

A transcript-based annotation of transposable elements allows for reduced bioinformatic complexity and direct hypothesis testing of previously challenging regions of the genome.
Abstract
Transcript-based annotations of genes facilitate both genome-wide analyses and detailed single-locus research. In contrast, transposable element (TE) annotations are rudimentary, consisting of information only on TE location and type. The repetitiveness and limited annotation of TEs prevent the ability to distinguish between potentially functional expressed elements and degraded copies. To improve genome-wide TE bioinformatics, we performed long-read sequencing of cDNAs from Arabidopsis (Arabidopsis thaliana) lines deficient in multiple layers of TE repression. These uniquely mapping transcripts were used to identify the set of TEs able to generate polyadenylated RNAs and create a new transcript-based annotation of TEs that we have layered upon the existing high-quality community standard annotation. We used this annotation to reduce the bioinformatic complexity associated with multimapping reads from short-read RNA sequencing experiments, and we show that this improvement is expanded in a TE-rich genome such as maize (Zea mays). Our TE annotation also enables the testing of specific standing hypotheses in the TE field. We demonstrate that inaccurate TE splicing does not trigger small RNA production, and the cell more strongly targets DNA methylation to TEs that have the potential to make mRNAs. This work provides a transcript-based TE annotation for Arabidopsis and maize, which serves as a blueprint to reduce the bioinformatic complexity associated with repetitive TEs in any organism.
INTRODUCTION
A consistent problem with the analysis of eukaryotic genomes is the complexity introduced by transposable elements (TEs). Thousands to millions of TEs are present in eukaryotic genomes, often nested in convoluted organizations. Analysis of these regions is cumbersome due to their repetitive nature (the number of similar or identical elements) and the fact that current TE annotations only describe minimal information. Even the best TE annotations consist of only three features: position on the chromosome, strand, and TE type (for the reference plant Arabidopsis [Arabidopsis thaliana], see Buisine et al., 2008). In contrast, gene annotations are regularly based on transcript information, which is missing for TEs. These gene annotations describe the transcriptional start sites (TSSs), polyadenylation site, direction, and splicing pattern, which provide genes with higher resolution in bioinformatic experiments compared with TEs. The lack of transcript information for TEs hampers downstream bioinformatic analyses, leading many researchers to ignore these regions of the genome altogether.
A TE transcript-based annotation would enable research on TE expression: which elements are expressed, the transcript forms they generate, and when they are differentially expressed. However, two shortcomings have prevented the use of RNA sequencing (RNA-seq) information to generate a transcript-based annotation of TEs. First, short reads generated by Illumina sequencing perfectly map to many TE locations, creating ambiguity regarding which TE is expressed (Teissandier et al., 2019). Second, TEs are subject to overlapping mechanisms that repress their expression, such as maintenance of epigenetic transcriptional silencing, small RNA-based chromatin modification, and posttranscriptional silencing mediated by RNA interference (RNAi; reviewed by Deniz et al., 2019; Ozata et al., 2019). Without TE expression (due to TE silencing), TE transcripts are generally not included in efforts to characterize the transcriptome.

We overcame both of these technical difficulties by producing a long-read transcriptome annotation in Arabidopsis plants that are deficient in multiple layers of TE repression. Previous research in Arabidopsis has identified key proteins that function in TE silencing. The DECREASED DNA METHYLATION1 (DDM1 [AT5G66750]) protein acts to condense chromatin and maintain the silencing of TEs, and subsequently the ddm1 mutant results in a broad activation of TE expression (Hirochika et al., 2000; Miura et al., 2001; Lyons and Zilberman, 2017). The RNA Polymerase V (Pol V) protein complex acts in the RNA-directed DNA methylation (RdDM) pathway to reinforce DNA methylation at short TEs and resilence active TEs (Lahmy et al., 2009; Zhong et al., 2012; Panda et al., 2016). The RNA-DEPENDENT RNA POLYMERASE6 (RDR6 [AT3G49500]) protein generates double-stranded RNA, triggering TE mRNA degradation and small interfering RNA (siRNA) production via RNAi (Nuthikattu et al., 2013; Baeg et al., 2017). We combined mutations in these three proteins (including NUCLEAR RNA POLYMERASE E1 ([AT2G40030], the catalytic subunit of the Pol V complex that is essential for RdDM [Wierzbicki et al., 2008]) to activate and stabilize TE transcripts. We performed Oxford Nanopore Technology (ONT) full-length cDNA sequencing on the triple mutant line to generate a genome-wide transcript annotation for TEs. Our annotation increases bioinformatic resolution of TE regions of the genome, allows focus on the potentially functional TE copies, and opens testing of long-standing hypotheses on the regulation of TE expression.
RESULTS
Long-Read Transcriptome Sequencing of TE-Activated Lines
We began by isolating total RNA, purifying polyadenylated RNAs, and performing ONT sequencing of full-length cDNAs from five Arabidopsis genotypes (Figure 1A; sequencing statistics are shown in Supplemental Table 1). We chose to analyze the poly(A)+ RNA fraction because these RNAs have the potential to produce TE proteins. The genotypes selected included wild-type reference strain Columbia (wild-type Col), which has transcriptionally silent TEs, and the ddm1 mutant with transcriptionally reactivated TEs (Hirochika et al., 2000; Miura et al., 2001; Zemach et al., 2013). We also combined the ddm1 mutation with either rdr6 or pol V, or both rdr6 and pol V, in the ddm1 rdr6 pol V triple mutant (the triple mutant phenotype is shown in Figure 1B). We found that in wild-type Col, very few reads (3.7%) overlap known TE annotations (Figure 1C), and the majority of these are genic transcripts that read-through a TE annotation (∼84%). Removing the read-through of genic transcripts into TEs, we find that only 2719 reads are bona fide TE-initiated transcripts (0.59%; Figure 1D). Overall, only 0.26% of TE bases are covered by TE-initiated reads in wild-type Col (Figure 1E), confirming the efficient epigenetic and posttranscriptional suppression of TE mRNA accumulation in wild-type Col plants. The few expressed TEs in wild-type Col include AT2TE16945 and AT4TE03410 (Sadhu family non-long terminal repeat [LTR] retrotransposons; Figure 1F), AT3TE63065 (a Copia family LTR retrotransposon), AT5TE72200 (a TSCL LTR retrotransposon), and AT5TE72580 (an AtSINE2A nonautonomous element). Our data confirm previous findings that Sadhu family TEs and the TSCL element are expressed in wild-type Col (Chye et al., 1997; Rangwala et al., 2006).
Figure 1.
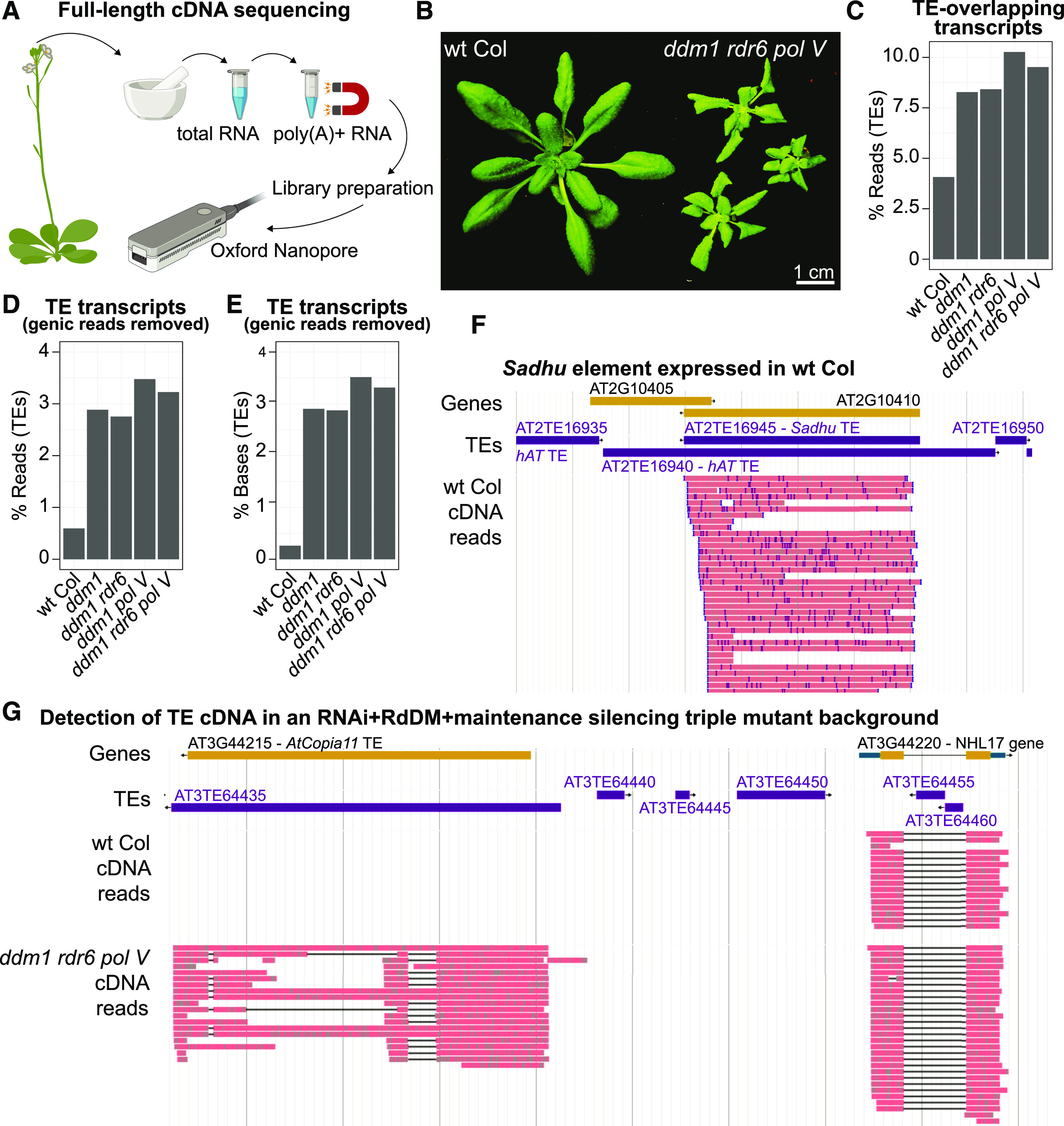
TE Expression Captured by Long-Read cDNA Sequencing.
(A) Experimental workflow of ONT cDNA sequencing. See Methods for additional experimental details. The cartoon was created with BioRender.
(B) Phenotype of the ddm1 rdr6 pol V mutant line.
(C) Percentage of reads that overlap a TE annotation in each genotype.
(D) Percentage of reads that overlap a TE annotation, but with genic transcripts that read-through a TE removed to focus on TE-initiated transcripts.
(E) Percentage of TE bases covered by reads, with genic transcripts that read-through TEs removed.
(F) Genome browser image of the expression of the Sadhu TE AT2TE16945 in wild-type (wt) Col.
(G) Genome browser image of the representative AT3TE64435 AtCopia11 TE (left), which is only expressed in TE-activated mutants, compared with a gene expressed in both genotypes (right).
In (F) and (G), genic exons are yellow, untranslated regions are blue, TEs are purple, and ONT cDNA reads are pink. Gene and TE annotations are from TAIR10.
In the TE-activated mutants, we see a roughly threefold increase in the percentage of TE reads and a fivefold increase in the percentage of TE bases covered, and roughly half of these are not read-through transcripts initiated by genes (Figures 1C to 1E). In the ddm1 TE-activated mutant, we now detect transcripts originating from 31 Gypsy LTR retrotransposons, 24 EnSpm DNA transposons, 18 Mutator DNA transposons, and many others (Supplemental Table 2). As an example, transcripts from the AtCopia11 element AT3TE64435 are only detected in TE-activated mutants (Figure 1G). Together, our findings demonstrate that epigenetic reactivation is required to expose TE transcripts for annotation.
TE Transcript Annotation
We performed additional sequencing on the ddm1 rdr6 pol V triple mutant to increase depth, as this genotype lacks three overlapping layers of TE suppression: transcriptional silencing via heterochromatin condensation, posttranscriptional mRNA degradation by RNAi, and retargeting of chromatin modifications via RdDM (Cuerda-Gil and Slotkin, 2016). We combined all 5,208,896 reads from all genotypes to generate a new transcript annotation for Arabidopsis, which was filtered specifically for TE-containing transcript models (see Methods). This provided 2188 transcript models of 1292 distinct TE annotations in the Arabidopsis genome, which include TE TSSs, transcript direction, splicing patterns, and polyadenylation sites (Figure 2A). We did not perform de novo TE discovery, but rather layered whether a TE was expressed, transcript features, and our TE annotation onto the existing high-quality The Arabidopsis Information Resource (TAIR10) TE list (Lamesch et al., 2012), which is broadly used by the community. Of the 31,189 TEs in the reference Arabidopsis genome, 24,431 (78.3%) showed no evidence of polyadenylated RNA accumulation, 5466 (17.5%) had at least one read but not enough to annotate a transcript, and 1292 (4.1%) were expressed and transcripts were annotated. Most expressed and annotated TEs are Gypsy LTR retrotransposons (33%), followed by Mutator (20%) and EnSpm (14%) DNA transposons, Helitron rolling-circle elements (9%), Copia LTR retrotransposons (9%), followed by fewer numbers of other TE families (Supplemental Table 2). The annotated TEs are both euchromatic and pericentromeric, showing the same overall positional distribution as all TEs in the Arabidopsis genome (Figure 2B). To the community-standard TAIR10 TE list, we have added TE length, copy number, distance from the centromere, distance to the nearest gene, RdDM type (from Panda et al., 2016), whether or not the TE is expressed, TSS position, polyadenylation site, direction of transcription, transcript ID, and transcript models (separate GFF file). These annotation files are given in Supplemental Files 1 and 2 and are version controlled on GitHub (see Accession Numbers).
Figure 2.
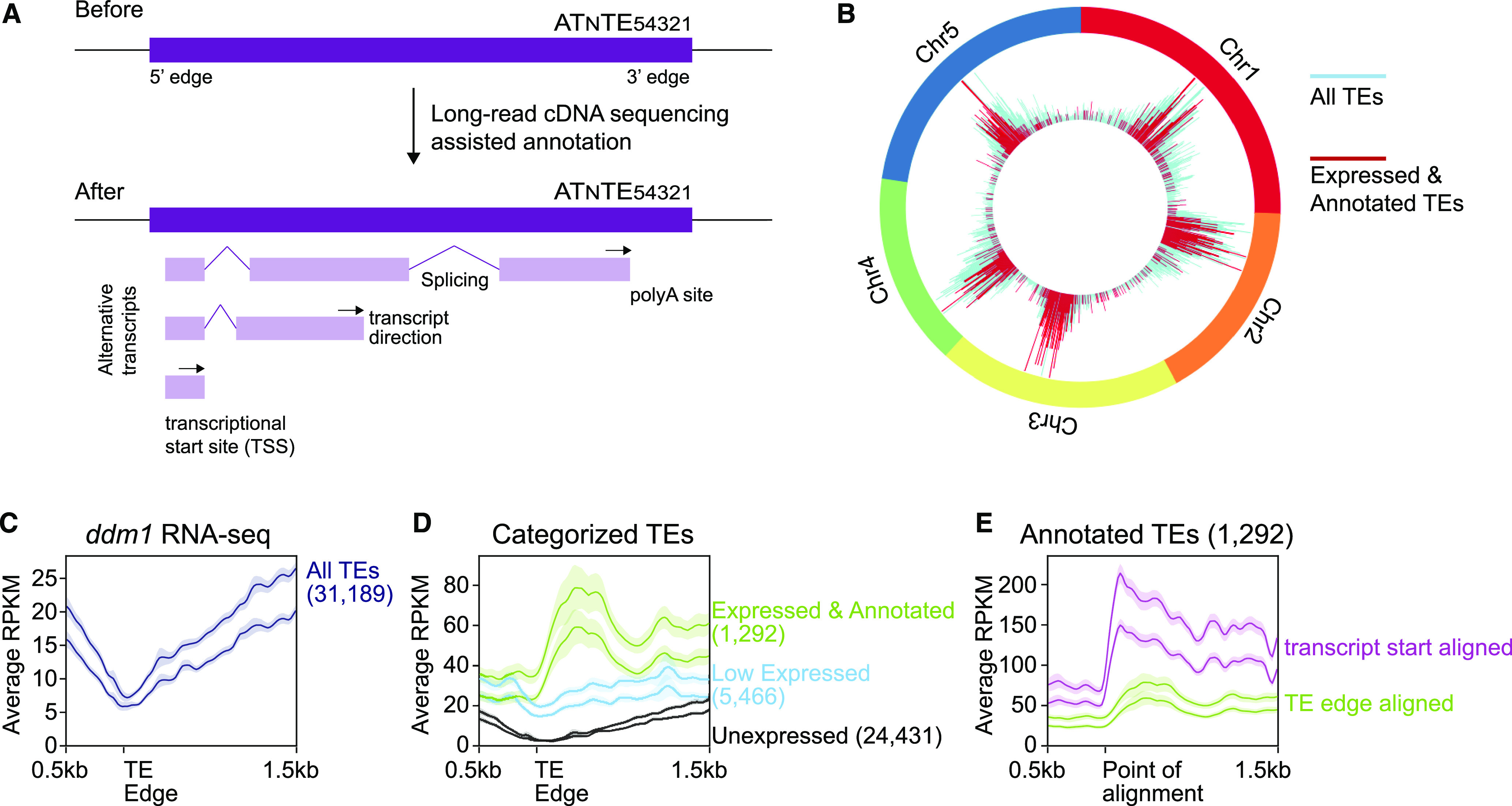
Genome-Wide Annotation of TE Transcripts.
(A) Cartoon of TE annotated features before and after this work.
(B) Visualization of the chromosomal location of the TEs that are expressed and have been annotated in this work.
(C) Metaplot alignment of Illumina RNA-seq reads from ddm1 to 5′ edges for all TEs. The y axis represents the average reads per kilobase per million mapped reads (RPKM).
(D) The same as in (C), but the TEs are divided into expression categories based on our ONT cDNA sequencing. Low expressed TEs did not produce enough reads for transcript annotation.
(E) The same as in (D), but the Expressed and Annotated TE class is shown twice, once aligned by the TE edge (green line, same as in [D]) and once aligned by the now annotated TE TSS (purple line).
In (C) to (E), the two lines in each group represent distinct biological replicates (pools of nonoverlapping plants), the solid line indicates the moving average with a bin size of 20 bases, and the translucent region is the 95% confidence interval. The 5′ edges of the TEs are defined by the TAIR10 TE annotation. Numbers in parentheses denote the number of TEs analyzed in each category.
We next aimed to assay the quality of our TE transcript annotation. We used the quantitative nature of high-depth short-read Illumina RNA-seq from TE-activated ddm1 plants (Oberlin et al., 2017) to measure the accuracy of regions we annotated as TE TSSs. Without our TE transcript annotation, TEs cannot be aligned by their TSS and have therefore been previously aligned by their edges (Figure 2C). The expression of all TEs in ddm1 aligned by their 5′ edge shows a reduction in transcripts at this point (Figure 2C), due to the large number of unexpressed short TEs. Dividing TEs into the expression categories defined by our transcript annotation shows that the “Expressed and Annotated” classification now displays a peak of expression close to the 5′ edge (Figure 2D). Importantly, our TE transcript annotation enables improved centering of TEs by their TSS rather than their edge. When the short-read RNA-seq data are mapped to the same set of TEs, either aligned by their 5′ edges or their now-annotated TSSs, we observe a sharper increase of expression and a higher density of mapped reads for the TEs aligned by their TSS (Figure 2E). Therefore, the independent Illumina RNA-seq data type validates our annotation of TE TSSs. We repeated this analysis for different TE types and found consistent results among terminal inverted repeat DNA TEs, Helitron elements, and LTR retrotransposons (Supplemental Figure 1), likely due to the fact that even though they have distinct transposition mechanisms, each must produce a polyadenylated mRNA for protein production prior to transposition.
Improved TE Annotations Provide Higher Bioinformatic Resolution
A major problem in TE bioinformatics is the frequent inability to differentiate distinct individual TE copies from closely related subfamily members (Shahid and Slotkin, 2020). The limited length of short-read sequencing generates multimapping reads that perfectly map to two or more locations in the genome. Several approaches have been taken to handle these multimapping short reads, while the long reads generated by ONT are unique to a single TE copy in the genome. To illustrate this point, we focused on the low-copy AtCopia93 Evadé (EVD) subfamily of TEs (Mirouze et al., 2009). Our ONT data demonstrate that the EVD5 element generates three transcript models, the two LTRs of EVD2 are transcribed, while EVD1, EVD3, and EVD4 are not expressed (Figure 3A).
Figure 3.
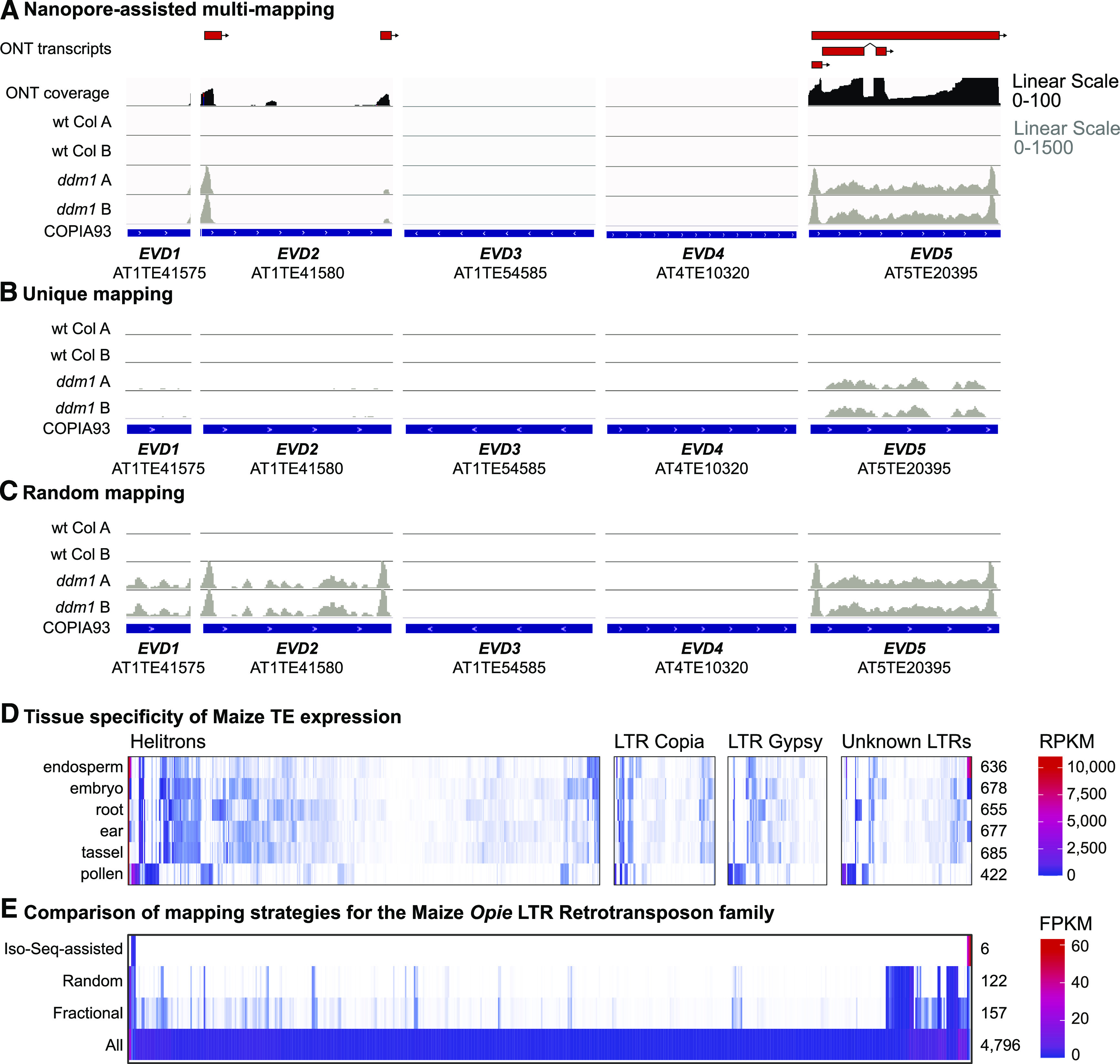
Increase in the Resolution of TE Bioinformatics with Improved TE Annotation.
(A) ONT TE annotation of five Arabidopsis Evadé/AtCopia93 TE copies (red) and ONT-assisted mapping of wt Col and ddm1 Illumina RNA-seq reads (PE125) to the TE transcripts. The peaks of expression on the ends of EVD2 and EVD5 are their LTRs, which can generate LTR-only poly(A) RNAs.
(B) Genome mapping of uniquely mapping reads only.
(C) Genome mapping with multimapping reads distributed randomly.
(D) Maize (B73) TE expression across six tissues as assayed by PacBio Iso-Seq. The number of elements with TE-initiated transcription that had ≥1 reads per kilobase per million mapped reads (RPKM) in each tissue is shown on the right.
(E) Illumina short-read (PE100) RNA-seq mapped to the B73 v4 genome with four different informatic approaches: multimapping reads assigned one location at random, multimapping reads assigned to all locations, and multimapping reads assigned to all locations followed by fractional counting based on how many other positions that read was mapped to. In addition, the same RNA-seq reads were mapped to the TE transcriptional annotation from Supplemental Files 3 and 4 (Iso-Seq assisted). Only 572 of the >12,000 Opie family LTR retrotransposons with a fragments per kilobase per million mapped fragments (FPKM) > 1 for at least one mapping strategy are shown in the heat map. The number of elements with FPKM > 1 for each mapping strategy is shown on the right.
When we map short-read Illumina RNA-seq data, multimapping reads can be handled four different ways. First, we can use our TE transcript annotation to guide the reads, which accurately represents the transcripts that ONT detected (Figure 3A). Second, we can use only unique-mapping reads, but these report only the interior of EVD5 and miss the EVD2 expression of LTR-only transcripts (Figure 3B). Third, the same results as in Figure 3B are obtained if we use the uniquely mapping reads to guide the multimapping reads (Jin et al., 2015). Fourth, we can randomly distribute multimapping reads among the locations they perfectly match, but this falsely identifies EVD1 and the interior of EVD2 as expressed (Figure 3C). Of the most commonly used approaches to handle multimapping reads, unique and guided mapping underestimate the number of elements expressed, while the random mapping overestimates it. Therefore, our TE transcript annotation helps guide multimapping reads to the correctly expressed individual elements.
Expressed and Annotated TEs in the Maize Genome
The problem of placing multimapping short reads is exacerbated with high-copy TE families and large genomes. To demonstrate the overall utility of our approach to reduce bioinformatic complexity, we repeated our computational analysis on publicly available long-read cDNA sequencing from maize (Zea mays inbred line B73). Maize represents an example of the bioinformatic complexity associated with studying repetitive DNA in a crop with a large genome: it has a complex genome 18.5 times larger than Arabidopsis and has a TE content of 85% (Schnable et al., 2009).
Without the depth of mutant resources available in Arabidopsis to reactivate TEs, we turned to the developmental relaxation of TE silencing (DRTS; Martínez and Slotkin, 2012) events known to occur in pollen and endosperm tissues (Slotkin et al., 2009; Wang et al., 2016; Anderson et al., 2019b; Warman et al., 2020). We combined PacBio Iso-Seq long reads from B73 embryo, endosperm, root, tassel, and pollen tissue (from Wang et al., 2016) and detected 1028 TE transcripts from 745 unique TE elements. The majority of these TEs expressed in B73 are Helitrons and LTR retrotransposons, and the distribution of TE families detected is shown in Supplemental Table 2.
To determine how genome-wide TE expression changes on the single-element level using long reads, we compared the six maize tissues (Figure 3D). We found the lowest TE expression level in endosperm and pollen (Figure 3D), but this is likely due to the lower depth of Iso-Seq sequencing in these tissues. Nevertheless, our data demonstrate that even during these DRTS events, only a small fraction of elements are expressed (Figure 3D). For example, in all tissues combined, we detected poly(A)+ RNA expression from only 6 Opie family LTR retrotransposon copies from more than 12,000 annotated in the B73 v4 genome (Figure 3E). In addition, we noted that many of the same TEs are expressed in endosperm, embryo, root, ear, and tassel tissue, while the TEs expressed in pollen represent different elements that are not expressed at other points in development (Figure 3D). This suggests that the DRTS event in pollen acts on a specific subset of TEs that are efficiently silenced for the rest of the maize developmental program. Our transcriptional annotation of maize B73 TEs is found as Supplemental Files 3 and 4.
To illustrate why it is important to understand which TEs are capable of poly(A) RNA expression, we compared our recent analysis of maize mature pollen TE expression using Illumina short reads (PE100; Warman et al., 2020) with the set of maize expressed and annotated TEs (all tissues combined). Similar to our analysis of the Evadé low-copy family of TEs from Arabidopsis in Figures 3A to 3C, we investigated the range of results one would observe based on how they map Illumina short-read RNA-seq to the high-copy Opie family in maize. We used “random” assignment of multimapping reads as well as mapping of “all” positions of multimapping reads either with or without “fractional” counting. The fractional approach splits the count of each read into different values based on how many other positions that read was mapped to. We compared these approaches with mapping the Illumina RNA-seq reads directly to only the maize TEs that we detected by Iso-Seq and were annotated. The results demonstrate that including multimapping reads (either randomly, fractionally, or assigned at all positions) overestimates TE expression compared with the six elements detected by Iso-Seq (Figure 3E). Together, the Arabidopsis and maize data demonstrate that producing transcriptional annotations of the repetitive fractions of genomes is a mechanism by which bioinformatic complexity can be reduced during RNA-seq analyses.
Expressed and Annotated TEs Are Targeted for Higher DNA Methylation
In addition to mapping short-read RNA-seq data, our TE transcript annotation provides resolution in other bioinformatic experiments such as genome-wide DNA methylation assayed by Illumina (methylC-seq). This improved resolution can be used to test standing hypotheses in the TE field that were previously inaccessible. Plant TEs have a known peak of CHH context DNA methylation (H = A, T, or C) at their edges (Zemach et al., 2013), which is thought to reinforce the boundary between the TE and neighboring chromatin (Li et al., 2015). When TEs are transcriptionally silenced in wild-type Col plants, we show that our expressed and annotated TEs that have the potential to create polyadenylated RNAs and our unexpressed TE category both have this peak of methylation at their edge (Figure 4A). This peak of CHH methylation remains in ddm1 mutants (Figure 4B), demonstrating that the DDM1 gene is not responsible for maintaining the edge of TE chromatin boundaries. We previously hypothesized that TEs that create mRNAs would be targeted for higher levels of CHH methylation through the expression-dependent RdDM pathway (Panda et al., 2016). We found that the expressed and annotated TE class defined by our transcript annotation has higher methylation at their edge in both wild-type Col and ddm1 mutants (Figures 4A and 4B) compared with the unexpressed TE class or random TEs. This observation is consistent between terminal inverted repeat DNA transposons, Helitron elements, and LTR retrotransposons (Supplemental Figure 2).
Figure 4.

Expressed and Annotated TEs Are Methylated at a Higher Level.
(A) Genome-wide CHH context DNA methylation in wild-type (wt) Col assayed with methylC-seq. TEs are aligned by their 5′ edge. Numbers in parentheses denote the number of TEs analyzed in each category. The random category represents 1292 TEs randomly chosen and assayed, a total of three times.
(B) Same as in (A), but with ddm1 mutant plants.
(C) Distribution of TE CHH methylation at TE 5′ edges (for each TE averaged and shown in [A] and [B]). The two box plots represent the two bins (0 to 20 bp and 20 to 40 bp) immediately 3′ of the TE 5′ edge (shown in [A] and [B]). Box plots represent 25th and 75th quartile values with a line at the median, the means are shown as diamonds, and the whisker lengths represent the 10th to 90th percentiles.
To determine if the concentration of CHH DNA methylation on expressed elements is due to a large effect generated by just a few TEs, we plotted the distribution of the percentage of CHH methylation at the TE edge. We found that the median of TE methylation of expressed TEs is higher than the median for unexpressed or random TEs (Figure 4C). This shows that the overall population has increased methylation at the TE edges and that the analysis is not biased by a few TEs. This finding supports our hypothesis and suggests that the cell can identify potentially mutagenic TEs and more strongly target them for repression. Thus, our transcript annotation provides a distinction between TEs based on their transcriptional potential, which is important to test hypotheses on how TEs are targeted for repression.
Analysis of TE Splicing
Our TE transcript annotation allows the transcriptome-wide investigation of TE splicing in plants. We compared splicing from TE-initiated transcripts with a set of genes that do not overlap TEs. There are 1050 TE-annotated transcripts that do not have introns, while we focused on the 1138 that have at least one intron and compared these with intron-containing genes. We found that the average number of introns is lower in TEs compared with genes (Figure 5A) and that the TEs have on average larger exons (Figure 5B) and introns (Figure 5C). This suggests that TEs are subject to less overall splicing and potentially less splicing-based RNA quality control compared with genes.
Figure 5.

TEs Have Fewer Introns and Are Spliced Less Accurately Than Genes.
(A) Number of introns in genes and TEs with at least one intron. Box plots represent 25th and 75th quartile values with a line at the median, means are shown as diamonds, and the whisker lengths represent the 10th to 90th percentiles. P values are calculated by Welch’s two-sample t test.
(B) Length of exons in genes and TEs.
(C) Length of introns in genes and TEs.
(D) Accuracy of splicing defined by comparing Illumina RNA-seq reads with our ONT-generated TE transcript annotation.
(E) Splicing accuracy of TEs ordered based on the amount of siRNAs generated in ddm1 pol IV double mutants.
(F) Same as in (E), but using only primary siRNAs generated in the ddm1 rdr6 pol IV triple mutant.
(G) Binned splicing accuracy of the expressed and annotated TEs with at least one intron compared with their primary siRNA production in the ddm1 rdr6 pol IV triple mutant. RPKM, reads per kilobase per million.
In (E) to (G), splicing accuracy is based on Illumina reads from (D).
In two nonplant organisms, stalled spliceosomes and low accuracy of splicing have been shown to affect the quality-control surveillance of TE mRNAs, specifically pushing TE transcripts into RNAi to generate primary small RNAs (Dumesic et al., 2013; Yu et al., 2019). The mechanism by which plant TE mRNAs are first degraded into primary siRNAs is currently enigmatic, so therefore we sought to test TE splicing accuracy in plants. We used the more quantitative and lower error rate per base Illumina RNA-seq reads and splice sites identified using our ONT-based transcript annotation to calculate the accuracy of splicing (see Methods). We found reduced splicing accuracy for TEs compared with genes (Figure 5D) and therefore conclude that TE transcript splicing occurs less often and at a lower accuracy compared with genes.
To determine if the reduction in splicing accuracy found for TEs is correlated with the entry of TE RNAs into RNAi, we examined siRNA production via small RNA sequencing. We first used a ddm1 pol IV double mutant to assay siRNAs because of the abundant RNAi of some TE RNAs that occurs in this mutant (Panda et al., 2016). We found no correlation between splicing accuracy and the entry of specific TE RNAs into RNAi (Figure 5E). Second, we performed the same analysis using small RNA sequencing data from a ddm1 rdr6 pol IV triple mutant, in which no secondary siRNAs are formed, allowing for the investigation of TE-specific primary siRNAs (Panda et al., 2016). We again found no correlation between TE splicing accuracy and the propensity of a TE to generate primary siRNAs (Figure 5F). Likewise, we detect no correlation in primary siRNA production (in the ddm1 rdr6 pol IV triple mutant) when compared across TEs binned by their splicing accuracy (Figure 5G). These data demonstrate that TE RNA splicing accuracy does not trigger posttranscriptional degradation via RNAi and overall establishes the utility of a transcript-based TE annotation.
DISCUSSION
By performing long-read transcriptomics on TE-expressed samples, we have captured full-length polyadenylated TE transcripts and used them to produce an improved TE annotation of Arabidopsis and maize. We have demonstrated two consequences of these improved annotations. First, this method identifies the relatively small number (1292 in Arabidopsis and 745 in maize) of individual TE copies per genome capable of the expression required to potentially make a protein, reducing the complexity of TE analyses. These transcripts can be used in future experiments to unambiguously map short-read RNA-seq data to identify the individual TE copies responsible for expression (as in Figure 3) and better calculate the differential expression of individual elements. Second, transcript-based TE annotation also enables current and future research testing hypotheses that require information on specific transcript features, such as TSSs or splicing patterns.
We used the new Arabidopsis TE transcript annotation to test two standing hypotheses in TE biology. First, we found that TEs capable of expression and therefore potentially mutagenic are targeted more strongly by the cell for repression via DNA methylation. Second, we showed that inaccurate TE splicing does not trigger RNAi on plant TEs. However, a limitation of our analysis is that it only tests the splicing accuracy on polyadenylated RNAs. Nevertheless, with our added transcript annotation based on the existing high-quality TAIR10 TE list, Arabidopsis now may have the best TE annotation of any multicellular organism.
In Arabidopsis, we used mutations as one way to activate TE transcription. In maize, we used the DRTS events that are developmentally programmed. In the future, different sources of TE activation can be combined to further improve these TE annotations and add elements that were missed. Additional mutations, different tissues, and/or abiotic and biotic stresses that are known to activate some TEs (reviewed by Horváth et al., 2017) can all be used to trigger TE transcript accumulation. In addition, we chose to analyze poly(A)+ RNA because of available protocols for ONT library production and to enrich for Pol II-derived mRNAs. This approach will miss other non-poly(A) TE RNAs. We have also not taken into account the presence/absence variation of TEs at specific positions not present in the Arabidopsis Columbia ecotype or maize B73 standard inbred, which may be under locus-specific control (Quadrana et al., 2016; Anderson et al., 2019a).
The higher number of TEs identified as expressed and annotated in Arabidopsis compared with maize is not a reflection of overall TE activity but rather a technical feature highlighting the higher strength of mutations in activating TEs compared with DRTS events and deeper long-read sequencing. In Arabidopsis, our annotation is based on over 5 million ONT long reads that pass our quality-control filter, while the maize Iso-Seq data are based on only 0.75 million long reads. Coupling the DRTS and stress-induced activation of TEs will be particularly useful for improving TE annotations in plants that do not have available mutants, where TE complexity poses a greater challenge. Future research should pair biotic and abiotic stresses with DRTS events and increased sequencing depth to maximize the breadth of the TE transcripts detected.
METHODS
Plants and Materials
All Arabidopsis (Arabidopsis thaliana) plants used were grown at 22°C on Pro-Mix EPX soil fertilized once or twice per week (with 15-16-17 at ∼150 ppm N) in Conviron MTPS-120 growth chambers with 16 h of 200 µmol m−2 s−1 light generated by Sylvania L58W/840 bulbs. The specific alleles of all the mutants used are listed in Supplemental Table 1. Inflorescence tissue, which has known high levels of TE expression in ddm1 mutants (McCue et al., 2012), was collected, flash-frozen, and stored at −80°C before RNA isolation. The ddm1 rdr6 and ddm1 pol V double mutants have been described by McCue et al. (2012). The ddm1 rdr6 pol V triple mutant was constructed by crossing a DDM1/−; rdr6 plant to a DDM1/−; pol V plant. We selected an F2 individual that was DDM1/−; rdr6; pol V to self-fertilize and generate the triple mutant.
Oxford Nanopore cDNA Sequencing
Total RNA was extracted using TRIzol reagent, quality controlled for RNA integrity number score, and enriched for poly(A)+ RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module. The cDNA-PCR Sequencing kit by Oxford Nanopore (SQK-PCS108) was used to prepare cDNA libraries from the poly(A)+ RNA. Briefly, 50 ng of poly(A)+ RNA, as measured by Qubit RNA HS assay (Thermo Fisher Scientific), was reverse transcribed using SuperScript IV reverse transcriptase (Thermo Fisher Scientific). The cDNA was PCR amplified for 13 to 14 cycles with specific barcoded adapters from the Oxford Nanopore PCR Barcoding Kit (SQK-PBK004). The amplified cDNA was purified using AMPure XP beads (Beckman Coulter). Finally, the 1D sequencing adapter was ligated to the DNA before loading onto a SpotON flow cell (FLO-MIN 106D R9 version) in a MinION sequencer. MinKNOW 3.1.19 was used to run the sequencing.
Read Processing
Raw fast5 reads were basecalled and demultiplexed using Guppy version 3.1.5+781ed57. Reads were mapped to the Arabidopsis genome (TAIR10 annotation) using Minimap2 (Li, 2018). Only primary (uniquely mapping) alignments (∼95%) were retained for analysis. A local instance of JBrowse v1.12.5 (Buels et al., 2016) was used to upload TAIR10 gene and TE annotation along with the nanopore primary alignments for the browser shots shown in Figures 1F and 1G. Overlap of the reads with TAIR10-annotated TEs was used to calculate read percentage in Figure 1C. To facilitate identifying reads initiated within genes, Pychopper2 was used to orient reads. All the reads that initiated within annotated genic exons or 5′ untranslated regions and matching the annotated transcript direction were marked as “genic reads,” and the remaining reads were overlapped with annotated TEs for calculation of TE-initiated reads (Figure 1D) and bases (Figure 1E).
Transcript Annotation
All Arabidopsis reads from all genotypes were pooled including the higher depth sequencing of the triple mutant (ddm1 rdr6 pol V). The Pinfish pipeline by Oxford Nanopore was used to call de novo transcripts using long reads and default parameters. The pipeline was run twice, first with a minimum cluster size of 3 and then again with a cluster size of 5. The reason we ran the pipeline additionally with a lower cluster size was to capture transcripts from TEs that did not have high expression. These two transcript files were merged, and duplicate transcripts were removed. A total of 34,254 transcripts were identified. Similar to the analysis of individual reads, transcripts that originated within genic exons or 5′ untranslated regions and in the matching orientation were annotated as “gene” transcripts. Of the remaining transcripts, only those transcripts were annotated as TE transcripts if at least 25% of an exon overlapped with an annotated TE. The TE superfamilies represented by the transcripts for each genotype are noted in Supplemental Table 2.
Transcript Assignment to Genes and TEs
First, the genes in the TAIR10 list annotated as TEs were removed to retain a pure “gene-only” list. Gene-initiated transcripts were removed as described above. Of the remaining transcripts, the ones that had at least one exon that overlapped at least 25% with an annotated TE were assigned as a “TE transcript.” A total of 2188 transcripts representing 1292 TEs were annotated as TE transcripts. There were many transcripts that overlapped with multiple TEs due to nested TE configurations. To assign transcripts to individual TEs, the following criteria were followed preferentially: (1) if a transcript overlapped with only one TE, the transcript was assigned to that specific TE; (2) if there was more than one TE that overlaps with the transcript, the TE that overlaps with a significantly longer section of the transcript (at least threefold greater than other overlapping TEs) was annotated to the specific transcript; (3) finally, if more than one TE continues to be assigned to one transcript, then the TE with the matching strand to the transcript direction was assigned the transcript. Following these criteria, a total of 1936 transcripts were assigned to one TE each, and 252 transcripts were assigned to more than one TE.
Updating the TAIR10 TE Annotation
The columns added to the TAIR10 TE list for this new version of the TE annotation are not only based on transcript annotation (this study) but also on TE characterization (from Panda et al., 2016): TE length, length category, subfamily copy number, copy number category, distance from the centromere, position category (euchromatic/pericentromeric), distance to the nearest gene, and RdDM type when TEs are silent (wild-type Col) and when TEs are active (ddm1). For transcript annotation, four columns were added. First, the expression categories of TEs: Expressed and Annotated if the transcripts are annotated as mentioned above; Low Expressed if no defined transcripts but at least one TE-initiating read was found in any of the genotypes; and. if no TE-initiating read was detected in any of the genotypes. For the Expressed and Annotated TE category, three additional columns were added: transcription start site, polyadenylation site, and transcript_ID. These columns may include comma-separated values if more than one transcript is found for a specific TE. This TE annotation is given in Supplemental File 1. The transcript_IDs can be used to cross-reference with Supplemental File 2, which is a GFF transcript annotation file that contains the exon structure for each TE transcript. All TEs and expressed and annotated TEs are used in the circular BioCircos plot (Cui et al., 2016) shown in Figure 2B.
RNA-Seq Validation
The ddm1 RNA-seq raw reads (Illumina PE125) from GSE93584 were trimmed for adapters using Trimmomatic (Bolger et al., 2014) and mapped to the Arabidopsis genome (TAIR10 annotation) using STAR (Dobin et al., 2013) with the following parameters: --runMode alignReads --outMultimapperOrder Random --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 50 --outFilterMatchNmin 30 --alignIntronMax 10000 --alignSJoverhangMin 3. DeepTools2 (Ramírez et al., 2016) was used to calculate the normalized read count for each TE using a bin size of 20 bp and to generate the metaplots in Figures 2C to 2E.
Evadé Mapping Strategies
Adapter-trimmed RNA-seq reads for wild-type Col and ddm1 (as mentioned above) were mapped to AtCopia93 (Evadé) copies using STAR with either a unique strategy for multiple mapping reads (Figure 3B) or a random strategy (Figure 3C). For nanopore-assisted multimapping, only the regions of AtCopia93 that produce transcripts were included as reference instead of all AtCopia93 copies (Figure 3A). The bam alignment file from all three mapping strategies was uploaded to IGV for the browser images shown in Figures 3A to 3C.
Analysis of Maize Iso-Seq Data
Maize PacBio Iso-Seq data were downloaded from SRP067440 (Wang et al., 2016). Raw reads of all size categories from all six tissues (endosperm, embryo, root, ear, tassel, and pollen) were combined. The B73 v4 genome (Jiao et al., 2017) and gene annotation were used in the Pinfish pipeline by Oxford Nanopore (same as the one used for Arabidopsis TE transcript annotation above). A minimum cluster size of five transcripts was used to call consensus transcripts. Only those transcripts that initiated within 5 bp of already annotated TEs (as per v4) and had at least one exon that overlapped at least 25% with an annotated TE were counted as TE-initiated transcripts and included for TE transcript annotation. The transcripts were assigned to TEs using the same approach as that of Arabidopsis (see Transcript Assignment to Genes and TEs). The TE transcript annotation was layered on top of the already available B73 v4 TE annotation and included as Supplemental File 3. The TE transcripts themselves are included as Supplemental File 4. Both the B73 v4 TE transcripts files are version controlled on GitHub (https://github.com/KaushikPanda1/AthalianaTETranscripts).
To assess tissue specificity, the raw reads from individual tissues were mapped to the B73 v4 genome using Minimap2 (-ax splice:hq -uf --secondary=no), and the TE transcripts annotated above were counted in each individual tissue sample using featureCounts (parameters: -L --ignoreDup -M -O; Liao et al., 2014). These counts were normalized to reads per kilobase per million mapped reads for each TE and displayed in a heat map (Figure 3D).
To compare mapping strategies, we used processed Illumina RNA-seq reads from mature pollen generated in a previous study (Warman et al., 2020). The processed reads were mapped to the maize B73 v4 genome using STAR aligner with multimapping reads assigned either randomly (--outMultimapperOrder Random --outSAMmultNmax 1) or to all (up to a 1000 best-matching) loci (--outMultimapperOrder Random --outSAMmultNmax -1 --outSAMprimaryFlag AllBestScore --outFilterMultimapNmax 1000 --winAnchorMultimapNmax 1000). We chose to focus on a single LTR retrotransposon family, Opie (RLC00004), to illustrate the differences between four mapping strategies. featureCounts was used to count the reads for Opie using these common parameters (-C -B -p -O --largestOverlap).
For the “random” mapping strategy, the “randomly placed multi-mapping reads” mapped file was used for featureCounts with additional parameters (-M --primary) to count multimapped reads randomly chosen from the primary reads. For the “all” strategy, the “all loci multi-mapped reads” mapped file was used with the additional featureCounts parameters (-M --primary) to count all multimapped primary reads. For the “fractional” strategy, the same mapped file as for “all loci” was used but with the featureCounts parameters (-M --fraction) to ensure the fractional count of all multimapped reads. For Iso-Seq-assisted mapping, the whole genome was masked except for the annotated TE transcript regions (with a 300-bp flank on either side). The Illumina reads were mapped to this masked genome and the transcripts were counted using featureCounts (-C -B -p -O --largestOverlap -M --fraction). These transcript counts were then assigned to their corresponding TEs as per Supplemental File 3. The fragments per kilobase per million mapped fragments values for all mapping strategies are displayed in a heat map (Figure 3E).
Methylation Metaplot Analysis
The wild-type Col and ddm1 methylC-seq processed files were used from GSE79746 to generate bigwig files using methylpy Schultz et al., 2015. These bigwig files were used in deepTools to generate the methylation metaplots (Figure 4) similar to the RNA-seq metaplots described above. deepTools generated a matrix of percentage CHH DNA methylation for all TE sets (random, unexpressed, or expressed TEs) where each row is an individual TE element and each column corresponds to a 20-bp bin along the element. Using this matrix, we determined that the peak of DNA methylation is in the second bin 3′ of the TE 5′ edge (20 to 40 bp after the TE 5′ edge). The distribution of percentage CHH DNA methylation was determined at the two 20-bp bins closest to the TE peak (20 to 40 bp and 40 to 60 bp) for all elements and is displayed in Figure 4C.
Splicing Accuracy
Splicing accuracy is defined as the to-the-nucleotide precision of a splice event compared with the consensus splice product. Intron retention via alternative splicing is not considered in this analysis. The number of reads that support the consensus splice product are counted and compared with reads that support splicing at 1, 2,. . . up to 25 nucleotides away from the consensus splice site. This 3′ splice site ratio method (from Herzel and Neugebauer, 2015) was adapted to calculate both the 5′ and 3′ splice site ratios of each intron and averaged across all introns for a particular transcript. To calculate the splicing accuracy of Illumina reads (GSE93584), the read depth of each position in the genome was calculated using the SAMtools depth function (Li et al., 2009). For any specific transcript (TE or gene), 5′ splice site accuracy was defined as the ratio of all of the depths at the exon/intron boundary and the depth of the position 25 bp upstream of the splice site (25 bp is the resolution of splicing efficiency calculation by Herzel and Neugebauer (2015). The 5′ and 3′ splicing accuracies were averaged for all splicing events (multiple introns) in a transcript, and the transcript splicing accuracy was defined as the average of 5′ and 3′ splicing accuracies. All individual transcript accuracy data are plotted in Figures 5D to 5G. Custom scripts to analyze splicing accuracy are available at https://github.com/KaushikPanda1.
Correlation of Splicing Accuracy and Small RNA Production
To capture the TEs that produce siRNAs, we first identified TEs that produced at least one siRNA read per million in a ddm1 pol IV mutant. We used this mutant as it has the highest levels of TE RNAi (Panda et al., 2016). The TEs were sorted by the number of normalized siRNAs produced (averaged across two biological replicates [nonoverlapping pools of plants]) and divided into 10 categories. As a control, all TEs and the TEs that do not produce at least one siRNA read per million were compared. Since RDR6 is involved in amplifying RNAi by generating small RNAs in a feedback loop, we aimed to combine the rdr6 mutation to remove secondary siRNAs and only investigate primary small RNAs; therefore, we repeated the siRNA correlation analysis using siRNAs from ddm1 rdr6 pol IV. All siRNAs used were downloaded from GSE79780. Raw reads were trimmed for adapters and mapped to the Arabidopsis genome using ShortStack (Axtell, 2013; fractional seeded strategy for multimapping reads). The SAMtools bedcov function was used to calculate the normalized siRNA read count of each TE.
Accession Numbers
All the raw data generated in this study are deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (GSE145066). The Panda TE transcript annotation v1.0 files are given as Supplemental Files 1 to 4 and version controlled available at GitHub (https://github.com/KaushikPanda1/AthalianaTETranscripts).
Supplemental Data
Supplemental Figure 1. Expression level peaks at the TSS of different TE types (Supports Figure 2).
Supplemental Figure 2. DNA methylation level peaks at the TSS of different TE types (Supports Figure 4).
Supplemental Table 1. Sequencing statistics and alleles used (Supports Figure 1).
Supplemental Table 2. TE super-family expression detected in each sample (Supports Figures 2 and 3).
Supplemental File 1. Panda version 1.0 of the Arabidopsis TAIR10 TE annotation with 14 added feature categories per element.
Supplemental File 2. Panda version 1.0 of the Arabidopsis TE transcript annotation GFF file.
Supplemental File 3. Panda version 1.0 of the Maize TE annotation (B73 v4) with 6 added feature categories per element.
Supplemental File 4. Panda version 1.0 of the Maize TE transcript annotation GFF file.
DIVE Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We thank Meredith Sigman for production of the ddm1 rdr6 pol V triple mutant line, Hayden Payne for plant images, and Alex Harkness for technical assistance with ONT sequencing. This work was supported by NSF | BIO | Division of Molecular and Cellular Biosciences (grant MCB-1608392 to R.K.S.).
AUTHOR CONTRIBUTIONS
K.P. and R.K.S. designed the research; K.P. performed the research, data analyses, and created all figures; K.P. and R.K.S. wrote the article.
Footnotes
Articles can be viewed without a subscription.
References
- Anderson S.N., Stitzer M.C., Brohammer A.B., Zhou P., Noshay J.M., O’Connor C.H., Hirsch C.D., Ross-Ibarra J., Hirsch C.N., Springer N.M.(2019a). Transposable elements contribute to dynamic genome content in maize. Plant J. 100: 1052–1065. [DOI] [PubMed] [Google Scholar]
- Anderson S.N., Stitzer M.C., Zhou P., Ross-Ibarra J., Hirsch C.D., Springer N.M.(2019b). Dynamic patterns of transcript abundance of transposable element families in maize. G3 (Bethesda) 9: 3673–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell M.J.(2013). ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 19: 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeg K., Iwakawa H.O., Tomari Y.(2017). The poly(A) tail blocks RDR6 from converting self mRNAs into substrates for gene silencing. Nat. Plants 3: 17036. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B.(2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L., Holmes I.H.(2016). JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 17: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisine N., Quesneville H., Colot V.(2008). Improved detection and annotation of transposable elements in sequenced genomes using multiple reference sequence sets. Genomics 91: 467–475. [DOI] [PubMed] [Google Scholar]
- Chye M.-L., Cheung K.-Y., Xu J.(1997). Characterization of TSCL, a nonviral retroposon from Arabidopsis thaliana. Plant Mol. Biol. 35: 893–903. [DOI] [PubMed] [Google Scholar]
- Cuerda-Gil D., Slotkin R.K.(2016). Non-canonical RNA-directed DNA methylation. Nat. Plants 2: 16163. [DOI] [PubMed] [Google Scholar]
- Cui Y., Chen X., Luo H., Fan Z., Luo J., He S., Yue H., Zhang P., Chen R.(2016). BioCircos.js: An interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics 32: 1740–1742. [DOI] [PubMed] [Google Scholar]
- Deniz Ö., Frost J.M., Branco M.R.(2019). Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 20: 417–431. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.(2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic P.A., Natarajan P., Chen C., Drinnenberg I.A., Schiller B.J., Thompson J., Moresco J.J., Yates J.R. III, Bartel D.P., Madhani H.D.(2013). Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152: 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L., Neugebauer K.M.(2015). Quantification of co-transcriptional splicing from RNA-Seq data. Methods 85: 36–43. [DOI] [PubMed] [Google Scholar]
- Hirochika H., Okamoto H., Kakutani T.(2000). Silencing of retrotransposons in Arabidopsis and reactivation by the ddm1 mutation. Plant Cell 12: 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth V., Merenciano M., González J.(2017). Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 33: 832–841. [DOI] [PubMed] [Google Scholar]
- Jiao Y., et al. (2017). Improved maize reference genome with single-molecule technologies. Nature 546: 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Tam O.H., Paniagua E., Hammell M.(2015). TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 31: 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahmy S., Pontier D., Cavel E., Vega D., El-Shami M., Kanno T., Lagrange T.(2009). PolV(PolIVb) function in RNA-directed DNA methylation requires the conserved active site and an additional plant-specific subunit. Proc. Natl. Acad. Sci. USA 106: 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamesch P., et al. (2012). The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 40: D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.(2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.(2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., et al. (2015). RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 112: 14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W.(2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Lyons D.B., Zilberman D.(2017). DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife 6: e30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez G., Slotkin R.K.(2012). Developmental relaxation of transposable element silencing in plants: Functional or byproduct? Curr. Opin. Plant Biol. 15: 496–502. [DOI] [PubMed] [Google Scholar]
- McCue A.D., Nuthikattu S., Reeder S.H., Slotkin R.K.(2012). Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8: e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouze M., Reinders J., Bucher E., Nishimura T., Schneeberger K., Ossowski S., Cao J., Weigel D., Paszkowski J., Mathieu O.(2009). Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461: 427–430. [DOI] [PubMed] [Google Scholar]
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T(2001). Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411: 212–214. [DOI] [PubMed] [Google Scholar]
- Nuthikattu S., McCue A.D., Panda K., Fultz D., DeFraia C., Thomas E.N., Slotkin R.K.(2013). The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 nucleotide small interfering RNAs. Plant Physiol. 162: 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlin S., Sarazin A., Chevalier C., Voinnet O., Marí-Ordóñez A.(2017). A genome-wide transcriptome and translatome analysis of Arabidopsis transposons identifies a unique and conserved genome expression strategy for Ty1/Copia retroelements. Genome Res. 27: 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozata D.M., Gainetdinov I., Zoch A., O’Carroll D., Zamore P. D.(2019). PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20: 89–108. [DOI] [PubMed] [Google Scholar]
- Panda K., Ji L., Neumann D.A., Daron J., Schmitz R.J., Slotkin R.K.(2016). Full-length autonomous transposable elements are preferentially targeted by expression-dependent forms of RNA-directed DNA methylation. Genome Biol. 17: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrana L., Bortolini Silveira A., Mayhew G.F., LeBlanc C., Martienssen R.A., Jeddeloh J.A., Colot V.(2016). The Arabidopsis thaliana mobilome and its impact at the species level. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.(2016). deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44: W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala S.H., Elumalai R., Vanier C., Ozkan H., Galbraith D.W., Richards E.J.(2006). Meiotically stable natural epialleles of Sadhu, a novel Arabidopsis retroposon. PLoS Genet. 2: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable P.S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. [DOI] [PubMed] [Google Scholar]
- Schultz Matthew D, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523: 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahid S., Slotkin R.K.(2020). The current revolution in transposable element biology enabled by long reads. Curr. Opin. Plant Biol. 54: 49–56. [DOI] [PubMed] [Google Scholar]
- Slotkin R.K., Vaughn M., Borges F., Tanurdzić M., Becker J.D., Feijó J.A., Martienssen R.A.(2009). Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136: 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissandier A., Servant N., Barillot E., Bourc’his D.(2019). Tools and best practices for retrotransposon analysis using high-throughput sequencing data. Mob DNA 10: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Tseng E., Regulski M., Clark T.A., Hon T., Jiao Y., Lu Z., Olson A., Stein J.C., Ware D.(2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 7: 11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warman C., Panda K., Vejlupkova Z., Hokin S., Unger-Wallace E., Cole R.A., Chettoor A.M., Jiang D., Vollbrecht E., Evans M.M.S., Slotkin R.K., Fowler J.E.(2020). High expression in maize pollen correlates with genetic contributions to pollen fitness as well as with coordinated transcription from neighboring transposable elements. PLoS Genet. 16: e1008462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki A.T., Haag J.R., Pikaard C.S.(2008). Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135: 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Koppetsch B.S., Pagliarani S., Johnston S., Silverstein N.J., Luban J., Chappell K., Weng Z., Theurkauf W.E.(2019). The piRNA response to retroviral invasion of the koala genome. Cell 179: 632–643.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A., Kim M.Y., Hsieh P.-H., Coleman-Derr D., Eshed-Williams L., Thao K., Harmer S.L., Zilberman D.(2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X., Hale C.J., Law J.A., Johnson L.M., Feng S., Tu A., Jacobsen S.E.(2012). DDR complex facilitates global association of RNA polymerase V to promoters and evolutionarily young transposons. Nat. Struct. Mol. Biol. 19: 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]