

Abstract
Duchenne muscular dystrophy (DMD), one of the most common neuromuscular disorders of children, is caused by the absence of dystrophin protein in striated muscle. Deletions of exons 43, 45, and 52 represent mutational “hotspot” regions in the dystrophin gene. We created three new DMD mouse models harboring deletions of exons 43, 45, and 52 to represent common DMD mutations. To optimize CRISPR-Cas9 genome editing using the single-cut strategy, we identified single guide RNAs (sgRNAs) capable of restoring dystrophin expression by inducing exon skipping and reframing. Intramuscular delivery of AAV9 encoding SpCas9 and selected sgRNAs efficiently restored dystrophin expression in these new mouse models, offering a platform for future studies of dystrophin gene correction therapies. To validate the therapeutic potential of this approach, we identified sgRNAs capable of restoring dystrophin expression by the single-cut strategy in cardiomyocytes derived from human induced pluripotent stem cells (iPSCs) with each of these hotspot deletion mutations. We found that the potential effectiveness of individual sgRNAs in correction of DMD mutations cannot be predicted a priori, highlighting the importance of sgRNA design and testing as a prelude for applying gene editing as a therapeutic strategy for DMD.
Keywords: dystrophin, CRISPR-Cas9, single guide RNA, myopathy, AAV9, human iPSCs
Graphical Abstract
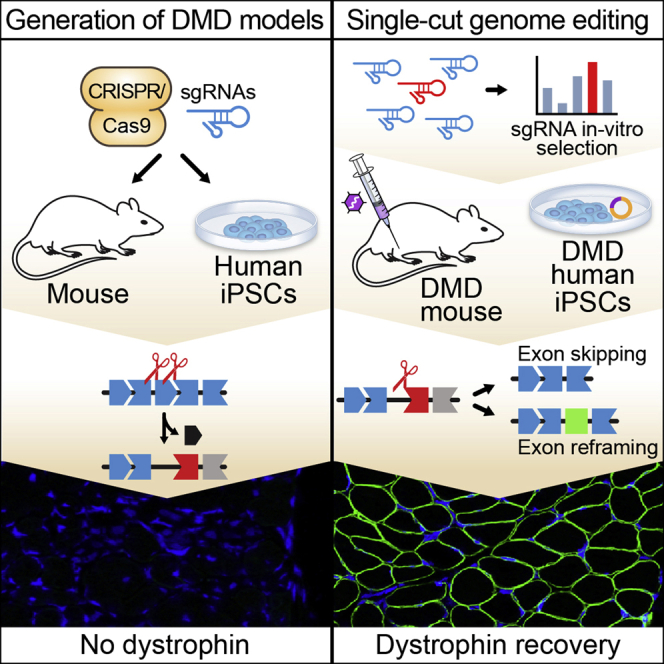
Olson and colleagues generated new mouse and human iPSC models of Duchenne muscular dystrophy (DMD), harboring common exon deletions found in DMD patients. A CRISPR-Cas9 single-cut genome editing approach enabled exon skipping and exon reframing to efficiently restore dystrophin expression in the DMD models.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder caused by mutations in the DMD gene, a massive gene spanning ~2.3 Mb of DNA, encoding the dystrophin protein.1 Dystrophin stabilizes muscle membranes by tethering the actin cytoskeleton to laminin on the inner surface of the sarcolemma.2,3 The incidence of DMD is estimated at 1:5,000 boys worldwide. More than 7,000 mutations have been identified in DMD patients,4 leading to the generation of a premature stop codon in the transcript and, consequently, to the absence of dystrophin protein. Many of these mutations lie within one of the hotspot regions spanning exons 43 to 53 of the dystrophin gene.4,5 Lack of dystrophin in skeletal muscle results in fragility of the sarcolemma, myocyte necrosis, and eventual replacement of muscle with fibrotic and fatty tissue.6 The symptoms of DMD are initially manifested by muscle weakness and a loss of ambulation but ultimately culminate in cardiomyopathy, respiratory failure, and premature death. Thus far, there has been no effective cure for the disease, despite numerous therapeutic efforts.
Full restoration of normal levels of dystrophin is not essential for an effective therapy. It has been estimated that as little as ~15% of normal levels of dystrophin protein could confer substantial therapeutic benefits.7 However, the minimal required level likely depends on the form of dystrophin being expressed. In a canine model of DMD, expression of micro-dystrophin (a truncated form of dystrophin) at ~50% of wild-type (WT) levels was shown to confer therapeutic benefit.8, 9, 10
Clustered regularly interspaced short palindromic repeat (CRISPR)-mediated gene editing represents a means of removing disease-causing mutations in the genome.11, 12, 13 With this approach, single guide RNAs (sgRNAs) are used to direct Cas9 or other endonucleases to specific sequences in the genome. In the presence of an exogenous DNA template, Cas9 can promote homology-directed DNA repair (HDR), whereas in the absence of a template, DNA double-strand breaks (DSBs) are repaired by non-homologous end joining (NHEJ) with various insertions or deletions (INDELs).14 Another pathway for repair of CRISPR-mediated DNA DSBs is microhomology-mediated end-joining (MMEJ), which uses regions with 5–25 bp of microhomology flanking a DSB for repair and results in deletion of the region between the microhomology.15
Recent studies by our group and others have explored the potential of gene editing as a means of correcting DMD mutations in vivo.16, 17, 18, 19, 20, 21, 22 Because HDR and MMEJ are restricted to proliferating cells, and mature myocytes are post-mitotic, CRISPR-Cas9 gene correction of DMD mutations requires the NHEJ pathway.23,24 NHEJ gene editing by double-cut or single-cut has been deployed to correct the open reading frame (ORF) of dystrophin and restore protein expression from mutant DMD alleles. Each editing approach has unique advantages and challenges.
Double-cut editing offers the potential to excise large regions of the DMD gene, producing truncated but still functional dystrophin.20,22,25,26 This approach allows correction of multiple types of mutations in a consolidated manner. However, it requires simultaneous DNA cutting with two sgRNAs separated by extended genomic distances and is relatively inefficient. Double-cutting can also introduce a relatively high frequency of unpredictable genomic rearrangements.21
To date, more than 60 spontaneous or engineered DMD animal models have been reported.27,28 CRISPR-Cas9 has been used to restore dystrophin expression in mouse, dog, pig, and human cells harboring the most common mutations in DMD patients, such as deletion of exon 50 or exon 44.16,18,19 Human exon 45 was deleted in the hDMD mouse model, which is a mouse containing the human dystrophin gene.29,30 This mouse model allows testing of possible genetic interventions designed to treat human patients.
With single-cut CRISPR editing, a sgRNA can be used to introduce INDELs that eliminate splice acceptor or donor sites or out-of-frame exons, thereby allowing exon skipping and restoration of the correct ORF.16,18,19 Alternatively, INDELs can be introduced within out-of-frame exons to restore the ORF by shifting the reading frame triplet forward (3n − 1 nt) or backward (3n + 1 nt). Single-cut editing has several advantages, including requiring only one sgRNA, relatively high efficiency, and minimal modification of the genome. However, it requires accurate optimization of sgRNAs for each mutational hotspot. For example, deletion of exon 50,16,18 exon 44,19,31 or exon 52 can be corrected by reframing the triplet codon reading frame of one of the flanking exons backward (3n + 1 nt), but deletion of exon 43 or exon 45 necessitates reframing of the triplet reading frame forward (3n − 1 nt) to achieve correction.
In addition to gene editing approaches based on the generation and repair of DSBs, CRISPR base editors have been developed that allow precise conversion of a single nucleotide base into another base.32 These genome editing tools have been used to correct the point mutation in the mdx mouse model of DMD. However, the large size of base editors necessitates splitting into two halves in vivo delivery, decreasing editing efficiency and dystrophin restoration.33
By packaging sgRNA and Cas9 expression cassettes into adeno-associated viruses (AAVs), efficient gene editing and dystrophin restoration has been achieved in mice and other large animal models, such as dogs and pigs.16, 17, 18, 19, 20,22,25,26,34 One sgRNA or two sgRNAs have been used to permanently correct different mdx mouse models, as well as mice lacking exons 44 and 50. Interestingly, a recent study demonstrated the correction of an exon 52 deletion in a pig model of DMD by double-cutting sequences flanking exon 51 with CRISPR-Cas9.26 In this study, we addressed the potential of single-cut gene editing in the exon-52-deleted mouse model.
For single-cut editing, a highly efficient sgRNA with the preferential generation of the correct INDEL patterns at the DSB site is required. However, it is not currently possible to predict a priori what sgRNAs might be most effective in allowing gene editing at different sites, necessitating experimental validation and identification of optimal sgRNAs for each type of mutational hotspot.
In this study, we generated three new DMD mouse models: Δ43 (exon 43 deletion), Δ45 (exon 45 deletion), and Δ52 (exon 52 deletion) DMD mice, representing three common exon deletions in DMD patients. For each of these mouse models, we developed single-cut CRISPR editing strategies to restore dystrophin expression. We used a dual-AAV delivery system with single-stranded AAV (ssAAV) to express SpCas9 and self-complementary AAV (scAAV) to express sgRNAs to deliver the CRISPR components and reframe the mutant Dmd gene to correct dystrophin expression in skeletal muscle. Additionally, to assess the therapeutic potential of the single-cut strategy, we generated cardiomyocytes from human DMD induced pluripotent stem cells (iPSCs) and tested the sgRNAs for effective CRISPR editing to restore dystrophin expression in these cells. This single-cut CRISPR gene editing strategy could potentially be effective as a therapy for ~18% of the DMD patient population. Our findings highlight the potential of gene editing to permanently correct a wide range of mutations that cause DMD and represent a significant step toward potential therapeutic translation.
Results
Generation of Mice with Deletions of Dystrophin Exon 52, 43, or 45
To extend gene editing as a means of correcting common DMD mutations that have not been previously addressed in vivo, we generated mice with a deletion of exon 52 (Δ52 DMD), exon 43 (Δ43 DMD), or exon 45 (Δ45 DMD) using CRISPR-Cas9 directed by two sgRNAs flanking each exon (Figure 1A; Table S1). C57BL/6 zygotes were co-injected with in-vitro-transcribed Cas9 mRNA and sgRNAs and then re-implanted into pseudo-pregnant females, yielding offspring that transmitted the mutant Dmd alleles through the germline. Deletion of Dmd exon 52, exon 43, or exon 45 was confirmed by RT-PCR analysis (Figure 1A). Deletion of each exon placed the dystrophin gene out of frame, leading to the absence of dystrophin protein in skeletal muscle and heart (Figure 1B). Mice lacking each exon showed pronounced dystrophic muscle at 1 month of age (Figure 1C). Serum analysis of the Δ52, Δ43, and Δ45 DMD mice showed elevated creatine kinase (CK) activity, a hallmark of muscle damage (Figure 1D). Overall, the severity and progression of disease in these mice, as marked by the absence of dystrophin protein expression, muscle histology, and serum CK (Figures 1B–1D), were comparable to those in other mouse models of DMD, such as mdx mice that were described previously.35,36
Figure 1.

Generation and Characterization of DMD Mouse Models with Deletion of Exon 52, 43, or 45
(A) CRISPR-Cas9 editing using two sgRNAs flanking an exon was used to delete exon 52, 43, or 45 and generate DMD mouse model Δ52, Δ43, or Δ45. Length of deleted exon is indicated (118 bp for Δ52, 173 bp for Δ43, and 176 bp for Δ45). PCR products generated by primers flanking the deleted exons are indicated beneath each set of exons. Shapes of intron-exon junctions denote complementarity that maintains the ORF upon splicing. (B) Dystrophin staining of TA, diaphragm, and heart of WT and of Δ52, Δ43, and Δ45 DMD mice. Dystrophin is indicated in green. Nuclei are marked by DAPI stain in blue. Scale bar, 50 μm. (C) H&E staining of TA, diaphragm, and heart of WT and of Δ52, Δ43, and Δ45 DMD mice. Note extensive inflammatory infiltrate and centralized myonuclei in Δ52, Δ43, and Δ45 DMD mice. Scale bar, 100 μm. (D) Serum creatine kinase (CK), a marker of muscle damage and membrane leakage, was measured in WT1 (C57BL/6) and WT2 (C57BL/10), mdx, Δ52, Δ43, and Δ45 DMD mice. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001 (n = 5). Data are presented as means ± SEM.
Correction of DMD Exon 52 Deletion in Mice by Intramuscular (IM) AAV9 Delivery of Gene Editing Components
In Δ52 DMD mice, the absence of exon 52 causes exon 53 to be out of frame with preceding exons (Figures 2A and S1A). Skipping of exon 53 or introduction of 3n + 1 INDELs before the stop codon in exon 53 has the potential to restore the correct ORF of dystrophin. Destroying the splice acceptor site for exon 53 by targeting SpCas9 using a specific sgRNA to that sequence will cause exon 53 skipping, allowing splicing of exon 51 to exon 54 and restoration of dystrophin expression (Figure 2A). Following NHEJ, repair of the DSB at the splice acceptor site may also introduce 3n + 1 INDELs in exon 53, allowing reframing and restoration of dystrophin expression (Figure 2A).
Figure 2.

Intramuscular AAV9 Delivery of Gene Editing Components to Δ52 DMD Mice Rescues Dystrophin Expression
(A) Diagram for sgRNA targeting strategy of exon 53 (212 bp) and potential products after editing. (B) RT-PCR analysis of TA muscles from WT and Δ52 DMD mice 3 weeks after intramuscular injection of ssAAV-Cas9 and scAAV-mE53 g2. Restoration of the ORF is obtained by reframing (middle bands, 379 bp) or skipping of exon 53 (lower bands, 167 bp). The proportion of the different products is illustrated in Figure S2A. (C) Western blot analysis indicates restoration of dystrophin expression in TA muscle of Δ52 DMD mice after AAV-Cas9 and AAV-mE53 g2 treatment. Relative dystrophin intensity (%) was calibrated with vinculin internal control before normalizing to the untreated WT control. (D) Immunohistochemistry indicates restoration of dystrophin in TA muscle of Δ52 DMD mice 3 weeks after intramuscular injection of gene editing components sgRNA-mE53 g2 and SpCas9, carried by AAV9. Dystrophin is indicated in green. Nuclei are marked by DAPI stain in blue. Scale bar, 100 μm.
For CRISPR single-cut editing of the splice acceptor site for exon 53, we designed 17 mouse sgRNAs (marked with “m”) in the proximity of the 5′ end of mouse exon 53 (Figure S1B). The cutting sites of the sgRNAs with reframing potential were designed to be located upstream of the premature stop codon in exon 53 that results from the deletion of exon 52 (Figure S1B). Using mouse N2a cells to screen genomic editing of sgRNAs that target exon 53, we found that sgRNA mE53 g2 showed superior total INDEL efficiency by Tracking of INDELs by Decomposition (TIDE) analysis (Figure S1C). Based on these in vitro findings, we selected mouse sgRNA mE53 g2 for evaluating in vivo exon skipping and/or exon reframing in the Δ52 DMD mouse model.
We used recombinant AAV9 to deliver the CRISPR components to the DMD mouse models, because AAV9 is a DNA virus that displays tropism to both skeletal muscle and heart and has been used in numerous clinical trials.37, 38, 39, 40 To test the genomic editing efficiency of the sgRNA in vivo, we used a dual-AAV9 viral system due to the cargo size limitation of AAV9 delivery system.18 We packaged an SpCas9 expression cassette in a single-stranded AAV9 (ssAAV9) vector and packaged the mouse sgRNA mE53 g2 expression cassette in a different self-complementary AAV9 (scAAV9) vector. Based on our previous studies, we discovered that scAAV9 expresses higher levels of sgRNA and increases the efficiency of genomic editing.31 To enhance muscle-specific gene editing and ensure SpCas9 expression primarily in skeletal and cardiac muscle, the CK8e regulatory cassette that combines enhancer and promoter regions of the muscle CK gene was utilized to drive SpCas9 expression in skeletal muscle.41,42 For expression of sgRNA, we used three RNA polymerase III promoters (U6, H1, and 7SK) to express three copies of each sgRNA, as described previously.43
To validate the efficacy of the single-cut genomic editing strategy in the Δ52 DMD mouse model, we performed localized IM injection of ssAAV9 encoding SpCas9 (ssAAV-Cas9) and scAAV9 encoding sgRNA (scAAV-mE53 g2) in the tibialis anterior (TA) muscle of postnatal day 12 (P12) mice. As a control group, WT and Δ52 DMD mice were injected with ssAAV-Cas9 without scAAV-sgRNA. In this study, 50 μL AAV9 was injected per leg, containing equal doses of ssAAV-Cas9 (5 × 1010 vg per leg) and scAAV-sgRNAs (5 × 1010 vg per leg). Three weeks after IM injection, we collected the TA muscles for analysis.
RT-PCR showed a distinct exon skipping band at 167 bp below the Δ52 DMD band at 379 bp (Figure 2B). TIDE analysis of the RT-PCR product also revealed that, in addition to exon skipping (10.3%), sgRNA mE53 g2 generated INDELs that allowed the reframing of the ORF (3n + 1, 4.3%; Figure S2A).
To evaluate dystrophin protein restoration after IM injection, we performed western blot analysis on the TA muscles of ssAAV-Cas9/scAAV-mE53 g2 treated Δ52 DMD mice. In ssAAV-Cas9/scAAV-mE53 g2-treated TA muscle, we observed ~50% of dystrophin protein restoration (Figure 2C). Immunostaining and whole-muscle scanning also revealed that ssAAV-Cas9/scAAV-mE53 g2-treated muscles restored ~72% of dystrophin-positive fibers (Figures 2D and S3A). Histological whole-muscle scanning and hematoxylin and eosin (H&E) staining showed that injection of ssAAV-Cas9/scAAV-mE53 g2 rescued the dystrophic phenotype, reducing necrotic cells and centralized nuclei (Figures S3B and S3C).
Correction of DMD Exon 52 Deletion in Human iPSCs by Delivery of Gene Editing Components
Correction of DMD exon 52 deletion by gene editing of exon 53 can potentially benefit 10% of DMD patients.5 To test whether the gene editing strategy that we validated in mice also works in human cells from DMD patients, we generated exon 52-deleted human iPSCs (human Δ52 DMD) by reprogramming the peripheral blood mononuclear cells (PBMCs) from a DMD patient with an exon 52 deletion. Similar to the mouse Δ52 DMD correction strategy, we designed 17 human sgRNAs (marked with “h”) in the proximity of the 5′ end of mouse exon 53, upstream of the premature stop codon (Figure S1D). By screening the sgRNAs in human 293T cells, we found that sgRNA hE53 g10 showed superior editing activity, based on total INDEL efficiency as measured by TIDE analysis (Figure S1E). Based on these findings, human sgRNA hE53 g10 was tested for its ability to restore dystrophin expression in the human Δ52 iPSC model.
We found that sgRNA hE53 g10 generated 26% of 3n + 1 genomic INDELs in human Δ52 DMD iPSCs (Figure S2B). Thus, this human sgRNA can potentially restore the dystrophin ORF by reframing exon 53. Human Δ52 DMD iPSCs were subjected to editing with sgRNA hE53 g10, and the cells were differentiated into cardiomyocytes. Single-clone characterization by RT-PCR and sequencing showed an 8-nt deletion, consistent with the reframing strategy (Figures 3A and 3B). Western blot and immunocytochemistry of iPSC-derived cardiomyocytes edited with sgRNA hE53 g10 showed restoration of dystrophin expression (Figures 3C and 3D).
Figure 3.

Human Δ52 DMD iPSC-Derived Cardiomyocytes Express Dystrophin after CRISPR-Cas9-Mediated Genome Editing
(A) RT-PCR analysis of WT and Δ52 DMD iPSC-derived cardiomyocytes after CRISPR-Cas9-mediated genome editing using hE53 g10 sgRNA. Restoration of the ORF is obtained by 8-nt deletion (band size, 502 bp). (B) Sequence of the restored ORF obtained by 8-nt deletion of exon 53 using sgRNA hE53 g10 and SpCas9 gene editing. (C) Western blot analysis indicates restoration of dystrophin protein expression in human Δ52 DMD iPSC-derived cardiomyocytes with hE53 g10 sgRNA, as indicated. Vinculin is the loading control. WT, iPSC-derived cardiomyocytes from a healthy control. (D) Immunostaining indicates restoration of dystrophin expression in edited Δ52 DMD iPSC-derived cardiomyocytes using hE53 g10 sgRNA and SpCas9. Dystrophin is indicated in red. Cardiac troponin I is indicated in green. Nuclei are marked by DAPI stain in blue. Scale bar, 50 μm.
Correction of DMD Exon 43 and 45 Deletions in Mice by Intramuscular AAV9 Delivery of Gene Editing Components
In Δ43 DMD mice, the lack of exon 43 generates a premature stop codon in the proximity of the 5′ region of exon 44. In Δ45 DMD mice, the absence of exon 45 causes exon 46 to be out of frame with preceding exons. Skipping of exon 44 can potentially restore dystrophin expression for both Δ43 DMD mice and Δ45 DMD mice. Moreover, 3n − 1 reframing of exon 44 can potentially restore the ORF and correct exon 45 deletion. However, the 3n − 1 reframing event in the 5′ region of exon 44 results in the introduction of a new stop codon in the context of the Δ45 DMD deletion (Figures 4A, S4A, and S4B). Due to these considerations, an editing strategy that aims to use the same sgRNA to correct both Δ43 and Δ45 DMD models needs to target the proximity of the splice donor site of exon 44 in the 3′ region of the exon.
Figure 4.

Intramuscular AAV9 Delivery of Gene Editing Components to Δ43 and Δ45 DMD Mice Rescues dystrophin Expression
(A) Diagram for exon 44 (148 bp) targeting strategy and potential products after editing of Δ43 and Δ45 DMD mice. The same sgRNA (mE44 g7) has been used to correct both mutations. (B and E) RT-PCR analysis of TA muscles from WT, Δ43, and Δ45 DMD mice 3 weeks after intramuscular injection of ssAAV-Cas9 and scAAV-mE44 g7. (B) For Δ43 DMD mice, restoration of the ORF is obtained by skipping of exon 44 (lower bands, 460 bp). The proportion of the different products is illustrated in Figure S5A. (E) For Δ45 DMD mice, restoration of the ORF is obtained by reframing (middle bands, 466 bp) or skipping of exon 44 (lower bands, 318 bp). The proportion of the different products is illustrated in Figure S5C. (C and F) Western blot analysis indicates restoration of dystrophin expression in TA muscle of (C) Δ43 and (F) Δ45 DMD mice after injection of AAV-Cas9 and AAV-mE44 g7. Relative dystrophin intensity (%) was calibrated with vinculin internal control before normalizing to the untreated WT control. (D and G) Immunohistochemistry indicates restoration of dystrophin in TA muscle of (D) Δ43 and (G) Δ45 DMD mice 3 weeks after intramuscular injection of gene editing components sgRNA-mE44 g7 and SpCas9, carried by AAV9. Dystrophin is shown in green. Nuclei are marked by DAPI stain in blue. Scale bars, 100 μm.
To target the splice donor site of exon 44, allowing for exon skipping of Δ43 and Δ45 deletions, or to introduce 3n − 1 INDELs in exon 44, allowing for reframing Δ45 deletions, we designed 13 mouse sgRNAs at the 3′ end of mouse exon 44 (Figure S4B). By screening these sgRNAs in mouse N2a cells, we found that sgRNA mE44 g7 showed superior total INDEL efficiency by TIDE analysis (Figure S4C). Due to its localization, sgRNA mE44 g7 has the potential to induce both exon skipping and reframing. Therefore, mouse sgRNA mE44 g7 was selected for testing exon skipping and/or exon reframing capability in both the Δ43 and Δ45 DMD mouse models. The dual viral delivery strategy (ssAAV9-SpCas9 and scAAV-sgRNA) of the gene editing components was the same as described earlier for the correction of the Δ52 DMD mouse model.
In Δ43 DMD muscle edited with sgRNA mE44 g7 and SpCas9, RT-PCR analysis showed a distinct exon-skipping band at 460 bp below the Δ43 DMD band at 608 bp (Figure 4B). TIDE analysis of the RT-PCR product revealed that sgRNA mE44 g7 generated ~16% of the average total INDELs following IM injection and that 3.7% of the transcripts successfully skipped exon 44; none of the transcripts were the product of 3n − 1 reframing (Figure S5A). These results demonstrated that our predicted strategy for editing with sgRNA mE44 g7 in Δ43 DMD is robust. In fact, sgRNA mE44 g7 targets the 3′ end of exon 44 and destroys the splice donor site of exon 44. Correction of the exon 43 deletion using sgRNA mE44 g7 with SpCas9 induces exon skipping, but not exon reframing, in exon 44.
In ssAAV-Cas9- and scAAV-mE44 g7-treated Δ43 DMD muscle, the restoration of dystrophin was about 14% of WT levels (Figure 4C). Immunostaining and whole-muscle scanning also revealed that scAAV-mE44 g7-treated muscles restored ~36% of dystrophin-positive fibers (Figures 4D and S6A). H&E staining and whole-muscle scanning showed that scAAV-mE44 g7 slightly improved the dystrophic phenotype (Figures S6B and S6C). Using the same scAAV-mE44 g7 to treat Δ45 DMD muscle, RT-PCR revealed a distinct exon-skipping band at 318 bp below the Δ45 DMD band at 466 bp (Figure 4E). TIDE analysis of the RT-PCR product showed a similar total INDEL percentage of scAAV-mE44 g7-treated Δ43 DMD muscles (~21%) (Figure S5C). Specifically, TIDE analysis of the RT-PCR product revealed that 3.4% of the transcripts successfully skipped exon 44 and that 6.3% of the transcripts were the product of 3n − 1 reframing (Figure S5C). These findings confirmed the accuracy of the strategy for correction of Δ45 DMD mice with sgRNA mE44 g7. As a sgRNA that targets the 3′ end of exon 44, mE44 g7 can correct exon 45 deletion by inducing both exon skipping and exon reframing in exon 44.
Dystrophin protein expression was restored to 23% of the WT level with scAAV-mE44 g7 (Figure 4F). Immunostaining and whole-muscle scanning also revealed that scAAV-mE44 g7-treated Δ45 DMD TA muscles showed ~60% of dystrophin-positive fibers (Figures 4G and S7A). H&E staining and whole-muscle scanning showed that scAAV-mE44 g7 improved the dystrophic phenotype of the injected muscle (Figures S7B and S7C). Although scAAV-mE44 g7 with ssAAV-Cas9 restores dystrophin expression in both Δ43 and Δ45 DMD mouse models, the higher efficiency achieved in Δ45 DMD muscles is likely due to the combination of exon skipping and reframing events.
Correction of DMD Exon 43 and 45 Deletions in Human iPSCs by Delivery of Gene Editing Components
As shown in vivo, DMD mice with a deletion of exon 43 or deletion of exon 45 can be corrected using the same sgRNA (mE44 g7) by editing the 3′ region of exon 44. Gene editing and correction of exon 44 can potentially benefit 7.6% of DMD patients. To test whether the gene editing strategy that we validated in Δ43 and Δ45 DMD mice also works in human cells, we generated exon-45- and exon-43-deleted human iPSCs (human Δ43 DMD and human Δ45 DMD) by removing exon 43 or exon 45 in a normal (WT) human iPSC line. Similar to the mouse correction strategy, we designed 15 human sgRNAs at the 3′ end of human exon 44 (Figure S4D) to target the splice donor site for exon 44 to allow exon skipping for human Δ43 and human Δ45 DMD iPSCs or to introduce 3n-1 INDELs in exon 44, allowing for reframing of human Δ45 DMD iPSCs. By screening these sgRNAs in human 293T cells, we found that sgRNA hE44 g4 with SpCas9 showed superior total INDEL efficiency by TIDE analysis (Figure S4E). Due to its localization, hE44 g4 has the potential to induce both exon skipping and reframing and shares the same sequence of the tested sgRNA mE44 g7. Therefore, human sgRNA hE44 g4 was tested for its ability to restore dystrophin expression in the human Δ43 and Δ45 DMD iPSC models.
We found that sgRNA hE44 g4 and SpCas9 generated 8% of genomic INDELs, resulting in the destruction of the exon 44 splice donor site in human Δ43 DMD iPSCs (Figure S5B). Therefore, this human sgRNA can potentially restore the dystrophin ORF by skipping exon 44. Human Δ43 DMD iPSCs were subjected to editing with sgRNA hE44 g4, and the cells were differentiated into cardiomyocytes. Single-clone characterization by RT-PCR and sequencing showed the deletion of exon 44, consistent with the exon-skipping strategy (Figures 5A and 5B). Western blot analysis and immunocytochemistry of iPSC-derived cardiomyocytes edited with sgRNA hE44 g4 confirmed restoration of dystrophin expression (Figures 5C and 5D).
Figure 5.

Human Δ43 and Δ45 DMD iPSC-Derived Cardiomyocytes Express Dystrophin after CRISPR-Cas9-Mediated Genome Editing
(A and E) RT-PCR analysis of WT, Δ43, and Δ45 DMD iPSC-derived cardiomyocytes after CRISPR-Cas9-mediated genome editing using hE44 g4 sgRNA and SpCas9. (A) For Δ43 DMD iPSC-derived cardiomyocytes, restoration of the ORF is obtained by exon skipping of exon 44 (148 bp; band size, 371 bp). (E) For human Δ45 DMD iPSC-derived cardiomyocytes, restoration of the ORF is obtained by 1-nt deletion within exon 44 (band size, 515 bp). (B and F) Sequences of the restored ORF obtained by (B) skipping of exon 44 (for Δ43) or by (F) 1-nt deletion editing exon 44 (for Δ45) with hE44 g4 sgRNA. (C and G) Western blot analysis indicates restoration of dystrophin protein expression in (C) Δ43 and (G) Δ45 DMD iPSC-derived cardiomyocytes with hE44 g4 sgRNA, as indicated. Vinculin is the loading control. WT, iPSC-derived cardiomyocytes from a healthy control. (D and H) Immunostaining indicates restoration of dystrophin expression in edited (D) Δ43 and (H) Δ45 DMD iPSC-derived cardiomyocytes. Dystrophin is indicated in red. Cardiac troponin I is indicated in green. Nuclei are marked by DAPI stain in blue. Scale bars, 50 μm.
Next, we tested sgRNA hE44 g4 in human Δ45 DMD iPSCs for its ability restore the dystrophin ORF by reframing exon 44. Addition of sgRNA hE44 g4 and SpCas9 to Δ45 DMD iPSCs generated 10% of 3n − 1 genomic INDELs (Figure S5D). To assess restoration of dystrophin expression, human Δ45 DMD iPSCs were subjected to editing with sgRNA hE44 g4 and SpCas9, followed by differentiation to cardiomyocytes. Single-clone characterization by RT-PCR and sequencing showed a 1-nt deletion, consistent with the reframing strategy (Figures 5E and 5F). Western blot analysis and immunocytochemistry of iPSC-derived cardiomyocytes edited with sgRNA hE44 g4 showed restoration of dystrophin expression (Figures 5G and 5H). Together, these data demonstrate that sgRNA hE44 g4 is a good candidate for future therapeutic applications to correct both exon 43 and exon 45 human DMD deletions.
Discussion
Deletions of exon 43, 45, or 52 represent three prominent human DMD mutations, and targeting exon 44 or 53 with single-cut correction could potentially benefit ~18% of the DMD patient population. Despite the prevalence of these mutations, there are no DMD mouse models with deletions of these key exons, hindering that ability to test different therapeutic strategies to rescue dystrophin expression. In this study, we generated three new mouse models with the deletion of dystrophin exon 43, 45, or 52, which recapitulate the pathologic hallmarks of DMD. These mice represent an important resource not only for testing possible gene editing therapies, as shown here, but also for investigating other therapeutic modalities either alone or in combination with gene editing.
To optimize single-cut genome editing correction of these DMD mutations, we systematically tested different sgRNAs that target DMD exons 44 and 53, with the potential to restore dystrophin expression through exon skipping and/or reframing in these new DMD mouse models. We demonstrated dystrophin protein recovery following IM delivery of AAV9 encoding selected sgRNAs and SpCas9. To ascertain the therapeutic potential of our strategies for correction of these three exon deletions, we generated human iPSC lines bearing similar exon deletion mutations. We then identified the most efficient sgRNAs capable of restoring dystrophin in human iPSC-derived cardiomyocytes from our DMD models.
Our in vivo results demonstrate that single-cut gene editing using a sgRNA that permits both exon skipping and exon reframing (instead of only exon skipping) confers the highest efficiency of dystrophin restoration. The highest efficacy of dystrophin recovery was obtained with CRISPR-Cas9 genome editing using sgRNA mE53 g2 for correcting the Δ52 mouse model and sgRNA mE44 g7 for correcting the Δ45 mouse model. Interestingly, we found that sgRNA mE44 g7 can correct both Δ43 and Δ45 DMD mutations. Genome editing using sgRNA mE44 g7 generated a similar frequency of INDELs in Δ43 and Δ45 DMD muscles. However, the dystrophin transcripts generated from these different corrected genes reflected the differences of the strategy. In particular, sgRNA mE44 g7 generated both exon-reframed and exon-skipped transcripts in Δ45 DMD muscle, but in Δ43 DMD muscle, sgRNA mE44 g7 generated only exon-skipped transcripts. The dystrophin protein recovery reflects the differences of the strategy, so that restoration of dystrophin in Δ45 DMD muscle was more efficient than in Δ43 DMD muscle when using the same sgRNA mE44 g7 for genomic editing.
As observed with Δ45 DMD mutations, different genomic editing strategies can be deployed, resulting in skipping or reframing different exons to correct a single type of DMD mutation. When selecting the genomic strategy to correct a specific mutation, consideration should be given to the possible functional outcomes obtained by the various truncated forms of dystrophin produced (e.g., editing exon 44 versus 46). For example, it has been reported that deletion of exons 45 and 46 is associated with a severe DMD phenotype, although deleting these two exons produces an in-frame dystrophin.44 Interestingly, when searching multiple DMD databases, very few patients lacking exons 44 and 45 were found, suggesting a bias of such mutations toward a very mild BMD phenotype.4,44 Such considerations may have significant consequences for eventual therapeutic outcomes.
Overall, our results highlight several key considerations in defining the choice of genomic editing strategy by selecting optimal sgRNAs for DMD gene correction. (1) 3n + 1 and 3n-1 reframing can be equally efficient when the top selected sgRNA can create desired INDEL types. (2) The INDEL types that a particular sgRNA creates in cultured cells is extremely important for predicting dystrophin protein restoration in vivo. (3) Dystrophin restoration by exon reframing is more efficient than exon skipping. sgRNAs that only induce exon skipping are less efficient and require further testing in mice to ensure dystrophin restoration efficiency.
Several previous studies indicated that no significant off-target effects were detected following single-cut gene editing in mice lacking exons 44 and 50 or following double-cut gene editing in mdx mice and a pig model of DMD lacking exon 52.18,19,21,26,31 Although these studies failed to detect off-target cutting by deep sequencing, further assessment of potential off-target effects with other optimized sgRNAs should be performed when new tools for off-target analysis become available.
Finally, numerous gene editing strategies are being developed for a variety of clinical applications. The continuous development of high-fidelity gene editing tools to reduce potential off-target activity may facilitate progress toward the possible clinical utilization of the AAV9-Cas9 genome editing approach. Going forward, it will also be important to test multiple different sgRNAs for each genomic target so as to ensure optimal editing efficiency.
Materials and Methods
Study Design
This study was designed with the primary aim of identifying the most efficient strategy to correct exon 52, exon 43, and exon 45 mutations in corresponding DMD mouse models and human DMD iPSCs. Secondary objectives were to investigate and compare the amounts of exon skipping/reframing, expression of dystrophin protein, and histological phenotype in corrected DMD mice. Animal work described in this article has been approved and conducted under the oversight of the University of Texas (UT) Southwestern Institutional Animal Care and Use Committee. Animals were allocated to experimental groups based on genotype; we did not use exclusion, randomization, or blinding approaches to assign the animals for the experiments. AAV injection and dissection experiments were conducted in a nonblinded fashion. Blinding approaches were used during histology validation and immunostaining analysis. PBMCs from healthy individuals and DMD patients were generated at the UT Southwestern Wellstone Myoediting Core. Male donors’ PBMCs were used in all experiments. PBMCs were collected based on the mutation of the patients; we did not use exclusion, randomization, or blinding approaches to select the donors. For each experiment, sample size reflects the number of independent biological replicates.
Plasmids
The pSpCas9(BB)-2A-GFP (PX458) plasmid contained the human codon-optimized SpCas9 gene with 2A-EGFP. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138).14 Cloning of sgRNA was done using Bbs I sites. The sgRNAs in this study, listed in Table S2, were selected using prediction of https://zlab.bio/guide-design-resources. sgRNA sequences were cloned into PX458 and then tested in tissue culture using HEK293 and N2a cells, as previously described.45
The AAV TRISPR-sgRNAs-CK8e-GFP plasmid contained three sgRNAs driven by the U6, H1, or 7SK promoter. The expression cassette was synthesized (GenScript), digested with restriction enzymes, and subcloned into the pSJG self-complementary AAV plasmid, a gift from S. Gray (UT Southwestern).
Mice
Mice were housed in a barrier facility with a 12-h:12-h light:dark cycle and maintained on standard chow (2916 Teklad Global). Δ52, Δ43, and Δ45 DMD mice were generated in the C57/BL6N background using the CRISPR-Cas9 system. The sgRNAs for generating the mouse models are listed in Table S1.
Genomic DNA Isolation, PCR Amplification, and TIDE Analysis of PCR Products
Genomic DNA of mouse N2a cells and of human HEK293T cells and human iPSCs was isolated using DirectPCR (cell) lysis reagent (Viagen) according to the manufacturer’s protocol. Genomic DNA of mouse muscle tissues was isolated using the DNeasy Blood and Tissue Kit (QIAGEN) according to the manufacturer’s protocol. Genomic DNA was PCR amplified using GoTaq DNA polymerase (Promega) or with primers. RT-PCR products were sequenced and analyzed by TIDE analysis.46 Primer sequences are listed in Table S1.
AAV Vector Production
AAVs were prepared by the Boston Children’s Hospital Viral Core, as previously described.47 AAV vectors were purified by discontinuous iodixanol gradients (Cosmo Bio, AXS-1114542-5) and then concentrated with a Millipore Amicon filter unit (UFC910008, 100 kDa). AAV titers were determined by quantitative real-time PCR assays. Briefly, 4 μL of the AAV vector was treated with DNase I (New England Biolabs, M0303S) and 2 M NaOH, followed by neutralization. The mixture was serially diluted, and Droplet Digital PCR (ddPCR) (Bio-Rad Laboratories) was performed according to the manufacturer’s protocol.
AAV9 Delivery to Δ52, Δ43, and Δ45 DMD Mice
Before intramuscular injection, the Δ52, Δ43, and Δ45 DMD mice were anesthetized. For AAV9 IM injection, the TA muscle of P12 male Δ52, Δ43, and Δ45 DMD mice was injected using an ultrafine needle (31G) with 50-μL AAV9 preparations or with saline solution.
Dystrophin Western Blot Analysis
For western blot of skeletal muscles, tissues were crushed into fine powder using a liquid-nitrogen-frozen crushing apparatus. For western blot of iPSC-derived cardiomyocytes, 2 × 106 cardiomyocytes were harvested and lysed with lysis buffer (10% SDS, 62.5 mM Tris [pH 6.8], 1 mM EDTA, and protease inhibitor). Cell or tissue lysates were passed through a 25G syringe and then a 27G syringe, 10 times each. Protein concentration was determined by bicinchonic acid assay (BCA) assay, and 50 μg total protein was loaded onto a 4%–20% acrylamide gel. Gels were run at 100 V for 15 min and switched to 200 V for 45 min followed by 1 h, 20 min transfer to a polyvinylidene fluoride (PVDF) membrane at 100 V at 4°C. The blot was incubated with mouse anti-dystrophin antibody (MANDYS8, Sigma-Aldrich, D8168) at 4°C overnight and then with goat anti-mouse horseradish peroxidase (HRP) antibody (Bio-Rad Laboratories) at room temperature for 1 h. The blot was developed using Western Blotting Luminol Reagent (Santa Cruz Biotechnology, sc-2048). The loading control was determined by blotting with mouse anti-vinculin antibody (Sigma-Aldrich, V9131).
Histological Analysis of Muscles
Skeletal muscles from WT and from Δ52, Δ43, and Δ45 DMD mice were individually dissected and cryo-embedded in a 1:2 volume mixture of gum tragacanth powder (Sigma-Aldrich) to tissue-freezing medium (TFM) (Triangle Bioscience Science). All embeds were snap frozen in isopentane heat extractant supercooled to −155°C. Resulting blocks were stored at −80°C prior to sectioning. Eight-micron transverse sections of skeletal muscle and frontal sections of heart were prepared on a Leica CM3050 cryostat and air dried prior to staining on the same day. H&E staining was performed according to established staining protocols,17 and dystrophin immunohistochemistry was performed using MANDYS8 monoclonal antibody (Sigma-Aldrich), with modifications to the manufacturer’s instructions. In brief, cryostat sections were thawed and rehydrated/delipidated in 1% Triton/phosphate-buffered-saline (PBS; pH 7.4). Following delipidation, sections were washed free of Triton, incubated with mouse immunoglobulin G (IgG) blocking reagent (M.O.M. Kit, Vector Laboratories), washed, and sequentially equilibrated with M.O.M. protein concentrate/PBS and MANDYS8 diluted 1:1,800 in M.O.M. protein concentrate/PBS. Following overnight primary antibody incubation at 4°C, sections were washed, incubated with M.O.M. biotinylated anti-mouse IgG, and washed, and detection was completed with incubation of Fluorescein Avidin DCS (Vector Labs). Nuclei were counterstained with propidium iodide (Molecular Probes) prior to coverslipping with Vectashield.
Human iPSC Maintenance and Nucleofection
Human iPSCs were cultured in mTeSR1 media (catalog no. 05850, StemCell Technologies) and passaged approximately every 3–4 days (1:6–1:18 split ratio). One hour before nucleofection, iPSCs were treated with 10 μM ROCK inhibitor, Y-27632 (catalog no. S1049, Selleckchem), and dissociated into single cells using Accutase (catalog no. A6964, Innovative Cell Technologies). iPSCs (8 × 105) were mixed with 5 μg total of pSpCas9(BB)-2A-GFP (PX458) from Feng Zhang (MIT, Cambridge, MA, USA; Addgene plasmid 48138),14 which contains gRNA as indicated, and then nucleofected using the P3 Primary Cell 4D-Nucleofector X Kit (catalog no. V4XP-3024, Lonza) according to the manufacturer’s protocol. After nucleofection, iPSCs were cultured in mTeSR1 media supplemented with 10 μM ROCK inhibitor and 100 μg/mL Primocin (InvivoGen), and the next day, the media were switched to fresh mTeSR1. Two days after nucleofection, GFP(+) and GFP(−) cells were sorted by fluorescence-activated cell sorting (FACS) and subjected to genotyping by PCR. Single clones derived from GFP(+) iPSCs were picked, expanded, genotyped, and sequenced.
Human iPSC-Cardiomyocyte Differentiation
To differentiate the iPSCs into cardiomyocytes, cells were cultured in CDM3 media48 supplemented with 4–6 μM CHIR99021 (catalog no. S2924, Selleckchem) for 2 days (days 1–2), followed by CDM3 supplemented with 2 μM WNT-C59 (catalog no. S7037, Selleckchem) for 2 days (days 3–4). Starting from day 5, cells were cultured in basal media (RPMI-1640, catalog no. 11875-093, GIBCO, supplemented with B-27 Supplement, catalog no. 17504044, Thermo Fisher Scientific) for 6 days (days 5–10). On day 10 after differentiation initiation, media were changed to selective media (RPMI-1640, no glucose, catalog no. 11879-020, GIBCO, supplemented with B-27 Supplement) for 10 days (days 11–20) and, last, by basal media for 2 to 6 days. Then, the cardiomyocytes were dissociated using TrypLE Express media (catalog no. 12605-028, GIBCO) and replated at 2 × 106 cells per well in a six-well dish. Cardiomyocytes were used for experiments on days 30–40 after initiation of differentiation.
Statistics
All data are presented as means ± SEM. Unpaired two-tailed Student’s t tests were performed for comparison between the respective two groups (WT and DMD mice) in serum CK activities of the mouse models. Data analyses were performed with statistical software (GraphPad Prism Software, San Diego, CA, USA). The p values that were less than 0.05 were considered statistically significant.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Information. Additional data related to this paper may be requested from the authors.
Author Contributions
Y.-L.M., F.C., H.L., R.B.-D., and E.N.O. wrote and edited the manuscript. Y.-L.M. generated the ΔEx52, -43, and -45 dystrophic mouse models and designed the gene editing strategy; performed AAV injection and animal studies; designed experiments; and performed sgRNA screening, western blots, imaging, and data analysis. H.L. cloned sgRNAs and performed sgRNA screening, RT-PCR, and TIDE analysis. F.C. performed animal studies, sgRNA screening, and data analysis. C.R.-C. and F.C. performed iPSC culture and nucleofection experiments. E.S.-O. performed western blot, immunohistochemistry, and imaging. A.A.M. performed tissue-processing experiments. J.M.S. performed immunohistochemistry and H&E staining. J.R.M. performed the zygote injection to generate the mice. Y.Z. provided the modified scAAV backbone.
Conflicts of Interest
R.B.-D. and E.N.O. are consultants for Exonics Therapeutics/Vertex Genetic Therapies. Y.-L.M. is currently an employee of Vertex Genetic Therapies. The other authors declare no competing interests.
Acknowledgments
We thank J. Cabrera for graphics; C. Wang and the Boston Children’s Hospital Viral Core for AAV production; the Metabolic Phenotyping Core for serum CK analysis; the Sanger Sequencing Core and Next Generation Sequencing Core for sequencing services; the Flow Cytometry Core for cell sorting; B. Johnson, K. Moulton, and A. Espejo for ddPCR-based AAV titer analysis; and J. Gromada, N. Jones, A. Mcvie-Wylie, and L. Amoasii for constructive advice. We are grateful to S. Hauschka (University of Washington) for providing the muscle-specific CK8e promoter, D. Grimm (University Hospital Heidelberg, Heidelberg, Germany) for providing TRISPR-sgRNA expression plasmid, and S. Gray (UT Southwestern Medical Center) for providing the self-complementary AAV plasmid. Funding: This work was supported by funds from NIH (HL130253 and AR-067294), the Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (U54 HD 087351), Vertex Genetic Therapies, and the Robert A. Welch Foundation (grant 1-0025 to E.N.O.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.024.
Supplemental Information
References
- 1.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Ahn A.H., Kunkel L.M. The structural and functional diversity of dystrophin. Nat. Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 3.Campbell K.P., Kahl S.D. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 4.Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aartsma-Rus A., Van Deutekom J.C.T., Fokkema I.F., Van Ommen G.J.B., Den Dunnen J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 6.Allen D.G., Whitehead N.P., Froehner S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016;96:253–305. doi: 10.1152/physrev.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman E.P., Kunkel L.M., Angelini C., Clarke A., Johnson M., Harris J.B. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39:1011–1017. doi: 10.1212/wnl.39.8.1011. [DOI] [PubMed] [Google Scholar]
- 8.Ohshima S., Shin J.-H., Yuasa K., Nishiyama A., Kira J., Okada T., Takeda S. Transduction efficiency and immune response associated with the administration of AAV8 vector into dog skeletal muscle. Mol. Ther. 2009;17:73–80. doi: 10.1038/mt.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum. Gene Ther. Clin. Dev. 2015;26:57–69. doi: 10.1089/humc.2015.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Guiner C., Servais L., Montus M., Larcher T., Fraysse B., Moullec S., Allais M., François V., Dutilleul M., Malerba A. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017;8:16105. doi: 10.1038/ncomms16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakuma T., Nakade S., Sakane Y., Suzuki K.T., Yamamoto T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016;11:118–133. doi: 10.1038/nprot.2015.140. [DOI] [PubMed] [Google Scholar]
- 16.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R., Olson E.N. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5:eaav4324. doi: 10.1126/sciadv.aav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallagher D.N., Haber J.E. Repair of a Site-Specific DNA Cleavage: Old-School Lessons for Cas9-Mediated Gene Editing. ACS Chem. Biol. 2018;13:397–405. doi: 10.1021/acschembio.7b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Refaey M., Xu L., Gao Y., Canan B.D., Adesanya T.M.A., Warner S.C., Akagi K., Symer D.E., Mohler P.J., Ma J. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circ. Res. 2017;121:923–929. doi: 10.1161/CIRCRESAHA.117.310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGreevy J.W., Hakim C.H., McIntosh M.A., Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wasala N.B., Chen S.J., Duan D. Duchenne muscular dystrophy animal models for high-throughput drug discovery and precision medicine. Expert Opin. Drug Discov. 2020;15:443–456. doi: 10.1080/17460441.2020.1718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.’t Hoen P.A.C., de Meijer E.J., Boer J.M., Vossen R.H.A.M., Turk R., Maatman R.G.H.J., Davies K.E., van Ommen G.J., van Deutekom J.C., den Dunnen J.T. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem. 2008;283:5899–5907. doi: 10.1074/jbc.M709410200. [DOI] [PubMed] [Google Scholar]
- 30.Young C.S., Mokhonova E., Quinonez M., Pyle A.D., Spencer M.J. Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy. J. Neuromuscul. Dis. 2017;4:139–145. doi: 10.3233/JND-170218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y., Li H., Min Y.L., Sanchez-Ortiz E., Huang J., Mireault A.A., Shelton J.M., Kim J., Mammen P.P.A., Bassel-Duby R., Olson E.N. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020;6:y6812. doi: 10.1126/sciadv.aay6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 34.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dangain J., Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve. 1984;7:700–704. doi: 10.1002/mus.880070903. [DOI] [PubMed] [Google Scholar]
- 36.Carnwath J.W., Shotton D.M. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- 37.Wang J.-Z., Wu P., Shi Z.-M., Xu Y.-L., Liu Z.-J. The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev. 2017;39:547–556. doi: 10.1016/j.braindev.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 38.Lau C.-H., Suh Y. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res. 2017;6:2153. doi: 10.12688/f1000research.11243.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 40.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 41.Büning H., Perabo L., Coutelle O., Quadt-Humme S., Hallek M. Recent developments in adeno-associated virus vector technology. J. Gene Med. 2008;10:717–733. doi: 10.1002/jgm.1205. [DOI] [PubMed] [Google Scholar]
- 42.Martari M., Sagazio A., Mohamadi A., Nguyen Q., Hauschka S.D., Kim E., Salvatori R. Partial rescue of growth failure in growth hormone (GH)-deficient mice by a single injection of a double-stranded adeno-associated viral vector expressing the GH gene driven by a muscle-specific regulatory cassette. Hum. Gene Ther. 2009;20:759–766. doi: 10.1089/hum.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt F., Beaudouin J., Börner K., Grimm D. 117. AAV-TRISPR – A Novel Versatile AAV Vector Kit for Combinatorial CRISPR and RNAi Expression. Mol. Ther. 2015;23(Suppl. 1):S48–S49. [Google Scholar]
- 44.Findlay A.R., Wein N., Kaminoh Y., Taylor L.E., Dunn D.M., Mendell J.R., King W.M., Pestronk A., Florence J.M., Mathews K.D., United Dystrophinopathy Project Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015;77:668–674. doi: 10.1002/ana.24365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long C., Li H., Tiburcy M., Rodriguez-Caycedo C., Kyrychenko V., Zhou H., Zhang Y., Min Y.-L., Shelton J.M., Mammen P.P.A. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018;4:eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 48.Burridge P.W., Matsa E., Shukla P., Lin Z.C., Churko J.M., Ebert A.D., Lan F., Diecke S., Huber B., Mordwinkin N.M. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental Information. Additional data related to this paper may be requested from the authors.