

Abstract
Immunosuppression posttransplantation exposes patients to an increased risk for refractory viral infections as an important cause of morbidity and mortality. Protective T cell immunity can be restored by adoptive T cell transfer, but ongoing immunosuppression limits efficacy of T cell responses. In order to deliver protection against viral pathogens and allow at the same time necessary steroid therapy, we generated glucocorticoid-resistant T cells by CRISPR-Cas9-mediated knockout of the glucocorticoid receptor in primary human virus-specific T cell products. Characterization of the T cell product revealed high efficiency of glucocorticoid receptor knockout and high purity of virus-specific T cells. This tandem T cell engineering preserved protective T cell functionality, such as cytotoxicity, CD107a degranulation, proliferative capacity, and cytokine release patterns. Virus-specific T cells with glucocorticoid receptor knockout were resistant to the suppressive effect of dexamethasone treatment on lymphocyte proliferation and cytokine secretion (tumor necrosis factor alpha [TNF-α], interleukin-4 [IL-4], IL-6, and sFas). Additionally, glucocorticoid receptor knockout cells remained sensitive to cyclosporine A treatment, thereby providing a rescue approach for patients in case of safety issues. This novel approach provides a therapeutic option for the treatment of patients with viral infections after transplantation who are receiving glucocorticoid therapy.
Keywords: virus-specific T cells, glucocorticoid receptor, CRISPR/Cas9, T cell therapy, genetic engineering, stem cell transplantation
Graphical Abstract
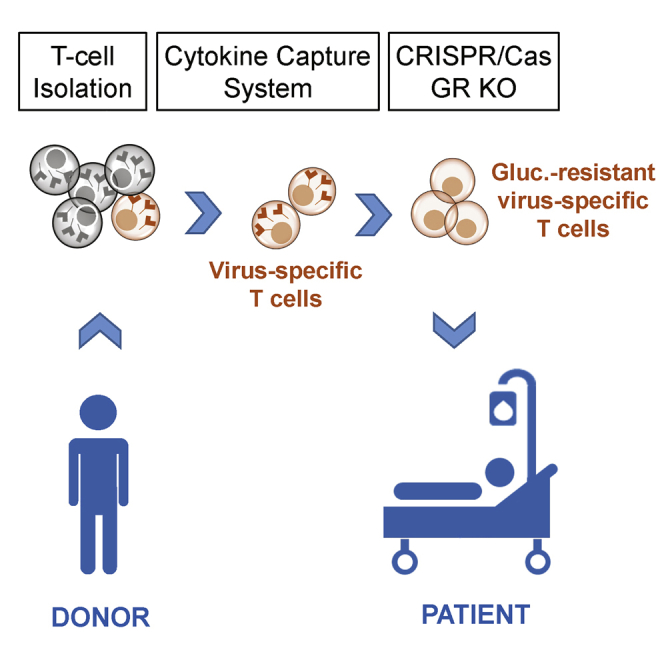
Immunosuppression posttransplantation exposes patients to an increased risk for refractory viral infections as an important cause of morbidity and mortality. Kaeuferle et al. generated glucocorticoid-resistant T cell products by CRISPR-Cas-mediated genetic engineering of virus-specific T cells to restore protective T cell immunity under steroid treatment via adoptive T cell therapy.
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) and solid organ transplantation (SOT) are curative treatments for a variety of diseases. However, refractory viral infections cause life-threatening conditions because of the deficient T cell response posttransplantation, a situation for which effective treatment options are urgently needed.1, 2, 3, 4 Protective T cell immunity can be restored by means of adoptive T cell transfer, generating virus-specific T cell products from healthy donor cells.5 Over the past 30 years it has been demonstrated that this approach can be successfully applied for patients with refractory viral infections after stem cell transplantation.6, 7, 8, 9 In evidence-based treatment guidelines, T cell therapy has become a second-line treatment recommendation.10,11 Virus-specific T cell products can be generated by in vitro stimulation and expansion12,13 or selection techniques. Selection has become possible by efforts made to directly isolate virus-specific T cells from peripheral blood of seropositive donors in order to induce efficient proliferation under physiological conditions in vivo. For direct selection of virus-specific T cells, cytokine capture technique could be applied, leading to a combination of virus-specific T helper (Th) and cytotoxic T cells.6 Isolation based on Streptamer technology leads to high purity of peptide-specific CD8+ virus-specific T cell products.14 Thus, virus-specific T cells are obtained in small amounts and are infused into the patient, where they can expand effectively upon the presence of viral antigen in vivo. This physiological re-stimulation and T cell response induce viral clearance and sustained protection. However, this treatment undergoes immediate inactivation in vivo in those patients receiving relevant immunosuppression, such as patients with graft-versus-host disease (GvHD) or patients after SOT. In many cases the immunosuppression itself is the major cause of the uncontrolled viral infection. Patients with significant GvHD after stem cell transplantation are at highest risk for recurrent and refractory viral infections, largely because of high-dose glucocorticoid therapy.15 Because glucocorticoids exert potent immunosuppressive effects, they remain the most effective and favored initial therapy for the treatment of acute and chronic GvHD. However, steroid treatment provides a major obstacle to adoptive T cell therapy, because glucocorticoids suppress activation and expansion16 of transferred T cells. T cells are inhibited after specific antigen-mediated activation by binding to their glucocorticoid receptor (GR).17 Thus, the most definitive way of preventing this adverse effect is the genetic knockout (KO) of the endogenous GR.
Clustered regularly interspaced short palindromic repeats-Cas (CRISPR-Cas) is the most advanced genetic engineering technology, which is site specific and highly effective combined with low-toxicity and off-target cleavage.18 The CRISPR RNA (crRNA)-guided Cas9 protein induces site-specific double-strand breaks in target DNA. The cleavage site is determined via base pairing between crRNA and target sequence, as well as a protospacer adjacent motif (PAM).19,20 Double-strand breaks can be repaired via non-homologous end joining (NHEJ) inducing insertions and deletions (InDels) at the cut site, and thus leading to gene KO.
In the present project, we genetically knocked out the endogenous GR via CRISPR-Cas9 technology in primary human, virus-specific T cells to induce resistance to suppressive effects of glucocorticoids. This approach provides a novel therapeutic option for the treatment of refractory viral infections after stem cell transplantation or SOT in patients receiving glucocorticoid therapy.
Results
CRISPR-Cas9-Mediated KO of GR Gene
In order to generate virus-specific glucocorticoid-resistant T cells, we enriched virus-specific T cells via cytokine capture technique followed by CRISPR-Cas9-mediated KO of the gene NR3C1 (GenBank: NG_009062.1) encoding the GR (Figure 1A). Two crRNAs were designed targeting exon 2 of the GR gene coding for the receptor’s N-terminal domain (Figure 1B). The KO efficiency of the GR was evaluated at days 5 and 8 postelectroporation on genetic level via PCR and Tracking of Indels by Decomposition (TIDE) analysis, as well as on protein level via western blot. KO was highly efficient (crRNA1: 59% on genetic and 84% on protein level at day 5) and remained stable over time (59% on genetic and 79% on protein level at day 8; Figures 1C and 1E). Furthermore, the KO efficiency did not differ significantly between the virus-specific T cell fraction (VST) and negative fraction of virus-specific T cell enrichment (bulk T cells: 59% ± 7% versus 62% ± 10% on genetic level and 84% ± 17% versus 91% ± 15% on protein level; Figures 1C and 1E). Investigating CRISPR-Cas9-induced mutation patterns of the crRNAs revealed donor-independent characteristic InDel patterns for the respective crRNAs (Figure 1D), although the overall KO efficiency did not differ significantly. Immunofluorescence (IF) microscopy confirmed the GR KO on protein level determined via western blot (Figures 1E and 1F).
Figure 1.

CRISPR-Cas9-Mediated Knockout (KO) of Glucocorticoid Receptor (GR) Gene
(A) Schematic overview of adoptive transfer of virus-specific T cells from a healthy donor to a patient receiving glucocorticoid therapy in order to treat refractory viral infections. Virus-specific T cells were isolated via cytokine capture technique ex vivo from peripheral blood of a seropositive donor, and GR was KO via CRISPR-Cas9. (B) Location of guide RNAs crRNA1 and crRNA2 binding sites within GRα gene. Chromatograms were obtained via Sanger Sequencing. (C) Confirmation of GR KO efficiency on genetic level. Total frequency of insertions and deletions (InDels) at the CRISPR cut site determined by TIDE webtool on days 5 and 8 after electroporation. InDels with a p value <0.05 in TIDE are depicted. Mean + SD (n = 3 donors). (D) crRNA1 InDel patterns of three different donors (left) and InDel patterns of crRNA1 and crRNA2 each of one representative donor (right). (E) GR KO efficiency on protein level evaluated via western blot and ImageJ-based digital image analysis. Protein levels were normalized to those of GAPDH. Mean + SD (n = 3 donors). (F) Immunofluorescence and light microscopy images of GR wild-type (WT) and GR KO T cells stained with GR (G-5) primary and Alexa Fluor 488 secondary antibody.
Enrichment of Virus-Specific T Cells
For generation of virus-specific T cells, primary human peripheral blood mononuclear cells (PBMCs) were stimulated with cytomegalovirus (CMV) pp65, and interferon γ (IFNγ)-secreting cells were enriched via cytokine capture technique. Using CMV pp65 Peptivator, a 15-mer peptide pool covering the immune-dominant protein pp65 of CMV, we enriched virus-specific T cells significantly (p = 0.031) and highly efficiently (163-fold) from mean initial frequencies of 0.3% in the starting material (PBMCs) to 49% in the VST (Figures 2A and 2B). Additionally, we analyzed enrichment of single-epitope-specific T cell clones. Specificity against the single peptide pp65 NLVPMVATV was detected using Streptamer staining, confirming an enrichment from 0.04% in the original fraction (PBMCs) to 1.97% in the VST. This enrichment of NLVPMVATV-specific T cell clones confirmed virus specificity of the IFNγ-based enrichment using cytokine capture technique (Figure 2B).
Figure 2.

Isolation of Virus-Specific T Cells and Phenotypic Characterization of the Final T Cell Product
(A and B) Enrichment of IFNγ-secreting T cells from primary human PBMCs via cytokine capture technique after stimulation with CMV pp65 or SEB (positive control). (A) Frequency of IFNγ+ T cells in the starting material (PBMCs), as well as in the negative (bulk T cells) and virus-specific T cell fraction (VST) after magnetic enrichment. Mean + SD (n = 3 donors). (B) Exemplary flow cytometry plots of IFNγ and NLV-multimer staining. (C–E) Characterization of the starting material (PBMCs) and the final GR KO virus-specific T cell product (GR KO VST). (C) Cellular composition and viability analyzed by flow cytometry. Mean: n = 4 donors. (D) T cell phenotype before GR KO (VST), in the final T cell product (GR KO VST) and in WT control (GR WT VST) during T cell production process (days −2 and 0) and after 3 (d3) and 5 days (d5) of in vitro expansion. Mean: n = 3 donors. (E) Characterization of flow-cytometric-sorted IFNγ+ T cells from the final T cell product. Western blot and respective ImageJ-based digital image analysis of the KO efficiency (mean + SD, n = 2 donors) and (F) flow-cytometric-based determination of the T cell phenotype (right, mean: n = 3 donors). ∗p < 0.05, ∗∗∗p < 0.001. n.s., not significant.
For the generation of a glucocorticoid-resistant T cell product, these virus-specific T cells were further processed by KO of the GR via CRISPR-Cas9. In order to characterize the final T cell product, cellular composition and viability were determined by flow cytometry in the starting material (PBMCs) and the T cell product (GR KO VST). Figure 2C shows that the starting material consisted of 44% T cells (30% CD4+ Th cells and 14% CD8+ cytotoxic T cells) and other leukocyte subsets, such as monocytes, B cells, and granulocytes. The virus-specific GR KO product consisted mainly of T cells (87%) and low frequencies of natural killer (NK) T cells (2%) and B cells (0.9%). Viability of the starting material and the T cell product was high (98% and 95%, respectively).
To investigate the influence of the manufacturing procedure on T cell maturation phenotype, we determined frequencies of naive T cells, stem cell-like memory T cells, central memory T (Tcm) cells, effector memory T cells, and effector T cells, according to the expression of CD62L and CD45RO and CD95. T cells of freshly isolated PBMCs consisted of high frequencies of naive T cells (58%) and lower numbers of effector memory (19%), central memory (15%), and effector cells (7%; Figure 2D). After re-stimulation and isolation of virus-specific T cells using cytokine capture technique, the enriched T cell fraction (VST) showed a mature phenotype with significantly higher frequencies of effector memory (52%) and effector T cells (22%) and significantly lower numbers of naive T cells (7.6%). GR KO did not influence the T cell phenotype significantly, confirming that the CRISPR engineering did not alter physiological maturation of virus-specific T cells (Figures 2D and 2E).
In order to confirm the GR absence in the virus-specific T cell population of the cell product, we further sorted IFNγ-positive T cells using fluorescence-activated cell sorting (FACS). Subsequent western blot analysis confirmed that the KO efficiency was 97.4% among virus-specific T cells (Figure 2E).
Functionality of Virus-Specific GR KO T Cells
In order to assess functionality of the virus-specific GR KO T cells, we investigated cytotoxicity, degranulation marker expression, proliferation, and cytokine release patterns upon re-stimulation. Upon re-stimulation with CMV, using donor-derived pp65 peptide-pulsed phytohemagglutinin (PHA) blasts, GR KO virus-specific T cells showed similar effector-to-target (E:T) ratio-dependent cytotoxic effects as GR wild-type (WT) virus-specific T cells (59% versus 57% specific target cell lysis; Figure 3A). Strong increase of degranulation marker CD107a expression on cytotoxic T cells upon re-stimulation with CMV-specific peptides confirmed the cytotoxicity results and showed donor dependency (4- to 98-fold; Figure 3A). Furthermore, upon re-stimulation, 78% of GR KO virus-specific T cells and 79% of GR WT virus-specific T cells proliferated compared with 29% and 22% in the respective unstimulated control samples (Figures 3B and 3C). Intracellular cytokine staining revealed highly significant upregulation of IFNγ and tumor necrosis factor alpha (TNF-α) secretion (1.5%–11% and 0.6%–6%, respectively) upon CMV re-stimulation of GR KO T cells comparable with the upregulation in GR WT cells (1.6%–14% and 0.7%–6%, respectively; Figure 3C).
Figure 3.

Functional Characterization of Virus-Specific GR KO T Cells
(A) Cytotoxic capacity of CMV-specific GR KO and CMV-specific GR WT T cells. Cytotoxic capacity against autologous CMV pp65 peptide-pulsed PHA blasts upon co-culture in effector-to-target ratios (E:T) of 4:1; 2:1, and 1:1 (left). Unpulsed PHA blasts served as negative control (unpulsed ctrl). Mean ± SD; n = 3 donors. Fold changes of cytotoxic marker CD107a+ T cell frequencies upon re-stimulation with CMV pp65 peptides in three donors (right). (B) Overlay of representative CTV-based proliferation assay histograms on day 3 after CD3/CD28 re-stimulation and unstimulated control. (C) Frequency of proliferating, IFNγ+, and TNF-α+ T cells in CMV pp65 peptide-stimulated virus-specific GR KO and GR WT cells, as well as the respective unstimulated controls. IFNγ+ and TNF-α+ determined by intracellular cytokine staining. Mean + SD; n = 3 donors. (D) Representative IFNγ and TNF-α histograms of cell culture supernatant analysis upon CMV pp65 peptide stimulation. (E) Correlation of absolute cytokine concentrations determined via multiplex analysis in cell culture supernatants of GR KO and GR WT cells upon CMV pp65 peptide re-stimulation and in the unstimulated setting (n = 4 independent experiments of two donors). Spearman’s r. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001.
In order to compare functionality of GR KO and GR WT cells upon CMV re-stimulation in terms of cytokine release, we analyzed a whole pattern of Th1 and Th2 cytokines and markers (interleukin-2 [IL-2], IL-4, IL-6, IL-10, TNF-α, IL-17a, sFas, sFasL, IFNγ, granzyme A, granzyme B, perforin, and granulysin) in the cell culture supernatants via multiplex assays. First, strong increase of Th1 cytokines IFNγ and TNF-α (Figure 3D) upon re-stimulation, as well as high absolute concentrations of IFNγ and TNF-α in the supernatant of CMV-stimulated T cells, confirmed results from intracellular cytokine staining (Figures 3C and 3D). Second, highly significant correlation of absolute cytokine/marker levels in GR KO and GR WT cells upon CMV re-stimulation (r = 0.99, p < 0.0001), as well as in the unstimulated situation (r = 0.98, p < 0.0001), confirmed that the GR KO did not influence functionality of the virus-specific T cells in terms of released cytokine patterns (Figure 3E). The highest concentrations were observed for cytoplasmic granule proteases and cytolytic proteins perforin, granzyme A and granzyme B, and granulysin (Figure 3E).
Glucocorticoid Resistance of Virus-Specific GR KO T Cells
In order to investigate glucocorticoid resistance of GR KO T cells, we analyzed cytokine expression, proliferation, and levels of GR KO cells during expansion upon high-dose glucocorticoid treatment. GR KO rescued the inhibitory effect of dexamethasone treatment on virus-specific upregulation of Th1 cytokines (TNF-α, IFNγ), confirmed by intracellular cytokine staining and absolute quantification of cytokine levels in the cell culture supernatant (Figures 4A, 4C, and 4D). Intracellular cytokine staining showed that Th1 cytokine expression in T cells such as TNF-α was upregulated 9-fold upon dexamethasone treatment in GR KO cytotoxic T cells and 7-fold in GR KO total T cells, whereas upregulation was strongly impaired by dexamethasone treatment in GR WT cells (2-fold upregulation in WT cytotoxic and total T cells; Figures 4A and 4C). Quantification of absolute cytokine levels in the supernatant confirmed the results of intracellular cytokine staining (Figure 4D). Additionally, physiological upregulation of Th2-driving IL-4, pro-inflammatory IL-6, and T cell activation marker Fas secretion was rescued by GR KO upon dexamethasone treatment (Figure 4D). As expected, under dexamethasone, Th1 cytokine response in WT T cells was strongly reduced, but not completely eliminated, whereas acute pro-inflammation (IL-6) and activation (Fas) were unresponsive to antigen stimulation under dexamethasone (Figure 4D). Expansion of lymphocytes in the T cell product was highly significantly increased by GR KO during dexamethasone treatment compared with GR WT cells (p = 0.002; Figure 4B). Western blot analyses showed that dexamethasone treatment during expansion led to elimination of GR WT cells, and thus enrichment of GR KO cells (Figure 4B). As expected, cytotoxicity of GR KO and GR WT T cells was not affected by dexamethasone treatment in our experimental setting (data not shown).
Figure 4.

Glucocorticoid Resistance of Virus-Specific GR KO T Cells
CMV-specific GR KO and GR WT T cells were treated with 200 μM dexamethasone. (A) TNF-α+ T cell frequencies were determined via intracellular cytokine staining in cytotoxic and total T cells (n = 3 donors). (B) Lymphocyte counts were assessed via flow cytometry using absolute quantification mode during 5 days of dexamethasone treatment (n = 3 donors), and GR KO rates were determined via western blot on day 5 of dexamethasone treatment. (C) Exemplary flow cytometry plots of TNF-α intracellular cytokine staining. (D) Absolute cytokine concentrations analyzed in cell culture supernatants. n = 2 technical duplicates of one donor. (E) Proliferative capacity with and without 400 ng/mL cyclosporine A treatment (n = 3 donors). Overlay of representative CTV-based proliferation assay histograms showing CD8+ T cell proliferation on day 3 after re-stimulation. ∗p < 0.05, ∗∗p < 0.001. Mean ± SEM.
In order to ensure safety in patients with high risk for GvHD, we investigated the effect of GR-independent immunosuppressive drugs on GR KO cells. Cyclosporine A is a commonly used drug in the posttransplantation setting, exerting its immunosuppressive effect by suppressing T cell proliferation. Treatment with low-dose cyclosporine A led to suppressed functionality of GR KO cells comparable with the suppression of WT cells, thereby providing a rescue treatment in case of safety issues (Figure 4E).
Discussion
Our results demonstrate an efficient CRISPR-Cas9-mediated KO of the GR in virus-specific T cells, leading to glucocorticoid resistance and preserved high antiviral functionality. The processes described here are in line with Good Manufacturing Practice (GMP) scale procedures, thereby providing a therapeutic option for the treatment of multiple refractory viral infections after transplantation in patients after SOT or in patients receiving ongoing glucocorticoid therapy due to significant GvHD after HSCT.
Enrichment of virus-specific T cells based on cytokine-capture technique has been already used in clinical practice for decades.6 Menger et al.21 provided a proof-of-concept study for the generation of virus-specific, glucocorticoid-resistant T cells by genetic KO of the GR using transcription activator-like effector nuclease (TALEN). However, Menger et al.21 performed the genetic KO in Streptamer-selected virus-specific T cells, an approach with the following restrictions: (1) it is HLA restricted, (2) it is available only for known immunodominant epitopes, and (3) generated T cell products are restricted to CD8 T cells only. Consequently, this approach has not reached clinical routine application to date. In this study, we combine a clinically available strategy of specific T cell selection (cytokine-capture technique) with fast and feasible CRISPR-Cas9 electroporation. This combinatorial cell engineering overcomes the hurdles of HLA restriction, limited immunodominant epitopes, and is compatible with requirements for GMP grade engineering in clinical studies. Inducing GR KO T cell responses opens a counterintuitive clinical scenario in which immunosuppression could be switched transiently from any other drug to dexamethasone until recovery of the viral infection.
Transient transfection of the CRISPR-Cas9-guide RNA (gRNA) ribonucleoprotein (RNP) complex leads to a reduction of off-target effects, as well as a reduction of long-term derangement of the cell compared with stable integration of extrinsic protein complexes. Furthermore, the delivery of a fully functional RNP complex via electroporation leads to early detectable genetic mutations and more stable efficacies over time, as previously described.22 KO efficiencies did not differ between virus-specific T cells and bulk T cells. The InDel patterns have been shown to be donor independent, but highly dependent on the gRNA sequence. Thus, the mutation pattern can be influenced by the design of the respective gRNA.
As previously described,23 dexamethasone treatment suppresses immune responses but did not influence cytotoxic capacity of single T cells. Thus, GR KO did not affect cytotoxic capacity of WT T cells upon dexamethasone treatment in the present study. In fact, it was previously described that dexamethasone treatment inhibits activation and proliferation rather than T cell apoptosis.23 Accordingly, lymphocyte counts did not decrease upon dexamethasone treatment, but CMV-induced increase of lymphocyte counts was inhibited by dexamethasone in WT cells. However, lymphocyte counts, as well as physiological regulation of TNF-α, IFNγ, IL-4, IL-6, and sFas, could be rescued by GR KO under high-dose dexamethasone treatment in the present study, confirming the induction of steroid resistance through GR KO in T cells. Clinical efficacy of virus-specific T cells with a GR KO will depend on the capacity of in vivo expansion. Animal models have limited predictive value for T cell persistence because of differences between human and animal pathogens. The phenotype of Tcm cells has been associated with in vivo persistence (Figure 2D). Nevertheless, clinical studies will have to clarify kinetics of protection after adoptive transfer of GR KO T cells.
Inducing steroid resistance in T cells may carry a risk for uncontrolled T cell activation. Therefore, a safety concern before clinical application is the sensitivity of the adoptively transferred T cells to other immunosuppressive compounds. Calcineurin inhibitors such as cyclosporine A act independent of the GR on the T cells, thereby presenting a potential rescue treatment. Our results show that cyclosporine A results in a robust suppression of proliferation in GR KO T cells. General safety issues concerning infusion of CRISPR KO cells remain to be investigated in prospective clinical studies. However, recently, first-in-human data have been generated using the same technique in another gene locus treating refractory cancer.24
In conclusion, the generated virus-specific GR KO T cell product was found to have high cytolytic activity, specific stimulating properties, and high proliferation capacity, while remaining resistant to glucocorticoid treatment. Characterization and identification of the product showed a stable effector-memory phenotype and high purities in terms of KO, as well as virus specificity. Taken together, CRISPR-Cas9-mediated GR engineering of virus-specific T cells is a novel approach for induction of protective T cell responses against persistent viruses under ongoing steroid-mediated immunosuppression.
Materials and Methods
Isolation of Virus-Specific T Cells via Cytokine Capture Technique
PBMCs were stimulated for 4 h with CMV pp65 Peptivator or Staphylococcus enterotoxin B (SEB) as positive control. IFNγ-secreting cells were isolated via IFNγ Secretion Assay kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. In brief, stimulated cells were coated with a CD45-IFNγ bispecific antibody. Bound IFNγ was labeled with an anti-IFNγ phycoerythrin (PE) antibody, and cells were magnetically enriched via an anti-PE antibody conjugated to magnetic beads (Miltenyi Biotec). PBMCs were obtained from healthy individuals after informed consent. Ethics approval was obtained from the local Research Ethics Committee, and the study was performed in accordance with the Declaration of Helsinki.
CRISPR-Cas9-Mediated GR KO
Virus-specific T cells were activated using Dynabeads (anti-CD3/anti-CD28) in a 1:2 bead-to-cell ratio (Thermo Fisher Scientific, Waltham, MA, USA). Beads were removed using the DynaMAG-2 magnet and, depending on cell counts, 2–8 days after isolation, 1 × 106 cells were electroporated in Buffer 1M (KCl 5 mM, MgCl2 15 mM, Na2HPO4/NaH2PO4 120 mM, mannitol 50 mM),25 mixed with RNP complex containing the respective guide RNA (crRNA 1: 5′-AACCAAAAGUCUUCGCUGCU-3′, crRNA 2: 5′-CUUUAAGUCUGUUUCCCCCG-3′), Cas9 enzyme, and enhancer according to the manufacturer’s instructions (all Integrated DNA Technologies, Skokie, IL, USA). After electroporation using Nucleofector II (Amaxa Biosystems, Cologne, Germany), cells were cultured in TexMACS medium supplemented with 2.5% human AB serum. GR WT cells were cultured and electroporated as GR KO cells, except electroporation without CRISPR-Cas9 reagents (Mock control).
Evaluation of KO Efficiency: TIDE
To evaluate KO efficiency on a genetic level, we isolated DNA using the QIAamp DNA Mini Kit and performed PCR using forward primer 5′-TCCTGGTAGAGAAGAAAACCCC-3′, reverse primer 5′-CCTGCAAAATGTCAAAGGTGC-3′, and AccuPrime Taq DNA high-fidelity Polymerase (Thermo Fisher Scientific). PCR products were Sanger sequenced, and KO rate was determined using TIDE as described previously.26
Western Blot
20 μg protein lysates generated using standard radioimmunoprecipitation assay (RIPA) buffer and supplemented with 6× Laemmli was separated and subsequently blotted onto a nitrocellulose membrane. The membrane was blocked and stained with anti-GR antibody 1:500–1,000 and anti-GAPDH antibody 1:10,000 (both Santa Cruz Biotechnology, Dallas, TX, USA). Using secondary goat anti-mouse immunoglobulin G (IgG) horseradish peroxidase (HRP) antibody (Santa Cruz Biotechnology) and Immun-Star HRP Chemiluminescence Kit (BioRad, Hercules, CA, USA), we immunodetected proteins. Blots were quantified using an ImageJ-based digital image analysis approach.
IF Microscopy
GR KO cells were fixed with 1% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100 (both Sigma-Aldrich, Merck, Darmstadt, Germany). Subsequently, cells were labeled with GR (G-5) antibody 1:200 (Santa Cruz Biotechnology) and stained with secondary goat anti-mouse Alexa Fluor 488 antibody 1:100 (BioLegend, San Diego, CA, USA). Stained cells were applied onto a poly-L-lysine-coated slide (Sigma-Aldrich, Merck, Darmstadt, Germany) under a coverslip with DAPI solution (Thermo Fisher Scientific).
Immunosuppressive Treatment
Dexamethasone (Sigma-Aldrich, Merck) was added at 200 μM. After 5 days, cells were stained with 7-aminoactinomycin D (7-AAD; BioLegend) and quantified using MACSQuant flow cytometer. Cyclosporin A (CsA; Novartis, Basel, Switzerland) was added at 400 ng/mL for 5 days. Subsequently, cells were labeled with CellTrace Violet Proliferation Kit (Thermo Fisher Scientific) and re-stimulated with TransAct (Miltenyi Biotec). Cells were analyzed 24, 48, and 72 h after re-stimulation via flow cytometry.
Intracellular IFNγ Staining
Cells were co-cultured with CD3- and CD56-depleted PBMCs as feeder cells in a 1:1 ratio and stimulated with CMV pp65 Peptivator (Miltenyi Biotec). After 2 h of incubation, 10 μg/mL brefeldin A (Sigma-Aldrich, Merck) was added. Six hours after re-stimulation, cells were stained for viability and CD3-allophycocyanin (APC)-Vio770, CD4-VioBright fluorescein isothiocyanate (FITC), CD8-VioBlue, CD56-PE-Vio615, CD45RO-PE-Vio770, and CD62L-APC (Miltenyi Biotec). Subsequently, cells were stained intracellularly with IFNγ-PE (Miltenyi Biotec) using FIX & PERM Cell Permeabilization Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions and were measured via flow cytometry.
TNF-α, CD107a, and CD154 Staining
In vitro expanded cells were cultured without any cytokines for 1 day before addition of brefeldin A and CD107a-APC antibody (Miltenyi Biotec). After 6 h of incubation, cells were extracellularly stained with CD4-VioGreen, CD3-FITC, and CD8-APC/Vio770 antibodies. Subsequently, the respective samples were fixated, permeabilized, and intracellularly stained using Inside Stain Kit and TNF-α-PE-Vio770 and CD154-VioBlue antibodies (all reagents were obtained by Miltenyi Biotec). Cells were analyzed via flow cytometry.
T Cell Phenotype Staining
Cells were stained using CD62L-VioBlue, CD45RO-PE-Vio770, and CD95-APC antibodies (all Miltenyi Biotec) at different time points and analyzed flow cytometrically to distinguish naive T cells (CD45RO− CD62L+ CD95−), stem-cell like memory T cells (CD45RO− CD62L+ CD95+), Tcm cells (CD45RO+ CD62L+ CD95+), effector memory T cells (CD45RO+ CD62L− CD95+), and effector T cells (CD45RO− CD62L− CD95+).
Streptamer Staining
Cells were stained with Strep-Tactin PE backbone-labeled Streptamer, either CMV p65 NLVPMVATV or NY-ESO-1 SSLMWITQV as a control (both IBA, Göttingen, Germany), according to the manufacturer’s instructions. Cells were washed and additionally stained for CD3-APC-Vio770, CD4-VioBright FITC, CD8-VioBlue, CD56-PE-Vio615 (Miltenyi Biotec), and viability and analyzed via flow cytometry.
Cytokine Release in Supernatant
Supernatants were harvested, and secreted cytokines were analyzed in a bead-based immunoassay (LegendPlex, BioLegend) according to the manufacturer’s instructions.
Proliferation
Cells were labeled with 1 μM CellTrace Violet (CTV) for 5 min at 37°C using the CellTrace Violet Proliferation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. After removal of unbound dye and washing according to the manufacturer’s instructions, 2 × 105 cells/well were seeded in a 96-well round-bottom plate and re-stimulated with 10 μL/mL CD3/CD28 stimulant TransAct. At 24, 48, and 72 h after re-stimulation, CTV intensity was measured via flow cytometry. For evaluation of T cell proliferation, cells with reduced CTV intensity compared with the 24-h measurement were determined as proliferating cells.
Cytotoxicity Assay
Autologous PBMCs were incubated in RPMI medium supplemented with 1% L-glutamine, 10% FCS, and 1 μg/mL PHA for 3 days to induce blast transformation. Subsequently, IL-2 was added for 3–5 days. Blasts were then pulsed with 20 μL/mL CMV pp65 Peptivator and labeled with CTV (Miltenyi Biotec). Virus-specific GR KO cells were enriched using EasySep Human T Cell Enrichment Kit (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Blasts and effector T cells were co-cultured in TexMACS GMP medium (Miltenyi Biotec) at different E:T ratios for 24 h. Propidium iodide was added 1:100 directly before flow cytometric measurement. For calculation of cytotoxic capacity, living target cell count in co-cultured wells was divided by the mean of living target cell count in control wells (“target cells only”).
Author Contributions
The concept was set up by T.F. Design and approach of experiments was done by T.K. and T.F. Experiments and setup of methods were done by T.K., L.D., L.J., T.A.S., F.B., and S.W. Data analysis was done by T.K., L.D., and T.F. T.K. and T.F. designed and drafted the manuscript. The manuscript was reviewed by all authors.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors thank all donors for participating in the study and Tanja Roßmann-Bloeck, Anita Popko-Scibor, and Tanja Weißer for excellent technical assistance. FACS was kindly supported by Raffaele Conca and Susanne Wullinger. This work was supported by Föfole–Förderung für Forschung und Lehre–Medizinische Fakultät der LMU München, Kinderkrebshilfe Ebersberg e.V., Gertrud und Hugo Adler-Stiftung, Hermine und Gottfried Kieser-Stiftung, Otto-Hellmeier-Stiftung and the German Center for Infection Research (DZIF).
References
- 1.Bahceci E., Epperson D., Douek D.C., Melenhorst J.J., Childs R.C., Barrett A.J. Early reconstitution of the T-cell repertoire after non-myeloablative peripheral blood stem cell transplantation is from post-thymic T-cell expansion and is unaffected by graft-versus-host disease or mixed chimaerism. Br. J. Haematol. 2003;122:934–943. doi: 10.1046/j.1365-2141.2003.04522.x. [DOI] [PubMed] [Google Scholar]
- 2.Small T.N., Papadopoulos E.B., Boulad F., Black P., Castro-Malaspina H., Childs B.H., Collins N., Gillio A., George D., Jakubowski A. Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood. 1999;93:467–480. [PubMed] [Google Scholar]
- 3.Federmann B., Hägele M., Pfeiffer M., Wirths S., Schumm M., Faul C., Vogel W., Handgretinger R., Kanz L., Bethge W.A. Immune reconstitution after haploidentical hematopoietic cell transplantation: impact of reduced intensity conditioning and CD3/CD19 depleted grafts. Leukemia. 2011;25:121–129. doi: 10.1038/leu.2010.235. [DOI] [PubMed] [Google Scholar]
- 4.Hill J.A., Mayer B.T., Xie H., Leisenring W.M., Huang M.L., Stevens-Ayers T., Milano F., Delaney C., Sorror M.L., Sandmaier B.M. The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood. 2017;129:2316–2325. doi: 10.1182/blood-2016-10-748426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riddell S.R., Watanabe K.S., Goodrich J.M., Li C.R., Agha M.E., Greenberg P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 6.Kaeuferle T., Krauss R., Blaeschke F., Willier S., Feuchtinger T. Strategies of adoptive T -cell transfer to treat refractory viral infections post allogeneic stem cell transplantation. J. Hematol. Oncol. 2019;12:13. doi: 10.1186/s13045-019-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feuchtinger T., Opherk K., Bethge W.A., Topp M.S., Schuster F.R., Weissinger E.M., Mohty M., Or R., Maschan M., Schumm M. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. 2010;116:4360–4367. doi: 10.1182/blood-2010-01-262089. [DOI] [PubMed] [Google Scholar]
- 8.Feucht J., Opherk K., Lang P., Kayser S., Hartl L., Bethge W., Matthes-Martin S., Bader P., Albert M.H., Maecker-Kolhoff B. Adoptive T-cell therapy with hexon-specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood. 2015;125:1986–1994. doi: 10.1182/blood-2014-06-573725. [DOI] [PubMed] [Google Scholar]
- 9.Icheva V., Kayser S., Wolff D., Tuve S., Kyzirakos C., Bethge W., Greil J., Albert M.H., Schwinger W., Nathrath M. Adoptive transfer of epstein-barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013;31:39–48. doi: 10.1200/JCO.2011.39.8495. [DOI] [PubMed] [Google Scholar]
- 10.Matthes-Martin S., Feuchtinger T., Shaw P.J., Engelhard D., Hirsch H.H., Cordonnier C., Ljungman P., Fourth European Conference on Infections in Leukemia European guidelines for diagnosis and treatment of adenovirus infection in leukemia and stem cell transplantation: summary of ECIL-4 (2011) Transpl. Infect. Dis. 2012;14:555–563. doi: 10.1111/tid.12022. [DOI] [PubMed] [Google Scholar]
- 11.Ljungman P., de la Camara R., Robin C., Crocchiolo R., Einsele H., Hill J.A., Hubacek P., Navarro D., Cordonnier C., Ward K.N., 2017 European Conference on Infections in Leukaemia group Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European Conference on Infections in Leukaemia (ECIL 7) Lancet Infect. Dis. 2019;19:e260–e272. doi: 10.1016/S1473-3099(19)30107-0. [DOI] [PubMed] [Google Scholar]
- 12.Leen A.M., Myers G.D., Sili U., Huls M.H., Weiss H., Leung K.S., Carrum G., Krance R.A., Chang C.C., Molldrem J.J. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat. Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 13.Gerdemann U., Vera J.F., Rooney C.M., Leen A.M. Generation of multivirus-specific T cells to prevent/treat viral infections after allogeneic hematopoietic stem cell transplant. J. Vis. Exp. 2011;51:2736. doi: 10.3791/2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neuenhahn M., Albrecht J., Odendahl M., Schlott F., Dössinger G., Schiemann M., Lakshmipathi S., Martin K., Bunjes D., Harsdorf S. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia. 2017;31:2161–2171. doi: 10.1038/leu.2017.16. [DOI] [PubMed] [Google Scholar]
- 15.Nichols W.G., Corey L., Gooley T., Drew W.L., Miner R., Huang M., Davis C., Boeckh M. Rising pp65 antigenemia during preemptive anticytomegalovirus therapy after allogeneic hematopoietic stem cell transplantation: risk factors, correlation with DNA load, and outcomes. Blood. 2001;97:867–874. doi: 10.1182/blood.v97.4.867. [DOI] [PubMed] [Google Scholar]
- 16.Cain D.W., Cidlowski J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017;17:233–247. doi: 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levinson B.B., Baxter J.D., Rousseau G.G., Tomkins G.M. Cellular site of glucocorticoid-receptor complex formation. Science. 1972;175:189–190. doi: 10.1126/science.175.4018.189. [DOI] [PubMed] [Google Scholar]
- 18.Osborn M.J., Webber B.R., Knipping F., Lonetree C.L., Tennis N., DeFeo A.P., McElroy A.N., Starker C.G., Lee C., Merkel S. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. 2016;24:570–581. doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menger L., Gouble A., Marzolini M.A., Pachnio A., Bergerhoff K., Henry J.Y., Smith J., Pule M., Moss P., Riddell S.R. TALEN-mediated genetic inactivation of the glucocorticoid receptor in cytomegalovirus-specific T cells. Blood. 2015;126:2781–2789. doi: 10.1182/blood-2015-08-664755. [DOI] [PubMed] [Google Scholar]
- 22.Kosicki M., Rajan S.S., Lorenzetti F.C., Wandall H.H., Narimatsu Y., Metzakopian E., Bennett E.P. Dynamics of Indel Profiles Induced by Various CRISPR/Cas9 Delivery Methods. Prog. Mol. Biol. Transl. Sci. 2017;152:49–67. doi: 10.1016/bs.pmbts.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Strauss G., Osen W., Debatin K.M. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin. Exp. Immunol. 2002;128:255–266. doi: 10.1046/j.1365-2249.2002.01777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chicaybam L., Sodre A.L., Curzio B.A., Bonamino M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS ONE. 2013;8:e60298. doi: 10.1371/journal.pone.0060298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]