

Abstract
Adeno-associated virus (AAV) vectors are a leading platform for gene-based therapies for both monogenic and complex acquired disorders. The success of AAV gene transfer highlights the need to answer outstanding clinical questions of safety, durability, and the nature of the human immune response to AAV vectors. Here, we present longitudinal follow-up data of subjects who participated in the first trial of a systemically delivered AAV vector. Adult males (n = 7) with severe hemophilia B received an AAV2 vector at doses ranging from 8 × 1010 to 2 × 1012 vg/kg to target hepatocyte-specific expression of coagulation factor IX; a subset (n = 4) was followed for 12–15 years post-vector administration. No major safety concerns were observed. There was no evidence of sustained hepatic toxicity or development of hepatocellular carcinoma as assessed by liver transaminase values, serum -fetoprotein, and liver ultrasound. Subjects demonstrated persistent, increased AAV neutralizing antibodies (NAbs) to the infused AAV serotype 2 (AAV2) as well as all other AAV serotypes tested (AAV5 and AAV8) for the duration of follow-up. These data represent the longest available longitudinal follow-up data of subjects who received intravascular AAV and support the preliminary safety of intravascular AAV administration at the doses tested in adults. Data demonstrate, for the first time, the persistence of high-titer, multi-serotype cross-reactive AAV NAbs for up to 15 years post- AAV vector administration. Our observations are broadly applicable to the development of AAV-mediated gene therapy.
Keywords: AAV, adeno-associated virus, neutralizing antibody, hemophilia, clinical gene therapy, gene therapy
Graphical Abstract
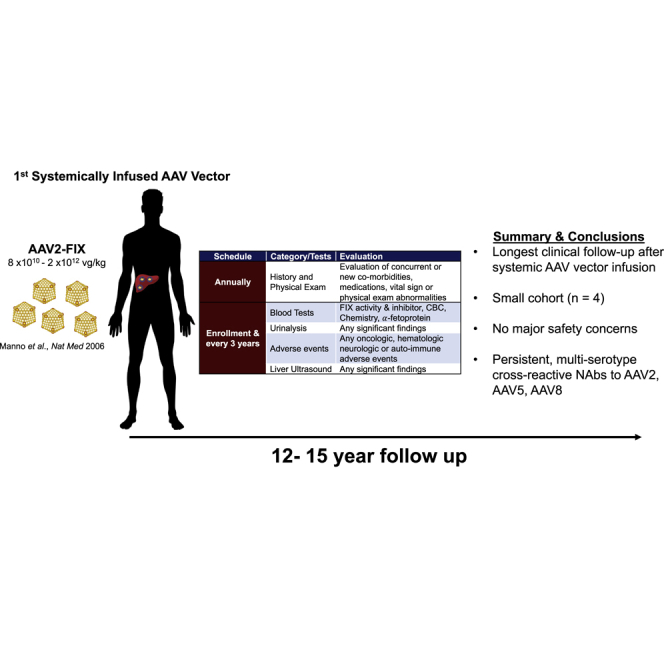
Presented here are the longest available longitudinal follow-ups of human subjects following intravascular adeno-associated virus (AAV) vector administration. These data support the preliminary long-term safety of systemic AAV delivery and demonstrate the persistence of high-titer, multi-serotype, cross-reactive AAV NAbs for up to 15 years after vector infusion.
Introduction
Recombinant adeno-associated virus (AAV)-mediated gene therapy has emerged as a promising treatment for a variety of inherited and acquired disorders.1,2 Recent achievements include the regulatory approval of 2 AAV-based therapeutics and an increasing number of clinical trials with encouraging results.3,4 However, these successes also highlight a number of unanswered questions with this approach, including: (1) AAV long-term safety, including the risk of tumorigenesis, and (2) the persistence of the humoral immune response to AAV, neutralizing antibodies (NAbs). To help address these two issues, we report the long-term follow-up of the first subjects to receive intravascular delivery of an AAV vector. Hemophilia B subjects were enrolled from 2001 to 2004 and received an AAV serotype 2 (AAV2) vector at doses of 8 × 1010 to 2 × 1012 vg/kg for hepatocyte-directed coagulation factor IX (FIX) gene transfer.5
Since our dosing study was initiated, nearly 2 decades of clinical trial investigation have refined and expanded intravascular AAV gene therapy.1,2,6 For F8 or F9 transfer for hemophilia, there are now 3 ongoing pivotal trials and an approximate log-fold more early-phase clinical trials.7, 8, 9, 10 In the broader context, successes in the clinical development of gene transfer for hemophilia have direct applicability to an estimated >400 genetic disorders that could potentially be targeted by liver-directed AAV gene therapy.11 In addition to targeting the liver, AAV vectors can efficiently transduce multiple tissue types in vivo, including cardiac muscle, skeletal muscle, the retina, and the central nervous system (CNS). Thus far, a single intravascularly administered AAV product, Zolgensma (AveXis, Bannockburn, IL, USA), is approved for infants with spinal muscular atrophy. Nearly all current clinical efforts in in vivo gene transfer or gene editing rely on AAV vectors to deliver a genetic payload.2,6
Recombinant AAV-vector-transduced cells maintain donor DNA predominantly as a stable episome. While AAV is nonpathogenic and predominantly non-integrating, direct sequencing data from animals and humans have demonstrated AAV integration events after vector administration or natural infection.12, 13, 14, 15, 16 Available data suggest that integration events occur in a dose-dependent manner, preferentially at sites of active transcription.12,13,17 Experiments in mice suggest that the use of tissue-specific promoters (as opposed to housekeeping promoters) reduces the risk of tumor formation as a consequence of integration.13 Thus, although AAV vectors have a low rate of genomic integration, genotoxicity is an identified theoretical safety risk with, as yet, no available long-term longititudinal clinical data to support or refute this concern.
In addition to the dearth of long-term safety data, there are limited clinical trial data on the duration of AAV NAb persistence following vector infusion. Pre-clinical study and the majority of available clinical trial data support that pre-existing high-titer AAV NAbs prevent successful transduction following systemic AAV delivery.5,7,18,19 Concurrently, published clinical trial data report that patients universally develop high-titer AAV NAbs after systemic AAV vector administration, though persistence and cross-reactivity of these AAV NAbs remain undefined.3,5,7,18 If AAV NAbs persist indefinitely, available data suggest that patients will likely only be able to receive a single administration of an AAV gene therapy; however, if NAbs wane, vector re-administration, with current practices, may be feasible. Thus, it is critical to understand the long-term persistence of AAV NAbs to assess their therapeutic implications.
Here, we present 12–15 years of longitudinal follow-up data in the first cohort of patients to receive intravascular administration of an AAV vector. As previously reported, expression from the vector was subtherapeutic or limited in duration due to a cellular immune response to the AAV capsid, which prevented analysis of expression durability and long-term efficacy.5,20 However, this cohort provides the longest longitudinal safety data after systemic AAV administration.5 Additionally, we demonstrate, for the first time, the persistence of high-titer AAV NAbs for up to 15 years following vector administration. Notably, the AAV NAbs are against the infused serotype (AAV serotype 2; AAV2) and also against alternative vector serotypes (AAV5 and AAV8), likely reflecting cross-reactivity of antibodies across serotypes. Given the nature of hepatic blood flow and AAV’s ubiquitous liver tropism, our overall safety data and the persistence of AAV NAbs following vector infusion reported here have implications for all current gene therapy efforts in which AAV is administered systemically.
Results
Four of 7 subjects with severe hemophilia B who received AAV2-hFIX16 were enrolled with this long-term follow-up protocol and followed for 12–15 years post-vector infusion. Among the 3 subjects who received vector but were not followed with this longitudinal follow-up protocol, 1 subject deferred enrollment, 1 subject was lost to follow-up, and, as previously reported, 1 subject expired following a myocardial infarction related to underlying atherosclerotic cardiovascular disease.5
Safety
There were no observed adverse events attributed to the viral vector during the course of study follow-up (years 6–15 after vector infusion). No subject developed an oncologic, hematologic, or auto-immune co-morbidity over the course of the study. A single subject, subject D, developed bilateral carpal tunnel syndrome at year 12 post-vector that was diagnosed by symptoms (bilateral positive Tinel’s sign and hand elevation test) and resolved with splinting. The same subject reported progression of baseline migraine headaches with associated dizziness at year 13 post-vector that returned to baseline symptomatology at his year-14 visit. A MRI brain analysis performed after his year-13 visit demonstrated no significant pathology and was unchanged from prior imaging completed before vector infusion due to a long-standing history of migraine headaches (Tables 1 and 2). Given the neurologic nature of the symptomatology, both were classified as adverse events. The etiologies were deemed related to underlying wrist hemarthropathy and overuse injury or an exacerbation of underlying migraine pathology, unrelated to AAV administration.
Table 1.
Summary of Protocol Schedule of Events
| Schedule and Category/Tests | Endpoint |
|---|---|
| At Entry | |
| Informed consent | signed by subject for study enrollment |
| Annually | |
| History and physical exam | evaluation of concurrent or new co-morbidities, concomitant medications, or vital sign or physical exam abnormalities |
| Blood tests | FIX activity, FIX inhibitor, aPTT, CBC, chemistry, α-fetoprotein |
| Urinalysis | any significant findings |
| Adverse events | any oncologic, hematologic neurologic or auto-immune adverse events |
| At Entry and Every 3 Years | |
| Liver ultrasound | any clinically significant findings |
FIX, factor IX; aPTT, activated partial thromboplastin time; CBC, complete blood count with differential.
Table 2.
Subject Pre- and Post-vector Co-morbidities, All Deemed Unrelated to Vector Administration
| Study ID (and Vector Dose in vg/kg) | Age at Vector Infusion/Follow-Up Duration (Years) | Previously Described Co-morbidities Prior to Vector Infusion5 | Co-morbidities Post-vector |
|---|---|---|---|
| Subject B (8 × 1010) | 48/14 | HCV | atrial fibrillation |
| HIV | cataracts | ||
| hemophilic arthropathya | HIV | ||
| liver fibrosis (F1)b | hypertension | ||
| non-Hodgkin’s lymphomac | kidney disease, stage 4 | ||
| NIDDM | |||
| hemophilic arthropathy, progressivea | |||
| Subject D (4 × 1011) | 20/13 | F1 liver fibrosisb | carpal tunnel, bilaterala |
| Gilbert’s disease | HCV | ||
| HCV | headaches, migraine with dizziness | ||
| headaches, migraine | HIV | ||
| hemophilic arthropathya | Gilbert’s disease | ||
| HIV | hemophilic arthropathy, progressivea | ||
| lipodystrophy | |||
| Subject G (4 × 1011) | 27/12.5 | hemophilic arthropathya | hemophilic arthropathy, progressivea |
| Subject E (2 × 1012) | 31/15 | hemophilic arthropathya | hemophilic arthropathy, progressivea |
| fatty liver infiltration |
vg, vector genomes; HCV, hepatitis C virus; HIV, human immunodeficiency virus; F1, fibrosis stage 1; NIDDM, non-insulin-dependent diabetes mellitus.
Co-morbidity deemed related to underlying hemophilia.
Metavir score determined by liver biopsy, repeat liver biopsies post-vector were not completed due to ethical concerns of performing a procedure in severe hemophilia B subjects with attendent hemorrhagic risk and no direct benefit.
Subject B was diagnosed with non-Hodgkin’s lymphoma and treated with chemotherapy and radiation prior to vector infusion and remained in remission throughout the duration of follow-up.
Subject baseline viral serologies and baseline liver fibrosis, as well as pre- and post-vector infusion co-morbidities are outlined in Table 2. Despite prophylactic exogenous FIX replacement therapy, and consistent with the natural history of hemophilia joint disease, all subjects had progression of their underlying hemophilic arthropathy resulting in decreased joint range of motion.21 Subject D developed lipodystrophic changes in the face, trunk, and abdomen that were deemed related to underlying HIV and antiretroviral medications. Because most subjects saw short-term or undetectable FIX expression, subjects continued treatment per their primary hematologist’s recommendation with exogenous FIX protein replacement either prophylactically or on demand. No subjects developed a FIX inhibitor at any point over the course of follow-up.
Specific to measures of hepatoxicity, no subject had an α-fetoprotein (AFP) or sustained liver transaminase elevation, as outlined in Figure 1. A single subject, subject B, had isolated liver transaminase elevations. Subject B experienced single-measurement alanine aminotransferase (ALT) and aspartate aminotransferase (AST) elevations that were 1.7-fold and 1.8-fold the upper limit of normal, respectively, at his 10-year post-vector administration visit; liver transaminases returned to normal values at subsequent follow-up visits. While no definitive etiology was identified for his isolated liver transaminase evaluations, subject B had a known history of active hepatitis C virus (HCV) infection at the time of these abnormalities. He acquired HCV prior to vector infusion and was successfully treated with direct-acting antiviral medications within a year after the described isolated elevated transaminase values. Serial liver ultrasound evaluation of all subjects showed no evidence of tumor. On ultrasound evaluation, subject E had fatty liver infiltration at years 9 and 12 following vector infusion that resolved at year 15. The etiology of subject E’s fatty liver infiltration was not definitely identified; however, he was obese (BMI = 31.6) at the time when fatty liver infiltration was identified and overweight (BMI = 28.9) at the time of resolution, suggesting non-alcoholic fatty liver disease as a likely cause. Subject longitudinal ALT, AST, and AFP values are outlined in Figure 1.
Figure 1.

Subject Longitudinal Serum Liver Transaminases and α-Fetoprotein Measurements
Blue dashed line indicates aspartate aminotransferase (AST) upper limit of normal. Red dashed line indicates ALT upper limit of normal. Normal ranges at individual subject clinical laboratories are as follows: Subjects B and D: alanine aminotransferase (ALT), 3–54 IU/dL; AST, 2–44 IU/dL; α-fetoprotein (AFP), <10 ng/mL. Subject E: ALT, 5–55 IU/dL; AST, 5–55 IU/dL; AFP, <10 ng/mL. Subject G: ALT, 9–46 IU/dL; AST, 10–35 IU/dL; AFP, <10 ng/mL.
Persistence of AAV NAbs
As outlined in Table 3 and Figure 2, subjects B, G, and E demonstrated persistent high-titer NAbs (1:100–1:1,000 to >1:3,000) to the infused AAV2 vector serotype for up to 15 years post-vector. Further, these antibodies cross-reacted with other naturally occurring AAV serotypes currently in clinical trial for hemophilia, AAV5 (1:2–1:20 to 1:100–1:1,000) and AAV8. All AAV NAbs persisted throughout the duration of their follow-up. Subjects B and E had sustained high-titer AAV5 and AAV8 NAb titers, while subject G had 1:20-1:200 1:2 AAV8 NAb titers and 1:2–1:20 AAV5 NAb titers throughout the duration of the study follow-up. Subject D did not consent for future research of banked specimens and, therefore, did not undergo AAV NAb analysis.
Table 3.
Persistence of AAV NAbs
| Study ID (with Vector Dose in vg/kg) and Years Post-vector | AAV2 Titer | AAV5 Titer | AAV8 Titer |
|---|---|---|---|
| Subject B (8 × 1010) | |||
| 9 | 1:100–1:1,000 | 1:20–1:200 | 1:20–1:200 |
| 10 | 1:100–1:1,000 | 1:20–1:200 | 1:20–1:200 |
| 11 | 1:100–1:1,000 | 1:20–1:200 | 1:20–1:200 |
| 12 | 1:100–1:1,000 | 1:20–1:200 | 1:20–1:200 |
| 13 | 1:100–1:1,000 | 1:20–1:200 | 1:20–1:200 |
| 14 | 1:1,000–1:2,000 | 1:20–1:200 | 1:20–1:200 |
| Subject G (4 × 1011) | |||
| 6 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 7 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 8 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 9 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 10 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 11 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| 12 | >1:3,000 | 1:2–1:20 | 1:20–1:200 |
| Subject E ( 2 × 1012) | |||
| 10 | >1:3,000 | 1:100–1:1,000 | 1:100–1:1,000 |
| 11 | >1:3,000 | 1:100–1:1,000 | 1:100–1:1,000 |
| 12 | >1:3,000 | 1:100–1:1,000 | 1:100–1:1,000 |
| 13 | >1:3,000 | 1:100–1:1,000 | 1:100–1:1,000 |
| 14 | >1:3,000 | 1:100–1:1,000 | 1:1,000–1:3,000 |
| 15 | >1:3,000 | 1:100–1:1,000 | 1:100–1:1,000 |
NAbs, neutralizing antibodies; vg, vector genomes.
Figure 2.

Subject Longtitudinal AA2, AAV, and AAV8 NAb Titers
Subject AAV NAbs are represented in the range of sera dilutions required for inhibition of ≥50% AAV transduction. AAV, adeno-associated virus; NAb, neutralizing antibody.
Discussion
With up to 15 years of follow-up of the first-in-human trial of intravascular infusion of an AAV vector, our data in adult males with severe hemophilia B represent the longest clinical follow-up after systemic AAV vector administration. Our results demonstrate (1) no major safety concerns following intravascular AAV administration and (2) the persistence of multi-serotype AAV NAbs for the duration of follow-up. These results are likely to be applicable to similarly dosed (8 × 1010 to 2 × 1012 vg/kg) systemically administered AAV products currently in clinical development.
There are currently 2 US Food and Drug Administration (FDA)-approved AAV products and multiple additional trials underway for a variety of disorders.3,4 As a now novel therapeutic drug class, AAV vectors have demonstrated remarkable efficacy, with no major short-term safety concerns; the risk-benefit profile of AAV vectors is undoubtedly optimistic. However, pre-clinical studies have outlined hepatotoxicity and genotoxicity as theoretical long-term AAV safety concerns with, to date, no published long-term clinical trial safety data extending beyond 3 years post-intravascular vector delivery.22 Although there are disease-, transgene-, and tissue-specific safety considerations, the theoretical risk of hepatic toxicity is common to all efforts incorporating intravascular AAV vector administration. This point was demonstrated in the licensing trial of the only FDA-approved intravascularly delivered AAV vector, Zolgensma (AveXis, Bannockburn, IL, USA), which is an AAV9 vector consisting of a ubiquitous promoter (CMV early enhancer/chicken β-actin promoter) driving expression of the gene encoding survival motor neuron-1 protein for spinal muscular atrophy (SMA). Investigators demonstrated remarkable efficacy but, prior to addition of prophylactic steroids, observed transient hepatic transaminase elevations after vector infusion with peak ALT and AST values >30 times the upper limit of normal resulting in a boxed safety warning of acute serious liver injury following vector infusion.3,23 Similarly, intravascular delivery of AAV vectors targeting hepatocytes for hemophilia, at doses (vector genomes per kilogram) that are 3- to 400-fold lower than for Zolgensma, has also demonstrated ALT and AST elevations >2 times the upper limit of normal.7,10,24 These observations are consistent with the collective clinical trial experience of intravascular AAV delivery that has demonstrated a dose-dependent, thus far transient, hepatotoxity risk.7,10
In addition to hepatoxicity, tumorigenesis remains a theoretical long-term safety concern with systemic AAV use. Dose-dependent AAV integration events have been demonstrated in murine models and remain the major theoretical long-term systemic AAV safety risk. Initial observations in neonatal murine models treated with high vector doses implicated AAV insertional mutagenesis in the development of hepatocellular carcinoma (HCC).14,25 Later work demonstrated that disruption of the murine RNA imprinted and accumulated in the nucleus (Rian) locus, which is not present is humans, led to near 100% HCC phenotype penetrance.14,26,27 A subsequent carefully conducted murine study demonstrated that the risk of HCC correlated directly with AAV vector dose and the degree of underlying cellular division and that the nature of the enhancer and promoter elements of the transgene were crucial for tumorigenesis; non-hepatocyte-specific elements increased HCC risk, while tissue-specific promoters were protective against oncogenesis.13 Specific to this clinical trial, preclinical murine studies of AAV2-hFIX16 documented low-frequency integration events in the albumin locus, but not in the AFP gene or Rian loci previously implicated in HCC, and no increased risk of HCC development relative to controls.16
To date, available human, canine, and non-human primate (NHP) studies have not supported genotoxicity-related AAV integration events.15,28 Evaluation of human liver specimens demonstrated wild-type AAV integration from natural infection.12 Integration was most commonly observed in liver-specific genes, such as albumin and AFP, with fewer integration events in HCC tissue than in healthy tissue.12 Analysis of NHP and human subject liver specimens following intravascular infusion of an AAV2/5 vector with a liver-specific promoter driving porphobilinogen synthesis demonstrated low-frequency, random integration events that were not clustered in HCC-associated genes, including absence of clustered integration events in the human ortholog of the rian locus (delta-like homolog 1-deiodinase type 3).15 More recently, analysis of canine specimens following AAV-mediated, hepatocyte-directed gene transfer reported integration events with evidence of clonal expansion without tumorigenesis.29
Our study is limited by the absence of direct hepatic sequencing data and detectable persistent transgene expression; subjects either lost expression following a capsid immune response (subject E) or had subtherapeutic expression (subjects B, D, and G). However, a prior study of subjects who received intramuscular-AAV-mediated F9 gene transfer demonstrated the persistence of AAV vector sequences in both injected muscle and liver for up to 10 years post-vector administration, despite the absence of circulating transgene-derived protein; thus, the absence of detectable transgene expression does not necessarily equate with the absence of AAV vector in transduced hepatocytes.30 Nonetheless, with cautious interpretation in light of the outlined limitations, the absence of sustained liver function study abnormalities and liver tumorigenesis with normal AFP values support no long-term clinical hepatotoxicity or HCC development. These results may then support the preliminary long-term liver safety of hepatocyte-directed intravascular AAV vector infusion in adults at the range of doses administered, 8 × 1010 to 2 × 1012 vg/kg. Current hemophilia gene therapy efforts use up to 30-fold higher vector doses, and some incorporate the use of corticosteroids to theoretically prevent loss of transduced hepatocytes.8,10 It remains unclear but possible that use of higher vector doses and steroids to maintain transduced hepatocytes may carry greater long-term vector-related safety concerns; indeed, this would be consistent with data previously published in mouse models.13 Alternatively, use of higher vector doses may reflect less efficient liver tropism of the capsid used and not translate to increased hepatocyte transduction events or resultant theoretical safety concerns. Further, collectively, available preclinical data across murine models suggest an age-dependent risk, with injection of neonatal mice resulting in the greatest risk of AAV-related genotoxicity, presumably due to the rapid growth of the neonatal liver, leading to chromosomal breaks that favor vector integration. It is unclear how well our adult data inform the safety of intravascular delivery of AAV vectors in the pediatric population, particularly neonates.14,25,31 Long-term pediatric safety data from SMA infants and toddlers treated with Zolgensma are best positioned to provide the first available long-term safety of systemic AAV in the pediatric population.3 Ultimately, definitive proof of integration event frequency, location, and genotoxicity risk will require direct sequencing data and larger cohort longitudinal data in subjects with sustained transgene expression.
Importantly, the hemophilia population is a provocative model for the clinical study of the theoretical risk of genotoxicity-related HCC following intravascular AAV vector administration. Approximately 90% of severe hemophilia patients >35 years old contracted iatrogenic HCV, resulting in an inherent elevated baseline HCC risk in the hemophilia population.32 Generally, after HCV-related cirrhosis onset, the annual risk of developing HCC is 1%–4%, resulting in an overall HCC incidence of approximately 30% at 25 years after HCV-related cirrhosis onset, with some data to suggest that HCV+ hemophilia patients have a greater risk of progressing to HCC.33, 34, 35, 36 Two of the 4 subjects followed were HCV+ for >25 years total and >10 years post-vector infusion, and neither developed HCC. Importantly, the >90% cure rate of direct-acting antiviral treatments for HCV is altering the natural history of HCV-related cirrhosis and HCC, making it difficult to predict the future risk of HCC in the hemophilia population that is expected to have access to licensed AAV vectors.37 Nonetheless, there is likely an increased background risk of HCC within the hemophilia population with a history of or current HCV; it will be critically important to investigate thoroughly any cases of HCC in AAV-treated hemophilia patients to determine the role of AAV integration events. Updated FDA industry guidance reduced recommended follow-up post-AAV gene transfer from 15 years to 5 years that, in practice, will limit the investigation of potential genotoxicity events to post-marketing surveillance.38
Lastly, with respect to safety outside evaluation of liver toxicity, there were no observed co-morbidities that could not be ascribed to underlying pre-vector infusion subject medical conditions. Thus, our data do not provide evidence of liver toxicity or any other unexpected AAV long-term safety concerns but are limited by small cohort size and lack of detectable FIX expression of >1%, suggesting that the number of persistant AAV-transduced cells is modest. Consistent with the general principle that the identification of rare events requires a large sample size, establishing long-term safety data will require large-cohort, mature clinical data that may best be achieved by cross-disciplinary study of all disorders for which intravascular AAV vectors are in clinical trial or practice.
Whether developing following natural infection or AAV vector administration, the existence of AAV NAbs has profound therapeutic implications; AAV NAbs preclude the efficacy of systemic AAV administration by interfering with cell-surface binding and/or causing reticuloendothelial vector clearance, preventing vector transduction of the target tissue.5,39, 40, 41 Although it is known that patients who receive systemic AAV vector universally develop high-titer NAbs in the short term, the durability of this humoral response is unknown.7,18,42 Canine studies demonstrated resolution of NAbs by an in vivo transduction-based assay 8 years following AAV infusion.43 In contrast, here, we demonstrate long-term persistence of high-titer NAbs to the infused AAV capsid (AAV2) for up to 15 years after vector administration.
In addition to the persistence of NAbs to the infused AAV2 serotype, our data support a polyclonal antibody response with shared epitopes among tested serotypes following systemic AAV administration. This is demonstrated by the presence and persistence of high-titer NAbs to the infused vector serotype (AAV2) as well as AAV8 and low- or high-titer NAbs to AAV5 in all subjects tested, including in a single subject with co-morbid HIV infection (subject B). However, the interpretation is limited, because baseline samples were not available to assess the presence of AAV5 or AAV8 NAbs. While the true incidence of NAbs to individual AAV serotypes due to natural infection is unclear, multiple studies have found the seroprevelance of AAV2 NAbs to be higher than that of AAV5 and AAV8, including studies conducted specifically in hemophilia cohorts.44, 45, 46 It is, therefore, unlikely that the subject’s baseline AAV5 or AAV8 NAb titers were higher than baseline AAV2 NAb titers, which were <1:1 or 1:2 for all subjects tested here.5 Additionally, and analogous to our observations following AAV2 vector infusion, analysis of NHP and human sera following environmental AAV2 exposure demonstrated the induction of broadly cross-reactive NAbs to multiple AAV serotypes (including AAV5 and AAV8 in human samples).47,48 Further, our data are consistent with rabbit and NHP studies that also demonstrated the development of AAV NAbs that cross-react with multiple AAV serotypes following systemic AAV vector infusion.49,50 Collectively, our data support that, as is the case for environmental AAV exposure in humans and infusion of AAV vectors in animal models, systemic AAV vector administration induces multi-serotype cross-reactive AAV NAbs.
Although the observed AAV5 NAbs were lower than for AAV2, our observations are perhaps most surprising, as they relate to the induction of a persistent AAV5 NAb response following AAV2 vector infusion, because AAV5 is the most distinct in phylogeny relative to other described natural AAV serotypes.51 However, structural data suggest that AAV antigenic regions are common, consistent with the principle that tertiary structure rather than sequence alone dictates the host’s humoral immune response to AAV.52 Consistent with structural observations, and like AAV NAbs that develop following environmental AAV2 exposure, our data suggest that AAV NAbs that develop following vector infusion cross-react with other serotypes.53,54 The current data support the persistence of AAV NAbs with multi-serotype cross-reactivity for up to 15 years following AAV vector infusion. Thus, in contrast to some murine, canine, and non-human primate data that have demonstrated successful repeat AAV administration using different serotypes, our data suggest that alternative serotypes are unlikely to overcome AAV NAbs developed post-AAV vector infusion in humans.55 Given that the therapeutic transgene is episomally maintained following AAV gene transfer, the loss or decrease of transgene expression in dividing or apoptotic cells over time may necessitate repeat administration; this is particularly relevant in pediatric patients. Our data suggest that repeat intravascular vector infusion of any serotype is unlikely to be efficacious without measures taken to overcome AAV NAbs. While we speculate that this is universal to all AAV serotypes, it is unknown whether these observations are specific to intravascular administration of AAV2 only.
In summary, our data are the longest available longitudinal clinical follow-up of intravascular AAV administration, and they support the preliminary long-term safety of systemic AAV delivery and demonstrate the persistence of high-titer, multi-AAV serotype NAbs after vector infusion.
Materials and Methods
Ethics Statement and Clinical Protocol
After an initial dosing study of the intrahepatic artery infusion of AAV2-hFIX16 in severe hemophilia B, subjects were enrolled in this international, multi-site, non-randomized, non-interventional, observational, long-term follow-up study (ClinicalTrials.gov; NCT00515710).5 The dosing study was originally sponsored by Avigen until 2005. The institutional review board at each study site approved the protocol. Written informed consent was obtained from all subjects at enrollment. Subjects were followed for 12–15 years after AAV vector infusion on an approximately annual basis. Evaluations consisted of annual monitoring for adverse events and co-morbidities, physical examinations, blood tests, urinalysis, and, every 3 years, a liver ultrasound, as outlined in the protocol (available with the full text of this article online) summarized in Table 1. In brief, laboratory evaluation included serum chemistry studies and liver function studies (ALT, AST, and bilirubin), as well as AFP, complete blood count with differential, and coagulation studies (FIX activity and FIX inhibitor). AFP measurements were conducted as a screening evaluation for HCC. Liver ultrasounds were conducted to screen for liver abnormalities, including tumorigenesis. Subject clinical laboratory studies were conducted at individual enrolling sites’ clinical laboratories. As advised by guidance from the FDA Center for Biologics Evaluation and Research (CBER) in November 2006, adverse event reporting for the long-term follow-up period focused on the development of oncologic, hematologic, neurologic, and auto-immune events following gene transfer.56 The study was sponsored by the Perelman Center for Cellular and Molecular Therapeutics at Children’s Hospital of Philadelphia (Philadelphia, PA, USA) from 2009 to 2013 and, thereafter, Spark Therapeutics (Philadelphia, PA, USA). Study oversight was provided by clinical principal investigators, the study sponsor, a data safety and monitoring board, and governing regulatory agencies, including the FDA. Data analysis was descriptive in nature. Subject nomenclature was maintained from the dosing study.5,20
AAV NAb Assay
Subject sera were analyzed for AAV NAbs; analysis was performed only for those subjects who consented for banked specimen availability for future research. Tests for AAV NAbs were performed using a cell-based assay as previously described and modified for AAV2, AAV5, and AAV8.57 In brief, the assay utilizes an AAV-luciferase reporter vector with an AAV2, AAV5, or AAV8 capsid. The target 2V6.11 cells are derived from a human embryonic kidney (HEK) 293 cell line that stably expresses the adenovirus E4 gene, which ensures efficient transduction by AAV vectors. Tested serum is diluted and mixed with the reporter vector. This preparation is then used to transduce 2V6.11 cells seeded in a 96-well plate, in triplicate. After 24 h, luciferase expression is measured using the Renilla Luciferase Assay System and a GloMax Navigator plate reader (Promega, Madison, WI, USA). Reported AAV NAb titers represent the lowest dilution for which luciferase expression is inhibited by ≥50%. Titers >1:5 are considered high titer, a threshold used clinically to exclude patients from enrollment on clinical trials.7,8,24
AAV2-hFIX16 Vector
The antecedent dosing study (ClinicalTrials.gov: NCT00076557; also referred to as IND9398) enrolled subjects in 3-dose cohorts of AAV2-hFIX16 (8 × 1010 vg/kg, 4 × 1011 vg/kg, and 2 × 1012 vg/kg) for targeted hepatocyte expression of F9.5 Subjects were enrolled in the dosing study between 2001 and 2004 and were followed for at least a year after vector infusion. As previously reported, the AAV2-hFIX16 construct consisted of an AAV2 capsid, human α-1 anti-tryspin promoter, APOE enhancer, hepatic control region, and human F9 cDNA interrupted by a 1.4-kb fragment of intron 1.5,58,59 The vector was manufactured by Avigen (Alameda, CA, USA) in an adherent HEK293 mammalian cell-culture system. Downstream purification included a density gradient step to remove the majority of empty capsids.5
Author Contributions
All co-authors designed the study or experiments, analyzed the data, vouch for the data and analysis, and decided to publish the data. L.A.G., M.V.R., J.E.J., L.R., A.R., Y.C., and K.K. collected data. L.A.G. and K.A.H. wrote the manuscript without editorial support, and all co-authors provided critical content revisions. L.A.G. wrote the first draft of the manuscript without editorial assistance.
Conflicts of Interest
B.J.S.-J., M.H., and A.R.R. declare no competing interests. L.A.G. has served as a consultant for Pfizer and serves on the DSMB for Avrobio. M.V.R. has received research funding from Alnylam, Biomarin, Bioverativ, Sangamo, Spark, and Shire/Takeda and has participated in advisory boards for Alnylam, Biomarin, Bioverativ, and Spark. J.E.J.R. has received honoraria or speakers’ fees from GlaxoSmithKline, Miltenyi, Takeda, Gilead, Pfizer, Spark, Novartis, Celgene, and Bluebird Bio; has equity ownership in Genea and RareCyte; and is a consultant for RareCyte and Imago. L.J.R. has participated in advisory boards for Roche/Genentech, Bayer, CSL Behring, and Hema-Biologics. J.A.W., K.W., X.M.A., Y.C., K.K., F.M., and K.A.H. are employees or former employees of Spark Therapeutics.
Acknowledgments
This work was additionally supported by National Institutes of Health grants K08 HL 146991-01 (to L.A.G.) and P01 HL64190 (to K.A.H., program director). We gratefully acknowledge the bravery of the study subjects whose contributions greatly contributed to current clinical development efforts of AAV-based therapeutics.
References
- 1.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 3.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 4.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 6.High K.A., Roncarolo M.G. Gene Therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 7.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 9.Bunting S., Zhang L., Xie L., Bullens S., Mahimkar R., Fong S., Sandza K., Harmon D., Yates B., Handyside B. Gene Therapy with BMN 270 Results in Therapeutic Levels of FVIII in Mice and Primates and Normalization of Bleeding in Hemophilic Mice. Mol. Ther. 2018;26:496–509. doi: 10.1016/j.ymthe.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 11.Fagiuoli S., Daina E., D’Antiga L., Colledan M., Remuzzi G. Monogenic diseases that can be cured by liver transplantation. J. Hepatol. 2013;59:595–612. doi: 10.1016/j.jhep.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Nault J.C., Datta S., Imbeaud S., Franconi A., Mallet M., Couchy G., Letouzé E., Pilati C., Verret B., Blanc J.F. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015;47:1187–1193. doi: 10.1038/ng.3389. [DOI] [PubMed] [Google Scholar]
- 13.Chandler R.J., LaFave M.C., Varshney G.K., Trivedi N.S., Carrillo-Carrasco N., Senac J.S., Wu W., Hoffmann V., Elkahloun A.G., Burgess S.M., Venditti C.P. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donsante A., Miller D.G., Li Y., Vogler C., Brunt E.M., Russell D.W., Sands M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 15.Gil-Farina I., Fronza R., Kaeppel C., Lopez-Franco E., Ferreira V., D’Avola D., Benito A., Prieto J., Petry H., Gonzalez-Aseguinolaza G., Schmidt M. Recombinant AAV Integration Is Not Associated With Hepatic Genotoxicity in Nonhuman Primates and Patients. Mol. Ther. 2016;24:1100–1105. doi: 10.1038/mt.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H., Malani N., Hamilton S.R., Schlachterman A., Bussadori G., Edmonson S.E., Shah R., Arruda V.R., Mingozzi F., Wright J.F. Assessing the potential for AAV vector genotoxicity in a murine model. Blood. 2011;117:3311–3319. doi: 10.1182/blood-2010-08-302729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakai H., Montini E., Fuess S., Storm T.A., Grompe M., Kay M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003;34:297–302. doi: 10.1038/ng1179. [DOI] [PubMed] [Google Scholar]
- 18.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spronck E.A., Liu Y.P., Lubelski J., Ehlert E., Gielen S., Montenegro-Miranda P., de Haan M., Nijmeijer B., Ferreira V., Petry H., van Deventer S.J. Enhanced Factor IX Activity following Administration of AAV5-R338L “Padua” Factor IX versus AAV5 WT Human Factor IX in NHPs. Mol. Ther. Methods Clin. Dev. 2019;15:221–231. doi: 10.1016/j.omtm.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E., Ragni M.V., Manno C.S., Sommer J., Jiang H. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 21.Manco-Johnson M.J., Soucie J.M., Gill J.C., Joint Outcomes Committee of the Universal Data Collection, US Hemophilia Treatment Center Network Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129:2368–2374. doi: 10.1182/blood-2016-02-683169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Food and Drug Administration Highlights of Prescribing Information [Zolgensma] 2019. https://www.fda.gov/media/126109/download
- 24.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwable J., Bonig H. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donsante A., Vogler C., Muzyczka N., Crawford J.M., Barker J., Flotte T., Campbell-Thompson M., Daly T., Sands M.S. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 26.Wang P.R., Xu M., Toffanin S., Li Y., Llovet J.M., Russell D.W. Induction of hepatocellular carcinoma by in vivo gene targeting. Proc. Natl. Acad. Sci. USA. 2012;109:11264–11269. doi: 10.1073/pnas.1117032109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong L., Malani N., Li M., Brady T., Xie J., Bell P., Li S., Jones H., Wilson J.M., Flotte T.R. Recombinant adeno-associated virus integration sites in murine liver after ornithine transcarbamylase gene correction. Hum. Gene Ther. 2013;24:520–525. doi: 10.1089/hum.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niemeyer G.P., Herzog R.W., Mount J., Arruda V.R., Tillson D.M., Hathcock J., van Ginkel F.W., High K.A., Lothrop C.D., Jr. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. 2009;113:797–806. doi: 10.1182/blood-2008-10-181479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen G.N., Everett J.K., Raymond H., Kafle S., Merricks E.P., Kazazian H.H., Nichols T.C., Bushman F.D., Sabatino D.E. Long-Term AAV-Mediated Factor VIII Expression in Nine Hemophilia A Dogs: A 10 Year Follow-up Analysis on Durability, Safety and Vector Integration. Blood. 2019;134(Suppl. 1):611. [Google Scholar]
- 30.Buchlis G., Podsakoff G.M., Radu A., Hawk S.M., Flake A.W., Mingozzi F., High K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. 2012;119:3038–3041. doi: 10.1182/blood-2011-09-382317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell P., Wang L., Lebherz C., Flieder D.B., Bove M.S., Wu D., Gao G.P., Wilson J.M., Wivel N.A. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol. Ther. 2005;12:299–306. doi: 10.1016/j.ymthe.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Mazepa M.A., Monahan P.E., Baker J.R., Riske B.K., Soucie J.M., US Hemophilia Treatment Center Network Men with severe hemophilia in the United States: birth cohort analysis of a large national database. Blood. 2016;127:3073–3081. doi: 10.1182/blood-2015-10-675140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodgame B., Shaheen N.J., Galanko J., El-Serag H.B. The risk of end stage liver disease and hepatocellular carcinoma among persons infected with hepatitis C virus: publication bias? Am. J. Gastroenterol. 2003;98:2535–2542. doi: 10.1111/j.1572-0241.2003.07678.x. [DOI] [PubMed] [Google Scholar]
- 34.Freeman A.J., Dore G.J., Law M.G., Thorpe M., Von Overbeck J., Lloyd A.R., Marinos G., Kaldor J.M. Estimating progression to cirrhosis in chronic hepatitis C virus infection. Hepatology. 2001;34:809–816. doi: 10.1053/jhep.2001.27831. [DOI] [PubMed] [Google Scholar]
- 35.Lu M., Li J., Zhang T., Rupp L.B., Trudeau S., Holmberg S.D., Moorman A.C., Spradling P.R., Teshale E.H., Xu F. Serum Biomarkers Indicate Long-term Reduction in Liver Fibrosis in Patients With Sustained Virological Response to Treatment for HCV Infection. Clin. Gastroenterol. Hepatol. 2016;14:1044–1055.e3. doi: 10.1016/j.cgh.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thalappillil A., Ragni M.V., Comer D.M., Yabes J.G. Incidence and risk factors for hepatocellular cancer in individuals with haemophilia: A National Inpatient Sample Study. Haemophilia. 2019;25:221–228. doi: 10.1111/hae.13668. [DOI] [PubMed] [Google Scholar]
- 37.Conti F., Buonfiglioli F., Scuteri A., Crespi C., Bolondi L., Caraceni P., Foschi F.G., Lenzi M., Mazzella G., Verucchi G. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016;65:727–733. doi: 10.1016/j.jhep.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 38.Food and Drug Administration Long Term Follow-Up After Administration of Human Gene Therapy Products. 2020. https://www.fda.gov/media/113768/download
- 39.Louis Jeune V., Joergensen J.A., Hajjar R.J., Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods. 2013;24:59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun J., Yuan Z., Abajas Y.L., Szollosi D.E., Hu G., Hua B., Xiao X., Li C. A Retrospective Study of the Cytokine Profile Changes in Mice with FVIII Inhibitor Development After Adeno-Associated Virus-Mediated Gene Therapy in a Hemophilia A Mouse Model. Hum. Gene Ther. 2018;29:381–389. doi: 10.1089/hum.2017.094. [DOI] [PubMed] [Google Scholar]
- 41.Wang M., Sun J., Crosby A., Woodard K., Hirsch M.L., Samulski R.J., Li C. Direct interaction of human serum proteins with AAV virions to enhance AAV transduction: immediate impact on clinical applications. Gene Ther. 2017;24:49–59. doi: 10.1038/gt.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manno C.S., Chew A.J., Hutchison S., Larson P.J., Herzog R.W., Arruda V.R., Tai S.J., Ragni M.V., Thompson A., Ozelo M. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 43.Sun J., Shao W., Chen X., Merricks E.P., Wimsey L., Abajas Y.L., Niemeyer G.P., Lothrop C.D., Monahan P.E., Samulski R.J. An Observational Study from Long-Term AAV Re-administration in Two Hemophilia Dogs. Mol. Ther. Methods Clin. Dev. 2018;10:257–267. doi: 10.1016/j.omtm.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 45.Li C., Narkbunnam N., Samulski R.J., Asokan A., Hu G., Jacobson L.J., Manco-Johnson M.J., Monahan P.E., Joint Outcome Study Investigators Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19:288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 46.Kruzik A., Fetahagic D., Hartlieb B., Dorn S., Koppensteiner H., Horling F.M., Scheiflinger F., Reipert B.M., de la Rosa M. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019;14:126–133. doi: 10.1016/j.omtm.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calcedo R., Wilson J.M. AAV Natural Infection Induces Broad Cross-Neutralizing Antibody Responses to Multiple AAV Serotypes in Chimpanzees. Hum. Gene Ther. Clin. Dev. 2016;27:79–82. doi: 10.1089/humc.2016.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aronson S.J., Veron P., Collaud F., Hubert A., Delahais V., Honnet G., de Knegt R.J., Junge N., Baumann U., Di Giorgio A. Prevalence and Relevance of Pre-Existing Anti-Adeno-Associated Virus Immunity in the Context of Gene Therapy for Crigler-Najjar Syndrome. Hum. Gene Ther. 2019;30:1297–1305. doi: 10.1089/hum.2019.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L., Bell P., Somanathan S., Wang Q., He Z., Yu H., McMenamin D., Goode T., Calcedo R., Wilson J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mingozzi F., Chen Y., Murphy S.L., Edmonson S.C., Tai A., Price S.D., Metzger M.E., Zhou S., Wright J.F., Donahue R.E. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B. Mol. Ther. 2012;20:1410–1416. doi: 10.1038/mt.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiorini J.A., Kim F., Yang L., Kotin R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tseng Y.S., Gurda B.L., Chipman P., McKenna R., Afione S., Chiorini J.A., Muzyczka N., Olson N.H., Baker T.S., Kleinschmidt J., Agbandje-McKenna M. Adeno-associated virus serotype 1 (AAV1)- and AAV5-antibody complex structures reveal evolutionary commonalities in parvovirus antigenic reactivity. J. Virol. 2015;89:1794–1808. doi: 10.1128/JVI.02710-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halbert C.L., Miller A.D., McNamara S., Emerson J., Gibson R.L., Ramsey B., Aitken M.L. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2006;17:440–447. doi: 10.1089/hum.2006.17.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Q., Huang W., Zhang H., Wang Y., Zhao J., Song A., Xie H., Zhao C., Gao D., Wang Y. Neutralizing antibodies against AAV2, AAV5 and AAV8 in healthy and HIV-1-infected subjects in China: implications for gene therapy using AAV vectors. Gene Ther. 2014;21:732–738. doi: 10.1038/gt.2014.47. [DOI] [PubMed] [Google Scholar]
- 55.Majowicz A., Salas D., Zabaleta N., Rodriguez-Garcia E., Gonzalez-Aseguinolaza G., Petry H., Ferreira V. Successful Repeated Hepatic Gene Delivery in Mice and Non-human Primates Achieved by Sequential Administration of AAV5(ch) and AAV1. Mol. Ther. 2017;25:1831–1842. doi: 10.1016/j.ymthe.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.US Food and Drug Administration (2006). Guidance for Industry: Gene Therapy Clinical Trials – Observing Subjects for Delayed Adverse Events, November 2006. https://www.fda.gov/media/72225/download.
- 57.Meliani A., Leborgne C., Triffault S., Jeanson-Leh L., Veron P., Mingozzi F. Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum. Gene Ther. Methods. 2015;26:45–53. doi: 10.1089/hgtb.2015.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miao C.H., Ohashi K., Patijn G.A., Meuse L., Ye X., Thompson A.R., Kay M.A. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol. Ther. 2000;1:522–532. doi: 10.1006/mthe.2000.0075. [DOI] [PubMed] [Google Scholar]
- 59.Kurachi S., Hitomi Y., Furukawa M., Kurachi K. Role of intron I in expression of the human factor IX gene. J. Biol. Chem. 1995;270:5276–5281. doi: 10.1074/jbc.270.10.5276. [DOI] [PubMed] [Google Scholar]