use of similar methods, the CCD was found to have an IC subtype with transport characteristics similar to the OMCD (α type) and also has a second IC subtype characterized by stilbene-insensitive apical Cl−/ exchange (39, 40, 195, 237, 240). This latter cell type, which we refer to as β-type ICs, secretes rather than H+ and absorbs Cl− vis-à-vis apical plasma membrane, Na+-independent, electroneutral, Cl−/ exchange (39, 40, 195, 237, 240), which acts in series with net H+ efflux across the basolateral plasma membrane mediated by the H+-ATPase (36, 38, 140). While the Na+-K+-ATPase provides the active step for ion movement in most cells, Na+ pump abundance and activity are much lower in ICs than in principal cells (43, 85, 168, 170, 204). As such, the active step that generates the driving force for β-type IC apical anion exchange is provided largely by the H+-ATPase, which localizes to the basolateral plasma membrane of type B ICs (22, 140). Apical Cl−/ exchange of the β-type IC requires CO2 as a proton source and is abolished with the application of carbonic anhydrase inhibitors (120). Thus CO2 likely generates H+ that exit the cell across the basolateral plasma membrane via the H+-ATPase.
Aldosterone or NaHCO3 administration have been widely used as rodent models of metabolic alkalosis (46, 121, 217, 219, 227). These treatment models stimulate apical Cl−/ exchange, which increases secretion and Cl− absorption by the β-type IC (48, 99, 165, 231). As such, the β-type IC was first thought to participate in the correction of acid-base balance during metabolic alkalosis by stimulating anion exchange-mediated secretion (165, 174, 217). While it has been known for some time that this exchanger mediates Cl− absorption, its role in Cl− balance was not appreciated until the gene encoding this exchanger was cloned and mice lacking this protein were developed and studied (see sect. IV).
A third IC subtype has been identified, which we call non-A, non-B ICs. These cells have numerous mitochondria and have vesicles coated with studs throughout the cell (205). Because non-A, non-B ICs localize primarily to the CNT in the mouse (230) and because mouse CNT has not been successfully perfused in vitro, the transport properties of the CNT, and hence the properties of non-A, non-B ICs, are less well understood than those of type B ICs, which are abundant in mouse CCD. While apical Na+-independent Cl−/ exchange is likely present in non-A, non-B ICs (211), the H+-ATPase localizes to the apical plasma membrane in this cell type, rather that the basolateral plasma membrane localization seen in type B ICs (205). Another distinction between type B and non-A, non-B ICs is that the ammonia transporter Rhbg is expressed in non-A, non-B ICs, but is undetectable in type B ICs (225). H+-ATPase, AE1, pendrin, and Rhbg labeling in mouse CNT and CCD are shown in FIGURE 2.
The distribution of Cl− and H+/OH− transporters within non-A, non-B ICs is consistent with experimental data showing that the CNT absorbs Cl− at least under most physiological conditions (188). In the CNT, type A ICs should secrete H+, whereas non-A, non-B and type B ICs should secrete H+ and in parallel (211), which would result in the net addition of CO2 and H2O into the luminal fluid. Whether non-A, non-B ICs are primarily acid- or base-secreting cells is not entirely clear. However, studies in CNTs perfused in vitro taken from untreated rabbits have shown that these segments secrete (211). Therefore, if the distribution of IC subtypes in the rabbit cortex is similar to that of rat and mouse, non-A, non-B ICs should secrete relatively more than H+ at least under basal conditions. Collectively, these studies suggest that transport across the CNT should result in the net addition of CO2 and H2O and the net loss of Cl− from the luminal fluid.
III. CHANGES IN OUR UNDERSTANDING OF INTERCALATED CELL ACID-BASE PHYSIOLOGY THAT FOLLOWED THE IDENTIFICATION AND CLONING OF INTERCALATED CELL TRANSPORTERS
The primary IC markers currently used to identify intercalated cell subtypes are the H+-ATPase, AE1, and the Cl−/ exchanger pendrin. This section therefore describes the studies that identified the genes encoding these proteins and how ablation of these genes impact acid-base balance. H+-ATPases are made up of two primary domains, i.e., a H+-translocating, Vo domain and a peripheral, V1 domain that mediates ATP hydrolysis (45). The V1 domain has three A and three B subunits. In humans and in cows, the B1 and B2 subunits are encoded by two separate genes (45). While the B2 isoform is ubiquitous, B1 subunit expression is much more cell-specific. Because the B1 subunit is found almost exclusively within renal ICs, it is widely used as a renal intercalated cell marker (129). The initial studies described above, which identified an H+-ATPase in the apical domains of intercalated cells and its role in luminal acidification, greatly facilitated the identification and cloning of the gene(s) encoding this H+ pump. In people, sequence variants of the B1 subunit of the H+-ATPase, ATP6V1B1, are associated with autosomal recessive deafness and distal renal tubular acidosis (83). After determining the mouse B1 subunit gene structure (Atp6v1b1) (45), Atp6v1b1 null mice were developed, which, like their human counterparts, were found to have impaired ability to fully acidify their urine (44). Moreover, while these mutant mice maintain a normal serum concentration under basal conditions, they have a lower serum than their wild-type littermates in rodent models of metabolic acidosis (44).
Band 3 protein, which we now call AE1, is a membrane-spanning, integral protein that is highly expressed in erythrocytes. Because band 3 makes up ~25% of total erythrocyte protein by weight (157), early studies were able to raise antibodies to purified band 3 protein. By immunoblot, this band 3 antibody detected a protein in red cell lysates and also a related protein in kidney (30). However, because the mobility of the band detected in kidney lysates was higher than that of red blood cells, size of the former was less than that of the latter protein. By immunofluorescence and immunogold cytochemistry, this band 3-related protein was observed on the basolateral membrane of renal type A ICs (74, 175, 222). Because band 3 was known to mediate Cl−/ exchange in erythrocytes (79), and because AE1 colocalizes with Cl−/ exchange in the type A ICs (15, 68, 117, 200), AE1 was assumed to mediate this basolateral anion exchange. This model was later confirmed after the gene encoding AE1 in kidney was identified (100), and is now called Slc4a1 in rodents and SLC4A1 in humans. While AE1 is encoded by the same gene as the red cell AE1, mouse kidney AE1 represents a truncated splice variant of the erythrocyte AE1, with the former lacking the first 79 amino acids of the red cell AE1 NH2-terminal cytoplasmic domain, which explains the lower size of kidney AE1 relative to red cell AE1 when detected by immunoblot.
AE1 most likely contributes substantially to the electroneutral exchange of chloride and bicarbonate across the basolateral membrane of type A IC. People with loss-of-function AE1 sequence variants develop hyperchloremic metabolic acidosis, variable hypokalemia, growth retardation, osteomalacia, nephrocalcinosis, and progressive renal insufficiency, all of which is consistent with distal renal tubular acidosis (84). Similarly, AE1-null mice have a severe metabolic acidosis with a low serum bicarbonate, a high serum chloride, and low net acid excretion (196). Following an oral acid load, differences between wild-type and knockout (KO) in acid-base balance mice are magnified. These AE1 KO mice also have a nephrogenic diabetes insipidus in that they have a high plasma osmolarity and a hypotonic urine (196). The diabetes insipidus observed in these AE1-null mice occurs in large part from a defect in aquaporin (AQP) 2 trafficking within the inner medulla, which prevents this water channel from reaching the apical plasma membrane. This impaired AQP2 trafficking may occur from the hypercalciuria observed in these KO mice. However, because the renal medulla is almost completely destroyed in the AE1-null kidney due to anemia-induced renal ischemia, the renal phenotype observed in these mice is difficult to interpret.
Identification of the gene encoding the type B IC apical anion exchanger was discovered serendipitously in studies that followed the identification of the gene responsible for Pendred syndrome. Without the benefit of hindsight, this syndrome would appear to be completely unrelated to what was then known of this type B IC apical anion exchanger's renal physiology. In 1896, Dr. Vaughan Pendred reported a single family in which 2 of 10 children suffered from congenital deafness and goiter, a disorder now called Pendred syndrome (148). This condition is inherited in an autosomal recessive fashion and seen in 7.5 per 100,000 persons (42). Over the century that followed this first report, Pendred syndrome was thought to be a disease specific to ear and thyroid, since abnormalities in other organ systems, such as kidney, had not been appreciated (148). However, after Everett et al. (42) identified and cloned the gene responsible for Pendred syndrome, a number of tools became available, such as anti-pendrin antibodies and pendrin-null mice, which enabled better study of Pendred syndrome’s renal phenotype. In this first report (42), pendrin (Slc26a4) mRNA expression was examined in a variety of tissues by Northern blot. As expected, high levels of Slc26a4 mRNA expression were observed in thyroid and then later in inner ear (42, 164, 235). However, much higher Slc26a4 mRNA expression was detected in kidney relative to many other tissues examined (42). This unexpected result prompted these investigators to examine renal pendrin protein localization by immunohistochemistry (165) and observed pendrin to be highly expressed in the apical region of a subset of intercalated cells within the human, rat, and mouse kidney cortex (165).
Further studies explored the IC subtype(s) that express pendrin, since intercalated cell subtypes have very different functions. Colocalization and ultrastructural studies showed that pendrin is expressed in the apical domains, including the apical plasma membrane and apical cytoplasmic vesicles, of type B and non-A, non-B intercalated cells of the mouse CNT and the CCD (95, 165, 230) and within the distal portion of the distal convoluted tubule (230). Because pendrin localizes to the apical membrane of a cell type (type B IC) that secretes and absorbs Cl− following aldosterone administration in vivo (165, 192, 231), and because pendrin is an electroneutral, Na+-independent, Cl−/ exchanger (181, 184, 192), we hypothesized that pendrin represents the apical, electroneutral anion exchanger observed in the β- or B-type IC of the CCD. We therefore examined Cl− and total CO2 () transport in CCDs from wild-type and pendrin-null mice given NaHCO3 and an aldosterone analogue to upregulate type B intercalated cell apical Cl−/ exchange (99, 165, 195, 217, 231). While significant Cl− absorption and secretion were observed in CCDs from wild-type mice, this anion exchange was absent in pendrin-null mice (165, 231). Later studies showed that pendrin gene ablation nearly eliminated the increment in IC intracellular pH that follows luminal Cl− removal (4a). Thus, in mouse CCD, pendrin mediates Cl− absorption and secretion through Cl−/ exchange. However, we speculate that pendrin-mediated Cl− absorption is greater in mouse CNT than CCD because apical plasma membrane pendrin protein abundance is greater in the former than in the latter segment under both basal and stimulated conditions (217, 230), and because total segment length in vivo is much greater for the CNT than the CCD (242).
Pendrin is not likely the only IC Na+-independent, apical Cl−/ exchanger in mouse CCD. While pendrin-positive ICs make up ~50% of all ICs in mouse CCD (93), apical Na+-independent, Cl−/ exchange is observed in roughly 86% of the ICs within this segment (35). These data suggest that apical Cl−/ exchange is also present in the type A IC of mouse CCD. One potential candidate is Slc26a11, which acts as either a Cl−/ exchanger or a Cl− channel and is expressed in the apical regions of type A ICs (246). What other transporters mediate this apical anion exchange, particularly within type A ICs, requires further study.
Apical Cl−/ exchange increases in a number of treatment models associated with metabolic alkalosis. Therefore, our laboratory and others have explored how pendrin abundance and/or subcellular distribution change in rodent models of metabolic acidosis or alkalosis. Following the administration of NaHCO3 or an aldosterone analogue, both of which are conditions associated with metabolic alkalosis, apical plasma membrane pendrin abundance increases in the type B IC, primarily through subcellular redistribution (217, 219). In these treatment models, pendrin label per cell also increases in non-A, non-B ICs (217, 219). Similarly, when quantified by immunoblot, pendrin total protein abundance increases following NaHCO3 administration (46). Conversely, pendrin total protein abundance falls in rodent models associated with metabolic acidosis, such as following AE1 gene ablation (196), H+-ATPase B1 (ATP6V1B1) subunit gene ablation (14), NH4+ administration (46, 60, 155, 227), or following carbonic anhydrase inhibition (60). Lastly, pendrin abundance also falls in rodent models of respiratory acidosis (29).
Because pendrin mediates renal tubular secretion, we hypothesized that during metabolic alkalosis, pendrin-mediated secretion increases, which attenuates the alkalosis. To test this hypothesis, we measured arterial pH and Pco2 in wild-type and pendrin-null mice given aldosterone or NaHCO3. Following either treatment, pendrin KO mice develop a greater alkalosis and have a lower urinary pH than do wild-type mice (217, 219). These data indicate an impaired ability of these mutant mice to either increase excretion of OH− equivalents or to appropriately reduce net acid excretion, as is needed to correct an alkalosis. As such, the physiological role of the type B intercalated cell was first thought to involve restoration of acid-base balance during metabolic alkalosis through pendrin-mediated secretion. These initial studies were also consistent with earlier models positing that intercalated cells regulate renal net acid excretion.
Whether pendrin regulates acid-base balance in humans is unresolved. A single case report described an individual with Pendred syndrome who developed a severe metabolic alkalosis following thiazide treatment, which was corrected with the administration of KCl and the mineralocorticoid receptor antagonist spironolactone (147). In another case report, severe metabolic alkalosis and hypokalemia were observed in a woman with Pendred syndrome after vomiting, which corrected with K+ and Mg2+ supplementation (82). In other single case reports, changes in acid-base balance have been shown to modulate renal pendrin abundance. Either very low or undetectable pendrin immunolabel was observed in kidney biopsy samples taken from people with distal renal tubular acidosis relative to normal controls (92, 215). The effect of acid-base balance on renal pendrin expression in people has also been assessed by quantifying pendrin abundance in urinary exosomes following changes in acid-base balance. Pathare et al. (138) observed that urinary pendrin exosome abundance falls with the acute oral administration of NH4Cl and rises with NaHCO3, consistent with observations in mice.
While ICs clearly participate in the regulation of acid-base balance, their critical role in Na+, K+ and Cl− balance was revealed after the genes were cloned that encode the transporters that mediate or modulate apical Cl−/ exchange and then after the development of mice with ablation of these respective genes. The role of pendrin-positive ICs in NaCl and K+ balance is given in sections V and VII.
Pendrin remains localized to the distal portion of the distal convoluted tubule (DCT), the CNT, and the CCD under most physiological conditions, even when stimulated. Following a NaCl-replete diet plus furosemide, renal pendrin total protein abundance increases (155), as does pendrin label in the region of the apical plasma membrane region (140). Pendrin-positive cells also appear much larger and bulge into the lumen, as is typical of conditions that increase apical plasma membrane length per cell (FIGURE 3) (140). However, while furosemide increases pendrin abundance in the cortex, it does not induce pendrin expression within the renal medulla to a significant extent (FIGURE 3). Thus, under most conditions examined to date, pendrin localizes primarily to the DCT, CNT, and CCD within the renal cortex.
FIGURE 3.
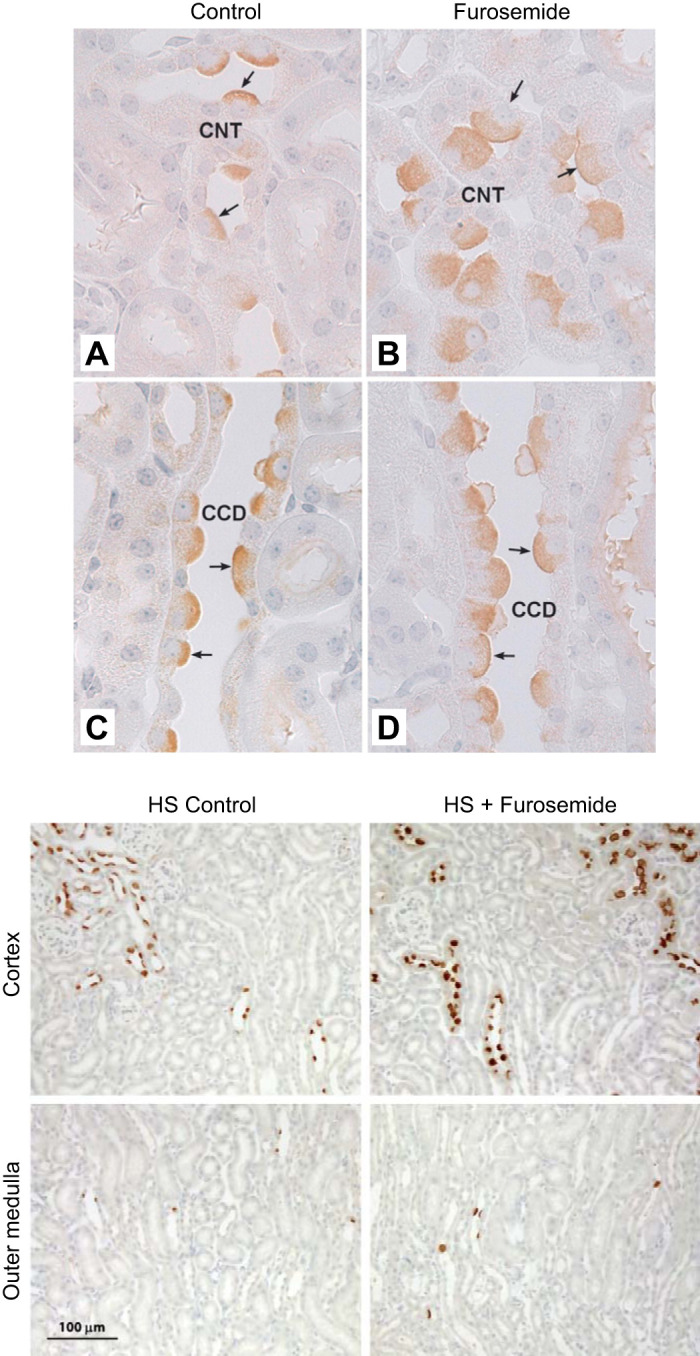
Effect of furosemide on pendrin abundance and distribution in the cortex and medulla. Top panel shows renal cortical sections labeled for pendrin from mice that received 7 days of a NaCl-replete diet or diet and furosemide. As shown, in both the cortical collecting duct (CCD) (C and D) and connecting tubule (CNT) (A and B), cells that label for pendrin are much larger with more pendrin label in each cell in sections taken from the furosemide-treated mice. [From Pech et al. (140).] Bottom panel shows pendrin label in sections from the same mice at lower magnification. As shown, pendrin label is much more prominent in the cortex of furosemide-treated than of vehicle-treated mice. In the medulla, few pendrin-positive cells are observed in mice from either group.
Under some treatment conditions, renal pendrin expression is observed beyond the renal cortex (72). For example, Himmel et al. (72) have observed that Li2+ treatment induces pendrin expression in the rat outer and inner medullary collecting duct and increases the abundance of intercalated cells relative to principal cells in these collecting duct segments. Moreover, in lithium-treated animals, pendrin is expressed in cells that do not have the classical markers of type B or non-A, non-B intercalated cells. In this treatment model, pendrin colocalizes with the principal cell marker AQP4 (72), indicating pendrin expression in “hybrid cells.” Chen et al. (23) employed single-cell RNA-seq to characterize the complete transcriptome of isolated type A and B ICs as well as principal cells of untreated mice. While most ICs and principal cells display the expected, classical markers, he observed a small fraction of what appear to be “hybrid cells” that express both AQP2 and either AE1 or pendrin transcripts (23). These immature precursor cells or hybrid cells constitute less than 3% of all epithelial cells in the collecting duct of the adult mouse kidney (23). Therefore, while “hybrid” cells are observed in rodents under basal conditions, they are much more abundant in lithium-treated animals. These observations are consistent with previous studies demonstrating that intercalated and principal cells either interconvert or that collecting duct precursor cells can terminally differentiate into mature ICs or principal cells upon demand (2, 163).
IV. ONTOGENY OF INTERCALATED CELL DIFFERENTIATION AND PENDRIN EXPRESSION
How collecting duct cells differentiate into mature principal and intercalated cells has been reviewed elsewhere (2, 163, 177). This section summarizes what is known. The adult kidney originates from two mesodermal embryonic structures, the metanephric mesenchyme and the ureteric bud (183). These two structures interact, giving rise to nephron development. The most proximal structures of the nephron, i.e., the glomerulus, proximal tubule, loop of Henle, and DCT, develop from the metanephric mesenchyme, whereas collecting ducts originate from the ureteric bud. The CNT develops from the union of the metanephric mesenchyme with the ureteric bud (12), while the collecting duct develops when the surrounding mesenchyme induces the growth and branching of the ureteric bud. The immature cells of the ureteric bud that line the collecting duct differentiate to form “mature” collecting duct epithelial cells (125) that we know of as principal and intercalated cells. Existing data indicate that principal and intercalated cells have a common cellular origin (13, 136).
As collecting duct cells differentiate and mature, they acquire markers typical of either principal or intercalated cells. In the mouse, the H+-ATPase appears at embryonic day 13 (E.13) (193), whereas pendrin-positive and AE1-positive cells appear by E.14. Throughout the remainder of gestation, pendrin- and AE1-positive cells increase in number (193).
Trepiccione et al. (210) observed that principal cells (PCs) and ICs share a common cell progenitor, although other investigators have challenged this notion (31). While ICs and PCs likely share a common precursor, the stimuli that induce these precursor cells to differentiate into one of these mature cell types are not well understood (193). Collectively, these data suggest that the origin of epithelial cells in the CCD is complex.
Immature precursor cells are thought to mature into differentiated PCs and ICs through a mechanism that depends on the forkhead transcription factor Foxi1 (FIGURE 4). In the adult, Foxi1-null kidney, the normal collecting duct epithelium is replaced by a single cell type that expresses both carbonic anhydrase II, an IC marker, and AQP2, a PC cell marker (13). These Foxi1-null mice develop a distal renal tubular acidosis (13), which is reversed with the induction of Foxi1 expression, thereby increasing AE1 and pendrin expression (80).
FIGURE 4.
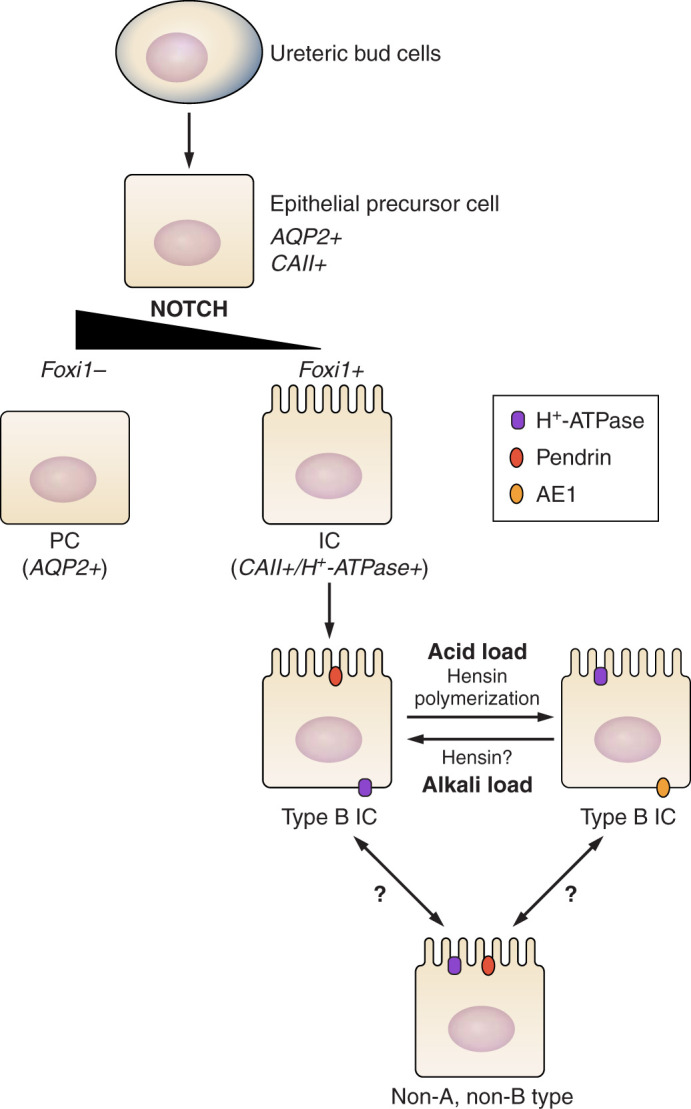
Ontogeny of intercalated cell (IC) differentiation. ICs and principal cells (PCs) are derived from a common immature precursor (epithelial precursor cell) that is governed by Notch and Foxi1 signaling. In the absence of Notch signaling, the transcription factor Foxi1 is suppressed. However, the “terminal” phenotype of the various IC subtypes may interconvert under certain experimental conditions.
Notch signaling is a ubiquitous signaling pathway that participates in the differentiation of many cell types (87, 130). Through a process called lateral inhibition, neighboring cells interact through a mechanism that is dependent on Notch signaling, which gives rise to a random, mosaic pattern of cells with a “salt and pepper”-like appearance (169) due to the different optical characteristics of the various cell types in kidney (5). With changes in Notch signaling, the relative abundance of ICs and PCs is altered. In the absence of Notch activation, as seen with either Mind bomb-1 (Mib1) or Adam10 gene ablation, IC cell abundance increases, whereas PC abundance decreases (59, 80). Notch signaling acts through transcription factor Foxi1 to regulate the differentiation of these precursor cells into either ICs or PCs (80). These observations are consistent with other studies showing that changes in Notch/Foxi1 expression result in the interconversion of ICs and PCs (123). In humans and in mouse models of chronic kidney disease, the abundance of ICs falls, whereas PC abundance rises, which occurs through Notch-dependent signaling (136). This IC-PC transition is thought to preserve the ability of the organism to maintain NaCl and water homeostasis, but at the expense of acid-base balance (136).
In addition to the PC-IC interconversion, some have suggested that IC subtypes might interchange phenotype upon demand, such as following changes in acid-base balance. Al-Awqati and collaborators observed that following an acid load, the proportion of cells within isolated CCDs with functional characteristics of type A ICs increases (178, 179), whereas with alkali excess cells with characteristics of type B ICs increases (47). These investigators observed that the interconversion of type B to type A ICs occurs through a mechanism that depends on hensin polymerization. Following acid exposure, hensin is released from ICs as a monomer (177). When secreted hensin is deposited as a polymer in the extracellular matrix of immortalized cultured cells, type B ICs convert to type A ICs through a β1 integrin-mediated mechanism (FIGURE 4). In mice with hensin gene ablation, type B ICs predominate in the renal cortex, despite the metabolic acidosis observed in these mice (47). Thus hensin expression is necessary for the normal development of type A ICs. However, other groups have not observed a change in the abundance of the various intercalated cell subtypes with perturbations in acid-base balance (223, 224) and have therefore challenged the conclusion that fully differentiated type B and type A ICs interconvert upon demand. Nevertheless, following pharmacological treatments, such as with the administration of lithium or carbonic anhydrase inhibitors, distal nephron cell remodeling occurs, which may explain changes in the relative abundance of principal cells and the various intercalated cell subtypes observed in vivo in response to these treatment conditions (9, 123).
In summary, ICs and PCs are most likely derived from a common immature precursor. Differentiation of this precursor into mature ICs or PCs occurs through a mechanism that involves Notch-Foxi1 signaling. Activating Notch signaling through lateral inhibition may explain the random presence of ICs and PCs. The “terminal” phenotypes of the various IC subtypes may interconvert, depending on the experimental conditions, although aspects of this hypothesis remain controversial.
V. CONTRIBUTION OF PRINCIPAL AND INTERCALATED CELLS TO RENAL NaCl ABSORPTION AND BLOOD PRESSURE REGULATION
A. Contribution of the CCD and CNT to Renal NaCl Absorption and the Mechanism of NaCl Absorption by the Cells in These Segments
During volume contraction, angiotensin II and aldosterone release increases, which raises renal NaCl absorption, thereby helping to correct the volume deficit. Conversely, during volume overload, the release of angiotensin II and aldosterone are suppressed, which reduces distal nephron NaCl absorption. Most distal NaCl transport occurs within the DCT, the CNT, and, to a lesser extent, the CCD (26, 27, 112, 197). NaCl absorption by the DCT has been quantified in micropuncture studies. NaCl delivery to the DCT is ~10% of filtered load (241). Of the NaCl delivered to the DCT, roughly 75% is absorbed before the CCD, which occurs equally along the DCT and CNT (27, 32, 86, 112, 197). Because rat and mouse CCDs can be perfused in vitro, while attempts to perfuse CNTs from these animals have been unsuccessful, transport in the CCD is much better understood than in the CNT. The transport properties of these segments are not likely identical, however, since the cell types found in the CNT and CCD differ (230, 232). The CNT has, however, been perfused in vitro in a limited number of studies, all of which were done in rabbit (3, 211). Based on these studies, we know that the rabbit CNT absorbs Na+ and Cl− (3, 188) and secretes (211). Because net Na+ absorption per millimeter tubule length is about three to four times higher in the CNT that in the CCD (3) and because the total length of the CNT in vivo is about sixfold greater than the CCD (33, 77, 150), cumulative Na+ absorption in vivo is about one order of magnitude greater in the CNT than in CCD. However, the CCD is still important in the final regulation of salt balance.
Studies that employed CCDs perfused in vitro have generated a large body of data, which has enabled characterization of the transport properties of this segment. Between 58 and 66% of the cells within mouse CCD are principal cells, while 33–42% are intercalated cells (25, 59, 93, 205), with type A and type B ICs being about equally prevalent (25). In contrast, within mouse CNT, 60% are CNT cells, while 10% are type A and 30% are non-type A ICs (25). As such, type A ICs are much less common in the CNT than are non-type A ICs.
In the CCD, principal cells absorb Na+ (144, 202), although there is evidence that intercalated cells also absorb Na+ (104). In the CCD, Cl− absorption occurs either through intercalated cell-mediated transcellular transport or through paracellular transport (172). The transporters that mediate this NaCl transport in principal and intercalated cells are shown in FIGURE 1.
Cl− absorption by the CCD occurs through two separate pathways, which differ in diuretic sensitivity. The first of these pathways mediates the electroneutral absorption of NaCl in rat and mouse CCD and is inhibited by thiazide diuretics (104, 206). While thiazides strongly inhibit the NaCl cotransporter of the DCT (NCC, encoded by Slc12a3) (34), NCC does not mediate thiazide-sensitive Cl− absorption in the CCD because it is not expressed in the collecting duct (91). Instead, Leviel et al. (104) reported that the thiazide-sensitive component of Na+ absorption observed in the mouse CCD occurs through a Na+-dependent Cl−/ exchanger (NDCBE) encoded by Slc4a8. They went on to show that pendrin-mediated Cl−/ exchange and NDCBE-mediated Na+-dependent Cl−/ exchange act in parallel to mediate electroneutral NaCl absorption in the mouse CCD.
Following dietary NaCl restriction, NDCBE-null mice have a higher renin activity, a higher urinary aldosterone, and lower blood pressure than do their wild-type littermates (190). However, these parameters were similar in wild-type and mutant mice following a NaCl-replete diet. Thus, NDCBE contributes to the maintenance of NaCl balance and blood pressure during dietary NaCl restriction (190).
The electroneutral, sodium-dependent chloride and bicarbonate exchanger (NDCBE) was cloned in 2001 (55) and represents a 1,044-amino acid protein that is found in brain, testis, kidney, and ovary. NDCBE has several splice variants, which are referred to as A, B, C, and D (55, 137, 234). In heterologous expression systems, NDCBE mediates the influx of Na+ and and the efflux of Cl− (55). While functional analysis indicates that NDCBE acts in concert with pendrin to mediate electroneutral NaCl absorption in type B intercalated cells of the mouse CCD (104), other studies have questioned this model and, in particular, whether NDCBE localizes to type B ICs (23, 247).
Electroneutral pendrin/NDCBE-mediated Na+ and Cl− absorption is thiazide sensitive, although the mechanism by which thiazides inhibit NDCBE is unclear. Since thiazides inhibit carbonic anhydrase (151) and since apical anion exchange in the type B IC is very sensitive to carbonic anhydrase inhibition (120), these drugs might reduce Cl− absorption in the CCD, at least in part, by inhibiting this enzyme rather than only through direct inhibition of a Na+ or Cl− transporter, such as NDCBE.
The transporters that mediate NaCl absorption and secretion by the type B IC are displayed in FIGURES 1 and 2. Following the electroneutral uptake of NaCl across the apical plasma membrane of type B ICs, NaCl exits across the basolateral plasma membrane of this cell type through the Na+- cotransporter AE4 (22) and through ClC-K2/barttin Cl− channels (70, 152), the latter of which is regulated by pH and membrane potential (70, 152).
Na+-K+-ATPase abundance is much lower in intercalated than in principal cells (43, 85, 168, 170, 204). Nonetheless, Cl− absorption in CCDs from aldosterone-treated mice is eliminated with the application of the Na+ pump inhibitor ouabain to the bath (143). Moreover, Na+-K+-ATPase inhibitors increase intercalated cell intracellular Na+, although to a lesser extent than in principal cells (170). Therefore, the Na+ pump likely affects active transport in the intercalated cell, at least to some degree. However, the active step that provides the driving force for Cl− absorption by the type B intercalated cell occurs largely through electrogenic, basolateral plasma membrane H+ exit, vis-à-vis the H+-ATPase, independently of the Na+-K+-ATPase, and occurs in parallel with AE4-mediated efflux (22, 36, 140). Pendrin may change intracellular Cl− concentration, thereby modulating ion flux mediated by these other type B and non-A, non-B IC transporters or channels (160).
The second mechanism of Cl− absorption in the CCD is electrogenic and eliminated with the application of ENaC inhibitors, such as amiloride (206). The magnitude of the amiloride- and the thiazide- sensitive components of NaCl absorption varies between studies and probably depends on the species being tested and the treatment model being used (104, 143, 206). While the epithelial Na+ channel, ENaC, represents the major mechanism of Na+ absorption by principal cells (144, 202), ENaC does not transport Cl− and thus modulates Cl− absorption in the CCD indirectly (143). Blockade of ENaC changes Cl− transport or channel activity either through paracrine signaling or by altering the driving force for Cl− movement. ENaC-mediated Na+ absorption generates a lumen-negative transepithelial voltage that augments the driving force for electrogenic, Cl− absorption. Thus the amiloride-sensitive component of Cl− absorption may involve paracellular Cl− transport or transcellular transport mediated by a Cl− channel or an electrogenic Cl− exchanger. While the mechanism by which ENaC inhibition changes Cl− transport is not known, previous studies have shown that it does not involve pendrin, ClC-5, or cystic fibrosis transmembrane conductance regulator (126, 142–144).
Claudin-mediated paracellular Cl− transport is important in both blood pressure regulation and in the maintenance of vascular volume. Following claudin 4 gene ablation, a natriuresis, a chloriuresis, and reduced blood pressure are observed particularly when dietary NaCl intake is restricted (53). Since claudin 4 acts as a Cl− channel and localizes to tight junctions between intercalated cells and principal cells (53, 75), ENaC blockade may alter electrogenic Cl− transport in the CCD by eliminating the driving force for claudin 4-mediated paracellular Cl− transport.
The contribution of transcellular relative to paracellular Cl− absorption, and the possible contrasting physiological roles of each, are not well understood. Pei et al. (146) have shown that under conditions where energy conservation is needed, the kidney is more dependent on paracellular than transcellular Cl− absorption. Since the lumen-negative voltage generated by ENaC-dependent Na+ absorption provides the driving force for paracellular Cl− absorption, ENaC-dependent Cl− absorption might be of more importance during energy depletion. Conversely, transcellular, electroneutral Cl− transport mediated by type B intercalated cell apical Cl−/ exchange might be most important when cellular energy is abundant. However, the relative role of these electrogenic and electroneutral pathways may depend instead on the need for K+ conservation. The role of pendrin-positive ICs in K+ homeostasis is reviewed in section V.
B. Role of Pendrin in Human and Rodent NaCl Balance and in Blood Pressure Regulation
Since pendrin mediates renal Cl− absorption, we explored its role in NaCl balance (96, 217, 219, 231). Following a NaCl-replete diet, apparent vascular volume is similar in wild-type and in global, pendrin-null mice (219, 231), although Cl− clearance is greater in the pendrin-null than in the wild-type mice (97). Subsequent experiments explored the role of pendrin in salt balance following dietary Na+ and Cl− restriction. After 4 days of a Na+, K+, and Cl− replete diet, Na+ and Cl− excretion are similar in wild-type and pendrin-null mice (96, 219). However, after introduction of a NaCl-restricted diet, NaCl excretion was initially greater in the pendrin-null than in wild-type mice, but were similar by about the second day of NaCl restriction (96, 219, 231). Because of this natriuresis and chloriuresis, pendrin-null mice have a higher blood urea nitrogen (BUN) and lost more weight over the treatment period than their wild-type littermates, which is consistent with greater apparent volume contraction in the former relative to the latter (219, 231).
Following dietary NaCl restriction, NaCl excretion is greater, while blood pressure and apparent vascular volume are lower in pendrin-null than in wild-type mice (96, 141, 219, 231). Therefore, the pendrin-null kidney has an impaired ability to fully conserve NaCl, which likely contributes to the lower blood pressure observed in these mutant mice (96, 219, 231). During dietary NaCl restriction or following an aldosterone infusion, both of which upregulate pendrin, the difference in blood pressure observed between pendrin-null and wild-type mice is enhanced (141, 217, 219, 231). Blood pressure falls either when pendrin gene ablation is induced in utero or when induced in adulthood (209).
Whereas blood pressure is reduced in pendrin-null mice, hypertension that is very sensitive to dietary Cl− intake is observed in mice that overexpress pendrin (78). It is notable that ICs have such an impact on blood pressure and NaCl balance since they represent only ~1% of total kidney volume (150) and ~2% of kidney mass (25).
Since blood pressure is the product of systemic vascular resistance and cardiac output, we explored the effect of pendrin gene ablation on vascular tone (203). Although pendrin expression is either low or absent in vascular tissue, thoracic aorta contractile force in response to either α-adrenergic agonists (phentolamine) or angiotensin II application is greater in pendrin-null than in wild-type mice (203). This greater contractile force is associated with increased myosin light chain kinase 20 abundance and eliminated with the administration of an angiotensin type 1a receptor inhibitor, i.e., candesartan, in vivo (203). This increase in vascular contractility may limit the hypotension that follows pendrin gene ablation. However, because pendrin expression in vascular tissue is very low, the changes in vascular tone we observed with pendrin gene ablation occurs from an indirect effect of pendrin gene ablation, such as through changes in circulating levels of angiotensin II and/or catecholamines (203).
Following our observation that pendrin regulates blood pressure in mice, other groups have explored whether pendrin regulates human blood pressure and/or salt balance. In the first of these human studies, a retrospective chart review examined the incidence of hypertension in persons with Pendred syndrome and in their unaffected family members (109). While this study was inadequately powered, it suggested that eliminating pendrin could be protective against the development of hypertension. Later studies measured blood pressure and NaCl excretion in a larger cohort of people with biallelic, inactivation mutations in SLC26a4 and in unaffected controls (89). This report demonstrated that pendrin gene ablation reduces blood pressure and induces a natriuresis and chloriuresis, similar to our earlier studies in mice.
Since up to ~50% of patients with Pendred syndrome are hypothyroid (243), one could speculate that the reduced blood pressure observed in these patients is due to changes in thyroid function. However, hypothyroidism is associated with increased diastolic blood pressure (28). As such, hypothyroidism should mitigate the fall in blood pressure observed in people with inactivating mutations in Slc26a4 (28). Moreover, the hypothyroidism and goiter associated with Pendred syndrome is highly dependent on modifying genetic and environmental factors, particularly dietary iodine content (243). Therefore, in people with Pendred syndrome, the incidence of goiter and hypothyroidism is very low in developed countries with adequate dietary iodide intake (135), such as South Korea where the study mentioned above was conducted (89).
C. Effect of Pendrin-Positive ICs on ENaC
The natriuresis observed in pendrin-null mice following dietary Na+ restriction (96) was a surprising observation since pendrin does not transport Na+. We therefore explored whether pendrin gene ablation changes the abundance of a major renal Na+ transporter using a “targeted proteomics” approach (16, 96). In mice consuming a NaCl-replete diet, where circulating plasma renin and aldosterone are suppressed, pendrin gene ablation did not change the abundance of the major renal Na+ transporters and channels, such as NHE3, NKCC2, α1 Na+-K+-ATPase, or ENaC (96). However, following an aldosterone infusion or dietary NaCl restriction, α, β and γ ENaC subunit abundance was lower in kidneys from pendrin-null relative to wild-type mice (96, 141, 144).
Other experiments explored the effect of pendrin gene ablation on ENaC function. In normal rats and mice, aldosterone administration markedly increases ENaC-mediated Na+ absorption in the CCD (132). Because the aldosterone-induced increment in Na+ absorption is nearly eliminated with ENaC inhibitor application (benzamil) (144, 201, 202), ENaC is the primary mediator of Na+ absorption in aldosterone-treated mice. This benzamil-sensitive component of Na+ absorption is, however, ~60% lower in CCDs from aldosterone-treated pendrin-null relative to wild-type mice (144). Therefore, the fall in ENaC abundance and function observed in pendrin-null mice likely contributes to the fall in blood pressure seen with pendrin gene ablation.
Aldosterone increases ENaC activity (NPo) by augmenting the frequency at which the channel is open (open probability, Po) (49), by increasing ENaC subunit total protein abundance (113), and by increasing the relative abundance of ENaC subunits in the region of the apical plasma membrane through subcellular redistribution (113). However, assembly of all three ENaC subunits is needed for aldosterone to efficiently deliver ENaC to the apical plasma membrane (76, 122, 213).
To determine how pendrin changes ENaC activity, we performed single-channel recordings in principal cells from split open mouse CCDs taken from aldosterone-treated wild-type and pendrin-null mice (144) and observed that pendrin gene ablation lowers channel activity due to the reduced channel surface density that occurs from both a fall in ENaC subunit total protein abundance as well as from ENaC subunit subcellular redistribution (96, 141, 144). In addition, pendrin gene ablation reduces channel open probability (144).
How pendrin changes ENaC abundance and function has been explored by us and others (58, 141). Because pendrin and ENaC localize to different cell types (62, 95, 165, 230) (FIGURE 1), these proteins cannot directly associate. In addition, pendrin does not change ENaC by altering the circulating level of a hormone that regulates ENaC, such as corticosterone, vasopressin, renin, aldosterone, or thyroid hormone (96, 97). Moreover, while pendrin gene ablation downregulates ENaC in kidney, pendrin gene ablation does not change ENaC abundance in thyroid and colon (96) and paradoxically increases ENaC subunit mRNA expression in the inner ear (88). As such, the fall in ENaC subunit abundance and function observed in pendrin-null mice appears restricted to the kidney.
Because ENaC subunit abundance and function is sensitive to pH and/or acid-base balance (90, 133, 141, 189), we hypothesized that pendrin-mediated secretion stimulates ENaC by increasing luminal concentration and/or pH (FIGURE 5). To test this hypothesis, we employed two separate treatment models, each of which increases luminal concentration within the CNT and CCD in vivo. In the first, we gave mice NaHCO3 and aldosterone to stimulate pendrin-mediated secretion (99, 141), thereby increasing luminal concentration in the CNT and CCD. In the second, we added a carbonic anhydrase inhibitor (acetazolamide) to this treatment protocol to increase distal delivery of from upstream segments, while downregulating pendrin expression and function (120, 141). When mice were treated with aldosterone and NaHCO3, pendrin gene ablation magnified the metabolic alkalosis and reduced renal ENaC subunit abundance and function (141). However, adding acetazolamide to this treatment protocol eliminated the differences between pendrin-null and wild-type mice in renal ENaC subunit abundance and function as well as differences in acid-base balance (141). One explanation of these findings is that increasing distal delivery from upstream segments rescues pendrin-null mice from the enhanced alkalosis and the subsequent fall in renal ENaC abundance and function.
FIGURE 5.
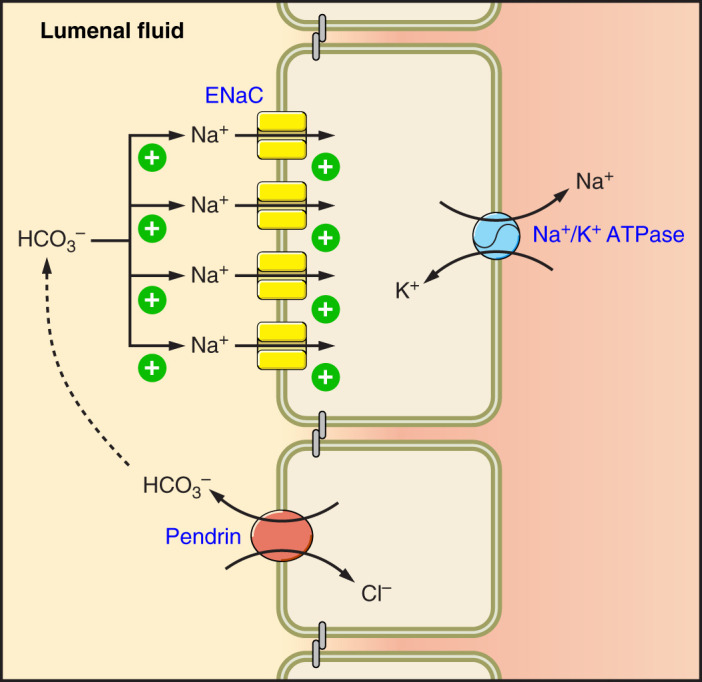
Pendrin-mediated HCO3− secretion modulates epithelial Na+ channel (ENaC) abundance and function. Pendrin mediates the secretion of HCO3−, which stimulates ENaC-mediated Na+ absorption as well as ENaC subunit abundance.
In other experiments, we used mouse principal cells (mpkCCD) to examine ENaC abundance and function when concentration was varied on the apical or basolateral side of the cell monolayer (141). With an increase in the concentration on the apical side of the monolayer, ENaC abundance and function rose, independently of substituting anion. Therefore, pendrin modulates ENaC, at least in part, either through changes in luminal or luminal pH.
Since H+-ATPase gene ablation reduces ENaC abundance and function in kidney (58) and since pendrin gene ablation significantly reduces H+-ATPase abundance in type B intercalated cells (98), pendrin gene ablation may reduce ENaC function from the fall in type B intercalated cell H+-ATPase activity seen in these mutant mice. Because B1-H+-ATPase gene ablation increases urinary excretion of prostaglandin E2 (PGE2), Gueutin et al. (58) hypothesized that type B cell H+-ATPase gene ablation reduces ENaC abundance and function through a PGE2-mediated mechanism (FIGURE 6). With B1 ATPase gene ablation, increased luminal ATP secretion was seen in the CCD, which acts through purinergic receptors on the apical plasma membrane of principal cells to stimulate principal cell Ca2+ release, thereby increasing PGE2 production, which reduces ENaC abundance and function (58). These studies are in keeping with a large body of work demonstrating that luminal ATP acts through principal cell apical plasma membrane purinergic receptors to reduce phosphatidylinositol 4,5-bisphosphate (PIP2) through a phospholipase C (PLC)-dependent pathway, which lowers ENaC subunit abundance and function (108, 153, 251).
FIGURE 6.

Pendrin gene ablation modulates luminal ATP concentration, which changes epithelial Na+ channel (ENaC) abundance and function. With pendrin gene ablation, H+-ATPase abundance falls in the type B intercalated cell, thereby increasing intercalated cell ATP content. Luminal ATP concentration then rises through enhanced connexin 30-mediated ATP secretion. Luminal ATP acts on apical membrane purinergic receptors to stimulate calcium release, which increases prostaglandin E2 production (PGE2). PGE2 acts through a receptor-mediated process to reduce ENaC abundance and function.
In summary, pendrin gene ablation reduces ENaC abundance and function by changing luminal and ATP concentration. However, other pathways may also contribute to this pendrin-ENaC interaction.
D. Compensatory Mechanisms Limiting the NaCl Loss that Follows Pendrin Gene Ablation: the Pendrin-NCC Interaction
Under basal conditions, i.e., following a diet replete in Na+, K+, and Cl−, pendrin-null mice have a mild phenotype, which is limited to hearing loss and a small reduction in blood pressure (96, 102, 217, 231). Under these basal conditions, arterial pH as well as serum Na+, K+, Cl−, , and BUN concentrations are similar in the wild-type and pendrin-null mice (96, 217, 231). As with pendrin-null mice, NCC (Slc12A3) knockout mice also have a very mild phenotype under similar basal conditions, since blood pressure as well as NaCl balance are similar in NCC-null and their wild-type littermates (173, 191). However, following dietary NaCl restriction, NCC-null (173) and pendrin-null mice (231) have significantly lower blood pressure than their wild-type littermates. These studies have led to the conclusion that a renal phenotype is unmasked in these mutant mice during vascular volume contraction, such as following dietary NaCl restriction.
Because NCC, NDCBE, and pendrin are electroneutral Cl− transporters, various investigators have explored whether these transporters compensate for the absence of one another. These studies have shown that pendrin and NDCBE are upregulated in kidneys from mice with reduced or absent NCC expression, such as in NCC-null or SPAK-null mice (56, 214). Conversely, NCC protein abundance is likely increased in kidneys from pendrin-null or NDCBE-null mice, at least under basal conditions (96, 190). These observations prompted Sinning et al. (190) to ask if ablation of both NDCBE and NCC produce a more severe phenotype than ablation of either of these genes alone. They observed that apparent vascular volume contraction is more severe in mice with gene ablation of both NCC and NDCBE relative to ablation of either NCC or NDCBE alone. Thus they concluded that during NaCl restriction, vascular volume is maintained in either NCC or NDCBE-null mice, through upregulation of the other transporter.
In related studies, Soleimani et al. (191) explored whether NCC or pendrin is upregulated to maintain NaCl balance the absence of the other. To do so, they compared blood pressure as well as acid-base and fluid and electrolyte balance in wild-type mice, NCC-null mice, pendrin-null mice, and in mice that are both NCC and pendrin null. While NCC-null mice and pendrin-null mice were again found to have a mild phenotype, mice that were both NCC and pendrin null were profoundly hypotensive with significant polyuria and salt wasting (191). Thus the increment in pendrin total protein abundance seen in NCC-null mice appears to be a compensatory mechanism by which NaCl balance and blood pressure are maintained in the absence of the other.
The mechanism by which NCC gene ablation increases pendrin total protein abundance, or vice versa, is unclear. Because pendrin and the thiazide-sensitive cotransporter NCC both colocalize to the apical region of the distal portion of the DCT (230), these transporters might directly associate in this segment, thereby changing the properties of the other. As discussed in section X, transporters that localize to different cell types communicate, at least in part, through paracrine mechanisms that involve signaling molecules, such as α-ketoglutarate (56). However, the more severe phenotype observed with ablation of two of these genes, versus ablation of one alone, may occur because removing two separate mechanisms of renal Cl− absorption compromises Cl− absorption more than removing one mechanism alone, rather than from a true linkage of these transporters. However, it is interesting that pendrin gene ablation downregulates ENaC, which should reduce electrogenic Cl− absorption. Further studies are needed to determine if ablation of both an electrogenic and an electroneutral mechanism of Cl− absorption produce a more severe phenotype than with ablation of only a single electroneutral pathway, such as pendrin.
Mice with gene Slc26a4 (pendrin) gene ablation have no known defect in urinary concentrating ability. However, mice with ablation of both Slc26a4 (pendrin) and Slc12a3 (NCC) (191) have a vasopressin-resistant defect in urinary concentrating ability, which is consistent with nephrogenic diabetes insipidus (191). How pendrin modulates urinary concentrating ability is not clear. If pendrin were critical for urinary concentration and the maintenance of water balance during water restriction, one would expect water restriction to upregulate pendrin, while water excess should downregulate pendrin. We were prompted therefore to explore the effect of H2O intake on pendrin abundance. We observed however that high water intake, achieved by raising water consumption from ~4 to ~11 ml/day, increased the ratio of pendrin label on apical plasma membrane relative to cytoplasm by ~75% in type B ICs, although circulating renin, aldosterone, and serum osmolality were unchanged (97). Since pendrin abundance fell with reduced H2O intake or with vasopressin administration, we explored the role of arginine vasopressin in this response by examining the effect of water intake on pendrin abundance when circulating vasopressin levels were clamped. We observed that raising water intake increased apical plasma membrane pendrin abundance in either the presence or the absence of changes in circulating vasopressin (97). Thus H2O intake modulates apical plasma membrane pendrin abundance, although not likely through a direct, vasopressin V2R-dependent mechanism. Pendrin is probably upregulated with increased water intake due to the increment in luminal fluid flow.
VI. REGULATION OF PENDRIN BY ALDOSTERONE AND ANGIOTENSIN II, THEIR RECEPTORS, AND OTHER DOWNSTREAM SIGNALING MOLECULES
A. Regulation of Pendrin by Angiotensin II
1. Effect of angiotensin II in vitro
Angiotensin II plays a critical role in NaCl balance and blood pressure regulation (11, 16, 149). Since angiotensin II increases apical Cl−/ exchange in rabbit CCD (239), we explored the mechanism by which it occurs (140). In CCDs perfused in vitro that were taken from furosemide-treated wild-type mice, we observed that angiotensin II addition to the bath increased Cl− absorption twofold, although transepithelial voltage (VT) was unchanged. However, in CCDs from pendrin knockout mice, angiotensin II changed neither Cl− absorption nor VT (140). These data suggest that this peptide hormone increases Cl− absorption through transcellular rather than paracellular transport (140).
Further experiments explored how angiotensin II modulates Cl− transport in vitro in mouse CCD (145). We hypothesized that angiotensin II increases CCD Cl− absorption either by increasing apical plasma membrane pendrin abundance or by stimulating another transporter, thereby increasing the driving force for apical anion exchange. Since the type B IC H+-ATPase provides the active transport step for apical anion exchange (22), angiotensin II might augment apical Cl−/ exchange by increasing basolateral plasma membrane H+-ATPase abundance. To discriminate between these possibilities, we perfused mouse CCDs in vitro in the presence or absence of angiotensin II and then fixed and labeled these tubules for pendrin and the H+-ATPase. Transporter subcellular distribution was quantified by immunogold cytochemistry with morphometric analysis. We observed, however, no effect of angiotensin II application on either pendrin or H+-ATPase subcellular distribution in type B ICs (145). Instead, we observed that in type A ICs from mouse CCD, angiotensin II increased the ratio of H+-ATPase abundance on the apical plasma membrane relative to the cytoplasm threefold through subcellular redistribution. Moreover, angiotensin II markedly increased absorption in mouse CCD, consistent with enhanced apical H+-ATPase-mediated H+ secretion (145) and with previous observations in the OMCD (162).
The increase in ENaC-mediated Na+ absorption seen in the CCD following angiotensin II application (149) should increase the lumen-negative voltage. However, we observed instead that VT in that segment is unchanged with angiotensin II application (145). Our observation that angiotensin II application stimulates H+ secretion provides a potential explanation for the absence of angiotensin II-induced changes in VT. By carrying a positive charge, angiotensin II-stimulated H+ secretion may attenuate the lumen-negative VT generated by ENaC-mediated Na+ absorption (127, 144, 149). In support of this hypothesis, we observed that application of the H+-ATPase inhibitor bafilomycin to the luminal fluid increases the lumen-negative transepithelial voltage in CCDs from aldosterone-treated mice (127). Since angiotensin II application in vitro increases both H+ secretion and Na+ absorption in the mouse CCD, movement of these two ions may shunt the voltage generated by the other.
These data show that angiotensin II does not increase pendrin-dependent Cl− absorption in vitro through pendrin subcellular redistribution or by increasing basolateral plasma membrane H+-ATPase abundance. Thus the mechanism of angiotensin II-stimulated Cl− absorption remains unexplained. This peptide hormone might activate a plasma membrane Cl− channel, thereby lowering intracellular Cl− concentration within the type B intercalated cell, which augments apical Cl−/ exchange. However, other mechanisms are also possible.
2. Effect of angiotensin II in vivo
Angiotensin II increases apical plasma membrane pendrin abundance in vivo through a mechanism that differs from its effects in vitro. In adrenalectomized mice, we observed that angiotensin II administered in vivo increases pendrin abundance in the most apical 10% of non-A, non-B ICs within the CNT through subcellular redistribution. Similarly, in adrenal-intact mice, we observed that angiotensin II acts on the angiotensin type 1 receptor of this cell type to increase apical plasma membrane pendrin abundance through subcellular redistribution, independently of aldosterone (218). While angiotensin II had no effect on pendrin label per cell in non-A, non-B ICs, in the type B IC, however, angiotensin II acts through the angiotensin type 1a receptor to increase pendrin label per cell. Conversely, angiotensin II acts through the angiotensin type 1a receptor to increase pendrin’s relative abundance in the region of the apical membrane through subcellular redistribution in non-A, non-B, but not in type B, ICs (218).
Other studies have found that angiotensin II upregulates pendrin total protein abundance entirely through the increment in aldosterone release that occurs in response to angiotensin II (73). Consistent with this study, we did not observe an increase in pendrin total protein abundance in kidney lysates from angiotensin II-treated mice whether taken from adrenal intact or adrenalectomized mice (218). Because angiotensin II increases pendrin total protein abundance in type B, but not non-A, non-B ICs (218), and because the former cell type is less abundant than the latter in mouse kidney, angiotensin II’s effect on pendrin total protein abundance should be small when quantified by immunoblot.
The absence of an angiotensin II effect on total renal pendrin protein abundance detected by immunoblot may be due to compensatory mechanisms that follow the administration of this peptide hormone. We observed that angiotensin II application in vivo stimulates the angiotensin type 2 receptor, which acts through nitric oxide to reduce pendrin protein abundance without changing pendrin subcellular distribution (218). As such, the angiotensin type 2 receptor provides a feedback loop, which attenuates the increase in apical plasma membrane pendrin abundance that follows angiotensin type 1a receptor activation by angiotensin II by blunting pendrin protein synthesis or increasing its degradation.
B. Regulation of Pendrin by Aldosterone, the Mineralocorticoid Receptor, and the Interaction of K+, Aldosterone, and Angiotensin II with This Receptor
Since aldosterone increases apical anion exchange in the CCD (48, 195), further studies asked if aldosterone changes renal pendrin abundance or subcellular distribution. Although apical plasma membrane pendrin abundance in the type B intercalated cell is low under basal conditions (217, 230), it rises about sixfold in response to aldosterone, primarily through subcellular redistribution (217). Moreover, the aldosterone-induced increment in apical anion exchange is virtually eliminated with pendrin gene ablation. Thus the increment in apical Cl−/ exchange observed in the type B intercalated cell of aldosterone-treated mice is primarily pendrin mediated.
Upon aldosterone binding to the MR, a series of downstream signaling events are triggered, which lead to increased abundance and function of transporters and channels, such as ENaC in principal cells, which represent the cell type in which aldosterone/MR signaling is best understood. When aldosterone binds to the principal cell MR, the receptor undergoes a conformational change, resulting in its translocation to the nucleus, thereby stimulating the transcription of genes, such as the subunits of ENaC (194). In principal cells, aldosterone as well as cortisol (or corticosterone in rodents) are MR ligands. However, if a cell expresses 11β-hydroxysteroid dehydrogenase type 2, these glucocorticoids are oxidized and inactivated, making aldosterone the preferred receptor ligand (101).
Like principal cells, the MR is highly expressed in ICs (1, 187). Moreover, MR inhibitors reduce the abundance of IC transporters such as pendrin and the H+-ATPase (187). However, a number of observations indicate that the mechanism of MR activation differs in principal cells and in ICs. While 11β-hydroxysteroid dehydrogenase type 2 is clearly expressed in principal cells, its expression is either low or absent in intercalated cells (1, 23), which implies that there is little aldosterone binding to the IC MR. While aldosterone acts as a ligand to the IC MR in heterologous expression systems, whether aldosterone is a ligand to the IC MR in vivo is not fully understood. The fall in intercalated cell pendrin abundance observed with chemical inhibitors of the MR, such as spironolactone (187), might occur from an indirect effect of MR blockade, such as through changes in serum K+ (207). However, the effect of serum K+ and K+ balance on pendrin abundance has varied between studies (121, 154, 155, 248).
Shibata et al. (187) reported that the IC MR is phosphorylated at S843, which represents a phosphorylation site unique to this cell type. When phosphorylated at S843, the mouse intercalated cell MR binds aldosterone poorly, thereby blunting aldosterone-induced IC MR activation (FIGURE 7). Conversely, IC MR S843 dephosphorylation enhances aldosterone binding to and activation of the receptor. This dephosphorylation event is enhanced with either angiotensin II or with aldosterone-induced hypokalemia. Therefore, in treatment models associated with vascular volume contraction, such as dietary NaCl restriction, angiotensin II production increases, which dephosphorylates the IC MR at S843, thereby augmenting aldosterone binding to, and thus activation of, the IC MR, which increases pendrin and H+-ATPase total protein abundance. Because aldosterone-induced S843 MR dephosphorylation is greater with angiotensin II, but reduced with hyperkalemia (187), angiotensin II augments, whereas hyperkalemia attenuates, aldosterone-induced intercalated cell MR activation. WNK4 also modulates this IC MR S843 phosphorylation. Gain-of-function WNK4 mutations, such as seen in mouse models of pseudohypoaldosteronism type II (PHA II), increase IC MR S843 phosphorylation, whereas WNK4 gene ablation promotes receptor dephosphorylation and thus its activation (187). Whether WNK4 modulates IC MR phosphorylation through changes in angiotensin II or serum K+ is, however, unknown.
FIGURE 7.

Mechanism of intercalated cell (IC) mineralocorticoid receptor (MR) activation. When the IC MR is phosphorylated at S843, aldosterone binding to the MR is inhibited. However, angiotensin II acts through the angiotensin type 1 receptor (AT1R) and mammalian target of rapamycin (mTOR) to phosphorylate, and thereby inactivate, ULK1, a kinase that mediates MR S843 phosphorylation. By inhibiting ULK1, S843 phosphorylation does not occur, which facilitates aldosterone binding to and thus activation of the IC MR. Upon aldosterone binding, the IC MR is translocated to the nucleus, which promotes transcription of IC transporters such as pendrin.
To ascertain the kinase responsible for IC MR S843 dephosphorylation, Shibata et al. (186) used a high-throughput screening assay to test 197 candidate kinases and observed that the principal kinase mediating this MRS843 phosphorylation is unc-51-like kinase 1 (ULK1), which is highly expressed in intercalated cells. They went on to show that angiotensin II acts through the angiotensin type 1 receptor to activate mammalian target of rapamycin (mTOR), which phosphorylates, and thereby inactivates, ULK1 (FIGURE 7). As such, angiotensin II acts through the angiotensin type 1 receptor to inactivate ULK1, and thereby dephosphorylate the IC MR at S843, which facilitates aldosterone binding to and thus activation of the IC MR (186).
C. Regulation of Pendrin by Nedd4-2
The signaling mechanism by which aldosterone regulates ENaC in principal cells is quite well understood (FIGURE 8). In this cell type, when ENaC associates with the ubiquitin ligase Nedd4-2, the channel is ubiquitylated and then endocytosed and degraded (139). However, this association is blunted when Nedd4-2 is phosphorylated, which occurs following phosphorylation of the kinase, Sgk1 (139). Aldosterone increases Sgk1 transcription, whereas insulin and possibly angiotensin II phosphorylate, and thereby activate Sgk1 (139).
FIGURE 8.
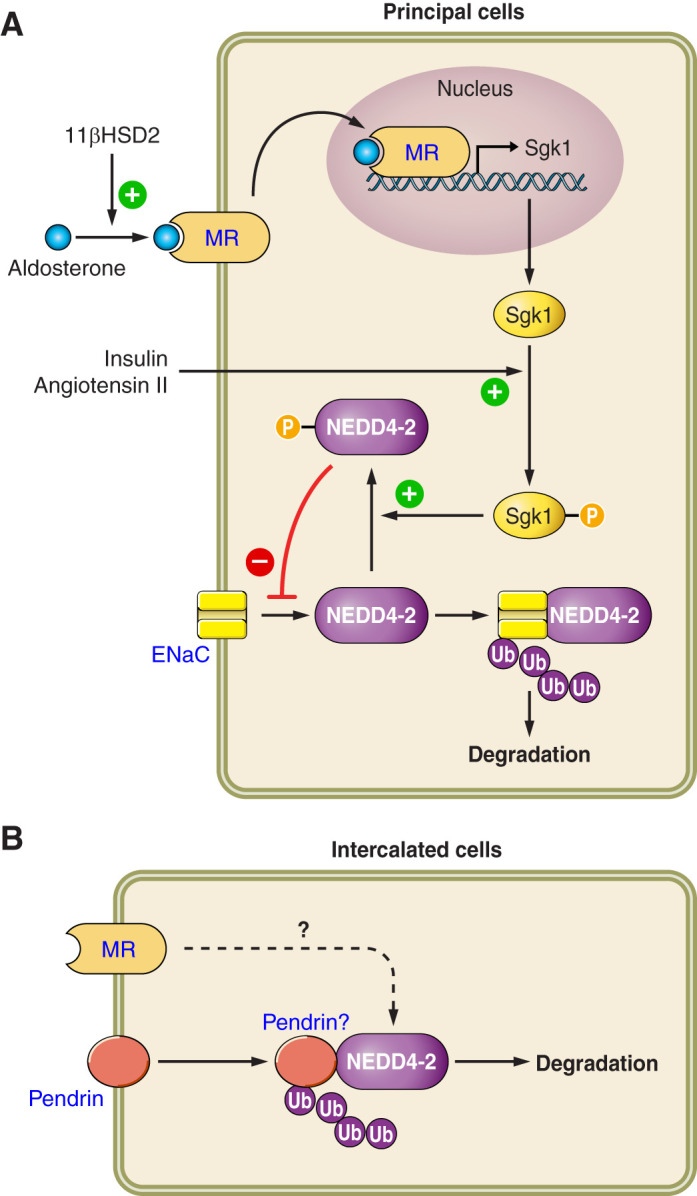
The regulation of intercalated cell (IC) and principal cell function by Nedd4-2. In principal cells, the ubiquitin ligase Nedd4-2 associates with epithelial Na+ channel (ENaC), which promotes the ubiquitylation and thus degradation of this Na+ channel. Nedd4-2 is activated through dephosphorylation, which enables it to associate with target proteins such as ENaC. Conversely, when phosphorylated, which occurs with the phosphoactivation of Sgk1 that follows insulin or angiotensin II application, Nedd4-2 cannot associate with these target proteins. When a cell expresses 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2), aldosterone can bind to the mineralocorticoid receptor (MR), resulting in its translocation to the nucleus, which induces gene transcription. In ICs, Nedd4-2 regulates pendrin subcellular distribution. Whether this occurs through a direct association of Nedd4-2 and pendrin, and whether Nedd4-2 acts downstream of the MR, remains to be determined.
Because intercalated cell function is markedly stimulated by aldosterone and because Nedd4-2 represents a step in aldosterone’s signaling cascade, we explored the effect of intercalated cell Nedd4-2 gene ablation on intercalated cell transporter abundance and function (FIGURE 7) (128). In NaCl-replete mice, IC Nedd4-2 gene ablation markedly stimulated Cl− absorption and secretion, consistent with apical anion exchange stimulation, although both the benzamil- and the thiazide-sensitive components of Cl− absorption were unchanged (128). Because apical Cl−/ exchange occurs through the action of AE4, the H+-ATPase and ClC-K2/barttin Cl− channels on the basolateral plasma membrane, which act in series with pendrin and NDCBE on the apical plasma membrane, we examined the effect of IC Nedd4-2 gene ablation on the abundance of each of these transporters (128). While IC Nedd4-2 gene ablation did not affect AE4, H+-ATPase, ClC-K2/barttin, or NDCBE abundance, it increased apical plasma membrane pendrin abundance by about twofold.
Nedd4-2 modulates both rodent and human blood pressure (81, 185, 244). We therefore asked if ICs contribute to Nedd4-2-dependent changes in blood pressure (128) and observed that Nedd4-2 gene ablation within intercalated cells increased blood pressure by ~10 mmHg (128). Further experiments confirmed the role of Nedd4-2 within ICs in blood pressure regulation using an independent experimental approach (128). We hypothesized that if Nedd4-2 gene ablation increases blood pressure, in part, by upregulating pendrin, then pendrin gene ablation should lower blood pressure more in mice that are global Nedd4-2 null than in mice harboring wild-type Nedd4-2. To test this hypothesis, we measured blood pressure in wild-type, global pendrin-null, global Nedd4-2-null, and mice that were null for both pendrin and Nedd4-2. In mice expressing wild-type Nedd4-2, blood pressure was similar in mice harboring wild-type pendrin (Nedd4-2+/+; Slc26a4+/+) relative to mice that were pendrin null (Nedd4-2+/+; Slc26a4−/−). However, in mice that were Nedd4-2 null, blood pressure was significantly lower in those that were pendrin null (Nedd4-2−/−; Slc26a4 −/−) relative to mice expressing wild-type pendrin (Nedd4-2−/−; Slc26a4+/+). We conclude that pendrin abundance and function are regulated either directly or indirectly by IC Nedd4-2 and that Nedd4-2 expressed within intercalated cells modulates blood pressure. What remains to be determined is if the increment in apical Cl−/ exchange observed with IC Nedd4-2 gene ablation occurs, at least in part, through stimulation of another IC transporter and what IC transporters directly associate with and are ubiquitinylated by Nedd4-2.
VII. ROLE OF PENDRIN-POSITIVE INTERCALATED CELLS IN K+ BALANCE AND THE IMPACT OF WNK KINASES IN THIS RESPONSE
In CCDs from mice receiving a NaCl-replete diet and aldosterone, Cl− absorption is unaffected by thiazides, but nearly obliterated with the application of an ENaC inhibitor (benzamil) (143). Since ENaC stimulates K+ secretion by generating a lumen-negative transepithelial voltage (233), NaCl absorption and K+ secretion increase when ENaC is upregulated, such as following a NaCl-restricted, high-K+ diet.
In CCDs from mice given a NaCl-deficient diet, thiazide application in vitro markedly reduces NaCl absorption, although transepithelial K+ transport is unchanged (104). This thiazide-sensitive pathway has been attributed to pendrin and NDCBE acting in tandem to mediate electroneutral NaCl absorption (104). As such, the pendrin/NDCBE transporter complex is felt to mediate K+-sparing NaCl absorption (104). In support of this hypothesis, Xu et al. (248) observed that serum K+ is lower in aldosterone-treated pendrin-null relative to wild-type mice. They concluded that pendrin attenuates the hypokalemia that follows aldosterone administration. However, further study is required since other studies have not shown a difference in K+ concentration between wild-type and pendrin-null mice after treatment with an aldosterone analogue (217).
Pendrin- and NDCBE-mediated NaCl absorption is thought to be significant during volume contraction where renal conservation of Na+, K+, and Cl− are needed. Sinning et al. (190) have shown that following dietary NaCl restriction, serum K+ is much lower in mice that are both NDCBE and NCC null relative to mice that are only NCC null. They concluded that in the absence of pendrin/NDCBE-mediated electroneutral NaCl absorption, vascular volume contraction is observed, which stimulates ENaC, thereby increasing the driving force for K+ secretion, which generates hypokalemia. Whether mice with NDCBE gene ablation develop hypokalemia from an inability of the kidney to fully conserve K+ remains unknown.
In response to either hyperkalemia or hypovolemia, the adrenal cortex increases aldosterone production. However, the body’s need for renal NaCl absorption versus K+ secretion differs dramatically in these two situations and is thus referred to as the “aldosterone paradox” (6). The kidney responds to these differing demands through the regulated release of angiotensin II and through the action of WNK kinases (6). The interaction of pendrin and WNK kinases has been the topic of great interest in the recent literature (107). Insight into the pathophysiology of this aldosterone paradox has been provided by the study of patients with PHA II (245), a rare genetic disorder characterized by hypertension, hyperchloremic metabolic acidosis, and hyperkalemia, which is corrected with thiazide diuretics (245). Schambelan et al. (171) observed that the pathogenesis of PHA II involves increased Cl− absorption in the distal nephron. In 2001, PHA II was found to occur from mutations in genes encoding 2 serine-threonine kinases in the WNK family, i.e., WNK1 and WNK4 (245). WNK1 and WNK4 are expressed in the DCT and also in type B ICs (131, 245). Subsequent studies showed that the increase in distal Cl− absorption that occurs in PHA II occurs in large part through the thiazide-sensitive NaCl cotransporter of the DCT (115). However, the observation that WNK4 is expressed in type B ICs prompted Lopez-Cayuqueo et al. (107) to explore the role of this IC subtype in the pathogenesis of PHA II. WNK4 expression is greatly increased in mouse models of PHA II (TgWnk4PHAII mice), which harbor the disease-producing WNK4 missense mutation (WNK4Q562E) seen in human PHA II (107). Moreover, this mouse model recapitulates the human phenotype by displaying hyperkalemia, hypertension, and metabolic acidosis. In the CCD of these mice, increased electroneutral NaCl absorption and increased apical Cl−/ exchange are observed, although K+ flux is unchanged, as expected with increased electroneutral, pendrin-NDCBE-mediated NaCl transport. Moreover, the hyperkalemia, metabolic acidosis, and hypertension observed in the TgWnk4PHAII mice are abolished when these mutant mice are rendered pendrin null. Thus the PHA II phenotype observed in the TgWnk4PHAII mice is pendrin-dependent. The interaction of pendrin and WNK1, however, is unexplored.
VIII. ROLE OF PENDRIN-POSITIVE INTERCALATED CELLS IN I− BALANCE
In heterologous expression systems, pendrin acts as an electroneutral Cl−/I−, Cl−/, and I−/I− exchanger (181, 184, 192). In these cell systems, pendrin-mediated Cl−/I− exchange results primarily in Cl− absorption and I− secretion (50, 250). Similarly, in the parotid gland, pendrin-mediated Cl−/I− exchange brings about the secretion of I− (184). While the mechanism by which pendrin modulates I− transport in the thyroid gland is unresolved, the hallmark of Pendred syndrome is inappropriate I− discharge upon stimulation with perchlorate or thiocyanate (164).
In humans, most iodine elimination occurs through the urinary excretion of inorganic I− (236). I− is filtered at the glomerulus and absorbed along the nephron through both active and passive mechanisms (236). While renal I− clearance changes in tandem with the glomerular filtration rate (GFR), I− clearance is lower than GFR due to renal I− absorption (236). However, transepithelial I− transport has not been measured directly in any renal segment. Instead, I−/ and Cl−/ exchange were quantified in rabbit CCDs perfused in vitro, by measuring changes in intercalated cell pHi in the presence or absence of I− or Cl− in the lumen or bath (37, 114). These studies demonstrate apical Cl−/ and I−/ exchange within intercalated cells of rabbit CCD (37, 114).
Because pendrin is an apical anion exchanger that acts as an I− porter (37, 114, 164), we asked if renal pendrin abundance and I− balance regulate one or the other (97). We observed no change in pendrin total protein abundance or subcellular distribution when daily dietary I− intake was varied between 0.9 and 13 μg/day, which represents the physiological range of dietary I− intake for a mouse (97). However, pendrin gene ablation markedly altered I− balance. Both total and inorganic serum I− concentration were lower in the pendrin-null than in wild-type mice, likely due to the greater I− clearance observed in the pendrin-null than in the wild-type mice. Moreover, with increased water consumption, a greater difference between pendrin-null and wild-type mice in I− clearance was observed (97). These data show that pendrin modulates I− balance, likely through pendrin-mediated renal I− absorption, and that as water intake rises, pendrin becomes increasingly critical in the maintenance of I− balance.
While pendrin-null mice develop marked hypoiodinemia during dietary I− restriction, they do not become hypothyroid, nor do they develop goiter (21, 41, 97). In contrast, ~50% of people with Pendred syndrome develop goiter, which is exacerbated with low dietary I− intake (243). Thus, while inactivation mutations in Slc26a4 in mice and in people produce not dissimilar changes in ear and kidney function, these mouse models do not recapitulate the thyroid disease seen in people with Pendred syndrome.
IX. PENDRIN REGULATION OF AND BY THE SYMPATHETIC NERVOUS SYSTEM
The sympathetic nervous system is well known to participate in the pathogenesis of hypertension, including the pressor response to aldosterone (8). While pendrin has not been detected in sympathetic nerves, pendrin expression and function in kidney is regulated by catecholamines and their receptors. β2 Adrenergic receptor transcript is highly expressed in type B ICs (23). Moreover, in CCDs perfused in vitro, NaCl absorption (8) and apical Cl−/ exchange increase (66) with application of the β agonist isoproterenol (1 μM) to the bath, which are accompanied by increased pendrin abundance in the region of the apical plasma membrane (8). These results indicate that β agonists increase type B IC apical Cl−/ and increase pendrin expression in the region of the apical plasma membrane. However, the role of IC β adrenergic receptors in blood pressure regulation is not known.
Blood pressure is regulated by an interaction between the brain, the adrenal gland, the kidney, and the cardiovascular system by way of the sympathetic nervous system. Although we have not observed pendrin expression either in the central nervous system or in sympathetic nerves, we did observe pendrin protein expression in both epinephrine- and norepinephrine-producing chromaffin cells of the rat and mouse adrenal medulla, where it modulates catecholamine release (102). While plasma epinephrine and norepinephrine concentrations were similar in pendrin-null and wild-type mice under basal, unstressed conditions, after 20 min of immobilization stress, catecholamine concentrations were higher in pendrin-null than in wild-type mice (102). Plasma norepinephrine (NE) concentration remained higher in the pendrin-null relative to wild-type mice 30 min after relief of stress (134). Moreover, while blood pressure was lower in pendrin-null than in wild-type mice before and during immobilization stress, blood pressure was similar in the wild-type and mutant mice 30 min after relief of stress (102). Thus it took longer for norepinephrine concentration to return to basal levels in the pendrin-null than in wild-type mice following relief from stress, which likely attenuated the blood pressure difference between the wild-type and mutant mice (134). Thus the increase in plasma catecholamine concentration seen in pendrin-null mice in models of stress may limit the fall in blood pressure that occurs in the absence of renal pendrin-mediated Cl− absorption. Moreover, there may be an interaction of pendrin in different tissues by way of the sympathetic nervous system.
X. PARACRINE REGULATION OF PENDRIN-POSITIVE INTERCALATED CELLS: ROLE OF α-KETOGLUTARATE
Blood pressure is regulated by renal transporters, such as ENaC, the thiazide-sensitive NCC, and pendrin. As mentioned above, however, ablation of the genes encoding pendrin (Slc26a4), NDCBE (Slc4a8), or NCC (Slc12a3) produces either no change or a relatively small change in blood pressure, at least under basal conditions (102, 173, 190). Moreover, under basal conditions, relatively little change in salt balance or blood pressure is observed with ablation of the gene encoding the serine/threonine kinase SPAK, which phosphorylates and thereby activates NCC (116, 249). Similarly, the effect of SPAK sequence variants on blood pressure in people is debated (124). This mild phenotype can occur through compensatory mechanisms that involve coordinated induction of transport systems from multiple genes and their respective signaling pathways. These networks may involve paracellular signaling between neighboring cell types or between different nephron segments. For example, the proximal tubule produces and then releases α-ketoglutarate (αKG) into the luminal fluid, which stimulates transporter expression downstream in the collecting duct (56).
αKG is produced in the proximal tubule through a highly regulated process. Urinary αKG concentration thus varies over several orders of magnitude, depending on the treatment model employed. During metabolic acidosis, achieved by the administration of NH4Cl, urinary αKG concentration is 3 μM, which is roughly similar to the αKG concentration of ~12 μM found in plasma (208). However, in models of metabolic alkalosis, such as with NaHCO3 administration, urinary αKG concentration rises ~3 orders of magnitude to 36 mM, although plasma αKG concentration remains much lower at ~30 μM (208). Thus models of metabolic alkalosis increase αKG excretion (208).
αKG is the natural ligand for the OXGR1 receptor (69), which localizes to the apical plasma membrane of type B and non-A, non-B ICs (208). Since the EC50 of the OXGR1 receptor for αKG is 32–69 μM (69), the αKG concentration expected in the luminal fluid of the mouse CCD is high enough to activate the receptor in vivo (103). Tokonami et al. (208) explored the effect of αKG application in vitro on IC function in the mouse CCD. In mouse CCDs perfused in vitro, Cl− absorption and apical Cl−/ exchange increase with the application of αKG to the luminal fluid at physiological concentrations, i.e., ~1 mM. However, this αKG-induced increment in Cl− absorption and Cl−/ exchange are eliminated with OXGR1 ablation (208).
NaHCO3 administration increases distal delivery of αKG, which in turn stimulates type B IC apical Cl−/ exchange. By increasing αKG production by the proximal tubule, metabolic alkalosis may stimulate type B IC anion exchange-mediated secretion, thereby helping to correct the alkalosis (57).
Because NaCl balance is similar in SPAK-null and wild-type mice, at least under basal conditions, Grimm et al. (56) were prompted to explore the compensatory mechanisms that occur in vivo, which helps these mutant mice maintain NaCl balance. With SPAK gene ablation they observed an increase in type B IC transporter abundance, particularly pendrin, AE4, NDCBE, and the H+-ATPase in the SPAK KO mice. Moreover, jagged 1/NOTCH signaling was induced in kidneys from these mutant mice, which contributed to the to the fall in type A ICs abundance and to the increased abundance of principal cells as well as type B and non-A, non-B ICs seen in these mice. Since αKG increases Cl− absorption in mouse CCDs in vitro (56, 208), SPAK gene ablation might increase type B IC transporter abundance and function by increasing the distal delivery of αKG.
Further studies by our laboratory sought to determine the signaling mechanism by which αKG increases apical Cl−/ exchange (FIGURE 9) (103). In particular, we asked if the increment in apical anion exchange observed with αKG application is pendrin-mediated. We therefore compared the effect of αKG on Cl− absorption in CCDs from wild-type and from pendrin-null mice and observed that while αKG increased Cl− absorption in CCDs from wild-type mice, it was without effect in CCDs from pendrin-null mice (103), indicating that αKG-induced Cl− absorption is pendrin-dependent. Since SPAK gene ablation increases the abundance of type B intercalated cell transporters and increases Cl− absorption in the CCD, we asked if SPAK gene ablation magnifies the effect of αKG on Cl− absorption in the mouse CCD. We observed, however, that while SPAK gene ablation upregulates pendrin and increases Cl− absorption in the CCD (56), it does not magnify the effect of αKG on Cl− absorption in this segment (103).
FIGURE 9.
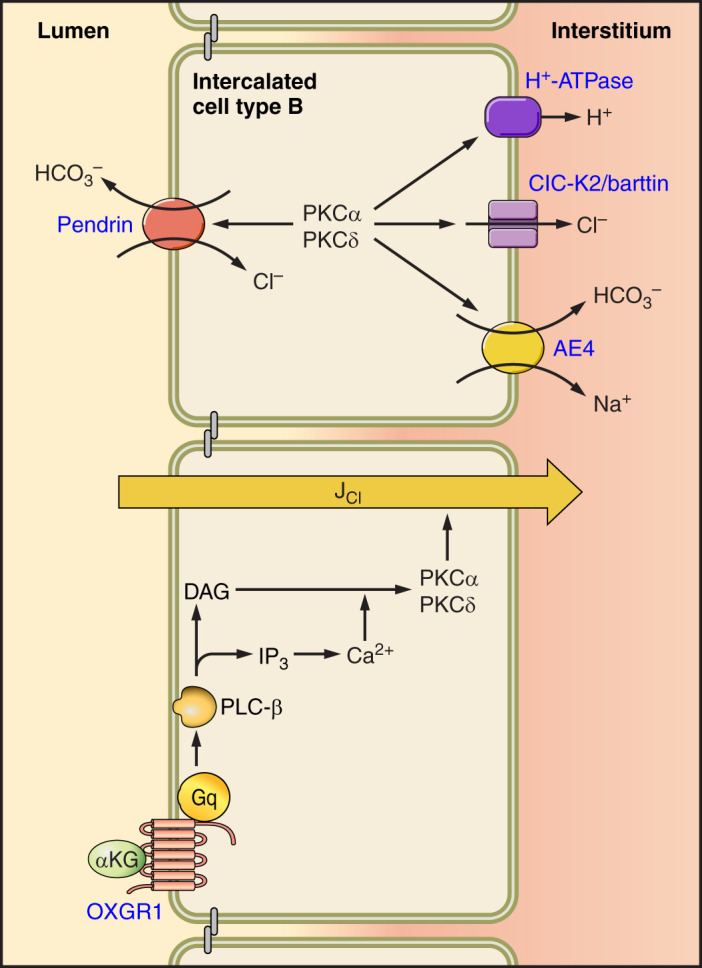
α-Ketoglutarate (αKG) signal transduction pathway in intercalated cells (ICs). αKG acts on the OXGR1 receptor, which stimulates protein kinase C (PKC)-α through a Ca2+-dependent mechanism and stimulates PKC-δ to increase pendrin-dependent Cl− absorption. The role of other type B IC transporters, known to modulate apical anion exchange, in αKG-induced Cl− absorption is unknown. DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; PLC, phospholipase C. [Modified from Lazo-Fernandez et al. (103).]
Since the OXGR1 receptor is a G protein-coupled receptor (GPCR) and since GPCRs are often stimulated by protein kinase C (PKC) (103), we asked if αKG stimulates Cl− absorption in the mouse CCD through a PKC-dependent mechanism (103). If so, the αKG-induced increment in Cl− absorption should be mimicked by PKC agonist application, but blocked with either a PKC inhibitor or with ablation of one or more of the genes encoding the PKC isoforms expressed in ICs, i.e., PKC-α, -β1, or -δ (94, 156). Like αKG, PKC agonist application increased Cl− absorption in the mouse CCD (103). However, in the presence of a PKC inhibitor or with ablation of the genes encoding either PKC-α or PKC-δ, αKG did not increase Cl− absorption (103). In addition, αΚG had no effect on Cl− absorption in the presence of the Ca2+ chelator BAPTA. We conclude that αKG stimulates Cl− absorption through a PKC-dependent mechanism that depends on PKC-α and -δ. FIGURE 9 shows the signaling cascade through which αKG stimulates apical Cl−/ exchange in the type B IC.
XI. CONCLUSIONS
Pendrin is an electroneutral, Na+-independent, Cl−/, Cl−/I−, and I−/I− exchanger that localizes to several tissues including inner ear, kidney, thyroid, and adrenal gland. In kidney, pendrin localizes to type B and non-A, non-B intercalated cells, where it mediates Cl− absorption and secretion. Pendrin-positive ICs are stimulated in many rodent treatment models in which metabolic alkalosis is observed, such as following an aldosterone infusion or the administration of NaHCO3. In so doing, pendrin-dependent secretion increases, thereby helping to correct the alkalosis. However, likely of more significance is the role that these cells play in renal Na+ and Cl− absorption and blood pressure regulation. Angiotensin II and aldosterone increase renal NaCl absorption in part by increasing apical plasma membrane pendrin abundance. Aldosterone upregulates pendrin, possibly by activating the IC MR, which has properties unique to ICs. Aldosterone stimulates pendrin, which then increases ENaC abundance and function, in part through pendrin-dependent changes in luminal and ATP. Thus aldosterone stimulates ENaC through both direct as well as through indirect pendrin-dependent mechanisms. IC transporters, such as pendrin, are also regulated by more proximal nephron segments, through mechanisms that involve paracrine signaling. Pendrin modulates blood pressure, particularly the pressor response to aldosterone. Whether pendrin changes blood pressure, at least in part, through its action outside kidney, such as in the adrenal medulla, remains to be determined.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK119793 (to S. M. Wall) and by a minority supplement to DK110375 (to C. A. Romero).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: S. M. Wall, Renal Division, Emory University School of Medicine, WMB Rm. 338, 1639 Pierce Dr. NE, Atlanta, GA 30322 (e-mail: smwall@emory.edu).
REFERENCES
- 1.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–F1485, 2010. doi: 10.1152/ajprenal.00437.2010. [DOI] [PubMed] [Google Scholar]
- 2.Al-Awqati Q, Gao XB. Differentiation of intercalated cells in the kidney. Physiology (Bethesda) 26: 266–272, 2011. doi: 10.1152/physiol.00008.2011. [DOI] [PubMed] [Google Scholar]
- 3.Almeida AJ, Burg MB. Sodium transport in the rabbit connecting tubule. Am J Physiol Renal Physiol 243: F330–F334, 1982. doi: 10.1152/ajprenal.1982.243.4.F330. [DOI] [PubMed] [Google Scholar]
- 4.Alper SL, Natale J, Gluck S, Lodish HF, Brown D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci USA 86: 5429–5433, 1989. doi: 10.1073/pnas.86.14.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, Soleimani M. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl−/ exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol 299: C33–C41, 2010. doi: 10.1152/ajpcell.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appel B, Givan LA, Eisen JS. Delta-Notch signaling and lateral inhibition in zebrafish spinal cord development. BMC Dev Biol 1: 13, 2001. doi: 10.1186/1471-213X-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arroyo JP, Gamba G. Advances in WNK signaling of salt and potassium metabolism: clinical implications. Am J Nephrol 35: 379–386, 2012. doi: 10.1159/000337479. [DOI] [PubMed] [Google Scholar]
- 7.Atkins JL, Burg MB. Bicarbonate transport by isolated perfused rat collecting ducts. Am J Physiol Renal Physiol 249: F485–F489, 1985. doi: 10.1152/ajprenal.1985.249.4.F485. [DOI] [PubMed] [Google Scholar]
- 8.Azroyan A, Morla L, Crambert G, Laghmani K, Ramakrishnan S, Edwards A, Doucet A. Regulation of pendrin by cAMP: possible involvement in β-adrenergic-dependent NaCl retention. Am J Physiol Renal Physiol 302: F1180–F1187, 2012. doi: 10.1152/ajprenal.00403.2011. [DOI] [PubMed] [Google Scholar]
- 9.Bagnis C, Marshansky V, Breton S, Brown D. Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibition. Am J Physiol Renal Physiol 280: F437–F448, 2001. doi: 10.1152/ajprenal.2001.280.3.F437. [DOI] [PubMed] [Google Scholar]
- 10.Bastani B, Purcell H, Hemken P, Trigg D, Gluck S. Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. J Clin Invest 88: 126–136, 1991. doi: 10.1172/JCI115268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension 41: 1143–1150, 2003. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- 12.Blake J, Rosenblum ND. Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol 36: 2–12, 2014. doi: 10.1016/j.semcdb.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 13.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergström G, Enerbäck S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 113: 1560–1570, 2004. doi: 10.1172/JCI20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bourgeois S, Bettoni C, Baron S, Wagner CA. Haploinsufficiency of the Mouse Atp6v1b1 Gene Leads to a Mild Acid-Base Disturbance with Implications for Kidney Stone Disease. Cell Physiol Biochem 47: 1095–1107, 2018. doi: 10.1159/000490186. [DOI] [PubMed] [Google Scholar]
- 15.Breyer MD, Jacobson HR. Regulation of rabbit medullary collecting duct cell pH by basolateral Na+/H+ and Cl−/base exchange. J Clin Invest 84: 996–1004, 1989. doi: 10.1172/JCI114264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks HL, Allred AJ, Beutler KT, Coffman TM, Knepper MA. Targeted proteomic profiling of renal Na+ transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension 39: 470–473, 2002. doi: 10.1161/hy02t2.102959. [DOI] [PubMed] [Google Scholar]
- 17.Brown D, Gluck S, Hartwig J. Structure of the novel membrane-coating material in proton-secreting epithelial cells and identification as an H+ATPase. J Cell Biol 105: 1637–1648, 1987. doi: 10.1083/jcb.105.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown D, Hirsch S, Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature 331: 622–624, 1988. doi: 10.1038/331622a0. [DOI] [PubMed] [Google Scholar]
- 19.Brown D, Hirsch S, Gluck S. Localization of a proton-pumping ATPase in rat kidney. J Clin Invest 82: 2114–2126, 1988. doi: 10.1172/JCI113833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burg M, Grantham J, Abramow M, Orloff J. Preparation and study of fragments of single rabbit nephrons. Am J Physiol 210: 1293–1298, 1966. doi: 10.1152/ajplegacy.1966.210.6.1293. [DOI] [PubMed] [Google Scholar]
- 21.Calebiro D, Porazzi P, Bonomi M, Lisi S, Grindati A, De Nittis D, Fugazzola L, Marinò M, Bottà G, Persani L. Absence of primary hypothyroidism and goiter in Slc26a4 (-/-) mice fed on a low iodine diet. J Endocrinol Invest 34: 593–598, 2011. doi: 10.3275/7262. [DOI] [PubMed] [Google Scholar]
- 22.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hübner CA, Eladari D. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 110: 7928–7933, 2013. doi: 10.1073/pnas.1221496110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci USA 114: E9989–E9998, 2017. doi: 10.1073/pnas.1710964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cil O, Haggie PM, Phuan PW, Tan JA, Verkman AS. Small-Molecule Inhibitors of Pendrin Potentiate the Diuretic Action of Furosemide. J Am Soc Nephrol 27: 3706–3714, 2016. doi: 10.1681/ASN.2015121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA. Representation and relative abundance of cell-type selective markers in whole-kidney RNA-Seq data. Kidney Int 95: 787–796, 2019. doi: 10.1016/j.kint.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costanzo LS. Comparison of calcium and sodium transport in early and late rat distal tubules: effect of amiloride. Am J Physiol Renal Physiol 246: F937–F945, 1984. doi: 10.1152/ajprenal.1984.246.6.F937. [DOI] [PubMed] [Google Scholar]
- 27.Costanzo LS, Windhager EE. Calcium and sodium transport by the distal convoluted tubule of the rat. Am J Physiol Renal Physiol 235: F492–F506, 1978. doi: 10.1152/ajprenal.1978.235.5.F492. [DOI] [PubMed] [Google Scholar]
- 28.Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep 5: 513–520, 2003. doi: 10.1007/s11906-003-0060-7. [DOI] [PubMed] [Google Scholar]
- 29.De Seigneux S, Malte H, Dimke H, Frøkiaer J, Nielsen S, Frische S. Renal compensation to chronic hypoxic hypercapnia: downregulation of pendrin and adaptation of the proximal tubule. Am J Physiol Renal Physiol 292: F1256–F1266, 2007. doi: 10.1152/ajprenal.00220.2006. [DOI] [PubMed] [Google Scholar]
- 30.Drenckhahn D, Schlüter K, Allen DP, Bennett V. Colocalization of band 3 with ankyrin and spectrin at the basal membrane of intercalated cells in the rat kidney. Science 230: 1287–1289, 1985. doi: 10.1126/science.2933809. [DOI] [PubMed] [Google Scholar]
- 31.El-Dahr SS, Li Y, Liu J, Gutierrez E, Hering-Smith KS, Signoretti S, Pignon JC, Sinha S, Saifudeen Z. p63+ ureteric bud tip cells are progenitors of intercalated cells. JCI Insight 2: e89996, 2017. doi: 10.1172/jci.insight.89996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elalouf J-M, Roinel N, de Rouffignac C. Effects of antidiuretic hormone on electrolyte reabsorption and secretion in distal tubules of rat kidney. Pflugers Arch 401: 167–173, 1984. doi: 10.1007/BF00583877. [DOI] [PubMed] [Google Scholar]
- 33.Ellison DH, Velázquez H, Wright FS. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest 83: 113–126, 1989. doi: 10.1172/JCI113847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellison DH, Velázquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol Renal Physiol 253: F546–F554, 1987. doi: 10.1152/ajprenal.1987.253.3.F546. [DOI] [PubMed] [Google Scholar]
- 35.Emmons C. Functional characterization of anion exchange in mouse cortical collecting duct (CCD) intercalated cells (ICs). J Am Soc Nephrol 8: 5A, 1997. [Google Scholar]
- 36.Emmons C. H+ extrusion in mouse cortical collecting duct (CCD) intercalated cells. J Am Soc Nephrol 8: A0020, 1997. [Google Scholar]
- 37.Emmons C. Halide transport patterns of apical anion exchange in rabbit cortical collecting duct intercalated cells. Am J Physiol Renal Physiol 271: F799–F805, 1996. doi: 10.1152/ajprenal.1996.271.4.F799. [DOI] [PubMed] [Google Scholar]
- 38.Emmons C. New subtypes of rabbit CCD intercalated cells as functionally defined by anion exchange and H+-ATPase activity. J Am Soc Nephrol 4: 838, 1993. [Google Scholar]
- 39.Emmons C. Transport characteristics of the apical anion exchanger of rabbit cortical collecting duct β-cells. Am J Physiol Renal Physiol 276: F635–F643, 1999. doi: 10.1152/ajprenal.1999.276.4.F635. [DOI] [PubMed] [Google Scholar]
- 40.Emmons C, Kurtz I. Functional characterization of three intercalated cell subtypes in the rabbit outer cortical collecting duct. J Clin Invest 93: 417–423, 1994. doi: 10.1172/JCI116976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10: 153–161, 2001. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 42.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17: 411–422, 1997. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 43.Fejes-Tóth G, Náray-Fejes-Tóth A. Isolated principal and intercalated cells: hormone responsiveness and Na+-K+-ATPase activity. Am J Physiol Renal Physiol 256: F742–F750, 1989. doi: 10.1152/ajprenal.1989.256.4.F742. [DOI] [PubMed] [Google Scholar]
- 44.Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, Giebisch G, Geibel JP, Lifton RP. The B1-subunit of the H+ ATPase is required for maximal urinary acidification. Proc Natl Acad Sci USA 102: 13616–13621, 2005. doi: 10.1073/pnas.0506769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finberg KE, Wagner CA, Stehberger PA, Geibel JP, Lifton RP. Molecular cloning and characterization of Atp6v1b1, the murine vacuolar H+-ATPase B1-subunit. Gene 318: 25–34, 2003. doi: 10.1016/S0378-1119(03)00790-X. [DOI] [PubMed] [Google Scholar]
- 46.Frische S, Kwon T-H, Frøkiaer J, Madsen KM, Nielsen S. Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am J Physiol Renal Physiol 284: F584–F593, 2003. doi: 10.1152/ajprenal.00254.2002. [DOI] [PubMed] [Google Scholar]
- 47.Gao X, Eladari D, Leviel F, Tew BY, Miró-Julià C, Cheema FH, Miller L, Nelson R, Paunescu TG, McKee M, Brown D, Al-Awqati Q. Deletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. [Correction in Proc Natl Acad Sci USA 109: 18625, 2012.] Proc Natl Acad Sci USA 107: 21872–21877, 2010. doi: 10.1073/pnas.1010364107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Austt J, Good DW, Burg MB, Knepper MA. Deoxycorticosterone-stimulated bicarbonate secretion in rabbit cortical collecting ducts: effects of luminal chloride removal and in vivo acid loading. Am J Physiol Renal Physiol 249: F205–F212, 1985. doi: 10.1152/ajprenal.1985.249.2.F205. [DOI] [PubMed] [Google Scholar]
- 49.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 50.Gillam MP, Sidhaye AR, Lee EJ, Rutishauser J, Stephan CW, Kopp P. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem 279: 13004–13010, 2004. doi: 10.1074/jbc.M313648200. [DOI] [PubMed] [Google Scholar]
- 51.Gluck S, Cannon C, Al-Awqati Q. Exocytosis regulates urinary acidification in turtle bladder by rapid insertion of H+ pumps into the luminal membrane. Proc Natl Acad Sci USA 79: 4327–4331, 1982. doi: 10.1073/pnas.79.14.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gluck S, Kelly S, Al-Awqati Q. The proton translocating ATPase responsible for urinary acidification. J Biol Chem 257: 9230–9233, 1982. [PubMed] [Google Scholar]
- 53.Gong Y, Yu M, Yang J, Gonzales E, Perez R, Hou M, Tripathi P, Hering-Smith KS, Hamm LL, Hou J. The Cap1-claudin-4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc Natl Acad Sci USA 111: E3766–E3774, 2014. doi: 10.1073/pnas.1406741111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gottschalk CW, Lassiter WE, Mylle M. Localization of urine acidification in the mammalian kidney. Am J Physiol 198: 581–585, 1960. doi: 10.1152/ajplegacy.1960.198.3.581. [DOI] [PubMed] [Google Scholar]
- 55.Grichtchenko II, Choi I, Zhong X, Bray-Ward P, Russell JM, Boron WF. Cloning, characterization, and chromosomal mapping of a human electroneutral Na+-driven Cl-HCO3 exchanger. J Biol Chem 276: 8358–8363, 2001. doi: 10.1074/jbc.C000716200. [DOI] [PubMed] [Google Scholar]
- 56.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB, Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest 125: 2136–2150, 2015. doi: 10.1172/JCI78558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grimm PR, Welling PA. α-Ketoglutarate drives electroneutral NaCl reabsorption in intercalated cells by activating a G-protein coupled receptor, Oxgr1. Curr Opin Nephrol Hypertens 26: 426–433, 2017. doi: 10.1097/MNH.0000000000000353. [DOI] [PubMed] [Google Scholar]
- 58.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Cornière N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R. Renal β-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest 123: 4219–4231, 2013. doi: 10.1172/JCI63492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Q, Wang Y, Tripathi P, Manda KR, Mukherjee M, Chaklader M, Austin PF, Surendran K, Chen F. Adam10 mediates the choice between principal cells and intercalated cells in the kidney. J Am Soc Nephrol 26: 149–159, 2015. doi: 10.1681/ASN.2013070764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hafner P, Grimaldi R, Capuano P, Capasso G, Wagner CA. Pendrin in the mouse kidney is primarily regulated by Cl− excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol 295: C1658–C1667, 2008. doi: 10.1152/ajpcell.00419.2008. [DOI] [PubMed] [Google Scholar]
- 61.Hagége J, Gabe M, Richet G. Scanning of the apical pole of distal tubular cells under differing acid-base conditions. Kidney Int 5: 137–146, 1974. doi: 10.1038/ki.1974.18. [DOI] [PubMed] [Google Scholar]
- 62.Hager H, Kwon T-H, Vinnikova AK, Masilamani S, Brooks HL, Frøkiaer J, Knepper MA, Nielsen S. Immunocytochemical and immunoelectron microscopic localization of alpha-, beta-, and gamma-ENaC in rat kidney. Am J Physiol Renal Physiol 280: F1093–F1106, 2001. doi: 10.1152/ajprenal.2001.280.6.F1093. [DOI] [PubMed] [Google Scholar]
- 63.Haggie PM, Phuan PW, Tan JA, Zlock L, Finkbeiner WE, Verkman AS. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J 30: 2187–2197, 2016. doi: 10.1096/fj.201600223R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamilton KL, Moore AB. 50 Years of renal physiology from one man and the perfused tubule: Maurice B. Burg. Am J Physiol Renal Physiol 311: F291–F304, 2016. doi: 10.1152/ajprenal.00198.2016. [DOI] [PubMed] [Google Scholar]
- 65.Hanley MJ, Kokko JP. Study of chloride transport across the rabbit cortical collecting tubule. J Clin Invest 62: 39–44, 1978. doi: 10.1172/JCI109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hayashi M, Yamaji Y, Iyori M, Kitajima W, Saruta T. Effect of isoproterenol on intracellular pH of the intercalated cells in the rabbit cortical collecting ducts. J Clin Invest 87: 1153–1157, 1991. doi: 10.1172/JCI115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hays SR. Mineralocorticoid modulation of apical and basolateral membrane H+/OH-/ transport processes in the rabbit inner stripe of outer medullary collecting duct. J Clin Invest 90: 180–187, 1992. doi: 10.1172/JCI115834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hays SR, Alpern RJ. Basolateral membrane Na+-independent Cl-/ exchange in the inner stripe of the rabbit outer medullary collecting tubule. J Gen Physiol 95: 347–367, 1990. doi: 10.1085/jgp.95.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429: 188–193, 2004. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 70.Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Cornière N, Böhm D, Jentsch TJ, Chambrey R, Teulon J, Hübner CA, Eladari D. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J Am Soc Nephrol 28: 209–217, 2017. doi: 10.1681/ASN.2016010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Himmel NJ, Wang Y, Rodriguez DA, Sun MA, Blount MA. Chronic lithium treatment induces novel patterns of pendrin localization and expression. Am J Physiol Renal Physiol 315: F313–F322, 2018. doi: 10.1152/ajprenal.00065.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hirohama D, Ayuzawa N, Ueda K, Nishimoto M, Kawarazaki W, Watanabe A, Shimosawa T, Marumo T, Shibata S, Fujita T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J Am Soc Nephrol 29: 57–68, 2018. doi: 10.1681/ASN.2017030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holthöfer H, Schulte BA, Pasternack G, Siegel GJ, Spicer SS. Three distinct cell populations in rat kidney collecting duct. Am J Physiol Cell Physiol 253: C323–C328, 1987. doi: 10.1152/ajpcell.1987.253.2.C323. [DOI] [PubMed] [Google Scholar]
- 75.Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA 107: 18010–18015, 2010. doi: 10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem 278: 37073–37082, 2003. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 77.Imai M. The connecting tubule: a functional subdivision of the rabbit distal nephron segments. Kidney Int 15: 346–356, 1979. doi: 10.1038/ki.1979.46. [DOI] [PubMed] [Google Scholar]
- 78.Jacques T, Picard N, Miller RL, Riemondy KA, Houillier P, Sohet F, Ramakrishnan SK, Büsst CJ, Jayat M, Cornière N, Hassan H, Aronson PS, Hennings JC, Hübner CA, Nelson RD, Chambrey R, Eladari D. Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 24: 1104–1113, 2013. doi: 10.1681/ASN.2012080787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jennings ML. Structure and function of the red blood cell anion transport protein. Annu Rev Biophys Biophys Chem 18: 397–430, 1989. doi: 10.1146/annurev.bb.18.060189.002145. [DOI] [PubMed] [Google Scholar]
- 80.Jeong HW, Jeon US, Koo BK, Kim WY, Im SK, Shin J, Cho Y, Kim J, Kong YY. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J Clin Invest 119: 3290–3300, 2009. doi: 10.1172/JCI38416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin H-S, Hong KW, Lim J-E, Hwang S-Y, Lee S-H, Shin C, Park HK, Oh B. Genetic variations in the sodium balance-regulating genes ENaC, NEDD4L, NDFIP2 and USP2 influence blood pressure and hypertension. Kidney Blood Press Res 33: 15–23, 2010. doi: 10.1159/000275706. [DOI] [PubMed] [Google Scholar]
- 82.Kandasamy N, Fugazzola L, Evans M, Chatterjee K, Karet F. Life-threatening metabolic alkalosis in Pendred syndrome. Eur J Endocrinol 165: 167–170, 2011. doi: 10.1530/EJE-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CWRJ, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 84.Karet FE, Gainza FJ, Györy AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA 95: 6337–6342, 1998. doi: 10.1073/pnas.95.11.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kashgarian M, Biemesderfer D, Caplan M, Forbush B III. Monoclonal antibody to Na,K-ATPase: immunocytochemical localization along nephron segments. Kidney Int 28: 899–913, 1985. doi: 10.1038/ki.1985.216. [DOI] [PubMed] [Google Scholar]
- 86.Khuri RN, Strieder N, Wiederholt M, Giebisch G. Effects of graded solute diuresis on renal tubular sodium transport in the rat. Am J Physiol 228: 1262–1268, 1975. doi: 10.1152/ajplegacy.1975.228.4.1262. [DOI] [PubMed] [Google Scholar]
- 87.Kiernan AE. Notch signaling during cell fate determination in the inner ear. Semin Cell Dev Biol 24: 470–479, 2013. doi: 10.1016/j.semcdb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim BG, Kim JY, Kim HN, Bok J, Namkung W, Choi JY, Kim SH. Developmental changes of ENaC expression and function in the inner ear of pendrin knock-out mice as a perspective on the development of endolymphatic hydrops. PLoS One 9: e95730, 2014. doi: 10.1371/journal.pone.0095730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim BG, Yoo TH, Yoo JE, Seo YJ, Jung J, Choi JY. Resistance to hypertension and high Cl− excretion in humans with SLC26A4 mutations. Clin Genet 91: 448–452, 2017. doi: 10.1111/cge.12789. [DOI] [PubMed] [Google Scholar]
- 90.Kim G-H, Martin SW, Fernández-Llama P, Masilamani S, Packer RK, Knepper MA. Long-term regulation of renal Na-dependent cotransporters and ENaC: response to altered acid-base intake. Am J Physiol Renal Physiol 279: F459–F467, 2000. doi: 10.1152/ajprenal.2000.279.3.F459. [DOI] [PubMed] [Google Scholar]
- 91.Kim G-H, Masilamani S, Turner R, Mitchell C, Wade JB, Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95: 14552–14557, 1998. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim HY, Kim SS, Bae EH, Ma SK, Kim SW. Decreased Renal Expression of H+-ATPase and Pendrin in a Patient with Distal Renal Tubular Acidosis Associated with Sjögren’s Syndrome. Intern Med 54: 2899–2904, 2015. doi: 10.2169/internalmedicine.54.4821. [DOI] [PubMed] [Google Scholar]
- 93.Kim J, Kim Y-H, Cha J-H, Tisher CC, Madsen KM. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J Am Soc Nephrol 10: 1–12, 1999. [DOI] [PubMed] [Google Scholar]
- 94.Kim W-Y, Jung J-H, Park EY, Yang C-W, Kim H, Nielsen S, Madsen KM, Kim J. Expression of protein kinase C isoenzymes α, betaI, and delta in subtypes of intercalated cells of mouse kidney. Am J Physiol Renal Physiol 291: F1052–F1060, 2006. doi: 10.1152/ajprenal.00016.2006. [DOI] [PubMed] [Google Scholar]
- 95.Kim Y-H, Kwon T-H, Frische S, Kim J, Tisher CC, Madsen KM, Nielsen S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol 283: F744–F754, 2002. doi: 10.1152/ajprenal.00037.2002. [DOI] [PubMed] [Google Scholar]
- 96.Kim Y-H, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin W, Verlander JW, Sutliff RL, Wall SM. Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am J Physiol Renal Physiol 293: F1314–F1324, 2007. doi: 10.1152/ajprenal.00155.2007. [DOI] [PubMed] [Google Scholar]
- 97.Kim Y-H, Pham TD, Zheng W, Hong S, Baylis C, Pech V, Beierwaltes WH, Farley DB, Braverman LE, Verlander JW, Wall SM. Role of pendrin in iodide balance: going with the flow. Am J Physiol Renal Physiol 297: F1069–F1079, 2009. doi: 10.1152/ajprenal.90581.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim Y-H, Verlander JW, Matthews SW, Kurtz I, Shin W, Weiner ID, Everett LA, Green ED, Nielsen S, Wall SM. Intercalated cell H+/OH− transporter expression is reduced in Slc26a4 null mice. Am J Physiol Renal Physiol 289: F1262–F1272, 2005. doi: 10.1152/ajprenal.00206.2005. [DOI] [PubMed] [Google Scholar]
- 99.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol Renal Physiol 249: F870–F877, 1985. doi: 10.1152/ajprenal.1985.249.6.F870. [DOI] [PubMed] [Google Scholar]
- 100.Kudrycki KE, Shull GE. Primary structure of the rat kidney band 3 anion exchange protein deduced from a cDNA. J Biol Chem 264: 8185–8192, 1989. [PubMed] [Google Scholar]
- 101.Kyossev Z, Walker PD, Reeves WB. Immunolocalization of NAD-dependent 11 beta-hydroxysteroid dehydrogenase in human kidney and colon. Kidney Int 49: 271–281, 1996. doi: 10.1038/ki.1996.39. [DOI] [PubMed] [Google Scholar]
- 102.Lazo-Fernandez Y, Aguilera G, Pham TD, Park AY, Beierwaltes WH, Sutliff RL, Verlander JW, Pacak K, Osunkoya AO, Ellis CL, Kim YH, Shipley GL, Wynne BM, Hoover RS, Sen SK, Plotsky PM, Wall SM. Pendrin localizes to the adrenal medulla and modulates catecholamine release. Am J Physiol Endocrinol Metab 309: E534–E545, 2015. doi: 10.1152/ajpendo.00035.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lazo-Fernandez Y, Welling PA, Wall SM. α-Ketoglutarate stimulates pendrin-dependent Cl− absorption in the mouse CCD through protein kinase C. Am J Physiol Renal Physiol 315: F7–F15, 2018. doi: 10.1152/ajprenal.00576.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. [Correction in J Clin Invest 121: 1668, 2011.] J Clin Invest 120: 1627–1635, 2010. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lombard WE, Kokko JP, Jacobson HR. Bicarbonate transport in cortical and outer medullary collecting tubules. Am J Physiol Renal Physiol 244: F289–F296, 1983. doi: 10.1152/ajprenal.1983.244.3.F289. [DOI] [PubMed] [Google Scholar]
- 106.Lönnerholm G. Histochemical demonstration of carbonic anhydrase activity in the rat kidney. Acta Physiol Scand 81: 433–439, 1971. doi: 10.1111/j.1748-1716.1971.tb04920.x. [DOI] [PubMed] [Google Scholar]
- 107.López-Cayuqueo KI, Chavez-Canales M, Pillot A, Houillier P, Jayat M, Baraka-Vidot J, Trepiccione F, Baudrie V, Büsst C, Soukaseum C, Kumai Y, Jeunemaître X, Hadchouel J, Eladari D, Chambrey R. A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int 94: 514–523, 2018. doi: 10.1016/j.kint.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 108.Ma HP, Eaton DC. Acute regulation of epithelial sodium channel by anionic phospholipids. J Am Soc Nephrol 16: 3182–3187, 2005. doi: 10.1681/ASN.2005040434. [DOI] [PubMed] [Google Scholar]
- 109.Madeo AC, Manichaikul A, Pryor SP, Griffith AJ. Do mutations of the Pendred syndrome gene, SLC26A4, confer resistance to asthma and hypertension? J Med Genet 46: 405–406, 2009. doi: 10.1136/jmg.2008.063610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Madsen KM, Tisher CC. Cellular response to acute respiratory acidosis in rat medullary collecting duct. Am J Physiol Renal Physiol 245: F670–F679, 1983. doi: 10.1152/ajprenal.1983.245.6.F670. [DOI] [PubMed] [Google Scholar]
- 111.Madsen KM, Tisher CC. Response of intercalated cells of rat outer medullary collecting duct to chronic metabolic acidosis. Lab Invest 51: 268–276, 1984. [PubMed] [Google Scholar]
- 112.Malnic G, Klose RM, Giebisch G. Micropuncture study of distal tubular potassium and sodium transport in rat nephron. Am J Physiol 211: 529–547, 1966. doi: 10.1152/ajplegacy.1966.211.3.529. [DOI] [PubMed] [Google Scholar]
- 113.Masilamani S, Kim G-H, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matsushima Y, Muto S, Taniguchi J, Imai M. Mechanism of iodide transport in the rabbit cortical collecting duct. Clin Exp Nephrol 10: 102–110, 2006. doi: 10.1007/s10157-006-0417-8. [DOI] [PubMed] [Google Scholar]
- 115.McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol Rev 91: 177–219, 2011. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McKinney TD, Burg MB. Bicarbonate transport by rabbit cortical collecting tubules. Effect of acid and alkali loads in vivo on transport in vitro. J Clin Invest 60: 766–768, 1977. doi: 10.1172/JCI108830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McKinney TD, Davidson KK. Bicarbonate transport in collecting tubules from outer stripe of outer medulla of rabbit kidneys. Am J Physiol Renal Physiol 253: F816–F822, 1987. doi: 10.1152/ajprenal.1987.253.5.F816. [DOI] [PubMed] [Google Scholar]
- 119.Milton AE, Weiner ID. Intracellular pH regulation in the rabbit cortical collecting duct A-type intercalated cell. Am J Physiol Renal Physiol 273: F340–F347, 1997. doi: 10.1152/ajprenal.1997.273.3.F340. [DOI] [PubMed] [Google Scholar]
- 120.Milton AE, Weiner ID. Regulation of B-type intercalated cell apical anion exchange activity by CO2/. Am J Physiol Renal Physiol 274: F1086–F1094, 1998. [DOI] [PubMed] [Google Scholar]
- 121.Mohebbi N, Perna A, van der Wijst J, Becker HM, Capasso G, Wagner CA. Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. PLoS One 8: e55286, 2013. doi: 10.1371/journal.pone.0055286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mueller GM, Kashlan OB, Bruns JB, Maarouf AB, Aridor M, Kleyman TR, Hughey RP. Epithelial sodium channel exit from the endoplasmic reticulum is regulated by a signal within the carboxyl cytoplasmic domain of the alpha subunit. J Biol Chem 282: 33475–33483, 2007. doi: 10.1074/jbc.M707339200. [DOI] [PubMed] [Google Scholar]
- 123.Mukherjee M, deRiso J, Otterpohl K, Ratnayake I, Kota D, Ahrenkiel P, Chandrasekar I, Surendran K. Endogenous Notch Signaling in Adult Kidneys Maintains Segment-Specific Epithelial Cell Types of the Distal Tubules and Collecting Ducts to Ensure Water Homeostasis. J Am Soc Nephrol 30: 110–126, 2019. doi: 10.1681/ASN.2018040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Murthy M, Kurz T, O’Shaughnessy KM. WNK signalling pathways in blood pressure regulation. Cell Mol Life Sci 74: 1261–1280, 2017. doi: 10.1007/s00018-016-2402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nagalakshmi VK, Yu J. The ureteric bud epithelium: morphogenesis and roles in metanephric kidney patterning. Mol Reprod Dev 82: 151–166, 2015. doi: 10.1002/mrd.22462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nanami M, Lazo-Fernandez Y, Pech V, Verlander JW, Agazatian D, Weinstein AM, Bao HF, Eaton DC, Wall SM. ENaC inhibition stimulates HCl secretion in the mouse cortical collecting duct. I. Stilbene-sensitive Cl- secretion. Am J Physiol Renal Physiol 309: F251–F258, 2015. doi: 10.1152/ajprenal.00471.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nanami M, Pech V, Lazo-Fernandez Y, Weinstein AM, Wall SM. ENaC inhibition stimulates HCl secretion in the mouse cortical collecting duct. II. Bafilomycin-sensitive H+ secretion. Am J Physiol Renal Physiol 309: F259–F268, 2015. doi: 10.1152/ajprenal.00120.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nanami M, Pham TD, Kim YH, Yang B, Sutliff RL, Staub O, Klein JD, Lopez-Cayuqueo KI, Chambrey R, Park AY, Wang X, Pech V, Verlander JW, Wall SM. The Role of Intercalated Cell Nedd4-2 in BP Regulation, Ion Transport, and Transporter Expression. J Am Soc Nephrol 29: 1706–1719, 2018. doi: 10.1681/ASN.2017080826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nelson RD, Guo XL, Masood K, Brown D, Kalkbrenner M, Gluck S. Selectively amplified expression of an isoform of the vacuolar H+-ATPase 56-kilodalton subunit in renal intercalated cells. Proc Natl Acad Sci USA 89: 3541–3545, 1992. doi: 10.1073/pnas.89.8.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Noah TK, Shroyer NF. Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol 75: 263–288, 2013. doi: 10.1146/annurev-physiol-030212-183741. [DOI] [PubMed] [Google Scholar]
- 131.Ohno M, Uchida K, Ohashi T, Nitta K, Ohta A, Chiga M, Sasaki S, Uchida S. Immunolocalization of WNK4 in mouse kidney. Histochem Cell Biol 136: 25–35, 2011. doi: 10.1007/s00418-011-0827-x. [DOI] [PubMed] [Google Scholar]
- 132.Pácha J, Frindt G, Antonian L, Silver RB, Palmer LG. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102: 25–42, 1993. doi: 10.1085/jgp.102.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Palmer LG, Frindt G. Effects of cell Ca and pH on Na channels from rat cortical collecting tubule. Am J Physiol Renal Physiol 253: F333–F339, 1987. doi: 10.1152/ajprenal.1987.253.2.F333. [DOI] [PubMed] [Google Scholar]
- 134.Park AY, Plotsky PM, Pham TD, Pacak K, Wynne BM, Wall SM, Lazo-Fernandez Y. Blood collection in unstressed, conscious, and freely moving mice through implantation of catheters in the jugular vein: a new simplified protocol. Physiol Rep 6: e13904, 2018. doi: 10.14814/phy2.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, Moon SK, Lee SC, Chun YM, Lee HK, Choi JY, Jung SC, Griffith AJ, Koo SK. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet 67: 160–165, 2005. doi: 10.1111/j.1399-0004.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- 136.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Suszták K. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018. doi: 10.1126/science.aar2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Parker MD, Bouyer P, Daly CM, Boron WF. Cloning and characterization of novel human SLC4A8 gene products encoding Na+-driven Cl-/ exchanger variants NDCBE-A, -C, and -D. Physiol Genomics 34: 265–276, 2008. doi: 10.1152/physiolgenomics.90259.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pathare G, Dhayat N, Mohebbi N, Wagner CA, Cheval L, Neuhaus TJ, Fuster DG. Acute regulated expression of pendrin in human urinary exosomes. Pflugers Arch 470: 427–438, 2018. doi: 10.1007/s00424-017-2049-0. [DOI] [PubMed] [Google Scholar]
- 139.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pech V, Kim Y-H, Weinstein AM, Everett LA, Pham TD, Wall SM. Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. Am J Physiol Renal Physiol 292: F914–F920, 2007. doi: 10.1152/ajprenal.00361.2006. [DOI] [PubMed] [Google Scholar]
- 141.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim Y-H, Sutliff RL, Bao H-F, Eaton DC, Wall SM. Pendrin modulates ENaC function by changing luminal . J Am Soc Nephrol 21: 1928–1941, 2010. doi: 10.1681/ASN.2009121257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pech V, Thumova M, Dikalov SI, Hummler E, Rossier BC, Harrison DG, Wall SM. Nitric oxide reduces Cl− absorption in the mouse cortical collecting duct through an ENaC-dependent mechanism. Am J Physiol Renal Physiol 304: F1390–F1397, 2013. doi: 10.1152/ajprenal.00292.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pech V, Thumova M, Kim Y-H, Agazatian D, Hummler E, Rossier BC, Weinstein AM, Nanami M, Wall SM. ENaC inhibition stimulates Cl− secretion in the mouse cortical collecting duct through an NKCC1-dependent mechanism. Am J Physiol Renal Physiol 303: F45–F55, 2012. doi: 10.1152/ajprenal.00030.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pech V, Wall SM, Nanami M, Bao HF, Kim YH, Lazo-Fernandez Y, Yue Q, Pham TD, Eaton DC, Verlander JW. Pendrin gene ablation alters ENaC subcellular distribution and open probability. Am J Physiol Renal Physiol 309: F154–F163, 2015. doi: 10.1152/ajprenal.00564.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pech V, Zheng W, Pham TD, Verlander JW, Wall SM. Angiotensin II activates H+-ATPase in type A intercalated cells. J Am Soc Nephrol 19: 84–91, 2008. doi: 10.1681/ASN.2007030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pei L, Solis G, Nguyen MT, Kamat N, Magenheimer L, Zhuo M, Li J, Curry J, McDonough AA, Fields TA, Welch WJ, Yu AS. Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J Clin Invest 126: 2509–2518, 2016. doi: 10.1172/JCI83942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pela I, Bigozzi M, Bianchi B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clin Nephrol 69: 450–453, 2008. doi: 10.5414/CNP69450. [DOI] [PubMed] [Google Scholar]
- 148.Pendred V. Deaf-mutism and goitre. Lancet 148: 532, 1896. doi: 10.1016/S0140-6736(01)74403-0. [DOI] [Google Scholar]
- 149.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. J Am Soc Nephrol 13: 1131–1135, 2002. doi: 10.1097/01.ASN.0000013292.78621.FD. [DOI] [PubMed] [Google Scholar]
- 150.Pfaller W. Structure Function Correlation on Rat Kidney. New York: Springer-Verlag, 1982. [PubMed] [Google Scholar]
- 151.Pickkers P, Garcha RS, Schachter M, Smits P, Hughes AD. Inhibition of carbonic anhydrase accounts for the direct vascular effects of hydrochlorothiazide. Hypertension 33: 1043–1048, 1999. doi: 10.1161/01.HYP.33.4.1043. [DOI] [PubMed] [Google Scholar]
- 152.Pinelli L, Nissant A, Edwards A, Lourdel S, Teulon J, Paulais M. Dual regulation of the native ClC-K2 chloride channel in the distal nephron by voltage and pH. J Gen Physiol 148: 213–226, 2016. doi: 10.1085/jgp.201611623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem 283: 36599–36607, 2008. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Procino G, Milano S, Tamma G, Dossena S, Barbieri C, Nicoletti MC, Ranieri M, Di Mise A, Nofziger C, Svelto M, Paulmichl M, Valenti G. Co-regulated pendrin and aquaporin 5 expression and trafficking in Type-B intercalated cells under potassium depletion. Cell Physiol Biochem 32: 184–199, 2013. doi: 10.1159/000356638. [DOI] [PubMed] [Google Scholar]
- 155.Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, Paillard M, Aronson PS, Eladari D. The Cl-/ exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol 287: F1179–F1188, 2004. doi: 10.1152/ajprenal.00211.2004. [DOI] [PubMed] [Google Scholar]
- 156.Redling S, Pfaff IL, Leitges M, Vallon V. Immunolocalization of protein kinase C isoenzymes alpha, beta I, beta II, delta, and epsilon in mouse kidney. Am J Physiol Renal Physiol 287: F289–F298, 2004. doi: 10.1152/ajprenal.00273.2003. [DOI] [PubMed] [Google Scholar]
- 157.Reithmeier RA, Lieberman DM, Casey JR, Pimplikar SW, Werner PK, See H, Pirraglia CA. Structure and function of the band 3 Cl-/ transporter. Ann N Y Acad Sci 574, 1 Bicarbonate: 75–83, 1989. doi: 10.1111/j.1749-6632.1989.tb25137.x. [DOI] [PubMed] [Google Scholar]
- 158.Rhodin J. Electron microscopy of the kidney. Am J Med 24: 661–675, 1958. doi: 10.1016/0002-9343(58)90373-5. [DOI] [PubMed] [Google Scholar]
- 159.Ridderstrale Y, Kashgarian M, Koeppen B, Giebisch G, Stetson D, Ardito T, Stanton B. Morphological heterogeneity of the rabbit collecting duct. Kidney Int 34: 655–670, 1988. doi: 10.1038/ki.1988.230. [DOI] [PubMed] [Google Scholar]
- 160.Rodighiero S, Bottà G, Bazzini C, Meyer G. Pendrin overexpression affects cell volume recovery, intracellular pH and chloride concentration after hypotonicity-induced cell swelling. Cell Physiol Biochem 28: 559–570, 2011. doi: 10.1159/000335120. [DOI] [PubMed] [Google Scholar]
- 161.Rosen S. Localization of carbonic anhydrase activity in the vertebrate nephron. Histochem J 4: 35–48, 1972. doi: 10.1007/BF01005267. [DOI] [PubMed] [Google Scholar]
- 162.Rothenberger F, Velic A, Stehberger PA, Kovacikova J, Wagner CA. Angiotensin II stimulates vacuolar H+-ATPase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J Am Soc Nephrol 18: 2085–2093, 2007. doi: 10.1681/ASN.2006070753. [DOI] [PubMed] [Google Scholar]
- 163.Roy A, Al-bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141: 839–845, 2000. doi: 10.1210/endo.141.2.7303. [DOI] [PubMed] [Google Scholar]
- 165.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98: 4221–4226, 2001. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sabolić I, Brown D, Gluck SL, Alper SL. Regulation of AE1 anion exchanger and H+-ATPase in rat cortex by acute metabolic acidosis and alkalosis. Kidney Int 51: 125–137, 1997. doi: 10.1038/ki.1997.16. [DOI] [PubMed] [Google Scholar]
- 168.Sabolić I, Herak-Kramberger CM, Breton S, Brown D. Na/K-ATPase in intercalated cells along the rat nephron revealed by antigen retrieval. J Am Soc Nephrol 10: 913–922, 1999. [DOI] [PubMed] [Google Scholar]
- 169.Sampogna RV, Al-Awqati Q. Salt and pepper distribution of cell types in the collecting duct. J Am Soc Nephrol 24: 163–165, 2013. doi: 10.1681/ASN.2012121183. [DOI] [PubMed] [Google Scholar]
- 170.Sauer M, Flemmer A, Thurau K, Beck F-X. Sodium entry routes in principal and intercalated cells of the isolated perfused cortical collecting duct. Pflugers Arch 416: 88–93, 1990. doi: 10.1007/BF00370227. [DOI] [PubMed] [Google Scholar]
- 171.Schambelan M, Sebastian A, Rector FC Jr. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): role of increased renal chloride reabsorption. Kidney Int 19: 716–727, 1981. doi: 10.1038/ki.1981.72. [DOI] [PubMed] [Google Scholar]
- 172.Schlatter E, Greger R, Schafer JA. Principal cells of cortical collecting ducts of the rat are not a route of transepithelial Cl− transport. Pflugers Arch 417: 317–323, 1990. doi: 10.1007/BF00370998. [DOI] [PubMed] [Google Scholar]
- 173.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J Biol Chem 273: 29150–29155, 1998. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- 174.Schuster VL. Function and regulation of collecting duct intercalated cells. Annu Rev Physiol 55: 267–288, 1993. doi: 10.1146/annurev.ph.55.030193.001411. [DOI] [PubMed] [Google Scholar]
- 175.Schuster VL, Bonsib SM, Jennings ML. Two types of collecting duct mitochondria-rich (intercalated) cells: lectin and band 3 cytochemistry. Am J Physiol Cell Physiol 251: C347–C355, 1986. doi: 10.1152/ajpcell.1986.251.3.C347. [DOI] [PubMed] [Google Scholar]
- 176.Schwartz GJ, Al-Awqati Q. Carbon dioxide causes exocytosis of vesicles containing H+ pumps in isolated perfused proximal and collecting tubules. J Clin Invest 75: 1638–1644, 1985. doi: 10.1172/JCI111871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schwartz GJ, Al-Awqati Q. Role of hensin in mediating the adaptation of the cortical collecting duct to metabolic acidosis. Curr Opin Nephrol Hypertens 14: 383–388, 2005. doi: 10.1097/01.mnh.0000172727.82993.aa. [DOI] [PubMed] [Google Scholar]
- 178.Schwartz GJ, Barasch J, Al-Awqati Q. Plasticity of functional epithelial polarity. Nature 318: 368–371, 1985. doi: 10.1038/318368a0. [DOI] [PubMed] [Google Scholar]
- 179.Schwartz GJ, Tsuruoka S, Vijayakumar S, Petrovic S, Mian A, Al-Awqati Q. Acid incubation reverses the polarity of intercalated cell transporters, an effect mediated by hensin. J Clin Invest 109: 89–99, 2002. doi: 10.1172/JCI0213292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schwartz JH, Rosen S, Steinmetz PR. Carbonic anhydrase function and the epithelial organization of H+ secretion in turtle urinary bladder. J Clin Invest 51: 2653–2662, 1972. doi: 10.1172/JCI107083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21: 440–443, 1999. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- 182.Scott WN, Shamoo YE, Brodsky WA. Carbonic anhydrase content of turtle urinary bladder mucosal cells. Biochim Biophys Acta 219: 248–250, 1970. doi: 10.1016/0005-2736(70)90084-2. [DOI] [PubMed] [Google Scholar]
- 183.Seely JC. A brief review of kidney development, maturation, developmental abnormalities, and drug toxicity: juvenile animal relevancy. J Toxicol Pathol 30: 125–133, 2017. doi: 10.1293/tox.2017-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shcheynikov N, Yang D, Wang Y, Zeng W, Karniski LP, So I, Wall SM, Muallem S. Slc26a4 functions as an electroneutral Cl−/I-/ exchanger: role of Slc26a4 and Slc26a6 in I− and secretion and in regulation of CFTR in the parotid duct. J Physiol 586: 3813–3824, 2008. doi: 10.1113/jphysiol.2008.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB, Yang B. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol 295: F462–F470, 2008. doi: 10.1152/ajprenal.90300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shibata S, Ishizawa K, Wang Q, Xu N, Fujita T, Uchida S, Lifton RP. ULK1 Phosphorylates and Regulates Mineralocorticoid Receptor. Cell Rep 24: 569–576, 2018. doi: 10.1016/j.celrep.2018.06.072. [DOI] [PubMed] [Google Scholar]
- 187.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shimizu T, Nakamura M, Imai M. Effect of S-8666 on Cl− transport in the rabbit connecting tubule perfused in vitro. Tohoku J Exp Med 164: 293–298, 1991. doi: 10.1620/tjem.164.293. [DOI] [PubMed] [Google Scholar]
- 189.Shipway A, Danahay H, Williams JA, Tully DC, Backes BJ, Harris JL. Biochemical characterization of prostasin, a channel activating protease. Biochem Biophys Res Commun 324: 953–963, 2004. doi: 10.1016/j.bbrc.2004.09.123. [DOI] [PubMed] [Google Scholar]
- 190.Sinning A, Radionov N, Trepiccione F, López-Cayuqueo KI, Jayat M, Baron S, Cornière N, Alexander RT, Hadchouel J, Eladari D, Hübner CA, Chambrey R. Double Knockout of the Na+-Driven Cl-/ Exchanger and Na+/Cl− Cotransporter Induces Hypokalemia and Volume Depletion. J Am Soc Nephrol 28: 130–139, 2017. doi: 10.1681/ASN.2015070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, Amlal H. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 109: 13368–13373, 2012. doi: 10.1073/pnas.1202671109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE. Pendrin: an apical Cl−/OH-/ exchanger in the kidney cortex. Am J Physiol Renal Physiol 280: F356–F364, 2001. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
- 193.Song HK, Kim WY, Lee HW, Park EY, Han KH, Nielsen S, Madsen KM, Kim J. Origin and fate of pendrin-positive intercalated cells in developing mouse kidney. J Am Soc Nephrol 18: 2672–2682, 2007. doi: 10.1681/ASN.2006101076. [DOI] [PubMed] [Google Scholar]
- 194.Soundararajan R, Lu M, Pearce D. Organization of the ENaC-regulatory machinery. Crit Rev Biochem Mol Biol 47: 349–359, 2012. doi: 10.3109/10409238.2012.678285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Star RA, Burg MB, Knepper MA. Bicarbonate secretion and chloride absorption by rabbit cortical collecting ducts. Role of chloride/bicarbonate exchange. J Clin Invest 76: 1123–1130, 1985. doi: 10.1172/JCI112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Stehberger PA, Shmukler BE, Stuart-Tilley AK, Peters LL, Alper SL, Wagner CA. Distal renal tubular acidosis in mice lacking the AE1 (band3) Cl-/ exchanger (slc4a1). J Am Soc Nephrol 18: 1408–1418, 2007. doi: 10.1681/ASN.2006101072. [DOI] [PubMed] [Google Scholar]
- 197.Stein JH, Osgood RW, Boonjarern S, Cox JW, Ferris TF. Segmental sodium reabsorption in rats with mild and severe volume depletion. Am J Physiol 227: 351–359, 1974. doi: 10.1152/ajplegacy.1974.227.2.351. [DOI] [PubMed] [Google Scholar]
- 198.Stetson DL, Steinmetz PR. Alpha and beta types of carbonic anhydrase-rich cells in turtle bladder. Am J Physiol Renal Physiol 249: F553–F565, 1985. doi: 10.1152/ajprenal.1985.249.4.F553. [DOI] [PubMed] [Google Scholar]
- 199.Stetson DL, Steinmetz PR. Role of membrane fusion in CO2 stimulation of proton secretion by turtle bladder. Am J Physiol Cell Physiol 245: C113–C120, 1983. doi: 10.1152/ajpcell.1983.245.1.C113. [DOI] [PubMed] [Google Scholar]
- 200.Stone DK, Seldin DW, Kokko JP, Jacobson HR. Anion dependence of rabbit medullary collecting duct acidification. J Clin Invest 71: 1505–1508, 1983. doi: 10.1172/JCI110905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Stoner LC, Burg MB, Orloff J. Ion transport in cortical collecting tubule; effect of amiloride. Am J Physiol 227: 453–459, 1974. doi: 10.1152/ajplegacy.1974.227.2.453. [DOI] [PubMed] [Google Scholar]
- 202.Stoos BA, Garcia NH, Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol 6: 89–94, 1995. [DOI] [PubMed] [Google Scholar]
- 203.Sutliff RL, Walp ER, Kim YH, Walker LA, El-Ali AM, Ma J, Bonsall R, Ramosevac S, Eaton DC, Verlander JW, Hansen L, Gleason RL Jr, Pham TD, Hong S, Pech V, Wall SM. Contractile force is enhanced in Aortas from pendrin null mice due to stimulation of angiotensin II-dependent signaling. PLoS One 9: e105101, 2014. doi: 10.1371/journal.pone.0105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Takada T, Yamamoto A, Omori K, Tashiro Y. Quantitative immunogold localization of Na, K-ATPase along rat nephron. Histochemistry 98: 183–197, 1992. doi: 10.1007/BF00315877. [DOI] [PubMed] [Google Scholar]
- 205.Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J Am Soc Nephrol 7: 260–274, 1996. [DOI] [PubMed] [Google Scholar]
- 206.Terada Y, Knepper MA. Thiazide-sensitive NaCl absorption in rat cortical collecting duct. Am J Physiol Renal Physiol 259: F519–F528, 1990. doi: 10.1152/ajprenal.1990.259.3.F519. [DOI] [PubMed] [Google Scholar]
- 207.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, Ellison DH. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol 27: 2436–2445, 2016. doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A, Firsov D. α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest 123: 3166–3171, 2013. doi: 10.1172/JCI67562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Trepiccione F, Soukaseum C, Baudrie V, Kumai Y, Teulon J, Villoutreix B, Cornière N, Wangemann P, Griffith AJ, Byung Choi Y, Hadchouel J, Chambrey R, Eladari D. Acute genetic ablation of pendrin lowers blood pressure in mice. Nephrol Dial Transplant 32: 1137–1145, 2017. doi: 10.1093/ndt/gfw393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Trepiccione F, Soukaseum C, Iervolino A, Petrillo F, Zacchia M, Schutz G, Eladari D, Capasso G, Hadchouel J. A fate-mapping approach reveals the composite origin of the connecting tubule and alerts on “single-cell”-specific KO model of the distal nephron. Am J Physiol Renal Physiol 311: F901–F906, 2016. doi: 10.1152/ajprenal.00286.2016. [DOI] [PubMed] [Google Scholar]
- 211.Tsuruoka S, Schwartz GJ. Mechanisms of secretion in the rabbit connecting segment. Am J Physiol Renal Physiol 277: F567–F574, 1999. doi: 10.1152/ajprenal.1999.277.4.F567. [DOI] [PubMed] [Google Scholar]
- 212.Tsuruoka S, Schwartz GJ. Metabolic acidosis stimulates H+ secretion in the rabbit outer medullary collecting duct (inner stripe) of the kidney. J Clin Invest 99: 1420–1431, 1997. doi: 10.1172/JCI119301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Valentijn JA, Fyfe GK, Canessa CM. Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J Biol Chem 273: 30344–30351, 1998. doi: 10.1074/jbc.273.46.30344. [DOI] [PubMed] [Google Scholar]
- 214.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschênes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 17: 2153–2163, 2006. doi: 10.1681/ASN.2005101054. [DOI] [PubMed] [Google Scholar]
- 215.Van den Wildenberg MJ, Hoorn EJ, Mohebbi N, Wagner CA, Woittiez AJ, de Vries PA, Laverman GD. Distal renal tubular acidosis with multiorgan autoimmunity: a case report. Am J Kidney Dis 65: 607–610, 2015. doi: 10.1053/j.ajkd.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 216.Verlander JW, Clapp WX. The Kidney. In: Brenner & Rector’s The Kidney, edited by Yu AS, Skorecki K, Chertow GM. New York: Elsevier, 2019. [Google Scholar]
- 217.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42: 356–362, 2003. doi: 10.1161/01.HYP.0000088321.67254.B7. [DOI] [PubMed] [Google Scholar]
- 218.Verlander JW, Hong S, Pech V, Bailey JL, Agazatian D, Matthews SW, Coffman TM, Le T, Inagami T, Whitehill FM, Weiner ID, Farley DB, Kim YH, Wall SM. Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. Am J Physiol Renal Physiol 301: F1314–F1325, 2011. doi: 10.1152/ajprenal.00114.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Verlander JW, Kim YH, Shin W, Pham TD, Hassell KA, Beierwaltes WH, Green ED, Everett L, Matthews SW, Wall SM. Dietary Cl− restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol 291: F833–F839, 2006. doi: 10.1152/ajprenal.00474.2005. [DOI] [PubMed] [Google Scholar]
- 220.Verlander JW, Madsen KM, Cannon JK, Tisher CC. Activation of acid-secreting intercalated cells in rabbit collecting duct with ammonium chloride loading. Am J Physiol Renal Physiol 266: F633–F645, 1994. doi: 10.1152/ajprenal.1994.266.4.F633. [DOI] [PubMed] [Google Scholar]
- 221.Verlander JW, Madsen KM, Galla JH, Luke RG, Tisher CC. Response of intercalated cells to chloride depletion metabolic alkalosis. Am J Physiol Renal Physiol 262: F309–F319, 1992. doi: 10.1152/ajprenal.1992.262.2.F309. [DOI] [PubMed] [Google Scholar]
- 222.Verlander JW, Madsen KM, Low PS, Allen DP, Tisher CC. Immunocytochemical localization of band 3 protein in the rat collecting duct. Am J Physiol Renal Physiol 255: F115–F125, 1988. doi: 10.1152/ajprenal.1988.255.1.F115. [DOI] [PubMed] [Google Scholar]
- 223.Verlander JW, Madsen KM, Tisher CC. Axial distribution of band 3-positive intercalated cells in the collecting duct of control and ammonium chloride-loaded rabbits. Kidney Int Suppl 57: S137–S147, 1996. [PubMed] [Google Scholar]
- 224.Verlander JW, Madsen KM, Tisher CC. Effect of acute respiratory acidosis on two populations of intercalated cells in rat cortical collecting duct. Am J Physiol Renal Physiol 253: F1142–F1156, 1987. doi: 10.1152/ajprenal.1987.253.6.F1142. [DOI] [PubMed] [Google Scholar]
- 225.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins RhBG and RhCG in mouse kidney. Am J Physiol Renal Physiol 284: F323–F337, 2003. doi: 10.1152/ajprenal.00050.2002. [DOI] [PubMed] [Google Scholar]
- 226.Wade JB. Membrane structural specialization of the toad urinary bladder revealed by the freeze-fracture technique. II. The mitochondria-rich cell. J Membr Biol 29: 111–126, 1976. doi: 10.1007/BF01868955. [DOI] [PubMed] [Google Scholar]
- 227.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP. Regulation of the expression of the Cl−/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int 62: 2109–2117, 2002. doi: 10.1046/j.1523-1755.2002.00671.x. [DOI] [PubMed] [Google Scholar]
- 228.Wall SM, Fischer MP. Contribution of the Na+-K+-2Cl− cotransporter (NKCC1) to transepithelial transport of H+, NH4+, K+, and Na+ in rat outer medullary collecting duct. J Am Soc Nephrol 13: 827–835, 2002. [DOI] [PubMed] [Google Scholar]
- 229.Wall SM, Fischer MP, Mehta P, Hassell KA, Park SJ. Contribution of the Na+-K+-2Cl− cotransporter NKCC1to Cl− secretion in rat OMCD. Am J Physiol Renal Physiol 280: F913–F921, 2001. doi: 10.1152/ajprenal.2001.280.5.F913. [DOI] [PubMed] [Google Scholar]
- 230.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol 284: F229–F241, 2003. doi: 10.1152/ajprenal.00147.2002. [DOI] [PubMed] [Google Scholar]
- 231.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension 44: 982–987, 2004. doi: 10.1161/01.HYP.0000145863.96091.89. [DOI] [PubMed] [Google Scholar]
- 232.Wall SM, Weinstein AM. Cortical distal nephron Cl− transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol 305: F427–F438, 2013. doi: 10.1152/ajprenal.00022.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 458: 157–168, 2009. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wang Z, Conforti L, Petrovic S, Amlal H, Burnham CE, Soleimani M. Mouse Na+: cotransporter isoform NBC-3 (kNBC-3): cloning, expression, and renal distribution. Kidney Int 59: 1405–1414, 2001. doi: 10.1046/j.1523-1755.2001.0590041405.x. [DOI] [PubMed] [Google Scholar]
- 235.Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med 2: 30–45, 2004. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wayne EJ, Koutras DA, Alexander WD. Excretion of iodine. In: Clinical Aspects of Iodine Metabolism. Philadelphia, PA: F.A. Davis Co, 1964, p. 73–82. [Google Scholar]
- 237.Weiner ID, Hamm LL. Regulation of intracellular pH in the rabbit cortical collecting tubule. J Clin Invest 85: 274–281, 1990. doi: 10.1172/JCI114423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Weiner ID, Hamm LL. Use of fluorescent dye BCECF to measure intracellular pH in cortical collecting tubule. Am J Physiol Renal Physiol 256: F957–F964, 1989. doi: 10.1152/ajprenal.1989.256.5.F957. [DOI] [PubMed] [Google Scholar]
- 239.Weiner ID, New AR, Milton AE, Tisher CC. Regulation of luminal alkalinization and acidification in the cortical collecting duct by angiotensin II. Am J Physiol Renal Physiol 269: F730–F738, 1995. doi: 10.1152/ajprenal.1995.269.5.F730. [DOI] [PubMed] [Google Scholar]
- 240.Weiner ID, Weill AE, New AR. Distribution of Cl-/ exchange and intercalated cells in rabbit cortical collecting duct. Am J Physiol Renal Physiol 267: F952–F964, 1994. doi: 10.1152/ajprenal.1994.267.6.F952. [DOI] [PubMed] [Google Scholar]
- 241.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol 289: F699–F720, 2005. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- 242.Weinstein AM. A mathematical model of rat distal convoluted tubule. II. Potassium secretion along the connecting segment. Am J Physiol Renal Physiol 289: F721–F741, 2005. doi: 10.1152/ajprenal.00044.2005. [DOI] [PubMed] [Google Scholar]
- 243.Wémeau JL, Kopp P. Pendred syndrome. Best Pract Res Clin Endocrinol Metab 31: 213–224, 2017. doi: 10.1016/j.beem.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 244.Wen H, Lin R, Jiao Y, Wang F, Wang S, Lu D, Qian J, Jin L, Wang X. Two polymorphisms in NEDD4L gene and essential hypertension in Chinese Hans - a population-based case-control study. Clin Exp Hypertens 30: 87–94, 2008. doi: 10.1080/10641960801949909. [DOI] [PubMed] [Google Scholar]
- 245.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 246.Xu J, Barone S, Li H, Holiday S, Zahedi K, Soleimani M. Slc26a11, a chloride transporter, localizes with the vacuolar H+-ATPase of A-intercalated cells of the kidney. Kidney Int 80: 926–937, 2011. doi: 10.1038/ki.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Xu J, Barone S, Zahedi K, Brooks M, Soleimani M. Slc4a8 in the Kidney: Expression, Subcellular Localization and Role in Salt Reabsorption. Cell Physiol Biochem 50: 1361–1375, 2018. doi: 10.1159/000494596. [DOI] [PubMed] [Google Scholar]
- 248.Xu N, Hirohama D, Ishizawa K, Chang WX, Shimosawa T, Fujita T, Uchida S, Shibata S. Hypokalemia and Pendrin Induction by Aldosterone. Hypertension 69: 855–862, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08519. [DOI] [PubMed] [Google Scholar]
- 249.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yoshida A, Hisatome I, Taniguchi S, Sasaki N, Yamamoto Y, Miake J, Fukui H, Shimizu H, Okamura T, Okura T, Igawa O, Shigemasa C, Green ED, Kohn LD, Suzuki K. Mechanism of iodide/chloride exchange by pendrin. Endocrinology 145: 4301–4308, 2004. doi: 10.1210/en.2004-0048. [DOI] [PubMed] [Google Scholar]
- 251.Zhang Y, Listhrop R, Ecelbarger CM, Kishore BK. Renal sodium transporter/channel expression and sodium excretion in P2Y2 receptor knockout mice fed a high-NaCl diet with/without aldosterone infusion. Am J Physiol Renal Physiol 300: F657–F668, 2011. doi: 10.1152/ajprenal.00549.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]