

Abstract
Objective
Stromal cells and immune cells have important clinical significance in the microenvironment of colorectal cancer (CRC). This study is aimed at developing a CRC gene signature on the basis of stromal and immune scores.
Methods
A cohort of CRC patients (n = 433) were adopted from The Cancer Genome Atlas (TCGA) database. Stromal/immune scores were calculated by the ESTIMATE algorithm. Correlation between prognosis/clinical characteristics and stromal/immune scores was assessed. Differentially expressed stromal and immune genes were identified. Their potential functions were annotated by functional enrichment analysis. Cox regression analysis was used to develop an eight-gene risk score model. Its predictive efficacies for 3 years, 5 years, overall survival (OS), and progression-free survival interval (PFI) were evaluated using time-dependent receiver operating characteristic (ROC) curves. The correlation between the risk score and the infiltering levels of six immune cells was analyzed using TIMER. The risk score was validated using an independent dataset.
Results
Immune score was in a significant association with prognosis and clinical characteristics of CRC. 736 upregulated and two downregulated stromal and immune genes were identified, which were mainly enriched into immune-related biological processes and pathways. An-eight gene prognostic risk score model was conducted, consisting of CCL22, CD36, CPA3, CPT1C, KCNE4, NFATC1, RASGRP2, and SLC2A3. High risk score indicated a poor prognosis of patients. The area under the ROC curves (AUC) s of the model for 3 years, 5 years, OS, and PFI were 0.71, 0.70, 0.73, and 0.66, respectively. Thus, the model possessed well performance for prediction of patients' prognosis, which was confirmed by an external dataset. Moreover, the risk score was significantly correlated with immune cell infiltration.
Conclusion
Our study conducted an immune-related prognostic risk score model, which could provide novel targets for immunotherapy of CRC.
1. Introduction
CRC, as a heterogeneous disease, is a common cause of cancer-related deaths worldwide [1]. TNM staging is usually considered to be one of the main tools for CRC prognosis [2]. However, the prognosis varies greatly among CRC patients with the same TNM stage, suggesting that the current TNM stage does not well provide complete prognostic information for CRC patients. Therefore, it is necessary to adopt a new strategy to increase the predictive efficiency of prognosis and survival outcomes of CRC patients.
Due to the considerable heterogeneity between CRCs, determination of the optimal treatment strategy at the individual level faces the large challenges. Thus, it is an urgent need to conduct robust models to identify high-risk CRC patients and to find novel molecular targets. In the tumor microenvironment (TME), stromal and immune cells are involved in the development of CRC [3, 4]. Increasing evidence suggests that stromal and immune cells possess critical clinical significance for CRC. It has been reported that stromal cells can contribute to transcriptome and clinical features of CRC subtype [5]. Furthermore, stromal gene expression can more robustly predict the prognosis of CRC subtypes compared to epithelial tumor cells [6]. In a large cohort of CRC patients, infiltrating immune cell data could better predict patients' survival than histopathological methods [7]. Growing studies have found that infiltrating immune cells are involved in chemoresistance [8] and metastasis [9]. Thus, it is essential to further analyze the biological characteristics of stromal and immune genes and to determine their prognostic value for CRC patients. However, there is a lack of stromal and immune scores that can predict CRC patients' prognosis based on multiple clinical factors. Moreover, robust prognostic models on the basis of stromal and immune scores are also lacking.
In this study, we established a reliable prognostic immune-related risk score for CRC. Our results could offer novel insights for prediction of CRC patients' prognosis and development of individualized immunity therapy strategies.
2. Materials and Methods
2.1. CRC Datasets
TCGA RNA-seq data (including Counts and FPKM data) of GDC CRC (including 469 CRC tissue samples and 41 adjacent normal tissue samples) were downloaded from the xenabrowser website (https://xenabrowser.net/). Among all CRC samples, 433 samples contained complete clinical information, including gender, age, TNM stage, tumor grade, microsatellite instability (MSI), and mismatch repair (MMR). The clinical information of 433 CRC patients is listed in Table 1. Survival information including OS status, OS time, progression-free survival (PFS) status, and PFS time was derived from the pan-cancer on the GDC website (https://gdc.cancer.gov/about-data/publications/PanCan-Clinical-2018). Furthermore, mutation data (including BRAF, KRAS, and TP53) were from CRC MuTect. An overview of the workflow is shown in Figure 1.
Table 1.
Clinical characteristics of CRC patients in TCGA datasets (overall = 433).
| Characteristics | Groups | N (%) |
|---|---|---|
| Age (%) | ≤60 | 136 (30.7) |
| >60 | 297 (68.6) | |
|
| ||
| Gender (%) | Female | 200 (46.2) |
| Male | 233 (53.8) | |
|
| ||
| Status (%) | Died | 338 (78.1) |
| Alive | 95 (21.9) | |
|
| ||
| Pathologic T (%) | T1 | 11 (2.5) |
| T2 | 75 (17.3) | |
| T3 | 296 (68.4) | |
| T4 | 51 (11.8) | |
|
| ||
| Pathologic N (%) | N0 | 254 (58.7) |
| N1 | 102 (23.6) | |
| N2 | 77 (17.8) | |
|
| ||
| Pathologic M (%) | M0 | 320 (75.1) |
| M1 | 61 (14.3) | |
| Mx | 45 (10.6) | |
|
| ||
| Tumor stage (%) | I | 73 (17.3) |
| II | 165 (39.1) | |
| III | 123 (29.1) | |
| IV | 61 (14.5) | |
Figure 1.
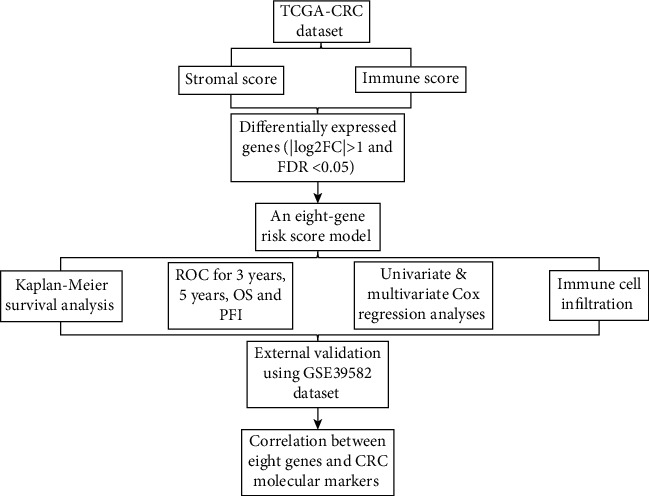
An overview of the workflow.
2.2. Estimation of Stromal/Immune Scores
ESTIMATE algorithm was used to calculate the stromal/immune scores on the basis of unique expression profiles of stromal/immune cells by the ESTIMATE package in R (https://bioinformatics.mdanderson.org/estimate/) [10].
2.3. Kaplan-Meier Survival Analysis
According to the optimal cutoff of stromal/immune scores, CRC samples were classified into high and low stromal/immune score groups. Kaplan-Meier plot of overall survival between the two groups was analyzed, and the results were evaluated by log-rank test.
2.4. Correlation between Clinical Characteristics and Stromal/Immune Scores
To probe into the clinical significance of stromal/immune scores, we analyzed the correlation between clinical characteristics (including pathologic T, pathologic N, pathologic M, and tumor stage) and stromal/immune scores.
2.5. Differential Expression Analysis
Differential expression analysis between high and low stromal/immune score groups was carried out using the edgeR package in R, following the screening criteria of ∣log2 fold change (FC) | >1 and FDR (adjusted p value) < 0.05. Then, up- or downregulated stromal/immune genes were intersected by the VennDiagram package in R, respectively.
2.6. Functional Enrichment Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of differentially expressed stromal and immune genes were carried out through the clusterProfiler package in R [11]. GO analysis contains three terms, cellular component (CC), molecular function (MF), and biological process (BP). p value after adjustment < 0.05 was significantly enriched.
2.7. Protein-Protein Interaction (PPI) Analysis
PPI analyses of differentially expressed stromal and immune genes were carried out via The Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/; version 11) [12]. Then, the PPI network was visualized through Cytoscape (version 3.7.2) [13].
2.8. Univariate and Multivariate Cox Regression Analyses
Univariate cox regression analysis of differentially expressed stromal and immune genes was carried out via the survival package in R. Genes with p value < 0.05 were screened for multivariate cox regression analysis. To validate the sensitivity and accuracy of the risk score for prediction of CRC, an ROC curve was drawn to evaluate the predictive performance of the risk core for 3 years, 5 years, OS, and PFI using the “tdROC” package in R. The results were visualized with the “ggplot2” package in R. The AUC was then calculated. The GSE39582 dataset from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov) was used to validate the prognostic value of the risk score. The dataset was composed of 566 CRC samples.
2.9. Immune Infiltration Analysis
The tumor-immune infiltration cells including B cells, CD4+T cells, CD8+T cells, macrophages, neutrophils, and dendritic cells were estimated via the TIMER (https://cistrome.shinyapps.io/timer/) [14]. Spearman's correlation between the risk score and the infiltrating levels of immune cells was evaluated through the psych package in R. Furthermore, we also assessed the correlation between the genes in the risk score and marker genes of immune cells. The strength of correlation followed the criteria: 0.7 ≤ ∣ r | ≥1 suggested a high correlation, 0.3 ≤ ∣ r | <0.7 suggested a moderate correlation, and 0 < ∣ r | <0.3 suggested a weak correlation [15].
3. Results
3.1. Immune Score Is in Significant Association with Prognosis and Clinical Features of CRC Patients
According to the optimal cutoff of stromal/immune scores, the CRC patients were divided into two groups. Kaplan-Meier OS analysis results showed that patients with high stromal score had shorter OS time than those with low stromal score; however, it was not statistically significant (Figure 2(a); p value = 0.057). As depicted in Figure 2(b), we found that patients with low immune score implied a poor prognosis (p value = 0.0094). Furthermore, we analyzed the correlation between stromal/immune scores and clinical features. As depicted in the results, stromal score was not significantly associated with pathologic T (Figure 3(a); p value = 0.61), pathologic N (Figure 3(b); p value = 0.28), pathologic M (Figure 3(c); p value = 0.63), tumor stage (Figure 3(d); p value = 0.68), and age (Figure 3(e); p value = 0.76). Similarly, we also found that there was no statistical significance between immune score and pathologic T (Figure 3(f); p value = 0.88) and pathologic N (Figure 3(g); p value = 0.17). As expected, immune score was in significant association with pathologic M (Figure 3(h); p value = 0.0045) and tumor stage (Figure 3(i); p value = 0.0093). However, no significant correlation between immune score and age was found in Figure 3(j) (p value = 0.29). Furthermore, ESTIMATE scores were not correlated with pathologic T (Figure 3(k); p value = 0.98), pathologic N (Figure 3(l); p value = 0.73), pathologic M (Figure 3(m); p value = 0.095), tumor stage (Figure 3(n); p value = 0.28), and age (Figure 3(o); p value = 0.74). These findings indicated that immune score was in significant association with CRC patients' prognosis and clinical features.
Figure 2.
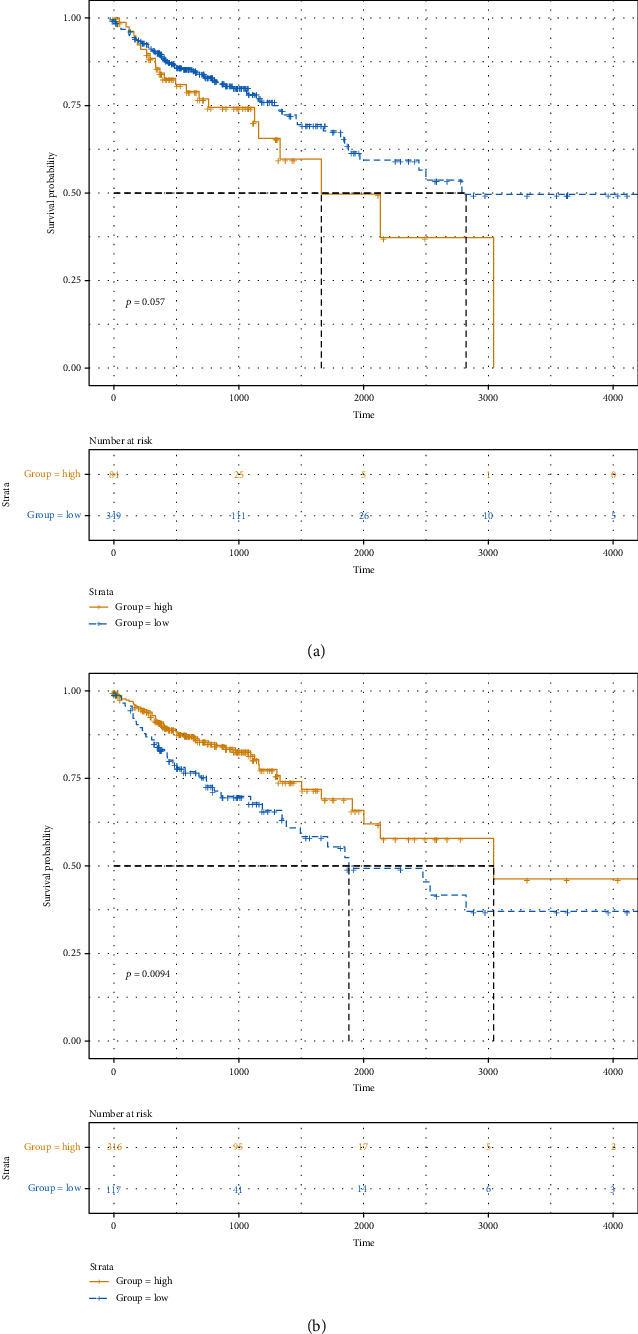
The correlation between stromal/immune scores and CRC patients' survival outcomes. (a) Stromal score. (b) Immune score. The x-axis suggests overall survival time and y-axis represents survival probability.
Figure 3.
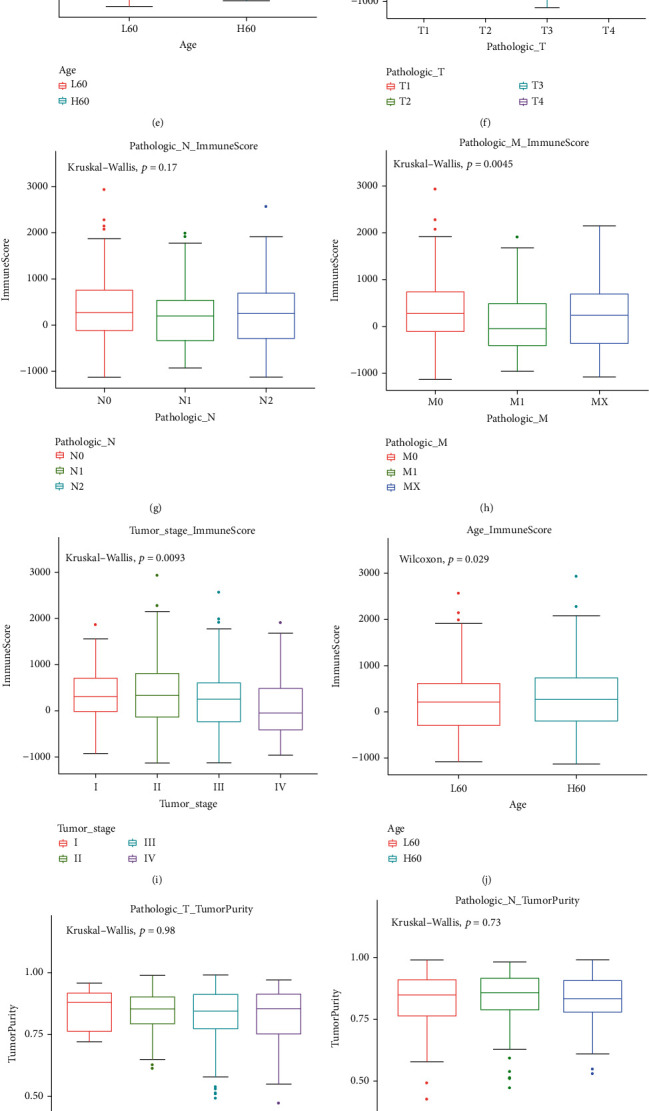
The correlation between stromal/immune/ESTIMATE scores and CRC patients' clinical features. Stromal score is not associated with (a) pathologic T (p value = 0.61), (b) pathologic N (p value = 0.28), (c) pathologic M (p value = 0.63), (d) tumor stage (p value = 0.68), and (e) age (p value = 0.76). Immune score is not correlated with (f) pathologic T (p value = 0.88) and (g) pathologic N (p value = 0.17). Immune score significantly associated with (h) pathologic M (p value = 0.0045) and (i) tumor stage (p value = 0.0093). Immune score is not correlated with (j) age (p value = 0.29). ESTIMATE scores are not correlated with (k) pathologic T (p value = 0.98), (l) pathologic N (p value = 0.73), (m) pathologic M (p value = 0.095), (n) tumor stage (p value = 0.28), and (o) age (p value = 0.74).
3.2. Identification of Differentially Expressed Stromal and Immune Genes for CRC
We analyzed differentially expressed genes (DEGs) with ∣log2FC | >1 and FDR < 0.05 between the high and low stromal/immune score groups. As volcano plots, there were 1197 up- and 28 downregulated stromal genes in the high stromal score group (Figure 4(a)). Furthermore, 899 immune genes were upregulated and eight immune genes were downregulated in the high immune score group (Figure 4(b)). Hierarchical clustering analysis results showed that both stromal DEGs and immune DEGs could distinguish high stromal/immune score from low stromal/immune score (Figures 4(c) and 4(d)). 736 genes were upregulated both in high stromal and immune scores (Figure 4(e)). Moreover, among eight downregulated immune genes, two genes were also downregulated in the high stromal score group (Figure 4(f)). We further performed functional enrichment analysis of these common stromal and immune genes. These genes were significantly correlated with inflammatory biological processes like regulation of inflammatory response and pathways such as cytokine-cytokine receptor interaction and chemokine signaling pathway (Figures 5(a)–5(d)). As shown in the PPI network, COL6A2, COL6A1, COL5A2, C1S, and C1R were the top five genes, which were considered hub genes (Figure 5(e)).
Figure 4.
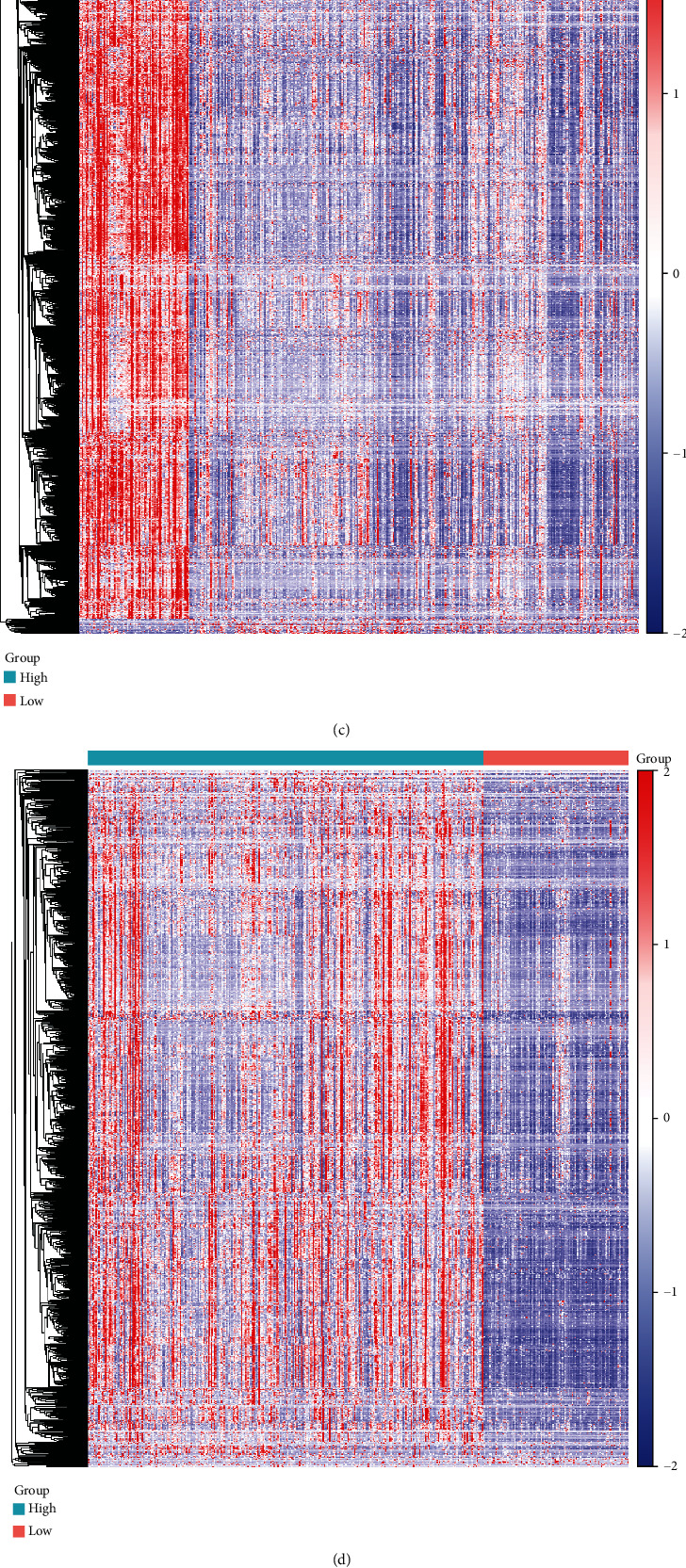
Identification of differentially expressed stromal and immune genes for CRC. Volcano plot showing up- (red) and downregulated (blue) stromal/immune genes in the high stromal score group (a) and immune score group (b). Heat maps depicting all DEGs in the high/low stromal score group (c) and high/low immune score group (d). Venn diagram showing common upregulated (e) and downregulated (f) stromal and immune genes.
Figure 5.

Functional enrichment and PPI analyses of common stromal and immune DEGs. (a) The top the BP terms of GO enrichment analysis. (b) The top ten CC terms of GO enrichment analysis. (c) The top ten MF terms of GO enrichment analysis. (d) The top ten KEGG pathway analysis. (e) PPI network construction.
3.3. Construction of an Eight-Gene Prognostic Signature for CRC
Among 738 differentially expressed stromal and immune genes, 23 genes were significantly associated with CRC patients' prognosis according to univariate Cox regression analysis results. Of them, 20 genes were risk factors, and the remaining three (CCL22, CPA3, and MMP1) were protective factors (Table 2). These genes were used for multivariate Cox regression analysis. Finally, an eight-gene signature was constructed for CRC. The risk score was calculated on the basis of the coefficients and expression values of the eight genes. All CRC patients were divided into two groups in accordance with the median value of risk score (Figure 6(a)). Heat maps depicted the difference in expression patterns of the eight genes (including CD36, KCNE4, CPT1C, SLC2A3, RASGRP2, NFATC1, CCL22, and CPA3) between the high and low risk scores (Figure 6(b)). As shown in Figure 6(c), the risk score was capable of predicting CRC patients' prognosis. High risk score implied a poor prognosis (p value < 0.0001). Among the eight genes, KCNE4 and CCL22 were protective factors of CRC, while CD36, CPT1C, SLC2A3, RASGRP2, NFATC1, and CPA3 were risk factors of CRC, as shown in the forest diagram (Figure 6(d)). We further validated the sensitivity and accuracy of the model. AUCs of the model for 3 years, 5 years, OS, and PFI were 0.71, 0.70, 0.73, and 0.66, respectively (Figures 6(e) and 6(f)). Thus, the risk score model could well predict CRC patients' prognosis, with high sensitivity and accuracy. As shown in the multivariate Cox regression analysis, the model and MMR could become independent factors for CRC prognosis after adjustment of other clinical characteristics (Table 3).
Table 2.
Univariate Cox regression analysis results of 23 differentially expressed stromal and immune genes.
| Variables | HR | Lower 95% CI | Upper 95% CI | p value |
|---|---|---|---|---|
| CD36 | 1.381 | 1.046 | 1.823 | 0.02284 |
| KCNE4 | 1.346 | 1.004 | 1.805 | 0.047303 |
| VEGFC | 1.39 | 1.011 | 1.912 | 0.042907 |
| PDE1B | 2.242 | 1.361 | 3.693 | 0.001518 |
| BGN | 1.166 | 1.003 | 1.355 | 0.046135 |
| CPT1C | 2.561 | 1.476 | 4.446 | 8.26E-04 |
| GPX3 | 1.231 | 1.03 | 1.471 | 0.022285 |
| NGFR | 1.344 | 1.014 | 1.779 | 0.039388 |
| SERPINE1 | 1.173 | 1.01 | 1.362 | 0.037066 |
| CHST1 | 1.448 | 1.02 | 2.055 | 0.038579 |
| FBLN7 | 2.648 | 1.003 | 6.994 | 0.049317 |
| KCNJ8 | 1.372 | 1.003 | 1.876 | 0.047805 |
| SLC2A3 | 1.225 | 1.007 | 1.49 | 0.041957 |
| CD72 | 1.714 | 1.127 | 2.606 | 0.011724 |
| APLP1 | 1.631 | 1.031 | 2.58 | 0.036535 |
| SIGLEC1 | 1.348 | 1.019 | 1.783 | 0.036725 |
| RASGRP2 | 1.641 | 1.044 | 2.579 | 0.031925 |
| SPHK1 | 1.218 | 1.002 | 1.481 | 0.048069 |
| NFATC1 | 1.674 | 1.157 | 2.421 | 0.006228 |
| LRRN2 | 1.653 | 1.139 | 2.398 | 0.008126 |
| CCL22 | 0.686 | 0.51 | 0.923 | 0.012756 |
| CPA3 | 0.81 | 0.68 | 0.966 | 0.019315 |
| MMP1 | 0.902 | 0.816 | 0.996 | 0.042396 |
Figure 6.
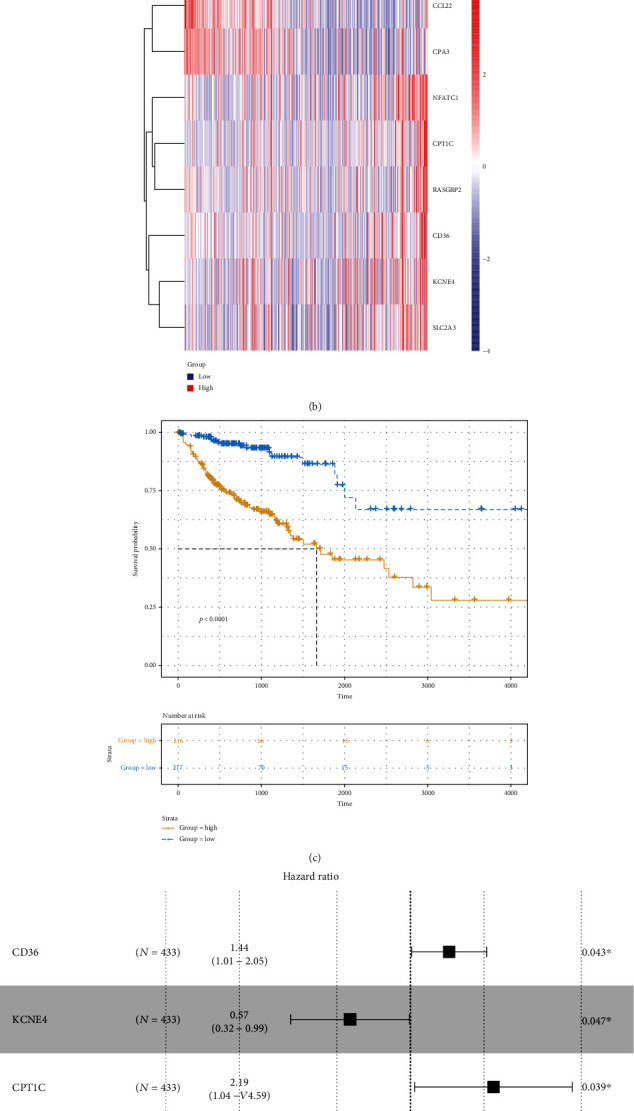
Construction of an eight-gene prognostic signature for CRC. (a) Risk score distribution and survival status. (b) Heat maps showing the expression patterns of the eight genes between high and low risk score. (c) Kaplan-Meier survival analysis of the model. (d) Forest plot of the eight genes for CRC. (e, f) ROC curve of the model for 3-year, 5-year, OS, and PFI.
Table 3.
Univariate and multivariate Cox regression analyses in a TCGA-CRC cohort.
| Characteristics | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Stromal score | 1 (1-1) | 0.653 | NA | NA |
| Immune score | 1 (1-1) | 0.941 | NA | NA |
| Age | 1.396 (0.878-2.22) | 0.158 | NA | NA |
| Gender | 1.127 (0.751-1.692) | 0.564 | NA | NA |
| Tumor stage | 3.064 (1.986-4.726) | <0.0001 | 3.320 (0.870-12.640) | 0.079 |
| Pathologic T | 3.204 (1.398-7.345) | 0.006 | 4.914 (0) | 0.996 |
| Pathologic N | 2.581 (1.705-3.909) | <0.0001 | 0.920 (0.290-2.890) | 0.880 |
| Pathologic M | 3.519 (2.312-5.356) | <0.0001 | 1.620 (0.84-3.13) | 0.151 |
| MMR | 0.181 (0.044-0.751) | 0.019 | 0.070 (0.010-0.500) | 0.009 |
| BRAF | 1.108 (0.620-1.980) | 0.729 | NA | NA |
| KRAS | 0.912 (0.580-1.434) | 0.691 | NA | NA |
| TP53 | 1.461 (0.884-2.417) | 0.139 | NA | NA |
| MSI | 0.907 (0.522-1.575) | 0.728 | NA | NA |
| Risk score | 2.718 (2.063-3.581) | <0.0001 | 2.420 (1.590-3.700) | <0.0001 |
NA: not available.
3.4. Eight Genes in the Risk Score Model Are Significantly Associated with CRC Patients' Prognosis
Box plot depicted the difference in expression patterns of CCL22 (Figure 7(a)), CD36 (Figure 7(b)), CPA3 (Figure 7(c)), CPT1C (Figure 7(d)), KCNE4 (Figure 7(e)), NFATC1 (Figure 7(f)), RASGRP2 (Figure 7(g)), and SLC2A3 (Figure 7(h)) between the high risk score and low risk score. Among them, CCL22 (p value < 2.22e-16), CPA3 (p value < 2.22e-16), CPT1C (p value = 0.00078), KCNE4 (p value = 0.023), NFATC1 (p value = 0.00062), and SLC2A3 (p value = 0.00081) were differentially expressed between the high and low risk scores. Furthermore, the expression levels of these genes between CRC samples and normal samples were visualized (Figures 8(a)–8(g)). CD36 (p value < 2.22e-16), CPA3 (p value < 2.22e-16), NFATC1 (p value = 9.1e-08), and RASGRP2 (p value < 2.22e-16) were highly expressed and SLC2A3 (p value = 0.0015) was lowly expressed in tumor samples. As shown in Figures 9(a)–9(h), low expression of CCL22 (p value = 0.0047) and CPA3 (p value = 0.035) indicated shorter OS time than high expression. Moreover, we found that highly expressed CPT1C (p value = 0.0017), KCNE4 (p value = 0.002), and SLC2A3 (p value = 0.0048) was significantly correlated with poor PFS (Figures 9(i)–9(p)).
Figure 7.
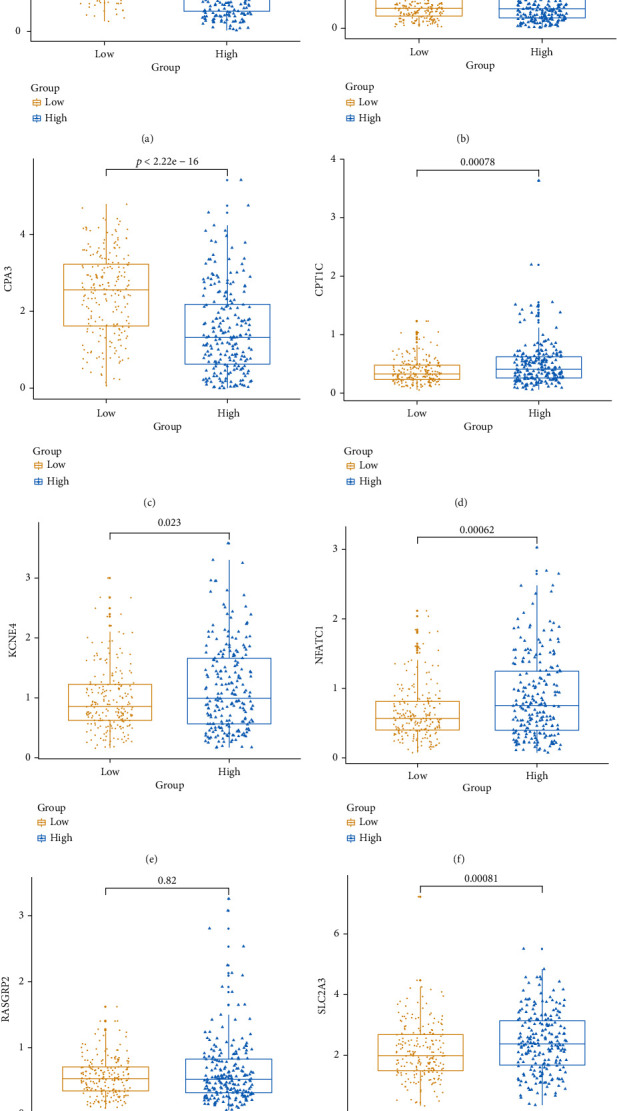
Box plots depicting the expression patterns of the eight genes in the risk score model between high and low risk score. (a) CCL22. (b) CD36. (c) CPA3. (d) CPT1C. (e) KCNE4. (f) NFATC1. (g) RASGRP2. (h) SLC2A3.
Figure 8.

Box plots showing the expression patterns of the eight genes in the risk score model between CRC samples and normal samples. (a) CCL22. (b) CD36. (c) CPA3. (d) CPT1C. (e) KCNE4. (f) NFATC1. (g) RASGRP2. (h) SLC2A3.
Figure 9.
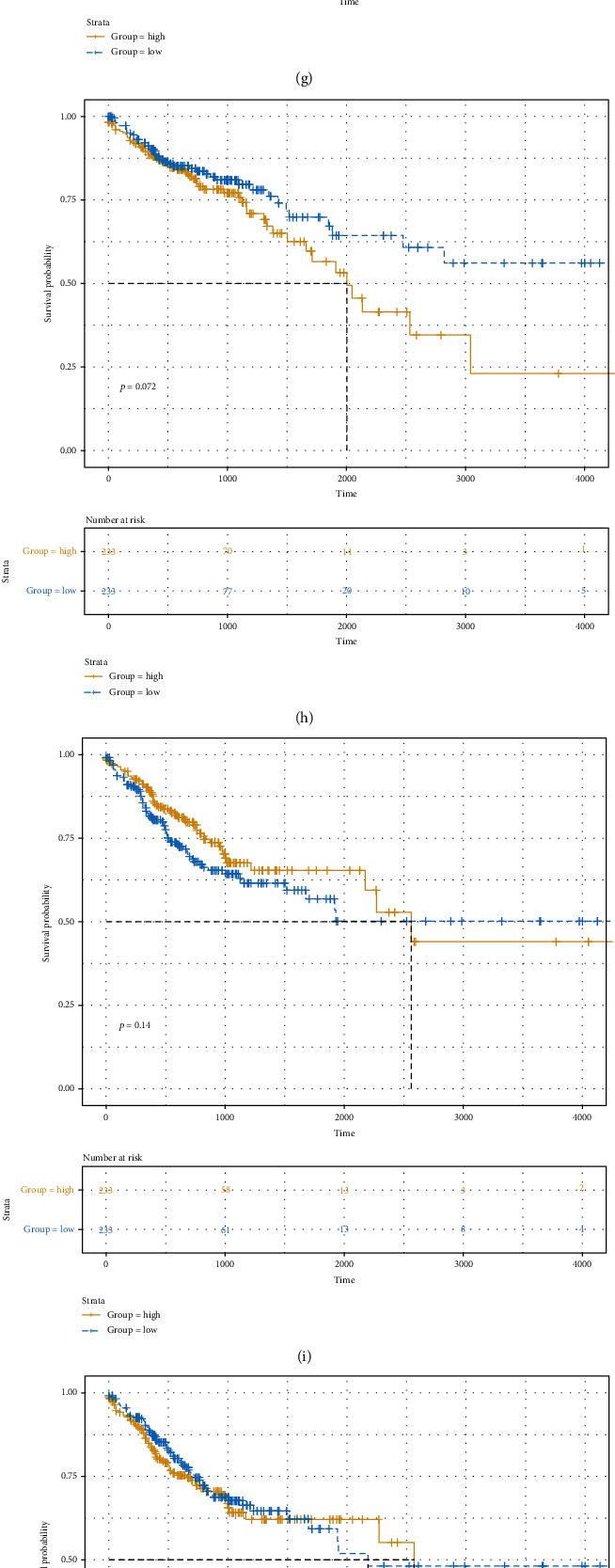
Kaplan-Meier plot of OS and PFS for the eight genes in the risk score model between high and low risk score. Kaplan-Meier OS results for (a) CCL22, (b) CD36, (c) CPA3, (d) CPT1C, (e) KCNE4, (f) NFATC1, (g) RASGRP2, and (h) SLC2A3. Kaplan-Meier PFS results for (i) CCL22, (j) CD36, (k) CPA3, (l) CPT1C, (m) KCNE4, (n) NFATC1, (o) RASGRP2, and (p) SLC2A3.
3.5. The Eight-Gene Model Is in Significant Correlation with Immune Cell Infiltration
The correlation between the model and the infiltrating levels of six immune cells was analyzed. We found that the model was in significant association with the infiltrating levels of six immune cells, including B cell (Figure 10(a); R = 0.13, p value = 0.0064) and CD4+T cell (Figure 10(b); R = 0.21, p value =4.3e-06). However, no significant correlation between the model and CD8+T cell was found in Figure 10(c) (R = 0.045, p value = 0.34). Furthermore, there were distinct correlations between the model and dendritic cell (Figure 10(d); R = 0.12, p value = 0.0072), macrophage (Figure 10(e); R = 0.19, p value = 3.3e-05), neutrophil (Figure 10(f); R = 0.18, p value = 9.4e-05). We also found that the expression levels of the eight genes in the model were significantly correlated with the infiltrating levels of six immune cells, including CCL22 (Figure 11(a)), CD36 (Figure 11(b)), CPA3 (Figure 11(c)), CPT1C (Figure 11(d)), KCNE4 (Figure 11(e)), NFATC1 (Figure 11(f)), RASGRP2 (Figure 11(g)), and SLC2A3 (Figure 11(h)). Moreover, the eight genes were in significant association with markers of immune cells (Supplementary Table 1). These results suggested that the model could be in association with immune cell infiltration.
Figure 10.
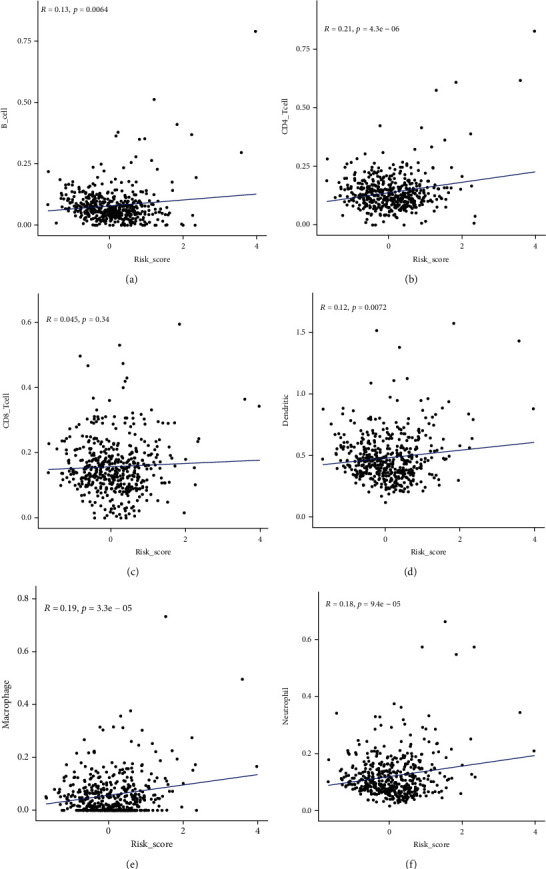
The correlation between the eight-gene risk score and immune cell levels. (a) B cell. (b) CD4+T cell. (c) CD8+T cell. (d) Dendritic cell. (e) Macrophage. (f) Neutrophil.
Figure 11.
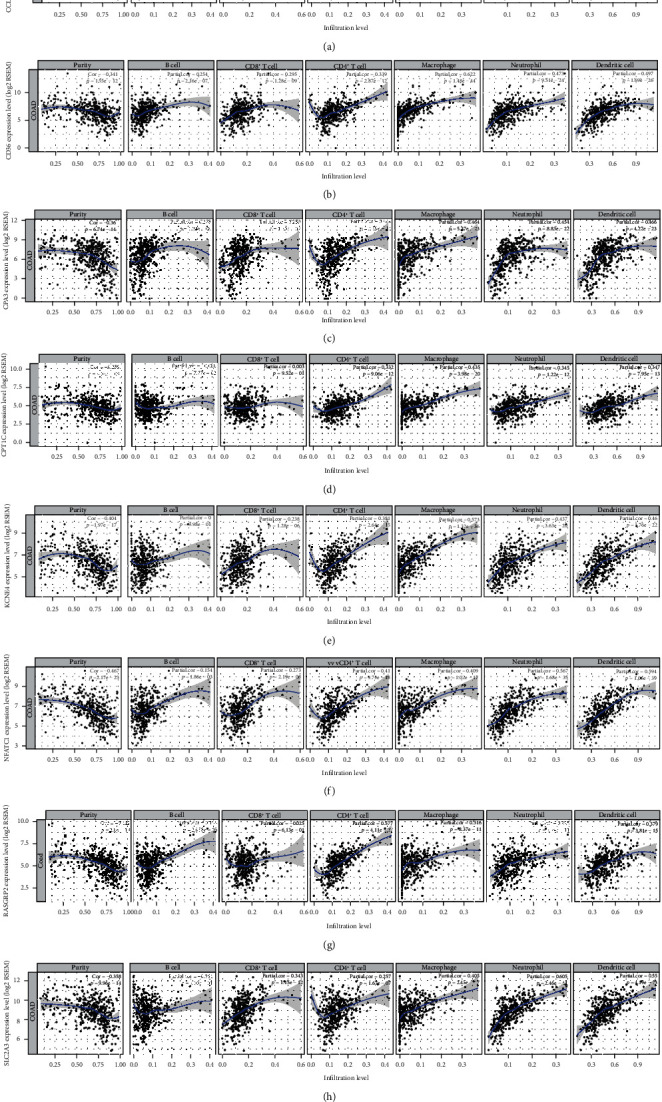
The correlation between the eight genes in the risk score model and immune cell levels. (a) CCL22. (b) CD36. (c) CPA3. (d) CPT1C. (e) KCNE4. (f) NFATC1. (g) RASGRP2. (h) SLC2A3.
3.6. Validation of the Risk Score Using an Independent Dataset
Based on 566 CRC samples from the GSE39582 dataset, the prognostic value of the risk score was validated. The risk score distribution and survival status of CRC patients are shown in Figure 12(a). Heat maps showed the expression differences of CD36, KCNE4, CPT1C, SLC2A3, RASGRP2, NFATC1, CCL22, and CPA3 between the high and low risk scores (Figure 12(b)). As expected, CRC patients with high risk score had a poorer prognosis than those with low risk score (Figure 12(c)). Among the eight genes, CD36, NFATC1, and CCL22 were significantly associated with prognosis of CRC patients (Figure 12(d)). AUCs of the model for 3 years and 5 years were 0.65 and 0.66, respectively (Figure 12(e)), indicating that the risk score could well predict CRC patients' prognosis. The expression levels of CCL22 (Figure 13(a)), CD36 (Figure 13(b)), CPA3 (Figure 13(c)), CPT1C (Figure 13(d)), KCNE4 (Figure 13(e)), NFATC1 (Figure 13(f)), RASGRP2 (Figure 13(g)), and SLC2A3 (Figure 13(h)) between the high risk score and low risk score were validated based on the 566 CRC samples. Univariate Cox regression analysis results showed that age, KRAS mutation, pathologic T, pathologic N, pathologic M, tumor stage, and risk score were notably associated with CRC patients' prognosis. After multivariate Cox regression analysis, we found that age, KRAS mutation, pathologic M, and risk score could be independent prognostic factors for CRC (Table 4).
Figure 12.
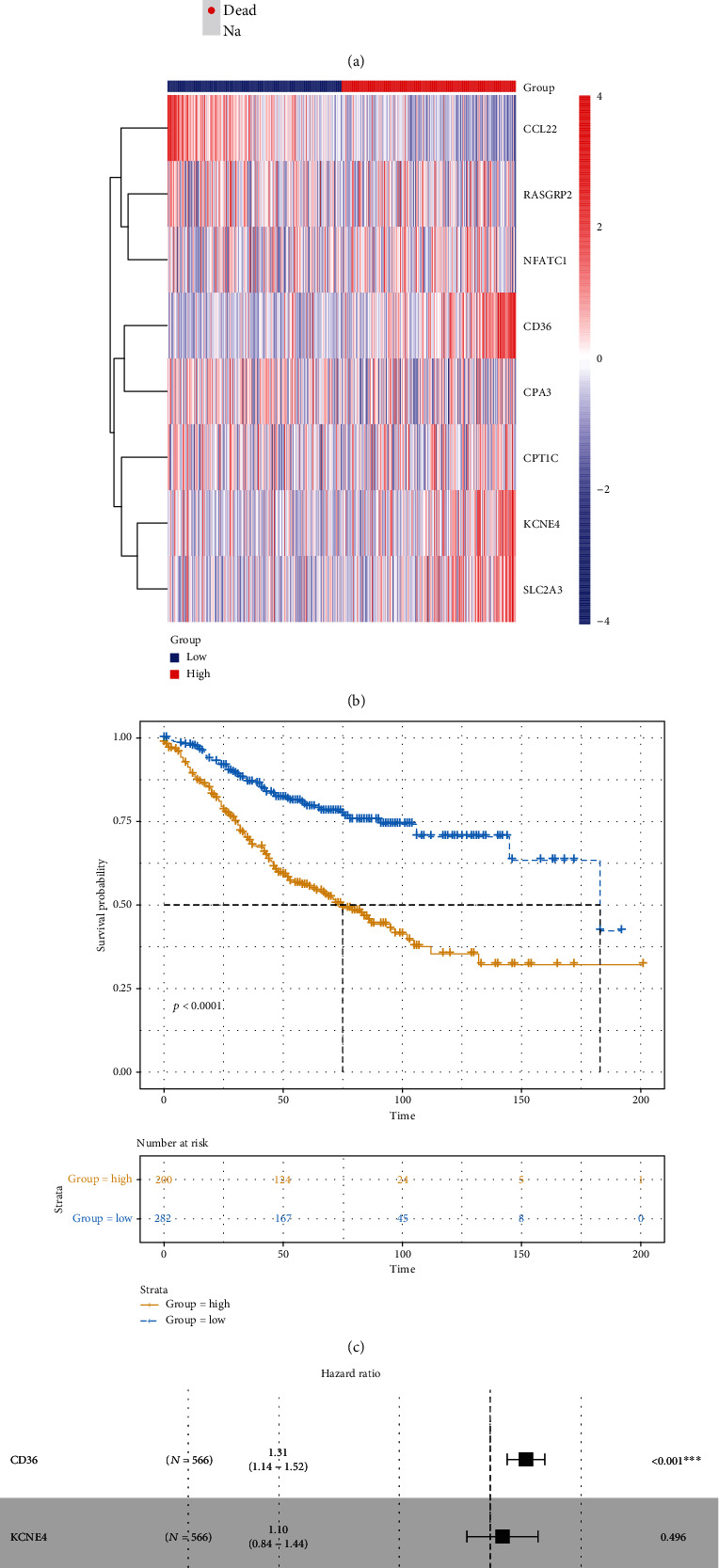
Validation of the eight-gene prognostic signature for CRC using an independent dataset. (a) Risk score distribution and survival status. (b) Heat maps showing the expression patterns of the eight genes between high- and low risk score. (c) Kaplan-Meier survival analysis of the model. (d) Forest plot of the eight genes for CRC. (e) ROC curve of the model for 3 years and 5 years.
Figure 13.
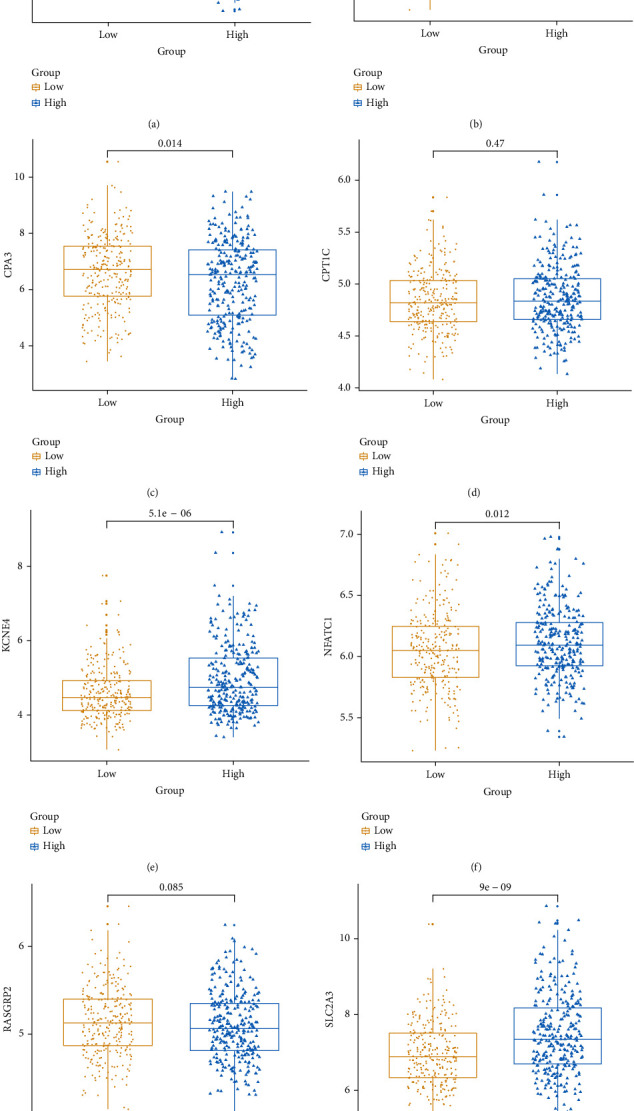
The expression patterns of the eight genes in the risk score model between high and low risk score were validated using an independent dataset. (a) CCL22. (b) CD36. (c) CPA3. (d) CPT1C. (e) KCNE4. (f) NFATC1. (g) RASGRP2. (h) SLC2A3.
Table 4.
Univariate and multivariate Cox regression analyses in the GSE39582 dataset.
| Characteristics | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Age | 1.455 (1.033-2.051) | 0.031 | 1.590 (1.110-2.260) | 0.010 |
| Sex | 1.310 (0.980-1.750) | 0.068 | NA | NA |
| BRAF mutation | 1.111 (0.664-1.861) | 0.688 | NA | NA |
| KRAS mutation | 1.361 (1.018-1.818) | 0.037 | 1.420 (1.040-1.930) | 0.025 |
| TP53 mutation | 1.197 (0.844-1.697) | 0.312 | NA | NA |
| MMR status | 0.768 (0.471-1.252) | 0.290 | NA | NA |
| Pathologic T | 2.005 (1.025-3.921) | 0.042 | 1.510 (0.770-2.970) | 0.229 |
| Pathologic N | 1.648 (1.228-2.213) | 0.0008 | 1.090 (0.500-2.380) | 0.822 |
| Pathologic M | 5.175 (3.621-7.395) | <0.0001 | 3.880 (2.510-5.990) | <0.0001 |
| Tumor stage | 1.767 (1.326-2.354) | 0.0001 | 1.200 (0.520-2.800) | 0.670 |
| Risk score | 2.718 (2.056-3.595) | <0.0001 | 2.420 (1.800-3.260) | <0.0001 |
NA: not available.
3.7. The Eight Genes in the Risk Score Are Distinctly Correlated with Molecular Markers of CRC Prognosis
In Figure 14(a), CCL22 was significantly correlated with BRAF mutation (p value = 0.014) and KRAS mutation (p value = 0.041). For CD36, there was a distinct correlation between its expression and KRAS mutation (p value = 0.00034) and MMR (p value = 0.025) in Figure 14(b). CPA3 (p value = 0.0066; Figure 14(c)) and CPT1C (p value = 0.005; Figure 14(d)) had higher expression levels in KRAS mutation. As shown in Figure 14(e), KCNE4 expression was in significant correlation with BRAF mutation (p value = 0.0014), KRAS mutation (p value = 0.049), and MSI (p value = 0.05). NFATC1 expression was prominently correlated with BRAF mutation (p value = 2.2e-11), KRAS mutation (p value = 0.00051) and MSI (p value = 1.1e-13) in Figure 14(f). In Figure 14(g), RASGRP2 expression was significantly decreased in KRAS mutation. For SLC2A3, we found that there was a distinct correlation between its expression and BRAF mutation (p value = 0.0011) and MSI (p value = 0.00013) in Figure 14(h).
Figure 14.
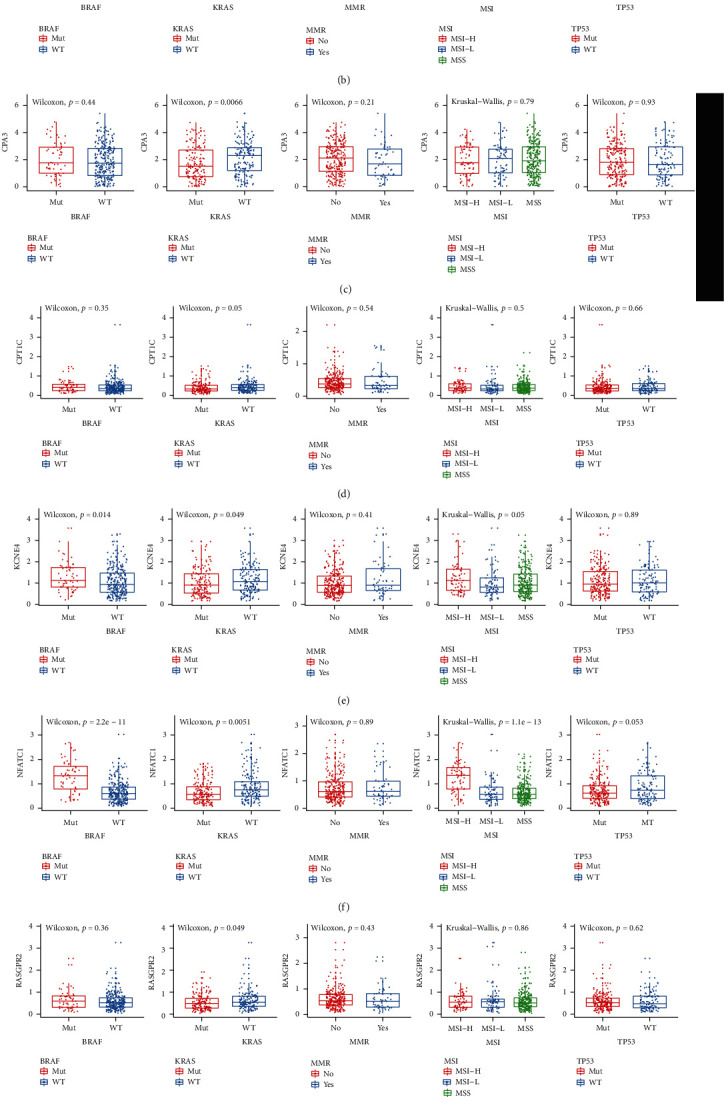
The eight genes in the risk score are distinctly correlated with molecular markers of CRC prognosis including BRAF mutation, KRAS mutation, MMR, MSI, and TP53. (a) CCL22. (b) CD36. (c) CPA3. (d) CPT1C. (e) KCNE4. (f) NFATC1. (g) RASGRP2. (h) SLC2A3.
4. Discussion
In TME, stromal and immune cells are involved in the development of CRC. In this study; using the ESTIMATE algorithm, stromal and immune scores were calculated. A significant correlation between the immune score and CRC patients' prognosis was observed. Both the stromal score and immune score were in significant correlation with clinical characteristics of CRC patients. Furthermore, we identified differentially expressed stromal and immune genes for CRC. Functional enrichment analysis results suggested that these genes were positively related with immune-related pathways like cytokine-cytokine receptor interaction [16, 17] and chemokine signaling pathway [18, 19].
Individual prognosis for CRC patients varies widely. Individual genes often cannot accurately predict the prognosis of patients with CRC. Genes in most prognostic risk scores are screened via differential expression analyses [20–22]. Yet, there are few prognostic models associated with CRC immune infiltration. Therefore, in this study, we selected eight differentially expressed stromal and immune genes related to prognosis for constructing a risk score model. However, focusing only on CRC-related immune-related genes may limit its clinical value. For this reason, through multivariate regression analysis, after adjustment of the clinical characteristics of CRC, we assessed the association between the risk score and CRC prognosis. The results showed that the model may become an independent prognostic factor for CRC. Our risk score exhibited well efficiency in predicting CRC patients' prognosis. Therefore, the risk score model possessed potential prognostic value, which was confirmed using an independent dataset. Among the eight genes in the model, both in the discovery and independent datasets, CCL22 was a protective factor of CRC, while CD36 and NFATC1 were two risk factors of CRC. However, other genes exhibited inconsistent results in the two datasets. This is partly due to the heterogeneity of the samples in the two datasets. Patients in the same pathological stage have different prognosis. Both in the discovery and independent datasets, CCL22 and CPA3 were lowly expressed and KCNE4, NFATC1, and SLC2A3 were highly expressed in the high-risk samples compared to the low-risk samples. However, there were inconsistent results about other genes between the high- and low-risk samples in the two datasets, partly due to the heterogeneity of the samples, different sequencing platforms, different background correction and normalization methods and so on. Thus, it is unreliable to predict CRC patients' prognosis by an individual gene. However, our risk score composed of these genes may accurately suggest the patient's prognosis.
As described in a previous study, high CCL22 expression was found in CRC tissues [23]. Recent study has found that CCL22 secreted by M2 macrophages could mediate CRC 5-FU-mediated chemoresistance [24]. Furthermore, it has been reported that CCL22 was in significant correlation with the infiltrating levels of different T cell subsets for CRC [25]. Our results showed that CD36 was significantly downregulated in CRC tissues compared to normal tissues, which was validated in vitro and in vivo [26]. Genome-wide DNA methylation analysis revealed that hypermethylation of CD36 could contribute to its low expression [27]. Fang et al. found that CD36 expression gradually decreased from adenoma to cancer and CD36 loss implied a poor prognosis in patients with CRC [28]. NFATC1 was deregulated in CRC tissues, which was consistent with previous findings [29]. In vitro, its overexpression significantly promoted CRC cell invasion and metastasis [30]. Kumar et al. reported that NFATC1 indicated poor survival outcomes of CRC patients [31]. High SLC2A3 expression was observed in CRC tissues and its high expression indicated a poor prognosis, consistently with previous research [32, 33]. Furthermore, downregulated CPA3 and RASGRP2 and upregulated CPT1C and KCNE4 were found in CRC tissues, which implied poor prognosis.
As for immune cell infiltration, we found that the eight genes in the risk score model were moderately correlated with the infiltering levels of CD4+T cell, dendritic cell, macrophage, and neutrophil. It has been confirmed that TME affects the efficacy of immunotherapy, and immune cells in TME possess predictive value for immunotherapy treatment [34–36]. Increasing genes have been shown to participate in the regulation of immune cells [37–40]. Therefore, our risk score model could possess potential value to predict CRC patients' prognosis, and the eight genes could become promising immunotherapeutic targets, which deserve further study.
Our correlation analysis results confirmed that the eight genes in the risk score were distinctly correlated with molecular markers of CRC prognosis. However, our study has several limitations. First, our retrospective study limited the application of this risk score. Second, the heterogeneity of the immune microenvironment would inevitably contribute to result bias. Therefore, it is necessary to validate our findings in a prospective clinical study.
5. Conclusion
In this study, we conducted an immune-related prognostic model for CRC on the basis of stromal and immune scores. The model had well predictive efficacy for CRC patients' prognosis. Our findings could provide novel biomarkers for predicting the prognosis of CRC patients and developing individualized immunity therapy strategies.
Abbreviations
- CRC:
Colorectal cancer
- TCGA:
The Cancer Genome Atlas
- ROC:
Receiver operating characteristic
- OS:
Overall survival
- PFI:
Progression-free survival interval
- AUC:
Area under the ROC
- TME:
Tumor microenvironment
- FC:
Fold change
- GO:
Gene Ontology
- KEGG:
Kyoto Encyclopedia of Genes and Genomes
- CC:
Cellular component
- MF:
Molecular function
- BP:
Biological process
- PPI:
Protein-protein interaction
- STRING:
The Search Tool for the Retrieval of Interacting Genes.
Data Availability
The (data type) data used to support the findings of this study are included within the supplementary information file(s).
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
Supplementary Table 1: the correlation between the eight genes in the risk score model and markers of immune cells.
References
- 1.Siegel R. L., Miller K. D., Jemal A. Cancer statistics, 2018. CA: a Cancer Journal for Clinicians. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Becht E., de Reyniès A., Giraldo N. A., et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clinical Cancer Research. 2016;22(16):4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Gomez A., Rodriguez-Ubreva J., Ballestar E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clinical Immunology. 2018;196:64–71. doi: 10.1016/j.clim.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Lambrechts D., Wauters E., Boeckx B., et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nature Medicine. 2018;24(8):1277–1289. doi: 10.1038/s41591-018-0096-5. [DOI] [PubMed] [Google Scholar]
- 5.Isella C., Terrasi A., Bellomo S. E., et al. Stromal contribution to the colorectal cancer transcriptome. Nature Genetics. 2015;47(4):312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- 6.Calon A., Lonardo E., Berenguer-Llergo A., et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nature Genetics. 2015;47(4):320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 7.Galon J., Costes A., Sanchez-Cabo F., et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 8.Zhu X., Chen L., Liu L., Niu X. EMT-mediated acquired EGFR-TKI resistance in NSCLC: mechanisms and strategies. Frontiers in Oncology. 2019;9:p. 1044. doi: 10.3389/fonc.2019.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van den Eynde M., Mlecnik B., Bindea G., et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell. 2018;34(6):1012–1026.e3. doi: 10.1016/j.ccell.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Yoshihara K., Shahmoradgoli M., Martínez E., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Communications. 2013;4(1, article 2612) doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu G., Wang L. G., Han Y., He Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szklarczyk D., Gable A. L., Lyon D., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research. 2019;47(D1):D607–d613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shannon P., Markiel A., Ozier O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li T., Fan J., Wang B., et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Research. 2017;77(21):e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akoglu H. User's guide to correlation coefficients. Turkish Journal of Emergency Medicine. 2018;18(3):91–93. doi: 10.1016/j.tjem.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landskron G., de la Fuente M., Thuwajit P., Thuwajit C., Hermoso M. A. Chronic inflammation and cytokines in the tumor microenvironment. Journal of Immunology Research. 2014;2014:19. doi: 10.1155/2014/149185.149185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West N. R., McCuaig S., Franchini F., Powrie F. Emerging cytokine networks in colorectal cancer. Nature Reviews Immunology. 2015;15(10):615–629. doi: 10.1038/nri3896. [DOI] [PubMed] [Google Scholar]
- 18.Frick V. O., Rubie C., Keilholz U., Ghadjar P. Chemokine/chemokine receptor pair CCL20/CCR6 in human colorectal malignancy: an overview. World Journal of Gastroenterology. 2016;22(2):833–841. doi: 10.3748/wjg.v22.i2.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itatani Y., Kawada K., Inamoto S., et al. The role of chemokines in promoting colorectal cancer invasion/metastasis. International Journal of Molecular Sciences. 2016;17(5):p. 643. doi: 10.3390/ijms17050643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun G., Li Y., Peng Y., et al. Identification of a five-gene signature with prognostic value in colorectal cancer. Journal of Cellular Physiology. 2019;234(4):3829–3836. doi: 10.1002/jcp.27154. [DOI] [PubMed] [Google Scholar]
- 21.Wang X., Zhou J., Xu M., et al. A 15-lncRNA signature predicts survival and functions as a ceRNA in patients with colorectal cancer. Cancer Management and Research. 2018;10:5799–5806. doi: 10.2147/CMAR.S178732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., Liu Q., Wang P., et al. Development and internal validation of a nine-lncRNA prognostic signature for prediction of overall survival in colorectal cancer patients. PeerJ. 2018;6, article e6061 doi: 10.7717/peerj.6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y. H., Cao Y. F., Jiang Z. Y., Zhang S., Gao F. Th22 cell accumulation is associated with colorectal cancer development. World Journal of Gastroenterology. 2015;21(14):4216–4224. doi: 10.3748/wjg.v21.i14.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei C., Yang C., Wang S., et al. M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. Oncotargets and Therapy. 2019;12:3051–3063. doi: 10.2147/OTT.S198126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cremonesi E., Governa V., Garzon J. F. G., et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut. 2018;67(11):1984–1994. doi: 10.1136/gutjnl-2016-313498. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X., Yao J., Shi H., Gao B., Zhang L. LncRNA TINCR/microRNA-107/CD36 regulates cell proliferation and apoptosis in colorectal cancer via PPAR signaling pathway based on bioinformatics analysis. Biological Chemistry. 2019;400(5):663–675. doi: 10.1515/hsz-2018-0236. [DOI] [PubMed] [Google Scholar]
- 27.Li M., Chen W., Sun X., et al. Metastatic colorectal cancer and severe hypocalcemia following irinotecan administration in a patient with X-linked agammaglobulinemia: a case report. BMC Medical Genetics. 2019;20(1):p. 157. doi: 10.1186/s12881-019-0880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang Y., Shen Z. Y., Zhan Y. Z., et al. CD36 inhibits β-catenin/c-myc-mediated glycolysis through ubiquitination of GPC4 to repress colorectal tumorigenesis. Nature Communications. 2019;10(1, article 3981) doi: 10.1038/s41467-019-11662-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pudova E. A., Kudryavtseva A. V., Fedorova M. S., et al. HK3 overexpression associated with epithelial-mesenchymal transition in colorectal cancer. BMC Genomics. 2018;19(Supplement 3):p. 113. doi: 10.1186/s12864-018-4477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tripathi M. K., Deane N. G., Zhu J., et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Research. 2014;74(23):6947–6957. doi: 10.1158/0008-5472.CAN-14-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R., Raman R., Kotapalli V., et al. Ca2+/nuclear factor of activated T cells signaling is enriched in early-onset rectal tumors devoid of canonical Wnt activation. Journal of Molecular Medicine (Berlin, Germany) 2018;96(2):135–146. doi: 10.1007/s00109-017-1607-4. [DOI] [PubMed] [Google Scholar]
- 32.Kim E., Jung S., Park W. S., et al. Upregulation of SLC2A3 gene and prognosis in colorectal carcinoma: analysis of TCGA data. BMC Cancer. 2019;19(1):p. 302. doi: 10.1186/s12885-019-5475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Romero J., Bueno-Fortes S., Martín-Merino M., Ramirez de Molina A., de Las Rivas J. Survival marker genes of colorectal cancer derived from consistent transcriptomic profiling. BMC Genomics. 2018;19(Supplement 8):p. 857. doi: 10.1186/s12864-018-5193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng B., Zhou F., Hou B., et al. Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Advanced Materials. 2018;30(38, article 1803001) doi: 10.1002/adma.201803001. [DOI] [PubMed] [Google Scholar]
- 35.Song W., Tiruthani K., Wang Y., et al. Trapping of lipopolysaccharide to promote immunotherapy against colorectal cancer and attenuate liver metastasis. Advanced Materials. 2018;30(52, article 1805007) doi: 10.1002/adma.201805007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tauriello D. V. F., Palomo-Ponce S., Stork D., et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 37.Zhao J., Ou B., Han D., et al. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3β/β-catenin pathways. Molecular Cancer. 2017;16(1):p. 70. doi: 10.1186/s12943-017-0629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin Y., Yao S., Hu Y., et al. The immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived IL6. Clinical Cancer Research. 2017;23(23):7375–7387. doi: 10.1158/1078-0432.CCR-17-1283. [DOI] [PubMed] [Google Scholar]
- 39.Gil J., Ramsey D., Pawlowski P., et al. The influence of tumor microenvironment on ATG4D gene expression in colorectal cancer patients. Medical Oncology. 2018;35(12):p. 159. doi: 10.1007/s12032-018-1220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L., Wang G., Qiao X., et al. Downregulated miR-524-5p participates in the tumor microenvironment of ameloblastoma by targeting the Interleukin-33 (IL-33)/suppression of tumorigenicity 2 (ST2) Axis. Medical Science Monitor. 2020;26, article e921863 doi: 10.12659/msm.921863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: the correlation between the eight genes in the risk score model and markers of immune cells.
Data Availability Statement
The (data type) data used to support the findings of this study are included within the supplementary information file(s).