

Abstract
Pompe disease is a neuromuscular disorder caused by disease-associated variants in the gene encoding for the lysosomal enzyme acid α-glucosidase (GAA), which converts lysosomal glycogen to glucose. We previously reported full rescue of Pompe disease in symptomatic 4-month-old Gaa knockout (Gaa−/−) mice by adeno-associated virus (AAV) vector-mediated liver gene transfer of an engineered secretable form of GAA (secGAA). Here, we showed that hepatic expression of secGAA rescues the phenotype of 4-month-old Gaa−/− mice at vector doses at which the native form of GAA has little to no therapeutic effect. Based on these results, we then treated severely affected 9-month-old Gaa−/− mice with an AAV vector expressing secGAA and followed the animals for 9 months thereafter. AAV-treated Gaa−/− mice showed complete reversal of the Pompe phenotype, with rescue of glycogen accumulation in most tissues, including the central nervous system, and normalization of muscle strength. Transcriptomic profiling of skeletal muscle showed rescue of most altered pathways, including those involved in mitochondrial defects, a finding supported by structural and biochemical analyses, which also showed restoration of lysosomal function. Together, these results provide insight into the reversibility of advanced Pompe disease in the Gaa−/− mouse model via liver gene transfer of secGAA.
Keywords: gene therapy, Pompe disease, AAV, secretable GAA, Pompe mouse, transcriptomics, advanced disease, liver gene transfer
Graphical Abstract
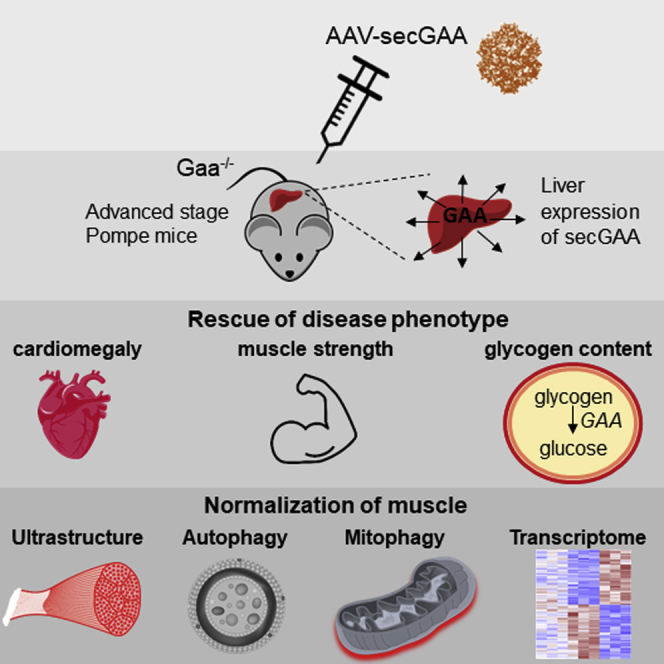
AAV vector-mediated hepatic expression of secretable GAA drives clearance of glycogen from multiple tissues, resulting in rescue of muscle function in Pompe mice with advanced pathology. Reversal of the Pompe phenotype in mice includes normalization of lysosomal parameters and mitochondrial homeostasis, which is also reflected in the muscle transcriptomics.
Introduction
Glycogen storage disease type II, also known as Pompe disease (PD), is an autosomal recessive disorder caused by mutations in the enzyme acid α-glucosidase (GAA) gene, which encodes for the lysosomal enzyme acid GAA.1 This enzyme converts glycogen into glucose within lysosomes, and its deficiency leads to the accumulation of the substrate in virtually all of the tissues of the body.2 Infantile-onset PD (IOPD) is the most severe form of PD, characterized by cardiomegaly, respiratory failure, muscle weakness, and cognitive impairment.3 IOPD subjects show absent or barely detectable residual GAA activity and, in the absence of any treatment, die within the first years of life.4 On the other hand, late-onset PD (LOPD) patients present a variable range of residual GAA activity and usually present a milder phenotype with progressive respiratory and muscle impairments with no signs of cardiac defects.1,5
Nowadays, the only approved treatment for PD is enzyme replacement therapy (ERT), based on high doses of recombinant human GAA (rhGAA), infused intravenously weekly in classic infantile patients and 2 two weeks in LOPD patients.6 ERT relies on a cross-correction mechanism by which circulating rhGAA binds to the mannose-6-phosphate receptors (MPRs) on the cell surface and trafficked to the lysosome.1 However, despite having a major impact on the course of the disease in both IOPD and LOPD patients, ERT presents important shortcomings due to the short half-life in plasma of rhGAA and its inability to cross the blood-brain barrier (BBB), resulting in limited efficacy in skeletal muscle and brain.3,7 Low expression of MPRs and the autophagy block in skeletal muscle are also among the factors limiting ERT efficacy.1,8 Finally, rhGAA exhibits high immunogenicity, particularly in IOPD patients, with consequent development of neutralizing antibodies that can reduce significantly the efficacy of ERT.9
Likely due to these limitations, emerging clinical data appear to indicate that ERT has more of a stabilizing effect on LOPD patients who generally experience improvements of muscle and respiratory function only during the first months after initiation.10,11 Similarly, early intervention with ERT appears to be crucial for a positive treatment outcome in IOPD patients,12 possibly highlighting the inability of the current treatment to reverse PD.
Development of gene therapy approaches may be a promising alternative to ERT for the treatment of PD. Adeno-associated virus (AAV) vectors have been proven as a safe and promising platform for in vivo gene transfer in humans, as demonstrated in numerous clinical trials.13, 14, 15, 16, 17, 18, 19, 20 In the context of PD, one reported trial was carried out in pediatric subjects by intra-diaphragmatic injection of an AAV1 encoding the human GAA transgene.21 The procedure was demonstrated safe with evidence of increase in respiratory function.21,22
Several preclinical studies of gene therapy have been published (reviewed in Ronzitti et al.23) using a well-established Gaa knockout mouse model (Gaa−/− hereafter).24,25 Among these studies, we recently showed full correction of PD in symptomatic 4-month-old Gaa−/− mice with AAV vector-mediated liver gene transfer of an engineered secretable GAA (secGAA) transgene. The high and stable circulating GAA plasma activity levels achieved with secGAA rescued muscle and central nervous system (CNS) pathology without development of humoral immune responses to the engineered transgene.26 However, questions about the approach remain, including the long-term efficacy of secGAA at limiting vector doses and its ability to correct PD in severely affected mice. Published studies suggest that rescue of advanced PD in mice with gene transfer is challenging, as an only partial amelioration of the disease phenotype was observed even at relatively high vector doses.27,28 One possible explanation of these findings relies on the impairment of lysosomal function characteristic of PD. Lysosomes play a key role in regulating many cellular processes, and recent studies showed significant mitochondrial defects in Pompe skeletal muscle, which have been associated with autophagic block,29, 30, 31 and may explain why advanced PD is poorly reversible.
Here, we first performed a study in 4-month-old Gaa−/− mice to demonstrate that secGAA can drive enhanced therapeutic efficacy and survival compared to the native version of the transgene following long-term hepatic gene transfer with low doses of AAV8 vectors. Next, given the superior efficacy of secGAA compared to its native form, we tested the reversibility of the PD phenotype by treating 9-month-old Gaa−/− mice with advanced disease by liver gene transfer with an AAV8-secGAA vector. AAV8-secGAA-mediated gene transfer resulted in high and sustained circulating plasma activity of GAA, driving body-wide rescue of glycogen accumulation and normalization of muscle function. Further investigation of muscle ultrastructure by electron microscopy (EM) revealed that pathological features were partially corrected by AAV8-secGAA treatment. RNA sequencing revealed normalization of expression levels of a majority of genes involved in bioenergetics homeostasis, along with partial normalization of a large part of transcriptomics alterations associated with PD. This correlated with functional, biochemical, and structural findings, suggesting that liver gene therapy with secGAA could potentially represent an efficient therapeutic strategy for the treatment at advanced stages of PD.
Results
Liver Expression of Secretable GAA Rescues PD at Low Vector Doses
We previously showed that liver-targeted expression of secGAA via AAV8 vectors drives superior therapeutic efficacy compared to the native version of the enzyme.26 However, long-term follow-up (10 months post-treatment) of animals injected with relatively high vector doses (2 × 1012 vector genome [vg]/kg) showed no clear difference between treatment groups.26 To compare more carefully the therapeutic efficacy of secGAA with that of the native form of the enzyme (GAAco), we injected 4-month-old Gaa−/− mice at a dose of 5 × 1011 vg/kg and followed the animals for 10 months (Figure 1A). At this limiting vector dose, GAAco failed to extend the lifespan of animals, whereas secGAA-treated animals had identical survival to wild-type (Gaa+/+) littermates (Figure 1B). Accordingly, secGAA expression in liver fully rescued (FR) cardiomegaly (Figure 1C) and muscle strength (Figure 1D). Conversely, liver expression of GAAco did not affect cardiomegaly (Figure 1C) and only partially rescued (PR) muscle strength (Figure 1D). We next looked at tissue GAA enzymatic activity and glycogen content. GAA activity (Figure 1E) and glycogen content (Figure 1F) in the diaphragm of animals treated with the secGAA or GAAco transgenes did not differ significantly. Similar results were observed in triceps (Figures S1A and S1B) and heart (Figures S1C and S1D). The lack of statistical significance between secGAA and GAAco cohorts was likely due to the low number of animals surviving to the end of the study in the GAAco cohort. A limited effect was observed in the brain of secGAA-treated animals (Figures S1E and S1F), whereas no effect was observed in the spinal cord of animals from either secGAA or GAAco cohorts (Figures S1G and S1H). Vector genome copy number in liver was not different across cohorts of treated animals, confirming the equivalent efficiency of liver transduction of the vectors used in the study (Figure S1I). These findings confirm that, at limiting vector doses, secGAA drives superior rescue of survival and muscle strength compared to GAAco in Gaa−/− mice.
Figure 1.

Liver Expression of Secretable GAA Rescues Pompe Disease at Low Vector Doses
(A–F) 4-month-old Gaa−/− mice were injected with an AAV8-GAAco (GAAco-Gaa−/−) or AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 5 × 1011 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 10 months after treatment (n = 7/8 per cohort). (A) Graphical representation of the study design (m.o., months old). (B) Survival curve. The numbers in parentheses show the number of animals survived at the end of the study. (C) Hypertrophy of cardiac muscle, reported as heart weight percentage of body mass. (D) Muscle strength tested by grip test at 3, 6, and 9 months post-treatment, presented as percentage of PBS-Gaa+/+. (E and F) GAA enzymatic activity (E) and glycogen content (F) in diaphragm. Statistical analyses: (B) Kaplan-Meier log rank test. ∗p < 0.05; (C, E, and F) one-way ANOVA with Tukey’s post hoc. ns, not significant. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; (D) two-way ANOVA time per treatment. +p < 0.05; ∗∗, ++, or ##p < 0.01; +++p < 0.001; ++++ or ∗∗∗∗p < 0.0001. In all graphs, error bars represent the standard deviation of the mean.
Hepatic Expression of secGAA Results in Correction of Muscle Phenotype in Gaa−/− Mice with Advanced Disease
Previous reports of liver gene transfer with AAV vectors expressing native forms of GAA showed only marginal rescue of established PD phenotype in Gaa−/− mice.27,28,32 Based on the results obtained (Figure 1), we therefore wanted to test whether secGAA expression could provide therapeutic benefit in Gaa−/− mice with advanced disease. We first measured the amount of glycogen in various tissues of naive Gaa−/− and Gaa+/+ mice at 4, 9, and 14 months of age. Gaa−/− mice showed greater glycogen accumulation compared to Gaa+/+ mice in all tissues (Figures S2A–S2D), with significantly higher amounts of glycogen measured at 9 months in triceps, brain, and diaphragm (Figures S2A–S2C). Lower glycogen content was observed in the tissues of 14-month-old Gaa−/− mice (Figures S2A–S2D), consistent with previous findings in Pompe mice.33 Survival of Gaa−/− mice was less than 50% at 14 months of age (Figure S2E). Grip-test measurement showed a significant decrease in Gaa−/− mice (Figure S2F), with loss of muscle strength slightly more pronounced at 9 and 14 months (average −32%) compared to 4 months (average −26%) (Figure S2F).
Based on this natural history study, we intravenously injected 9-month-old Gaa−/− mice with an AAV8-secGAA vector at a dose of 2 × 1012 vg/kg (Figure 2A). PBS-treated, age-matched Gaa−/− and Gaa+/+ male mice were used as the control in all of the experiments. Liver-mediated gene transfer of secGAA in Gaa−/− mice resulted in high and stable enzyme activity in plasma throughout the 9 months of follow-up (Figure 2B). This resulted in detection of supraphysiologic enzyme activity in muscle (Figure 2C) and liver (Figure S3A). Muscle uptake of secGAA resulted in the complete clearance of glycogen in heart, diaphragm, and triceps (Figures 2D and S3B).
Figure 2.

secGAA Normalizes Muscle Phenotype in Advanced Stages of Pompe Disease
(A–G) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (A) Graphical representation of the study design (m.o., months old). (B) GAA enzymatic activity measured in plasma at 1, 3, 6, and 9 months postinjection. (C) GAA enzymatic activity in heart, diaphragm, and triceps muscles, reported as percentage of PBS-Gaa+/+ activity. (D) Glycogen content in heart, diaphragm, and triceps muscles, reported as percentage of PBS-Gaa−/− glycogen. (E) Hypertrophy of cardiac muscle, reported as heart weight percentage of body mass. (F) Grip-test analysis reported as percentage of PBS-Gaa+/+ muscle strength. (G) Tidal volume measured by whole-body plethysmography. Statistical analyses: (B, E, and G) one-way ANOVA with Tukey’s post hoc. ∗∗p < 0.01; (C and D) two-way ANOVA with Tukey’s post hoc (treatment, tissue). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001; (F) two-way ANOVA with Tukey’s post hoc (treatment, time). #p < 0.05; ∗∗ or ##p < 0.01; ###p < 0.001; ∗∗∗∗ or ####p < 0.0001. In all graphs, error bars represent the standard deviation of the mean.
In agreement with the correction of the disease phenotype at the biochemical level, we observed the normalization of cardiomegaly and body weight (Figures 2E and S3C) in AAV8-secGAA-treated animals to an extent undistinguishable from PBS-Gaa+/+ mice. Mice were also subjected to grip test to assess muscle strength before AAV vector injection and at 4, 6, and 9 months thereafter (Figure 2F). Gaa−/− mice treated with the AAV8-secGAA vector showed a steady increase in muscle strength over time, achieving full recovery by the end of the study (Figure 2F). Improvement in muscle strength was accompanied by a normalization of the skeletal muscle weight (Figure S3D), whereas a significant loss of weight in triceps and gastrocnemii was detected in PBS-treated Gaa−/− mice (Figure S3D).
Plethysmography measurement of respiratory function34 did not show any significant difference among treatment cohorts, due to the low number of mice in each group, although a tendency to improve tidal volume was noted in Gaa−/− mice treated with the AAV8-secGAA vector (Figure 2G). Altogether, these results highlight rescue of advanced muscle pathology at the biochemical and functional level in Gaa−/− mice upon AAV8-secGAA treatment.
secGAA Partially Corrects Glycogen Accumulation in the CNS of 18-Month-Old Gaa−/− Mice
We next measured GAA enzymatic activity in the CNS of mice, specifically in the brain and spinal cord (Figure 3). Although detectable, significant, lower enzyme activity was found in Gaa−/− mice treated with the AAV8-secGAA vector compared to PBS-treated Gaa+/+ mice (Figure 3A). The residual GAA activity detected was reflected in the partial correction of glycogen content in brain and spinal cord (about 30% lower than the PBS-Gaa−/− cohort) (Figure 3B). Western blot analysis of brain and spinal cord (Figures 3C and 3D) allowed detection of the precursor (110-kDa) and lysosomal (76-kDa) forms of human GAA in Gaa−/− mice injected with the AAV8-secGAA vector, documenting the presence of functionally active GAA in the lysosomes in the CNS. Finally, representative histological sections of the brain (Figure 3E) stained with periodic acid-Schiff (PAS) confirmed the partial reduction of glycogen content in the brain of AAV8-secGAA-treated Gaa−/− mice. Together, these results confirm that sustained levels of circulating GAA can, at least partially, ameliorate the CNS pathology in Gaa−/− mice.
Figure 3.

Hepatic Expression of secGAA Partially Normalizes Glycogen Content in the Central Nervous System
(A–E) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (A) GAA enzymatic activity in brain and spinal cord reported as percentage of PBS-Gaa+/+ activity. (B) Glycogen content in brain and spinal cord reported as percentage of PBS-Gaa−/− glycogen. (C and D) Western blot analysis of brain (C) and spinal cord (D) lysates, 9 months after treatment using anti-GAA monoclonal antibody. An anti-tubulin antibody was used as a loading control. (E) Representative images of periodic acid-Schiff (PAS) staining of brain sections. Insets represent the caudate putamen (striatum) region of the brain. Statistical analysis: (A and B) two-way ANOVA with Tukey’s post hoc (treatment, tissue). ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. In all graphs, error bars represent the standard deviation of the mean.
Hepatic Expression of secGAA Restores Muscle Structure and Lysosomal Function in Old Gaa−/− Mice
In order to assess reversal of muscle pathology at the structural level, we carried out hematoxylin and eosin (H&E) staining and EM analysis on muscle sections. Histological sections revealed structural abnormalities in affected Gaa−/− mice treated with PBS, whereas wild-type littermates (Gaa+/+) and secGAA-treated Gaa−/− mice had similar muscle structure (Figure 4A). Quantification of p62, Lamp1, Beclin1, and LC3b, known markers of lysosomal homeostasis and autophagic buildup in PD,35, 36, 37 showed significant phenotype reversal in secGAA-treated animals for most markers analyzed (Figures 4B–4H). High-resolution EM also revealed the presence of enlarged lysosomes throughout the muscle of Gaa−/− mice (Figure 4I). Quantification of EM findings showed widespread presence of enlarged lysosomes in PBS-treated Gaa−/− mice, which were undetectable in wild-type animals (Figure 4I). Significantly smaller lysosomes (∼75% of reduction compared to PBS-treated Gaa−/− mice) were observed in AAV8-secGAA vector-treated Gaa−/− mice (Figure 4J).
Figure 4.

AAV-Mediated secGAA Expression Restores Histology and Partially Reverses Autophagy Block in Muscle
(A–E) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (A) Representative images of hematoxylin and eosin (H&E) staining of diaphragm, heart, and triceps. Scale bar, 10 μm. (B) Western blot analysis of triceps lysates using anti-p62/sequestosome-1 (SQSTM1) monoclonal antibody. An anti-GAPDH antibody was used as a loading control. (C) Western blot analysis of triceps lysates using anti-Lamp1 polyclonal and anti-Beclin1 polyclonal antibodies. An anti-GAPDH antibody was used as a loading control. (D) Western blot analysis of triceps lysates using anti-LC3b monoclonal antibody. An anti-GAPDH antibody was used as a loading control. (E) Quantification of p62 levels normalized to GAPDH levels. (F) Quantification of Lamp1 levels normalized to GAPDH levels. (G) Quantification of Beclin1 levels normalized to GAPDH levels. (H) Quantification of LC3b-I levels normalized to GAPDH levels. (I) Electron microscopy analysis of tibialis anterior. Arrows indicate enlarged lysosomes. Scale bars, 2 μm. (J) Quantification of lysosome length from the sections in electron microscopy. Statistical analyses: (E–H) one-way ANOVA with Tukey’s post hoc. ∗p < 0.05; ∗∗p < 0.01. (J) unpaired t test. ∗p < 0.0001. In all graphs, error bars represent the standard deviation of the mean.
Alterations of lysosomal function have been associated with mitochondrial defects in the skeletal muscle of Gaa−/− mice.30,31 EM analysis of skeletal muscle showed the expected ultrastructure with mitochondria homogenously distributed along the muscle fibers in Gaa+/+ mice (Figure 5A). Muscle from the PBS-Gaa−/− cohort exhibited areas of clustering of mitochondria and glycogen-filled lysosomes, which were less evident in Gaa−/− mice treated with the AAV8-secGAA vector (Figure 5A). Western blot analysis was then performed to assess mitochondrial content using the three known mitochondrial markers: CoxIV (a subunit of complex IV), ATP-b (beta subunit of complex V), and SDHA (succinate dehydrogenase A, a subunit of complex II). This analysis showed that PBS-treated Gaa−/− mice had a slight but not statistically significant increase in the amounts of CoxIV compared to Gaa−/− mice treated with AAV8-secGAA and Gaa+/+ mice (Figures 5B and 5C). Significantly higher amounts of both ATP-b (Figures 5D and 5E) and SDHA (Figures 5F and 5G) were detected in Gaa−/− mice compared to both AAV8-secGAA-treated Gaa−/− mice and wild-type littermates (PBS-Gaa+/+) (Figures 5E and 5G).
Figure 5.

AAV8-secGAA Restores Physiological Mitochondrial Content in Skeletal Muscle
(A–G) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (A) Mitochondria analysis in tibialis anterior using electron microscopy. Arrows indicate regions with accumulated mitochondria. Squares indicate zoomed areas that are presented in the lower panels. Scale bars, 2 μm. (B) Western blot analysis of triceps lysates using anti-CoxIV monoclonal antibody. An anti-GAPDH antibody was used as a loading control. (C) Quantification of CoxIV levels from the corresponding western blots shown in (B). (D) Western blot analysis of triceps using anti-ATP-b monoclonal antibody. An anti-GAPDH antibody was used as a loading control. (E) Quantification of ATP-b levels from the corresponding western blots shown in (D). (F) Western blot analysis of triceps using anti-SDHA monoclonal antibody. An anti-GAPDH antibody was used as a loading control. (G) Quantification of SDHA levels from the corresponding western blots shown in (F). Statistical analysis: (C, E, and G) one-way ANOVA with Tukey’s post hoc. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. In all graphs, error bars represent the standard deviation of the mean.
These results demonstrate that hepatic expression of secGAA can normalize autophagy, lysosome size, and mitochondrial content.
Liver-Targeted Expression of secGAA Largely Normalizes the Transcriptome in Skeletal Muscle
In order to better understand the molecular signatures of diseased muscle in advanced PD and analyze the effect of AAV8-secGAA at the molecular level, we performed transcriptome profiling by RNA sequencing using quadriceps of animals at the end of the study (18 months of age, 9 month postvector infusion). Principal-component analysis (PCA) revealed that the global transcriptome of secGAA-treated animals was intermediate between wild-type littermates (PBS-Gaa+/+) and untreated Gaa−/− animals (PBS-Gaa−/−) (Figure S4A). Differential expression analysis was then performed to identify subsets of genes with significant expression changes across the experimental groups. Pairwise comparative analysis yielded in the highest number of differentially expressed genes (DEGs) between PBS-treated Gaa+/+ and PBS-treated Gaa−/− animals (2,802 DEGs) (Figure S4B). AAV8-secGAA-treated Gaa−/− mice displayed a lower number of DEGs when compared to Gaa−/− and Gaa+/+ animals–434 and 371, respectively (Figure S4B). The decrease in DEGs in secGAA-Gaa−/− versus PBS-Gaa+/+ mice, as compared to PBS-Gaa−/− versus PBS-Gaa+/+, is in agreement with the functional and biochemical data (Figures 2, 4, and 5) and is consistent with a phenotype of secGAA- Gaa−/− mice intermediate between PBS-Gaa+/+ and PBS-Gaa−/−. Hierarchical clustering using the expression profile confirmed that muscle from AAV8-secGAA-treated Gaa−/− animals was more similar to PBS-treated Gaa+/+ animals than to PBS-treated, affected Gaa−/− animals (Figure S4C). Functional enrichment analysis performed with Ingenuity Pathway Analysis (IPA) on the list of DEGs (2,802 genes from the comparison of PBS-Gaa−/− versus PBS-Gaa+/+) identified 257 upstream transcription factors (TFs), including nuclear factor κB (NF-κΒ), signal transducer and activator of transcription 1 (STAT1), and RELA Proto-Oncogene (REL-A) (Figure S5A). These TFs, which are mainly involved in inflammatory responses and in the muscle tissue homeostasis pathways,38, 39, 40, 41 were found to be highly activated in Gaa−/− animals (Figures S5A and S5B).
After having identified genes with altered expression in the Gaa−/− background (relative to Gaa+/+), we focused on those for which expression had been restored to wild-type levels upon liver-targeted expression of secGAA. For this purpose, we grouped the complete collection of genes detected as differentially expressed in any of the three comparisons (Figure S4B) into six clusters using k-means. Each cluster contained around 450 genes with similar expression profiles. We grouped clusters with complementary profiles into three metaclusters that represented genes with different degrees of normalized expression toward Gaa+/+ (wild-type) levels: full, partial, or no rescue (Figure 6A). After AAV8-secGAA treatment, gene expression was either partially or fully normalized in approximately 75% of the identified DEGs, whereas the expression of 25% of the DEGs was not rescued (NR). Functional enrichment analysis with IPA indicated that metaclusters containing genes with FR or PR expression levels were associated to pathways involved in bioenergetics homeostasis, such as sirtuin signaling, mitochondrial dysfunction, and integrin signaling (Figure 6B). Pathways related to glucose metabolism (i.e., glycolysis I, gluconeogenesis I, glycogen degradation II, and glycogen degradation III) were also associated to metaclusters with FR or PR expression levels after hepatic expression of secGAA (Figure S6).
Figure 6.

AAV Liver Gene Therapy Largely Normalizes Transcriptome in Muscle
(A and B) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice, injected with PBS, served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (A) Gene expression in quadriceps muscle tissue from PBS-Gaa−/−, PBS-Gaa+/+, and secGAA-Gaa−/− mice was analyzed by RNA sequencing. The expression profiles of 2,864 genes, detected as differentially expressed in any of the three possible pairwise contrasts (Figure S4B), were assigned to clusters using k-means. Three metaclusters, containing genes in which their expression levels had been fully rescued (FR; 23.3%, 666 genes), partially rescued (PR; 51.3%, 1,470 genes), or not rescued (NR; 25.4%, 728 genes) by the expression of secGAA in the Gaa−/− background, were defined by combining gene clusters with complementary expression profiles. Normalized expression counts, averaged for each condition and scaled for each gene, are represented on the y axis. Gray lines represent the expression profile of individual genes. Black lines represent average expression profiles for each of the combined clusters. (B) Significant associations in 9 of total 41 canonical pathways were found for the three metacluster gene lists (FR, PR, and NR) after Ingenuity Pathway Analysis (IPA) enrichment analyses. Bar plots represent Fisher’s test p values for nine of the pathways and the three metaclusters. Red lines indicate the significance threshold (p < 0.05). Results for the remaining 33 pathways are presented in Figure S6.
As defects in muscle regeneration have been recently documented in Gaa−/− mice,33,42 we analyzed the expression pattern of several genes of interest. We observed partial normalization of the expression levels of genes involved in muscle regeneration and plasticity, such as Capn3, Myoz1, and Dysf43, 44, 45 (Figure S7). Interestingly, the expression level of the dystrophin gene (Dmd), a key gene for structure and function of muscle cells, in which mutations can result in Duchenne and Becker muscular dystrophy, was normalized46 (Figure S7). Along the same line, we also evaluated fibrosis in muscle. At the histological level, sirius red staining showed limited changes in signal intensity in the three cohorts (PBS-Gaa+/+, PBS-Gaa−/−, and secGAA-Gaa−/−) (Figure S8). Interestingly, at the transcriptomics levels, approximately 75% of genes involved in fibrosis and fibrogenesis, which were dysregulated in PBS-Gaa+/+ mice, were FR or PR upon AAV-secGAA treatment (Figure S8). Together, these results highlight a significant amelioration of PD at the transcriptional level.
AAV8-secGAA Liver Gene Transfer Partially Re-establishes Mitochondria Homeostasis in 18-Month-Old Gaa−/− Mice
Deeper analysis of mechanistic differences was performed using the transcriptome data from skeletal muscle of Gaa−/− compared to Gaa+/+ mice to better evaluate the bioenergetics defects. Upstream regulator analysis (IPA) identified various TFs involved in oxidative stress, which were associated to genes with altered expression in PBS-Gaa−/− mice relative to PBS-Gaa+/+ mice, such as hypoxia-inducible factor 1 alpha (Hif1α), nuclear factor erythroid 2-like 2 (NFE2L2; also known as Nrf2), and mitochondrial homeostasis, such as peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PPARGC1α), peroxisome proliferator-activated receptor gamma coactivator 1 beta (PPARGC1β), and sirtuin1 (Sirt1), which were enriched in PBS-Gaa−/− mice (Figure 7A). Interestingly, PPARGC1α and PPARGC1β, central players of mitochondrial biogenesis,47 were predicted as inhibited (negative Z score) in Gaa−/− mice (Figure 7A). This was consistent with the high mitochondrial content detected in skeletal muscle (Figure 5), linked to defective mitophagy and consequent inhibition of the mitochondrial biogenesis machinery as a compensatory mechanism.48 Given that the oxidative stress-related TFs (including Nrf2 and Hif1α) were predicted to be more active in PBS-Gaa−/− than PBS-Gaa+/+ (with a positive Z score), we decided to asses global reactive oxygen species (ROS) production in skeletal muscle. We observed slightly elevated amounts of ROS in PBS-Gaa−/− animals, which where normalized upon treatment with secGAA (Figure 7B).
Figure 7.

AAV8-secGAA Administration Partially Normalizes Mitochondrial Stress Signaling in Skeletal Muscle
(A) Significant associations to 257 upstream transcriptional regulators were found for the set of 2,802 genes detected as differentially expressed in the comparison of PBS-Gaa−/− versus PBS-Gaa+/+ after IPA enrichment analyses. The circular plot represents a subset of 16 regulators involved in metabolic processes and a selection of their connected functions retrieved from IPA’s knowledge base with the “grow” function. Circular crown sectors, representing transcriptional regulators, are colored according to activation Z score. Red and blue colors indicate a higher activation state in PBS-Gaa−/− or PBS-Gaa+/+ mice, respectively. (B–D) 9-month-old mice were injected with an AAV8-secGAA (secGAA-Gaa−/−) vector at a dose of 2 × 1012 vg/kg. Gaa+/+ (PBS-Gaa+/+) and Gaa−/− (PBS-Gaa−/−) mice injected with PBS served as controls in the study. Animals were followed for 9 months after treatment (n = 3/4 per cohort). (B) ROS production measured in snap-frozen tissues by EPR spectroscopy. (C) Western blot analysis of triceps lysates using monoclonal antibody anti-AMPK. An anti-GAPDH antibody was used as a loading control. (D) Quantification of AMPK levels normalized to GAPDH levels from the corresponding western blot shown in (C). Statistical analysis: one-way ANOVA with Tukey’s post hoc. ∗∗p < 0.01. In all graphs, error bars represent the standard deviation of the mean.
To test whether the correction of mitochondrial content in skeletal muscle of AAV-treated Gaa−/− mice was accompanied by a recovery of mitochondrial function, we performed a western blot analysis of key players in mitochondrial homeostasis in skeletal muscle. We looked at the “energy sensor” serine/threonine kinase AMP-activated protein kinase (AMPK) complex,49 based on the fact that AMPK signaling was significantly enriched in Gaa−/− mice compared to Gaa+/+ mice and that genes regulated by AMPK signaling were rescued in AAV8-secGAA-treated animals (Figure 6B). Interestingly, we observed increased levels, which were normalized levels upon AAV8-secGAA treatment, of AMPK in PBS-treated Gaa−/− mice (Figures 7C and 7D). In summary, transcriptomic data and biochemical analysis suggest that long-term exposure to secGAA, following hepatic gene transfer, results in partial normalization of mitochondrial homeostasis in skeletal muscle of Gaa−/− mice with advanced disease.
Discussion
PD is a progressive neuromuscular disease caused by accumulation of lysosomal glycogen, which results in tissue disruption, autophagy block, and metabolic impairments.1 Introduction of ERT with rhGAA represented a major advance in the treatment of PD, being a lifesaving treatment for infantile patients3,50,51 and providing benefit to the adult Pompe population.11,52,53 In vivo gene delivery with AAV vectors is a promising alternative to ERT for PD, owing to the promising results in preclinical models of the disease26,54,55 and the excellent safety and efficacy profile in human trials for a variety of diseases.14,16,20,56,57 We previously demonstrated the full rescue of PD in 4-month-old Gaa−/− mice using a secretable form of the GAA enzyme expressed in hepatocytes via AAV vectors.26 Here, we further defined the therapeutic potential of this approach by comparing the long-term effect of low vector doses of AAV8 vectors expressing secretable GAA or its native counterpart in Gaa−/− mice treated at 4 months of age. Despite the small increase in GAA activity, detectable in tissues of animals treated with secretable GAA, survival data clearly demonstrate that, at 5 × 1011 vg/kg, native GAA is unable to provide any therapeutic benefit, whereas Gaa−/− mice treated with secretable GAA present a lifespan undistinguishable from that of wild-type littermates. These results are of fundamental importance in the prospective of translating these results to the clinic, as the use of transgenes with enhanced bioavailability, like secretable GAA, could potentially provide superior therapeutic benefit to patients, while reducing the AAV doses administered. Importantly, the use of transgenes with an improved therapeutic index in the context of hemophilia B gene-therapy trials allowed a decrease in the therapeutic vector dose, thus reducing capsid immune-mediated toxicities16 and potentially lowering manufacturing requirements. Future side-by-side studies of gene transfer of secretable GAA with the current standard of care for PD1 will help highlight differences in pharmacokinetics and bioavailability between gene therapy and ERT.
An open question about ERT, which also concerns all novel therapies for PD, is whether providing a source of GAA enzyme could reverse an already-established disease phenotype. Experience with ERT in IOPD patients shows clear beneficial effects, including the resolution of hypertrophic cardiomyopathy, amelioration of muscle functions, and, importantly, survival.3,50,51 However, in this patient population, early intervention is crucial for a positive treatment outcome,12 suggesting that the effects of glycogen accumulation are poorly reversible with ERT. Similarly, LOPD patients receiving ERT in the long term tend to achieve, at best, stable muscle and respiratory functions,11,52,53 possibly due to the fact that ERT is started when patients become symptomatic, thus with relatively advanced disease progression.58 The presence of vacuolated fibers and autophagy impairment in muscle has also been demonstrated to hamper the trafficking of rhGAA in the context of ERT.8,29
In the context of gene transfer, two studies attempted to correct PD in Gaa−/− mice with an advanced pathology. A first study described 10-month-old Gaa−/− mice treated with an AAV vector expressing the GAA transgene in the liver.27 Despite the high vector dose used (1 × 1012/mouse), 6 months after treatment, the animals showed only partial glycogen clearance in skeletal muscle and no improvements of muscle function.27 In a second study, 12-month-old Gaa−/− mice received an AAV vector expressing a GAA transgene carrying a heterologous signal peptide to enhance secretion from hepatocytes.28 Also, in this case, AAV-treated mice exhibited incomplete reduction of glycogen in muscle.28 Additional published data in Gaa−/− mice with advanced phenotype show lack of correction of glycogen accumulation with muscle-directed expression of GAA.59 Results presented here clearly show that hepatic expression of secretable GAA results in clearance of glycogen from multiple tissues, including a partial clearance in CNS, and restoration of muscle function to wild-type levels in Gaa−/− mice with advanced pathology. Although this is somewhat in contrast with published findings,27,28,59 the higher bioavailability of secretable GAA compared with the native form of the enzyme or with enzymes carrying heterologous signal peptides26 could explain our findings. Alternatively, the difference in time of follow-up and age of inclusion of the animals in the study, as well as genetic background of the Gaa−/− colonies used in the study,24,25,60 could explain the observed differences across studies. The fact that animals presented a clear, severe phenotype at the time of treatment, both in terms of glycogen accumulation and muscle strength, would argue that hepatic expression of secretable GAA can efficiently reverse an advanced disease phenotype in Gaa−/− mice. Transcriptomics analysis of genes involved in muscle regeneration and remodeling observed in our study further supports our findings, although similar studies in the context of gene therapy trials will be necessary to elucidate the effects of secretable GAA expression on human muscle plasticity. Morphological changes observed in skeletal muscle of Pompe patients, followed by magnetic resonance imaging (MRI),61,62 show gradual replacement of skeletal muscle mass with fat tissue, a hallmark of muscle degeneration.63 This phenomenon has not been documented in Gaa−/− mice, which could indicate that the animal model fails to fully recapitulate the human condition when it comes to muscle pathophysiology. Results in our study show that, at 18 months of age, untreated Gaa−/− mice show a marked decrease of skeletal muscle mass, and secretable GAA treatment appears to restore both skeletal muscle and body weight to wild-type levels.
Another possible shortcoming of the current study is the small number of animals per cohort. Whereas a larger study could address this point and help us to understand the reversibility of more variable endpoints of therapeutic efficacy, like those reflecting respiratory function,34,64,65 limited, long-term survival of untreated Gaa−/− mice26 limits the ability to conduct larger studies with aged Pompe mice. Nevertheless, statistical analysis and correlation of multiple endpoints at the biochemical, functional and morphological level and the changes in the transcriptional signature of skeletal muscle support the robustness of the current findings on the reversibility of the advanced Pompe phenotype in mice.
Consistent with previous findings in PD26 and other lysosomal storage disorders (LSDs),66 mice treated with secretable GAA showed a significant glycogen reduction in the CNS, although a full evaluation of CNS rescue, previously reported by our lab,26 was not presented in the current study. The mechanism underlying transport of GAA from the systematic circulation to the CNS is currently unknown and could involve transcytosis, transport via exosomes,67 or lysosomal exocytosis at neuromuscular junctions.68 Although encouraging, these results suggest that high plasma levels of GAA derived from the hepatic expression of secGAA are likely to be needed to fully correct the CNS manifestations characteristic of IOPD.34,69 Alternatively, direct targeting of neurons has also been explored,70 although this strategy achieved full correction of CNS glycogen accumulation with little effect on peripheral tissues and muscle function. Notably, a GAA-expressing vector based on the recently developed AAV-PHP.B serotype71 delivered at high doses (5 × 1012 vg/kg) completely cleared heart and brain glycogen in Pompe mice, while reducing glycogen content in quadriceps and gastrocnemius.72
Lysosomal defects have been linked to severe autophagy impairment in skeletal muscle of both Pompe patients and Gaa−/− mice.29,36 In addition, studies in primary cell cultures and muscle tissue from Gaa−/− mice show secondary defects in mitochondria and mitophagy,30 consistent with recent findings on the link between autophagy block and mitochondria impairment in juvenile Gaa−/− mice.31 Clearance of defective mitochondria via mitophagy is essential to maintain a healthy mitochondrial network, whereas accumulation of dysfunctional mitochondria can have detrimental consequences in various diseases, including LSDs.73 Here, we showed that liver expression of secretable GAA normalized both autophagy and mitophagy, as documented by EM and western blot analyses, with normalization of lysosomal length and markers of autophagy, such as p62, Lamp1, and LC3b, as well as normalization of mitochondria content and improvements in mitochondria metabolism in skeletal muscle, as suggested by changes in AMPK levels. Impaired mitophagy at 18 months of age resulted in accumulation of mitochondria and modestly increased levels of ROS, which were normalized by clearance of mitochondria. At the transcriptomics level, untreated Gaa−/− showed profound alterations of mitochondrial biogenesis and upregulation of inflammatory pathways in skeletal muscle, similar to findings in other LSDs.74 Large transcriptional alterations were observed in the muscle of untreated Pompe mice at the end of the observational study—18 months. Liver expression of secretable GAA resulted in either full or partial rescue of the expression pattern of about two-thirds of the genes studied. Signaling pathway analysis confirmed partial rescue of the alterations observed in Pompe mice. Those alterations were possibly related to the deep modifications of the muscle physiology, and their partial rescue after gene transfer was likely expected. As Pompe patients have access to ERT, it is conceivable that the muscle of humans would present a somewhat less altered expression pattern. Work by Koeberl and colleagues75 reported a short list of genes for which their expression level was normalized after clenbuterol treatment in Pompe patients. Interestingly, NFE2 is one of the genes with normalized expression, showing the importance of our findings on normalization of oxidative stress-related transcriptomic changes and ROS levels.75
Characterization of satellite cells in muscle biopsies from both IOPD and LOPD patients suggests that satellite cells found in muscle are not altered in number but are less functional.76 Two independent studies reported that the Gaa−/− mouse has functionally normal satellite cells; however, they are not capable of repairing disease damage, probably due to an activation signal defect.33,42 Here, we observed partial rescue of genes related to muscle plasticity after gene transfer, although the activation state of satellite cells in old mice may be affected by age-related changes unrelated to PD.77 Additional studies in Pompe mice and in PD patients will be required to further understand differences in plasticity of the muscle across species and in particular, whether gene transfer with secretable GAA will result in rescue of PD in older patients.
Further work is also needed to assess the therapeutic potential of our approach in IOPD patients. Because of the nonintegrative nature of AAV vectors, treatment of newborn IOPD patients with liver-expressed secretable GAA might result in transient expression of the donated transgene. This would require the readministration of the therapeutic vector78 to counteract vector genome dilution and loss of transgene expression due to liver growth.79
In summary, this work confirms the superior therapeutic efficacy of secretable GAA compared to the native form of the enzyme. It also demonstrates that gene transfer with secretable GAA drives efficient rescue of the advanced Pompe phenotype in Gaa−/− mice and in particular, has a profound impact on the restoration of autophagy, mitophagy, and overall bioenergetic and inflammatory homeostasis of muscle. Future translation of the therapeutic modality based on secretable GAA to the clinic will help address similar mechanistic questions on the reversibility of the Pompe phenotype in humans.
Materials and Methods
GAA Expression Cassettes and AAV Vectors
The GAA transgene expression cassettes containing the codon-optimized coding sequence encoding for the native and secretable forms of human GAA were previously described.26
AAV vectors used in this study were produced using an adenovirus-free transient transfection method, as described earlier.80 Titers of AAV vector stocks were determined using quantitative real-time PCR and confirmed by SDS-PAGE, followed by SYPRO Ruby protein gel stain and band quantification using ImageJ software. All vector preparations used in the studies were quantified side by side at least 3 times before use.
In Vivo Studies
Mouse studies were performed according to the French and European legislation on animal care and experimentation (2010/63/EU) and approved by the local Institutional Ethical Board (protocol number 2015-008).
The Gaa−/− mice were purchased from Jackson Laboratory (B6; 129-Gaatm1Rabn/J, stock number 004154, 6neo). This mouse model was originally generated by Raben et al.24 Gaa+/+ are derived from the same colony and breed separately.
For the comparative analysis of GAAco with secGAA, AAV8-GAAco or AAV8-secGAA26 was administered intravenously to 4-month-old male mice. Gaa−/− mice were treated at a vector dose of 5 × 1011 vg/kg and followed for 10 months (n = 7 per treatment group). Gaa−/− and Gaa+/+ mice were injected with PBS and used as controls (n = 8 per group).
For the treatment of mice at advanced stages of PD, an AAV8-secGAA vector26 was administered intravenously via the tail vein to 9-month-old male mice. Gaa−/− mice were treated at a vector dose of 2 × 1012 vg/kg and followed for 9 months (n = 3/4 per treatment group). Age-matched Gaa−/− and Gaa+/+ mice were injected with PBS and used as controls. All of the animals were perfused at the time of sacrifice.
Vector Genome Copy Number Analysis
Vector genome copy number was determined using quantitative real-time PCR, as previously described.26 The PCR primers used in the reaction were located on the human alpha-1 antitrypsin promoter sequence within the expression cassette: forward primer, 5′-GGCGGGCGACTCAGATC-3′; reverse primer 5′-GGGAGGCTGCTGGTGAATATT-3′.
Measurement of GAA Activity and Glycogen Content
GAA activity was measured as previously described.81 Briefly, 10 μL of sample, either tissue homogenate or plasma, was incubated for 1 h at 37°C with 20 μL of 4-methylumbelliferyl α-d-glucopyranoside (3 mM), diluted in acetate buffer solution (pH 4.65) (Sigma-Aldrich). The reaction was stopped using the carbonate solution, and the fluorescence (λex 360 nm/λem 449 nm) was read with an EnSpire alpha plate reader (PerkinElmer, Waltham, MA). A standard curve was prepared using 4-methylumbelliferone, diluted in 0.5 M of carbonate solution (pH 10.5).
Glycogen content was measured indirectly in tissue homogenates as the glucose released after total digestion with Aspergillus niger amyloglucosidase (Sigma-Aldrich, St. Louis, MO, USA). Samples were incubated for 5 min at 95°C and then cooled at 4°C; 25 μL of amyloglucosidase diluted 1:50 in 0.1 M potassium acetate (pH 5.5) was then added to each sample. A control reaction without amyloglucosidase was prepared for each sample. Both sample and control reactions were incubated at 37°C for 90 min. The reaction was stopped by incubating samples for 5 min at 95°C. The glucose released was determined using a glucose assay kit (Sigma-Aldrich, St. Louis, MO, USA) and by measuring resulting absorbance at the EnSpire alpha plate reader (PerkinElmer, Waltham, MA, USA) at 540 nm.
Western Blot Analyses
Mouse tissues were mechanically homogenate in H2O using lysing matrix tubes (MP Biomedicals). Protein concentration was determined using the bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).
SDS-PAGE was performed in a 4%–15% gradient polyacrylamide gel. After transfer, the membrane was blocked with Odyssey buffer (LI-COR Biosciences, Lincoln, NE, USA) and incubated with an anti-GAA antibody (rabbit monoclonal; Abcam, Cambridge, MA, USA), anti-p62 (mouse monoclonal; Abcam, Cambridge, MA, USA), anti-LAMP1 (rabbit polyclonal; Abcam, Cambridge, MA, USA), anti-Beclin1 (rabbit polyclonal; Abcam, Cambridge, MA, USA), anti-LC3B (rabbit polyclonal; Novus Biologicals), anti-tubulin (mouse monoclonal; Sigma), anti-actin (rabbit polyclonal; Sigma), anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (rabbit polyclonal; Thermo Fisher Scientific), anti-AMPK (rabbit polyclonal; Cell Signaling Technology), anti-CoxIV (rabbit polyclonal; Abcam, Cambridge, MA, USA), anti-ATP-b (mouse monoclonal; Abcam, Cambridge, MA, USA), and anti-SDHA (mouse monoclonal; Invitrogen) . The membrane was washed and incubated with the appropriate secondary antibody (LI-COR Biosciences), conjugated with fluorescent probes with extended linear range, and visualized by the Odyssey imaging system (LI-COR Biosciences), which allows identification and exclusion of saturated signals during band quantification.
Histological Evaluation
For histology, immediately after euthanasia, heart, triceps brachii, diaphragm, and brain were snap frozen in isopentane (−160°C), previously chilled in liquid nitrogen. Serial 8 μm cross-sections were cut in a Leica CM3050 S cryostat (Leica Biosystems). To minimize sampling error, 3 sections of each specimen were obtained and stained with H&E, sirius red, and PAS, according to standard procedures.
Electron Microscopy Analyses
Muscle samples were fixed with 2.5% of glutaraldehyde (Sigma-Aldrich) in 0.1 M PHEM (pH 7.2). Postfixation was done with 1% osmium tetroxide (Merck) and 1.5% ferrocyanide (Sigma-Aldrich) in 0.1 M HEPES. After dehydration by a graded series of ethanol, the tissue samples were infiltrated with epoxy resin. Polymerized resin blocks were thin sectioned longitudinally using a Leica UC 7 microtome (Leica Microsystems).The 70-nm sections were collected on Formvar-coated slot grids (Electron Microscopy Sciences [EMS]) and were contrasted with 4% uranyl acetate and Reynolds lead citrate. Stained sections were observed with a Tecnai spirit Field Electron and Ion (FEI) operated at 120 kV. Images were acquired with FEI Eagle digital camera. Quantification of lysosome length was performed using ImageJ software.
Functional Assessment
Respiratory function during quiet breathing was evaluated as already reported.26,34 Briefly, a flow-through (0.5-L/min) plethysmograph (Emka Technologies, Paris, France) was used to measure the breathing pattern in treated Gaa−/− mice and untreated Gaa−/− and wild-type littermates. Before the assessment, the instrument was calibrated with known airflow and pressure signals for optimal data collection. Signals were analyzed by using IOX2 software (Emka Technologies). Tidal volume was evaluated over a period of 5 min. Animals were allowed for acclimation into the plethysmograph chamber before testing. During both acclimation and data acquirement, mice were breathing normoxic air (21% O2, 79% N2).
Grip strength was measured, as already reported, by the TREAT-NMD Network (https://treat-nmd.org). The test was performed on AAV-treated Gaa−/− mice and untreated Gaa−/− and wild-type littermates. With the use of a grip-strength meter (Columbus Instruments, San Diego, CA), three independent measurements of the four limbs’ strength were calculated. Mean values of the grip strength were reported.
Electron Paramagnetic Resonance (EPR) Spectroscopy
Free radical production was measured by EPR spectroscopy using 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl pyrrolidine hydrochloride (CMH; Noxygen, Elzach, Germany) as a spin probe. In brief, muscles were frozen in liquid nitrogen and kept at −80°C until assay. Tissues were weighed and homogenized in Krebs-HEPES buffer (pH 7.4). Then, samples were incubated in Krebs-HEPES buffer containing 5 mM diethyldithiocarbamic acid silver salt (DETC), 25 mM deferoxamine (DF), and 50 mM of CMH in 24-well plates at 37°C. Blank wells without tissue were prepared in parallel. After a 60-min incubation, samples were immediately frozen in liquid nitrogen to stop the reaction. Oxidized CMH spectra were recorded with a MiniScope MS-200 (Magnettech, Berlin). Acquisition EPR parameters were the following: Bo-field 3325.96 G; microwave power 1 mW; modulation amplitude 5 G; sweep time 60 s. Spectra intensity was expressed in arbitrary units, normalized per milligram of protein.
RNA Isolation and Transcriptome Profiling
The RNAs were isolated from snap-frozen quadriceps tissues preserved at −80°C. Samples were initially homogenized in Trizol, and total RNA was column purified by PureLink RNA Mini Kit (Invitrogen). Next-generation sequencing (NGS) experiments were performed in the Genomics Unit of the Centro Nacional de Investigaciones Cardiovasculares (CNIC; Madrid, Spain). 200 ng of total RNA was used to generate barcoded RNA sequencing (RNA-seq) libraries using the NEBNext Ultra RNA Library Prep Kit (New England Biolabs). Briefly, poly A+ RNA was purified using poly-T oligo-attached magnetic beads, followed by fragmentation, and then first and second cDNA strand synthesis. Next, cDNA 3′ ends were adenylated, and the adapters were ligated followed by PCR library amplification. Finally, the size of the libraries was checked using the Agilent 2100 Bioanalyzer DNA 1000 chip, and their concentration was determined using the Qubit fluorometer (Life Technologies). Libraries were sequenced on a HiSeq 2500 (Illumina) to generate 60 bases of single reads. FastQ files for each sample were obtained using CASAVA version (v.)1.8 software (Illumina). Sequencing data discussed here have been deposited in NCBI’s Gene Expression Omnibus and accessible through GEO: GSE150935.
Bioinformatics Analysis of RNA Sequencing Data
RNA-seq data analysis was performed by the Bioinformatics Unit of CNIC (Madrid, Spain). Sequencing reads were processed with a pipeline that used FastQC to evaluate their quality (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/; developed by the Barbraham Institute) and cutadapt to trim sequencing reads, eliminating Illumina adaptor remains, and to discard reads that were shorter than 30 bp. Resulting reads were mapped against mouse transcriptome GRCm38.91, and gene-expression levels were estimated with RNA-Seq by Expectation-Maximization (RSEM).82 Around 91% of the reads from any sample participated in at least one reported alignment. Expression count matrices were then processed with an analysis pipeline that used Bioconductor package linear models for microarray data (limma) for normalization (using the trimmed mean of M-values normalization [TMM] method) and differential expression testing.83 Changes in gene expression were considered significant if associated to a Benjamini and Hochberg adjusted p value < 0.05.
The expression profiles of 2,864 genes detected as differentially expressed in any of the three performed contrasts (Figure S4C) were clustered using k-means with n = 6. Clusters with complementary expression profiles were combined to define three metaclusters (Figure 7A). Each metacluster contained genes with expression levels that had been FR, PR, or NR in animals receiving the AAV8-secGAA vector.
Differentially expressed and metacluster gene collections were processed with IPA (QIAGEN) to identify associations with canonical pathways, diseases and biofunctions, and upstream transcriptional regulators. Functional associations provided by IPA are characterized by two parameters: p values to describe enrichment test significance and Z scores to indicate predicted activation state. Enrichments were considered significant if associated to a p value < 0.05. The calculation of Z score for a given pathway, or regulator, is based on the fold-change value of associated genes and can only be applied, by definition, to pairwise contrast results. Positive and negative Z scores suggest higher activity in the first or the second condition being compared, respectively. Absolute Z score (abs(Z score)) values higher than 2 are considered relevant. Circular plots, representing the association between enriched transcriptional regulators and functional terms, were produced with the GOplot package.84
Statistical Analysis
All of the data shown in the present manuscript are reported as mean ± standard deviation. The number of sampled units (n), upon which we reported statistics, is the single mouse for the in vivo experiments (one mouse is n = 1). GraphPad Prism 7 software (GraphPad Software) was used for statistical analyses. p values <0.05 were considered significant. For all of the datasets, data were analyzed by parametric tests, alpha = 0.05 (one-way and two-way ANOVA with Tukey’s post hoc correction and multiple t tests with Sidak-Bonferroni post hoc correction). The survival of mice was compared by Kaplan-Meier log rank test. The statistical analysis performed for each dataset is indicated in the figure legends.
Author Contributions
F.P., P.C., O.B., G.R., and F.M. directed the study. U.C. and F.P. performed most of the experiments and data analysis. U.C., F.P., and F.M. wrote the manuscript. U.C. and M.J.G. analyzed the RNA sequencing data. F.P., P.C., U.C., G.R., and F.M. contributed to the interpretation of results and provided critical insights into the significance of the work. P.S. contributed to the biochemical analyses. L.V.W. and N.D. performed the delivery of AAV vectors in mice and managed the harvesting of mouse samples. N.G. performed grip-test and plethysmography experiments. B.G. performed histological staining of mouse muscle. U.C., M.M.-N., and J.K.-L. performed EM analysis on mice skeletal muscle. C.A. performed the ROS measurements. F.C., S.C., and M.S.S. produced the AAV vectors.
Conflicts of Interest
F.P., P.C, G.R., and F.M. are inventors in patents describing the secGAA technology. F.M. is an employee and equity holder of Spark Therapeutics, Inc. All other authors declare no competing interests.
Acknowledgments
This work was supported by Genethon, the French Muscular Dystrophy Association (AFM), and Spark Therapeutics. It was also supported by the European Union’s Research and Innovation Program under grant agreement number 667751 (to F.M.), the European Research Council Consolidator Grant under grant agreement number 617432 (to F.M.), and Marie Skłodowska-Curie Actions-Individual Fellowship (MSCA-IF) grant agreement number 797144 (to U.C.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.05.025.
Supplemental Information
References
- 1.van der Ploeg A.T., Reuser A.J. Pompe’s disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- 2.Hoefsloot L.H., Hoogeveen-Westerveld M., Kroos M.A., van Beeumen J., Reuser A.J., Oostra B.A. Primary structure and processing of lysosomal alpha-glucosidase; homology with the intestinal sucrase-isomaltase complex. EMBO J. 1988;7:1697–1704. doi: 10.1002/j.1460-2075.1988.tb02998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebbink B.J., Poelman E., Plug I., Lequin M.H., van Doorn P.A., Aarsen F.K., van der Ploeg A.T., van den Hout J.M. Cognitive decline in classic infantile Pompe disease: An underacknowledged challenge. Neurology. 2016;86:1260–1261. doi: 10.1212/WNL.0000000000002523. [DOI] [PubMed] [Google Scholar]
- 4.van den Hout H.M., Hop W., van Diggelen O.P., Smeitink J.A., Smit G.P., Poll-The B.T., Bakker H.D., Loonen M.C., de Klerk J.B., Reuser A.J., van der Ploeg A.T. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 5.Güngör D., Reuser A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. A. 2013;161A:399–400. doi: 10.1002/ajmg.a.35662. [DOI] [PubMed] [Google Scholar]
- 6.Kishnani P.S., Nicolino M., Voit T., Rogers R.C., Tsai A.C., Waterson J., Herman G.E., Amalfitano A., Thurberg B.L., Richards S. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim J.A., Li L., Raben N. Pompe disease: from pathophysiology to therapy and back again. Front. Aging Neurosci. 2014;6:177. doi: 10.3389/fnagi.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nascimbeni A.C., Fanin M., Tasca E., Angelini C., Sandri M. Impaired autophagy affects acid α-glucosidase processing and enzyme replacement therapy efficacy in late-onset glycogen storage disease type II. Neuropathol. Appl. Neurobiol. 2015;41:672–675. doi: 10.1111/nan.12214. [DOI] [PubMed] [Google Scholar]
- 9.de Vries J.M., van der Beek N.A., Kroos M.A., Ozkan L., van Doorn P.A., Richards S.M., Sung C.C., Brugma J.D., Zandbergen A.A., van der Ploeg A.T., Reuser A.J. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol. Genet. Metab. 2010;101:338–345. doi: 10.1016/j.ymgme.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 10.van der Ploeg A.T., Clemens P.R., Corzo D., Escolar D.M., Florence J., Groeneveld G.J., Herson S., Kishnani P.S., Laforet P., Lake S.L. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 11.Schoser B., Stewart A., Kanters S., Hamed A., Jansen J., Chan K., Karamouzian M., Toscano A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J. Neurol. 2017;264:621–630. doi: 10.1007/s00415-016-8219-8. [DOI] [PubMed] [Google Scholar]
- 12.Yang C.F., Yang C.C., Liao H.C., Huang L.Y., Chiang C.C., Ho H.C., Lai C.J., Chu T.H., Yang T.F., Hsu T.R. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016;169:174–180.e1. doi: 10.1016/j.jpeds.2015.10.078. [DOI] [PubMed] [Google Scholar]
- 13.Mingozzi F., High K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 14.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 15.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 18.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bainbridge J.W., Smith A.J., Barker S.S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G.E., Stockman A., Tyler N. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 20.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith B.K., Collins S.W., Conlon T.J., Mah C.S., Lawson L.A., Martin A.D., Fuller D.D., Cleaver B.D., Clément N., Phillips D. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum. Gene Ther. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corti M., Liberati C., Smith B.K., Lawson L.A., Tuna I.S., Conlon T.J., Coleman K.E., Islam S., Herzog R.W., Fuller D.D. Safety of Intradiaphragmatic Delivery of Adeno-Associated Virus-Mediated Alpha-Glucosidase (rAAV1-CMV-hGAA) Gene Therapy in Children Affected by Pompe Disease. Hum. Gene Ther. Clin. Dev. 2017;28:208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ronzitti G., Collaud F., Laforet P., Mingozzi F. Progress and challenges of gene therapy for Pompe disease. Ann. Transl. Med. 2019;7:287. doi: 10.21037/atm.2019.04.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raben N., Nagaraju K., Lee E., Kessler P., Byrne B., Lee L., LaMarca M., King C., Ward J., Sauer B., Plotz P. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 25.Sidman R.L., Taksir T., Fidler J., Zhao M., Dodge J.C., Passini M.A., Raben N., Thurberg B.L., Cheng S.H., Shihabuddin L.S. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J. Neuropathol. Exp. Neurol. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puzzo F., Colella P., Biferi M.G., Bali D., Paulk N.K., Vidal P., Collaud F., Simon-Sola M., Charles S., Hardet R. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci. Transl. Med. 2017;9:eaam6375. doi: 10.1126/scitranslmed.aam6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler R.J., Bercury S.D., Fidler J., Zhao M.A., Foley J., Taksir T.V., Ryan S., Hodges B.L., Scheule R.K., Shihabuddin L.S., Cheng S.H. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum. Gene Ther. 2008;19:609–621. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 28.Sun B., Zhang H., Bird A., Li S., Young S.P., Koeberl D.D. Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression. J. Gene Med. 2009;11:913–920. doi: 10.1002/jgm.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raben N., Wong A., Ralston E., Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am. J. Med. Genet. C. Semin. Med. Genet. 2012;160C:13–21. doi: 10.1002/ajmg.c.31317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim J.A., Li L., Kakhlon O., Myerowitz R., Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy. 2015;11:385–402. doi: 10.1080/15548627.2015.1009779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim J.A., Li L., Shirihai O.S., Trudeau K.M., Puertollano R., Raben N. Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO Mol. Med. 2017;9:353–370. doi: 10.15252/emmm.201606547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franco L.M., Sun B., Yang X., Bird A., Zhang H., Schneider A., Brown T., Young S.P., Clay T.M., Amalfitano A. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol. Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 33.Schaaf G.J., van Gestel T.J.M., In ’t Groen S.L.M., de Jong B., Boomaars B., Tarallo A., Cardone M., Parenti G., van der Ploeg A.T., Pim Pijnappel W.W.M. Satellite cells maintain regenerative capacity but fail to repair disease-associated muscle damage in mice with Pompe disease. Acta Neuropathol. Commun. 2018;6:119. doi: 10.1186/s40478-018-0620-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr., Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raben N., Hill V., Shea L., Takikita S., Baum R., Mizushima N., Ralston E., Plotz P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nascimbeni A.C., Fanin M., Masiero E., Angelini C., Sandri M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII) Cell Death Differ. 2012;19:1698–1708. doi: 10.1038/cdd.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCall A.L., Stankov S.G., Cowen G., Cloutier D., Zhang Z., Yang L., Clement N., Falk D.J., Byrne B.J. Reduction of Autophagic Accumulation in Pompe Disease Mouse Model Following Gene Therapy. Curr. Gene Ther. 2019;19:197–207. doi: 10.2174/1566523219666190621113807. [DOI] [PubMed] [Google Scholar]
- 38.Cai D., Frantz J.D., Tawa N.E., Jr., Melendez P.A., Oh B.C., Lidov H.G., Hasselgren P.O., Frontera W.R., Lee J., Glass D.J., Shoelson S.E. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 39.Lu A., Proto J.D., Guo L., Tang Y., Lavasani M., Tilstra J.S., Niedernhofer L.J., Wang B., Guttridge D.C., Robbins P.D., Huard J. NF-κB negatively impacts the myogenic potential of muscle-derived stem cells. Mol. Ther. 2012;20:661–668. doi: 10.1038/mt.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pijet B., Pijet M., Litwiniuk A., Gajewska M., Pająk B., Orzechowski A. TNF- α and IFN-s-dependent muscle decay is linked to NF-κB- and STAT-1α-stimulated Atrogin1 and MuRF1 genes in C2C12 myotubes. Mediators Inflamm. 2013;2013:171437. doi: 10.1155/2013/171437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H.N., Li L., Gao P., Chen H.Z., Zhang R., Wei Y.S., Liu D.P., Liang C.C. Involvement of the p65/RelA subunit of NF-kappaB in TNF-alpha-induced SIRT1 expression in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2010;397:569–575. doi: 10.1016/j.bbrc.2010.05.160. [DOI] [PubMed] [Google Scholar]
- 42.Lagalice L., Pichon J., Gougeon E., Soussi S., Deniaud J., Ledevin M., Maurier V., Leroux I., Durand S., Ciron C. Satellite cells fail to contribute to muscle repair but are functional in Pompe disease (glycogenosis type II) Acta Neuropathol. Commun. 2018;6:116. doi: 10.1186/s40478-018-0609-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hauerslev S., Sveen M.L., Duno M., Angelini C., Vissing J., Krag T.O. Calpain 3 is important for muscle regeneration: evidence from patients with limb girdle muscular dystrophies. BMC Musculoskelet. Disord. 2012;13:43. doi: 10.1186/1471-2474-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frey N., Richardson J.A., Olson E.N. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc. Natl. Acad. Sci. USA. 2000;97:14632–14637. doi: 10.1073/pnas.260501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu Y.H., Hornsey M.A., Klinge L., Jørgensen L.H., Laval S.H., Charlton R., Barresi R., Straub V., Lochmüller H., Bushby K. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum. Mol. Genet. 2009;18:1976–1989. doi: 10.1093/hmg/ddp121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dumont N.A., Wang Y.X., von Maltzahn J., Pasut A., Bentzinger C.F., Brun C.E., Rudnicki M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015;21:1455–1463. doi: 10.1038/nm.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 48.Palikaras K., Lionaki E., Tavernarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015;22:1399–1401. doi: 10.1038/cdd.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parini R., De Lorenzo P., Dardis A., Burlina A., Cassio A., Cavarzere P., Concolino D., Della Casa R., Deodato F., Donati M.A. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J. Rare Dis. 2018;13:32. doi: 10.1186/s13023-018-0771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prater S.N., Banugaria S.G., DeArmey S.M., Botha E.G., Stege E.M., Case L.E., Jones H.N., Phornphutkul C., Wang R.Y., Young S.P., Kishnani P.S. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet. Med. 2012;14:800–810. doi: 10.1038/gim.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Ploeg A.T., Barohn R., Carlson L., Charrow J., Clemens P.R., Hopkin R.J., Kishnani P.S., Laforêt P., Morgan C., Nations S. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol. Genet. Metab. 2012;107:456–461. doi: 10.1016/j.ymgme.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 53.Park J.S., Kim H.G., Shin J.H., Choi Y.C., Kim D.S. Effect of enzyme replacement therapy in late onset Pompe disease: open pilot study of 48 weeks follow-up. Neurol. Sci. 2015;36:599–605. doi: 10.1007/s10072-014-2000-5. [DOI] [PubMed] [Google Scholar]
- 54.Colella P., Sellier P., Costa Verdera H., Puzzo F., van Wittenberghe L., Guerchet N., Daniele N., Gjata B., Marmier S., Charles S. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol. Ther. Methods Clin. Dev. 2018;12:85–101. doi: 10.1016/j.omtm.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mah C., Pacak C.A., Cresawn K.O., Deruisseau L.R., Germain S., Lewis M.A., Cloutier D.A., Fuller D.D., Byrne B.J. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol. Ther. 2007;15:501–507. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]
- 56.Bennett J., Wellman J., Marshall K.A., McCague S., Ashtari M., DiStefano-Pappas J., Elci O.U., Chung D.C., Sun J., Wright J.F. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388:661–672. doi: 10.1016/S0140-6736(16)30371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bainbridge J.W., Mehat M.S., Sundaram V., Robbie S.J., Barker S.E., Ripamonti C., Georgiadis A., Mowat F.M., Beattie S.G., Gardner P.J. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015;372:1887–1897. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan J., Desai A.K., Kazi Z.B., Corey K., Austin S., Hobson-Webb L.D., Case L.E., Jones H.N., Kishnani P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017;120:163–172. doi: 10.1016/j.ymgme.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Raben N., Jatkar T., Lee A., Lu N., Dwivedi S., Nagaraju K., Plotz P.H. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol. Ther. 2002;6:601–608. [PubMed] [Google Scholar]
- 60.Raben N., Nagaraju K., Lee E., Plotz P. Modulation of disease severity in mice with targeted disruption of the acid alpha-glucosidase gene. Neuromuscul. Disord. 2000;10:283–291. doi: 10.1016/s0960-8966(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 61.Figueroa-Bonaparte S., Segovia S., Llauger J., Belmonte I., Pedrosa I., Alejaldre A., Mayos M., Suárez-Cuartín G., Gallardo E., Illa I., Díaz-Manera J., Spanish Pompe Study Group Muscle MRI Findings in Childhood/Adult Onset Pompe Disease Correlate with Muscle Function. PLoS ONE. 2016;11:e0163493. doi: 10.1371/journal.pone.0163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Figueroa-Bonaparte S., Llauger J., Segovia S., Belmonte I., Pedrosa I., Montiel E., Montesinos P., Sánchez-González J., Alonso-Jiménez A., Gallardo E., Spanish Pompe group Quantitative muscle MRI to follow up late onset Pompe patients: a prospective study. Sci. Rep. 2018;8:10898. doi: 10.1038/s41598-018-29170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pichiecchio A., Rossi M., Cinnante C., Colafati G.S., De Icco R., Parini R., Menni F., Furlan F., Burlina A., Sacchini M. Muscle MRI of classic infantile pompe patients: Fatty substitution and edema-like changes. Muscle Nerve. 2017;55:841–848. doi: 10.1002/mus.25417. [DOI] [PubMed] [Google Scholar]
- 64.Falk D.J., Soustek M.S., Todd A.G., Mah C.S., Cloutier D.A., Kelley J.S., Clement N., Fuller D.D., Byrne B.J. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol. Ther. Methods Clin. Dev. 2015;2:15007. doi: 10.1038/mtm.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turner S.M., Hoyt A.K., ElMallah M.K., Falk D.J., Byrne B.J., Fuller D.D. Neuropathology in respiratory-related motoneurons in young Pompe (Gaa(-/-)) mice. Respir. Physiol. Neurobiol. 2016;227:48–55. doi: 10.1016/j.resp.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruzo A., Garcia M., Ribera A., Villacampa P., Haurigot V., Marcó S., Ayuso E., Anguela X.M., Roca C., Agudo J. Liver production of sulfamidase reverses peripheral and ameliorates CNS pathology in mucopolysaccharidosis IIIA mice. Mol. Ther. 2012;20:254–266. doi: 10.1038/mt.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzales P.A., Pisitkun T., Hoffert J.D., Tchapyjnikov D., Star R.A., Kleta R., Wang N.S., Knepper M.A. Large-scale proteomics and phosphoproteomics of urinary exosomes. J. Am. Soc. Nephrol. 2009;20:363–379. doi: 10.1681/ASN.2008040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andrews N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000;10:316–321. doi: 10.1016/s0962-8924(00)01794-3. [DOI] [PubMed] [Google Scholar]
- 69.Korlimarla A., Lim J.A., Kishnani P.S., Sun B. An emerging phenotype of central nervous system involvement in Pompe disease: from bench to bedside and beyond. Ann. Transl. Med. 2019;7:289. doi: 10.21037/atm.2019.04.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee N.C., Hwu W.L., Muramatsu S.I., Falk D.J., Byrne B.J., Cheng C.H., Shih N.C., Chang K.L., Tsai L.K., Chien Y.H. A Neuron-Specific Gene Therapy Relieves Motor Deficits in Pompe Disease Mice. Mol. Neurobiol. 2018;55:5299–5309. doi: 10.1007/s12035-017-0763-4. [DOI] [PubMed] [Google Scholar]
- 71.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lim J.A., Yi H., Gao F., Raben N., Kishnani P.S., Sun B. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid α-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2019;12:233–245. doi: 10.1016/j.omtm.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plotegher N., Duchen M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017;23:116–134. doi: 10.1016/j.molmed.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Yambire K.F., Fernandez-Mosquera L., Steinfeld R., Mühle C., Ikonen E., Milosevic I., Raimundo N. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. eLife. 2019;8:e39598. doi: 10.7554/eLife.39598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koeberl D.D., Case L.E., Smith E.C., Walters C., Han S.O., Li Y., Chen W., Hornik C.P., Huffman K.M., Kraus W.E. Correction of Biochemical Abnormalities and Improved Muscle Function in a Phase I/II Clinical Trial of Clenbuterol in Pompe Disease. Mol. Ther. 2018;26:2304–2314. doi: 10.1016/j.ymthe.2018.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schaaf G.J., van Gestel T.J., Brusse E., Verdijk R.M., de Coo I.F., van Doorn P.A., van der Ploeg A.T., Pijnappel W.W. Lack of robust satellite cell activation and muscle regeneration during the progression of Pompe disease. Acta Neuropathol. Commun. 2015;3:65. doi: 10.1186/s40478-015-0243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chakkalakal J.V., Jones K.M., Basson M.A., Brack A.S. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018;9:4098. doi: 10.1038/s41467-018-06621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bortolussi G., Zentillin L., Vaníkova J., Bockor L., Bellarosa C., Mancarella A., Vianello E., Tiribelli C., Giacca M., Vitek L., Muro A.F. Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum. Gene Ther. 2014;25:844–855. doi: 10.1089/hum.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ayuso E., Mingozzi F., Montane J., Leon X., Anguela X.M., Haurigot V., Edmonson S.A., Africa L., Zhou S., High K.A. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 81.Amalfitano A., McVie-Wylie A.J., Hu H., Dawson T.L., Raben N., Plotz P., Chen Y.T. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc. Natl. Acad. Sci. USA. 1999;96:8861–8866. doi: 10.1073/pnas.96.16.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walter W., Sánchez-Cabo F., Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi: 10.1093/bioinformatics/btv300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.