

Autophagy is critical for recycling amino acids and nitrogen-rich nucleotides, adjusting respiratory substrates, and the retention of assimilated nitrogen during fixed-carbon starvation.
Abstract
Autophagic recycling of proteins, lipids, nucleic acids, carbohydrates, and organelles is essential for cellular homeostasis and optimal health, especially under nutrient-limiting conditions. To better understand how this turnover affects plant growth, development, and survival upon nutrient stress, we applied an integrated multiomics approach to study maize (Zea mays) autophagy mutants subjected to fixed-carbon starvation induced by darkness. Broad metabolic alterations were evident in leaves missing the core autophagy component ATG12 under normal growth conditions (e.g., lipids and secondary metabolism), while changes in amino acid-, carbohydrate-, and nucleotide-related metabolites selectively emerged during fixed-carbon starvation. Through combined proteomic and transcriptomic analyses, we identified numerous autophagy-responsive proteins, which revealed processes underpinning the various metabolic changes seen during carbon stress as well as potential autophagic cargo. Strikingly, a strong upregulation of various catabolic processes was observed in the absence of autophagy, including increases in simple carbohydrate levels with a commensurate drop in starch levels, elevated free amino acid levels with a corresponding reduction in intact protein levels, and a strong increase in the abundance of several nitrogen-rich nucleotide catabolites. Altogether, this analysis showed that fixed-carbon starvation in the absence of autophagy adjusts the choice of respiratory substrates, promotes the transition of peroxisomes to glyoxysomes, and enhances the retention of assimilated nitrogen.
INTRODUCTION
Plants employ a variety of adaptive responses to maintain adequate supplies of nutrients needed for growth, development, reproduction, and stress protection, including dynamically modulating processes that underpin nutrient acquisition, the release of nutrient stores, and the recycling of existing macromolecules. One main recycling route involves the autophagic turnover of intracellular proteins, lipids, carbohydrates, and nucleic acids, which is essential for both the proper disposal of unwanted or dysfunctional material central to cellular homeostasis, and for the subsequent reuse of their constituent nutrients (Yang and Bassham, 2015; Havé et al., 2017; Marshall and Vierstra, 2018b). Accordingly, autophagy has been found to be crucial for the efficient recycling of assimilated nitrogen, fixed carbon, iron, manganese, phosphorous, sulfur, and zinc, with accelerated senescence often seen when autophagy-deficient plants are starved of these nutrients (Thompson et al., 2005; Li et al., 2015; Dong et al., 2017; Eguchi et al., 2017; Havé et al., 2017; Avin-Wittenberg et al., 2018; McLoughlin et al., 2018; Pottier et al., 2019; Lornac et al., 2020). In addition, autophagy contributes to metabolite mobilization from source to sink tissues. This is especially evident during reproduction, when plants concentrate energy-rich compounds and essential nutrients in seeds for subsequent use during germination (Masclaux-Daubresse et al., 2008; Li et al., 2015).

As in animals and fungi, plants engage several autophagic routes (Yang and Bassham, 2015; Marshall and Vierstra, 2018b). The best understood is macroautophagy, where an expanding cup-shaped membrane called the phagophore (or isolation membrane) emerges from the endoplasmic reticulum (ER) to engulf cytoplasmic material, which then seals to create a double-membrane-bound autophagosome. Autophagosomes fuse with vacuoles to release the inner core called the autophagic body, which is degraded along with its cargo by vacuolar hydrolases, followed by export of the breakdown products back to the cytoplasm for reuse. Microautophagy employs some of the same components as macroautophagy. This process involves invagination of the tonoplast membrane and eventual scission to release autophagic bodies and their cargo directly into the vacuole lumen for subsequent consumption (Goto-Yamada et al., 2019). The most extreme case is mega-autophagy, which initiates rupture of the tonoplast to release vacuolar enzymes directly into the cytoplasm, which then indiscriminately break down cellular material en masse as a final step in programmed cell death (van Doorn and Papini, 2013).
Macroautophagy (hereafter called autophagy) is orchestrated by a collection of over 40 AUTOPHAGY-RELATED (ATG) proteins that show strong conservation from animals and yeast to plants (Thompson and Vierstra, 2005; Yang and Bassham, 2015; Marshall and Vierstra, 2018b). Included are (1) the ATG1 protein kinase that responds to nutritional signals emanating from upstream kinases such as TARGET OF RAPAMYCIN (TOR) and SUCROSE-NON-FERMENTING-RELATED1 (SnRK1) to phosphorylate various downstream effectors; (2) the ATG2/9/18 complex that helps supply membrane during phagophore expansion; (3) the VPS34 phosphatidylinositol-3-kinase complex that decorates the autophagic membranes with a phosphatidylinositol-3-phosphate signature; (4) specific components of vesicular trafficking that deliver autophagosomes to vacuoles; and (5) an enzymatic cascade related to ubiquitylation that decorates phagophores and autophagosomes with ATG8 attached to the lipid phosphatidylethanolamine (PE). ATG8 is first energized via ATP hydrolysis by the activating enzyme ATG7, transferred to the conjugating enzyme ATG3, and finally added to PE using a dimeric ligase complex consisting of an ATG12-ATG5 conjugate bound to ATG16, which mediates the formation of an amide bond between the C-terminal Gly of ATG8 and the ethanolamine moiety in PE (Doelling et al., 2002; Thompson et al., 2005; Phillips et al., 2008; Chung et al., 2010).
Once inserted into autophagic membranes, the ATG8-PE adduct provides a docking platform for various autophagic adaptors that help sculpt and seal autophagosomes and fuse them with the tonoplast, and receptors that selectively recruit appropriate cargo before enclosure (Marshall and Vierstra, 2018b). Often, the adaptors/receptors work by exploiting ATG-Interacting Motif (AIM) or Ubiquitin-like-Interacting Motif (UIM) sequences that bind to hydrophobic pockets on ATG8 (Noda et al., 2008; Maqbool et al., 2016; Marshall et al., 2019). Through the action of specific AIM/UIM-based receptors, autophagy selectively removes various organelles, including mitochondria, peroxisomes, ER, chloroplasts and sections of the nuclear envelope, individual proteins, protein complexes such as ribosomes, proteasomes, and the CELL DIVISION CYCLE (CDC)-48 hexamer, dysfunctional protein aggregates, lipid droplets, complex carbohydrates, rRNA, and even invading intracellular pathogens (Khaminets et al., 2016; Marshall and Vierstra, 2018b; Fan et al., 2019; Marshall et al., 2019; Jung et al., 2020; Zhang et al., 2020).
Metabolomic studies combined with the analysis of mutants compromised in various autophagy components have confirmed the importance of this recycling system to nutrient homeostasis and for maintaining sufficient supplies needed for normal growth and development in various plant species. For example, Arabidopsis (Arabidopsis thaliana) lines defective in ATG8 lipidation (e.g., harboring atg5, atg7, or atg12 mutations) display a strong hypersensitivity to nitrogen and fixed-carbon starvation (Doelling et al., 2002; Thompson et al., 2005; Chung et al., 2010), which are connected to altered respiration, lipid metabolism, and starch content, in part through defects in ER function, peroxisomal β-oxidation, and lipid droplet and starch granule catabolism (Wang et al., 2013; Masclaux-Daubresse et al., 2014; Avin-Wittenberg et al., 2015; Barros et al., 2017; Fan et al., 2019; Havé et al., 2019). Mutants affected in the ATG1 kinase display a similar hypersensitivity to nutrient deprivation (Suttangkakul et al., 2011; Li et al., 2014) as well as reveal an alternative SnRK1-dependent route bypassing ATG1 that promotes autophagy during prolonged fixed-carbon starvation (Huang et al., 2019). Rice (Oryza sativa) atg7 mutants also have altered lipid and starch metabolism and plastid turnover, which ultimately impair anther development and seed maturation (Kurusu et al., 2014; Izumi et al., 2019; Sera et al., 2019).
To further define how autophagy contributes to plant metabolism, we have been studying maize (Zea mays) atg12 mutants compromised in ATG8-PE assembly, which effectively attenuate autophagic body formation (Chung et al., 2009; Li et al., 2015). Consistent with a role for autophagy in nutrient recycling, the mutant plants display premature leaf senescence, are hypersensitive to nitrogen deprivation, suppress nitrogen partitioning to seeds, and ultimately have compromised seed yield (Chung et al., 2009; Li et al., 2015). A follow-up multiomics study that combined deep metabolomic, proteomic, transcriptomic, and ionomic analyses with growth on nitrogen-rich and -deficient soils revealed dramatic alterations in maize leaf metabolism, even in the absence of nitrogen stress (McLoughlin et al., 2018). Strongly affected were secondary metabolism, antioxidant accumulation, and lipid profiles, the latter of which were underpinned by significantly altered levels of phospholipases, peroxisomal proteins, and other factors associated with lipid catabolism. Both the loss of intact phospholipids and the accumulation of breakdown products such as lysophospholipids, free fatty acids, oxylipins, and dicarboxylic acids were consistent with a disruption in lipid homeostasis and turnover. Cross-comparing mRNA and protein abundances then identified specific organelles, protein complexes, and individual proteins targeted for autophagic clearance, thereby revealing the importance of autophagy in sculpting plant proteomes under normal and nitrogen-deficit conditions (McLoughlin et al., 2018).
Here, we continued this system-wide omics approach with the analysis of maize atg12 leaves subjected to fixed-carbon deprivation induced by short-term darkness (Brouquisse et al., 1998; Weaver and Amasino, 2001). In addition to altered lipid metabolism and lipid droplet breakdown associated with the atg12 mutations, we uncovered roles for autophagy in amino acid, carbohydrate, and starch metabolism, together with a strong accumulation of nitrogen-rich catabolites. These responses were often distinct from those induced by nitrogen starvation and presumably reflected a strategy to maintain energy flow through the use of alternative respiratory substrates in the absence of photosynthesis combined with mechanisms to retain the assimilated nitrogen. By integrating the metabolomic, proteomic, and transcriptomic profiles, key points of metabolic regulation by autophagy emerged, as did the identification of autophagic substrates, which in turn could provide opportunities to exploit autophagy to modify the carbon/nitrogen economy of maize and other crop species.
RESULTS
Fixed-Carbon Starvation Substantially Affects the Maize Metabolome, Transcriptome, and Proteome
To better understand the relationship between fixed-carbon nutrition and autophagy, we exploited leaf shading to activate this recycling process (Brouquisse et al., 1998; Weaver and Amasino, 2001; Chung et al., 2009; Li et al., 2015; Hirota et al., 2018), whereby the middle section of the second leaf from 2-week-old maize plants (W22 inbred) grown in well-fertilized soil under a long-day photoperiod (16 h of light/8 h of dark) was partially covered with aluminum foil for 2 d while still attached to the plant, beginning at 6 h into the light period (Figure 1A).
Figure 1.
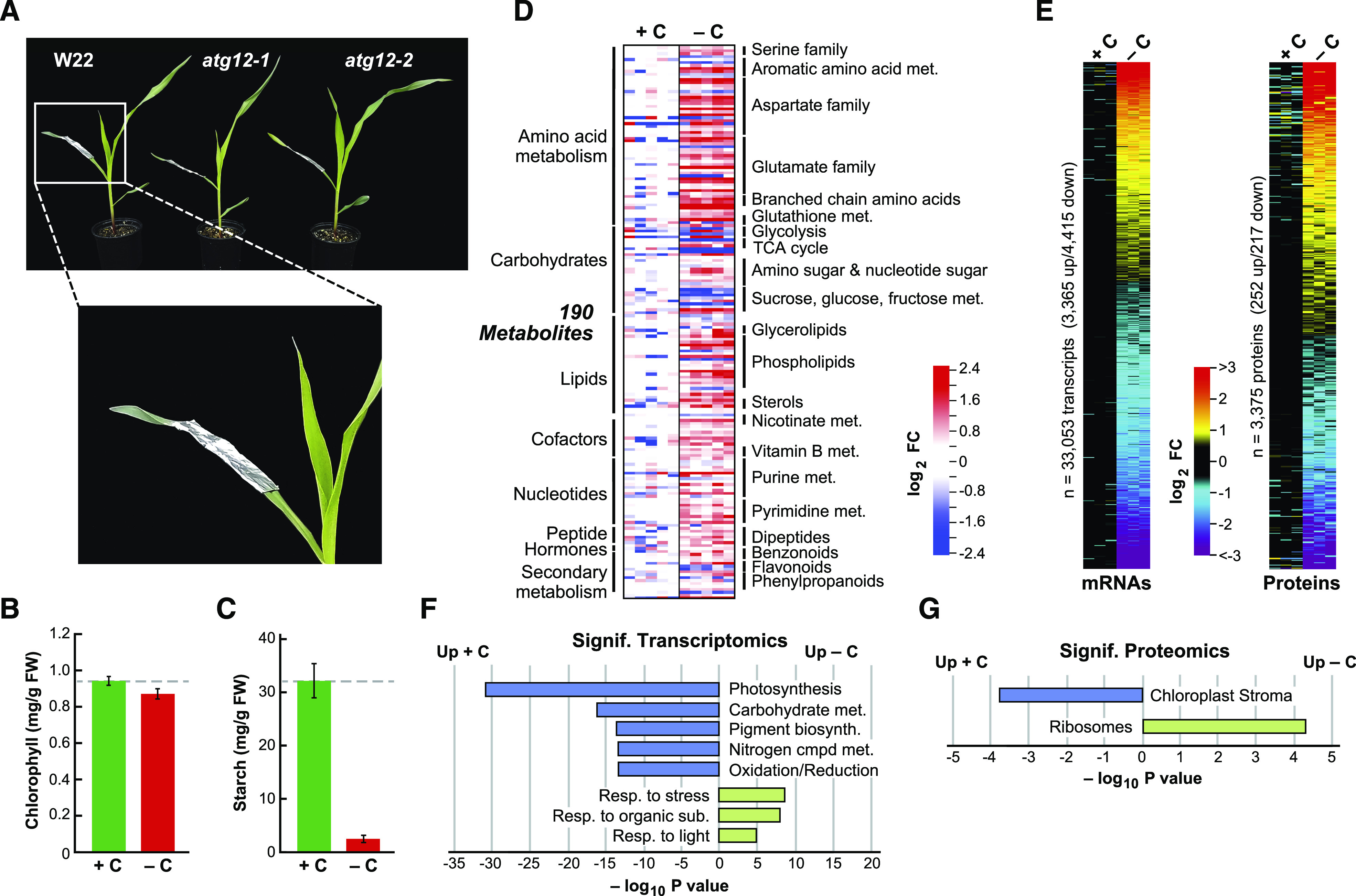
Fixed-Carbon Starvation Induced by Darkness Strongly Affects the Maize Leaf Metabolome, Transcriptome, and Proteome.
(A) Visualization of the fixed-carbon starvation regime. The middle sections of the second leaves from 2-week-old maize seedlings grown in long days were left alone (+C) or covered with aluminum foil for 2 more days (−C) starting 6 h into the light period. Shown are atg12-1 and atg12-2 seedlings alongside their W22 wild-type counterpart.
(B) Fixed-carbon starvation does not significantly affect leaf chlorophyll content. Shown are average chlorophyll levels ± sd quantified spectrophotometrically from five biological replicates (+/−C). FW, fresh weight.
(C) Fixed-carbon starvation dramatically reduces leaf starch content. Shown are average starch levels ± sd quantified spectrophotometrically from five biological replicates (+/−C).
(D) Fixed-carbon starvation profoundly affects the maize metabolome. In total, 405 metabolites were quantified from the wild type by LC-MS from five biological replicates, each containing five leaf sections (+/−C). The relative abundance of each metabolite was normalized to the mean generated with the +C samples. Shown are log2 fold change (FC) values in 190 metabolites that were deemed significant by two-sided ANOVA contrasts (P ≤ 0.05), with an additional false discovery rate (FDR) cutoff of ≤0.05 to correct for multiple comparisons (+/−C). Metabolite groupings and subgroupings are shown along the left and right sides of the heat maps, respectively.
(E) Fixed-carbon starvation induces broad alterations in the maize leaf transcriptome and proteome. mRNA (33,053) and protein (3375) profiles were quantified by RNA-seq and tandem MS, respectively, from the three mean wild-type biological replicates shown in (D). Shown are log2 FC heat maps ranked by the mean values that were significantly different in abundance, as determined by two-sided ANOVA contrasts (P ≤ 0.05), with an additional FDR cutoff of ≤0.05 to correct for multiple comparisons in +/−C samples.
(D) and (E) The lanes represent separate biological replicates, each containing five leaf sections. The analysis of all 405 metabolites and a comparison with that for nitrogen-starved leaves is shown in Supplemental Figure 2A.
(F) and (G) The most significant GO categories affected by fixed-carbon starvation in wild-type leaves calculated from the data in (E) using a singular enrichment analysis (+/−C). Shown are −log10 transformed P values of significantly affected transcripts (F) and proteins (G).
While such a short dark treatment did little to induce senescence or damage the photosynthetic machinery in the darkened section, as judged by the retention of chlorophyll and the minimal (F0) and maximal (Fmax) values of chlorophyll fluorescence (Figure 1B; Supplemental Figure 1; Supplemental Data Set 1), strong changes in the wild-type leaf metabolome were evident. Included were a substantial drop in starch levels (>10-fold; Figure 1C) and altered levels of 190 metabolites among a comprehensive list of 405 analytes (Figure 1D). This list was assayed by a suite of nontargeted liquid chromatography-mass spectrometry (LC-MS) methods (McLoughlin et al., 2018), using five biological replicates each containing five leaves, which compared equivalent darkened versus lit leaf sections (Supplemental Data Set 2). As can be gleaned from the heat maps in Figure 1D, extended darkness altered the levels of numerous metabolites, including noticeable increases in compounds linked to amino acid, carbohydrate, lipid, and nucleotide metabolism, suggestive of a broad increase in recycling. Strikingly, the metabolic alterations induced by fixed-carbon starvation were often different from those elicited by starving maize leaves for nitrogen (Supplemental Figure 2A; McLoughlin et al., 2018), implying that these nutritional stresses have both overlapping and distinct effects on maize metabolism.
Subsequent transcriptome analysis by Illumina-based RNA sequencing (RNA-seq) and proteome analysis by non-targeted tandem MS of peptides derived from trypsinized leaf protein also revealed changes in the mRNA and protein compositions of the darkened leaf sections (see below for methodological details). Of the 33,053 transcripts quantified, levels of 24% were significantly altered (P ≤ 0.05), with 3365 in greater abundance versus 4415 in lesser abundance, indicating a robust transcriptional response to leaf shading and subsequent fixed-carbon starvation (Figure 1E). As predicted for plants with stalled photosynthesis, Gene Ontology (GO) analysis showed that mRNA categories related to stress, light, and response to organic substances were elevated in response to darkness, while mRNA categories related to photosynthesis, pigmentation, energy, and carbohydrate and nitrogen metabolism were suppressed (Figure 1F). Of the 3375 proteins measured by MS, using the precursor ion intensities of the MS1 scans for quantification, levels of 14% were significantly affected (P ≤ 0.05), with a similar number up or down in abundance in the darkened leaves (252 versus 217; Figure 1E). Proteome analysis by GO revealed an increase in the ribosome category and a decrease in the chloroplast stroma category, with the latter enrichment suggesting that the darkened/fixed-carbon starved sections were beginning to dismantle their photosynthetic machinery as a prelude to senescence (Figure 1G).
The atg12 Mutants Show Substantially Altered Metabolomes both Before and during Fixed-Carbon Starvation
To dissect the relationship between autophagy and fixed-carbon availability, we compared the responses of darkened versus lit leaf sections from the wild type versus two UniformMu mutant alleles in the W22 background that compromised the single locus encoding ATG12, which is essential for assembling the ATG8-PE adduct. The atg12-1 allele behaves as a functional null mutant, while the atg12-2 allele behaves as a moderate knockdown mutant based on both genotypic and phenotypic analyses (Li et al., 2015; McLoughlin et al., 2018; this report). Indeed, principal component analysis (PCA) of darkened and lit sections from wild-type versus atg12-1 and atg12-2 leaves showed that both fixed-carbon starvation and genotype profoundly affected the maize leaf metabolome, transcriptome, proteome, and ionome, with the atg12-2 leaves invariably showing an intermediate response between the wild type and atg12-1 (Figure 2A).
Figure 2.
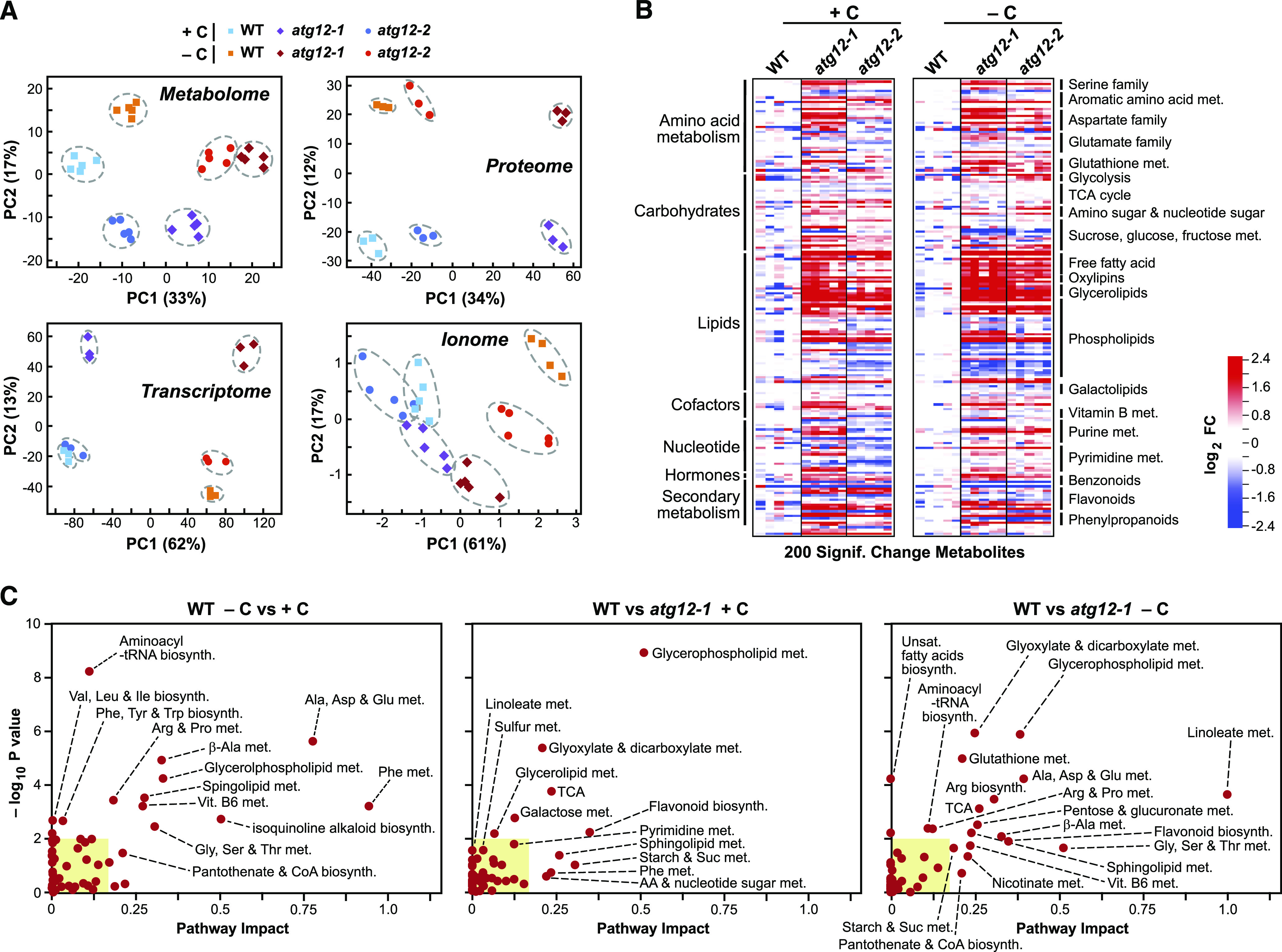
Fixed-Carbon Starvation and the atg12 Mutations Differentially Affect the Maize Leaf Metabolome.
(A) PCA of the metabolome, transcriptome, proteome, and ionome data sets for wild-type, atg12-1, and atg12-2 leaves (+/−C). The values were determined from log2 transformed ion counts for the metabolomic (n = 5 biological replicates) and ionomic (n = 5 biological replicates) data sets, median-normalized log2 transformed transcript counts for the transcriptomic data sets (n = 3 biological replicates), and log2 transformed MS1 precursor ion intensities for the proteomic data sets (n = 3 biological replicates), with the number of principal components (PCs) set at five. The amounts of variation explained by the first two components are indicated on the axes, and the numbers represent the PC scores (Z values × eigenvector). The dashed lines outline biological replicates associated with each genotype/treatment.
(B) Heat maps comparing the abundances of metabolites strongly altered by the atg12 alleles in the second leaf from 2-week-old plants grown for 2 d with or without fixed carbon (see Figure 1A). In total, 405 metabolites were quantified by LC-MS from five biological replicates, each containing five leaf sections from the atg12-1 and atg12-2 mutants and the wild type. The relative abundance of each metabolite (+/−C) was normalized to the mean generated with the wild type within each treatment (+/−C). Shown are log2 FC in 200 metabolites that were deemed significant in both atg12 alleles, as determined by two-sided ANOVA contrasts (P ≤ 0.05), with an additional FDR cutoff of ≤0.05 to correct for multiple comparisons (+/−C). Metabolite groupings and subgroupings are shown along the left and right sides of the heat maps, respectively. Each lane represents a biological replicate. The responses (wild type versus atg12-1) to fixed-carbon starvation for the full collection of 405 metabolites clustered in Cytoscape along with their chemical names are shown in Supplemental Figure 3.
(C) The metabolomes from atg12 leaves are differentially affected by fixed-carbon starvation compared with the wild type. Metabolic pathway overrepresentation and topology analysis are shown for metabolites that differ significantly in the wild type with or without leaf darkening (left) and in both atg12-1 and atg12-2 compared with the wild type grown with (middle) or without (right) fixed carbon. P values reflect the over-representation of each category, while the pathway impact weighs the importance of the affected metabolites within the pathway. Several significantly affected metabolic pathways/clusters (P < 0.05 and pathway impact > 0.2) are labeled. Yellow boxes highlight pathway impacts < 0.15 and −log10 P < 2.
Comparisons of the 405 metabolites analyzed revealed that the two homozygous atg12 lines have strongly modified metabolism compared with the wild type under both normal growth conditions and fixed-carbon starvation, with the levels of 200 metabolites significantly altered (FC ≥ 1.5 or ≤ 0.7, P ≤ 0.05; Figure 2B; Supplemental Figure 2B). Like that reported by McLoughlin et al. (2018) for nitrogen starvation, the atg12 alleles, with or without fixed-carbon starvation, had lower levels of intact phospholipids and higher levels of complex lipid breakdown products such as monoacyl lipids, free fatty acids, oxylipins, and dicarboxylic acids than the wild type, consistent with a general disruption in lipid homeostasis in the absence of autophagy, regardless of nutritional status (Figure 2B). Examples of breakdown products include a 28.1-fold increase in hydroxypalmitate levels, a 45.6-fold increase in levels of the oxylipin 13-hydroxyoctadeca-9,11,15-trienoic acid, and a 9-fold increase in levels of the dicarboxylic acid azelate (nonanedioate) in atg12-1 leaves (Supplemental Figure 3). Elevated levels of antioxidants such as rutin (17.3-fold), syringic acid (6.3-fold), and secoisolariciresinol (7.6-fold) were also seen.
In addition, we observed strong increases in the levels of amino acids, sugars, organic acids and other simple carbohydrates, nucleotide metabolites, and components of GSH metabolism when the atg12 leaves were subjected to fixed-carbon stress, as illustrated by both heat maps and Cytoscape representations of the metabolome (Figure 2A; Supplemental Figure 3). Most notable were a 6.5-fold increase in fructose-6-phosphate levels and a 40.5-fold increase in levels of the tetraglucosaccharide cellotetraose, presumably derived from cellulose and/or hemicellulose hydrolysis, concomitant with strong reductions in the levels of maltose, maltotriose, and maltotetraose derived from starch along with modest but significant reductions in Suc and Fru levels. Presumably, these changes arose as the atg12 plants attempted to maintain energy availability in the absence of photosynthesis and autophagic routes by catabolizing lipids, starch, and other glycans (Wang et al., 2013; Fan et al., 2019). Despite these changes, the atg12 mutants showed little effect on photosynthetic capacity, with the levels of chlorophyll and F0 and Fmax measures of chlorophyll fluorescence only marginally affected, even in darkened leaves (Supplemental Figure 1).
Many of the metabolic changes found here were distinct from those reported previously in response to nitrogen starvation (McLoughlin et al., 2018), which were readily apparent by comparing in Cytoscape atg12-1 versus wild-type leaves grown with or without fixed carbon to those grown with or without nitrogen (Figures 3A and 3B; Supplemental Figure 4). For example, levels of the nitrogen-rich compounds allantoin and allantoic acid rose 21.2- and 7.4-fold, respectively, after fixed-carbon starvation but were unaffected by nitrogen starvation, while the level of the ribonucleoside xanthosine rose almost 10-fold after nitrogen starvation but was unaffected by fixed-carbon starvation. Abscisic acid (ABA) also selectively hyperaccumulated in the atg12-1 plants under fixed-carbon starvation (Figure 3B), suggesting that this stress was stronger than nitrogen deprivation despite being shorter in duration (2 d versus 7 d). Such elevated stress might also explain why levels of the antioxidant GSH and its precursors γ-glutamyl-valine and γ-glutamyl-glutamine were selectively elevated in atg12 leaves exposed to fixed-carbon starvation (Figures 3A and 3B). Conversely, some metabolites were selectively upregulated by nitrogen starvation, including several flavonoids such as quercetin 3-O-glucoside, glucose-6-phosphate (involved in glycolysis and starch assembly), mannose-6-phosphate (important to Golgi function), and sucrose-6-phosphate (provides a regulatory point for Suc synthesis; Figures 3A and 3B; Supplemental Figure 4).
Figure 3.
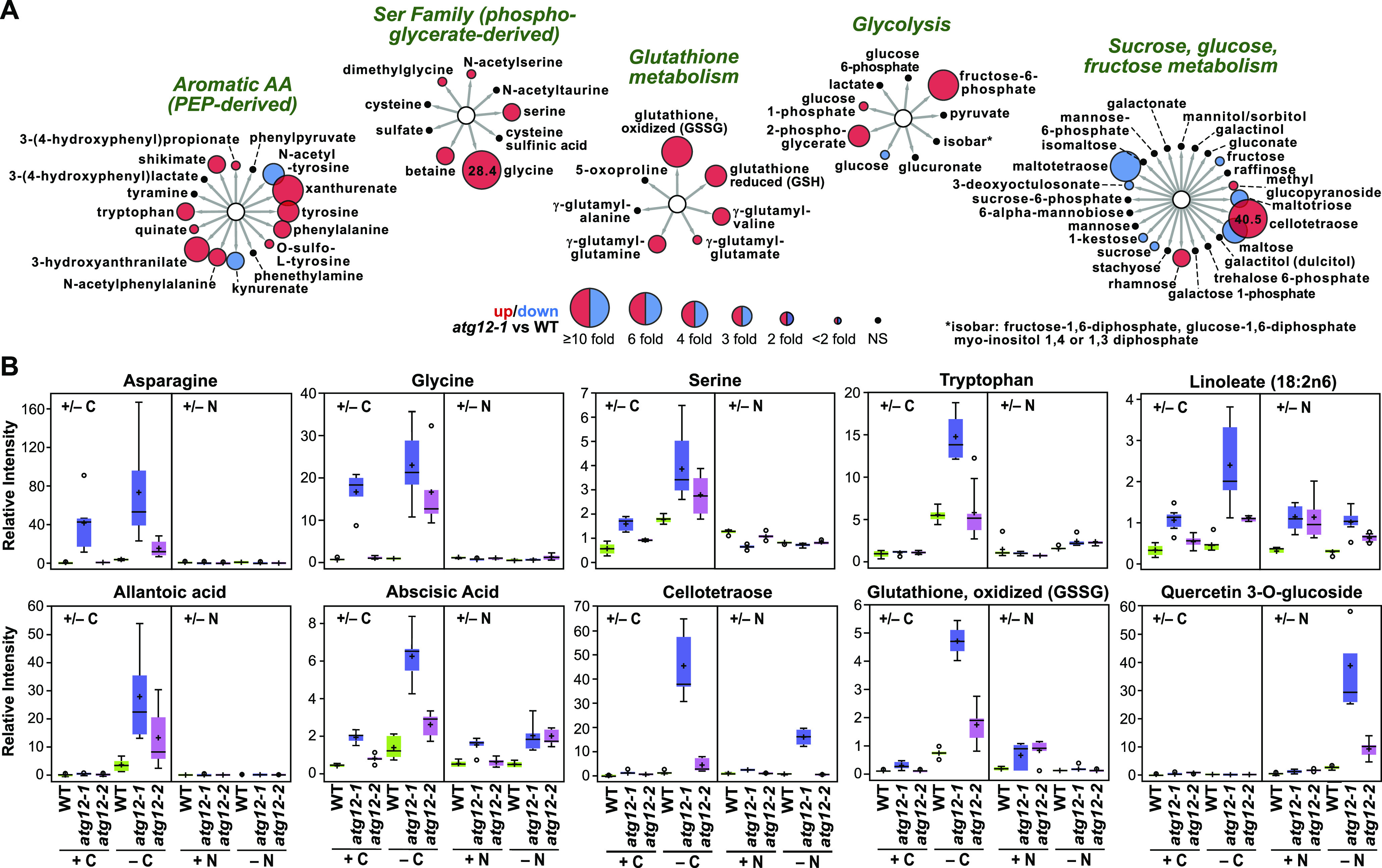
The Metabolome of Maize atg12 Leaves Is Strongly Influenced by Fixed-Carbon Starvation.
(A) Effects of the atg12-1 mutation on representative metabolites of the second leaf from plants that were starved of fixed carbon for 2 d. Metabolites were clustered using Cytoscape based on their metabolic function/pathway; representative subclusters that were differentially affected by fixed carbon are shown. The sizes of the circles reflect FC in abundance between the wild type and atg12-1; red indicates significantly higher in atg12-1, blue indicates significantly higher in the wild type, and black indicates no significant difference. For those metabolites with FC > 10, the actual values are included within the circles. Expanded Cytoscape views of metabolic clusters that are strongly influenced by fixed-carbon starvation are shown in Supplemental Figure 3. A comparison of the metabolic profiles affected by nitrogen starvation for 7 d (McLoughlin et al., 2018) can be found in Supplemental Figure 4.
(B) Examples of specific metabolites that differentially accumulated in atg12-1 and atg12-2 leaves in response to fixed-carbon and nitrogen starvation. In each box plot, the center line locates the median, + indicates the average, the box encompasses the upper and lower quartiles, error bars show the maximum and minimum of the distributions, and circles identify outlying data points (n = 5 biological replicates).
Particularly notable was the preferential effect of fixed-carbon starvation on amino acid levels, which were accentuated by impaired autophagy. The levels of most rose in the atg12-1 background, including Ala, Asn, Asp, Gln, Glu, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Trp, Tyr, and Val (1.4- to 28.4-fold; Figure 3; Supplemental Figure 3). Interestingly, no significant changes in abundance were seen for Arg, Met, or Cys, despite the connection between the latter two and autophagy, TOR, and sulfur metabolism (Dong et al., 2017; Lornac et al., 2020). The largest increases were measured for Asn and Gly, with respective fold increases of 26.4 and 28.4 measured for fixed-carbon starved atg12-1 versus wild-type leaves (Figure 3; Supplemental Figure 3). In addition, we noticed strong increases in xanthurenate, 3-hydroxyanthranilate, NΔ-acetylornithine, trans-4-hydroxylproline, 2-aminoadipate, N6-acetyl lysine, and S-adenosylmethionine levels (Figure 3A). As the first six compounds are catabolic intermediates specific to or shared among Trp, Arg, Pro,and Lys, their increased levels could reflect accelerated turnover of these amino acids as alternative respiratory sources. The increased levels of Leu, Val, and Ile were intriguing given the roles for these branched chain amino acids in suppressing autophagy in Arabidopsis through their activation of TOR (Cao et al., 2019). While fixed-carbon starvation strongly elevated Leu, Val, and Ile levels in wild-type leaves, additional modest but significant increases were seen (1.4- to 1.9-fold) in the atg12-1 mutant (Figure 3B; Supplemental Figure 3).
To help provide a context to the metabolome data, we binned the changes seen in the nonstarved versus fixed-carbon starved wild-type and atg12-1 leaves into specific metabolic pathways as defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) and assessed how individual pathways were affected using MetaboAnalyst (Chong et al., 2019). As shown in Figure 2C (left), hypergeometric tests revealed a number of KEGG metabolic pathways that were significantly altered (as defined by pathway impact and hypergeometric test P values) just by the darkening treatment alone. In line with metabolome changes described in Figure 1D, most of the affected categories for darkened versus lit wild-type samples were connected to various aspects of amino acid and lipid metabolism.
Upon comparing the atg12-1 mutant with the wild type, additional affected categories emerged. In non-starved samples, high impact and/or a P value of significance were seen for several lipid pathways (glycerophospholipid and sphingolipid metabolism), glyoxylate and dicarboxylate metabolism, Phe and flavonoid biosynthesis, the tricarboxylic acid (TCA) cycle, and starch and Suc metabolism (Figure 2C, middle). Notably, when we calculated this impact between fixed-carbon starved atg12-1 and wild-type leaves, several additional pathways emerged or became more prominent (Figure 2C). These included metabolic clusters related to GSH and amino acid metabolism (Ala, Asp and Glu, Arg and Pro, Gly, Ser and Thr metabolism, and Arg biosynthesis), both of which coincided with our observed increases in the levels of GSH and the respective amino acids and their precursors/products in these samples (Figures 3A and 3B). Emergent pathways also included the GSH, nicotinate, pantothenate and CoA, and β-Ala clusters, along with linoleate metabolism, which surfaced as the most significantly affected metabolic pathway in the darkened atg12-1 leaf sections, which is consistent with the increases seen for linoleate (Figure 2C). Together, these affected categories indicate that amino acid metabolism was severely compromised by darkness and became even more acute upon the loss of autophagy, while other processes, such as GSH and linoleate metabolism, were predominantly altered by the atg12 mutations.
Predicting that our observed effects on carbohydrate metabolism were linked to starch breakdown induced by shading and/or the lack of autophagy (Wang et al., 2013; Ren et al., 2014; Sulpice et al., 2014), we measured starch content in the leaf samples. As shown by iodine staining (Figure 4A), atg12-1 and atg12-2 leaves accumulated less starch compared with the wild type when kept in the light, with the atg12-1 leaves being more dramatically affected, while the starch levels in all three genotypes were reduced commensurately by darkness. Microscopic examination of bundle sheath cells surrounding the major and minor leaf veins, which are considered to be major storage points for leaf starch (Miyake and Maeda, 1978; Majeran et al., 2005), revealed strong reductions in starch content within these cells in atg12 versus wild-type leaves (Figure 4B). Both darkness and autophagy also affected starch content when measured spectrophotometrically. Whereas atg12-2 leaves had wild-type levels of starch under normal growth conditions, the amount in atg12-1 leaves was substantially depressed (4.1-fold; Figure 4C). Fixed-carbon starvation then induced a 12.9-fold decrease in the wild type and 32.3- and 27.2-fold decreases in atg12-1 and atg12-2, respectively, making the suppression now easily evident for both atg12-1 and atg12-2 leaves (Figure 4C).
Figure 4.
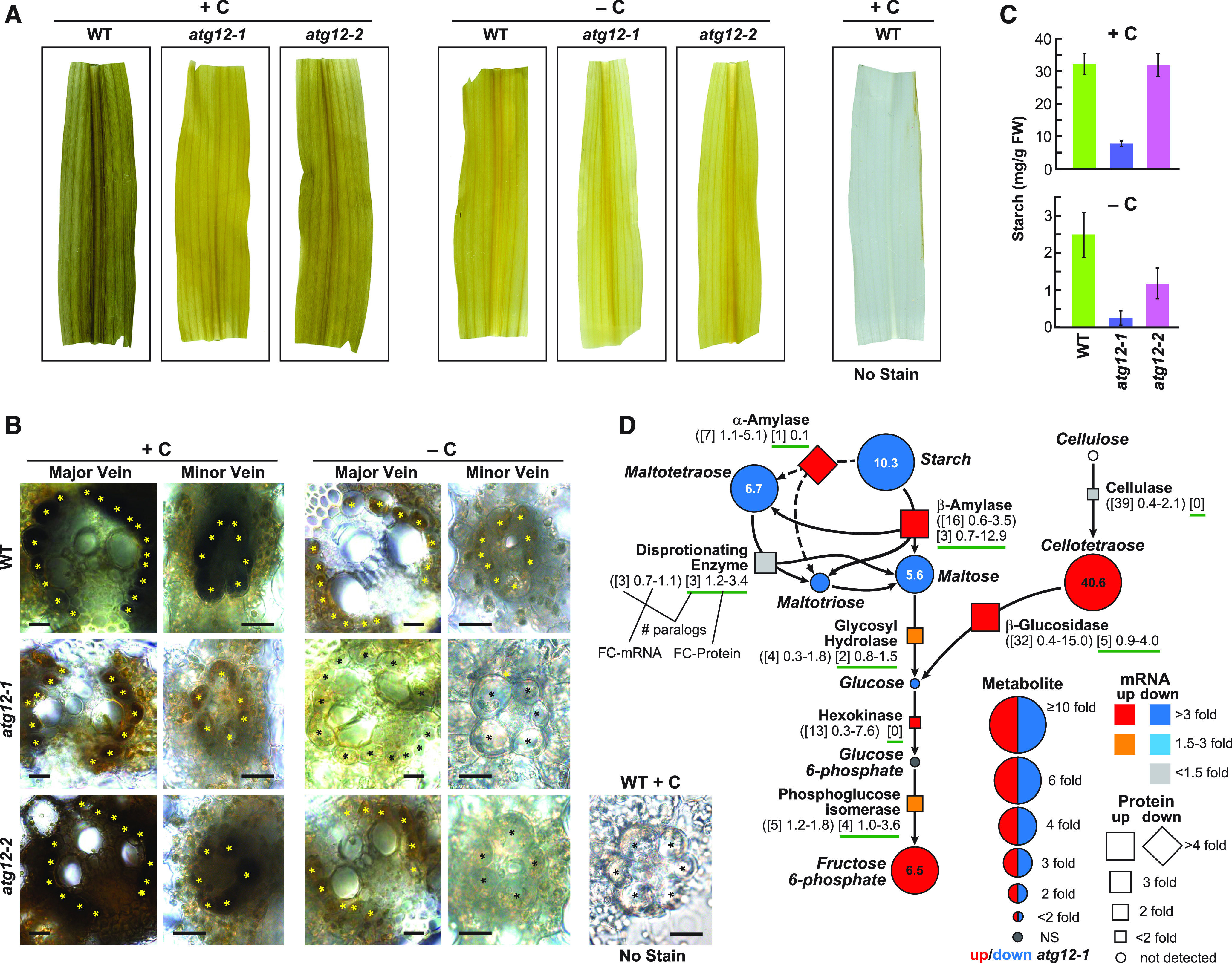
The atg12 Mutations and Fixed-Carbon Starvation Alter Starch/Cellulose Metabolism in Maize.
(A) Leaf starch content is lower in the atg12-1 and atg12-2 mutants than in the wild type (+/−C). Shown are leaf sections obtained from 2-week-old plants grown for 2 d with or without fixed carbon and stained for starch with Lugol’s iodine.
(B) Cross sections of wild-type, atg12-1, and atg12-2 leaves highlighting the reductions in starch levels in the major and minor leaf veins. Bundle sheath cells are indicated by asterisks. A minor vein without staining is shown to the right. Bars = 20 µm.
(C) Starch levels, as quantified spectrophotometrically, are depressed by the atg12-1 and atg12-2 mutations (+/−C). Each bar represents the average ± sd of five biological replicates. FW, fresh weight.
(D) Simplified flow chart for starch/cellulose breakdown to fructose-6-phosphate describing changes in associated metabolites, together with changes in relevant enzymes and their corresponding mRNAs in atg12 versus the wild type grown without fixed carbon. FC in each metabolite is indicated by the size of the circle. Red indicates significantly higher in atg12-1, and blue indicates significantly higher in the wild type. FC in the enzyme abundance is indicated by the size of the box (up) or diamond (down), whereas FC up or down in the transcript level is indicated by the color of the box/diamond. The numbers below each enzyme indicate the number of homologous genes or detected proteins in maize (brackets) and the fold changes in transcripts (parentheses) and detected protein levels (green underline) among the paralogs in atg12 versus the wild type (see example). NS, not significant.
Similarly, we predicted that changes in lipid homeostasis in the atg12 mutants, instigated by a block in membrane turnover and lipolysis, would ultimately affect cytoplasmic lipid droplet accumulation. Indeed, both the assembly of lipid droplets and their turnover have been connected to autophagy in Arabidopsis via the degradation of membranes and the conversion of the released fatty acids to triacylglycerides, which form the core of lipid droplets, and the subsequent autophagic recycling of lipid droplet triacylglycerides during starvation for use as respiratory substrates through β-oxidation (Avin-Wittenberg et al., 2015; Fan et al., 2019). Here, we found that the atg12-1 mutant accumulated significantly more cytoplasmic lipid droplets under normal growth conditions (1.45% of cell area) than the wild type (0.56%), as measured by Nile Blue A staining of leaf mesophyll cells (Jose and Burgess, 2006), while atg12-2 might have accumulated slightly more (0.85%), which was consistent with inhibited autophagic turnover (Figures 5A and 5B). This increase in atg12-1 leaves was further supported by the increased levels of the Plant UBX domain-containing10 (PUX10) protein (Figure 5C), which was recently connected to cytoplasmic lipid droplet turnover (Kretzschmar et al., 2018). Surprisingly, lipid droplet numbers were unaffected in wild-type leaves (0.54%) and only marginally reduced in atg12 leaves (1.16%) when subjected to darkness (Figures 5A and 5B), either suggesting that these lipid stores are not major respiratory substrates in the absence of fixed carbon or, more likely, that they are in a dynamic equilibrium as membranes are consumed and fed into respiration.
Figure 5.
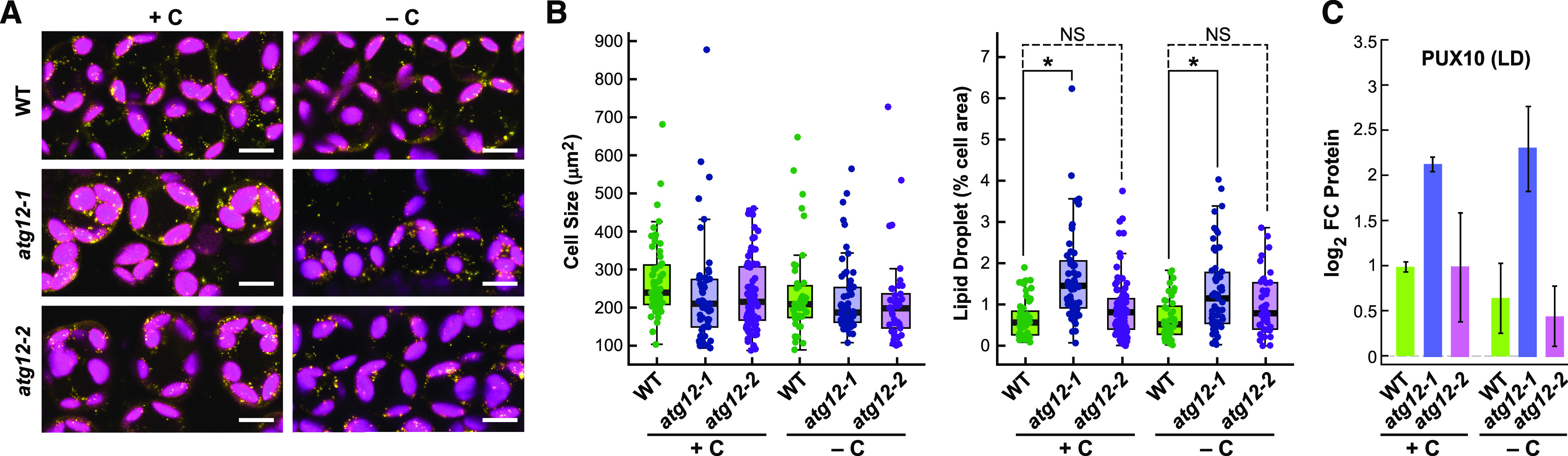
Lipid Droplets Hyperaccumulate in atg12-1 Leaves.
(A) Visualization of lipid droplets in wild-type, atg12-1, and atg12-2 leaves by Nile Blue A staining. Shown are mesophyll cells from 2-week-old maize plants grown for 2 d with or without fixed carbon. The fluorescence signals obtained from the Nile Blue A stain and chlorophyll are shown in yellow and magenta, respectively. Bars = 10 µm.
(B) Quantification of lipid droplet content. The area of the lipid droplets in 42 to 84 individual mesophyll cells from the second leaf from two separate plants was normalized to the size of the cell. In each box plot, the center line locates the median, the box encompasses the upper and lower quartiles, and the error bars show the maximum and minimum of the distributions. Asterisks indicate statistical significance based on P ≤ 0.05, as calculated by the Tukey-Kramer method. NS, not significant.
(C) Levels of the lipid droplet-associated protein PUX10. Protein levels were determined by tandem MS of total leaf protein, using the precursor ion intensities in the MS1 scans for quantification. Each bar represents the mean of three biological replicates ± sd, each analyzed in quadruplicate.
Autophagy Mutants and Fixed-Carbon Starvation Only Subtly Alter the Maize Leaf Ionome
Given the connections between autophagy and the availability of inorganic elements (Eguchi et al., 2017; Pottier et al., 2019; Lornac et al., 2020), we quantified the levels of 20 elements commonly found in plants by ion-coupled plasma MS. Analysis of the same set of five leaf samples assayed above for metabolites revealed subtle changes for individual elements, which became collectively evident by PCA when comparing the genotypes with or without fixed carbon (Figure 2A). Interestingly, abundances for arsenic, calcium, cobalt, iron, magnesium, and nickel were down in atg12 leaves, especially after fixed-carbon starvation, but were relatively unaffected for phosphorous and sulfur content (Supplemental Figure 5; Supplemental Data Set 3). Presumably, these slight effects connect autophagy to defects in vacuolar transport and/or storage, but the underlying mechanism was unclear.
Autophagy Mutants Have Altered Transcriptome Profiles
We surmised that changes in gene expression and protein composition underpinned the altered metabolomes seen in atg12 versus wild-type leaves with or without sufficient supplies of fixed carbon. To examine a transcriptional connection, we generated RNA-seq data sets from the same three mean biological replicates studied above by metabolomics and ionomics, which were condensed to only include ∼25,000 shared maize transcripts (Supplemental Data Set 4). For each genotype/condition, the replicates showed strong correlations in transcript abundances (0.98 to 0.99), thus confirming the reliability of the data (Supplemental Figure 6A). The affected genes (with or without fixed carbon) were similar for the atg12-1 and atg12-2 data sets, with strong correlations between the two (0.78 and 0.82 correlation values for the log2 transformed data), implying that the transcriptomes in both mutants responded in a similar fashion relative to the wild type (Figure 6A). This commonality was supported by PCA of the transcriptomes: the values in atg12-2 leaves were invariably intermediate to those of wild-type and atg12-1 leaves with or without fixed carbon (Figure 2A). Surprisingly, the overlap between the significantly affected transcripts (atg12 versus the wild type) in control and fixed-carbon starved leaves was low (15%), with only 74 shared transcripts, implying that light and/or starvation have a stronger effect on the mRNA profiles compared with genotype (Figure 6B).
Figure 6.
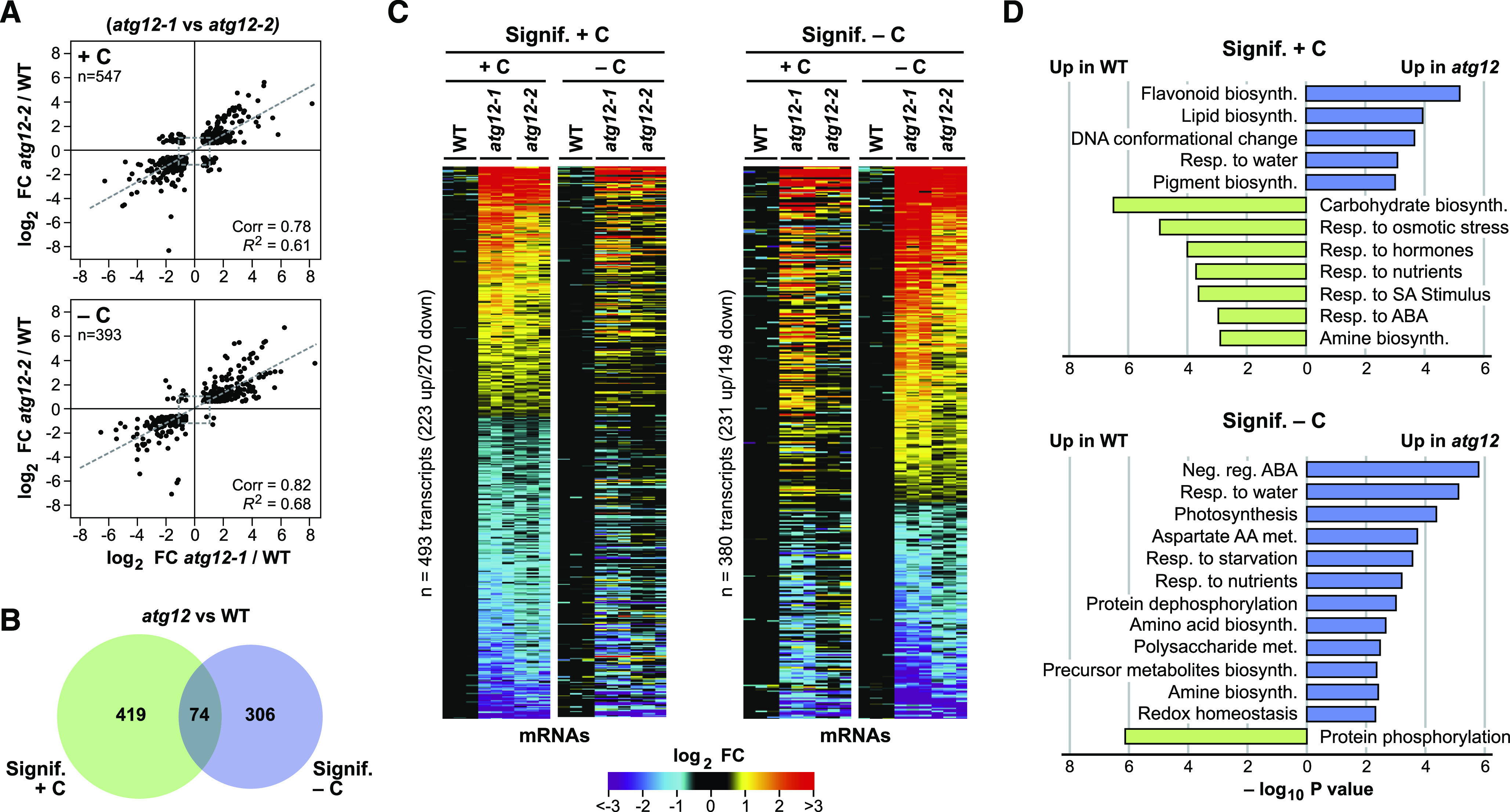
Fixed-Carbon Starvation Strongly Influences the Transcriptome of Maize atg12 Leaves.
(A) The atg12-1 and atg12-2 mutations similarly affect the maize leaf transcriptome. The mRNA profile of the second leaf sections was analyzed by RNA-seq from 2-week-old wild-type and atg12 plants grown for 2 d with or without fixed carbon (see Figure 1A). Shown are scatterplots of significantly affected transcripts comparing the log2 FC in the two atg12 alleles versus the wild type (+/−C). The average abundance of each transcript was generated from three biological replicates. The total number of significantly affected transcripts analyzed is indicated in each plot, along with Pearson’s correlation coefficient (Corr) and fit (R2) values. Dashed lines show the correlation within each comparison.
(B) Venn diagram showing the overlap of transcripts significantly affected in atg12 versus wild-type leaves (+/−C).
(C) Heat maps showing the log2 FC in abundance for leaf transcripts significantly affected by each atg12 allele (+/−C), as determined by the pairwise EBSeq algorithm, with significant hits having a posterior probability of equal expression < 0.05. Transcripts that were significantly affected in the same direction for both atg12-1 and atg12-2 were selected in the +C condition (left) or −C condition (right). The abundances were normalized to the average value obtained from the wild type for each treatment (+/−C).
(D) Log10 fold enrichment/depletion based on a singular enrichment analysis of specific GO terms for transcripts consistently altered in abundance for the atg12 mutants versus the wild type (+/−C).
Cross-comparisons identified 493 and 380 individual mRNAs with significantly altered levels in the atg12 mutants grown with or without fixed carbon relative to those in the wild type (FDR ≤ 0.05), respectively, with a similar number being upregulated or downregulated (Figure 6C). Interesting genes upregulated in atg12 leaves under fixed-carbon starvation included those encoding CoA synthases, apyrases, ureide permeases, α/β-amylases, and other carbohydrate-related enzymes, together with several aquaporins, thus further connecting autophagy to fatty acid, nucleotide, and carbohydrate metabolism and the control of water transport and/or osmolarity (Supplemental Data Set 4). We validated the changes in transcript abundance seen by RNA-seq for several loci using qRT-PCR. Included were mRNAs whose levels were higher in the wild type (e.g., encoding adenosine kinase), some that were higher in the atg12 backgrounds (e.g., encoding β-glucosidase, β-amylase, malate synthase, hexokinase-2, and ascorbate reductase), and some that were immune to both fixed-carbon starvation and the atg12 mutations, such as the mRNAs for ATG8c and the autophagy cargo receptor NEIGHBOR OF BRCA1 (NBR)-1a (Supplemental Figure 7).
In agreement with our prior analyses (McLoughlin et al., 2018), atg12 leaves kept in the light showed significant transcriptome differences compared with the wild type for a number of GO categories, consistent with impaired autophagy (Figure 6D). GO categories upregulated in the atg12 backgrounds include flavonoid, pigment, and lipid biosynthesis terms, which at least partially support the increased flavonoid accumulation, the enhanced breakdown of phospholipids, and the accumulation of lipid breakdown products seen in atg12 leaves, respectively. Conversely, those GO categories dampened in the atg12 mutants were consistent with attenuated nutrition and associated nutrient stress in the absence of autophagic recycling, including carbohydrate biosynthesis, response to nutrients, and response to hormones (including salicylic acid (SA) and ABA stimuli; Figure 6D).
When we analyzed changes to the transcriptome profiles under shading/fixed-carbon starvation, a number of new GO terms emerged that became prominent in the atg12 lines, which were consistent with photosynthetic deprivation and reduced starch content (Figure 6D). Included were categories associated with photosynthesis and redox homeostasis that likely reflected insufficient energy supply, several aspects of amino acid and carbohydrate metabolism (Asp, amino acid, and precursor metabolite biosynthesis, as well as polysaccharide metabolism), and more general stress-related terms such as negative regulation by ABA, response to starvation, and response to nutrients (Figure 6D). Collectively, the emergent terms pointed to a general transcriptional response to nutritional stress elicited by the lack of autophagy, which became more acute under fixed-carbon deficits.
Both Fixed-Carbon Starvation and Autophagy Mutants Globally Alter the Maize Proteome
Similar to the previous studies with Arabidopsis and maize (Avin-Wittenberg et al., 2015; McLoughlin et al., 2018; Havé et al., 2019), the maize atg12 leaves studied here consistently accumulated more total protein per gram of fresh weight both before and after fixed-carbon starvation, which was seen by SDS-PAGE analysis and by bicinchoninic acid (BCA)-based protein quantifications of soluble protein extracts (Figures 7A and 7B). In fact, atg12-1 leaves had ∼2.5 times more total soluble protein than wild-type leaves when exposed to either the darkened or lit condition. This increase further supports the notion that autophagy is critical for maintaining general proteostasis in plants, presumably through the recycling of dysfunctional and unwanted proteins/organelles.
Figure 7.
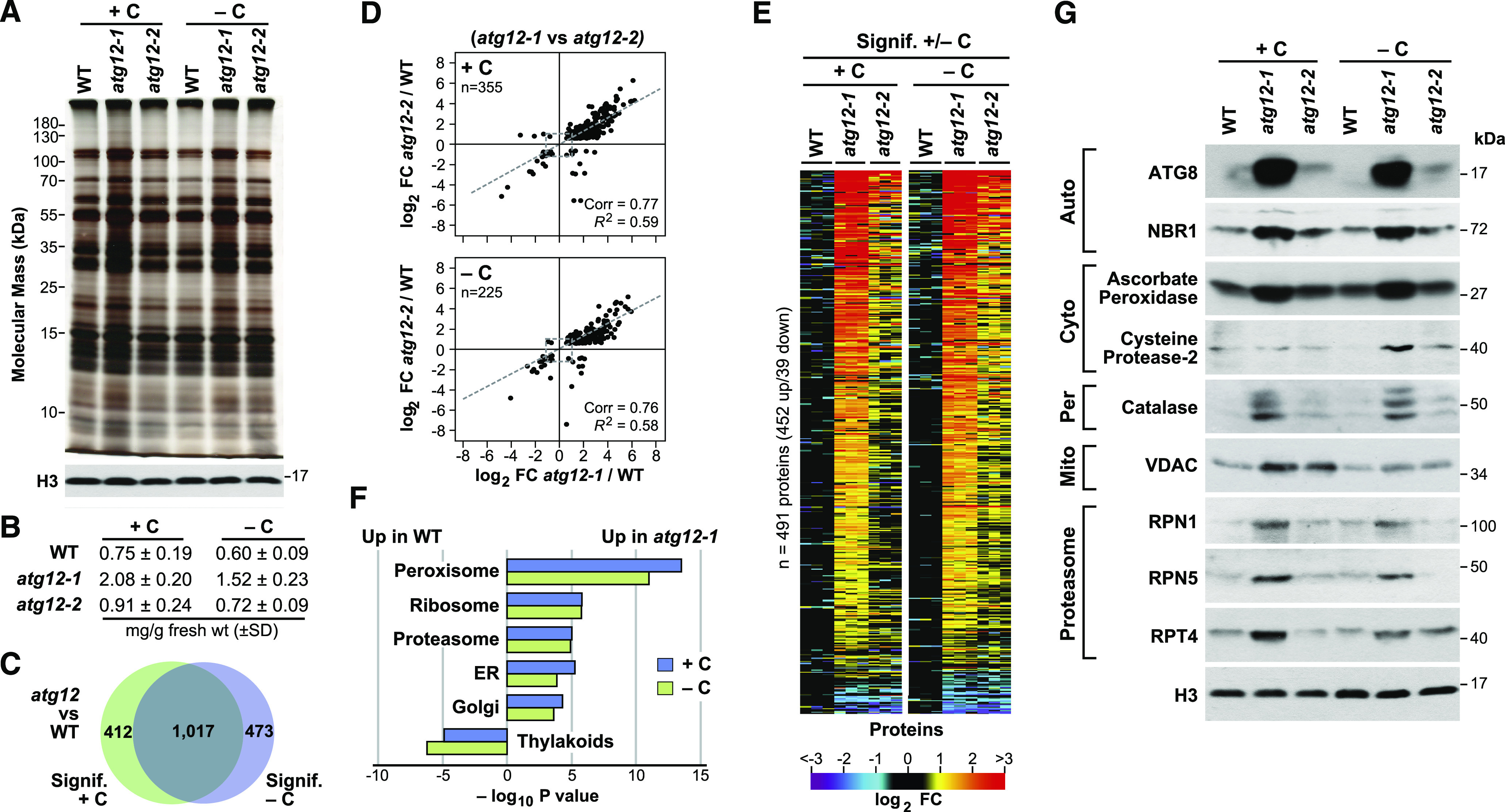
The Maize Leaf Proteome Is Strongly Influenced by the atg12 Mutations.
(A) and (B) The atg12-1 mutant accumulates more protein than the wild type. Total protein extracted from the second leaf sections from 2-week-old wild-type, atg12-1, and atg12-2 plants grown for 2 d with or without fixed carbon (see Figure 1A) was separated by SDS-PAGE and stained for protein with silver (A) or assayed for total protein by the BCA assay (B). Nearly equal tissue loading was confirmed by immunoblot analysis with anti-histone H3 antibodies. BCA assays reflect the mean ± sd of three biological replicates, each analyzed in duplicate.
(C) Venn diagram showing the overlap of proteins significantly affected in atg12 versus the wild type (+/−C).
(D) The atg12-1 and atg12-2 mutations have similar effects on the maize leaf proteome. Shown are scatterplots of significantly affected proteins comparing the log2 FC in the two atg12 alleles versus the wild type (+/−C). The average abundance of each protein was quantified from the MS1 precursor ion intensities from three biological replicates, each analyzed in quadruplicate. Significance was calculated using ANOVA contrasts (P ≤ 0.05), with an additional FDR cutoff of ≤0.05 to account for multiple comparisons. Proteins that were deemed significant in both atg12 alleles were plotted based on their FC compared with the wild type. The total number of proteins analyzed in each panel is indicated, along with Pearson’s correlation coefficient (Corr) and fit (R2) values. Dashed lines show the correlation within each comparison.
(E) Heat maps displaying the log2 FC in abundance for those proteins consistently and significantly affected in both atg12 mutants (+/−C). Each lane represents a biological replicate.
(F) Log10 fold enrichment/depletion using a singular enrichment analysis of specific GO terms for proteins consistently altered in abundance for the atg12 mutants versus the wild type (+/−C).
(G) Immunoblot detection of representative proteins whose abundance was affected by the atg12 mutations. Total protein extracts analyzed in (A) were subjected to SDS-PAGE and probed with the indicated antibodies. The location or specific function of each protein is indicated. Auto, autophagy-related; Cyto, cytoplasm; Mito, mitochondrion; Per, peroxisome; Proteasome, subunits of the 26S proteasome; Vac, vacuole.
To describe in-depth how the maize proteome was affected and to relate it to the observed metabolomic and transcriptomic changes, we applied a quantitative MS strategy to monitor protein abundances in bulk (McLoughlin et al., 2018). Here, the three mean biological replicates from each genotype/treatment described above were analyzed in quadruplicate, with the first two MS runs analyzed without exclusion and the second two runs analyzed with exclusion of the 5000 most prominent peptides to increase analysis depth. To help normalize the data sets, we compiled a list of 150 leaf proteins whose abundances were relatively consistent across all backgrounds/treatments (Supplemental Figure 8A; Supplemental Data Set 5). The average abundance of this list then provided a normalization factor for each protein in the sample (ranging from 0.79 to 1.09; Supplemental Figure 8B). This approach was validated by comparing the collection of abundantly detected histones, whose levels now also varied little among the samples after correction (Supplemental Figures 8C and 8D).
One concern was that the MS sampling of proteins would be biased compared with that for the corresponding transcripts, given the tremendous depth provided by RNA-seq and the expectation that some leaf proteins and their corresponding processes/locations/functions would be in greater abundance and thus dominate the MS analysis (e.g., proteins connected to chloroplasts/photosynthesis). Remarkably, using protein locations defined by GO as a criterion, we found that the profiles of detectable proteins versus transcripts were roughly similar (Figures 8A and 8B). As expected, an over-representation of chloroplast proteins relative to nuclear proteins was seen compared with those predicted based on mRNAs (chloroplast, 12.5% of mRNAs versus 21.3% of proteins [1.7-fold enrichment] relative to nuclear, 15.9% of mRNA versus 9.3% of proteins [0.58-fold depletion]; Figure 8D). However, for other compartments, reasonable protein coverages were seen that better agreed with their mRNA coverages. Overall, this congruity implies that the MS sampling provided a reasonably unbiased assessment of most cellular compartments.
Figure 8.
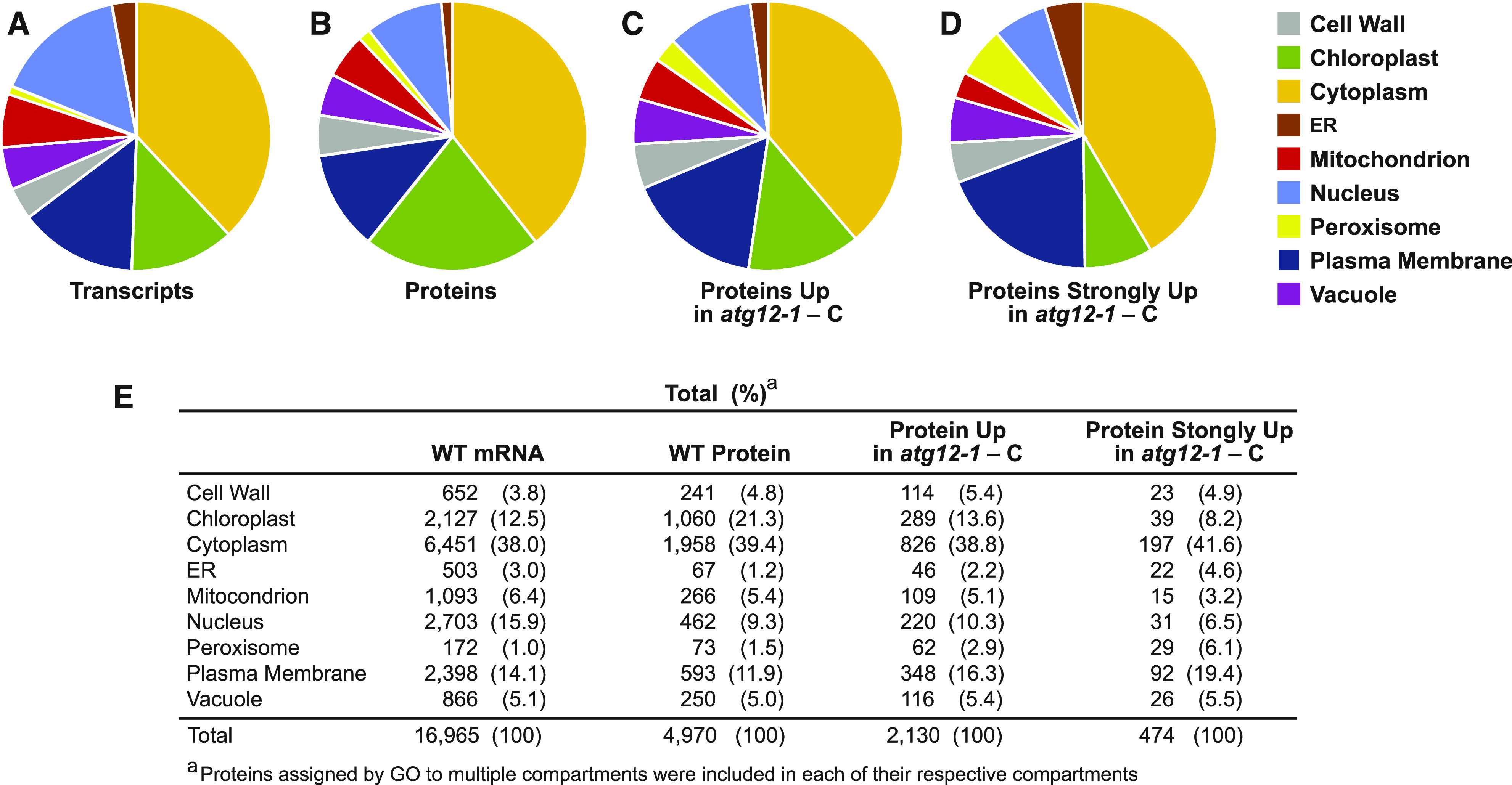
Relative Distribution of Leaf Proteins Predicted by RNA-Seq or Detected by MS Based on Cellular Location.
The protein products predicted by RNA-seq and the individual proteins detected by MS were assigned to specific cellular compartments using GO. For proteins with multiple locations, all possibilities were included in the analysis.
(A) Locations of proteins predicted from the RNA-seq data sets.
(B) Locations of the proteins detected in the MS data sets.
(C) Locations of MS-detectable proteins identified as being more abundant in atg12-1 versus wild-type leaves subjected to fixed-carbon starvation (FC ≥ 1.5, P ≤ 0.05).
(D) Locations of MS-detectable proteins found to be strongly enriched in atg12-1 versus wild-type leaves subjected to fixed-carbon starvation (FC ≥ 4, P ≤ 0.01).
(E) Table of proteins assigned to each cellular compartment as described in (A) to (D). The number of proteins assigned to each category and their percentage of the total are indicated.
In total, quantitative MS data were collected for 3375 leaf proteins, which showed strong correlations in normalized protein abundance among the three replicates (0.96 for the log2 transformed data; Supplemental Figure 6B; Supplemental Data Set 6), indicating that this label-free method provided consistent quantitative measurements. Cross-comparisons identified 491 proteins with significantly altered levels (FC ≥ 1.5 and ≤ 0.7) in both the atg12-1 and atg12-2 backgrounds relative to the wild type (FDR ≤ 0.05). The levels of most proteins (92%) were higher in the atg12-1 background, which was consistent with the observed increases in total protein levels (Figure 7E). When the abundances for the affected protein lists generated from the darkened and lit samples were compared, high correlations between the atg12-1 and atg12-2 data sets were seen (0.77 and 0.76 correlation values for the log2 transformed data), implying that the proteomes of both mutants responded in a similar fashion but at different strengths (Figures 7D and 7E). This commonality was again supported by PCA: the values for the proteomes of atg12-2 leaves were invariably intermediate to those for wild-type and atg12-1 leaves (Figure 2A). In addition, the heat maps revealed that the ranks for the proteins with increased or decreased abundance in the two atg12 backgrounds were similar in both non-starved and starved samples (Figure 7E). Within the PCA, the variation explained by PC1 strongly coincided with genotype (34%) while the variation explained by PC2 coincided with fixed-carbon availability (12%), implying that the two variables have largely independent effects on the proteome (Figure 2A). Further supporting the notion that the proteome was more strongly influenced by genotype (i.e., the absence of autophagy) than by fixed-carbon starvation was the considerable overlap in the differentially accumulating proteins (53%) between atg12 and the wild type regardless of fixed-carbon availability (Figure 7C).
To help confirm the effect of autophagy on the maize proteome, we performed immunoblot analysis of total leaf extracts to assess the levels of representative proteins assigned to specific compartments/functions that appeared to be upregulated based on the MS data. As shown in Figure 7G, strong increases in the levels of autophagy factors ATG8 and NBR1 and the RPN1, RPN5, and RPT4 subunits of the 26S proteasome (a confirmed autophagy substrate) were seen in the atg12-1 mutant; these results are consistent with a block in autophagy (Thompson et al., 2005; Marshall et al., 2015; Jung et al., 2020). Also increased were the levels of catalase (peroxisomes) and the voltage-dependent ion channel (VDAC; mitochondria), further validating the increase in various organellar proteins in the atg12 backgrounds. In addition, we assayed for ascorbate peroxidase and Cys protease-2, which might help counteract changes in redox potential and the lack of autophagic turnover, respectively. Levels of both proteins rose in atg12-1 leaves (Figure 7G), which is consistent with the increases in their corresponding mRNAs, implying that the increases were transcriptionally driven (Supplemental Data Set 4).
GO enrichment analysis of affected proteins identified a number of cellular process/component/location categories whose levels significantly increased or decreased in response to the atg12 mutations with or without fixed-carbon stress (Figure 7F). Particularly notable was a strong overrepresentation of proteins associated with peroxisomes, ER, Golgi, ribosomes, and proteasomes in atg12 leaves. These results are in agreement with the previous finding that these organelles/complexes are targets of autophagy and hence should be stabilized in the atg backgrounds (Hillwig et al., 2011; Liu et al., 2012; Farmer et al., 2013; Kim et al., 2013; Marshall et al., 2015; McLoughlin et al., 2018; Havé et al., 2019; Zhang et al., 2020). Conversely, the thylakoid category was under-represented in the atg12 mutants (Figure 7F), which is consistent with the premature senescence of chloroplasts in the absence of autophagy, whose disassembly could augment metabolism (Izumi et al., 2013; Avin-Wittenberg et al., 2015).
To better define how the maize proteome was influenced by the atg12 mutations and fixed-carbon starvation, we constructed volcano plots to compare the level of each of the 3375 shared proteins in the atg12-1 versus wild-type data sets based on both log2 FC and −log10 P value of significance. As shown in Figure 9A, the distribution of the collection was substantially skewed toward the atg12-1 ledger, with 44% (1492 total) of the proteins showing a significant FC increase but only 6% (195 total) showing a significant FC decrease. Again, these findings are consistent with the increased total protein levels in the atg12 backgrounds (volcano plots analyzing the behavior of all detected proteins under all genotype and growth conditions are shown in Supplemental Figures 8A, 8C, and 8D). The levels of a number of proteins increased substantially in atg12-1 (>16-fold), such as the autophagy components ATG8c, ATG8d, and NBR1, and various organellar proteins, including urate oxidase (UOX; peroxisome) and prenylated RAB Acceptor1 (ER/Golgi; Figure 9A). UOX, which helps direct the synthesis of allantoin from uric acid (which is subsequently converted to allantoic acid), rose almost 50-fold in atg12 leaves upon fixed-carbon starvation.
Figure 9.
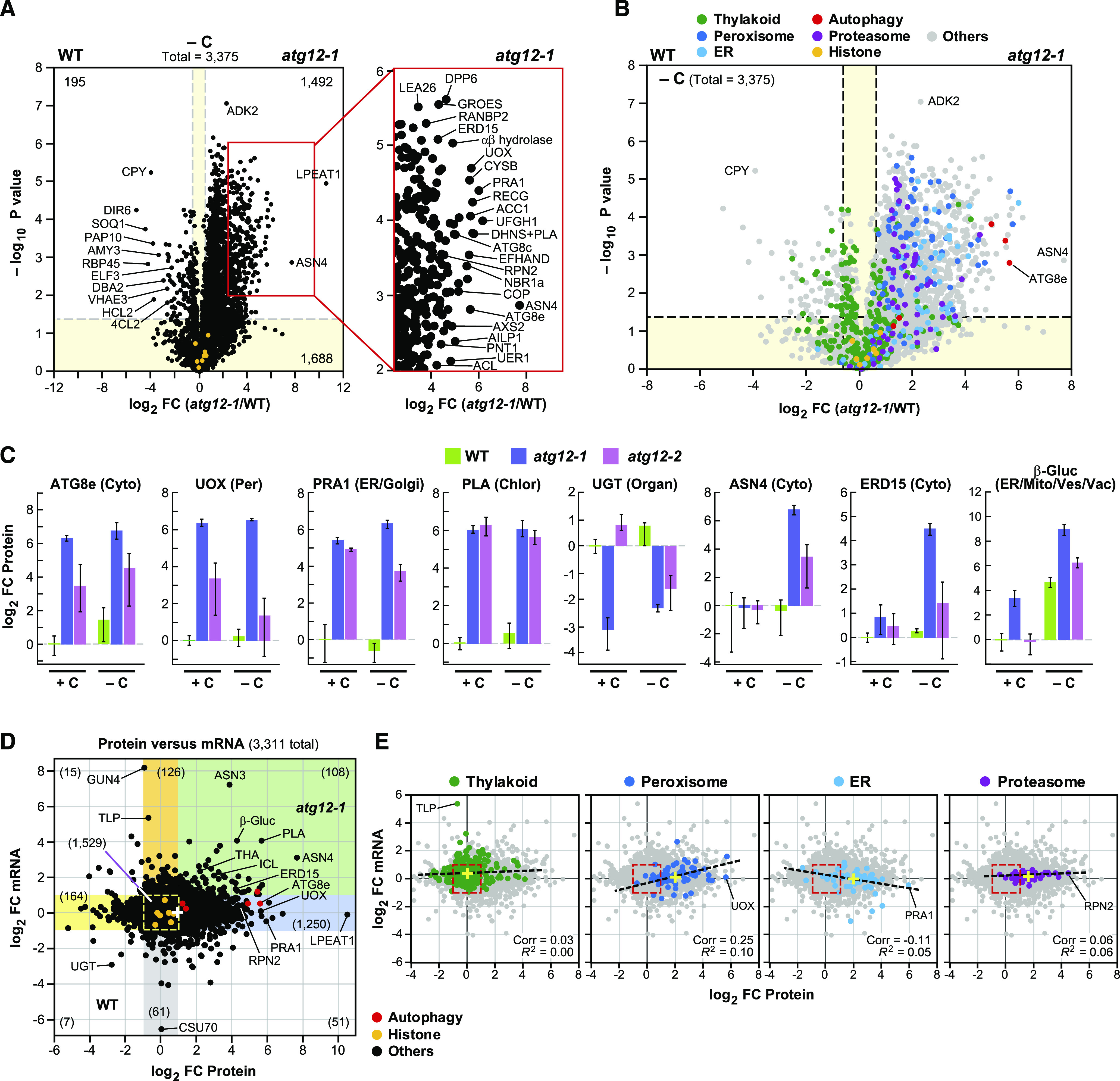
Plots Highlighting the Changes in Protein Abundance in Maize Caused by the atg12 Mutations.
(A) Volcano plot showing the preferential accumulation of individual proteins in the second leaf sections of 2-week-old atg12-1 versus wild-type plants grown for 2 d without fixed carbon (−C). Proteins were identified by LC-MS/MS and quantified by their MS1 precursor ion intensities, as in Figure 7. Each protein (3375 total) was plotted based on its log2 FC in abundance (atg12-1/wild type) and its −log10 P value of significance based on three biological replicates, each analyzed in quadruplicate. Dashed gray lines locate the significance threshold for either FC (≥1.5 fold) or −log10 P value of significance (≤0.05). The yellow sectors identify those proteins whose levels and mRNA abundances were not significantly affected (FC ≤ 1.5 and P ≤ 0.05). The numbers of proteins significantly different in the wild type versus atg12-1 (1492 up in atg12-1 and 195 up in the wild type) or unaffected (1688) are indicated. The right panel highlights the section of the volcano plot containing proteins that were at least fourfold more abundant in atg12 with −log10 P ≤ 0.01. Selected significantly affected proteins are indicated. Amply detected histones used to validate equivalent sample analysis are shown in orange.
(B) Volcano plot as in (A) showing the preferential accumulation of leaf proteins assigned by GO to specific compartments, processes, or protein complexes in atg12-1 versus wild-type leaves. Proteins assigned to the chloroplast thylakoid, peroxisome, ER, autophagy, and 26S proteasome are colored in green, blue, cyan, red, and purple, respectively. Amply detected histones and proteins with other GO classifications are colored in orange and gray, respectively.
(C) Levels of representative leaf proteins affected by the atg12 mutants and/or fixed-carbon starvation. Each bar represents the average ± sd of three biological replicates, each analyzed in quadruplicate by MS. Each value was normalized to the average value obtained from the wild type. The known or predicted location of each protein is indicated. Chloro, chloroplast; Cyto, cytoplasm; Mito, mitochondrion; Organ, organelle; Per, peroxisome; Ves, vesicles; Vac, vacuole.
(D) Scatterplot showing the relationship between changes in protein and mRNA abundances for 3311 proteins in atg12-1 versus wild-type leaves when grown under fixed-carbon starvation, as determined by plotting the log2 FC in mRNA abundance versus the log2 FC in protein abundance. The colored sectors indicate regions that contain proteins and/or mRNAs whose abundances were altered two-fold or greater by the atg12-1 mutation: yellow, proteins (but not mRNA) that were more abundant in the wild type; orange, mRNAs (but not corresponding proteins) that were more abundant in atg12-1; blue, proteins (but not corresponding mRNAs) that were more abundant in atg12-1; green, proteins and their corresponding mRNAs that were both more abundant in atg12-1; gray, mRNAs (but not corresponding proteins) that were more abundant in the wild type. The number of transcript/protein pairs in each sector is shown in parentheses. Differentially affected transcripts/proteins of interest are highlighted. The yellow dashed box outlines those transcripts/proteins that were not significantly affected in the wild type versus atg12-1 (1529). Amply detected histones and autophagy-related proteins are colored in orange and red, respectively. The average FC value for all proteins and mRNAs is shown by a white cross.
(E) Scatterplots highlighting proteins from specific cellular compartments/complexes. Pearson’s correlation coefficient (Corr) and fit (R2) values are given for each selection. The dashed lines show the correlation for the highlighted proteins. The entire collection of transcript/protein values as shown in (D) is shown in gray. A representative transcript/protein in each compartment is indicated to orient the scatterplots to that in (D). The average FC values for all highlighted proteins and mRNAs are shown by yellow crosses.
Visualization of the proteome by volcano plots based on functions and cellular compartment designations highlighted the global effect of autophagy (Figure 9B), which was then validated by individual assessments of specific proteins assigned to specific compartments (Figure 9C). Whereas the levels of proteins associated with thylakoids were evenly distributed between the wild type and atg12 with or without fixed-carbon starvation, proteins associated with peroxisomes, ER, and proteasomes were more abundant in atg12 leaves, consistent with their behavior as autophagy substrates (Liu et al., 2012; Farmer et al., 2013; Kim et al., 2013; Marshall et al., 2015). Additional volcano plots focusing on a range of GO categories continued to emphasize the importance of autophagy in maintaining the maize proteome even in the absence of fixed-carbon starvation (Supplemental Figure 9). Included were the increased levels of ribosome-associated proteins and factors connected to redox homeostasis and fatty acid, small molecule, amino acid, nucleotide, and GSH catabolism, whose turnover by autophagy was inferred by their preferential sorting to the atg12-1 ledger. By contrast, proteins associated with starch metabolism and plastoglobules showed little to no changes in atg12 relative to the wild type, suggesting that the breakdown of starch and chloroplast lipids is not regulated by the autophagic clearance of key effectors (Supplemental Figure 9).
This stabilization of peroxisomal, ER, and plasma membrane proteins in atg12-1 leaves starved for carbon was also evident from our assignment of total MS-detectable proteins to the main cellular compartments as described above (Figure 8). For example, the percentage of detectable proteins assigned to peroxisomes rose from 1.5 to 2.9% for those with ≥1.5-fold increases (P ≤ 0.05) in atg12-1 leaves and rose further to 6.1% if only the strongly elevated proteins were assessed (≥4-fold increases; P ≤ 0.01; Figures 8B to 8E). ER proteins saw similar enrichment percentages, rising from 1.2 to 2.2% and then to 4.6%. By contrast, chloroplast proteins became less prominent in the atg12 data sets, implying that chloroplasts were not strongly targeted for autophagy under our experimental conditions (i.e., going from 21.3% of the total MS-detectable proteins to 13.6% with ≥1.5-fold increases and to 8.2% with ≥4-fold increases in atg12-1 leaves under fixed-carbon stress; Figures 8B to 8E). Furthermore, cell wall proteins, which are inaccessible to autophagy, were unaffected by the atg12-1 background, with the number of detectable proteins staying constant when comparing total proteins with those strongly upregulated in fixed-carbon starved atg12-1 leaf sections (4.8 versus 4.9%; Figure 8E).
Roles for Autophagy and Synthesis in Controlling Protein Abundances
To obtain a fuller picture of how protein abundances were influenced by autophagic turnover versus synthesis, we compared the full transcriptome and proteome data sets for atg12-1 versus wild-type leaves starved of fixed carbon (McLoughlin et al., 2018). Strikingly, of the 3311 proteins present in both data sets, 54% (1782) had FC values at least twofold different for either mRNA or protein, implying strong transcriptional and/or post-transcriptional effects (Figure 9D; Supplemental Figures 10A and 10B). Although this comparison does not account for possible contributions from translation efficiency or circadian cycles in transcript accumulation, the median value for proteins showed an overall FC increase of 1.9 in atg12-1 leaves, while that for total mRNAs was 1.1, implying that the differences in protein values between the wild type and mutant were mainly driven by suppressed autophagy. To identify which cellular functions/locations were affected, we subdivided the scatterplot into sectors (using a two-fold cutoff) that showed significant FC increases in mRNA only (orange), both mRNA and protein (green), and protein only (blue) or FC decreases in protein only (yellow) and mRNA only (gray; Figure 9D). As predicted, the most populated sector described significant FC in protein but not mRNA (blue) and included 38% of the total (1250 of 3311 total).
Strikingly, as seen previously with maize leaves grown under nutrient-rich conditions (McLoughlin et al., 2018), GO analysis of the proteins that were upregulated on both the protein and transcript levels (green sector) and solely on the protein level (blue sector) in Figure 9D identified a number of protein function/cellular location categories that were significantly affected by the atg12 background and/or fixed-carbon starvation (Supplemental Figure 10C). In particular, the green sector housed GSH transferases as the sole significant GO term, indicating that a main transcriptional response was to direct the synthesis of enzymes related to GSH production and oxidative stress protection. Significant GO terms in the blue sector included categories related to the cytoplasm, proteasomes, ER, peroxisomes, Golgi bodies, and clathrin-mediated transport, strongly suggesting that these organelles/complexes/factors are autophagy targets whose levels rose in the atg12 backgrounds despite little to no increases in mRNA (Supplemental Figure 10C). Such roles became more obvious when we focused on individual proteins associated with selected GO categories in the mRNA versus protein scatterplots and determined correlation values (Figure 9E). Whereas proteins connected to thylakoids had nearly equivalent FC values for mRNA and protein, with average values of 1.3 and 1.1 for the atg12-1 mutant versus the wild type, respectively, the average values for peroxisomes, ER, and proteasomes increased to 4.0, 4.1, and 3.0, respectively, for protein but were relatively unchanged for mRNA (1.1, 0.9, and 1.1, respectively).
We also analyzed the gray sector, which focused on mRNAs downregulated in the wild type versus atg12-1 but had little influence on protein levels (Figure 9D). When we compared control versus fixed-carbon starved leaves from the wild-type and atg12-1 or atg12-2, numerous transcripts were collectively enriched that binned into GO categories related to photosynthesis, starch metabolism, and oxidation/reduction (Supplemental Figure 10D). Presumably, these reductions reflected the transcriptional down-regulation of photosynthetic potential, as the darkened leaves began to senescence prematurely (Weaver and Amasino, 2001; Chung et al., 2009; Brouwer et al., 2012).
High-Resolution Mapping of Maize Metabolism Influenced by Autophagy and Fixed-Carbon Starvation
Integration of the omics data provided us with the opportunity to map how autophagy and fixed-carbon stress collectively influenced maize metabolism. Using simplified pathway diagrams for starch metabolism, TCA/glyoxylate cycles, GSH synthesis, and nucleotide breakdown as examples, multiple control points regulated by autophagic turnover and/or transcription became apparent (Figures 4D and 10A; Supplemental Figure 11). The breakdown of starch, which presumably provided alternative respiratory substrates in the absence of photosynthesis, appeared to be affected by the steps that convert starch to free simple sugars, which could potentially contribute to the strong increase in fructose-6-phosphate levels even in the absence of photosynthesis (Figure 4D). The upregulated steps in this hydrolysis were found to be (1) β-amylases, whose mRNA and protein levels for the 16 isoforms rose up to 3.5- and 12.9-fold, respectively; (2) hexokinases, whose mRNA levels rose up to 7.6-fold; and (3) phosphoglucose isomerase, whose mRNA and protein levels for the cytoplasmic isoforms rose up to 1.8- and 3.6-fold, respectively, in atg12-1 leaves experiencing fixed-carbon starvation (Figure 4D). There was likely little contribution of α-amylases to transitory leaf starch breakdown given the dominance of β-amylases (Yu et al., 2005) and the finding that the level of the only detectable α-amylase isoform dropped 10-fold in atg12-1 leaves under carbon stress (Figure 4D).
Figure 10.
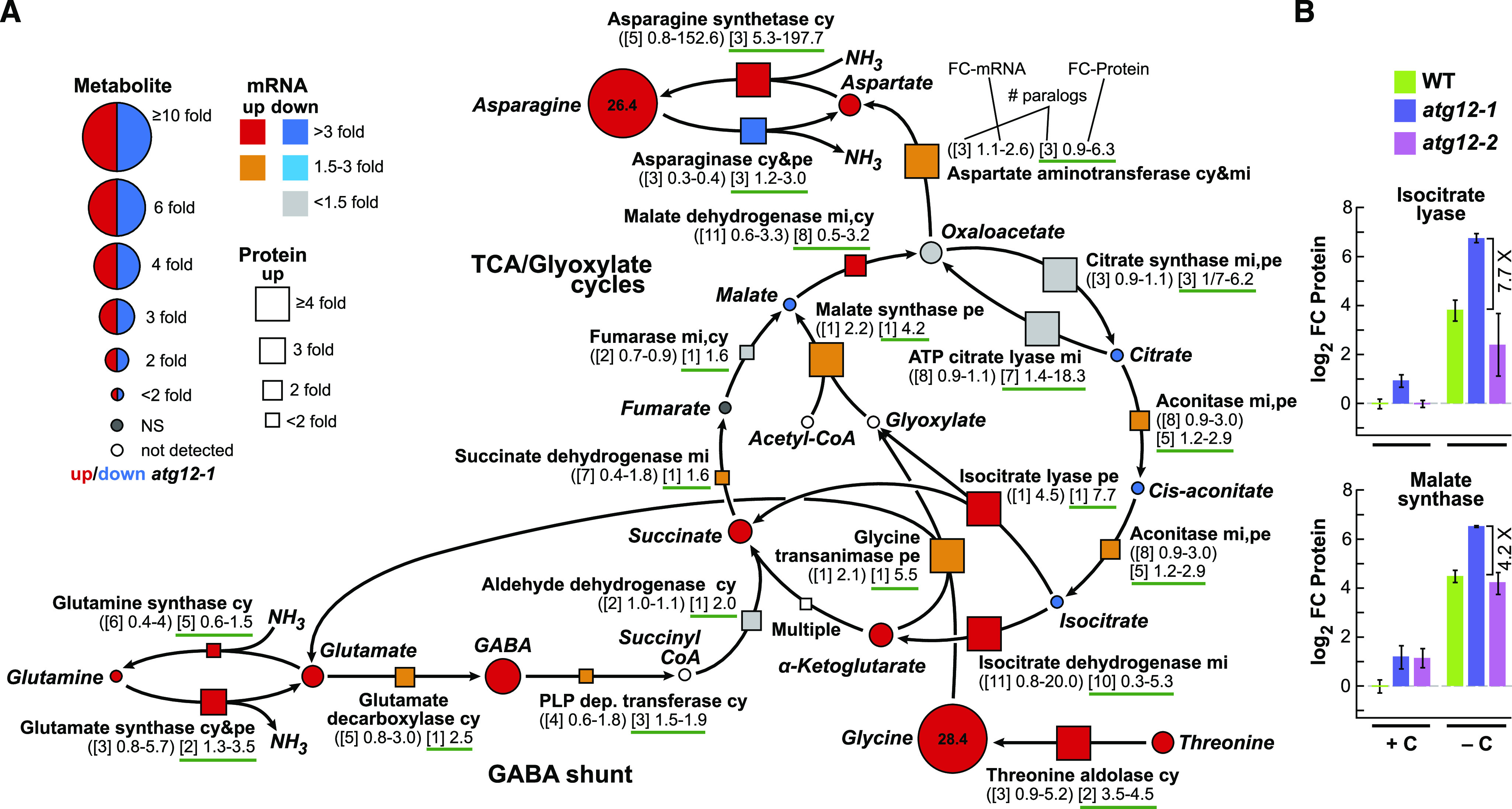
Maize Autophagy Mutants Upregulate the TCA and Glyoxylate Cycles under Fixed-Carbon Starvation.
(A) Simplified flow chart of the TCA cycle (including the glyoxylate cycle and Asn and Gln metabolism), describing changes in associated metabolites, together with changes in relevant enzymes and their corresponding mRNAs in atg12 versus the wild type grown without fixed carbon. FC in each metabolite is indicated by the size of the circles. Red indicates significantly higher in atg12-1, and blue indicates significantly higher in the wild type. The FC in enzyme abundance is indicated by the size of the box (up), whereas FC in the transcript level is indicated by the color of the box. The numbers below each enzyme indicate the number of paralogous genes in maize (brackets) and the FCs in transcripts (parentheses) and detected protein levels (green underlined) among the paralogs in atg12 versus the wild type (see example). GABA, γ-aminobutyric acid; NS, not significant. Predicted enzyme locations are as follows: cy, cytoplasm; mi, mitochondrion; pe, peroxisome.
(B) Effect of the atg12 mutants and/or fixed-carbon starvation on the abundance of isocitrate lyase and malate synthase within the glyoxylate cycle. Protein abundances were normalized to the average value obtained from wild-type samples (+C). Bars represent the average ± sd of three biological replicates, each analyzed in quadruplicate by MS.
A beneficial side reaction might also be the production of the β1-4-linked polysaccharide cellotetraose, which could arise from the partial breakdown of cellulose and/or hemicellulose through a family of cellulases (or other endo-β-glucosidases) and then be released as free glucose by a collection of β-glucosidases whose mRNA and protein levels rose by up to 15- and 4-fold, respectively (Figure 4D). The end result was vastly diminished starch and possibly free cellulose/hemicellulose levels, as the plants generated glycolytic substrates such as fructose-6-phosphate.
In a similar fashion, omics maps of the TCA/glyoxylate cycles and the pathways used to generate Asn and Gln located key points controlled by either autophagic turnover and/or transcription (Figure 10A). The glyoxylate cycle appears particularly influential. This cycle uses isocitrate lyase to convert isocitrate from the TCA cycle to glyoxylate and succinate, followed by malate synthase that generates malate from glyoxylate and acetyl-CoA for eventual mitochondrial respiration. Notably, both isocitrate lyase and malate synthase levels rose substantially during fixed-carbon starvation and then increased again by 7.7- and 4.2-fold, respectively, in the atg12-1 mutant (Figure 10B). Also intriguing were the robust increases in aspartate aminotransferase (up to 6.3-fold) and Asp synthetase (up to 198-fold) that ultimately increased the levels of Asn by over 26-fold in the atg12-1 background (Figure 10A). A similar, but not nearly as robust, effect was also seen for Gln synthesis, where modestly increased levels of succinate appeared to be converted to γ-aminobutyric acid and ultimately to Gln via the sequential action of glutamate decarboxylase and glutamine synthase (Figure 10A).
Ultimately, the upregulated TCA and glyoxylate cycles appear to terminate with increases in mainly Asn, whose concentrations are tightly correlated with the expression levels of ASPARAGINE SYNTHASE1 (ASN1) in Arabidopsis (Lam et al., 1994, 2003). We hypothesize that this increase provides a way to store assimilated nitrogen during the respiratory catabolism of amino acids, which could then be used as an amide nitrogen donor to restore amino acid pools or synthesize other nitrogenous compounds. Also surprising was the 28.4-fold increase in Gly levels (Figure 10A). Gly might be used to support C1 metabolism and to generate glyoxylate and Glu, as the mRNA and protein levels of the relevant enzyme, glycine transaminase, increased up to 2.1- and 5.5-fold, respectively, while the levels of the upstream enzyme threonine aldolase, which converts Thr to Gly, increased up to 5.2- and 4.5-fold, respectively, in fixed-carbon starved atg12-1 leaves (Figure 10A).
The enhanced levels of both GSH and GSH synthase in atg12-1 leaves upon fixed-carbon starvation prompted us to evaluate the routes that generate this tripeptide reductant, which is essential for oxidative stress protection (Hasanuzzaman et al., 2019). Important features appeared to be the increases in the levels of Ser and Glu, which are eventually used to generate the γ-linked Glu-Cys dipeptide intermediate, partially through increases in O-acetylserine lyase and oxoprolinase, whose protein levels rose up to 3.6- and 3.7-fold in atg12-1 versus wild-type leaves starved of fixed carbon (Supplemental Figure 11A). The γ-l-glutamyl-l-cysteine intermediate is then fused with Gly via an α-linked peptide bond by GSH synthase, whose level also rose 2.4-fold. Also of interest was the cycle of enzymes, namely dehydroascorbate reductase and GSH reductase, which increased in abundance by up to 2.4- and 2.8-fold, respectively. Dehydroascorbate reductase consumes reduced GSH to generate GSSG, and GSH reductase then regenerates GSH (Supplemental Figure 11A). GSH thereby provides the reducing power (through ascorbate) needed to consume hydrogen peroxide and related oxidants (Hasanuzzaman et al., 2019).
Consistent with the autophagic recycling of nucleotides, we detected increased levels of guanosine monophosphate and ultimately elevated amounts of the nitrogen-rich ureide allantoin and its acyclic metabolite allantoic acid, which stem from purine catabolism. Their levels rose 21.2- and 7.4-fold, respectively, in atg12-1 leaves without fixed carbon compared with the wild type (Supplemental Figure 11B). While it was plausible that these increases were simply driven by increases in the level of the peroxisomal enzyme UOX that converts urate to the allantoin precursor 5-hydroxyisourate, which rose 48.5-fold without a commensurate increase in its mRNA level, a similar increase in UOX protein level (47.3-fold) was seen in atg12-1 leaves under carbon-replete conditions but without commensurate increases in allantoin or allantoic acid levels (Figures 3B and 9C). Consequently, other mechanisms must exist to influence allantoin/allantoic acid levels beyond changes in detectable UOX protein levels. More modest increases in protein but not mRNA levels were also seen for xanthine dehydrogenase and hydroxyisourate hydrolase, which bracket UOX in the pathway starting from the purine xanthine (2.6- and 3.0-fold increases, respectively; Supplemental Figure 11B). UOX is abundant in glyoxysomes, suggesting that its elevated level is at least partially induced by the conversion of peroxisomes to glyoxysomes during shading combined with suppressed turnover of this organelle in the atg12 background (Hauck et al., 2014). As discussed below, the allantoin and allantoic acid products could represent a mechanism used to store assimilated nitrogen, as nucleotides are consumed by respiration in the absence of autophagy and photosynthesis, as well as providing a distinct stress signal (Takagi et al., 2016). Beyond these examples, we expect that further integration of the omics data sets will identify other aspects of maize metabolism that are influenced by autophagy and/or fixed-carbon starvation.
DISCUSSION
It is becoming increasingly evident that autophagy is central to plant cell homeostasis and metabolic regulation through its ability to recycle intracellular nutrients, maintain sufficient amino acid and fixed-carbon pools, and eliminate dysfunctional or unwanted proteins, lipids, and organelles (Yang and Bassham, 2015; Havé et al., 2017; Marshall and Vierstra, 2018b). Here, using leaf shading to induce fixed-carbon starvation combined with non-targeted metabolomic, transcriptomic, proteomic, and ionomic analyses, we provide compelling in-depth evidence for how autophagy influences numerous aspects of the maize metabolome and proteome. As with our previous studies on nitrogen starvation (McLoughlin et al., 2018), a strength of this approach was the use of the same samples for each of the omics profiles, which when combined with multiple biological and technical replicates enabled us to make statistically robust connections among the data sets. The proteomic strategy provided reasonably good coverage of most, it not all, cellular compartments and could be aligned with ∼30% of the total maize transcripts, suggesting that the method surveyed most cellular functions/processes. The results were also bolstered by exploiting a strong and weak allele affecting ATG12, which helped confirm the observed trends by PCA and other correlative comparisons.
Furthermore, our ability to match changes in metabolites with changes in protein and mRNA levels allowed us to identify processes directly influenced by autophagy, transcript levels, or a combination of the two. For example, the increased levels of proteasomes in the atg12 mutants appeared to derive from the lack of autophagy alone, while the increased levels of peroxisome-type organelles appeared to be driven both by a block in autophagy and by transcriptional mechanisms.
A central feature of our study was the use of partial leaf shading to assess fixed-carbon stress. While the 2-d treatment had little effect on the photosynthetic machinery of the darkened sections, as evidenced by minimal loss of chlorophyll and F0 and Fmax fluorescence measures of photosynthetic potential, they were clearly responding, based on substantially reduced levels of starch and altered metabolomes, transcriptomes, and proteomes. Similar to prior studies (Palenchar et al., 2004; Gutiérrez et al., 2007), GO analyses (+/− darkness) detected strong reductions in transcripts associated with photosynthesis and carbohydrate and nitrogen metabolism but increases in transcripts associated with stress and light, which were paralleled by significant reductions in chloroplast stromal proteins, all of which were consistent with the beginnings of dark-induced leaf senescence (Weaver and Amasino, 2001; Chung et al., 2009; Brouwer et al., 2012).
The atg12 mutants appear to revert to a skotomorphogenic habit (i.e., nonphotoautotrophic growth) involving the transformation of peroxisomes to glyoxysomes to enhance fatty acid β-oxidation, which was supported by dark-induced increases in the levels of the glyoxysomal enzymes isocitrate lyase and malate synthase along with increased levels of allantoin and allantoic acid in the darkened area. These changes were also paralleled by increased levels of free fatty acids such as linoleate, which could either stimulate lipid droplet assembly or reflect increased lipid droplet catabolism. Although further experiments are needed to validate this dynamic, we suspect the latter scenario, given that no changes in chloroplast-localized phospholipases were observed (e.g., PLA; Figure 9C), together with the upregulation of peroxisomal/glyoxysomal transcripts/proteins mentioned above. Altogether, these data strongly implicate the engagement of alternative protein and lipid catabolic processes in response to starvation in the absence of autophagy to help maintain sources of respiratory substrates.
Of all our omics analyses, the ionome was least affected by genotype and darkness, which probably reflected the use of mature leaves that were no longer expanding and had little control over ion availability. However, we did notice a strong drop in iron content in atg12 leaves after fixed-carbon starvation (Supplemental Figure 5), which might reflect a strategy to mitigate oxidative stress by reducing iron availability (Connolly and Guerinot, 2002). A decrease in magnesium content was also seen in atg12 leaves that could be related to the slight decrease in photosynthesis and chlorophyll content seen in the mutants (Supplemental Figure 1).
Prior studies on maize atg12 mutants grown with or without nitrogen revealed general roles for autophagy in lipid turnover, secondary metabolism, and the accumulation of antioxidants through the selective clearance of individual proteins, protein complexes, and organelles, as well as indirectly through transcriptional upregulation (McLoughlin et al., 2018). Our analysis of leaves undergoing fixed-carbon starvation induced by shading added to this notion by connecting autophagy to amino acid homeostasis, starch and lipid droplet metabolism, and nucleotide recycling. Particularly notable was the robust consumption of starch in atg12 leaves beyond that induced by darkness, presumably reflecting an alternative avenue to source respiratory substrates in the absence of photosynthesis and autophagy. Substantially elevated levels of glutathione (GSH and GSSG) and related antioxidants in the darkened atg12 section would provide further protection against energy deprivation. Also striking were the increased levels of nitrogen-rich compounds such as Asn, allantoin, and allantoic acid. These increases might reflect a mechanism to store assimilated nitrogen, as the carbon skeletons from various metabolites were consumed to maintain energy production through glycolysis and the TCA cycle. The increased levels of these nitrogen-rich compounds could also reflect accelerated senescence, as atg12 leaves begin to export nutrients to sink tissues. The overall picture seen here with maize was largely consistent with previous, less-in-depth studies of Arabidopsis and rice, thus reinforcing the notion that autophagy is central to nutrient, protein, lipid, and organelle recycling (Izumi et al., 2013; Kurusu et al., 2014; Masclaux-Daubresse et al., 2014; Avin-Wittenberg et al., 2015; Barros et al., 2017; Fan et al., 2019).
Previous omics studies identified a number of potential autophagy substrates based on their increased levels in atg backgrounds, which could not be explained by increased levels of the corresponding mRNAs (McLoughlin et al., 2018; Havé et al., 2019). Our work validates a number of these examples, such as peroxisomes, mitochondria, Golgi bodies, ER, ribosomes, and proteasomes, and adds new targets, including proteins connected to glycolysis and fatty acid and amino acid metabolism. Given that these metabolic routes likely associate into dynamic metabolon complexes to enhance substrate channeling and regulate flux (Sweetlove and Fernie, 2018), it is possible that one or more of the assembled subunits bear AIM or UIM sequences to help tether the organelles/complexes to ATG8 lining the engulfing autophagic vesicles (Marshall and Vierstra, 2018b). Possible examples could be PEX10, COPI coatomers, several clathrin adaptors, and VDAC, which might bear AIM or UIM sequences that could help dock peroxisomes, cytoplasmic lipid droplets, Golgi bodies, and mitochondria, respectively (Marshall et al., 2019). The strong increases in levels of the AIM-containing ubiquitin binding protein NBR1 in atg12 plants also support its role as an autophagic receptor for ubiquitylated protein aggregates (Svenning et al., 2011; Jung et al., 2020). The levels of both the CDC48 hexamer and its regulatory subunit UFD1 increased in atg12 leaves (3.5- and possibly up to 40-fold, respectively). This observation is consistent with the recent finding that autophagy controls the levels of this disaggregase complex, which is important for proteostasis (Marshall et al., 2019). Further studies on atg plants subjected to various stress treatments and chemical inhibitors should further validate the role(s) of these potential autophagic receptors.
A striking feature of our omics work was the distinctions seen for atg12 leaves undergoing fixed-carbon stress versus nitrogen stress, strongly implying that the two deficiencies have distinct physiological consequences in the absence of autophagy. For example, while nitrogen deficiency did little to affect amino acid pools, substantial rises in glutathione (GSH and GSSG) and most amino acid levels were evident in atg12 leaves starved for fixed carbon, especially for Asn, which increased over 28-fold. Allantoin and allantoic acid levels also rose selectively during fixed-carbon starvation, while the levels of glucose-6-phosphate and flavonoids such as quercetin-3-O-glucoside rose selectively during nitrogen starvation. Another compound that would have informed us on the influence of autophagy on the glyoxylate cycle would be its central metabolite glyoxylate; unfortunately, our MS-based profiling did not measure this compound, leaving us to presume its importance based on the profiling of closely related metabolites and their corresponding enzymes within the pathway.
The reasons underpinning these differences between nitrogen and fixed-carbon deficiencies are not completely clear. While they could arise from the severity of the stress (absence of light versus limiting amounts of nitrogen), likely explanations are related to how the two stresses affect energy availability, the carbon/nitrogen balance, and autophagy. Whereas nitrogen is often replenished from stored forms, thus allowing leaves to survive nitrogen stress by acquiring assimilated nitrogen from other tissues such as roots, fixed carbon in source tissues such as mature leaves is typically replenished locally, with photosynthesis and starch metabolism providing the daytime and nighttime supplies (Lattanzi et al., 2005; Graf et al., 2010; Wang et al., 2013). At the molecular level, a number of genes have been identified that selectively respond to nitrogen, fixed carbon, or a combination of the two, including factors involved in protein turnover (Palenchar et al., 2004; Gutiérrez et al., 2007; Zheng, 2009). How plants respond to carbon/nitrogen balance and which compounds are used for storage/transport might also differ among species. For example, whereas the transcriptional expression of the Gln-dependent Asp synthetase gene ASN1, which is central to nitrogen storage and transport, is suppressed by fixed carbon in Arabidopsis (Lam et al., 1994), homologous ASN transcripts and proteins were strongly upregulated in maize (this report). Autophagy is also differentially affected by available nitrogen and carbon. As Arabidopsis examples, whereas the autophagic turnover of proteasomes is robust under nitrogen stress, it is only modestly upregulated under fixed-carbon stress (Marshall and Vierstra, 2018a), while only prolonged fixed-carbon stress activates autophagy independently of the ATG1 kinase but through SnRK1 (Huang et al., 2019).
Previous studies showed that etiolated Arabidopsis atg5 seedlings undergoing fixed-carbon starvation after germination experienced a drop in amino acid levels (Avin-Wittenberg et al., 2015), while in the current study, we observed increased levels of most amino acids in mature maize leaves, suggesting that developmental differences exist with respect to the autophagic control of amino acid metabolism. Because fixed-carbon stress is expected to repress translation through the TOR/SnRK1 signaling pathways (Liu and Bassham, 2010) and might activate other proteolytic routes, the rise in free amino acid levels seen here might reflect repressed translation and/or activation of other proteolytic pathways needed to maintain sufficient amino acid pools and respiration in the absence of autophagy. The ubiquitin-proteasome system is thought to be a contributing proteolytic route (Brouquisse et al., 1998), but the increased levels of 26S proteasome subunits seen here in fixed-carbon starved atg12 leaves might be more connected to failed proteasome turnover via proteaphagy than to the upregulation of the complex (Marshall et al., 2015). Alternatively, myriad other plant proteases could become engaged in this process (van der Hoorn, 2008), but we failed to find a consistent upregulation of their corresponding mRNAs in atg12 leaves under fixed-carbon starvation, and saw few of the proteins that rose to detectable levels as determined by tandem MS. However, a few were significantly upregulated, including increased mRNA levels for specific subtilisin-, aspartyl-, and Cys-type proteases (Supplemental Figure 12) and increased protein levels for one or more Cys proteases (Figure 7G). It is also possible that the synthetic/catabolic pathways for amino acids are altered in atg backgrounds; unfortunately, we have yet to find convergent themes given their complexity and unrelatedness (Hildebrandt et al., 2015).
Despite our primitive understanding of how autophagy affects amino acid pools, a specific case can be made for the strongly increased levels of Asn and modest increases in Gln in fixed-carbon starved atg12 leaves. As these amino acids are often used for storage and long-distance transport (Hildebrandt et al., 2015), their elevated levels could reflect a strategy to both store assimilated nitrogen derived from other amino acids consumed by dark-induced respiration and to transport the fixed nitrogen out of the darkened section as it undergoes senescence, which were both accelerated by the atg12 mutations. Such incorporation would have the added benefit of capturing highly toxic free ammonia released as a by-product of amino acid breakdown (Esteban et al., 2016).
Like Asn and Gln, the dramatically increased levels of allantoin and allantoic acid in the atg12 mutants under fixed-carbon starvation might have an analogous purpose. Both ureides are generated in peroxisomes and essentially represent a mechanism to retain and mobilize assimilated nitrogen derived from purine catabolism (Takagi et al., 2016). In fact, legumes use allantoin and allantoic acid more globally as vehicles for transporting and storing symbiotically fixed nitrogen (Smith and Atkins, 2002). Recent studies on Arabidopsis plants treated with allantoin or blocked in allantoin synthesis or catabolism have also connected allantoin to seedling establishment by regulating peroxisome function and to stress protection by increasing the levels of osmoprotectants and the stress hormones ABA and jasmonic acid (Watanabe et al., 2014; Takagi et al., 2016). Consequently, it is possible that allantoin has hormone-like activity in maize, and thus the increased level seen in atg12 leaves experiencing fixed-carbon deficits might represent a direct stress signal. The increased ABA levels seen in atg12 leaves could in turn be a secondary effect triggered by heightened allantoin levels. Clearly, further analyses of maize mutants altered in allantoin biosynthesis and metabolism are needed to test this intriguing possibility.
We note that two other metabolites, trehalose-6-phosphate and shikimic acid, are thought to function as metabolic stress signals in Arabidopsis that influence sugar and amino acid turnover in response to carbon deficits. In our studies with maize, neither fixed-carbon starvation nor the atg12 mutations affected trehalose-6-phosphate levels and only modestly increased shikimate levels (approximately two-fold), suggesting that maize leaves do not rely on these metabolites as signals. By contrast, both fixed-carbon stress and the atg12 mutations synergistically increased ABA levels, which might be the key stress hormone signal under these conditions. SA-induced senescence might also function as a secondary response in Arabidopsis autophagy mutants subjected to nitrogen starvation (Masclaux-Daubresse et al., 2014; Havé et al., 2019). For maize, no changes in SA levels were observed in the atg12 mutants grown under either nutrient-rich or nitrogen- or fixed-carbon-deficient conditions (McLoughlin et al., 2018; this report). Nor were diagnostic SA-induced genes upregulated, such as PAD4 and PR1 involved in SA biosynthesis and signaling, respectively, even though GO analysis identified response to SA as a significant category (Figure 6D). Consequently, we are uncertain whether maize activates an SA-mediated stress response in the absence of autophagy.
Taken together, the maize omics analyses described here combined with prior studies (Wang et al., 2013; Masclaux-Daubresse et al., 2014; Avin-Wittenberg et al., 2015; Barros et al., 2017; Hirota et al., 2018; McLoughlin et al., 2018; Fan et al., 2019; Havé et al., 2019; Izumi et al., 2019) illustrate the importance of autophagy to plant physiology through its effect on energy flux, lipid and carbohydrate turnover, and the composition and housekeeping of plant proteomes. Nevertheless, it remains intriguing that mutants null for macroautophagy (and possibly microautophagy) are phenotypically normal and produce progeny (albeit at lower efficiency) when grown in nutrient-rich conditions (Doelling et al., 2002; Li et al., 2015; Barros et al., 2017; Sera et al., 2019), strongly implying the existence of robust metabolic networks that can overcome these deficiencies and backup mechanisms that can mitigate energy and proteotoxic stress when autophagy is blocked. Our proteomic analyses also provide a large data set of potential autophagic substrates that presumably extends the reach of autophagy to many cellular compartments and most, if not all, aspects of plant growth, development, and stress protection. Of the 605 proteins whose levels rose over four-fold in carbon-starved atg12 plants (P ≤ 0.01; Figure 9D), 86% displayed little change in the corresponding mRNA levels (0.5 < FC < 2) and thus represent candidate autophagy substrates for further study. Eventually, through such an integrated approach, a number of autophagy-dependent and energy-related processes should emerge whose manipulation could be exploited to modify crop metabolism and ultimately optimize yield and stress resilience, especially as they pertain to nutrient recycling.
METHODS
Plant Materials, Growth Conditions, and Phenotypic Analysis
The maize (Zea mays) atg12-1 (mu02975) and atg12-2 (mu02196) mutants generated by UniformMu transposon mutagenesis of the W22 inbred background were grown as described by Li et al. (2015) and McLoughlin et al. (2018). Plants used for omics analyses were grown in a randomized arrangement in Metromix 360 soil (SunGro), which was subsequently fertilized with modified Hoagland solution containing 15 mM nitrogen. After 2 weeks of growth in a controlled environment chamber at 25°C under a 16-h-light/8-h-dark long-day photoperiod provided by 300 μmol m−2 s−1 white light, the middle part of the second leaf was tightly covered with aluminum foil for 2 d (Figure 1A). In the early afternoon (between 1:00 and 2:00 pm), the leaf section 1 cm inside the covering was harvested; samples from five leaves were pooled for each biological replicate, ground to a fine powder in liquid nitrogen, and stored at −80°C. To visualize starch content, count lipid droplet numbers, and measure F0 and Fmax chlorophyll fluorescence, the plants were grown in a greenhouse for 14 d at 300 μmol m−2 s−1 light, and the middle part of the second leaf was covered as above for 2 d.
Metabolome and Ionome Profiling
Leaf sections from five biological replicates were subjected to unbiased metabolite profiling using four independent platforms in conjunction with Metabolon (www.metabolon.com), as previously described by McLoughlin et al. (2018). Each platform employed a Waters ACQUITY ultra-high-performance liquid chromatograph in combination with a Q-Exactive Plus high-resolution/accurate mass spectrometer (Thermo Fisher Scientific) interfaced with a heated electrospray ionization source and an Orbitrap mass analyzer operated at 35,000 mass resolution (Evans et al., 2009; Ohta et al., 2009). The tissues were extracted, dried, and reconstituted in solvents compatible to each of the four platforms, each of which was spiked with a collection of MS standards at fixed concentrations to assess injection and chromatographic consistency. Hydrophilic compounds were separated with a C18 column (Waters UPLC BEH C18; 2.1 × 100 mm, 1.7 µm) by a water/methanol gradient containing 0.05% (v/v) perfluoropentanoic acid and 0.1% formic acid and analyzed in positive ion mode. More hydrophobic compounds were separated with the same C18 column by a methanol/acetonitrile/water gradient containing 0.05% (v/v) perfluoropentanoic acid and 0.01% formic acid and analyzed in positive ion mode. More basic compounds were separated with a second C18 column by a methanol/water gradient containing 6.5 mM ammonium bicarbonate (pH 8.0) and analyzed in negative ion mode. Additional compounds were separated with a HILIC column (Waters UPLC BEH-amide; 2.1 × 150 mm, 1.7 µm) by a water/acetonitrile gradient containing 10 mM ammonium formate (pH 10.8) and analyzed in negative ion mode. All four MS analyses alternated between MS and data-dependent MS scans using dynamic exclusion.
Metabolites (405 total) were identified by automated comparisons with the retention times, ion features, and MS/MS fragmentation patterns of a reference chemical library (Supplemental Data Set 7; Dehaven et al., 2010). Metabolite abundances were normalized based on the values obtained from metabolites considered least variable among the samples (sd/average) to correct for technical variations. Compounds were clustered into subgroups based on function and/or metabolic pathway and plotted using Cytoscape (Shannon et al., 2003).
Elemental ion profiles were obtained from all five biological replicates by ion-coupled MS as described by McLoughlin et al. (2018) at the USDA-Agricultural Research Service Plant Genetics Facility at the Donald Danforth Plant Science Center. Lyophilized and pulverized leaf samples (∼75 mg) were digested in 2.5 mL of HNO3, mixed with 115In (BDH Chemicals) as an internal standard, and heated to 105°C for 2 h before analysis (Ziegler et al., 2013).
Transcriptome Profiling
Total RNA was extracted from 50 to 100 mg of powdered leaf tissue using an RNeasy Plant Mini kit (Qiagen). mRNA was then enriched by poly(A) selection and deep sequenced (>21 million reads per sample) as 100-mers using the Illumina HiSeq-2500 platform (DNA Sequencing Facility, University of Wisconsin Biotechnology Center). Raw data were processed as described by McLoughlin et al. (2018). Transcripts were aligned to the B73 reference files Zea_mays_AGPv3.31.gff3 and Zea_mays.AGPv3.31.dna.toplevel.fa from ENSEMBL Plants (http://plants.ensembl.org) using Bowtie 2 (Langmead and Salzberg, 2012), with the default settings set at very_sensitive. The fragment-length-mean and fragment-length-sd parameters are summarized in Supplemental Data Set 8.
qRT-PCR Analysis
One microgram of total RNA isolated as described above was treated with DNase I (Invitrogen), converted to cDNA using the SuperScript III first-strand synthesis system (Invitrogen) and oligo(dT)20 primers, and subjected to qRT-PCR using a CFX Connect Real-time PCR Detection System (Bio-Rad) in combination with the LightCycler 480 SYBR Green I master mix (Roche Diagnostics) as described by McLoughlin et al. (2018). The relative transcript abundance for each locus was determined by the comparative threshold cycle method (Pfaffl, 2001), using the Fpgs (GRMZM2G393334) and Lug (GRMZM2G425377) reference genes as internal controls (Manoli et al., 2012). Two technical replicates were analyzed for each of three biological replicates; all transcript measurements were then normalized to the values obtained from non-starved W22 leaves. Details of oligonucleotide primers are provided in Supplemental Data Set 9.
Proteome Profiling
Proteins were extracted at 4°C from pulverized leaf tissue into 50 mM HEPES (pH 7.5), 5 mM Na2EDTA, 2 mM DTT, and 1× plant protease inhibitor cocktail (Sigma-Aldrich; Aguilar-Hernández et al., 2017), using a Pyrex Potter-Elvehjem tissue grinder (Fisher Scientific). After clarification by centrifugation at 16,000g, 150 μL of the protein extract was precipitated in 4:1:3 (v/v) methanol:chloroform:water and collected by centrifugation (McLoughlin et al., 2018). The resulting pellet was lyophilized to dryness, resuspended in 100 μL of 8 M urea, and reduced at 22°C for 1 h with 6 μL of 200 mM DTT, followed by alkylation with 25 μL of 200 mM iodoacetamide for 1 h. The reaction was quenched with 25 μL of 200 mM DTT, diluted with 900 μL of 25 mM ammonium bicarbonate to reduce the urea concentration below 1.5 M, and digested overnight at 37°C with 0.5 µg of sequencing-grade modified porcine trypsin (Promega). The resulting peptides were lyophilized to a final volume of ∼250 µL, acidified with 0.5% (v/v) trifluoroacetic acid (pH < 3.0), and desalted and concentrated using a 100-μL Bond Elut OMIX C18 pipette tip (Agilent Technologies) according to the manufacturer’s instructions. Bound peptides were eluted in 50 μL of 75% (v/v) acetonitrile and 0.1% acetic acid, lyophilized, and resuspended in 20 μL of 5% (v/v) acetonitrile and 0.1% formic acid. Each sample was analyzed in quadruplicate; the first two runs were performed without an exclusion list, while the third and fourth runs were performed with an exclusion list containing the 5000 most abundant peptides that were detected in the first two runs, to increase sample coverage by maximizing the suppression of abundant peptides (McLoughlin et al., 2018). Raw MS2 files from all four runs were merged, resulting in two technical replicates per sample. Additional mass spectrometric details are described by McLoughlin et al. (2018).
The resulting MS data sets were queried by Proteome Discoverer (version 2.0.0.802; Thermo Fisher Scientific) against the maize B73 proteome database (Zea-mays.AGPv3.21.pep.all from www.maizegdb.org) and a list of common protein contaminants. Peptides were assigned by SEQUEST HT (Eng et al., 1994), allowing a maximum of two missed tryptic cleavages, a minimum peptide length of 6, a precursor mass tolerance of 10 ppm, and fragment mass tolerances of 0.02 D. Carbamidomethylation of Cys and oxidation of Met were specified as static and dynamic modifications, respectively. Protein abundances among genotypes/conditions were normalized based on the average values from 150 proteins considered least variable among the samples (sd/average; Supplemental Data Set 5), which was validated by comparing the abundances of amply detected histones (McLoughlin et al., 2018).
Immunoblot Detection
Abundances of representative proteins that were affected by the atg12 backgrounds were verified by immunoblot analysis using available antibodies at the indicated dilutions: ATG8 (1:1000; Thompson et al., 2005), NBR1 (1:3000; Jung et al., 2020), Cys protease2 (1:1000; Agrisera AS16 3110), catalase (1:1000; Agrisera AS09 501), ascorbate peroxidase (1:5000; Agrisera AS06 180), VDAC (1:500; Subbaiah et al., 2006), RPN1 (1:3000; Yang et al., 2004), RPN5 (1:3000; Smalle et al., 2002), and RPT4 (1:3000; Gemperline et al., 2019). Immunoblot detection of histone H3 with anti-histone H3 antibodies (1:100,000; Abcam AB1791) was used to confirm nearly equal protein loading. Silver staining of the crude protein extract was conducted as described by McLoughlin et al. (2018).
Starch, Chlorophyll, and Protein Quantification
Starch was assayed using the protocol described by Smith and Zeeman (2006). Pulverized leaf tissue (∼200 mg) was heat extracted for 3 min in 5 mL of 80% (v/v) ethanol and clarified by centrifugation at 5000g for 10 min. The pellet was rewashed twice with 80% ethanol, dried, resuspended in 3 mL of water, and heated to the boiling point for 10 min. The gelatinized starch granules (500 µL) were mixed with 500 μL of 200 mM sodium acetate (pH 5.5) and incubated at 37°C for 4 h, either alone or after mixing with 6 units of amyloglucosidase (Roche) and 1 unit of α-amylase (Roche) to convert starch to glucose. After clarification, glucose in the supernatant was measured spectrophotometrically at 340 nm based on the formation of NADH in the presence of 1 unit of hexokinase and 1 unit of glucose-6-phosphate dehydrogenase (Roche) in 1 mL (total volume) of 100 mM HEPES (pH 7.5), 0.5 mM ATP, 1 mM NAD, and 4 mM MgCl2. Reaction volumes were adjusted between 20 and 500 µL, depending on the treatment/genotype, to ensure signal linearity. OD340 values for control samples were subtracted from the values of digested samples to account for spurious hydrolysis. Starch content was calculated by dividing the change in OD340 by the millimolar extinction coefficient of NADH (6.22) and multiplied by the mass of anhydroglucose (162 D; Smith and Zeeman, 2006). Five independent biological replicates were measured for each genotype/treatment.
To detect starch by iodine staining, leaf sections were heated to the boiling point in 80% (v/v) ethanol for ∼20 min to remove chlorophyll, incubated for 10 min in a 1:10 dilution of Lugol’s iodine solution (Ward’s Science, Weigert formulation, 470301-328), and washed twice in water (Dinges et al., 2003). Cross sections were hand dissected and visualized using a BX60 epifluorescence microscope, a PlanApo 60× oil objective (numerical aperture 1.40), and a DP 72 camera (Olympus). Total protein content was determined using a Pierce BCA protein assay kit (Thermo Fisher Scientific). Protein from equal amounts of tissue (fresh weight) was extracted from pulverized tissue, precipitated with methanol/chloroform as described by McLoughlin et al. (2018), and assayed using BSA as the standard.
Chlorophyll and Photosynthesis Efficiency Assays
Chlorophyll content was measured spectrophotometrically from the second leaf of 14-d-old plants grown at 25°C, after extracting 300 mg of leaf tissue into 5 mL of 80% (v/v) acetone (McLoughlin et al., 2018). Photosynthetic efficiency was measured in situ based on the F0 and Fmax values of chlorophyll fluorescence as described by Tschiersch et al. (2017). Plants were dark adapted and fluorescence was measured in a closed FluorCam 800MF (Photon Systems Instruments) using the following settings: shutter = 1, sensitivity = 30, act1 = 100, act2 = 0, super = 100; F0: duration = 2 s, period = 200 ms; Fmax: pulse duration = 960 ms; a1 = F0 duration + 40 ms, a2 = a1 + 480 ms (n = 3).
Lipid Droplet Quantification
For the lipid droplet assays, plants were grown for 14 d and subjected to fixed-carbon starvation as above. The second leaf of each plant was harvested at 1:00 pm, stored on ice, and stained with 125 mg/L Nile Blue A for 15 min after the epidermis was peeled (Jose and Burgess, 2006). Lipid droplets were imaged with a 780 Zeiss laser-scanning microscope equipped with a 40× objective (numerical aperture 1.10), using excitation/emission wavelengths set at 488/550 to 620 nm, respectively. Z-stacks containing 10 slides were obtained in 1-μm increments starting at the cortical sides of the mesophyll cells (total thickness = 10 μm). Lipid droplet areas were calculated and normalized to the total area of each cell with NIH ImageJ (https://imagej.nih.gov/ij). For each genotype, 42 to 84 mesophyll cells from the second leaves of two separate plants were analyzed.
Mapping Metabolic Pathways
Changes in metabolite, protein, and/or transcript abundances were superimposed onto metabolic pathway maps obtained from Metabolon (www.metabolon.com) and the KEGG maps as modified for the maize metabolome (https://www.genome.jp/kegg-bin/show_organism?menu_type= pathway_mapsandorg=zma). Transcripts and proteins were associated with their corresponding metabolic conversions using KEGG; if multiple transcripts or proteins were identified, the FC range was indicated for each.
Statistical Analyses
Statistical analyses of the metabolites were conducted using the Qiagen Array Suite software package (www.omicsoft.com/array-studio/) and R (www.cran.r-project.com/). Following log2 transformation and imputation of missing values, ANOVA contrasts identified metabolites whose concentrations differed significantly among experimental groups (P ≤ 0.05; McLoughlin et al., 2018). FDRs were calculated for multiple comparisons to estimate the significant difference in a metabolite above the P value cutoff. Metabolites whose levels significantly differed in both atg12 alleles, or were significant in one and approached significance in the other (0.05 ≤ P ≤ 0.1), were normalized to their wild-type equivalent to generate heat maps in Perseus (Tyanova et al., 2016). A similar approach was applied to study the effect of fixed-carbon starvation on wild-type leaves where only the significantly affected metabolites (P ≤ 0.05) were selected and plotted. Comparisons between the effects of fixed-carbon and nitrogen starvation were conducted using the nitrogen-starvation data sets described by McLoughlin et al. (2018), which were obtained from the same leaf at a slightly older developmental stage (3 weeks versus 2 weeks). PCA among genotypes was calculated in Perseus using a Benjamini-Hochberg FDR cutoff of 0.05 without relative enrichment. Based on the composite Z-scores for each of the 405 metabolites, the three biological replicates with values closest to the mean response were further evaluated by transcriptomic and proteomic analyses.
Metabolic topology-based pathway over-representation analyses of significantly altered metabolites were conducted using MetaboAnalyst (Chong et al., 2019). Metabolites were assigned to pathways within the Arabidopsis (Arabidopsis thaliana) KEGG pathway library and tested for pathway over-representation using a hypergeometric test. Pathway impacts were calculated using relative-betweeness centrality, which accounts for the positions of the metabolites within the pathway to provide an estimate of pathway importance. P values reflected the over-representation of each category, while the pathway impact weighed the importance of the affected metabolites within the pathway.
Significant differences in starch levels, lipid droplet area, ion content, and transcript abundances (as measured by qRT-PCR) were determined using one-way ANOVA followed by Tukey’s posthoc test to correct for multiple comparisons using SPSS (http://www-01.ibm.com/software/analytics/spss/). Differentially expressed transcripts in the RNA-seq data sets were identified by EBSeq v1.12.0 (Leng et al., 2013), using pairwise comparisons between genotypes/conditions. Library sizes were median-normalized prior to 10 iterations of the EBtest algorithm set to a 5% FDR threshold. Only those genes showing consistent differential expression (posterior probability of equal expression ≤ 0.05) in both atg12-1 and atg12-2 versus the wild type were used for comparisons.
Protein identifications based on FDR ≤ 0.05% were included in the analysis and grouped according to the principle of parsimony (strict). Label-free MS1 quantification was performed as previously described by Silva et al. (2006) using Proteome Discoverer with a minimum Quan value threshold set to 0.0001 using unique peptides and “3 Top N” peptides used for area calculation. The resulting values were log2 transformed, and missing values were imputed assuming a normal distribution, a width distribution shrinkage of 0.3, and a downshift of 1.8 standard deviations, using the Perseus computational platform (Tyanova et al., 2016). Significant changes in protein abundance were calculated by ANOVA contrasts (P ≤ 0.05), excluding proteins with FDR > 0.05 in either or both atg12-1 or atg12-2 to account for multiple comparisons.
Enrichments of GO protein categories were identified by the AgriGO v2 analysis toolkit (Tian et al., 2017), using a singular enrichment analysis (Z. mays AGP v3.30), where the lists of identified proteins provided the reference to avoid biases toward abundantly detected GO categories. Displayed GO categories were selected based on their uniqueness, significance, and degree of completeness. Pearson’s correlation coefficient and fit (R2) values were calculated in Perseus (Tyanova et al., 2016). Cellular location(s) of individual proteins predicted from the RNA-seq data sets or identified by MS were assigned by GO. For proteins with multiple locations, all possibilities were included in the analysis.
Accession Numbers
The gene identifiers for the various genomic loci and corresponding proteins (GRMZM locators) derived from the genomic sequence of the reference B73 cultivar available at http://www.maizegdb.org can be found in the various Supplemental Data Sets. The raw data for the metabolomic, ionomic, transcriptomic, and proteomic data sets can be found in Supplemental Data Sets 3, 7, 10, and 11, respectively. RNA-seq data sets were deposited at the National Center for Biotechnology Information Sequence Read Archive database under the submission number PRJNA606338 (https://www.ncbi.nlm.nih.gov/sra). The .raw, .msf, and .xml files for the MS data sets are available in the ProteomeXchange database under accession number PXD017077 within the PRIDE repository (http://www.proteomexchange.org/). Protein identifiers and the corresponding gene accession numbers for the catalog of maize proteins identified here are provided in Supplemental Data Set 6. If any data sets are unavailable through the links stated above, they can be obtained from the corresponding author upon request.
Supplemental Data
Supplemental Figure 1. The fixed-carbon starvation regime has a minimal effect on photosynthesis.
Supplemental Figure 2. Autophagy defects and fixed-carbon and nitrogen starvation differentially affect the maize leaf metabolome.
Supplemental Figure 3. Cytoscape survey of metabolite changes in wild-type (WT) versus atg12-1 maize leaves after fixed-carbon starvation.
Supplemental Figure 4. Cytoscape surveys of metabolites in wild-type (WT) versus atg12-1 maize leaves that are differentially affected by fixed-carbon or nitrogen starvation.
Supplemental Figure 5. Autophagy and fixed-carbon starvation have modest effects on the maize leaf ionome.
Supplemental Figure 6. The maize leaf transcriptomic and proteomic data sets were consistent when comparing biological replicates.
Supplemental Figure 7. qRT-PCR verifying the effect of the atg12 mutations and fixed-carbon starvation on the abundance of representative maize transcripts.
Supplemental Figure 8. Volcano plots showing the effect of the atg12 mutations on the maize leaf proteome from samples grown with or without fixed carbon.
Supplemental Figure 9. Proteins from specific GO subcategories differentially accumulated in wild-type (WT) and atg12 maize leaves.
Supplemental Figure 10. Fold changes in mRNA and protein abundances comparing wild-type (WT) versus atg12-1 and atg12-2 maize leaves grown with or without fixed carbon.
Supplemental Figure 11. Maize autophagy mutants upregulate glutathione and purine metabolism during fixed carbon starvation.
Supplemental Figure 12. Changes in abundance for transcripts encoding several maize proteases in atg12 versus wild-type leaves.
Supplemental Data Set 1. ANOVA tables.
Supplemental Data Set 2. Metabolome analysis: WT versus atg12 +/− carbon.
Supplemental Data Set 3. Ionome raw data: WT versus atg12 +/− carbon.
Supplemental Data Set 4. Transcriptome analysis: WT versus atg12 +/− carbon.
Supplemental Data Set 5. Normalization protein list and values used for sample correction.
Supplemental Data Set 6. Proteome analysis: WT versus atg12 +/− carbon.
Supplemental Data Set 7. Metabolome raw data: WT versus atg12 +/− carbon.
Supplemental Data Set 8. Transcriptomics fragment length and correlation variation values.
Supplemental Data Set 9. Oligonucleotide primers used for qRT-PCR.
Supplemental Data Set 10. Transcriptome raw data: WT versus atg12 +/− carbon.
Supplemental Data Set 11. Proteome raw data: WT versus atg12 +/− carbon.
DIVE Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We thank Daniel Alexander and Lining Guo for the metabolome studies, Jennifer Barrett, Gregory Zeigler, and Ivan Baxter for help with the ionomics, Michelle Liberton and Himadri Pakrasi for help with the chlorophyll fluorescence measurements, and Michael Dyer for greenhouse assistance. This work was supported by the U.S. National Science Foundation (grants IOS-1339325 and IOS-1840687 to M.S.O. and R.D.V.), the U.S. National Institutes of Health, National Institute of General Medical Sciences (grant R01-GM124452 to R.D.V.), the U.S. Department of Agriculture, the National Institute of Food and Agriculture Hatch Act Formula Fund (grant WIS01791 to M.S.O.), and the Department of Botany at the University of Wisconsin (to X.D.).
AUTHOR CONTRIBUTIONS
F.M., F.L., M.S.O., and R.D.V. designed the research; F.M., R.S.M., X.D., L.D.K., and R.C.A. performed the research; F.M., R.S.M., X.D., E.C.C., R.C.A., M.S.O., and R.D.V. analyzed the data; F.M. and R.D.V. wrote the article with input from all authors.
Footnotes
Articles can be viewed without a subscription.
References
- Aguilar-Hernández V., Kim D.Y., Stankey R.J., Scalf M., Smith L.M., Vierstra R.D.(2017). Mass spectrometric analyses reveal a central role for ubiquitylation in remodeling the Arabidopsis proteome during photomorphogenesis. Mol. Plant 10: 846–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avin-Wittenberg T., Bajdzienko K., Wittenberg G., Alseekh S., Tohge T., Bock R., Giavalisco P., Fernie A.R.(2015). Global analysis of the role of autophagy in cellular metabolism and energy homeostasis in Arabidopsis seedlings under carbon starvation. Plant Cell 27: 306–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avin-Wittenberg T., et al. (2018). Autophagy-related approaches for improving nutrient use efficiency and crop yield protection. J. Exp. Bot. 69: 1335–1353. [DOI] [PubMed] [Google Scholar]
- Barros J.A.S., Cavalcanti J.H.F., Medeiros D.B., Nunes-Nesi A., Avin-Wittenberg T., Fernie A.R., Araújo W.L.(2017). Autophagy deficiency compromises alternative pathways of respiration following energy deprivation in Arabidopsis thaliana. Plant Physiol. 175: 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouquisse R., Gaudillere J.P., Raymond P.(1998). Induction of a carbon-starvation-related proteolysis in whole maize plants submitted to light/dark cycles and to extended darkness. Plant Physiol. 117: 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer B., Ziolkowska A., Bagard M., Keech O., Gardeström P.(2012). The impact of light intensity on shade-induced leaf senescence. Plant Cell Environ. 35: 1084–1098. [DOI] [PubMed] [Google Scholar]
- Cao P., Kim S.J., Xing A., Schenck C.A., Liu L., Jiang N., Wang J., Last R.L., Brandizzi F.(2019). Homeostasis of branched-chain amino acids is critical for the activity of TOR signaling in Arabidopsis. eLife 8: e50747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J., Wishart D.S., Xia J.(2019). Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics 68: e86. [DOI] [PubMed] [Google Scholar]
- Chung T., Phillips A.R., Vierstra R.D.(2010). ATG8 lipidation and ATG8-mediated autophagy in Arabidopsis require ATG12 expressed from the differentially controlled ATG12A and ATG12B loci. Plant J. 62: 483–493. [DOI] [PubMed] [Google Scholar]
- Chung T., Suttangkakul A., Vierstra R.D.(2009). The ATG autophagic conjugation system in maize: ATG transcripts and abundance of the ATG8-lipid adduct are regulated by development and nutrient availability. Plant Physiol. 149: 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E.L., Guerinot M.L.(2002). Iron stress in plants. Genome Biol. 3: 1024.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehaven C.D., Evans A.M., Dai H., Lawton K.A.(2010). Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinges J.R., Colleoni C., James M.G., Myers A.M.(2003). Mutational analysis of the pullulanase-type debranching enzyme of maize indicates multiple functions in starch metabolism. Plant Cell 15: 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doelling J.H., Walker J.M., Friedman E.M., Thompson A.R., Vierstra R.D.(2002). The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J. Biol. Chem. 277: 33105–33114. [DOI] [PubMed] [Google Scholar]
- Dong Y., et al. (2017). Sulfur availability regulates plant growth via glucose-TOR signaling. Nat. Commun. 8: 1174. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Eguchi M., Kimura K., Makino A., Ishida H.(2017). Autophagy is induced under Zn limitation and contributes to Zn-limited stress tolerance in Arabidopsis (Arabidopsis thaliana). Soil Sci. Plant Nutr. 63: 342–350. [Google Scholar]
- Eng J.K., McCormack A.L., Yates J.R.(1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5: 976–989. [DOI] [PubMed] [Google Scholar]
- Esteban R., Ariz I., Cruz C., Moran J.F.(2016). Mechanisms of ammonium toxicity and the quest for tolerance. Plant Sci. 248: 92–101. [DOI] [PubMed] [Google Scholar]
- Evans A.M., DeHaven C.D., Barrett T., Mitchell M., Milgram E.(2009). Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 81: 6656–6667. [DOI] [PubMed] [Google Scholar]
- Fan J., Yu L., Xu C.(2019). Dual role for autophagy in lipid metabolism in Arabidopsis. Plant Cell 31: 1598–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer L.M., Rinaldi M.A., Young P.G., Danan C.H., Burkhart S.E., Bartel B.(2013). Disrupting autophagy restores peroxisome function to an Arabidopsis lon2 mutant and reveals a role for the LON2 protease in peroxisomal matrix protein degradation. Plant Cell 25: 4085–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemperline D.C., Marshall R.S., Lee K.H., Zhao Q., Hu W., McLoughlin F., Scalf M., Smith L.M., Vierstra R.D.(2019). Proteomic analysis of affinity-purified 26S proteasomes identifies a suite of assembly chaperones in Arabidopsis. J. Biol. Chem. 294: 17570–17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Yamada S., Oikawa K., Bizan J., Shigenobu S., Yamaguchi K., Mano S., Hayashi M., Ueda H., Hara-Nishimura I., Nishimura M., Yamada K.(2019). Sucrose starvation induces microautophagy in plant root cells. Front. Plant Sci. 10: 1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf A., Schlereth A., Stitt M., Smith A.M.(2010). Circadian control of carbohydrate availability for growth in Arabidopsis plants at night. Proc. Natl. Acad. Sci. USA 107: 9458–9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R.A., Lejay L.V., Dean A., Chiaromonte F., Shasha D.E., Coruzzi G.M.(2007). Qualitative network models and genome-wide expression data define carbon/nitrogen-responsive molecular machines in Arabidopsis. Genome Biol. 8: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanuzzaman M., Bhuyan M.H.M.B., Anee T.I., Parvin K., Nahar K., Mahmud J.A., Fujita M.(2019). Regulation of ascorbate-glutathione pathway in mitigating oxidative damage in plants under abiotic stress. Antioxidants 8: e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck O.K., Scharnberg J., Escobar N.M., Wanner G., Giavalisco P., Witte C.P.(2014). Uric acid accumulation in an Arabidopsis urate oxidase mutant impairs seedling establishment by blocking peroxisome maintenance. Plant Cell 26: 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havé M., Luo J., Tellier F., Balliau T., Cueff G., Chardon F., Zivy M., Rajjou L., Cacas J.L., Masclaux-Daubresse C.(2019). Proteomic and lipidomic analyses of the Arabidopsis atg5 autophagy mutant reveal major changes in endoplasmic reticulum and peroxisome metabolisms and in lipid composition. New Phytol. 223: 1461–1477. [DOI] [PubMed] [Google Scholar]
- Havé M., Marmagne A., Chardon F., Masclaux-Daubresse C.(2017). Nitrogen remobilization during leaf senescence: Lessons from Arabidopsis to crops. J. Exp. Bot. 68: 2513–2529. [DOI] [PubMed] [Google Scholar]
- Hildebrandt T.M., Nunes Nesi A., Araújo W.L., Braun H.P.(2015). Amino acid catabolism in plants. Mol. Plant 8: 1563–1579. [DOI] [PubMed] [Google Scholar]
- Hillwig M.S., Contento A.L., Meyer A., Ebany D., Bassham D.C., Macintosh G.C.(2011). RNS2, a conserved member of the RNase T2 family, is necessary for ribosomal RNA decay in plants. Proc. Natl. Acad. Sci. USA 108: 1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T., Izumi M., Wada S., Makino A., Ishida H.(2018). Vacuolar protein degradation via autophagy provides substrates to amino acid catabolic pathways as an adaptive response to sugar starvation in Arabidopsis thaliana. Plant Cell Physiol. 59: 1363–1376. [DOI] [PubMed] [Google Scholar]
- Huang X., et al. (2019). Genetic analyses of the Arabidopsis ATG1 kinase complex reveal both kinase-dependent and independent autophagic routes during fixed-carbon starvation. Plant Cell 31: 2973–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi M., Hidema J., Makino A., Ishida H.(2013). Autophagy contributes to nighttime energy availability for growth in Arabidopsis. Plant Physiol. 161: 1682–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi M., Nakamura S., Li N.(2019). Autophagic turnover of chloroplasts: Its roles and regulatory mechanisms in response to sugar starvation. Front. Plant Sci. 10: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J., Burgess K.(2006). Benzophenoxazine-based fluorescent dyes for labeling biomolecules. Tetrahedron 62: 11021–11037. [Google Scholar]
- Jung H., Lee H.N., Marshall R.S., Lomax A.W., Yoon M.J., Kim J., Kim J.H., Vierstra R.D., Chung T.(2020). Arabidopsis cargo receptor NBR1 mediates selective autophagy of defective proteins. J. Exp. Bot. 71: 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A., Behl C., Dikic I.(2016). Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 26: 6–16. [DOI] [PubMed] [Google Scholar]
- Kim J., Lee H., Lee H.N., Kim S.H., Shin K.D., Chung T.(2013). Autophagy-related proteins are required for degradation of peroxisomes in Arabidopsis hypocotyls during seedling growth. Plant Cell 25: 4956–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar F.K., Mengel L.A., Müller A.O., Schmitt K., Blersch K.F., Valerius O., Braus G.H., Ischebeck T.(2018). PUX10 is a lipid droplet-localized scaffold protein that interacts with CELL DIVISION CYCLE48 and is involved in the degradation of lipid droplet proteins. Plant Cell 30: 2137–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurusu T., et al. (2014). OsATG7 is required for autophagy-dependent lipid metabolism in rice postmeiotic anther development. Autophagy 10: 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H.M., Peng S.S., Coruzzi G.M.(1994). Metabolic regulation of the gene encoding glutamine-dependent asparagine synthetase in Arabidopsis thaliana. Plant Physiol. 106: 1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H.M., Wong P., Chan H.K., Yam K.M., Chen L., Chow C.M., Coruzzi G.M.(2003). Overexpression of the ASN1 gene enhances nitrogen status in seeds of Arabidopsis. Plant Physiol. 132: 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L.(2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzi F.A., Schnyder H., Thornton B.(2005). The sources of carbon and nitrogen supplying leaf growth: Assessment of the role of stores with compartmental models. Plant Physiol. 137: 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M., Haag J.D., Gould M.N., Stewart R.M., Kendziorski C.(2013). EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29: 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Chung T., Pennington J.G., Federico M.L., Kaeppler H.F., Kaeppler S.M., Otegui M.S., Vierstra R.D.(2015). Autophagic recycling plays a central role in maize nitrogen remobilization. Plant Cell 27: 1389–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Chung T., Vierstra R.D.(2014). AUTOPHAGY-RELATED11 plays a critical role in general autophagy- and senescence-induced mitophagy in Arabidopsis. Plant Cell 26: 788–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Bassham D.C.(2010). TOR is a negative regulator of autophagy in Arabidopsis thaliana. PLoS One 5: e11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Burgos J.S., Deng Y., Srivastava R., Howell S.H., Bassham D.C.(2012). Degradation of the endoplasmic reticulum by autophagy during endoplasmic reticulum stress in Arabidopsis. Plant Cell 24: 4635–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lornac A., Havé M., Chardon F., Soulay F., Clément G., Avice J.C., Masclaux-Daubresse C.(2020). Autophagy controls sulphur metabolism in the rosette leaves of Arabidopsis and facilitates S remobilization to the seeds. Cells 9: e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W., Cai Y., Sun Q., van Wijk K.J.(2005). Functional differentiation of bundle sheath and mesophyll maize chloroplasts determined by comparative proteomics. Plant Cell 17: 3111–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli A., Sturaro A., Trevisan S., Quaggiotti S., Nonis A.(2012). Evaluation of candidate reference genes for qPCR in maize. J. Plant Physiol. 169: 807–815. [DOI] [PubMed] [Google Scholar]
- Maqbool A., Hughes R.K., Dagdas Y.F., Tregidgo N., Zess E., Belhaj K., Round A., Bozkurt T.O., Kamoun S., Banfield M.J.(2016). Structural basis of host autophagy-related protein 8 (ATG8) binding by the Irish potato famine pathogen effector protein PexRD54. J. Biol. Chem. 291: 20270–20282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.S., Hua Z., Mali S., McLoughlin F., Vierstra R.D.(2019). ATG8-binding UIM proteins define a new class of autophagy adaptors and receptors. Cell 177: 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.S., Li F., Gemperline D.C., Book A.J., Vierstra R.D.(2015). Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis. Mol. Cell 58: 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.S., Vierstra R.D.(2018a). Proteasome storage granules protect proteasomes from autophagic degradation upon carbon starvation. eLife 7: e34532. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Marshall R.S., Vierstra R.D.(2018b). Autophagy: The master of bulk and selective recycling. Annu. Rev. Plant Biol. 69: 173–208. [DOI] [PubMed] [Google Scholar]
- Masclaux-Daubresse C., Clément G., Anne P., Routaboul J.M., Guiboileau A., Soulay F., Shirasu K., Yoshimoto K.(2014). Stitching together the multiple dimensions of autophagy using metabolomics and transcriptomics reveals impacts on metabolism, development, and plant responses to the environment in Arabidopsis. Plant Cell 26: 1857–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masclaux-Daubresse C., Reisdorf-Cren M., Orsel M.(2008). Leaf nitrogen remobilisation for plant development and grain filling. Plant Biol (Stuttg) 10: 23–36. [DOI] [PubMed] [Google Scholar]
- McLoughlin F., Augustine R.C., Marshall R.S., Li F., Kirkpatrick L.D., Otegui M.S., Vierstra R.D.(2018). Maize multi-omics reveal roles for autophagic recycling in proteome remodelling and lipid turnover. Nat. Plants 4: 1056–1070. [DOI] [PubMed] [Google Scholar]
- Miyake H., Maeda E.(1978). Starch accumulation in bundle sheath chloroplasts during leaf development in C3 and C4 plants of Gramineae. Can. J. Bot. 56: 880–882. [Google Scholar]
- Noda N.N., Kumeta H., Nakatogawa H., Satoo K., Adachi W., Ishii J., Fujioka Y., Ohsumi Y., Inagaki F.(2008). Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 13: 1211–1218. [DOI] [PubMed] [Google Scholar]
- Ohta T., Masutomi N., Tsutsui N., Sakairi T., Mitchell M., Milburn M.V., Ryals J.A., Beebe K.D., Guo L.(2009). Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol. Pathol. 37: 521–535. [DOI] [PubMed] [Google Scholar]
- Palenchar P.M., Kouranov A., Lejay L.V., Coruzzi G.M.(2004). Genome-wide patterns of carbon and nitrogen regulation of gene expression validate the combined carbon and nitrogen (CN)-signaling hypothesis in plants. Genome Biol. 5: R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M.W.(2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A.R., Suttangkakul A., Vierstra R.D.(2008). The ATG12-conjugating enzyme ATG10 is essential for autophagic vesicle formation in Arabidopsis thaliana. Genetics 178: 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier M., Dumont J., Masclaux-Daubresse C., Thomine S.(2019). Autophagy is essential for optimal translocation of iron to seeds in Arabidopsis. J. Exp. Bot. 70: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C., Liu J., Gong Q.(2014). Functions of autophagy in plant carbon and nitrogen metabolism. Front. Plant Sci. 5: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sera Y., et al. (2019). Essential roles of autophagy in metabolic regulation in endosperm development during rice seed maturation. Sci. Rep. 9: 18544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.(2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J.C., Gorenstein M.V., Li G.Z., Vissers J.P., Geromanos S.J.(2006). Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteomics 5: 144–156. [DOI] [PubMed] [Google Scholar]
- Smalle J., Kurepa J., Yang P., Babiychuk E., Kushnir S., Durski A., Vierstra R.D.(2002). Cytokinin growth responses in Arabidopsis involve the 26S proteasome subunit RPN12. Plant Cell 14: 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.M., Zeeman S.C.(2006). Quantification of starch in plant tissues. Nat. Protoc. 1: 1342–1345. [DOI] [PubMed] [Google Scholar]
- Smith P.M.C., Atkins C.A.(2002). Purine biosynthesis: Big in cell division, even bigger in nitrogen assimilation. Plant Physiol. 128: 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaiah C.C., Palaniappan A., Duncan K., Rhoads D.M., Huber S.C., Sachs M.M.(2006). Mitochondrial localization and putative signaling function of sucrose synthase in maize. J. Biol. Chem. 281: 15625–15635. [DOI] [PubMed] [Google Scholar]
- Sulpice R., Flis A., Ivakov A.A., Apelt F., Krohn N., Encke B., Abel C., Feil R., Lunn J.E., Stitt M.(2014). Arabidopsis coordinates the diurnal regulation of carbon allocation and growth across a wide range of photoperiods. Mol. Plant 7: 137–155. [DOI] [PubMed] [Google Scholar]
- Suttangkakul A., Li F., Chung T., Vierstra R.D.(2011). The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis. Plant Cell 23: 3761–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenning S., Lamark T., Krause K., Johansen T.(2011). Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 7: 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove L.J., Fernie A.R.(2018). The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 9: 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H., Ishiga Y., Watanabe S., Konishi T., Egusa M., Akiyoshi N., Matsuura T., Mori I.C., Hirayama T., Kaminaka H., Shimada H., Sakamoto A.(2016). Allantoin, a stress-related purine metabolite, can activate jasmonate signaling in a MYC2-regulated and abscisic acid-dependent manner. J. Exp. Bot. 67: 2519–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.R., Doelling J.H., Suttangkakul A., Vierstra R.D.(2005). Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol. 138: 2097–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.R., Vierstra R.D.(2005). Autophagic recycling: Lessons from yeast help define the process in plants. Curr. Opin. Plant Biol. 8: 165–173. [DOI] [PubMed] [Google Scholar]
- Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z.(2017). agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45: W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschiersch H., Junker A., Meyer R.C., Altmann T.(2017). Establishment of integrated protocols for automated high throughput kinetic chlorophyll fluorescence analyses. Plant Methods 13: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J.(2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13: 731–740. [DOI] [PubMed] [Google Scholar]
- van der Hoorn R.A.(2008). Plant proteases: From phenotypes to molecular mechanisms. Annu. Rev. Plant Biol. 59: 191–223. [DOI] [PubMed] [Google Scholar]
- van Doorn W.G., Papini A.(2013). Ultrastructure of autophagy in plant cells: A review. Autophagy 9: 1922–1936. [DOI] [PubMed] [Google Scholar]
- Wang Y., Yu B., Zhao J., Guo J., Li Y., Han S., Huang L., Du Y., Hong Y., Tang D., Liu Y.(2013). Autophagy contributes to leaf starch degradation. Plant Cell 25: 1383–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Matsumoto M., Hakomori Y., Takagi H., Shimada H., Sakamoto A.(2014). The purine metabolite allantoin enhances abiotic stress tolerance through synergistic activation of abscisic acid metabolism. Plant Cell Environ. 37: 1022–1036. [DOI] [PubMed] [Google Scholar]
- Weaver L.M., Amasino R.M.(2001). Senescence is induced in individually darkened Arabidopsis leaves, but inhibited in whole darkened plants. Plant Physiol. 127: 876–886. [PMC free article] [PubMed] [Google Scholar]
- Yang P., Fu H., Walker J., Papa C.M., Smalle J., Ju Y.M., Vierstra R.D.(2004). Purification of the Arabidopsis 26 S proteasome: Biochemical and molecular analyses revealed the presence of multiple isoforms. J. Biol. Chem. 279: 6401–6413. [DOI] [PubMed] [Google Scholar]
- Yang X., Bassham D.C.(2015). New insight into the mechanism and function of autophagy in plant cells. Int. Rev. Cell Mol. Biol. 320: 1–40. [DOI] [PubMed] [Google Scholar]
- Yu T.-S., et al. (2005). α-Amylase is not required for breakdown of transitory starch in Arabidopsis leaves. J. Biol. Chem. 280: 9773–9779. [DOI] [PubMed] [Google Scholar]
- Zhang X., Ding X., Marshall R.S., Paez-Valencia J., Lacey P., Vierstra R.D., Otegui M.S.(2020). Reticulon proteins modulate autophagy of the endoplasmic reticulum in maize endosperm. eLife 9: e51918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z.-L.(2009). Carbon and nitrogen nutrient balance signaling in plants. Plant Signal. Behav. 4: 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler G., Terauchi A., Becker A., Armstrong P.R., Hudson K.A., Baxter I.(2013). Ionomic screening of field-grown soybean identifies mutants with altered seed elemental composition. Plant Genome 6: 2. [Google Scholar]