

Abstract
The successful implementation of chimeric antigen receptor (CAR)-T cell therapy in the clinical context of B cell malignancies has paved the way for further development in the more critical setting of acute myeloid leukemia (AML). Among the potentially targetable AML antigens, CD33 is insofar one of the main validated molecules. Here, we describe the feasibility of engineering cytokine-induced killer (CIK) cells with a CD33.CAR by using the latest optimized version of the non-viral Sleeping Beauty (SB) transposon system “SB100X-pT4.” This offers the advantage of improving CAR expression on CIK cells, while reducing the amount of DNA transposase as compared to the previously employed “SB11-pT” version. SB-modified CD33.CAR-CIK cells exhibited significant antileukemic activity in vitro and in vivo in patient-derived AML xenograft models, reducing AML development when administered as an “early treatment” and delaying AML progression in mice with established disease. Notably, by exploiting an already optimized xenograft chemotherapy model that mimics human induction therapy in mice, we demonstrated for the first time that CD33.CAR-CIK cells are also effective toward chemotherapy resistant/residual AML cells, further supporting its future clinical development and implementation within the current standard regimens.
Keywords: AML, CAR, CD33, non-viral gene transfer, Sleeping Beauty transposon, cytokine-induced killer cells, immunotherapy
Graphical Abstract
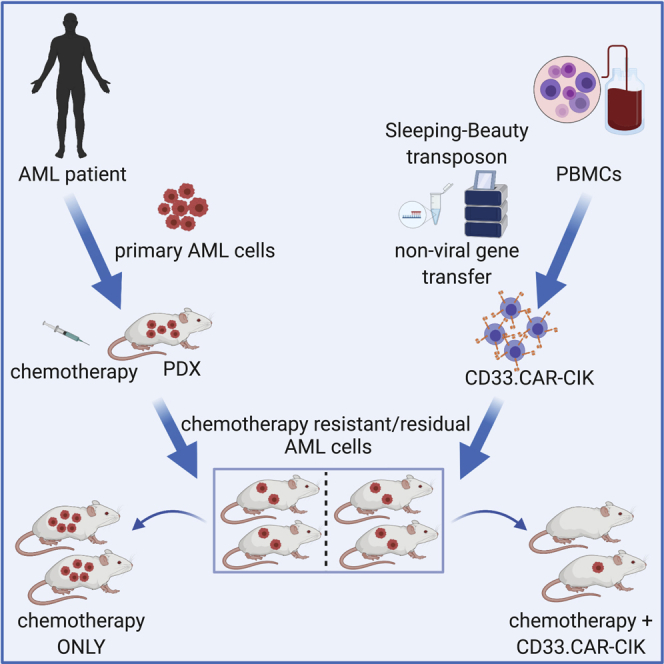
Rotiroti and colleagues exploited the latest version of the non-viral Sleeping Beauty (SB) transposon system “SB100X-pT4” to successfully generate CD33.CAR-redirected cytokine-induced killer (CIK) cells. By using patient-derived xenograft chemotherapy models that mimic induction therapy, they demonstrated for the first time that CD33.CAR-CIK cells are also effective toward chemotherapy-resistant/residual AML cells.
Introduction
Starting from their first laboratory characterization by Zelig Eshhar and coworkers in the late 1980s,1 chimeric antigen receptors (CARs) have now reached bedside applications, revolutionizing the field of cancer immunotherapy, particularly in the context of B cell malignancies.2,3 CARs are recombinant molecules joining an antigen-binding domain, usually derived from a monoclonal antibody (mAb), in the form of a single-chain fragment variable (scFv), to the T cell receptor (TCR) CD3ζ and additional costimulatory domains, thus allowing to redirect the T cell effector functions toward any surface antigen in a human leukocyte antigen (HLA)-independent manner. This potentially reflects a broad applicability for implementing the CAR strategy in other hematological malignancies, such as acute myeloid leukemia (AML), which is still associated with high relapse rates and represents a high unmet medical need. In this clinical setting, the use of CARs is still an open route of investigation due to intrinsic AML characteristics. This includes disease heterogeneity, absence of leukemia-restricted target antigens, and resistance mechanisms toward the immune responses. Among the lineage-specific antigens, the CD33 molecule has been extensively validated as an effective AML immunotherapeutic target in clinical studies.4 It is broadly expressed by leukemic blasts in the majority of AML patients,5 and it is present on leukemic stem cells (LSCs) in some patients.4 While investigating CD33 targeting using retrovirally modified CAR-cytokine-induced killer (CIK) cells, our group already demonstrated a specific anti-AML activity both in vitro and in vivo.6,7
However, the high costs and cumbersome manufacturing processes associated with the use of viral vectors limit the translation of the CAR technology to the clinic. Thus, the development of more sustainable protocols is warranted,8 and current efforts are mainly focused on solving the issues associated with the use of viral vectors.
Non-viral transposon-based gene transfer systems have recently been considered as an alternative to viral vectors for CAR gene transfer. These systems are usually composed of two elements: the transposon and the transposase. The transposon contains the transgene of interest flanked by inverted terminal repeats (ITRs). These are recognized by the transposase, which mediates the excision and integration of the transposon from the donor plasmid to the target genome through a so-called “cut and paste” mechanism. Among the different transposon systems exploited, Sleeping Beauty (SB) has been found to exhibit significantly higher activity in vertebrates compared to other transposons tested,9 with a close-to-random genomic integration pattern.10 Furthermore, the ease and reduced costs associated with its manufacturing combined with the increased biosafety compared to viral vectors make SB vectors appealing for clinical application. Moreover, the SB system has already been clinically employed to genetically modify T cells with a CD19.CAR.11 The transposition efficiency of the SB technology has shown a progressive improvement due to both the generation of hyperactive transposase mutants (SB10, SB11, SB100X) and the optimization of the transposon donor vector architecture (pT, pT2, pT3, pT4).12 Our group has pioneered the use of the SB11-pT platform version to redirect CIK cells with different CARs. With a single stimulation step, we have achieved a stable and efficient CAR expression while simultaneously preserving the phenotype, viability, and expansion of transduced CIK cells.13 Here, we coupled the CD33 anti-AML CAR strategy to the SB technology using the newest SB system to further optimize the CIK cell engineering for the treatment of AML. The new system is composed by the hyperactive SB100X transposase and the pT4 transposon (SB100X-pT4).14,15 This novel SB platform for CAR engineering clearly meets the crucial need of simplifying the production process, which is a prerequisite for a wide application of this technology. Furthermore, by using in vivo models mimicking different disease settings, we achieved a more accurate preclinical evaluation of our anti-AML CAR-T cell immunotherapy strategy. Since chemotherapy-resistant AML represents the main unmet clinical need, we evaluated for the first time the efficacy of SB-modified CD33.CAR-CIK cells in an AML xenograft chemotherapy model.16 This model mimics AML relapse after human induction therapy in mice. These results are fundamental in contributing to a better design of future CAR-based approaches for the treatment of relapsed/refractory (R/R) AML.
Results
Transposition Using SB100X-pT4 Combination Grants Higher Levels of CD33.CAR Expression on CIK Cells Compared to SB11-pT System
According to our previously optimized protocol,13 CIK cells were engineered with the third-generation CD33.CAR by using the SB11-pT and the SB100X-pT4 combinations. To establish the optimal amounts of SB100X transposase- and CD33.CAR pT4 transposon-encoding plasmids to use, different SB100X-pT4 ratios were tested (Figure S1A). We found that the 0.5 μg:15 μg transposase:transposon ratio resulted in a stable and efficient CD33.CAR expression on CIK cells without detectable cell toxicity. Additionally, at the end of the differentiation process, at day 21, we found that the novel SB100X-pT4 combination resulted in higher levels of gene transfer than the previously optimized SB11-pT platform, in terms of both percentage and mean fluorescence intensity (MFI) of the CAR+ cells (median 63.7% versus 51.9%, p = 0.0300; 2,872 versus 1,339, p = 0.0129; n = 9 for SB100X/pT4, n = 7 SB11/pT) (Figures 1A and 1B). Further, the CD33.CAR expression was similarly high in all the CIK cell subpopulations (data not shown). The typical CIK cell phenotype was minimally affected by the genetic modification as compared to the unmanipulated condition (NO DNA) (Figures S1B and S2A). The CD33.CAR-CIK cells showed efficient expansion after 3 weeks of culture (median fold increase of 38.89 for SB100X/pT4 versus 34.23 for SB11/pT, n = 4) (Figure S2B). The median of the vector copy number (VCN) per diploid genome in SB100X/pT4-transduced CIK cells was 8.00 (n = 7; Figure S3A), which falls within the range of VCNs typically seen with CAR-T cells manufactured with non-viral gene transfer systems.17, 18, 19 However, in view of a translation into clinic, a further optimization will be considered to lower this value.
Figure 1.

CAR Expression, Integration Site Analysis, and SB Transposase Detection in SB-Modified CD33.CAR-CIK Cells at Day 21
(A) Percentage of CAR expression. (B) MFI values. (C) The integration distribution of SB represented on the chromosome level for each healthy donor, where healthy donor 1’s 8,063 integrations are in red, donor 2’s 7,213 are in blue, and donor 3’s 6,856 integrations are in green. (D) Pie chart of SB integrations in relation to RefSeq genome annotations as a percentage of the total 22,132 integrations. The integrations within 1 kb upstream and downstream of TSS, 3′, and 5′ UTR were also considered. 94.5% of the integrations were intergenic or in introns. (E) SB transposase transcripts. (F) Western blotting for SB100X in three different CD33.CAR-CIK cell productions. The box contains data that fall between the first and third quartiles, the horizontal line indicates the median, the diamond indicates the mean, and the brackets delineate 1.5 times the interquartile range. Individual data are also shown. Comparisons between groups were done according to Wilcoxon rank sum test.
SB100X-pT4 Transposition Exhibits a Safe, Close-to-Random Integration Profile
The novel version of the SB transposon vector (pT4), optimized for the SB100X transposase, was chosen for its higher transposition efficiency as compared to previous versions.15 Although assumed to have the same random genome-wide integration pattern as previous versions, this has not yet been shown. The DNA of three different healthy donor-derived CD33.CAR-CIK cells was analyzed to determine the integration sites of the transposon and its distribution, as previously described.20,21 Following stringent filtering, 22,132 genome-wide integrations in total for the three samples were determined; donor 1 had 8,063 integrations, donor 2 had 7,213 integrations, and donor 3 had 6,856 integrations. These were compared to RefSeq annotations and to a randomized control, shown in Figure S3B. We found no detectable differences in integration pattern between the donor samples. All donor samples had integrations in all chromosomes (Figure 1C), and the integration pattern of SB was close to random (Figure S3B), with no detectable genomic preferences. The vast majority of integrations are intronic or intergenic, in accordance with previous publications of older SB systems (Figure 1D).13,21
By qPCR, we confirmed the absence of SB100X and SB11 transcripts in our genetically modified CD33.CAR-CIK cells at the end of the culture, which is an important release criterion for the non-viral SB CAR products. Notably and consistent with the lower DNA amount used and shown in Figure 1E, we found even lower transcript levels for SB100X than SB11, (median 0.31/10000 GUS copies, n = 3, versus 12.40/10000 GUS copies, n = 3, p = 0.0495). Furthermore, we confirmed by means of western blotting that our cellular product is free from SB100X protein at an early time point of the differentiation period (i.e., day 7; Figure 1F), while, concerning SB11, we have previously reported that it was still present at day 7 and fell below the limit of detection by qPCR only at day 21.13
SB-Modified CD33.CAR-CIK Cells Exhibit Potent In Vitro Antileukemic Ability
CD33.CAR-CIK cells modified with SB11-pT and SB100X-pT4 combinations were evaluated for their ability to mount effective antileukemic responses in vitro. Despite differences in CAR expression, we found that both transposase/transposon combinations conferred a specific killing of CD33+ AML target cell lines (THP-1 and MA9) as well as primary AML cells, as compared to the NO DNA control. A basal killing capacity toward the CD33− cell line MHH-CALL-4, similar to the NO DNA control, was also detected (Figure 2A). Furthermore, CD33.CAR-CIK cells were able to retain a significant cytotoxic activity when re-challenged with the target cells following a previous stimulation, demonstrating their ability to respond to repetitive antigen exposure (data not shown). Intracellular staining for interferon-γ (IFN-γ) and interleukin-2 (IL-2) revealed a CD33.CAR-specific cytokine production after co-culture with CD33+ target cells (Figures 2B and 2C). Furthermore, both sets of CD33.CAR-CIK cells extensively proliferate in a CD33.CAR-specific manner, as measured by Ki67 staining and carboxyfluorescein succinimidyl ester (CFSE) dilution (Figures 2D–2F).
Figure 2.

In Vitro Characterization of SB-Modified CD33.CAR-CIK Cells
(A) Short-term cytotoxic assay E:T 5:1 (THP-1, n = 7 for NO DNA and SB11/pT, n = 3 for SB100X/pT4; MA9, n = 7 for NO DNA and SB11/pT, n = 6 for SB100X/pT4; PRIMARY AML, n = 6; MHH-CALL-4, n = 7 for NO DNA and SB11/pT, n = 4 for SB100X/pT4). (B and C) Intracellular staining for IFN-γ (B) and IL-2 (C) after 5 h co-culture of CIK cells with the various targets E:T 1:3 (THP-1, n = 7 for NO DNA and SB11/pT, n = 3 for SB100X/pT4; MA9, n = 7 for NO DNA and SB11/pT, n = 4 for SB100X/pT4; PRIMARY AML, n = 7; MHH-CALL-4, n = 7 for NO DNA and SB11/pT, n = 3 for SB100X/pT4). (D) Intracellular staining for Ki67 after 72 h co-culture of CIK cells with the various targets, E:T 1:1 (THP-1, n = 7 for NO DNA and SB11/pT, n = 3 for SB100X/pT4; MA9, n = 3 for NO DNA and SB11/pT, n = 2 for SB100X/pT4; PRIMARY AML, n = 7 for NO DNA and SB11/pT, n = 2 for SB100X/pT4; MHH-CALL-4, n = 7 for NO DNA and SB11/pT, n = 3 for SB100X/pT4). (E and F) CFSE proliferation assay of SB11/pT- (E) and SB100X/pT4- (F) engineered CD33.CAR-CIK cells after 72 h co-culture with the various targets, E:T 1:1. The result of a representative experiment out of 3 is shown. Data represented are the result of mean ± SEM; comparisons between groups were done according to the Kruskal-Wallis test.
SB-Modified CD33.CAR-CIK Cells Are Able to Target AML In Vivo
Next, we sought to investigate the ability of CD33.CAR-CIK cells to control AML growth in vivo focusing on CD33.CAR-CIK cells generated by the new platform SB100X-pT4.
In order to assess the CD33.CAR-CIK cell antileukemic activity over time, we first performed a 1-week cytotoxic assay with a low effector-to-target (E:T) cell ratio of 1:10. Even at this low E:T ratio, CD33.CAR-CIK cells effectively killed the AML target as compared to the NO DNA control (Figure S4). As a result, we established patient-derived xenografts (PDXs) in immunodeficient NSG mice from two different AML samples at the onset of disease and subsequently used them as models for the preclinical examination of our CD33.CAR-CIK cell immunotherapeutic approach (see Table 1 for AML patients’ details).
Table 1.
Characteristics of AML Patients Used for PDX Generation
| Patient | Age | Sex | Fab | Karyotype | Molecular Status |
|---|---|---|---|---|---|
| AML1 | 1 | M | M5a | 46,XY,t(10;11)(p12;q23),der(14)t(1;14)(q?21;q11)[20] | MLL-AF10 |
| AML2 | 13 | F | M2 | 46,XX,t(3;7)(?q25;?q22),t(8;21)(q22;q22)[2]/46,idem,del(9)(q12q22)[18] | AML1-ETO, cKIT mut |
Three different settings to mimic different disease conditions were considered. First, we evaluated the effect of CD33.CAR-CIK cell immunotherapy specifically on leukemia initiating cells (LICs). We therefore administered CD33.CAR-CIK cells as an “early treatment” in mice transplanted with the MA9 cell line, which retains a high frequency of LICs.22 Starting on day 5, mice received once a week for 3 weeks an intravenous infusion of 107 CD33.CAR-CIK cells (Figure S5A). When sacrificed, mice untreated or treated with NO DNA cells exhibited a similar tumor burden. In contrast, and shown in Figure S5B, mice that received CD33.CAR-CIK cells showed a significant reduction in bone marrow (BM) engraftment (median 27.80 for the untreated group and 22.60 for the NO DNA control versus 6.45 for the CD33.CAR-CIK cell group, n = 4 mice per group, p = 0.0207). Moreover, the “early treatment” has been tested in OCI-AML3 Luc-GFP-cell-engrafted mice, to track the tumor burden by In Vivo Imaging System (IVIS) (Figures 3A and 3B). Disease control and significantly more favorable survival outcome were obtained upon CD33.CAR-CIK cell treatment as compared to the untreated mice (n = 4 mice per group, p = 0.0082, Mantel-Cox test; Figure 3C). Then, we moved on testing CD33.CAR-CIK cell “early treatment” in secondarily transplanted PDX (AML1; Table 1) (Figure 4A). This setting presumably reflects the typical LIC properties of self-renewal and leukemia reconstitution.16 A clear BM engraftment reduction was observed in the treated cohort and nearly undetectable in 2 out of 5 mice, whereas a high leukemic burden was detected in the untreated mice (up to 70% of BM engraftment) (Figures 4C and 4F). A significant leukemia reduction was also measured in the peripheral blood (PB) and spleen of treated mice (Figures 4B and 4D). This evidences the potential of CD33.CAR-CIK cells in reducing the dissemination of AML in the periphery. Moreover, the CD33 expression on residual AML cells was similar to the CD33.CAR-CIK-cell-treated and untreated mice (Figures 5A and 5B). When re-exposed ex vivo to CD33.CAR-CIK cells, residual AML cells were still sensitive to the treatment (Figures 5C and 5E), thus indicating that no resistance mechanisms occurred.
Figure 3.

In Vivo Antitumor Efficacy of SB100X/pT4 CD33.CAR-CIK Cells against OCI-AML3 Luc-GFP Cell Line
(A) NSG mice were inoculated with 106 OCI-AML3 Luc-GFP cells. Starting from day 3, mice received once weekly, for 3 weeks, 107 CD33.CAR-CIK cells. (B) Bioluminescence imaging was performed using the IVIS lumina III imaging system (Perkin-Elmer). Tumor burden visualized on days 3, 10, 17, and 24. (C) Kaplan-Meier survival curve representing survival outcomes. Statistical significance was calculated with a Mantel-Cox (log rank) test, p = 0.0082.
Figure 4.

In Vivo Antitumor Efficacy of SB100X/pT4 CD33.CAR-CIK Cells in an “Early Treatment” Model
(A) NSG mice were inoculated with 106 cells of a secondary PDX. Starting from day 5, mice received once weekly, for 3 weeks, 107 CD33.CAR-CIK cells. Mice were sacrificed 7 days after the last injection. (B–D) Percentage of leukemia engraftment in the peripheral blood (PB) (B), in the bone marrow (BM) (C), and spleen (D). (E) Percentage of CD3+ cells in the different districts detected in each of the treated mouse at the end of the experiment. (F) A representative plot of the BM from each group is shown. Comparisons between groups were done according to Wilcoxon rank sum test.
Figure 5.

CD33 Expression on AML Cells and Ex Vivo Treatment of Residual Leukemic Cells
(A and B) Representative dot plots (A) and histogram (B) showing CD33 antigen expression on AML-PDX in untreated (leukemia ONLY) and CD33.CAR-CIK cell-treated mice (BM). (C) Representative dot plots showing the staining with annexin V and 7AAD on ex vivo leukemic cells alone and in the presence of CD33.CAR-CIK cells. (D) Representative dot plots of cytokine production by CD33.CAR-CIK cells following target stimulation. (E) CD33.CAR-CIK cell proliferation by CFSE dilution following target stimulation.
CD33.CAR-CIK cells could be detected in all the analyzed organs with a preferential homing in the spleen (Figure 4E), mostly displaying an effector memory and central memory phenotype (Figure S6).
To determine whether CD33.CAR-CIK cells are also effective in treating established AML, we started the treatment when leukemia engraftment was clearly manifested in the BM (Figures 6A and 6B; AML1-PDX). Even in this setting, a significant decrease of the disease in the treated cohort was found, confirming the efficiency of the proposed CAR strategy (Figures 6C–6E). CIK cells were detectable in all the analyzed districts and maintained the CAR expression (Figures 6F and 6G).
Figure 6.

In Vivo Antitumor Efficacy of SB100X/pT4 CD33.CAR-CIK Cells in an “AML Treatment” Model
(A) NSG mice were inoculated with 106 cells of a secondary PDX. When a substantial tumor burden was evident in the BM, mice received once weekly, for 3 weeks, 107 CD33.CAR-CIK cells. Mice were sacrificed 7 days after the last injection. (B) Representative dot plot of BM engraftment before treatment. (C–E) Percentage of leukemia engraftment in the PB (C), the BM (D), and spleen (E). (F) Percentage of CD3+ cells in the different districts detected in each of the treated mouse at the end of the experiment. (G) A representative dot plot of CD3 and CAR expression from PB of a treated mouse is shown. Comparisons between groups were done according to Wilcoxon rank sum test.
Finally, we wondered if CD33.CAR-CIK cells are also effective in targeting chemotherapy R/R AML cells, which are the main contributors to AML relapse. Using the xenograft chemotherapy model established by Wunderlich et al.,16 we confirmed that the optimized “5+3” chemotherapy induction protocol was able to induce a remission-like status in the BM, producing only mild and temporary side effects. However, disease recurrence has been observed within a short time in both of the established AML PDX models (AML1 and AML2 PDX; Table 1). When adding CD33.CAR-CIK cell treatment on top of chemotherapy (Figure 7A and S7A), we observed a significant decrease of the disease in the CD33.CAR-CIK cell-treated group. It reached undetectable levels in half of the mice (Figures 7B–7D and S7B–S7D), whereas a high leukemic burden was detected in the chemotherapy-only-treated mice (up to 60% of engraftment in BM). In this setting, CD33.CAR-CIK cells were also detected in all the analyzed organs with a preferential homing to the spleen and PB (Figures 7E and S7E). Concomitantly, immune checkpoint molecule characterization revealed an upregulation of TIGIT, TIM-3, PD-1, and LAG-3 in BM-isolated CD33CAR.CIK cells as compared to the spleen-isolated ones and to the same cells before the injection (Figure S8).
Figures 7.

In Vivo Antitumor Efficacy of SB100X/pT4 CD33.CAR-CIK Cells in the Xenograft Chemotherapy Model
(A) NSG mice were inoculated with 106 cells of a secondary PDX. When a substantial tumor burden was evident in the BM (≈20%), mice received the “5+3” chemotherapy induction protocol resulting in a remission-like status with disease recurrence within short time. At the first evidence of disease recurrence, mice received once weekly, for 3 weeks, 107 CD33.CAR-CIK cells. Mice were sacrificed 7 days after the last injection. (B) Percentage of leukemia engraftment in the BM of chemotherapy-only-treated mice (CONTROL) and CD33.CAR-CIK cells treated mice before chemotherapy (CTX), after CTX and after CD33.CAR-CIK cell treatment. (C and D) Percentage of leukemia engraftment at sacrifice in PB (C) and spleen (D). (E) Percentage of CD3+ cells in the different districts detected in each of the treated mouse at the end of the experiment. Comparisons between groups were done according to Wilcoxon rank sum test.
Discussion
CAR-T cell therapy represents a real therapeutic option for certain types of B-ALL and large B cell lymphomas and holds great potential for treating other types of cancer.2,3 Nevertheless, several limitations need to be overcome before its widespread clinical use, including the development of simplified and cost-effective production protocols. Another challenging aspect is the safe and efficient translation of CAR-T cell immunotherapy to other tumor entities.
Currently, CAR-T cells are typically generated by employing viral gene transfer systems that are however associated with high costs and cumbersome manufacturing.23 To address these issues, we preclinically investigated the feasibility of engineering CIK cells with a CD33.CAR through the non-viral SB transposon system24 to target AML. The use of the relatively inexpensive SB vectors would bring down the costs and, in so doing, address the challenge of providing CAR-T cell therapy to a larger number of patients. Additionally, integration data clearly indicate the superior safety profile of SB vectors with respect to retroviral and lentiviral ones.25,26 However, major impediments to a broad diffusion of the SB transposon system has persisted, both due to the low gene transfer efficiency and significant T cell toxicity associated with electroporation and high plasmid doses. Repeated stimulations with feeder cells expressing the CAR target antigen, costimulatory molecules and cytokines were necessary to achieve a sufficient number of CAR-T cells.27 Our group has recently optimized the older SB platform (SB11-pT) in order to reach stable and efficient expression of different CARs on the surface of CIK cells by using γ-irradiated autologous peripheral blood mononuclear cells (PBMCs). This is done in a single stimulation step,13 thus greatly simplifying the culturing process. We also demonstrated that CAR expression on CIK cells can be further increased by using the latest optimized version of the SB transposon system SB100X-pT4.14,15 The new platform offers the advantage of reducing the amount of transposase DNA required for the transposition, further improving the safety of the SB system by limiting the unintended events of transposase integration or transgene remobilization. This should also make sufficient numbers of clinical-grade CAR-redirected CIK cells more easily achieved in a shorter time frame and at lower costs, with undetectable levels of SB100X. This aspect, together with the pT4 close-to-random integration profile and VCN within a range typically obtained with non-viral systems,17 makes the SB100X-pT4 system to be considered safe in a therapeutic context,28,29 with the great benefit of much higher transposition efficiency.
The use of the CIK cell effector population may offer additional advantages in the context of CAR-T cell immunotherapy due to their peculiar biological features. Indeed, CIK cells are heterogeneous polyclonal T effector cells with the phenotypic and functional properties of both T and natural killer (NK) cells.30 CIK cells are therefore able to mediate HLA-independent NK-like cytotoxicity against different types of tumors, thus overcoming the tumor immune escape mechanism of HLA downregulation. Additionally, they have a minimal capacity to cause graft versus host disease (GVHD) in the allogeneic settings.31 This increases the number of patients that could benefit from CIK cell-based therapy. Moreover, the protocol for expanding CIK cells is relatively easy and can be scaled up under GMP conditions in a straightforward and reproducible manner. It is furthermore associated with relatively low costs. A phase II trial has been performed by our group to evaluate the safety and efficacy of donor-derived CIK cells in patients with hematological malignancies relapsing after allogeneic HSCT. Low incidence of GVHD occurred, but a limited disease control has been achieved.32
To improve the antitumor activity of CIK cells, we and others have demonstrated the feasibility of engineering CIK cells to express CARs.33,34 Specifically, we obtained clinically sufficient numbers of donor-derived CD19.CAR-CIK cells with the SB system starting from 50 mL of PB.18
Moving beyond B cell malignancies, there are additional challenges facing the application of CAR-T cell therapy, especially with regards to AML, where the employment of CAR-T cells is still in its infancy. The first pioneering clinical trials have not produced the same impressive results experienced when treating B cell malignancies.35, 36, 37 The myeloid setting is more complex than the lymphoid setting due to the intrinsic characteristics of the disease. This includes heterogeneity, lack of tumor-specific antigens that can lead to off-target toxicity and tolerance, and/or T cell exhaustion induced by immunosuppressive factors within the AML microenvironment. Thus, the development of more predictive preclinical models that better recapitulate the AML complexity would greatly contribute to the ongoing efforts of designing an anti-AML CAR immunotherapy approach that matches efficacy with safety.
In this study, we targeted the CD33 molecule, which is one of the main validated target antigens in AML so far. It is broadly expressed on AML blasts in almost 90% of the patients, with a differential expression between leukemic cells and normal hematopoietic stem/progenitor cells (HSPCs).5,38 In agreement with our previous results obtained with retrovirally manipulated CD33.CAR-CIK cells,6,7 we found that SB-modified CD33.CAR-CIK cells exhibit a potent antileukemic activity in vitro, specifically killing, proliferating, and producing inflammatory cytokines in a target-specific fashion. The antileukemic activity of CD33.CAR-CIK cells in vivo has been demonstrated in NOD-SCID IL2Rγ null (NSG) mice grafted with MA9 and OCI-AML3 cell lines, and in two PDXs established by our group. In order to more accurately evaluate our approach, we established three different settings that resemble different AML disease conditions. Specifically, we found that CD33.CAR-CIK cells are effective against LICs when administered as an “early treatment” in a secondarily transplanted PDX, likely selected for dominant AML subclones. Furthermore, CD33.CAR-CIK cells prolong the survival of treated mice when administered as “early treatment” and significantly delay the progression of an established disease in mice. Moving toward a more translational perspective, efforts have been made to employ a preclinical in vivo model to address the unmet need of targeting drug-resistant AML cells. Therefore, we exploited an already established xenograft chemotherapy mouse model mimicking human induction therapy.16 Notably, induction chemotherapy has been shown to enhance AML aggressiveness, mainly in terms of increasing the disease clonality and activating resistance pathways.39,40 To the best of our knowledge, this is the first in vivo study characterizing CAR-T cells in models of patient-derived R/R AML cells, and it resembles one of the most relevant clinical settings in which CAR-T cells could be exploited. We found that CD33.CAR-CIK cells target chemotherapy R/R AML cells, delaying leukemia progression following chemotherapy treatment. Importantly, CD33.CAR-CIK cell treatment was well tolerated, and no CD33-negative AML cells were detected after up to three infusions of CD33.CAR-CIK cells. Indeed, when residual leukemic cells were ex vivo re-exposed to CD33.CAR-CIK cells, they were still sensitive to the treatment. Upregulation of some immune checkpoint molecules (i.e., PD-1 and TIM-3) has been detected on CD33CAR.CIK cells isolated from the BM of treated mice, suggesting a potential dampening effect exerted by the AML microenvironment on CD33.CAR-CIK cell effector functions.
Our findings do indicate the therapeutic potential of CD33.CAR CIK cells, but the AML cell persistence we observed after treatment clearly demands further efforts to address several obstacles that are known to be unique to this disease. Indeed, significant but incomplete AML control has been observed in other preclinical studies exploiting CAR-T cells,41, 42, 43 and this could explain the transient and limited clinical activity observed so far in first-in-human clinical applications of anti-AML CAR-T cell therapy.44 Understanding the factors underpinning AML survival concomitantly with the design of smarter CARs and/or the use of combinatorial strategies (i.e., with checkpoint inhibitors) could help in counteracting the immunosuppressive AML microenvironment, ultimately resulting in more effective CAR-T cell therapies. A careful evaluation of the clinical therapeutic study design is also essential. A potential clinical application of this currently proposed AML immunotherapeutic strategy would be the administration of donor-derived CD33.CAR-CIK cells to pediatric and young adult patients relapsing after allogeneic transplant. Autologous and/or off-the-shelf cells could be considered as starting sources for chemo-refractory patients or elderly patients who are not fit to receive aggressive chemo-radiation regimens. As the CD33 expression on normal HSPCs can cause myelotoxicity, safeguards to control the sustained myelosuppression are needed in clinical settings. This could entail the incorporation of a suicide gene, drug-mediated T cell depletion, or transplant of CD33-knockout HSPCs to allow persistent myelopoiesis during CD33 CAR treatment.45 Nevertheless, the few published results of early-phase clinical trials exploiting CAR-T cells for the treatment of R/R AML did not report long-term disease control, with a lack of T cell persistence likely contributing to this phenomenon.35,36
The bridge-to-transplant setting would offer a safe therapeutic window to evaluate the antileukemic and myeloablative properties of CD33.CAR-CIK cells, either from autologous and/or off-the-shelf sources. Nevertheless, to reach a complete remission MRD (minimal residual disease) negative status before the transplant, the effectiveness of CD33.CAR-CIK cells within the immunosuppressive AML microenvironment still needs to be fully assessed.
In conclusion, SB-based CIK cell engineering with CD33.CAR offers a unique alternative to current viral-based strategies to be explored in the setting of resistant forms of AML. A phase 1 clinical study at our institution is planned in the near future to address this largely unmet clinical need.
Materials and Methods
Plasmids
The high-affinity, humanized rat anti-human CD33, 113, in scFv generated using UCB’s selected lymphocyte antibody method (kindly provided by UCB Celltech, Slough, UK), was codon optimized and cloned in frame with CH2CH3-CD28 transmembrane-CD28-OX40-ζ6 in the SB transposon plasmid. The SB-pT vector and SB11-transposase were kindly provided by Dr. Cooper (MD Anderson Cancer Center, Houston, TX, USA). The SB-pT4 vector and SB100X-transposase were kindly provided by Dr. Izsvak (MDC, Berlin, Germany).
Generation of CD33.CAR Genetically Modified CIK Cells
CD33.CAR-CIK cells were generated as previously described by adding to peripheral blood mononuclear cells (PBMCs) IFN-γ (1,000 U/mL; Dompè Biotec, Milano, Italy) at day 0 and IL-2 (300 U/mL, Proleukin, Novartis, Basel, Switzerland) and anti-CD3 monoclonal antibody OKT-3 (50 ng/mL, Takara USA, Madison, WI, USA) at day 1.13 Cells were cultured for 21 days; fresh medium and IL-2 were added twice a week.
Cell Lines and Primary AML Cells
THP-1 and MHH-CALL4 cell lines were obtained from the American Type Culture Collection (ATCC). The THP-1 cell line was maintained in advanced RPMI medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Biosera, Ringmer, UK), 2 mM L-glutamine, 25 IU/mL of penicillin, and 25 mg/mL of streptomycin (Lonza, Basel, Switzerland). The MHH-CALL-4 cell line was maintained in 20% FBS advanced RPMI complete medium. MA9-NRas AML cells were kindly provided by Prof. Mulloy (University of Cincinnati, Cincinnati, OH, USA) and cultured in 20% IMDM (Iscove's Modified Dulbecco's Media, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) complete medium. OCI-AML3 Luc-GFP cells were kindly provided by Prof. Falini (University of Perugia, Italy). Primary AML cells were obtained from BM and PBMCs collected from AML patients. The Institutional Review Board of the Ethical Committee of San Gerardo Hospital approved this study, and informed consent was obtained from patients or their guardians.
Flow Cytometry
CIK cells were tested for the expression of CD3 (clone SK7), CD8 (clone RPA-T8), CD4 (clone SK3), CD56 (clone B159), CD62L (clone DREG-56), and CD45R0 (clone UCHL1) using specific antibodies (BD Bioscience, San Jose, CA, USA). CAR expression was detected using an anti-human immunoglobulin G (IgG) (H+L)-specific antibody (Jackson ImmunoResearch, Suffolk, UK). Target cells were assessed for CD45 (clone 2D1), CD33 (clone HIM3-4), CD123 (clone 7G3), and CD19 (clone HIB19) expression (BD). Anti-human IFN-γ (clone B27), IL-2 (clone MQ1-17H129), and Ki67 (clone B56) mAbs (BD) have been used to assess CIK cell cytokine production and proliferation. Cell death and apoptosis were detected using the GFP-certified apoptosis/necrosis detection kit (Enzo Life Sciences, Farmingdale, NY, USA), according to the manufacturer’s instructions. CIK cell proliferation was also assessed by measuring CFSE dilution (Invitrogen). Target cells were also labeled with fluorescein isothiocyanate (FITC) and phycoerythrin (PE) cell tracker (Invitrogen). Human grafts in mice were assessed using anti-human CD45, CD33, and CD123 and anti-mouse CD45 (clone 30-F11) (eBioscience, San Diego, CA, USA) mAbs. Samples were acquired using the FACS Canto II flow cytometer (BD), and data were analyzed using BD FACS DIVA software version 6.1.3.
VCN Analysis
DNA was extracted from CD33.CAR-CIK cells using QIAamp DNA mini kit (QIAGEN, Hilden, Germany) and quantitative real-time PCR was performed on 100 ng of genomic DNA as already described.18 Primers and probes were specific for the inverted repeat/direct repeat (IR/DR) sequences were as follows: forward primer 5′-CTCGTTTTTCAACTACTCCACAAATTTCT-3′; reverse primer 5′-GTGTCATGCACAAAGTAGATGTCCTA-3′; (FAM)-labeled probe 5′-CTGACTTGCCAAAACT-3′.
Integration Site Analysis
The DNA from three different healthy-donor-derived CD33.CAR CIK cells was sequenced on an Ilumina 150-bp single read. The resulting FASTQ file’s 5′ reads were trimmed, using Cutadapt,46 with SB to obtain the flanking human genome sequence. Those that did not match were discarded. As an extra quality filter, we utilized SB’s very strong preference for integrating in TA dinucleotide sites. We therefore checked that the first two basepairs of the trimmed sequences were indeed TA, using FASTQC. The resulting FASTQ file was then aligned to the human reference genome hg19 (UCSC, GRCh37/hg19) using Bowtie2 with local sensitive settings.47 Mapped sequences with less than five identical reads were discarded. The sequences mapped with five or more reads were considered genuine integrations. These were intersected using Bedtools,48 with genome annotations from RefSeq.49 The randomized data for Figure 2B were created using a Poisson regression insertion model (PRIM) to calculate the expected insertion rate for non-overlapping 20-kb windows along the length of each chromosome in the human reference genome (hg19). The PRIM algorithm generated a statistical model based on the number of TA dinucleotides within each window, the chromosome in which the window resides, and the total number of unique insertions. For each window, the expected number of insertions was calculated and compared to the observed number of insertions to produce a p value. Bonferroni correction was then applied to identify windows that showed enrichment for detection of inserted transposons. Random sequences from the reference genome containing TA were then generated, mapped using Bowtie2, and plotted against our real integration data. Calculations and plots were made using ggplot2 in R.50 Figures were drawn using HOMER and ChIPseeker.51,52
Quantitative Real-Time PCR for Detection of Transposase Enzyme
RNA was extracted from CD33.CAR-CIK cells using the QIAamp RNA mini kit (QIAGEN) and retrotrascribed with SuperScript IV VILO master mix (Invitrogen). Amplification by qPCRs of homologous regions within the SB100X and SB11 transcripts was performed using TaqMan gene expression master mix (Applied Biosystems, Thermo Fisher Scientific) and FastStart universal probe master (Roche Diagnostic, Mannheim, Germany). Primers were as follows: forward primer, 5′-AAGCCGAAGAACACCATCC-3′; reverse primer, 5′-AGCACCCCCACAACATGA-3′; UPL probe #87, 5′-CTGACTTGCCAAAACT-3′. The number of copies was quantified based on a seven-point standard curve of plasmidic DNA diltutions from 107 to 20 copies of transposase. β-glucuronidase gene (GUS) was used for data normalization,53 by quantitative RT-PCR using the Ipsogen GUS control gene kit (QIAGEN), and analyzing data in agreement with the statistical delta-delta CT (ddCT) method.54
Western Blotting for Detection of Transposase Enzyme
One million cells were lysed in 100 μL lysis buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 10 mM EDTA, 5% glycerine, 1% NP-40 supplemented by protease inhibitor Mini tablets, EDTA free (Pierce, Thermo Fisher Scientific) and benzonase nuclease (NOVAGEN) for 40 min at 4°C. Cell debris was removed by centrifugation at 14.,00 rpm for 5 min at 4°C. Protein concentration was measured by the bicinchoninic acid assay(BCA) . 30 μg clear lysates were resolved by Bio-Rad 4%–20% gradient mini-protean TGX SDS-PAGE gel and transferred to polyvinylidene fluoride (PVDF) membranes by the Bio-Rad trans-blot turbo transfer system. The membrane was blocked with TBS-T (TBS supplemented with 0.05% Tween-20) containing 5% non-fat dry milk and was then incubated overnight at 4°C with anti-SB antibody (goat; in 1:1,000) R&D Systems AF2798 and anti-actin antibody (mouse; in 1:2,500) (A7811, Sigma-Aldrich) in TBS-T containing 5% non-fat dry milk. After incubation, the membrane was washed intensively with TBS-T. To detect primary antibodies, the washed membrane was incubated for 1 h at room temperature with protein A conjugated to horseradish peroxidase (1:1,000; Merck Millipore, #18-160) in TBS-T containing 5% non-fat dry milk. The blots were subsequently washed with TBS-T, then the bands were detected by ECL prime western blotting detection reagent (Amersham), and images were then analyzed by the ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA).
Overproduction of EGFP-SB100X and SB100X
4 × 106 HEK293 cells were transfected by 1.1 μg pSB100X and pEGFP-SB100X (1:10) plasmid vectors. 1 day postransfection, cells were lysed as it was described before, and 30 μg clear lysate was loaded onto SDS-PAGE gel as positive control to detect SB transposase.
Short- and Long-Term Cytotoxicity Assays
In the short-term cytotoxic assay, CIK cells were co-cultured for 4 h with the target cells (previously labeled with PE-cell tracker) at an E:T ratio of 5:1. Target cell killing was measured through apoptosis detection by flow cytometry as previously described.55 In the long-term cytotoxic assay, CIK cells were co-cultured with THP-1 cells for 1 week at an E:T ratio of 1:10 without exogenous IL-2, on a human BM-derived stromal mesenchymal cell layer as previously described.56 Target cells were then stained with anti-CD33 mAb, and the percentage of viable target cells was detected by flow cytometry-based quantitative analysis.
Proliferation Assay
CD33.CAR CIK cells were co-cultured with the various irradiated target cells (E:T ratio of 1:1). After 72 h co-culture, cells were stained for CD3 and CAR and CAR-CIK cell proliferation was assessed by Ki67 (clone B56, BD) or CFSE (Invitrogen) staining by flow cytometry.
Intracellular Cytokine Staining
CD33.CAR CIK cells were co-cultured with the various target cells (E:T ratio of 1:3). After 2.5 h co-culture, BD GolgiStop was added. After an additional 2.5 h, the cells were stained for CD3 and CAR. IFN-γ (clone B27, BD) and IL-2 (clone MQ1-17H129, BD) were used for intracellular staining.
Mouse Models
For cell line experiments, 0.2 × 106 MA9 cells or 106 OCI-AML3 Luc-GFP cells were transplanted in NSG mice (The Jackson Laboratory, Bar Harbor, ME, USA). Human AML-PDX models were generated by transplanting NSG recipient mice with splenocytes and/or BM cells from primary NSG donors. Mice received an intravenous infusion of 107 CD33.CAR-CIK cells once a week for 3 weeks. CIK cell administration started on day 3 or 5 after transplantation, in the “early treatment approach,” or after clear leukemia engraftment in the BM for the “AML treatment model.” Bioluminescence imaging was performed using an IVIS Lumina III imaging system (PerkinElmer, USA). Whole-body imaging was performed 12 min after subcutaneous injection with 150 mg/kg firefly D-luciferin (PerkinElmer, USA) in isoflurane-anesthetized mice. Image processing was carried out with a background subtraction using Living Image software (PerkinElmer, USA) and expressed as photons/s/cm2/steradian (avg. radiance). The xenograft chemotherapy model was established as previously described.16 After leukemia engraftment (≈20% in the BM), mice were treated for with 50 mg/kg cytarabine (Sigma-Aldrich, Haverhill, UK) (day 1–5) together with 1.5 mg/kg doxorubicin (Sigma-Aldrich) (day 1–3). The study was approved by the Italian Ministry of Health (approval no. 102/2013-B). Procedures involving animal handling and care were conformed to protocols approved by the Milano-Bicocca University in compliance with national and international law and policies.
Statistical Analysis
Descriptions were based on mean ± SEM or median, as appropriate. Comparisons between groups were done according to non-parametric tests: the Kruskal-Wallis (three groups) or the Wilcoxon rank sum test (two groups). All the tests were two sided, with a level of significance of 0.05. The log-rank (Mantel-Cox) test was utilized in the survival studies analysis to calculate the statistical significance. Analyses were performed using R 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism (La Jolla, CA, USA) softwares.
Author Contributions
M.C.R. and S.T. designed the research, performed the experiments, analyzed the data, and wrote the paper. C.B. and V.M.P. contributed to performing the experiments, S.A., C.F.M., C.C., and G.A. provided technical support. Z.I. provided the SB vectors, and T.R., F.L., and A.P. performed the integration site analyses. S.G. and M.G.V. performed the statistical analyses. S.T. and A.B. coordinated the research. E.B., A.B., M.S., G.D., and M.I. revised the data and performed a final revision of the paper.
Conflicts of Interest
Fondazione Tettamanti submitted a PCT application on November 6, 2015 (PCT/EP2015/075980), “Improved method for the generation of genetically modified cells,” Biondi, A., Biagi, E., Magnani, C.F., Tettamanti, S.
Acknowledgments
The authors thank Dr. Grazia Fazio, Dr. Silvia Rigamonti, Beatrice Heuser, and Dr. Csaba Miskey for technical support. The authors also thank the parent committees Quelli che…con LUCA onlus, Comitato Maria Letizia Verga, Amici di Duccio, and Stefano Verri for their generous and constant support. M.C.R. was supported by fellowships from the Doctoral Program in Molecular and Translational Medicine (DIMET, University of Milano-Bicocca), the Beat Leukemia Foundation, and Società Italiana Ematologia Sperimentale (SIES). The work was supported by grants from AIRC IG 2015, “Novel leukemia treatment by the use of chimeric antigen receptors (CARs)” (grant 17248); Ricerca Finalizzata-Giovani Ricercatori (GR-2016-02363491); AIRC 5x1000 “Immunity in Cancer Spreading and Metastasis (ISM)” (grant 21147); and the Plagencell project by Fondazione Regionale per la Ricerca Biomedica. Gratitude goes to Quelli che…con LUCA onlus, which funded the project, as well as to the Paolo Belli AIL Bergamo section.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.021.
Supplemental Information
References
- 1.Gross G., Waks T., Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter R.B., Appelbaum F.R., Estey E.H., Bernstein I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehninger A., Kramer M., Röllig C., Thiede C., Bornhäuser M., von Bonin M., Wermke M., Feldmann A., Bachmann M., Ehninger G., Oelschlägel U. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marin V., Pizzitola I., Agostoni V., Attianese G.M.P.G., Finney H., Lawson A., Pule M., Rousseau R., Biondi A., Biagi E. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica. 2010;95:2144–2152. doi: 10.3324/haematol.2010.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pizzitola I., Anjos-Afonso F., Rouault-Pierre K., Lassailly F., Tettamanti S., Spinelli O., Biondi A., Biagi E., Bonnet D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser A.D., Assenmacher M., Schröder B., Meyer M., Orentas R., Bethke U., Dropulic B. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Ther. 2015;22:72–78. doi: 10.1038/cgt.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer S.E.J., Wienholds E., Plasterk R.H.A. Regulated transposition of a fish transposon in the mouse germ line. Proc. Natl. Acad. Sci. USA. 2001;98:6759–6764. doi: 10.1073/pnas.121569298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackett P.B., Largaespada D.A., Switzer K.C., Cooper L.J.N. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Transl. Res. 2013;161:265–283. doi: 10.1016/j.trsl.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kebriaei P., Singh H., Huls M.H., Figliola M.J., Bassett R., Olivares S., Jena B., Dawson M.J., Kumaresan P.R., Su S. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Invest. 2016;126:3363–3376. doi: 10.1172/JCI86721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kebriaei P., Izsvák Z., Narayanavari S.A., Singh H., Ivics Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet. 2017;33:852–870. doi: 10.1016/j.tig.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Magnani C.F., Turazzi N., Benedicenti F., Calabria A., Tenderini E., Tettamanti S., Giordano Attianese G.M., Cooper L.J., Aiuti A., Montini E. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget. 2016;7:51581–51597. doi: 10.18632/oncotarget.9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mátés L., Chuah M.K.L., Belay E., Jerchow B., Manoj N., Acosta-Sanchez A., Grzela D.P., Schmitt A., Becker K., Matrai J. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009;41:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Pryputniewicz-Dobrinska D., Nagy E.E., Kaufman C.D., Singh M., Yant S., Wang J., Dalda A., Kay M.A., Ivics Z., Izsvák Z. Regulated complex assembly safeguards the fidelity of Sleeping Beauty transposition. Nucleic Acids Res. 2017;45:311–326. doi: 10.1093/nar/gkw1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wunderlich M., Mizukawa B., Chou F.S., Sexton C., Shrestha M., Saunthararajah Y., Mulloy J.C. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–e97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miskey C., Amberger M., Reiser M., Prommersberger S., Beckmann J., Machwirth M., Einsele H., Hudecek M., Bonig H., Ivics Z., on behalf of the CARAMBA consortium Genomic Analyses of SLAMF7 CAR-T Cells Manufactured by Sleeping Beauty Transposon Gene Transfer for Immunotherapy of Multiple Myeloma. bioRxiv. 2019:675009. [Google Scholar]
- 18.Magnani C.F., Mezzanotte C., Cappuzzello C., Bardini M., Tettamanti S., Fazio G., Cooper L.J.N., Dastoli G., Cazzaniga G., Biondi A., Biagi E. Preclinical Efficacy and Safety of CD19CAR Cytokine-Induced Killer Cells Transfected with Sleeping Beauty Transposon for the Treatment of Acute Lymphoblastic Leukemia. Hum. Gene Ther. 2018;29:602–613. doi: 10.1089/hum.2017.207. [DOI] [PubMed] [Google Scholar]
- 19.van der Velden V.H.J., Cazzaniga G., Schrauder A., Hancock J., Bader P., Panzer-Grumayer E.R., Flohr T., Sutton R., Cave H., Madsen H.O., European Study Group on MRD detection in ALL (ESG-MRD-ALL) Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21:604–611. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 20.Moldt B., Miskey C., Staunstrup N.H., Gogol-Döring A., Bak R.O., Sharma N., Mátés L., Izsvák Z., Chen W., Ivics Z., Mikkelsen J.G. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol. Ther. 2011;19:1499–1510. doi: 10.1038/mt.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter M., Saydaminova K., Yumul R., Krishnan R., Liu J., Nagy E.E., Singh M., Izsvák Z., Cattaneo R., Uckert W. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood. 2016;128:2206–2217. doi: 10.1182/blood-2016-04-711580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulloy J.C., Wunderlich M., Zheng Y., Wei J. Transforming human blood stem and progenitor cells: a new way forward in leukemia modeling. Cell Cycle. 2008;7:3314–3319. doi: 10.4161/cc.7.21.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milone M.C., O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hackett P.B., Largaespada D.A., Cooper L.J.N. A transposon and transposase system for human application. Mol. Ther. 2010;18:674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field A.C., Vink C., Gabriel R., Al-Subki R., Schmidt M., Goulden N., Stauss H., Thrasher A., Morris E., Qasim W. Comparison of lentiviral and sleeping beauty mediated αβ T cell receptor gene transfer. PLoS ONE. 2013;8:e68201. doi: 10.1371/journal.pone.0068201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vargas J.E., Chicaybam L., Stein R.T., Tanuri A., Delgado-Cañedo A., Bonamino M.H. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J. Transl. Med. 2016;14:288. doi: 10.1186/s12967-016-1047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X., Wilber A.C., Bao L., Tuong D., Tolar J., Orchard P.J., Levine B.L., June C.H., McIvor R.S., Blazar B.R., Zhou X. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood. 2006;107:483–491. doi: 10.1182/blood-2005-05-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gogol-Döring A., Ammar I., Gupta S., Bunse M., Miskey C., Chen W., Uckert W., Schulz T.F., Izsvák Z., Ivics Z. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4+ T cells. Mol. Ther. 2016;24:592–606. doi: 10.1038/mt.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jong J., Akhtar W., Badhai J., Rust A.G., Rad R., Hilkens J., Berns A., van Lohuizen M., Wessels L.F., de Ridder J. Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet. 2014;10:e1004250. doi: 10.1371/journal.pgen.1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt-Wolf I.G., Negrin R.S., Kiem H.P., Blume K.G., Weissman I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991;174:139–149. doi: 10.1084/jem.174.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X., Mi Y., Guo N., Xu H., Xu L., Gou X., Jin W. Cytokine-Induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017;8:774. doi: 10.3389/fimmu.2017.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Introna M., Lussana F., Algarotti A., Gotti E., Valgardsdottir R., Micò C., Grassi A., Pavoni C., Ferrari M.L., Delaini F. Phase II Study of Sequential Infusion of Donor Lymphocyte Infusion and Cytokine-Induced Killer Cells for Patients Relapsed after Allogeneic Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017;23:2070–2078. doi: 10.1016/j.bbmt.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Marin V., Dander E., Biagi E., Introna M., Fazio G., Biondi A., D’Amico G. Characterization of in vitro migratory properties of anti-CD19 chimeric receptor-redirected CIK cells for their potential use in B-ALL immunotherapy. Exp. Hematol. 2006;34:1219–1229. doi: 10.1016/j.exphem.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Zuo S., Wen Y., Panha H., Dai G., Wang L., Ren X., Fu K. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol. Immunol. 2017;85:293–304. doi: 10.1016/j.molimm.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 35.Ritchie D.S., Neeson P.J., Khot A., Peinert S., Tai T., Tainton K., Chen K., Shin M., Wall D.M., Hönemann D. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013;21:2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q.S., Wang Y., Lv H.Y., Han Q.W., Fan H., Guo B., Wang L.L., Han W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baumeister S.H., Murad J., Werner L., Daley H., Trebeden-Negre H., Gicobi J.K., Schmucker A., Reder J., Sentman C.L., Gilham D.E. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019;7:100–112. doi: 10.1158/2326-6066.CIR-18-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haubner S., Perna F., Köhnke T., Schmidt C., Berman S., Augsberger C., Schnorfeil F.M., Krupka C., Lichtenegger F.S., Liu X. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33:64–74. doi: 10.1038/s41375-018-0180-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hackl H., Astanina K., Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017;10:51. doi: 10.1186/s13045-017-0416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeer N.A., Philip J., Geiger H., Ries R.E., Lavallée V.P., Walsh M., Shah M., Arora K., Emde A.K., Robine N. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia. 2019;33:1934–1943. doi: 10.1038/s41375-019-0402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Hear C., Heiber J.F., Schubert I., Fey G., Geiger T.L. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. 2015;100:336–344. doi: 10.3324/haematol.2014.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenderian S.S., Ruella M., Shestova O., Klichinsky M., Kim M.Y., Porter D.L., June C.H., Gill S.I. Identification of PD1 and TIM3 As Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Blood. 2015;126:852. [Google Scholar]
- 43.Petrov J.C., Wada M., Pinz K.G., Yan L.E., Chen K.H., Shuai X., Liu H., Chen X., Leung L.H., Salman H. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018;32:1317–1326. doi: 10.1038/s41375-018-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummins K.D., Gill S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin. Hematol. 2019;56:155–163. doi: 10.1053/j.seminhematol.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Kim M.Y., Yu K.R., Kenderian S.S., Ruella M., Chen S., Shin T.H., Aljanahi A.A., Schreeder D., Klichinsky M., Shestova O. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell. 2018;173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. [Google Scholar]
- 47.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hadley W. Springer; 2009. Ggplot2: Elegant Graphics for Data Anlysis. [Google Scholar]
- 51.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 53.Beillard E., Pallisgaard N., van der Velden V.H.J., Bi W., Dee R., van der Schoot E., Delabesse E., Macintyre E., Gottardi E., Saglio G. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) - a Europe against cancer program. Leukemia. 2003;17:2474–2486. doi: 10.1038/sj.leu.2403136. [DOI] [PubMed] [Google Scholar]
- 54.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 55.Arcangeli S., Rotiroti M.C., Bardelli M., Simonelli L., Magnani C.F., Biondi A., Biagi E., Tettamanti S., Varani L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. 2017;25:1933–1945. doi: 10.1016/j.ymthe.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marin V., Kakuda H., Dander E., Imai C., Campana D., Biondi A., D’Amico G. Enhancement of the anti-leukemic activity of cytokine induced killer cells with an anti-CD19 chimeric receptor delivering a 4-1BB-ζ activating signal. Exp. Hematol. 2007;35:1388–1397. doi: 10.1016/j.exphem.2007.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.