

Structural and functional studies of the C-glycosyltransferase UGT708C1 provide a basis for understanding the C-glycosylation mechanism and facilitate enzyme engineering to produce bioactive C-glycosides.
Abstract
C-Glycosyltransferases (CGTs) catalyze the formation of C-glycosidic bonds for the biosynthesis of C-glycosides, but the underlying mechanism is unclear. This process improves the solubility and bioavailability of specialized metabolites, which play important roles in plant growth and development and represent rich resources for drug discovery. Here, we performed functional and structural studies of the CGT UGT708C1 from buckwheat (Fagopyrum esculentum). Enzymatic analysis showed that UGT708C1 is capable of utilizing both UDP-galactose and UDP-glucose as sugar donors. Our structural studies of UGT708C1 complexed with UDP-glucose and UDP identified the key roles of Asp382, Gln383, Thr151, and Thr150 in recognizing the sugar moiety of the donor substrate and Phe130, Tyr102, and Phe198 in binding and stabilizing the acceptor. A systematic site-directed mutagenesis study confirmed the important roles of these residues. Further structural analysis combined with molecular dynamics simulations revealed that phloretin binds to the acceptor binding pocket in a bent state with a precise spatial disposition and complementarity. These findings provide insights into a catalytic mechanism for CGTs.
INTRODUCTION
Plant secondary metabolites exhibit high structural novelty and diversity and play important roles in protecting plants from UV irradiation (van de Staaij et al., 2002; Peng et al., 2017), reproduction and fertility (Mahajan et al., 2011), and plant defense against pathogens and herbivores (Cornell and Hawkins, 2003). Plant secondary metabolites possess a variety of bioactivities, such as antitumor, antibacterial, and antibiotic activities, and they represent an important source for drug discovery (Ogasawara et al., 2004; Newman and Cragg, 2012; Giddings and Newman, 2013; Paradkar, 2013; Katz and Baltz, 2016; Li, 2016). However, many plant secondary metabolites often have poor biological activity, low bioavailability, poor specificity, and toxicity. Glycosylation is a modification strategy that can improve the solubility and bioavailability of natural plant products (Muñoz-Antoli et al., 2014; Liang et al., 2015; Ilmberger and Rabausch, 2017). Uridine diphosphate glucosyltransferases (UGTs) catalyze glycosylation reactions to transfer sugar moieties from activated donors to various small molecular acceptors.
Glycosylation occurs on O-, C-, N-, and S-atoms to produce O-glycosides, C-glycosides, N-glycosides, and S-glycosides, respectively (Jones and Vogt, 2001). C-Glycosides are more stable in response to acid hydrolysis and glycosidase than the others. Plant-derived C-glycosides exhibit a variety of pharmacological properties, including antimicrobial, antioxidant, antinociceptive (for pain relief), α-glucosidase inhibitory, and antiinflammatory activities. For example, isovitexin is an antimicrobial bioactive compound found in ethanol extracts from Adesmia aegiceras (Agnese et al., 2001). Vitexin has anticonvulsant effects on rat brains (Abbasi et al., 2012). Gorzalczany et al. (2011) demonstrated the antinociceptive effects of vitexin and vicenin in an algesic chemical model. Vitexin and isovitexin from mung bean (Vigna radiata) seed coats play important roles in protecting plants from oxidative injury caused by heat stress (Cao et al., 2011).
The chemical synthesis of C-glycosides is always challenging due to stereoselectivity and harsh reaction conditions. The difficulty in preparing sugar precursors also restricts the synthesis of C-glycosides (Yang and Yu, 2017). By contrast, the biosynthesis of glycosides involves much milder conditions and has high stereospecificity. Therefore, research on C-glycosyltransferases (CGTs) is beneficial for studying the biosynthesis of C-glycosides, thereby facilitating the development and utilization of C-glycoside-based drugs.
CGTs have been identified in several plant species, including OsCGT from rice (Oryza sativa), which catalyzes the C-glycosylation of 2-hydroxyflavanones (Brazier-Hicks et al., 2009); UGT708G1 from kumquat (Fortunella crassifolia) and UGT708G2 from satsuma mandarin (Citrus unshiu), which recognize 2-hydroxyflavanones, dihydrochalcone, and their mono-C-glucosides as sugar acceptors (Ito et al., 2017); GtUF6CGT1 from Japanese gentian (Gentiana triflora), which utilizes flavonoids as substrates (Sasaki et al., 2015); PlUGT43 from kudzu (Pueraria lobata), which only exhibits activity with isoflavones, such as daidzein and genistein (Wang et al., 2017); and TcCGT1 from jin lian hua (Trollius chinensis), which recognizes 36 flavones and other flavonoids (He et al., 2019).
Little is known about the catalytic mechanism of UGTs. Thus far, the SN2-like displacement mechanism has been proposed for O-glycosyltransferases (OGTs), which was supported by various structural and biochemical studies (Lairson et al., 2008). However, the catalytic mechanism of CGTs remains controversial. Two catalytic mechanisms have been proposed for C-glycosylation. One mechanism is Lewis acid-mediated ortho-C-O rearrangement to produce α-glycoside (Matsumoto et al., 1988; Hosoya et al., 1996; Bililign et al., 2004; Dürr et al., 2004). The other mechanism is similar to the Friedel-Crafts aromatic substitution in stereochemistry; the main product of this mechanism is β-glycoside (Matsumoto et al., 1989; Hayashi et al., 2000; Bililign et al., 2004; Dürr et al., 2004). These two mechanisms are based on regiochemical catalysis and lack biological data. Gutmann and Nidetzky (2012) studied the important role of His as a Brønsted base in OGT-CGT interconversion using PcOGT from common pear (Pyrus communis) and OsCGT from rice as models and proposed a mechanism for aryl‐C‐glycosylation through direct nucleophilic displacement, but this mechanism lacks structural evidence. Understanding the substrate specificity and catalytic mechanism of C-glucosyltransferase is essential for engineering C-glucosyltransferase for drug development based on C-glycosides.
UGT708C1 from buckwheat (Fagopyrum esculentum) utilizes UDP-glucose as the sugar donor and catalyzes the C-glycosylation of 2-hydroxyflavanones (Nagatomo et al., 2014). Here, we further explored this mechanism, finding that UGT708C1 utilizes both UDP-glucose and UDP-galactose as sugar donors. We then determined the crystal structures of UGT708C1 and its complexes with UDP and UDP-glucose. Based on this analysis combined with structure-based mutagenesis analysis and molecular dynamics (MD) simulations, we identified key amino acids for substrate binding and stabilization and proposed a catalytic mechanism of UGT708C1.
RESULTS
Enzymatic Analysis of UGT708C1
To study the substrate specificity of UGT708C1, we expressed and purified UGT708C1 with an N-terminal His tag from Escherichia coli (Supplemental Figure 1) and performed substrate screening using four types of sugar donors and various acceptor candidates. To analyze acceptor specificity, we used UDP-glucose as the sugar donor and various candidate acceptor substrates, including flavones (icaritin, chrysin, luteolin, apigenin, scutellarein, and baicalein), flavonols (kaempferol and quercetin), flavanonols (hesperetin and taxifolin), flavonone (liquiritigenin), isoflavones (genistein and daidzein), dihydrochalcone (phloretin), and others (2′,4′,6′-trihydroxyacetophenone, 2,4,6-trihydroxybenzaldehyde, avobenzone, and berberrubine; Supplemental Figure 2). UGT708C1 only exhibited C-glycosylation activity toward 2′,4′,6′-trihydroxyacetophenone and phloretin, rather than flavones, flavonols, isoflavones, and flavanonol compounds, which is consistent with previous reports (Nagatomo et al., 2014). In addition, UGT708C1 showed higher catalytic efficiency (kcat/Km) to phloretin than to 2′,4′,6′-trihydroxyacetophenone, with an approximately fivefold higher kcat/Km (Table 1), and the binding affinity (Kd) of UGT708C1 was higher for phloretin than for 2′,4′,6′-trihydroxyacetophenone (Table 2).
Table 1. Statistics of the Kinetic Parameters of UGT708C1 and Mutants Each enzymatic reaction was performed with more than three replicates (n ≥ 3), and kinetic parameters are represented as means ± se.
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) | Vmax (μM/min) |
|---|---|---|---|---|
| Sugar acceptor phloretina | ||||
| Wild type | 3.73 ± 0.97 | 1.70 | 4.56 × 105 | 0.28 ± 0.02 |
| F130A | 7.82 ± 1.88 | 3.33 | 4.27 × 105 | 0.55 ± 0.05 |
| Y102F | 10.95 ± 1.98 | 0.41 | 3.74 × 104 | 0.68 ± 0.04 |
| F198A | 23.90 ± 4.78 | 0.50 | 2.08 × 104 | 1.37 ± 0.11 |
| T150A | 10.09 ± 1.55 | 0.29 | 2.85 × 104 | 0.79 ± 0.04 |
| T151A | 7.53 ± 2.41 | 0.49 | 6.53 × 104 | 0.27 ± 0.03 |
| D382E | 10.41 ± 1.88 | 0.13 | 1.29 × 104 | 1.11 ± 0.07 |
| Q383H | 12.90 ± 1.59 | 0.34 | 2.64 × 104 | 1.29 ± 0.05 |
| Q383A | 13.63 ± 1.64 | 1.38 | 1.01 ×105 | 0.75 ± 0.03 |
| Sugar acceptor 2′,4′,6′-trihydroxyacetophenonea | ||||
| Wild type | 14.66 ± 1.80 | 1.38 | 9.44 × 104 | 0.76 ± 0.03 |
| Sugar donor UDP-glucose | ||||
| Wild typeb | 56.95 ± 7.69 | 3.13 | 5.50 × 104 | 8.60 ± 0.37 |
| Wild typec | 42.55 ± 5.51 | 2.03 | 4.78 × 104 | 4.47 ± 0.15 |
| F130Ab | 85.4 ± 12.6 | 3.51 | 4.11 × 104 | 9.63 ± 0.35 |
| Y102Fb | 29.00 ± 6.27 | 0.57 | 1.96 × 104 | 9.38 ± 0.55 |
| F198Ab | 40.50 ± 4.02 | 0.45 | 1.11 × 104 | 7.33 ± 0.16 |
| T150Ab | 45.68 ± 3.86 | 0.35 | 7.66 × 103 | 6.71 ± 0.14 |
| T151Ab | 280 ± 80 | 0.53 | 1.90 × 103 | 7.24 ± 0.85 |
| D382Eb | 67.80 ± 8.74 | 0.10 | 1.47 × 103 | 7.98 ± 0.27 |
| Q383Hb | 120 ± 10 | 0.46 | 3.92 × 103 | 7.61 ± 0.21 |
| Q383Ab | 203 ± 41 | 1.33 | 6.56 × 103 | 8.76 ± 0.55 |
| Sugar donor UDP-galactose | ||||
| Wild typeb | 360 ± 30 | 1.35 | 3.80 × 103 | 14.8 ± 0.47 |
| Wild typec | 104 ± 15 | 0.63 | 6.12 × 103 | 4.18 ± 0.16 |
UDP-glucose was used as the sugar donor for determining the kinetic parameters.
Phloretin was used as the acceptor for determining the kinetic parameters.
2′,4′,6′-Trihydroxyacetophenone was used as the acceptor for determining the kinetic parameters.
Table 2. Statistics of Kd Values of Wild-Type UGT708C1 and Mutants for Binding of Different Substrates.
| Protein | Kd (for UDP-Galactose) | Kd (for 2′,4′,6′-Trihydroxyacetophenone) |
|---|---|---|
| Wild-type UGT708C1 | 181 ± 2.3 µM | 5.42 ± 2.00 mMa |
| Kd (for UDP-Glucose) | Kd (for Phloretin) | |
| Wild-type UGT708C1 | 47.88 ± 5.74 µM | 12.90 ± 2.53 µM |
| F130A | 130 ± 20 µM | 1.56 ± 0.33 µM |
| Y102F | 470 ± 90 µM | 12 ± 67 mMa |
| F198A | 130 ± 30 µM | 250 ± 150 µMa |
| T150A | 102 ± 32 µM | 25.76 ± 4.72 µM |
| T151A | 1.06 ± 0.30 mM | 3.48 ± 0.85 µM |
| D382E | 0.80 ± 0.23 mM | 1.71 ± 3.65 mMa |
| Q383H | 670 ± 80 µM | 22.46 ± 8.90 µM |
| Q383A | 630 ± 110 µM | 16.98 ± 2.92 µM |
Each kinetic parameter was calculated from three independent thermophoresis measurements. Data are shown as means ± sd (n = 3).
In these values, the Kd value between the corresponding protein and the substrate is too large to be measured more accurately, due to the solubility limitation of the substrate.
UGT708C1 utilizes UDP-glucose as a sugar donor and cannot use UDP-galactose (Nagatomo et al., 2014). To explore the substrate specificity of UGT708C1, we tested four sugar donors using phloretin and 2′,4′,6′-trihydroxyacetophenone as acceptors, namely UDP-glucose, UDP-galactose, UDP-glucuronic acid, and UDP-rhamnose. UGT708C1 exhibited catalytic activity toward both UDP-glucose and UDP-galactose (Figures 1A to 1D). We performed UPLC-tandem mass spectrometry (MS/MS) to identify the products obtained using UDP-galactose as a sugar donor (Figures 1E and 1F). Enzyme kinetic analysis indicated that the catalytic efficiency (kcat/Km) of UGT708C1 was ∼14-fold higher toward UDP-glucose than toward UDP-galactose using phloretin as the acceptor (Table 1). To further verify the ability of UGT708C1 to utilize UDP-galactose as a sugar donor, we measured the kinetic parameters of UGT708C1 on UDP-glucose and UDP-galactose using 2′,4′,6′-trihydroxyacetophenone as the sugar acceptor. The results were similar to the results obtained using phloretin as the sugar acceptor. These results indicate that UGT708C1 prefers UDP-glucose to UDP-galactose (Table 1).
Figure 1.

HPLC-MS Analysis of the Reaction Products of UGT708C1.
(A) HPLC elution profile of phloretin monitored at 300 nm. mAU, milli-absorbance unit.
(B) HPLC elution profile of 2′,4′,6′-trihydroxyacetophenone monitored at 300 nm.
(C) HPLC elution profile of the reaction product of UGT708C1 with UDP-galactose as the sugar donor and phloretin as the acceptor. The retention time of the product is 17.920 min at 300 nm.
(D) HPLC elution profile of the reaction product of UGT708C1 with UDP-galactose as the sugar donor and 2′,4′,6′-trihydroxyacetophenone as the acceptor. The retention time of the product is 13.393 min at 300 nm.
(E) UPLC-MS/MS profile of the product in (C).
(F) UPLC-MS/MS profile of the product in (D).
Overall Structure of UGT708C1
We determined the crystal structure of apo-UGT708C1 (residues 18 to 456 of 457 amino acids) from buckwheat at 2.1 Å. The crystal belongs to the P 21 21 2 space group, and each asymmetric unit contains two independent molecules. The structure of apo-UGT708C1 contains two distinct domains, the N-terminal and C-terminal domains, which form a long, deep groove for substrate binding (Figure 2A); the first 17 residues of the N terminus were not observed. The N-terminal domain consists of seven parallel β-strands surrounded by eight long α-helices and three short α-helices. The C-terminal domain contains a six-stranded β-sheet flanked by eight α-helices, and the structure exhibits a canonical GT-B fold (Lairson et al., 2008).
Figure 2.

Overall Structure of UGT708C1 and Complexes.
(A) Cartoon representation of the structure of apo-UGT708C1. The α-helices of the N-terminal domain and C-terminal domain are shown in violet and cyan, respectively, and the β-strands in the N-terminal and C-terminal domains are shown in yellow and green, respectively.
(B) Cartoon representation of the structure of UGT708C1-UDP-glucose. The sugar donor UDP-glucose is shown as sticks, and all atoms are colored according to element (carbon, green; nitrogen, dark blue; oxygen, red; phosphorus, orange). The colors of the α-helices and β-strands correspond to those in (A).
(C) Cartoon representation of the structure of UGT708C1-UDP. The ligand UDP is shown as sticks, and all atoms are colored according to element (carbon, green; nitrogen, dark blue; oxygen, red; phosphorus, orange). The colors of the α-helices and β-strands correspond to those in (A).
(D) Cartoon representation of the structural model UGT708C1-phloretin. The acceptor phloretin is shown as sticks, and all atoms are colored according to element (carbon, green; oxygen, red). The colors of the α-helices and β-strands correspond to those in (A).
We determined the structures of UGT708C1 bound with UDP-glucose and UDP at 2.0 and 2.25 Å, respectively (Figures 2B and 2C). The electron density of the first 17 residues (Met1 to Asn17) in the structures of the UGT708C1-UDP and UGT708C1-UDP-glucose complexes were not observed, as was the case for the structure of apo-UGT708C1. The electron density of residues Lys167 to Ser169 in the structure of complex UGT708C1-UDP-glucose was not observed, and the electron densities of residues Thr308 to Lys310 in the structure of complex UGT708C1-UDP and residues Thr308 to Ile311 in the structure of apo-UGT708C1 were not observed. The last residues (Arg457) in the structures of apo-UGT708C1 and complex UGT708C1-UDP were not visible. The detailed interactions between the sugar donor substrate and UGT708C1 are described below.
To explore the acceptor specificity of UGT708C1, we made extensive efforts to obtain a structure of UGT708C1 complexed with phloretin. Using a diffraction data set collected for a crystal of UGT708C1 soaked in phloretin, weak electron density was observed in a hydrophobic pocket formed by residues Phe99, Phe130, Phe155, Met159, Pro188, Pro190, Phe191, and Phe198, which corresponds to the binding position of quercetin in the structures of UGT89C1 (Protein Data Bank [PDB] code 6IJD; Zong et al., 2019) and kaempferol in VvGT1 (PDB code 2C1Z; Offen et al., 2006). The weak density likely represents phloretin with partial occupancy (0.82), and a phloretin molecule was fitted into the density. Therefore, we propose a structural model of the complex comprising UGT708C1 and phloretin (Figure 2D).
Interactions between the Enzyme and Sugar Donor
Sequence alignment of UGT708C1 with other plant UGTs showed that the plant secondary product glycosyltransferase (PSPG) motif is conserved. This motif is thought to be involved in the recognition and binding of the sugar donor (Supplemental Figure 3; Shao et al., 2005; Offen et al., 2006; Zong et al., 2019). In the complex structure of UGT708C1 with UDP-glucose, UDP-glucose is buried in a long, narrow pocket located in the C-terminal domain of UGT708C1 (Figure 3A), and UDP-glucose interacts with UGT708C1 primarily through the conserved residues of the PSPG motif.
Figure 3.
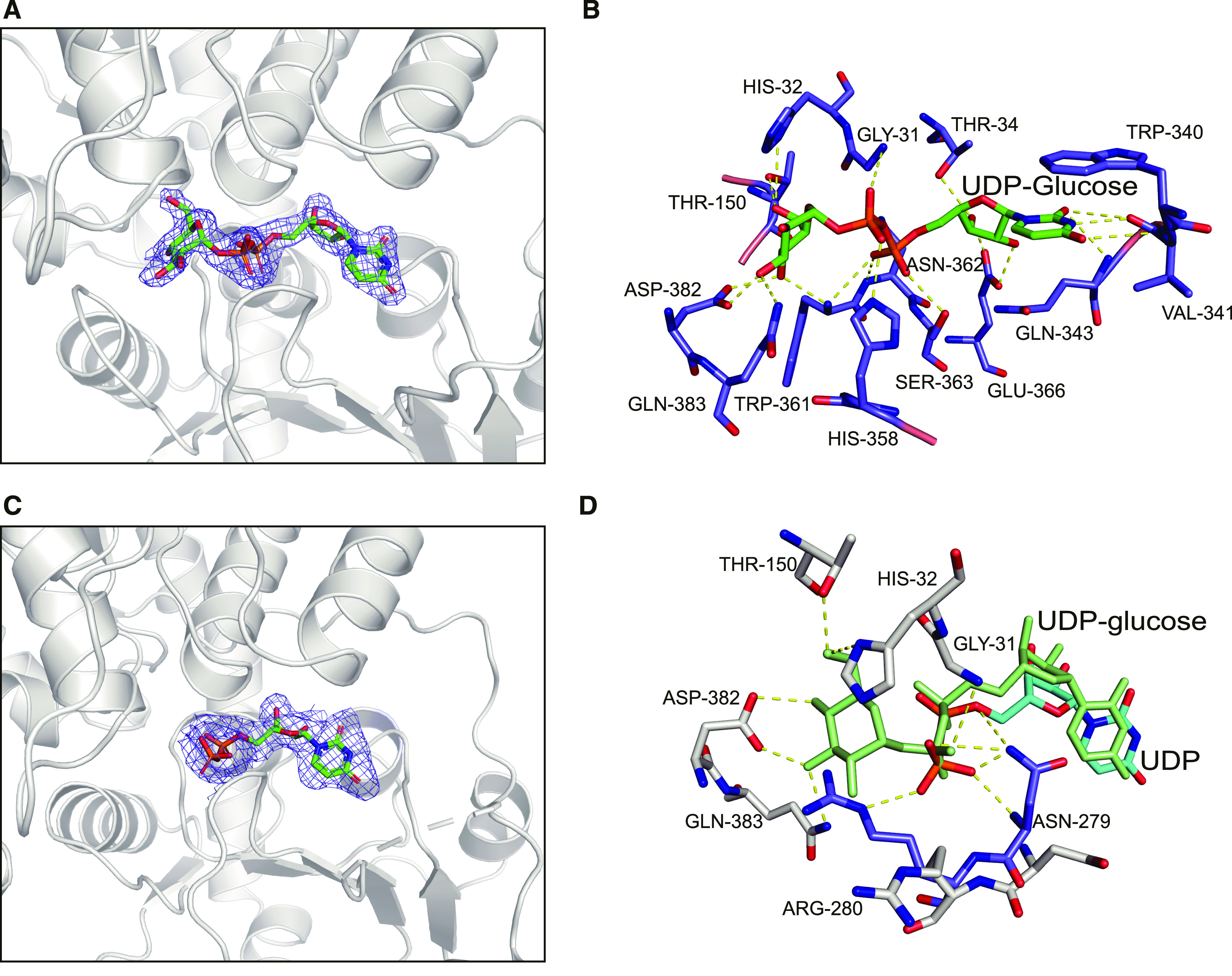
The Interactions between UGT708C1 and Donor Substrate.
(A) Fo-Fc omit electron density map of UDP-glucose in complex with UGT708C1. The structure of UGT708C1 is represented as a gray cartoon, UDP-glucose is shown as a colored stick, and all atoms are colored according to element (carbon, green; nitrogen, dark blue; oxygen, red; phosphorus, orange). The map is contoured at 3.0 σ.
(B) Interactions between UDP-glucose and the residues of UGT708C1. Hydrogen bonds are represented by yellow dashed lines. All atoms, shown in stick models, are colored according to element (carbon, green or violet; nitrogen, dark blue; oxygen, red; phosphorus, orange).
(C) Fo-Fc omit electron density map of UDP in complex with UGT708C1. The structure of UGT708C1 is represented as a gray cartoon, UDP is shown as a colored stick, and all atoms are colored according to element (carbon, green; nitrogen, dark blue; oxygen, red; phosphorus, orange). The map is contoured at 3.0 σ.
(D) Comparison of the interactions between donor substrate and UGT708C1 in structures of UGT708C1-UDP-glucose and UGT708C1-UDP. UDP-glucose is shown as a green stick, and residues that interact with UDP-glucose are shown as gray sticks. UDP is shown as a cyan stick; residues Arg280 and Asn279, which have a different conformation in the structure of UGT708C1-UDP and interact with UDP, are shown as violet sticks. Hydrogen bonds are represented by yellow dashed lines. All atoms, shown in UDP and residue stick models, are colored according to element (carbon, cyan, gray, or violet; nitrogen, dark blue; oxygen, red; phosphorus, orange).
The uracil ring of UDP-glucose interacts with the Val341 residue via hydrogen bonds between N3, O2, and O4 of the uracil ring and the main chain oxygen and nitrogen atoms of Val341 and the N atom of Gln343. In addition, the uracil ring interacts with the Trp340 residue through parallel π-stacking interactions. The ribose ring of UDP forms hydrogen bonds between its O3C and O2C atoms and the OE1 and OE2 atoms of Glu366 and the OG1 atom of Thr34. The α-phosphate establishes hydrogen bonds through its O2A atom and the OG atom of Ser363 and its O1A atom and the N atom of Asn362. The β-phosphate establishes hydrogen bonds through its O1B atom and the N atom of Gly31 and its O2B atom and the NE2 atom of His358.
The O3′ atom of the glucose ring forms hydrogen bonds with the NE2 atom of Gln383 and the OD1 atom of Asp382. The O4′ atom of the glucose ring forms hydrogen bonds with the OD2 atom of Asp382 and the N atom of Trp361; the O6′ atom of the glucose ring forms hydrogen bonds with the ND1 atom of His32 and the OG1 atom of Thr150 (Figure 3B). Mutagenesis confirmed the importance of these key residues, whose interactions with sugar donors are important for their binding and catalysis (see below).
In the structure of UGT708C1 with UDP, electron density was also clearly observed for UDP (Figure 3C). Hydrogen bonds were observed between UDP and Asn279, since the β-phosphate of UDP and Asn279 undergo a small rotation and shift compared with that in the structure of UGT708C1 complexed with UDP-glucose. The β-phosphate of UDP and Arg280 form a hydrogen bond, since Arg280 is in an alternative conformation different from that in the structure of UGT708C1 complexed with UDP-glucose (Figure 3D).
Acceptor Binding Site, Molecular Docking, and MD Simulations
In the structural model of the complex of UGT708C1 and phloretin, phloretin is in a bent state, and its monohydroxyl B ring extends into the hydrophobic pocket, while the trihydroxyl A ring is located near the entrance of the pocket (Figure 2D). The O4 atom of phloretin forms a hydrogen bond with the OH group of Tyr102, and the O3 atom of phloretin forms separate hydrogen bonds with the N atom of Phe130 and the O atom of Phe148 (Figure 4A).
Figure 4.

Acceptor Binding Site and Conformational Changes of UGT708C1.
(A) Binding site of the acceptor substrate phloretin in the structural model of UGT708C1-phloretin. The structure is shown as a violet cartoon, phloretin is shown as green sticks, and the residues in the acceptor binding pocket are shown as violet sticks. All atoms are colored according to element (carbon, green or violet; nitrogen, dark blue; oxygen, red; phosphorus, orange).
(B) Binding mode of phloretin-pose 1 predicted by MD simulations. The structure is shown as a gray cartoon, phloretin is shown as cyan sticks, and UDP-glucose is shown as gray sticks. All atoms are colored according to element (carbon, gray or cyan; nitrogen, dark blue; oxygen, red; phosphorus, orange).
(C) Binding mode of phloretin-pose 2 predicted by MD simulations. The structure is shown as a gray cartoon, phloretin is shown as yellow sticks, and UDP-glucose is shown as gray sticks. All atoms are colored according to element (carbon, gray or yellow; nitrogen, dark blue; oxygen, red; phosphorus, orange).
(D) Comparison of the structural model of UGT708C1-phloretin, the molecular docking model of UGT708C1-UDP-glucose-phloretin, and the final model of the MD simulations. The UGT708C1 structures are shown as gray cartoons, and the ligands of phloretin are shown as green, salmon, and cyan sticks. All atoms are colored according to element (carbon, green, cyan, or salmon; oxygen, red). The structural alignment shows that the binding positions and orientations of phloretin in the three structures are basically the same.
(E) Structural comparison of apo-UGT780C1 (green cartoon), the UGT708C1-UDP complex (cyan cartoon), the UGT708C1-UDP-glucose complex (salmon cartoon), and the UGT708C1-phloretin model complex (magenta cartoon). Arg280 exhibited two conformations. Arg280, Asp96, and phloretin are shown as sticks of the same color as the cartoon. All atoms are colored according to element (carbon, green, cyan, salmon, or magenta; oxygen, red; nitrogen, dark blue).
(F) Conformational change of Trp340 before and after sugar donor binding. The color scheme of the cartoon structure is consistent with that of (E). Trp340, UDP, and UDP-glucose are shown as sticks of the same color as the cartoon. All atoms are colored according to element (carbon, green, cyan, salmon, or magenta; nitrogen, dark blue; oxygen, red; phosphorus, orange).
UGT708C1 shows C-glycosylation activity toward 2-hydroxyflavanones, dihydrochalcone, and other related compounds (Nagatomo et al., 2014), while UGT71G1 exhibits O-glycosylation activity toward flavonoids and triterpene compounds (Shao et al., 2005). However, a structural comparison showed that the acceptor binding position of UGT708C1 is the same as that of UGT71G1 (PDB code 2ACV; Shao et al., 2005), and His32 and Asp129 of UGT708C1 are located in the corresponding positions of the proposed catalytic residues His22 and Asp121 in UGT71G1, suggesting that His32 and Asp129 might also act as catalytic residues for C-glycosylation catalyzed by UGT708C1. We also observed differences in their acceptor binding pockets. Specifically, helices Nα3, Nα5b, Nα5a, Cα5, and Nα4 of UGT71G1 are closer to the acceptor binding region than the corresponding helices Nα3, Nα8, Nα7, Cα7, and Nα4 of UGT708C1 (Supplemental Figure 4A). The novel structural features of the acceptor binding pocket and the bent conformation of the acceptor are likely the two key features required for the C-glycosylation activity of UGT708C1.
To investigate the binding mode between phloretin and UGT708C1-UDP-glucose, we docked the phloretin into the binding site of UGT708C1-UDP-glucose. The docking poses were chosen as pose 1 and pose 2 (Figures 4B and 4C), with the lowest docking score from the top two clusters given by AutoDock. We then performed MD simulations to study the binding stability of phloretin in UGT708C1-UDP-glucose. MD simulations revealed that the position of phloretin-pose 1 changed slightly, with an RMSlig (which is the RMSD of the ligand with respect to its docking pose coordinates) of 1.78 Å relative to the docking pose, while the position of phloretin-pose 2 changed significantly, with an RMSlig of 2.87 Å relative to the docking pose. The binding free energy values for phloretin-pose 1 and -pose 2 binding to UGT708C1-UDP-glucose were −28.93 and −21.52 kcal/mol, respectively. These results indicate that the binding mode phloretin-pose 1 is more stable and more reasonable than phloretin-pose 2. Both the position and orientation of phloretin-pose 1 in the models from the molecular docking and MD calculations are consistent with those in the structural model of UGT708C1-phloretin built from the weak density map (Figure 4D; Supplemental Figure 4B), further confirming the reliability of the phloretin model.
We performed the parallel docking and MD simulations on the structure of VvGT1 complexed with kaempferol (VvGT1-kaempferol; PDB code 2C1Z) and the structure of UGT708C1 complexed with phloretin (UGT708C1-phloretin model). The docking modes with the lowest free energy of binding from the top cluster given by AutoDock were chosen as the best binding modes for kaempferol and phloretin. The conformations of kaempferol and phloretin were similar to the binding states of kaempferol in the crystal structures of VvGT1 (PDB code 2C1Z) and phloretin in UGT708C1 (UGT708C1-phloretin model; Supplemental Figures 4C and 4D). The root-mean-square deviation of kaempferol and phloretin (relative to the pose coordinates in the crystal structures) were 1.10 and 1.66 Å, respectively. These docking studies reconstructed the accurate binding mode for the protein-ligand complex. We then performed 100-ns MD simulations for VvGT1-kaempferol and UGT708C1-phloretin starting from the crystal structures. We calculated the binding free energy values using the last 10-ns trajectory of the MD simulations. The binding free energies were −29.49 and −28.44 kcal/mol for the VvGT1-kaempferol and UGT708C1-phloretin complexes, respectively. These values are similar to the binding free energy in the UGT708C1-UDP-glucose and phloretin complex (−28.93 kcal/mol).
To further explore the conformational changes in UGT708C1 caused by substrate binding, we compared the structure of apo-UGT780C1 with the structures of the UGT708C1-UDP complex, UGT708C1-UDP-glucose complex, and UGT708C1-phloretin complex models. Arg280 near the active site has two conformations: the side chain of Arg280 extends toward the acceptor binding pocket or rotates clockwise and forms a salt bridge with Asp96. In the structure of the UGT708C1-UDP-glucose complex structure, Arg280 forms a salt bridge with Asp96, while in the other three structures, the side chain of Arg280 extends toward the acceptor binding pocket (Figure 4E). Moreover, both Arg280 and Asp96 are highly conserved in CGTs that utilize 2-hydroxyflavanone as acceptors (Supplemental Figure 4E) but are not conserved in other plant UGTs (Supplemental Figure 3). This interaction might play an important role in maintaining the catalytic activity of CGTs.
In addition, the side chain of Trp340 is located toward the outside of the sugar donor binding site in the structures of apo-UGT708C1 and UGT708C1-phloretin. By contrast, in the structures of the UGT708C1-UDP-glucose and UGT708C1-UDP complexes, the side chain of Trp340 is rotated counterclockwise, forming a parallel π-stacking interaction with the uracil ring of the sugar donors and participating in the binding of sugar donors (Figure 4F).
Enzymatic Analysis of Mutants of UGT708C1
To investigate the catalytic characteristics of UGT708C1, we performed structure-based site-directed mutagenesis of this protein. As structural analysis revealed that Arg280 and Asp96 form the salt bridge, substitution of Arg280 or Asp96 with Ala led to the complete loss of catalytic activity (Figure 5A), demonstrating that Arg280 and Asp96 play important roles in the catalytic activity of UGT708C1.
Figure 5.

The Relative Catalytic Activity of UGT708C1 and Its Mutants, and Results of MD Simulations.
(A) The relative catalytic activity of wild-type UGT708C1 and its mutants. UDP-glucose is the sugar donor and phloretin is the sugar acceptor. The relative catalytic activities are the statistical results of three replicates, and the bars represent means ± se.
(B) Predicted binding modes for phloretin in the acceptor binding site of UGT708C1-UDP-glucose after 100-ns MD simulations (phloretin is shown as cyan sticks and UDP-glucose as gray sticks) of docking pose 1 (phloretin is shown as salmon sticks and UDP-glucose as gray sticks). All atoms are colored according to element (carbon, cyan or salmon; oxygen, red).
(C) Predicted binding modes for kaempferol in the acceptor binding site of UGT708C1-UDP-glucose after 100-ns MD simulations (kaempferol is shown as cyan sticks and UDP-glucose as gray sticks) of the docking pose (kaempferol is shown as salmon sticks and UDP-glucose as gray sticks). All atoms are colored according to element (carbon, cyan or salmon; oxygen, red).
To explore the recognition mechanism of sugar donors, we generated a series of mutations of the amino acids Asp382, Gln383, Thr150, and Thr151 near the binding site of the sugar moiety. When Asp382 was changed to Asn or Ala, the relative activity was completely lost. When Asp382 was substituted with Glu, the binding affinity (Kd) with UDP-glucose and the catalytic efficiency (kcat/Km) decreased dramatically (Tables 1 and 2). In addition, the catalytic activity of mutants T150A, T151A, and Q383A was reduced to 10, 40, and 20%, respectively, of the wild-type activity, and the catalytic efficiency of Q383H decreased by ∼14-fold (Table 1).
We also generated various mutations of amino acids at the UDP binding region. The relative catalytic activity of mutants W340A, H358A, N362A, S363A, E366A, and H380A decreased to less than 20% of wild-type values (Figure 5A), indicating that the parallel π-stacking interactions between Trp340 and the uracil ring, the hydrogen bond between Glu366 and the uracil ring, and the hydrogen bonds formed by residues His358, Asn362, and Ser363 and the α-phosphate and β-phosphate contribute significantly to the stable binding of sugar donors, while the hydrogen bonds between residues Val341 and Gln343 and the uracil ring have less of an effect on the stable binding of the sugar donor (Figure 5A).
To study the recognition mechanism of the acceptor, we also mutated some residues in the acceptor binding region. Substituting Tyr102 with Phe significantly reduced the catalytic efficiency for phloretin (Table 1), and substituting Tyr102 with Ala or Thr caused a loss of catalytic activity (Figure 5A). These results indicate that the hydrogen bonding interaction between Tyr102 and phloretin is important for acceptor binding and that the structural support from the aromatic ring of Tyr102 and the precise spatial disposition and complementarity between the acceptor binding pocket of the enzyme and acceptor substrates are essential for the C-glycosylation activity of UGT708C1 (Figure 4A). The Phe198 and Phe155 residues are involved in the formation of the hydrophobic pocket near phloretin (Figure 4A). Disrupting the hydrophobic environment near an acceptor can result in a reduction in catalytic activity: mutation of the hydrophobic residue Phe198 to Ala reduced the catalytic efficiency (Table 1), and the catalytic activity of mutant F155A was reduced to 50%, highlighting the importance of the hydrophobic pocket for acceptor binding (Figure 5A). Moreover, the binding affinity (Kd) of the F130A mutant for UDP-glucose decreased to ∼30% compared with wild-type UGT708C1, indirectly indicating that the leaving group of the donor could leave more easily. The binding affinity of F130A to phloretin was significantly higher than that of the wild type (Table 2), indicating that the mutation made it difficult for the glycosylation product to leave. Therefore, we speculate that the departure of the glycosylation product is the rate-limiting step of the reaction. However, more comprehensive studies are needed to verify this hypothesis.
DISCUSSION
Donor Specificity of UGT708C1
The last two amino acids in the PSPG motif (i.e., Asp382 and Gln383 in UGT708C1) are critical for sugar recognition and specificity (Shao et al., 2005). The acidic residue Asp382 forms hydrogen bonds with O3′ and O4′ atoms of the glucose moiety, and the D382A mutation disrupted the hydrogen bonds and resulted in a loss of catalytic activity. When Asp382 was changed to Glu with a longer side chain, the binding affinity (Kd) with UDP-glucose and the catalytic efficiency (kcat/Km) decreased (Tables 1 and 2), indicating that Asp382 plays an important role in the binding of the sugar moiety. The loss of activity of the D382N mutant confirms the importance of an acidic residue at this position (Figure 5A).
While UDP-galactose binds to the sugar donor binding pocket, the O4′ atom of the galactose moiety points in a direction that prevents it from forming a hydrogen bond with Asp382. However, the O3′ atom of UDP-galactose can still form a hydrogen bond with Asp382, and enzymatic analysis revealed that UGT708C1 exhibited weak activity toward UDP-α-d-galactose (Supplemental Figure 4F).
The last residue in the PSPG motif is considered to define the sugar donor specificity, such as Gln for glucose specificity (e.g., Gln382 in UGT71G1) and His for rhamnose specificity (e.g., His357 in UGT89C1). The mutation H357Q allowed UGT89C1 to use UDP-rhamnose and UDP-glucose as sugar donors; however, residues Pro147 and Ile148 of Arabidopsis (Arabidopsis thaliana) UGT89C1 are also essential for the recognition of rhamnose (Zong et al., 2019). Residues Thr150, Thr151, and Gln383 of UGT708C1 correspond to those of UGT89C1 and form hydrogen bonds with the glucose moiety of the donor molecule. The catalytic efficiency of UGT708C1 with the T150A mutation exhibited an approximately sevenfold decrease and that of the T151A mutation exhibited an ∼30-fold decrease (Table 1). The relative activity of UGT708C1 with the T151A mutation was dramatically lower than that of the wild type (Figure 5A), and the relative activity of this enzyme with the Q383A mutation decreased to 20%. The mutation of Gln383 to His resulted in reduced catalytic activity but did not give the enzyme the ability to transfer rhamnose. Multiple mutations of these three residues would be required to alter sugar specificity, requiring extensive studies.
Acceptor Specificity of UGT708C1
In the UGT708C1-UDP-glucose and phloretin complex, the interactions between phloretin and UGT708C1-UDP-glucose were stable during the 100-ns MD simulations (Figure 5B). The position of phloretin changed slightly, with an RMSlig value of 1.78 Å relative to the docking pose 1. When we used kaempferol for the docking and MD simulations, the conformation of kaempferol changed dramatically, with an RMSlig value of 5.49 Å, indicating that the binding mode of this molecule in the acceptor binding pocket of UGT708C1 was unstable (Figure 5C). Moreover, we calculated the binding free energies of phloretin and kaempferol toward UGT708C1-UDP-glucose based on the last 10-ns trajectory of the MD simulations. The binding free energy of phloretin toward UGT708C1-UDP-glucose was −28.93 kcal/mol, which was lower than that of kaempferol (−23.65 kcal/mol). These results indicate that kaempferol had a lower affinity toward UGT708C1-UDP-glucose than phloretin. In addition, kaempferol dissociated from the binding area, with an RMSlig value of 5.49 Å during the MD simulations. These results are consistent with the results of substrate screening, in which UGT708C1 could not catalyze the C-glycosylation of kaempferol.
UGT708C1 shows C-glycosylation activity toward 2-hydroxyflavanones, dihydrochalcone, trihydroxyacetophenones, and other related compounds with chemical structures similar to that of 2′,4′,6′-trihydroxyacetophenone (Figure 6A; Nagatomo et al., 2014). These active substrates share similar structural features: 2-hydroxyflavanones have 5,7-hydroxyl groups on their benzene ring A, and dihydrochalcone and 2′,4′,6′-trihydroxyacetophenone have three hydroxyls on their benzene ring. 2-Hydroxyflavanones lacking 5-hydroxyl groups, 7-hydroxyl groups, or both were not substrates for the CGT OsCGT (Brazier-Hicks et al., 2009). This observation indicates that the hydroxyl groups at the C5 and C7 positions of 2-hydroxyflavanones and the hydroxyl groups at the C2′, C4′, and C6′ positions of dihydrochalcone are essential for CGT activity.
Figure 6.

The Effective Substrates of UGT708C1 and Comparison of the Substrate Binding States of UGT708C1 and VvGT1.
(A) The core structures and detailed information about the active substrates of UGT708C1.
(B) The combination mode of phloretin in our proposed model UGT708C1-phloretin. The structure of UGT708C1 is shown as a gray surface, and phloretin is shown as green sticks. All atoms are colored according to element (carbon, green; oxygen, red).
(C) The combination mode of kaempferol in VvGT1. The structure of VvGT1 is displayed as a cyan surface, and kaempferol and UDP-2FGlc are shown as violet sticks. All atoms are colored according to element (carbon, violet; oxygen, red).
(D) Comparison of the models UGT708C1-phloretin and VvGT1. Phloretin and kaempferol bind to the binding pocket in different modes. The red dotted circle marks the area exhibiting an obvious difference between the two binding pockets.
In the crystal structural model of the UGT708C1 and phloretin complex and the model from the MD simulations, phloretin was bound in the inner hydrophobic pocket in a bent form with a precise spatial disposition and complementarity (Figure 6B). By contrast, in the structure of the OGT VvGT1-kaempferol complex, rigid kaempferol was located near the entrance of the pocket (Figure 6C; Offen et al., 2006). The acceptor binding pocket of UGT708C1 is shallower and smaller than that of VvGT1, and rigid kaempferol cannot stably bind in the pocket (Figure 6D). Therefore, the morphology of the binding pocket of UGT708C1 determines the binding mode of the acceptor. The open-chain form of 2-hydroxyflavanone is more flexible than the closed-chain form and facilitates binding to UGT708C1. This spatial morphology of the acceptor binding pocket supports the open-chain form of 2-hydroxyflavanone, which is consistent with previous reports (Nagatomo et al., 2014). In addition, the open-chain form of 2-hydroxyflavanone contains three phenolic hydroxyl groups on the A ring. These phenolic hydroxyl groups might enhance the nucleophilicity of the acceptor for forming C-glycosides in the para- and ortho-positions of the phenolic hydroxyl group.
To further elucidate the acceptor specificity of UGT708C1, we compared the structures of UGT708C1-phloretin and CGT TcCGT1 (PDB code 6JTD; He et al., 2019). TcCGT1 exhibits substrate promiscuity toward different types of flavonoids, including active substrate 2-hydroxyflavanones of UGT708C1, while UGT708C1 shows substrate specificity toward 2-hydroxyflavanones, dihydrochalcone, trihydroxyacetophenones, and other related compounds with chemical structures similar to that of 2′,4′,6′-trihydroxyacetophenone. Comparing the acceptor binding pockets of UGT708C1 and TcCGT1, three obvious differences were observed (Figure 7A). First, at the top of the acceptor binding pocket, UGT708C1 shows an open conformation, while the long loop-1 of TcCGT1 and the helices of TcCGT1, which correspond to positions Nα3 and Nα7 of UGT708C1, form a compact conformation (Figure 7A). Second, in the middle of the acceptor binding pocket, a narrow cavity forms in UGT708C1, while the helix of TcCGT1 corresponding to Nα5 of UGT708C1 is biased toward the outside of the binding pocket, forming a more spacious cavity (Figure 7B). Finally, inside the acceptor binding pocket, the short loop-2 of TcCGT1 limits the depth of the binding pocket (Figure 7C). Overall, the acceptor binding pocket of UGT708C1 appears to exhibit a curved “L” shape (Figure 7D), while the acceptor binding pocket of TcCGT1 appears as a horizontal “I” shape (Figure 7E). This difference in spatial morphology determines the substrate specificity and promiscuity of these two enzymes.
Figure 7.

Comparison of the Structures of UGT708C1-Phloretin and CGT TcCGT1.
(A) Overall structural comparison of UGT708C1-phloretin (gray cartoon) and TcCGT1 (salmon cartoon). Loop-1 and loop-2 in the structure of TcCGT1 are shown in cyan and indicated by dotted boxes. The positions of Nα3, Nα5, and Nα7 in the structure of UGT708C1 are labeled.
(B) The helix of TcCGT1 (salmon cartoon) corresponding to the Nα5 of UGT708C1 (gray cartoon) is significantly shifted toward the outside of the binding pocket.
(C) Residue Val123 on loop-2 in the structure of TcCGT1 is located in the position of phloretin in the structural model of UGT708C1-phloretin. Phloretin is shown as green sticks, and all atoms are colored according to element (carbon, green; oxygen, red).
(D) The UGT708C1 acceptor binding pocket is shaped like a curved “L.” Phloretin binds to the pocket in a bent state. Phloretin is shown as green sticks, and all atoms are colored according to element (carbon, green; oxygen, red).
(E) The TcCGT1 acceptor binding pocket is shaped like a horizontal “I.” The substrate binds to the spacious pocket.
Implications for the Catalytic Mechanism of C-Glycosylation
The His32 and Asp129 residues of UGT708C1 are highly conserved in plant UGTs and lie in the corresponding positions of proposed catalytic residues in other UGTs (e.g., His22 and Asp121 in UGT71G1; Supplemental Figure 3; Shao et al., 2005). The catalytic activity was completely lost in UGT708C1 with the H32A mutation, and that with the D129S mutation was reduced to 10% (Figure 5A), suggesting that residues His32 and Asp129 are the key catalytic residues for UGT708C1. These two residues and acceptors form an “acceptor-His-Asp” triad, as observed in other plants UGTs (Wang, 2009; Ito et al., 2017; Rahimi et al., 2019), and the transfer of electrons is mediated by a water molecule, which is located between His32 and Asp129.
Moreover, mutagenesis studies revealed the complete loss of catalytic activity of the R280A and D96A mutants, suggesting that Arg280 might participate in the catalytic reaction via the swing of its long side chain. When a salt bridge forms between Arg280 and Asp96, it is essential for maintaining the stability of the active center of UGT708C1. When the side chain of Arg280 swings toward the acceptor binding pocket, Arg280 might enhance the release of the UDP moiety of sugar donors in a manner similar to that of Arg292 in UGT708D1 reported previously (Hirade et al., 2015).
Based on the studies described above, we propose that, like OGTs, the CGT UGT708C1 also utilizes the SN2-like catalytic mechanism (Figure 8). The acidic amino acid Asp129 helps the general base His32 deprotonate the phenolic hydroxyl group of the acceptor through conjugation effects, allowing the ortho-aromatic carbon to undergo nucleophilic attack to the anomeric carbon of the sugar donor and produce the C-glycoside products. The presence of three phenolic hydroxyl groups enhances the nucleophilicity of the aromatic carbon, and Arg280 likely enhances product release.
Figure 8.
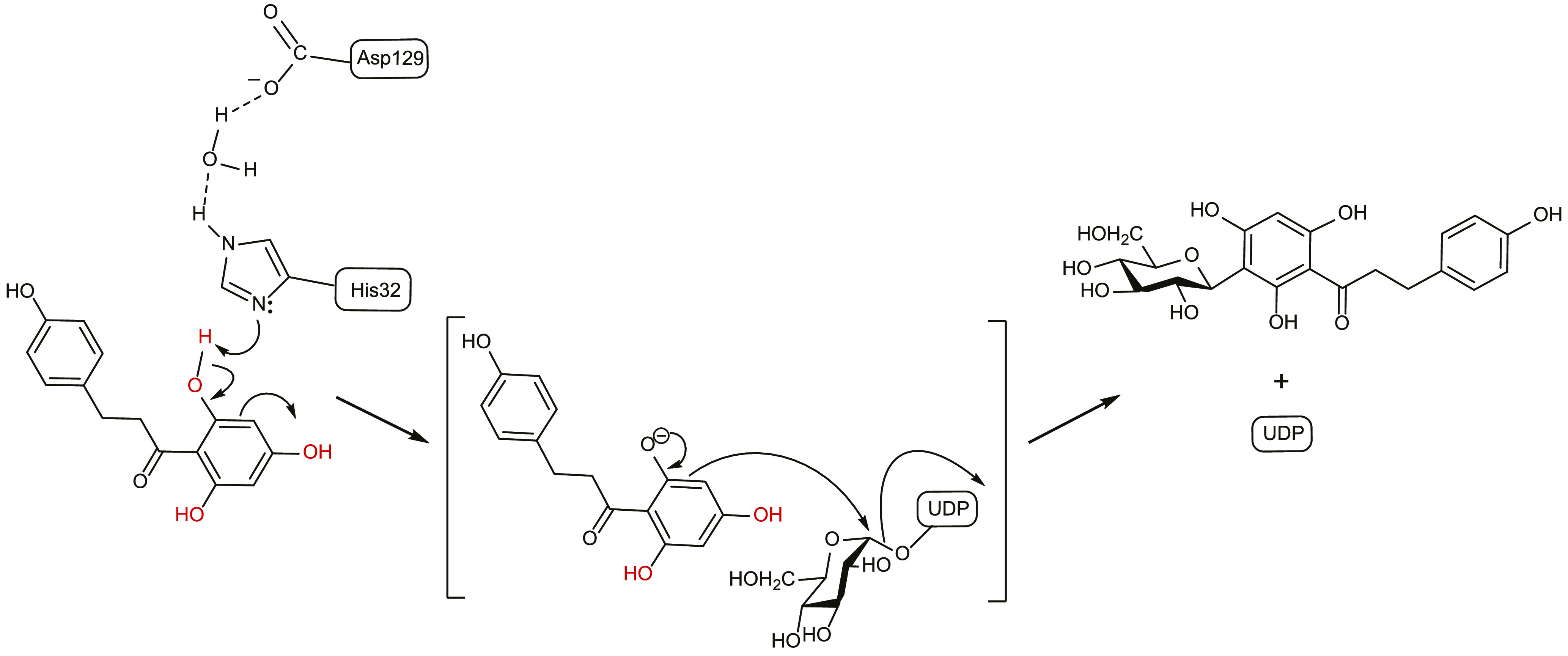
Proposed Catalytic Mechanism of UGT708C1.
The C-glycosylation reaction catalyzed by UGT708C1 is an SN2-like nucleophilic displacement. The three phenolic hydroxyl groups of the substrate are necessary for this reaction, and His and Asp are the key catalytic residues.
Our studies revealed the molecular and structural basis for understanding the catalytic mechanism of the CGT UGT708C1 at the molecular level, providing important insights into the formation of C-C-glycosidic bonds and the biosynthesis of C-glycosides. UGT708C1 is specifically expressed in buckwheat cotyledons (Nagatomo et al., 2014), suggesting that it might play an important role in regulating germination and cotyledon development (Zhang et al., 2016) and in plant defense, since many C-glycosides have antifungal activity by function as phytoalexins (McNally et al., 2003; Poppenberger et al., 2003; Subramanian et al., 2005). Our studies of the structure and mechanism of the CGT UGT708C1 provide a basis for enzyme engineering to regulate plant development and defense and improve plant health. Our findings could also facilitate the design of novel CGT biocatalysts to produce C-glycosides of phloretin, flavonoids, and other bioactive C-glycosides with health benefits for both plants and humans.
METHODS
Expression and Purification of UGT708C1 and Mutants
The full-length UGT708C1 gene from buckwheat (Fagopyrum esculentum) was cloned into the pET28a vector with an N-terminal hexa-His tag and a thrombin cleavage site. Escherichia coli BL21(DE3) CodonPlus cells were transformed with the plasmid and grown at 37°C in Luria-Bertani medium containing 30 µg/mL kanamycin until A600 = 0.6 to 0.8. The cultures were induced with 0.15 mM isopropyl 1-thio-β-galactopyranoside and grown for ∼16 to 18 h at 16°C. The cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, and 500 mM NaCl) supplemented with protease inhibitor (1 mM PMSF). The resuspended cells were subjected to ultrasonic decomposition on ice, and the lysate was centrifugation at 12,000 rpm at 4°C for 40 min. The supernatant containing the target proteins supplemented with 10 mM imidazole was incubated with Ni-NTA agarose for 2 h at 4°C. Following incubation, the resin was loaded onto a chromatographic column and washed with 4 column volumes of wash buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 20 mM imidazole) followed by 2 column volumes of wash buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 50 mM imidazole). The proteins were eluted from the affinity resin with 4 volumes of elution buffer C (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 250 mM imidazole), and the flow-through was collected and concentrated by centrifugation in ultrafiltration tubes at 4°C.
The concentrated proteins were subjected to anion-exchange chromatography on a Resource Q column equilibrated with Q buffer A (50 mM Tris-HCl, pH 8.0, 40 mM NaCl, and 2 mM DTT). The target protein was eluted with Q buffer B (50 mM Tris-HCl, pH 8.0, 1 M NaCl, and 2 mM DTT) using a 14% gradient, and fractions were concentrated for gel filtration. The gel filtration experiments were performed on a Superdex 200 Increase (10/300 GL) preparation grade column connected to an AKTA-purifier fast protein liquid chromatography system (GE Healthcare) and equilibrated with buffer E (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 2 mM DTT). The purified proteins were analyzed by SDS-PAGE.
Site-directed mutagenesis of UGT708C1 was performed by PCR with primers designed to generate the desired mutations (Supplemental Data Set). The wild-type UGT708C1-pET28a plasmid was used as the PCR template. PCR was performed using Fastpfu DNA polymerase (TransGen Biotech) with an extension temperature of 72°C and extension time of 4 min, followed by digestion of the unmutated template with the restriction enzyme DpnI and transformation into T1 Competent Cell. The mutants were confirmed by sequencing. The mutant proteins were purified with the same method used for the wild-type enzyme.
Substrate Screening of UGT708C1
Substrate screening of UGT708C1 was performed in a reaction mixture (200 μL) consisting of 100 μM different acceptor, 1 mM UDP-glucose, 100 mM Tris-HCl (pH 8.0), 1 mM DTT, and 2 μg of purified UGT708C1. The reaction was incubated at 30°C for 30 min and stopped by the addition of a final concentration of 1 M HCl. The reaction mixture was centrifugation at 14,000 rpm at 4°C for 10 min, and the supernatant was analyzed by HPLC (Agilent 1260 Infinity with a diode array detector). The sample (40 μL) was applied to a ZORBAX SB-Aq column (4.6 × 150 mm, 3.5 μm; Agilent) at a flow rate of 1 mL/min with solvent A (deionized water containing 0.1% [v/v] formic acid) and solvent B (methanol containing 0.1% [v/v] formic acid). The gradients of solvent B were 5% (v/v; 0 to 5 min), 60% (v/v; 5 to 17 min), 60% (v/v; 17 to 25 min), 5% (v/v; 25 to 26 min), and 5% (26 to 30 min). The reaction product was further confirmed by LC-QTOF-electrospray ionization/MS (ACQUITY UPLC I-Class/UPCC/M-Class/SYNAPT G2-SI; Waters).
Enzyme Kinetics Assay
The enzyme kinetic assay of UGT708C1 was performed in a reaction mixture (200 μL) with UDP-glucose or UDP-galactose as the sugar donor and phloretin or 2′,4′,6′-trihydroxyacetophenone as the sugar acceptor. The enzyme kinetic parameters of the mutants were measured with UDP-glucose and phloretin as the sugar donor and acceptor, respectively. Reactions were performed at 30°C for 10 min, and the other experimental conditions and procedures were the same as those used for substrate screening of UGT708C1. The ratio of product to substrate was determined based on the HPLC peak areas of the product and the remaining substrate, and the amount of product was obtained by multiplying the ratio by the initial amount of substrate. The formula used to derive the kinetic parameters was the Michaelis-Menten equation:
where [S] is the concentration of the substrate, Vmax is the maximum enzyme rate, and Km is the substrate concentration needed to achieve a half-maximum enzyme rate. The initial rates (v0) of a reaction were measured at saturating concentrations of one substrate and varying concentrations of the other substrate. The concentrations of the sugar acceptor ranged from 2 to 200 μM, and the concentrations of the sugar donor ranged from 8 μM to 2 mM. We used the Michaelis-Menten equation in the GraphPad Prism 5.0 software package to calculate the Km and Vmax values.
Microscale Thermophoresis Assay
The purified wild-type and mutant UGT708C1 proteins were exchanged into PBS buffer for the microscale thermophoresis (MST) experiments, and the proteins were diluted to a final concentration of 11 µM using PBS buffer. A total of 100 μL of 23.5 µM NT-647-NHS-ester dye solution was added to a 100-μL aliquot of 11 µM protein solution. The protein/dye mixture was mixed well and incubated at room temperature in the dark for 30 min. The excess unbound dye was removed using a gel filtration column (Sephadex G25; GE Healthcare), which was equilibrated with test buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, and 0.05% Tween 20). The labeled protein was eluted and collected using 600 μL of test buffer. The binding affinity between substrates and proteins was analyzed on a Monolith NT.115 instrument (Nanotemper Technologies). The labeled protein was diluted fivefold with test buffer. Serial dilutions of phloretin were prepared in test buffer containing 2% DMSO (the stock concentration was 25 mM in 100% DMSO). Serial dilutions of 2′,4′,6′-trihydroxyacetophenone were prepared in test buffer containing 2% DMSO (the stock concentration was 400 mM in 100% DMSO). Serial dilutions of UDP-glucose and UDP-galactose were prepared in deionized water (the stock concentration was 20 mM in deionized water). Various concentrations of substrate solutions were mixed at a 1:1 ratio with protein solution in a final volume of 20 µL. The reaction mixture was loaded into standard treated capillaries (Monolith NT.115 series capillary MO-K022) and analyzed by MST at medium MST power and auto-detect excitation power with a laser-on time of 1.5 s and a detection temperature of 25°C. The Kd values were calculated with MO.Affinity Analysis Software from three independent thermophoresis measurements. The Kd fit model describes a molecular interaction with a 1:1 stoichiometry according to the law of mass action. The Kd was estimated by fitting the equation:
where f(c) is the fraction bound at a given ligand concentration c; Unbound is the Fnorm signal of the target alone; Bound is the Fnorm signal of the complex; Fnorm refers to the normalized fluorescence; Kd is the dissociation constant or binding affinity; and ctarget is the final concentration of target in the assay.
Crystallization and Data Collection
Purified UGT708C1 was concentrated to 7 mg/mL and crystallized by the sitting-drop vapor diffusion method at 16°C at a 1:1 ratio in well solution, which comprised 0.2 M ammonium sulfate, 0.1 M MES, pH 7.0, 26.5% (w/v) polyethylene glycol 5000, and 1% (w/v) benzamidine hydrochloride. The crystals were harvested into nylon-fiber Mounted CryoLoops (Hampton Research) and cryoprotected in well solution supplemented with 30% (v/v) ethylene glycol. The data were collected to 2.1 Å resolution at beamline BL19U1 (Shanghai Synchrotron Radiation Facility) under cryogenic conditions at 100 K.
Crystals of UGT708C1 complexed with UDP-glucose and UDP were also obtained using the methods and conditions for growing crystals described above, except that 1.3% (w/v) benzamidine hydrochloride and 5 mM UDP-glucose and UDP, respectively, were placed in the well solution. The crystals were cooled as described previously, using cryoprotectant solution comprising well solution with 35% ethylene glycol. The data were collected at beamline BL19U1 (Shanghai Synchrotron Radiation Facility) to 2.0 and 2.25 Å resolution, respectively, under cryogenic conditions at 100 K. All data sets were processed using HKL2000 software (Otwinowski and Minor, 1997).
Structure Determination and Refinement
The structure of UGT708C1 was resolved by the molecular replacement method using the program BALBES (Long et al., 2008) using the UGT71G1 structure (Shao et al., 2005) as the search model. Model building was performed using COOT (Emsley and Cowtan, 2004), and structure refinement was performed with the PHENIX program (Adams et al., 2002). The orientations of the amino acid side chains and bound water molecules were modeled based on 2Fobs-Fcalc and Fobs-Fcalc difference Fourier maps. Detailed data collection and refinement statistics are presented in the Supplemental Table.
Molecular Docking and MD Simulations
Molecular docking studies were performed with the AutoDock4 program (Morris et al., 2009). We docked phloretin and kaempherol into the binding pocket of UGT708C1 and chose the docking mode with the lowest free energy of binding from the top cluster given by AutoDock4 (Morris et al., 2009) as the best docking mode to obtain the phloretin-UGT708C1-UDP-glucose and kaempherol-UGT708C1-UDP-glucose complexes. Two systems based on the complexes were subsequently built for MD simulations. Using the LEAP plugin in AMBER18 (Case et al., 2018), protein residues were set to the standard protonation states at the appropriate protonation states, which were calculated with the H++ program (Anandakrishnan et al., 2012). Each complex structure was solvated by TIP3P water with sodium ions neutralized. Each system included ∼50,200 atoms. The dimensions of each system were 80 Å × 95 Å × 80 Å.
MD simulations were performed using the PMEMD module of AMBER18 (Case et al., 2018). The AMBER FF14SB force field (Maier et al., 2015) was used for UGT708C1. We generated prmtop and inpcrd files for UDP-glucose, phloretin, and kaempherol using general amber force field (Wang et al., 2004) with the antechamber module of AMBER18 (Case et al., 2018). First, each system was minimized for 10,000 steps. Second, the thermalization of heating each system from 0 to 310 K was performed in 500 ps using a Langevin thermostat (Pastor et al., 1988), followed by a 1-ns equilibration using NVT ensemble. The protein and ligands were fixed with the constraint of 50 kcal mol−1 Å−2. Third, the systems were equilibrated for 2 ns using NPT ensemble, and the protein and ligands were constrained with the position restraints gradually reduced from 10 (1 ns) to 1 kcal mol−1 Å−2 (1 ns). Finally, we performed a 100-ns MD simulation for each system in a constant pressure (NPT) with no constraints. A 12 Å cutoff was set for the nonbonded interaction. The SHAKE (Ryckaert et al., 1977) algorithm integration was used to constrain the covalent bonds involving hydrogen atoms, and the particle mesh Ewald (Darden et al., 1993) algorithm was applied to treat long-range electrostatic interactions. The time step was set to 2 fs during all MD simulations. The frames were saved every 5000 steps for analysis. The trajectory were analyzed with the VMD (Humphrey et al., 1996) and CPPTRAJ tools in AMBER18 (Case et al., 2018). Binding free energies between the compounds and protein were calculated with the MM-PBSA module in AMBER18, and the Poisson-Boltzmann surface area method (Kuhn et al., 2005) was used.
Accession Numbers
Sequence data from this article can be found the UniProt KB database under the following accession numbers: A0A0A1HA03, Q5IFH7, P51094, A4F1R4, A6XNC5, Q9LNE6, Q7XT97, A0A0A1H7N4, A0A096SRM5, I1L3T1, A0A224AM54, A0A224AKZ9, and C3W7B0 for UGT708C1, MtUGT71G1, VvGT1, CtUGT78K6, MtUGT85H2, AtUGT89C1, Os79, UGT708C2, UGT708A6, UGT708D1, UGT708G1, UGT708G2, and OsCGT, respectively. The structures of apo-UGT708C1, UGT708C1-UDP, and UGT708C1-UDP-glucose have been deposited in the PDB under accession numbers 6LLG, 6LLW, and 6LLZ, respectively. The PDB codes of the UGT89C1, VvGT1, TcCGT1, and UGT71G1 structures used in this work are 6IJD, 2C1Z, 6JTD, and 2ACV, respectively.
Supplemental Data
Supplemental Figure 1. Size exclusion chromatography profile and SDS-PAGE of UGT708C1.
Supplemental Figure 2. The structures of substrates used in substrate screening in this study.
Supplemental Figure 3. Sequence alignment of UGT708C1 and other plant UGTs.
Supplemental Figure 4. Comparison of UGT708C1 and other UGTs.
Supplemental Table. Data collection and refinement statistics of UGT708C1 and its complexes.
Supplemental Data Set. PCR primers used in this study.
DIVE Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We thank W. Meng for technical assistance and the staffs of beamlines BL19U1 and BL18U1 at the Shanghai Synchrotron Radiation Facility and beamline BL-17A at Photon Factory (Tsukuba, Japan) for excellent technical assistance during the data collection. This work was supported by the National Key R&D Program of China (grants 2018YFA0901800 and 2017YFA0504801).
AUTHOR CONTRIBUTIONS
M.Z.L. and X.Q.W. designed the research; M.Z.L., D.D.W., X.M.L., and S.F. performed research; Y.L. and J.P.L. performed research and contributed computational tools; M.Z.L., X.Y., and G.N.Z. analyzed data; M.Z.L. wrote the article; X.Q.W. and Y.Q.S. reviewed and edited the article; X.Q.W., and Y.Q.S. acquired the funding for this study.
References
- Abbasi E., Nassiri-Asl M., Shafeei M., Sheikhi M.(2012). Neuroprotective effects of vitexin, a flavonoid, on pentylenetetrazole-induced seizure in rats. Chem. Biol. Drug Des. 80: 274–278. [DOI] [PubMed] [Google Scholar]
- Adams P.D., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Read R.J., Sacchettini J.C., Sauter N.K., Terwilliger T.C.(2002). PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58: 1948–1954. [DOI] [PubMed] [Google Scholar]
- Agnese A.M., Pérez C., Cabrera J.L.(2001). Adesmia aegiceras: Antimicrobial activity and chemical study. Phytomedicine 8: 389–394. [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R., Aguilar B., Onufriev A.V.(2012). H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40: W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bililign T., Hyun C.G., Williams J.S., Czisny A.M., Thorson J.S.(2004). The hedamycin locus implicates a novel aromatic PKS priming mechanism. Chem. Biol. 11: 959–969. [DOI] [PubMed] [Google Scholar]
- Brazier-Hicks M., Evans K.M., Gershater M.C., Puschmann H., Steel P.G., Edwards R.(2009). The C-glycosylation of flavonoids in cereals. J. Biol. Chem. 284: 17926–17934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D., Li H., Yi J., Zhang J., Che H., Cao J., Yang L., Zhu C., Jiang W.(2011). Antioxidant properties of the mung bean flavonoids on alleviating heat stress. PLoS One 6: e21071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D.A., et al. (2018). AMBER 2018.. (San Francisco: University of California; ). [Google Scholar]
- Cornell H.V., Hawkins B.A.(2003). Herbivore responses to plant secondary compounds: A test of phytochemical coevolution theory. Am. Nat. 161: 507–522. [DOI] [PubMed] [Google Scholar]
- Darden T., York D., Pedersen L.(1993). Particle mesh Ewald: An Nlog (N) method for Ewald sums in large systems. J. Chem. Phys. 98: 10089–10092. [Google Scholar]
- Dürr C., Hoffmeister D., Wohlert S.E., Ichinose K., Weber M., Von Mulert U., Thorson J.S., Bechthold A.(2004). The glycosyltransferase UrdGT2 catalyzes both C- and O-glycosidic sugar transfers. Angew. Chem. Int. Ed. Engl. 43: 2962–2965. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K.(2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Giddings L.A., Newman D.J.(2013). Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 40: 1181–1210. [DOI] [PubMed] [Google Scholar]
- Gorzalczany S., Marrassini C., Miño J., Acevedo C., Ferraro G.(2011). Antinociceptive activity of ethanolic extract and isolated compounds of Urtica circularis. J. Ethnopharmacol. 134: 733–738. [DOI] [PubMed] [Google Scholar]
- Gutmann A., Nidetzky B.(2012). Switching between O- and C-glycosyltransferase through exchange of active-site motifs. Angew. Chem. Int. Ed. Engl. 51: 12879–12883. [DOI] [PubMed] [Google Scholar]
- Hayashi M., Nakayama S.-z., Kawabata H.(2000). HF-pyridine promoted Friedel-Crafts type arylation of 2-acetoxy-d-glucal: Stereoselective synthesis of 1-arylhex-3-enopyranosiduloses. Chem. Commun. 1329–1330. [Google Scholar]
- He J.B., Zhao P., Hu Z.M., Liu S., Kuang Y., Zhang M., Li B., Yun C.H., Qiao X., Ye M.(2019). Molecular and structural characterization of a promiscuous C-glycosyltransferase from Trollius chinensis. Angew. Chem. Int. Ed. Engl. 58: 11513–11520. [DOI] [PubMed] [Google Scholar]
- Hirade Y., Kotoku N., Terasaka K., Saijo-Hamano Y., Fukumoto A., Mizukami H.(2015). Identification and functional analysis of 2-hydroxyflavanone C-glucosyltransferase in soybean (Glycine max). FEBS Lett. 589: 1778–1786. [DOI] [PubMed] [Google Scholar]
- Hosoya T., Ohashi Y., Matsumoto T., Suzuki K.(1996). On the stereochemistry of aryl C-glycosides: Unusual behavior of bis-TBDPS protected aryl C-olivosides. Tetrahedron Lett. 37: 663–666. [Google Scholar]
- Humphrey W., Dalke A., Schulten K.(1996). VMD: Visual molecular dynamics. J. Mol. Graph. 14: 33–38. [DOI] [PubMed] [Google Scholar]
- Ilmberger N., Rabausch U.(2017). Screening glycosyltransferases for polyphenol modifications. Methods Mol. Biol. 1539: 229–236. [DOI] [PubMed] [Google Scholar]
- Ito T., Fujimoto S., Suito F., Shimosaka M., Taguchi G.(2017). C-Glycosyltransferases catalyzing the formation of di-C-glucosyl flavonoids in citrus plants. Plant J. 91: 187–198. [DOI] [PubMed] [Google Scholar]
- Jones P., Vogt T.(2001). Glycosyltransferases in secondary plant metabolism: Tranquilizers and stimulant controllers. Planta 213: 164–174. [DOI] [PubMed] [Google Scholar]
- Katz L., Baltz R.H.(2016). Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 43: 155–176. [DOI] [PubMed] [Google Scholar]
- Kuhn B., Gerber P., Schulz-Gasch T., Stahl M.(2005). Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem. 48: 4040–4048. [DOI] [PubMed] [Google Scholar]
- Lairson L.L., Henrissat B., Davies G.J., Withers S.G.(2008). Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 77: 521–555. [DOI] [PubMed] [Google Scholar]
- Li R.(2016). Natural product-based drug discovery. Med. Res. Rev. 36: 3. [DOI] [PubMed] [Google Scholar]
- Liang D.M., Liu J.H., Wu H., Wang B.B., Zhu H.J., Qiao J.J.(2015). Glycosyltransferases: Mechanisms and applications in natural product development. Chem. Soc. Rev. 44: 8350–8374. [DOI] [PubMed] [Google Scholar]
- Long F., Vagin A.A., Young P., Murshudov G.N.(2008). BALBES: A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan M., Ahuja P.S., Yadav S.K.(2011). Post-transcriptional silencing of flavonol synthase mRNA in tobacco leads to fruits with arrested seed set. PLoS One 6: e28315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.(2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11: 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T., Katsuki M., Suzuki K.(1988). New approach to C-aryl glycosides starting from phenol and glycosyl fluoride: Lewis acid-catalyzed rearrangement of O-glycoside to C-glycoside. Tetrahedron Lett. 29: 6935–6938. [Google Scholar]
- Matsumoto T., Katsuki M., Suzuki K.(1989). Cp2ZrCl2-AgClO4: Efficient promoter for the Friedel-Crafts approach to C-aryl glycosides. Tetrahedron Lett. 30: 833–836. [Google Scholar]
- McNally D.J., Wurms K.V., Labbé C., Quideau S., Bélanger R.R.(2003). Complex C-glycosyl flavonoid phytoalexins from Cucumis sativus. J. Nat. Prod. 66: 1280–1283. [DOI] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J.(2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Antoli C., Cortés A., Sotillo J., Fried B., Esteban J.G., Toledo R.(2014). Differential expression and glycosylation of proteins in the rat ileal epithelium in response to Echinostoma caproni infection. J. Proteomics 101: 169–178. [DOI] [PubMed] [Google Scholar]
- Nagatomo Y., Usui S., Ito T., Kato A., Shimosaka M., Taguchi G.(2014). Purification, molecular cloning and functional characterization of flavonoid C-glucosyltransferases from Fagopyrum esculentum M. (buckwheat) cotyledon. Plant J. 80: 437–448. [DOI] [PubMed] [Google Scholar]
- Newman D.J., Cragg G.M.(2012). Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75: 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offen W., Martinez-Fleites C., Yang M., Kiat-Lim E., Davis B.G., Tarling C.A., Ford C.M., Bowles D.J., Davies G.J.(2006). Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 25: 1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara Y., Katayama K., Minami A., Otsuka M., Eguchi T., Kakinuma K.(2004). Cloning, sequencing, and functional analysis of the biosynthetic gene cluster of macrolactam antibiotic vicenistatin in Streptomyces halstedii. Chem. Biol. 11: 79–86. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W.(1997). Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Paradkar A.(2013). Clavulanic acid production by Streptomyces clavuligerus: Biogenesis, regulation and strain improvement. J. Antibiot. (Tokyo) 66: 411–420. [DOI] [PubMed] [Google Scholar]
- Pastor R.W., Brooks B.R., Szabo A.(1988). An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 65: 1409–1419. [Google Scholar]
- Peng M., et al. (2017). Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 8: 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppenberger B., Berthiller F., Lucyshyn D., Sieberer T., Schuhmacher R., Krska R., Kuchler K., Glössl J., Luschnig C., Adam G.(2003). Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 278: 47905–47914. [DOI] [PubMed] [Google Scholar]
- Rahimi S., Kim J., Mijakovic I., Jung K.H., Choi G., Kim S.C., Kim Y.J.(2019). Triterpenoid-biosynthetic UDP-glycosyltransferases from plants. Biotechnol. Adv. 37: 107394. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P., Ciccotti G., Berendsen H.J.C.(1977). Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of N-alkanes. J. Comput. Phys. 23: 327–341. [Google Scholar]
- Sasaki N., Nishizaki Y., Yamada E., Tatsuzawa F., Nakatsuka T., Takahashi H., Nishihara M.(2015). Identification of the glucosyltransferase that mediates direct flavone C-glucosylation in Gentiana triflora. FEBS Lett. 589: 182–187. [DOI] [PubMed] [Google Scholar]
- Shao H., He X., Achnine L., Blount J.W., Dixon R.A., Wang X.(2005). Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell 17: 3141–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S., Graham M.Y., Yu O., Graham T.L.(2005). RNA interference of soybean isoflavone synthase genes leads to silencing in tissues distal to the transformation site and to enhanced susceptibility to Phytophthora sojae. Plant Physiol. 137: 1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Staaij J., de Bakker N.V., Oosthoek A., Broekman R., van Beem A., Stroetenga M., Aerts R., Rozema J.(2002). Flavonoid concentrations in three grass species and a sedge grown in the field and under controlled environment conditions in response to enhanced UV-B radiation. J. Photochem. Photobiol. B 66: 21–29. [DOI] [PubMed] [Google Scholar]
- Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A.(2004). Development and testing of a general amber force field. J. Comput. Chem. 25: 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wang X.(2009). Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 583: 3303–3309. [DOI] [PubMed] [Google Scholar]
- Wang X., Li C., Zhou C., Li J., Zhang Y.(2017). Molecular characterization of the C-glucosylation for puerarin biosynthesis in Pueraria lobata. Plant J. 90: 535–546. [DOI] [PubMed] [Google Scholar]
- Yang Y., Yu B.(2017). Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 117: 12281–12356. [DOI] [PubMed] [Google Scholar]
- Zhang G.Z., Jin S.H., Jiang X.Y., Dong R.R., Li P., Li Y.J., Hou B.K.(2016). Ectopic expression of UGT75D1, a glycosyltransferase preferring indole-3-butyric acid, modulates cotyledon development and stress tolerance in seed germination of Arabidopsis thaliana. Plant Mol. Biol. 90: 77–93. [DOI] [PubMed] [Google Scholar]
- Zong G., Fei S., Liu X., Li J., Gao Y., Yang X., Wang X., Shen Y.(2019). Crystal structures of rhamnosyltransferase UGT89C1 from Arabidopsis thaliana reveal the molecular basis of sugar donor specificity for UDP-β-l-rhamnose and rhamnosylation mechanism. Plant J. 99: 257–269. [DOI] [PubMed] [Google Scholar]