

Abstract
Pyridine nucleotides, such as NADPH and NADH, are emerging as critical players in the regulation of heart and vascular function. Glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the pentose phosphate pathway, is the primary source and regulator of cellular NADPH. In the current study, we have identified two isoforms of G6PD (slow and fast migrating) and functionally characterized the slow migrating isoform of G6PD (G6PD545) in bovine and human arteries. We found that G6PD545 is eluted in the caveolae fraction of vascular smooth muscle (VSM) and has a higher maximum rate of reaction (Vmax: 1.65-fold) than its fast migrating isoform (G6PD515). Interestingly, caveolae G6PD forms a complex with the pore-forming α1C-subunit of the L-type Ca2+ channel, Cav1.2, as demonstrated by a proximity ligation assay in fixed VSMCs. Additionally, Förster resonance energy transfer (FRET) analysis of HEK293-17T cells cotransfected with red fluorescent protein (RFP)-tagged G6PD545 (C-G6PD545) and green fluorescent protein (GFP)-tagged Cav1.2-(Cav1.2-GFP) demonstrated strong FRET signals as compared with cells cotransfected with Cav1.2-GFP and C-G6PD515. Furthermore, L-type Ca2+ channel conductance was larger and the voltage-independent component of availability (c1) was augmented in C-G6PD545 and Cav1.2-GFP cotransfectants compared with those expressing Cav1.2-GFP alone. Surprisingly, epiandrosterone, a G6PD inhibitor, disrupted the G6PD-Cav1.2 complex, also decreasing the amplitude of L-type Ca2+ currents and window currents, thereby reducing the availability of the c1 component. Moreover, overexpression of adeno-G6PD545-GFP augmented the KCl-induced contraction in coronary arteries compared with control. To determine whether overexpression of G6PD had any clinical implication, we investigated its activity in arteries from patients and rats with metabolic syndrome and found that G6PD activity was high in this disease condition. Interestingly, epiandrosterone treatment reduced elevated mean arterial blood pressure and peripheral vascular resistance in metabolic syndrome rats, suggesting that the increased activity of G6PD augmented vascular contraction and blood pressure in the metabolic syndrome. These data suggest that the novel G6PD-Cav1.2 interaction, in the caveolae fraction, reduces intrinsic voltage-dependent inactivation of the channel and contributes to regulate VSM L-type Ca2+ channel function and Ca2+ signaling, thereby playing a significant role in modulating vascular function in physiological/pathophysiological conditions.
NEW & NOTEWORTHY In this study we have identified a novel isozyme of glucose-6-phosphate dehydrogenase (G6PD), a metabolic enzyme, that interacts with and contributes to regulate smooth muscle cell l-type Ca2+ ion channel function, which plays a crucial role in vascular function in physiology and pathophysiology. Furthermore, we demonstrate that expression and activity of this novel G6PD isoform are increased in arteries of individuals with metabolic syndrome and in inhibition of G6PD activity in rats of metabolic syndrome reduced blood pressure.
Keywords: diabetes, human, ion channels, NADPH
INTRODUCTION
Glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the pentose phosphate pathway (PPP), generates >60% of NADPH that is essential for the following: 1) maintaining redox poise; 2) anabolic/synthetic reactions; and 3) producing ribose sugars needed for de novo nucleic acid synthesis in the cell. After the discovery of the PPP and the initial work establishing the primary role of G6PD in glucose metabolism, the significance of G6PD in other aspects of cell physiology was relatively unexplored (27, 50). Typically, G6PD is thought of as a housekeeping enzyme, whose activity is detected in multiple locations within cells, including the sarco(endo)plasmic reticulum, cytosol, peroxisomes, and periphery (11). Furthermore, G6PD expression reportedly exhibits adaptive regulation, such that it is constitutively expressed in some tissues but transcriptionally and posttranslationally regulated in others (6, 31, 32, 52). Consistent with these findings, our laboratory demonstrated that increases in G6PD activity in coronary artery (CA) are PKC-, phosphatidylinositol 3-kinase (PI3K)-, and phosphatase and tensin homolog (PTEN) dependent (1, 13), whereas such increases in rat liver and endothelial cells are Src kinase dependent (14, 43). Other studies have suggested the presence of two or more G6PD isoforms in plants, yeast, and the bark beetle (8, 51, 53). However, the subcellular localization and the number of G6PD isoforms in vascular smooth muscle cells (VSMCs) have not been studied.
In addition to the traditional role of G6PD as an antioxidant, recent studies have suggested that G6PD-derived NADPH fuels NADPH oxidase-dependent superoxide oxide production: in cardiac myocytes and endothelial cells cultured in high-glucose media (3, 54); in vascular and cardiac tissue from obese-diabetics and diabetic rats (26, 48); and in adipose tissue and liver of diabetic mice (25) and humans (46). Consistently, previously we found that inhibition or knockdown of G6PD reduces superoxide generation and relaxes precontracted arteries by reducing the membrane depolarization-induced Ca2+ influx and increasing Ca2+ uptake by the sarcoplasmic reticulum (13, 16, 21). These observations suggest the existence of a membrane-bound G6PD in mammalian cells, which may account for membrane-dependent novel functions of G6PD, which may be critical in regulating the redox poise and the voltage-gated Ca2+ channel function in VSMCs.
This study was undertaken to identify the different G6PD isoforms and their subcellular location in the VSMCs and to elucidate the mechanism by which G6PD influences membrane function. We identified two isoforms of G6PD (slow and fast migrating) and found that their expression was compartmentalized in the CA and coronary artery smooth muscle cells (CASMCs). Furthermore, the slow migrating G6PD formed a complex with the pore-forming α1C subunit of L-type Ca2+ channel Cav1.2. Interestingly, epiandrosterone (EPI), a G6PD inhibitor, dissociated this complex and induced an increase of voltage-dependent inactivation as explained by the appearance of intrinsic inactivation of the Cav1.2 channel. Taken together, our data demonstrated the existence of two G6PD isoforms in the VSMCs, which play crucial but distinct roles in regulating Ca2+ influx and signaling, which evoke VSM contraction in the physiological and pathophysiological state.
MATERIALS AND METHODS
Tissue Preparation
Bovine.
The left anterior descending and circumflex coronary arteries [bovine coronary artery (BCA)] were harvested from hearts of male Angus cows purchased from a slaughterhouse (Stuckey's Meat Packer, Semmes, AL). After the heart was quickly excised from the animal, it was placed in normal Tyrode’s solution (in mmol/L: 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 5 HEPES, and 11 glucose; pH was adjusted to 7.40 with NaOH) and transported to the laboratory on ice. All animal work was approved by Institutional Biosafety Committee and Institutional Animal Care and Use Committee at University of South Alabama.
Human.
Surgical human internal mammary artery (HIMA) discards were collected from de-identified patients undergoing coronary artery bypass graft surgery at Westchester Medical Center, Valhalla, NY, between 2000 and 2004 as described previously (17, 23). These samples were transported to the laboratory in ice-cold normal Tyrode’s solution and immediately used for vascular function and biochemical studies. HIMA were collected based on Institutional Review Board policy and approval to use discarded de-identified patient material, including a waiver of informed consent, was obtained as described previously (17, 23). Therefore, the demographics, including sex, of the patients were unknown.
G6PD-Deficient (G6PDDef) Mice
G6PDDef mice (male, 25–30 g) that harbor a polymorphism within the 5′-UTR of G6pd were provided by Dr. Jane Leopold (Brigham’s Women Hospital, Harvard School of Medicine, Boston, MA). Metabolic syndrome (JCR) rats (males, 600–800 g) were provided by Dr. Spencer Proctor (University of Alberta, Edmonton, Alberta, Canada) to Dr. Petra Rocic (New York Medical College, NY). Sprague-Dawley (SD) rats were used as the physiological control for JCR.
Cells and Culture Media
Bovine and human coronary artery smooth muscle cells (BCASMCs and HCASMCs) were purchased from Cell Application, Inc. (San Diego, CA). Human embryonic kidney (HEK) cell line HEK293-17T cells were purchased from ATCC. BCASMCs and HEK293-17T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) and 10% fetal bovine serum. For serum-free studies, the BCASMCs were cultured in the absence or presence of serum (10%). HCASMCs were cultured in HSMC growth media from Cell Application (San Diego, CA).
Reagents
All reagents used in the present study were purchased from Sigma Chemical (St. Louis, MO) and Cayman Chemical (Ann Arbor, MI).
Western Blot Analysis
Western blot analyses were performed as described previously (20). Briefly, lysates were prepared from frozen tissue (20 mg) in lysis buffer (50 mM Tris·HCl pH 7.4, 150 M NaCl, and 0.5% Nonidet P-40) containing EDTA-free complete protease and phosphatase inhibitor (Sigma-Aldrich/Roche). Protein levels were measured using Bradford assays (Bio-Rad), after which 35-μg samples were run on 7–9% SDS-PAGE gels, transferred to nitrocellulose membranes and blocked with either 5% bovine serum albumin (BSA) or 4% milk. The blots were subsequently incubated with respective primary, and secondary antibodies as noted below and specific proteins were detected using SuperSignal West Pico Chemiluminescent Substrate kit (ThermoFisher Scientific).
Antibodies
Western blot analyses were performed using following antibodies mentioned below and in the respective figures. Polyclonal anti-G6PD (for Western blot), monoclonal anti-β-actin, monoclonal anti-LDH, and polyclonal anti-glucose kinase antibodies were purchased from Santa Cruz (Dallas, TX). A monoclonal anti-red fluorescent protein (RFP) antibody was purchased from Clontech (Mountain View, CA), monoclonal anti-caveolin-1 antibody was purchased from Cell Signaling Technology (Danvers, MA), and monoclonal anti-Cav1.2 antibody (NeuroMab clone N263/31) was purchased from University of California (UC) Davis (Sacramento, CA). Monoclonal anti-Na+-K+-ATPase, monoclonal anti-dihydropyridine (DHP)-α2, and polyclonal anti-G6PD (for immunoprecipitation) antibodies were purchased from Sigma and Santa Cruz. The specificity of the following: 1) anti-G6PD antibody was determined by our laboratory by knocking down G6PD in BCASMCs; and Western blot analyses of G6PD expression in nontransfected and human G6PD transfected HEK 293T whole cell lysate by Santa Cruz (sc-67165); 2) anti-Cav1.2 antibody was determined by transiently transfected COS cells with untagged Cav1.2, Cav1.3, or Kv2.1 plasmids and probed with N263/31 by the vendor (UC Davis NeuroMab Data Sheet); 3) anti-LDH and -glucose kinase antibodies was verified and published previously (33, 47); and 4) anti-Na+K+ATPase and -DHP, widely used antibodies, were characterized previously (2, 39).
Reverse Transcription and PCR
Total RNA was isolated from cultured HCASMCs and BCASMCs using RNeasy mini kits (Qiagen, Valencia, CA). First-strand cDNA was synthesized using a SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Random hexamers (RHx; 50 ng·μL−1·reaction−1) and OligodT20 (OdT; 50 μM/reaction) were used, as recommended by the manufacturer for reverse transcription. The following primers for G6PD545 gene (1,638 bp; NM_000402.4): forward: 5′-GTCGACATGGGCCGGCGGGG-3′ and reverse: 5′-AAAGCTTGAGCTTGTGGGGGTTCACCCAC-3′ were used for PCR amplification. The PCR products were run on 1% agarose gel along with a DNA marker: M; New England Biolabs, Ipswich, MA). Following subcloning, the pDNAs were send for DNA sequencing (Davis Sequencing, Davis, CA).
Construction of RFP-Tagged G6PD pDNAs and Overexpression in HEK293-17T Cells
Human full-length G6PD545 (1,638 bp; NM_000402.4) and G6PD515 (1,548 bp; NM_001042351.3) pDNAs cloned into the pCMV6-XL5 vector were purchased from Origene Technologies, Inc. G6PD545 and G6PD515 full-length cDNAs were isolated by PCR and cloned into the p-Ds-RedN1 expression vector (Clontech) between the Nhe I and Xho I sites such that the G6PD545/515 genes were ligated to the NH2-terminus of the RFP gene (C-G6PD545/515), respectively, leaving the NH2-terminus of G6PD545/515 genes free. Also, a full-length G6PD545 cDNA was cloned into p-Ds-RedC1 expression vector (Clontech) between the Xho I and Sal I sites, such that the G6PD545 genes were ligated to the COOH-terminus of RFP gene (N-G6PD545), leaving the COOH-terminus of G6PD545 gene free. HEK293-T17 cells (106) were incubated for 48 h in 24-well plates and then transfected with 0.8 µg of p-Ds-Red vector or G6PD545/515-RFP pDNAs, respectively, using 2 mg of FUGENE 6 reagent (Roche) for 48 h.
Construction of G6PD545 and G6PD515-Coral Green Fluorescent Protein Chimers in an Adenoviral Vector and Transfection in Bovine Coronary Artery
The full-length cDNA sequence of G6PD545 (1,638 bp) and G6PD515 (1,548 bp) was isolated by PCR and cloned into a pshuttle-CMV adenoviral (Ad) vector (Genscript) between the Sal I and Hind III restriction sites, such that the coral green fluorescent protein (GFP) gene was ligated at COOH-terminus of G6PD545 and G6PD515. Bovine CA rings (2 × 2 mm) were incubated for 48 h in 24-well plates and transfected with 5.0 µg of adenoviral vector alone (Ad-GFP-Ctrl) or adenoviral G6PD545-GFP (Ad-G6PD545-GFP) and G6PD515-GFP (Ad-G6PD515-GFP), respectively, using 20.0 µg of Lipofectamine 2000 for 48 h, as described previously (13).
Construction of Cav1.2-GFP pDNA and Förster Resonance Energy Transfer Analysis in HEK293-T17 Cells
Cav1.2-GFP pDNA was a gift from Dr. Nagomi Kuryabashi (Juntendo University School of Medicine, Tokyo, Japan). HEK293-T17 cells were plated in 24-well plates at a density of 106/well and incubated for 48 h. They were then cotransfected with 0.8 µg of 1) Cav1.2-GFP + N-G6PD545; 2) Cav1.2-GFP + C-G6PD545; and 3) Cav1.2-GFP + N-G6PD515, separately, using 2 mg of FUGENE 6 reagent (Roche), for 48 h. Förster resonance energy transfer (FRET) experiments were performed on a Leica TCS SP2 confocal Microscope (×63 oil objective) using the sensitized emission approach. Spectral bleed through was corrected for by measuring donor only and acceptor only samples. Regions of interest (ROIs) were selected and FRET efficiency calculated with Leica software using the formula EA (i) = [B − A × b − C × (c − a × b)]/C, where EA is the apparent FRET efficiency; A, B, and C are the donor, FRE, and acceptor channel intensities, respectively; and a, b, and c are correction factors. FRET signals determined using the sensitized emission approach were confirmed using acceptor photobleaching.
G6PD Knockdown
BCASMCs were cultured in DMEM, in a 24-well plate at 37°C for 72 h and were then transfected with 1) Smart pool-G6PD-siRNA (100 nmol/L; Dharmacon) targeting the four exons in the G6PD gene that are common to both the G6PD545 (slow migrating) and G6PD515 (fast migrating) isoforms; or 2) with siRNA (100 nmol/L) targeting only the G6PD545 (5′ CGGAAACGGUCGUACACUUCG 3′; Ambion/Life Technologies, Carlsbad, CA), using 10 mg of Lipofectamine 2000 reagent (Invitrogen), for 67 h. Control experiments were performed using a nontargeting/scrambled (NT) siRNA (negative control).
G6PD Activity
G6PD activity was measured in homogenates (50 μL) by following the reduction of NADP+ to NADPH, as published previously (20). Briefly, G6PD enzyme activity was determined by measuring the rate of conversion of NADP+ to NADPH. Substrate concentrations used were glucose-6-phosphate (G6P; 200 μM) and NADP+ (100 μM). NADPH fluorescence (excitation: 340 nm; emission: 460 nm) was detected using an Flx800 microplate and Synergy 2 fluorescence detectors (BioTek Instruments, Winooski, Vermont).
Cell Fractionation and Cholesterol Studies
Bovine coronary artery (BCA) homogenates were prepared by centrifuging a filtered homogenate suspension at 29,000 g and 5°C. The supernatant obtained was transferred to a Beckman ultracentrifuge equipped with a Ti50 rotor and centrifuged for 60 min at 100,000 g, as described previously (38). Following centrifugation, the membrane fraction was further purified on a Ficol gradient, and the cholesterol-rich caveolae fraction was isolated. Cholesterol was measured using a kit purchased from Cayman Chemical (MI) following the manufacturer’s instructions.
NADP(H) Levels
NADP(H) levels were measured by HPLC using an Elite LaChrom Chromatography System (Hitachi Corporation, Tokyo, Japan), as described previously (22).
Coimmunoprecipitation Assays
Bovine coronary rings were pretreated with contractile agents for various times indicated in the specific figures, after which they were homogenized in lysis buffer containing 1 mM Na3VO4 and 1 mM NaF, as described previously (15). The precleared lysates were incubated with the polyclonal anti-G6PD antibody (Sigma) for 3 h at 4°C. The immune complexes were then immobilized on agarose-A and G beads overnight at 4°C, followed by Western blot analyses using antibodies as mentioned in Fig. 3. Specific proteins for immunoblotting were detected using chemiluminescence, or the proteins were eluted and analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS; Applied Biomics, Hayward, CA). NanoLC fractionation and matrix-assisted laser desorption ionization-time of flight/time of flight (MALDI-TOF/TOF) were followed by a standard search of the National Center for Biotechnology Information and SwissProt databases, using MASCOT.
Fig. 3.

Glucose-6-phosphate dehydrogenase (G6PD) interacts with Cav1.2, a α1c-subunit of L-type Ca2+ channels. A: whole cell homogenates obtained from bovine coronary artery (BCA) were immunoprecipitated (IP) using anti-G6PD antibody and immunoblotted (IB) with anti-Cav1.2 (A,i) and anti-dihydropyridine (DHP)-α2 (A,ii) antibodies, respectively. B: in separate experiments, whole cell homogenates obtained from BCA were subjected to coimmunoprecipitation assays with anti-G6PD and anti-glucose kinase (GK) antibodies, respectively, and immunoblotted with anti-Cav1.2 antibody. C: G6PD-Cav1.2 interaction was detected in fixed vascular smooth muscle cells (VSMCs) using Duolink Proximity Ligation Assay (PLA). The pictures show a maximum intensity projection of the raw image. C,i and C,ii: the image was acquired in one z-plane. Assays were performed in bovine coronary artery smooth muscle cells (BCASMCs) without the presence (C,i) of the anti-G6PD antibody (without 1 Ab) and in the presence (C,ii) of anti-G6PD antibody (with 1°Ab). PLA signals are shown in red (positive reaction) and the nuclei in blue (negative reaction). C,iii and C,iv: BCASMCs were treated with epiandrosterone (EPI; 100 μM) for an hour, and G6PD-Cav1.2 interaction was assessed using Duolink PLA in the absence and presence of anti-G6PD antibody. C,v: summary data of pixel density-to-area ratio from PLA experiment are shown (No-Ab: n = 20 observation/cells from 5 separate experiments; Ab: 25 observation/cells from 5 separate experiments; and Ab + EPI: 20 observations/cells from 5 separate experiments). D: schematic representation of human Cav1.2-green fluorescent protein (GFP) and red fluorescent protein (RFP)-tagged G6PD constructs. E: G6PD activity was determined for C-G6PD545 and C-G6PD515, respectively. Enzyme kinetics demonstrated that G6PD545 had a higher Vmax than G6PD515; n = 5 experiments in each group. F: Förster resonance energy transfer (FRET) was performed using HEK293-17T cells that were cotransfected with Cav1.2-GFP and RFP-G6PD constructs. Graph represents the cumulative data from FRET analyses demonstrating energy transfer efficiency between Cav1.2-GFP + N-G6PD545 (left) (n = 17 observations from 5 separate experiments) Cav1.2-GFP + C-G6PD545 (middle) (n = 5 observations from 5 separate experiments), and Cav1.2-GFP+C-G6PD515 (right) (n = 8 observations from 5 separate experiments), respectively.
Isolation of BCASMCs
The endothelium was removed, and the medial layer of the BCA was finely cut in pieces and then digested with a collagenase (91 mg/mL) solution containing soybean trypsin inhibitor (0.25 mg/mL) and elastase (0.125 mg/mL) in 20 mM MOPS-KOH buffer (pH 7.4) containing 250 mM sucrose (1 g tissue/2 mL buffer) at 37°C for 15 min, as described previously (22). Freshly isolated BCASMCs were used for the Proximity Ligation Assay and to record L-type Ca2+ currents described below.
Proximity Ligation Assay for Detection of Protein-Protein Interaction
BCASMCs were cultured in tissue culture slides and incubated without or with epiandrosterone (100 μM) or control for 1 h. Protein-protein interaction between G6PD and Cav1.2 was detected using Duolink in situ Proximity Ligation Assay kit (Sigma-Aldrich). Briefly, cells were fixed in 10% formalin (in PBS, pH7.0) for 30 min at room temperature followed by three washes in PBS. After the postfixation PBS washes, cells were permeabilized with 0.25% Triton-X100 for 10 min at room temperature followed by three washings in PBS. Blocking was performed with Duolink Blocking Solution in a heated humidity chamber for 60 min at 37°C. Samples were stained with 2 μg/mL primary rabbit anti-G6PD antibody and mouse anti-Cav1.2 antibody diluted in Duolink Antibody Diluent and incubated and incubated in the humidity chamber for 60 min at 37°C. Next, cells were incubated in PLUS and MINUS Proximity Ligation Assay (PLA) secondary proximity probes diluted 1:5 in the Duolink Antibody Diluent and the slides were then incubated in a preheated humidity chamber for 1 h at 37°C followed by incubation in ligase (1:40 in 1× ligation buffer) in a preheated humidity chamber for 30 min at 37°C. Finally, polymerase (1:80 in 1× amplification buffer) was added and the slides were incubated in a preheated humidity chamber for 100 min at 37°C and washed two times for 10 min in 1× wash buffer at room temperature and in 0.01× Wash Buffer B for 1 min. Slides were mounted in Duolink in situ mounting medium with DAPI for 15 min before analyzing in a fluorescent or confocal microscope, using a ×20 objective.
Electrophysiology
L-type Ca2+ channel currents (ICa,L) were recorded and analyzed as described in detail previously (41). Briefly, cells were dispersed on a cover glass in a chamber mounted on an inverted microscope (IX72, Olympus, Tokyo, Japan). After attachment of the cells to the cover glass, the chamber was superfused, first with normal Tyrode’s solution, then with 10 mM Ba2+ solution for ~40 min before the first ICa,L was recorded. Patch pipettes were pulled from hard glass capillary tubing containing a glass filament using a micropipette puller (P-97 Sutter Instrument Co., Novato, CA), coated with silicone elastomer (Sylgard 184, Dow Corning Co., Midland, MI), and fire-polished using a microforge (MF-830, Narishige, Tokyo, Japan). The pipette resistance was ~10 MΩ when filled with pipette solution. The voltage-clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA) was driven by Clampex 10 software via a digital interface (Digidata 1400, Molecular Devices). Currents were filtered at 2 kHz using the amplifier's low-pass eight-pole Bessel filter and digitized at 10 kHz before being stored on a computer hard drive for later analysis. Membrane capacitance was obtained by applying a negative-going ramp step, as well as by using the built-in program in Clampex. The holding potential (HP) was −60 or −80 mV. The current-voltage (I-V) relationship for ICa,L was obtained by applying 500-ms depolarization steps at 0.2 Hz from −50 to 50 mV in 10-mV increments. I-V relationships of ICa,L were fitted with an equation adapted from the Boltzmann’s equation: ICa,L = Gmax(V − Erev)/[1 + exp(V − V0.5/k)], where V is the membrane potential, Gmax is the maximal conductance, Erev is the reversal potential, V0.5 is the half activation potential, and k is the slope factor (Boltzmann’s equation of activation). Quasi-steady-state inactivation curves (f∞-V) were obtained using a gapped double-pulse protocol at 0.1 Hz. A 2-s conditioning pulse to potentials between −100 and 20 mV from an HP of −80 mV was followed by a 50- or 70-ms step to −40 mV, then a 500-ms test pulse to 0 mV. f∞-V were fitted with the Boltzmann’s equation: f∞ = c0 + (c1-c0)/{1 + exp[−(V − V0.5)/k]}, where c0 is the maximal availability, c1 is the voltage-independent availability, c1-c0 is the maximal availability of the voltage-dependent inactivation; V is the membrane potential of the prepulse; and V0.5 is the voltage for 50% inactivation of the voltage-dependent component (Boltzmann’s equation of inactivation).
Arterial Contraction Measurements
Endothelium-intact bovine and mouse coronary artery and HIMA were studied for changes in isometric force as described previously (19).
Detection of Hydrogen Peroxide
Hydrogen peroxide levels were determined using luminol luminescence assay. In brief, arteries were homogenized in ice-cold buffer, followed by estimation of the protein concentrations using the Bradford method. Tissue homogenates (20 μL) were incubated at 37°C in the presence of 10 μM luminol plus 1 μM horseradish peroxidase for the detection of H2O2 in a final volume of 200 μL of air-equilibrated Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4). Chemiluminescence was determined using a microplate reader (Synergy 2 from BioTek Instruments, Winooski, VT).
Hemodynamics
All hemodynamic measurements were performed as described previously (29). Briefly, the adult rat was anesthetized with 4% isoflurane, and 2% isoflurane was used to maintain anesthesia for the entire duration of the surgery and data acquisition. The body temperature of the animal during the surgery was maintained using a heating pad. About 3 sq. cm area of skin over the ventral neck region was exposed to locate the right common carotid artery, which was then carefully isolated and a 2.0-F Millar microtip pressure-volume catheter was inserted to the carotid artery and pushed to the left ventricle to measure the systemic blood pressure and left ventricular hemodynamic parameter. Total peripheral resistance was calculated from mean arterial pressure (MAP)/cardiac output (CO).
Statistical Analysis
Values are reported as means ± SE (Table 1) or means ± SD (Tables 2–5). ANOVA and Tukey’s multiple comparison tests were used for analyses in all vascular contractility studies, biochemical assays, and electrophysiological studies with three or more groups. All enzyme activity, Western blot data, and vascular contractility were analyzed using Student’s t tests between two groups. Values of P < 0.05 were considered significant. In all cases, the number of experimental determinations (n) was equal to the number of individual cell culture wells, cells used to record ion currents, hearts from which CAs were harvested, or patients from which HIMAs were obtained.
Table 1.
List of calcium channel subunits and the PPP proteins in the immunocomplex with G6PD detected with LC-MALDI-TOFF
| Top Ranked Protein Name | Accession No. GI | Protein MW |
Protein PI | Peptide Count | Total Ion Score | Total Ion CI, % |
|---|---|---|---|---|---|---|
| Voltage-dependent L-type calcium channel subunit-α1D | 300795582 | 247,309 | 6.4 | 5 | 69 | 100 |
| Voltage-dependent L-type calcium channel subunit-α1D-isoform 2 | 296474884 | 242,190 | 6.3 | 5 | 65 | 100 |
| Voltage-dependent R-type calcium channel subunit-α1E | 300798325 | 256,162 | 8.6 | 5 | 49 | 100 |
| Glyceraldehyde-3-phosphate dehydrogenase | 77404273 | 35,845 | 8.5 | 4 | 200 | 100 |
| Voltage-dependent calcium channel subunit α-2/δ-2 | 358418205 | 135,507 | 5.2 | 3 | 43 | 98 |
| Voltage-dependent L-type calcium channel subunit-α1F | 296470773 | 192,279 | 5.8 | 4 | 34 | 86 |
| 6-Phosphoglucoronate | 296486083 | 27,514 | 5.6 | 2 | 32 | 81 |
LC-MALDI-TOFF, liquid chromatography-matrix-assisted laser desorption ionization-time of flight; PPP, pentose phosphate pathway; PI, protein isoelectric point; CI, confidence interval; MW, molecular weight; G6PD, glucose-6-phosphate dehydrogenase; GI, GenInfo Identifier.
Table 2.
Effect of G6PD on parameters of the I-V relationship
| n | Gmax, pS/pF | V0.5, mV | k, mV | Erev, mV | |
|---|---|---|---|---|---|
| Cav1.2 | 32 | 200 (20) | −13.6 (1.4) | 6.6 (0.8) | 47.3 (2.4) |
| C-G6PD545 | 33 | 262 (16)**** | −11.2 (0.9)*** | 6.9 (0.5) | 43.5 (1.2) |
| N-G6PD545 | 21 | 208 (16)#### | −6.4 (1.1)****,#### | 6.8 (0.6) | 45.4 (1.5) |
Values are means (SD); n, number of experiments (n was 9 for the simulation). Gmax, maximal conductance; V0.5, half activation potential; k, slope factor; Erev, the reversal potential; G6PD, glucose-6-phosphate dehydrogenase. Statistical comparison of the parameters was performed using one-way ANOVA, followed by Turkey’s multiple comparison test.
P < 0.05,
P < 0.0001, compared with Cav1.2;
P < 0.0001, compared with C-G6PD.
Table 5.
Effect of epiandrosterone on parameters of the f∞-V relationship
| n | c1 | c0 | V0.5, mV | k, mV | |
|---|---|---|---|---|---|
| Control | 7 | 0.19 (0.09) | 0.99 (0.01) | −31.4 (0.5) | 6.0 (0.4) |
| 10 µM EPI | 7 | 0.14 (0.01) | 0.98 (0.01) | −32.1 (0.4)*,#### | 7.0 (0.3)*** |
| 100 µM EPI | 7 | 0.08 (0.01)** | 0.99 (0.01) | −33.9 (0.2)****,#### | 7.0 (0.2)*** |
| Washout | 4 | 0.15 (0.01) | 0.97 (0.01) | −28.7 (0.6)**** | 7.5 (0.5)**** |
Values are means (SD); n, number of experiments. f∞-V, quasi-steady-state inactivation curve; V0.5, voltage to produce 50% inactivation of the voltage-dependent component; c1, voltage-independent component; c0, maximal current; k, slope factor; EPI, epiandrosterone. Statistical comparison of the parameters was performed using one-way ANOVA, followed by Turkey’s multiple comparison test.
P < 0.05,
P < 0.01,
P < 0.001,
P < 0.0001, compared with control.
P < 0.0001, compared with washout.
RESULTS
Vascular Smooth Muscle Cells Express Two Isoforms of G6PD
To determine whether the VSM expresses more than one isoform of G6PD and whether two isoforms are expressed in arteries and/or in cultured smooth muscle cells, we performed Western blot analyses using whole cell lysates obtained from the HIMAs, BCAs, HCASMCs, and BCASMCs, respectively. We observed two bands (fast and slow migrating) for G6PD, in the HIMAs, BCAs, HCASMCs, and BCASMCs lysates (Fig. 1A and Supplemental Fig. S1; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.12311807).
Fig. 1.

Vascular tissue and vascular smooth muscle cells (VSMCs) express 2 isoforms of glucose-6-phosphate dehydrogenase (G6PD). A: Western blot analyses were performed using homogenates obtained from the human internal mammary artery (HIMA), bovine coronary artery (BCA), cultured human coronary artery smooth muscle (HCASMC), and cultured bovine coronary artery smooth muscle (BCASMC) cells, respectively. Proteins were separated on 9% SDS-PAGE and immunoblotted using rabbit anti-G6PD antibody. Two distinct bands (a fast and slow migrating) of G6PD were observed as shown in the representative Western blot of 4 different experiments. B: 2-step RT and PCR were performed with total RNA isolated from HCASMC and BCASMC. OligodT (OdT) and random hexamers (RHx) were used for the RT, and specific primers for G6PD545 (1,638 bp; NM_000402.4) were used for the PCR amplification. The PCR products were run on 1% agarose gel along with a DNA marker (M). C: G6PD545 mRNA/cDNA isolated from HCASMCs and BCASMCs, respectively, exhibited 100% homology with the human (accession no. NM_000402.4; C,i) and bovine (accession No. NM_001244135.1; C,ii) G6PD545 mRNA, reported in GenBank. Alignment of the first 93 nucleotides (5′-end) of the human and bovine G6PD545 and G6pd545 genes showed the presence of 2 ATG/start codons, located 90 nucleotides apart. C,iii: alignment of the first 30, NH2-terminal amino acids between the human (NP_000393.4) and bovine (NP_001231064.1) orthologs of G6PD545 protein shows 63.3% homology across the 2 species. Hu, human; gb, Genbank; Bv, bovine. D and E: bovine coronary arteries smooth muscle cells (BCASMCs) were transfected with Smartpool-G6PD-siRNAs (D) or G6PD545-siRNA (E) separately and scramble/nontargeting (NT) siRNA sequence for 72 h. Western blot analyses were performed using rabbit anti-G6PD antibody. β-Actin (ACTB) was used as a loading control. UT, untransfected. A representative of 4 different Western blot experiments is shown. F: BCASMCs were cultured in serum-free or 10% serum containing cell culture media. Western blot analyses were performed using rabbit anti-G6PD antibody. The slow migrating G6PD isoform was not expressed in SMCs cultured in serum-free media; n = 3 in each group. G: basal G6PD activity was measured in SMCs cultured in serum-free and 10% serum containing media. The activity increased (*P < 0.05) by culturing the BCASMCs in 10% serum as compared to serum-free condition; n = 5 in each group; note overlapping circles represent number of experiments.
There are two different mRNAs reported for G6PD in GenBank (1,638 bp; NM_000402.4 and 1,548 bp; NM_001042351.3) that encodes for two different isoforms of G6PD, one harboring 545 amino acids (G6PD545; NP_000393.4) and the other harboring 515 amino acids (G6PD515; NP_001035810). We performed two-step reverse transcription (RT) and polymerase chain reaction (PCR) for the G6PD545-mRNA/cDNA (1,638 bp; NM_000402.4), using total RNA extracted from HCASMCs and BCASMCs. We obtained a band at 1,638 bp using random hexamers (RHx) primers for RT and G6PD545 specific primers for PCR (Fig. 1B). DNA sequencing data revealed that the G6PD545 mRNA/cDNA sequence was identical to that of G6PD515 mRNA (NM_001042351.3) with the exception of the first 90 nucleotides at the 5′-end of the transcript. Furthermore, G6PD545 mRNA/cDNA isolated from HCASMCs and BCASMCs exhibited 100% homology with the human (accession no. NM_000402.4) and bovine (accession no. NM_001244135.1) G6PD545 mRNA, respectively, reported in the GenBank. Alignment of the first 93 nucleotides (5′-end) of the human (Fig. 1C,i) and bovine (Fig. 1C,ii) G6PD545 mRNA/cDNA shows the presence of two ATG/start codons located 90 nucleotides apart. Consequently, G6PD545 protein contains 30 additional amino acids residues at its NH2-terminus as compared with the G6PD515 isoform. Furthermore. the human and bovine orthologs share 63.3% homology (Fig. 1C,iii), within these 30 residues.
To further substantiate the existence of the two G6PD isoforms in VSMCs, we used two sets of G6PD-siRNAs to perform knockdown assays in BCASMCs. The smart pool-G6PD-siRNA (Dharmacon, CO) that targeted four exons in the G6PD gene downregulated both the G6PD515 (fast migrating; 61 ± 15.4%) and G6PD545 (slow migrating; 25 ± 8.7%) as compared with nontargeting siRNA (Fig. 1D and Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.12311807). Interestingly, the second siRNA that we designed to target the first 90 nucleotides of G6PD545, specifically downregulated (56 ± 8.2%) the expression of the slow migrating G6PD isoform without affecting the expression of the fast migrating G6PD isoform compared with nontargeting siRNA (Fig. 1E and Supplemental Fig. S1), suggesting that two distinct isoforms of G6PD existed in BCASMCs, with G6PD545 isoform migrating slower than the G6PD515 isoform.
To determine whether the expression of the two G6PD isoforms is differentially regulated, we cultured the BCASMCs, in the presence and absence of serum. Interestingly, the expression of slow migrating G6PD545 was absolutely serum dependent (Fig. 1F), as compared with the fast migrating G6PD515, which was expressed even in the serum-free media. Consistently, we found basal G6PD activity in BCASMCs cultured in serum-free condition; however, this activity further increased by addition of serum to the media (Fig. 1G), coinciding with the expression of the slow migrating G6PD545.
G6PD Appeared in the Plasma Membrane Fraction and Exhibited Activity in the Caveolae Fraction
We performed a computational subcellular localization analysis of the G6PD protein sequence using the PSORT II prediction tool (28). In silico analysis predicted that G6PD would likely be a peripheral protein with a very high membrane localization score. To determine the subcellular localization of G6PD, ultracentrifugation was performed, using BCA homogenates and the plasma membrane fraction was separated from the microsomal fraction. Western blot analyses were performed on 4–12% gradient gels. G6PD that coeluted in the caveolae fraction along with Na+-K+ ATPase and caveolin-1 (CAV-1; Fig. 2A) migrated slower than the G6PD that appeared in the cytosolic fraction, suggesting that the two G6PD isoforms had distinct localizations in the cell. Next, we separated the cholesterol-enriched caveolae fraction and the cytoplasmic fraction and measured G6PD activity in both these fractions. Although we found G6PD activity in both the cholesterol-enriched caveolae and the cytoplasmic fraction (Fig. 2B), NADPH levels were higher (P < 0.05) in the caveolae than the cytoplasmic fraction (Fig. 2C).
Fig. 2.

Subcellular localization of glucose-6-phosphate dehydrogenase (G6PD) in the bovine coronary artery (BCA). A: ultracentrifugation of the endothelium and adventitia removed BCA homogenate was performed to separate microsomal and cytosolic fractions. The microsomal fraction was further separated on a Ficol gradient, and Western blot analyses of the caveolae fractions were conducted using 4–12% gel and monoclonal anti-Na+-K+-ATPase, anti-caveolin-1 (CAV1), and polyclonal anti-G6PD antibody, respectively. LDH was used as a cytosolic marker. The caveolae G6PD migrated slower than the cytosolic G6PD. Representative Western blot of 5 separate experiments is shown. B: the graph represents the G6PD activity measured in the nucleus (fraction 2), mitochondria plus large organelles (fraction 3), submitochondrial particles plus small organelles (fraction 4) and cholesterol-rich caveolae (in yellow box; fraction 5), and cytosolic (fraction 6) fractions. C: the graph represents NADPH levels in the caveolae (triangle) and cytosolic (square) fractions of CAs; n = 20 observation from 5 separate experiments in each group.
G6PD Interacts with Cav1.2, a α1C Subunit of L-type Ca2+ Channels
Coelution of G6PD with CAV-1 (Fig. 2A) implied that it might be associated with membrane proteins involved in regulating the intracellular signaling. To test this possibility, we performed immunoprecipitation experiments using BCA lysates with anti-G6PD antibody and IgG (control), followed by LC-MALDI-TOFF. In lysates immunoprecipitated with anti-G6PD antibody, but not IgG, G6PD was found to be associated with the subunits of voltage-gated Ca2+ channel (Table 1), which is the main gateway for Ca2+ entry in VSMCs and regulator of VSM contraction (7).
To confirm the LC-MALDI-TOFF results, coimmunoprecipitation experiments were performed using anti-G6PD antibody. We found that the pore-forming α1C-subunit of L-type Ca2+ channel, Cav1.2, formed a complex with G6PD (Fig. 3A,i and Supplemental Fig. S2). We further confirmed G6PD-Cav1.2 interaction using antibodies that recognized the dihydropyridine (DHP) binding site of Cav1.2 (DHPα2; Fig. 3A,ii and Supplemental Fig. S2). Interestingly, Cav1.2 did not coimmunoprecipitate with other metabolic enzymes, such as glucose kinase (GK; Fig. 3B), suggesting that the interaction between G6PD and Cav1.2 was specific.
To demonstrate the G6PD-Cav1.2 interaction in situ, we performed the Duolink Proximity Ligation Assay (PLA) in fixed BCASMCs. G6PD interacted with Cav1.2 (speckled red fluorescence seen on the cell surface and perinuclear regions) in the presence (Fig. 3C,ii) but not in the absence (Fig. 3C,i) of anti-G6PD antibody (Fig. 3C), further substantiating a direct interaction between G6PD and Cav1.2. To identify modulators of the G6PD-Cav1.2 interaction, we treated BCASMCs with epiandrosterone [EPI; an uncompetitive inhibitor of G6PD (12) that inhibits G6PD activity in VSMC (1, 13)] for an hour before performing the PLA. Surprisingly, EPI disrupted G6PD-Cav1.2 interaction endogenously, as demonstrated by the absence of speckled red fluorescence, in the cells, even in the presence of G6PD antibody (Fig. 3C).
To further confirm the G6PD-Cav1.2 interaction, we designed G6PD545 constructs with a RFP tag at the NH2-terminus (N-G6PD545; COOH-terminal free) or at the COOH-terminus (C-G6PD545; NH2-terminal free) of G6PD545; a G6PD515 construct with a RFP tag at the COOH-terminus (C-G6PD515; NH2-terminal free); and a Cav1.2 construct with a GFP tag at its COOH-terminus (Cav1.2-GFP; NH2-terminal free) (Fig. 3D). To determine whether the ectopically overexpressed RFP-tagged G6PD isoforms were enzymatically active, we overexpressed C-G6PD545 and C-G6PD515, in HEK293-T17 cells, respectively. The immunocomplexes (35 μL) obtained after immunoprecipitation of these cell lysates with an anti-RFP antibody were then used to estimate the G6PD activity. Both isoenzymes reduced NADP+ to NADPH. Interestingly, C-G6PD545 exhibited a 1.65-fold higher Vmax than C-G6PD515 (Fig. 3E), suggesting that both isoforms were enzymatically active. HEK293-17T cells were then separately cotransfected with Cav1.2-GFP + N-G6PD545; Cav1.2-GFP + C-G6PD545; or Cav1.2-GFP + C-G6PD515, respectively. FRET analyses of the cotransfected cells revealed stronger FRET signals in cells cotransfected with Cav1.2-GFP + C-G6PD545 than in cells cotransfected with Cav1.2-GFP + N-G6PD545 (Fig. 3F). Furthermore, we observed very weak FRET signals in cells cotransfected with Cav1.2-GFP + C-G6PD515, suggesting that G6PD515 did not (or weakly) interact with Cav1.2.
G6PD Modulated L-type Ca2+ Channel Function
To determine whether the G6PD-Cav1.2 interaction modulates L-type Ca2+ channel function, we ectopically overexpressed Cav1.2-GFP alone, Cav1.2-GFP + N-G6PD545, or Cav1.2-GFP + C-G6PD545, respectively, in HEK293-17T cells and recorded the ICa,L (Fig. 4). The I-V relationship revealed coexpression of C-G6PD545 with Cav1.2 induced a larger ICa,L,peak than that of N-G6PD545 (Fig. 4A). Moreover, Cav1.2-GFP+N-G6PD545 cotransfected cells exhibited a smaller ICa,L,peak compared with those expressing of Cav1.2-GFP alone (Fig. 4A). Simulation of I-V with the Botlzmann's equation of activation revealed a small but significant shift of V0.5 to the right by N-G6PD545 compared with Cav1.2 alone (Table 2). Coexpression of Cav1.2-GFP + N-G6PD545 and Cav1.2-GFP + C-G6PD545 significantly increased maximal conductance (Gmax) compared with the expression of Cav1.2-GFP alone, and the extent of the Gmax increase was significantly higher with C-G6PD545 compared with N-G6PD545 (Table 2). N-G6PD545 coexpression slightly shifted the quasi-steady-state inactivation curve (f∞-V) to the left compared with Cav1.2 and C-G6PD545 (Fig. 4B and Table 3); c1, noninactivating component of f∞-V, was significantly increased with C-G6PD545 than with Cav1.2 alone and N-G6PD545 (Table 3), suggesting that C-G6PD545 and Cav1.2 interaction downregulated voltage-dependent inactivation of the channel.
Fig. 4.

Effect of glucose-6-phosphate dehydrogenase (G6PD) on L-type Ca2+ channel current (ICa,L) recorded from HEK293-17T cells transfected with Cav1.2. HEK293-17T cells were transfected with Cav1.2 alone or cotransfected with 1) Cav1.2 + C-G6PD545 or 2) Cav1.2 + N-G6PD545, respectively. A: typical images of the transfected HEK293-17T cells are shown on the top. Calibration bar = 20 µm. Current-voltage (I-V) relationship. a–c: Specimen records obtained by 500-ms-voltage pulses. a: From transfection by Cav1.2 alone; b: Cav1.2 and C-G6PD545; c: Cav1.2 and N-G6PD545. d: I-V relationship of peak current density of ICa,L from cells transfected by Cav1.2 (n = 32), Cav1.2 and C-G6PD545 (n = 33), and Cav1.2 and N-G6PD545 (n = 21). Time calibration indicates 100 ms, and vertical calibration gives 2 pA/pF. As indicated in voltage-step protocols, black traces indicate current swipes from −40 mV to 0 mM and gray traces indicate current swipes from +10 mV to +40 mV (Aa–Ac), and black traces indicate current swipes from −80 mV to −30 mV and gray traces indicate current swipes from −20 mV to +20 mV (Ba–Bc). Means ± SE are plotted. *P < 0.05; **P < 0.01; ***P < 0.001, compared C-G6PD545 vs. N-G6PD545. #P < 0.05, compared Cav1.2 vs. N-G6PD545. Curve fittings were performed with the Boltzmann's equation of activation (see materials and methods) with parameters shown in Table 2. B: quasi-steady-state inactivation curve (f∞-V) relationships. a–c: Specimen records to obtain the availability curves with pulses shown at top. a: from cells with Cav1.2 alone; b: from cells with Cav1.2 and C-G6PD545; c: from cells with Cav1.2 and N-G6PD545. d: quasi-steady-state inactivation curves. Availability (f∞)-voltage (V) relationship from cells transfected by Cav1.2 alone (n = 13), Cav1.2 and C-G6PD545 (n = 7), and Cav1.2 and N-G6PD545 (n = 7). Curves were fitted to the Boltzmann's equation of inactivation. Table 3 gives actual parameters for the fitting. Arrows indicate the half activation potential (V0.5).
Table 3.
Effect of G6PD-binding on parameters of the f∞-V relationship
| n | c1 | c0 | V0.5, mV | k, mV | |
|---|---|---|---|---|---|
| Cav1.2 | 13 | 0.05 (0.02) | 0.99 (0.02) | −26.0 (0.9) | 14.0 (0.9) |
| C-G6PD545 | 7 | 0.12 (0.03)**** | 0.96 (0.03) | −27.1 (1.6) | 13.4 (1.7) |
| N-G6PD545 | 7 | 0.06 (0.04) ### | 1.01 (0.05) | −31.1 (2.3)****,#### | 16.0 (2.7)*,## |
Values are means (SD); n, number of experiments. G6PD, glucose-6-phosphate dehydrogenase; c1, voltage-independent component; c0, maximal availability; V0.5, voltage to produce 50% inactivation of the voltage-dependent component; k, slope factor. Statistical comparison of the parameters was performed using one-way ANOVA, followed by Turkey’s multiple comparison test.
P < 0.05,
P < 0.0001, compared with control.
P < 0.01,
P < 0.001,
P < 0.0001, compared with C-G6PD.
Next, we investigated whether the inhibition of G6PD activity and therefore disruption of the G6PD-Cav1.2 complex by EPI affected the properties of ICa,L in freshly isolated BCASMCs. EPI dose dependently and reversibly decreased ICa,L,peak and the terminal current (I500) (Fig. 5A) and Gmax (Table 4). Application of EPI (100 μM) shifted the f∞-V relationship to the left and decreased the noninactivating component (c1) (Fig. 5B and Table 5). The window current (IWD) of ICa,L appeared with marked current noises reflecting the random opening of the channels and was markedly depressed by EPI in a dose-dependent and reversible manner (Fig. 5C). Cd2+, an inorganic ICa,L inhibitor, completely suppressed IWD that appeared with a marked current noise.
Fig. 5.

Epiandrosterone (EPI) modulates L-type Ca2+ channel (ICa,L) function. EPI-induced inhibition of ICa,L and window current (IWD) in bovine coronary artery smooth muscle cells (BCASMCs). A: ICa,L. a–d: typical traces: a: control; b: 10 µM EPI; c: 100 µM EPI; d: washout (WO). e: Current-voltage (I-V) relationship of ICa,L of peak current (ICa,L,peak) and terminal current (ICa,L,500). Means ± SE are shown; n =7 except for 5 in WO. *P < 0.05; **P < 0.01, compared with control. #P < 0.05, compared with 100 µM EPI. Data were fitted by the Boltzmann’s equation of activation with parameters partly shown in Table 4. B: quasi-steady-state inactivation curve (f∞-V) relationships. a–d: Specimen records. Test pulse to 0 mV was applied following 2-s conditioning pulses as shown in the inset. Black sweeps were obtained after conditioning pulses to −30 mV. Means ± SE are shown; n = 7, except for 4 in WO. e: Quasi-steady-state inactivation data were plotted. *P < 0.05; **P < 0.01; ****P < 0.0001, compared with control. #P < 0.05; ##P < 0.01; ####P < 0.00001, compared with 100 µM EPI. Curves show fitting with the Boltzmann's equation of inactivation with parameters shown in Table 5. C: window current (IWD). a–d: Typical traces obtained a step-like increase of 20-s depolarization step from −80 mV to 20 mV. a: Control; b: 10 µM EPI; c: 100 µM EPI; d: WO; e: 0.5 mM Cd2+. Current traces were low-pass filtered at 200 Hz. f: IWD-V relationship. IWD values represent the time-averaged current density in each step. Means ± SE are shown; n = 7 for control and 100 µM EPI and 5 for 10 µM EPI and WO. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, compared with control. #P < 0.05; ###P < 0.001, compared with 100 µM EPI.
Table 4.
Effect of epiandrosterone on parameters of the I-V relationship
| Current | n | Gmax, pS/pF |
|---|---|---|
| ICa,L,peak | ||
| Control | 6 | 137 (11) |
| 10 µM EPI | 6 | 123 (11)* |
| 100 µM EPI | 6 | 99 (7)****,#### |
| Washout | 6 | 134 (8) |
| ICa,L,500 | ||
| Control | 6 | 55 (5) |
| 10 µM EPI | 6 | 42 (4)**** |
| 100 µM EPI | 6 | 27 (2)****,#### |
| Washout | 6 | 43 (3)**** |
Values are means (SD); n, number of experiments. Gmax, maximal conductance; EPI, epiandrosterone; I-V, current-voltage; ICa,L,peak, peak L-type Ca2+ channel current; ICa,L,500, terminal L-type Ca2+ channel current. Statistical comparison of the parameters was performed using one-way ANOVA, followed by Turkey’s multiple comparison test.
P < 0.05,
P < 0.0001, compared with control;
P < 0.0001, compared with WO.
Overexpression of G6PD545 Increased KCl-Induced BCA Contraction
Smooth muscle contraction is mediated by Ca2+ influx through L-type Ca2+ channels. Our findings that G6PD-Cav1.2 interaction increased L-type Ca2+ channel conductance raised the possibility that the level of G6PD protein/activity might influence smooth muscle contractions. To determine the effects of the G6PD on vascular contraction, we overexpressed adenoviral vector alone with a GFP tag (Ad-GFP-Ctrl) and adeno-GFP-tagged G6PD545 (Ad-G6PD545-GFP) and G6PD515 (Ad-G6PD515-GFP) in BCA, and we performed Western blot analyses and measured the activity of Ad-G6PD545-GFP and Ad-G6PD515-GFP, respectively (Fig. 6, A and B and Supplemental Fig. S3). Overexpression of Ad-G6PD545-GFP, but not Ad-G6PD515-GFP, significantly increased the KCl-induced contraction of BCA as compared with control (Fig. 6C).
Fig. 6.

Overexpression of Ad-G6PD545-green fluorescent protein (GFP) increased KCl-induced bovine coronary artery (BCA) contraction. A and B: BCAs were transfected with adenoviral vector (Ad) alone (Ad-Ctrl-GFP; n = 6); Ad-G6PD545-GFP (n = 8); and Ad-G6PD515-GFP (n = 5). Glucose-6-phosphate dehydrogenase (G6PD) expression was determined by Western blot analyses using anti-GFP antibody and its activity was measured in the BCA homogenates. GAPDH was used as loading control. Unt, untransfected. C: the graph summarizes the peak contractions evoked by 2 concentrations of KCl (30 mmol/L, 30 K and 120 mmol/L, 120 K) in BCAs overexpressing empty Ad-Ctrl-GFP (n = 9 in 30 KCl and 120 KCl each), Ad-G6PD545-GFP (n = 5 in 30 KCl and 6 in 120 KCl), and Ad-G6PD515-GFP (n = 5 in 30 KCl and 120 KCl each), respectively. *P < 0.05 vs. Ad-Ctrl-GFP.
Overexpression of G6PD515 Decreased Hydrogen peroxide Levels in HEK293–17T cells and BCA
G6PD is considered as a significant anti-oxidant that reduces hydrogen peroxide within cells. Hence, we investigated the effects of Ad-G6PD545-GFP and Ad-G6PD515-GFP overexpression on the levels of hydrogen peroxide, in BCA. Ad-G6PD515-GFP, but not Ad-G6PD545-GFP, decreased hydrogen peroxide (Table 6), suggesting that the cytosolic G6PD515 appears to control hydrogen peroxide levels in the cell.
Table 6.
Effect of G6PD545 and G6PD515 overexpression on hydrogen peroxide levels
| n | HEK293-17T, AU/mg protein | BCA, AU/mg protein | |
|---|---|---|---|
| Wild type | 4 | 683 ± 283 | 66 ± 9 |
| G6PD545 | 7 | 905 ± 281 | 96 ± 14 |
| G6PD515 | 3 | 164 ± 153*,# | 37 ± 5*,# |
Values are means (SD); n, number of experiments. BCA, bovine coronary artery; G6PD, glucose-6-phosphate dehydrogenase; AU, arbitrary units. Statistical comparison of the parameters was performed using one-way ANOVA, followed by Turkey’s multiple comparison test.
P < 0.05, compared with wild type;
P < 0.05, compared with G6PD545.
Inhibition of G6PD Activity Decreased KCl-induced BCA Contraction
To investigate the effect of EPI-induced inhibition of G6PD activity on contraction, we applied EPI, to BCA precontracted with KCl (30 mM). We found decreased (P < 0.05) G6PD activity in the microsome (Fig. 7A,i) and a dose-dependent relaxation (P < 0.05) of BCAs, precontracted with KCl (Fig. 7A,ii). To emulate this observation in an endogenously G6PD-deficient animal model, G6PDDef mice that harbor a polymorphism within the 5′-UTR of G6pd gene were used. Previous studies showed that G6PDDef mice develop less angiotensin-II-mediated hypertension (36). In the present study, G6PDDef mice expressed less G6PD protein than wild-type mice (Fig. 7B,i and Supplemental Fig. S4), and coronary arteries from G6PDDef mice generated less KCl-elicited force than wild-type mice (Fig. 7B,ii). Intriguingly, EPI elicited less relaxation of coronary arteries, precontracted with KCl (30 mM), from G6PDDef mice [EPI (30 μM): −4 ± 13% and EPI (100 μM): −40 ± 32%] compared with wild-type [EPI (30 μM): −79 ± 10% and EPI (100 μM): −96 ± 1%].
Fig. 7.
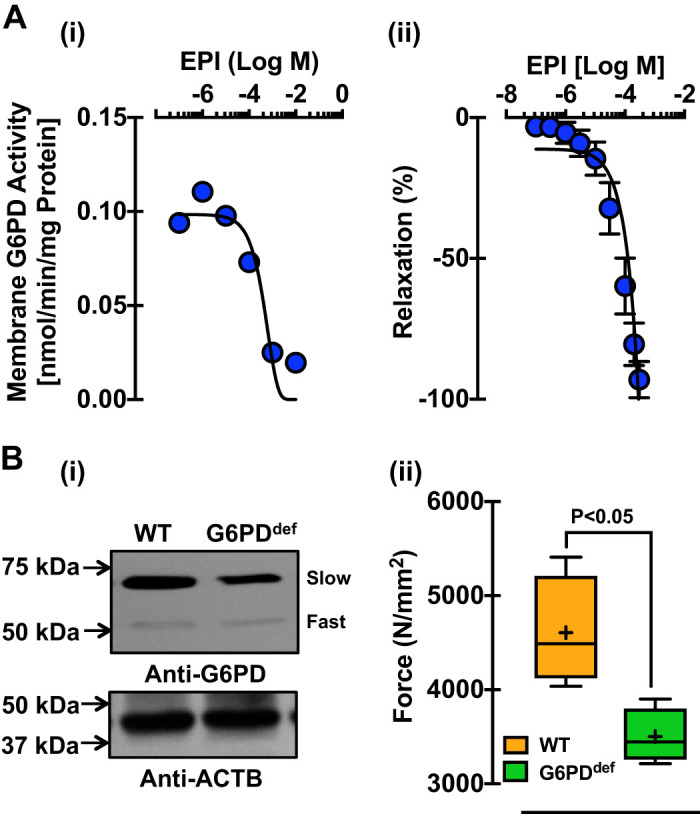
Glucose-6-phosphate dehydrogenase (G6PD) inhibition and deficiency decreased KCl-induced bovine coronary artery (BCA) contraction. A: the graphs represent G6PD activity (A,i) and relaxation (A,ii) of BCAs precontracted with KCl (30 mM) and treated with epiandrosterone (EPI) in a dose-dependent manner (n=10 experiments in each group). B,i: Western blot analyses were performed to demonstrate the expression of G6PD in the vascular tissue of G6PD-deficient (G6PDDef) mice using polyclonal anti-G6PD antibody (B,i; top). β-Actin (ACTB) was used as a loading control (B,i; bottom). B,ii: force generation to KCl (30 mmol/L; n = 5 experiments in each group) by the coronary artery of G6PDDef mice was measured as compared to wild-type (WT) mice.
Inhibition of G6PD Activity Decreased Blood Pressure in Metabolic Syndrome Rats
To determine whether increased G6PD expression and activity contributes to increased vascular contraction and elevated blood pressure observed in diseases, such as obesity and obesity-induced diabetes or metabolic syndrome, we measured the expression (Fig. 8A,i) and activity (Fig. 8A,ii) of G6PD in human internal mammary arteries (HIMA) from obese diabetics (metabolic syndrome) and obese patients without diabetes, who had undergone coronary artery bypass graft surgery. The expression of G6PD545 was augmented by 1.62 ± 0.18-fold and activity of G6PD was >2-fold higher (P < 0.05) in HIMAs from obese-diabetics than obese individuals without diabetes (Fig. 8A,ii). Correspondingly, HIMA from obese-diabetics produced (∼50%) more force than obese patients without diabetes (Fig. 8A,iii). Similarly, in the aorta of metabolic syndrome rats, G6PD activity was more than twofold higher (P < 0.05) compared with control rats (Fig. 8B,i). Furthermore, to investigate the possibility of whether increased G6PD, observed in arteries of metabolic syndrome rats, contributed to increased blood pressure, these rats were treated with EPI (30 mg·kg−1·day−1 sc, since EPI disrupted the G6PD-Cav1.2 interaction and relaxed precontracted arteries) for 28 days. Hemodynamic measurements revealed that EPI treatment significantly decreased the mean arterial pressure (MAP; Fig. 8B,ii) and total peripheral resistance (TPR; Fig. 8B,iii) in these rats, suggesting a correlation between elevated levels of G6PD and vasoconstriction.
Fig. 8.

Increased glucose-6-phosphate dehydrogenase (G6PD) expression and activity in arteries from metabolic syndrome patients and rats contributes to increasing blood pressure. A: homogenates from internal mammary arteries of obese patients with (Obese + T2D) and without (Obese − T2D) type 2 diabetes were subjected to Western blot analysis using polyclonal anti-G6PD antibody (A,i, top). β-Actin (ACTB) was used as a loading control (A,i, Bottom). Total G6PD activity (A,ii; n = 3 represents 2 individual patient samples combined out of total 6 individual patient samples in each group) and force generation by different concentrations of Ca2+ (A,iii; n = 6 represents 6 individual patient samples) was measured in the arteries from obese patients with and without T2D. B: metabolic syndrome rats (JCR) were treated with epiandrosterone (EPI; 30 mg·kg−1·day−1 sc) and the total G6PD activity [B,i; n = 5 in Sprague-Dawley (SD) and JCR + EPI group and 4 in JCR group], mean arterial pressure (MAP; B,ii; n = 5 in SD; 11 in JCR; and 6 JCR + EPI), and total peripheral resistance (TPR; B,iii; n = 5 in SD; 11 in JCR; and 6 JCR + EPI) was measured in these rats after 28 days.
DISCUSSION
The role of G6PD in regulating glucose metabolism has been well studied in plants, insects, and various mammalian tissues. However, expression of G6PD isozymes in VSMCs or any other excitable mammalian cells has not been identified and studied previously. In the present study, we have demonstrated that the two G6PD isozymes (slow and fast migrating) are expressed in HCASMCs, BCASMCs, HIMA, and BCA. We found that the slow moving G6PD isoform, harboring 545 amino acids (G6PD545), was encoded by a 1,638 bp mRNA, consistent with the previous findings (30). Ectopic overexpression of human G6PD545 and G6PD515 (fast migrating isoform, harboring 515 amino acids), in HEK293-17T cells, demonstrated that both isozymes were enzymatically active, and that G6PD545 exhibited higher Vmax than G6PD515. Identification of two G6PD isozymes raised the possibility that they are differentially localized and have distinct functions within VSMCs. Cell fractionation studies demonstrated that active G6PD545 was localized within caveolae fraction, whereas active G6PD515 was localized only in the cytosol of BCA. Consistently, Bublitz and Steavenson found two sets of enzymes catalyzing reactions of the PPP, one set in the microsomes (particulate fraction) and another one in cytoplasm from various rat organs (5). Taken together with these studies, our findings imply that compartmentalization or subcellular localization of active G6PD isozymes in VSMCs plays a crucial role in regulating key signaling processes that control different cellular functions.
Proteomic analyses of G6PD immunocomplexes, coimmunoprecipitation assays, and PLA demonstrated that G6PD formed a complex with the ion channel cluster, including α subunit (Cav1.2), the pore-forming subunit of the L-type Ca2+ channel (7), in BCASMCs and BCA. FRET analyses indicated that the C-G6PD545-RFP and Cav1.2-GFP fusion proteins directly associated with one another, potentially without involvement of a bridging protein. These findings suggested that localization of G6PD545 in the caveolae fraction, which is a signal trafficking hub, may play a crucial role in modulating the Ca2+ signal transduction within VSMCs. Consistently, coexpression of human Cav1.2-GFP and C- or N-G6PD545-RFP increased Gmax in HEK293-T17 cells compared with the expression of Cav1.2-GFP alone. The fact that C-G6PD545-RFP increased Gmax compared with the expression of Cav1.2-GFP alone and coexpression of Cav1.2-GFP and N-G6PD545-RFP suggests that the site of protein-protein interaction is potentially important to determine the conductance of Cav1.2. The small inhibition of ICa,L,peak by N-G6PD545-RFP in the presence of a significant increase of Gmax may be explained by the shift of V0.5 of activation to the right. L-type Ca2+ channels are the main gateway for Ca2+ entry into VSMCs. Along with other Ca2+ channels and Ca2+ stores, they function to increase intracellular Ca2+, which is a crucial regulator of vascular smooth muscle contraction and essential in controlling cellular function, including mitochondrial metabolism (7, 10). Therefore, our findings that G6PD545 interacts with Cav1.2 and, at least partly, regulates L-type Ca2+ channel function could have a potential impact on vascular function in physiological and pathophysiological states.
EPI is a lipophilic steroid like cholesterol, which intercalates in the membrane and has been previously shown to inhibit ICa,L (41). We found that EPI disrupted the interaction between G6PD and Cav1.2 in BCA and decreased ICa,L as well as IWD in BCASMCs, by augmenting voltage-dependent inactivation. Taken together, the data presented in this study, provides compelling evidence that augmentation of the voltage-dependent inhibition and thereby markedly decrease ICa,L and IWD elicited by EPI, is due to a novel mechanism involving the disruption of the G6PD-Cav1.2 complex. Furthermore, since the coexpression of C-G6PD545 and Cav1.2 produced higher c1, the voltage- and time-independent sustained component of availability of ICa,L that potentially underlies IWD, the binding of G6PD to the COOH-terminal of Cav1.2, as evident by FRET signals, is crucial for the maintenance of c1. We propose that G6PD prevents intrinsic voltage-dependent inactivation process by binding to the COOH-terminal of Cav1.2 protein, and EPI-induced the inactivation of ICa,L, decreased IWD by displacing G6PD from the binding site. G6PD binding to the C-terminal of Cav1.2 produced sustained increase of influx of Ca2+ and intracellular Ca2+ in BCASMCs, potentially mediating KCl-induced contraction of BCA. As EPI and other steroid hormone levels decline after the third decade of life (34), the propensity of cardiovascular diseases increases in men and women (45). It is therefore reasonable to propose that reduced steroid levels facilitate G6PD-Cav1.2 interaction, which may have paramount ramifications in the deterioration of cardiovascular health.
Ectopic overexpression of G6PD545 in BCA revealed the involvement of G6PD in augmenting KCl-induced smooth muscle contraction. Consistently, it has been reported that slow migrating G6PD is upregulated within the atherosclerotic plaques in African women (44), suggesting that increased levels of G6PD play a potential role in the pathogenesis of vascular diseases in humans. In contrast, G6PD deficiency decreased the contraction of mice arteries evoked by KCl. It has been observed that individuals who harbor a nonsynonymous single nucleotide polymorphism in the G6PD gene (e.g., the Mediterranean-type mutation) are less likely to develop vascular disease (18). Moreover, G6PDDef mice are less susceptible to angiotensin II-induced hypertension (36), and progeny from crossbreeding ApoE−/− and G6PDDef mice show less arteriosclerosis (37). Furthermore, G6PD inhibition relaxes arteries (19), reduces hypoxic pulmonary vasoconstriction and hypertension (4, 6, 16, 21, 42), and decreases proinflammatory cytokine levels (35), which promotes inflammation of blood vessels in hypertension (9, 24, 25). In this study, we found that G6PD activity was upregulated in the arteries of obese-diabetics compared with obese individuals without diabetes and metabolic syndrome rats and EPI (a G6PD inhibitor that disrupted G6PD-Cav1.2 complex to decrease ICa,L) decreased elevated blood pressure in metabolic syndrome rats (Fig. 8). These observations suggest that downregulation of G6PD activity could be beneficial in alleviating hypertension and other vasculopathy in humans, potentially by decreasing Ca2+ channel function, among other yet unidentified mechanisms.
Overexpression of human G6PD515 (a fast migrating protein), but not G6PD545 (a slow migrating protein), in BCA reduced hydrogen peroxide (Table 6), suggesting that cytosolic G6PD515-derived NADPH supports glutathione reductase system and other enzymes to remove intracellular hydrogen peroxide (49). Furthermore, our results are consistent with transgenic mice overexpressing human G6PD protein (migrating around 50–55 kDa) that show decreased lipid peroxidation, which is elicited by hydrogen peroxide and hydroxyl radicals, in the liver and brain (40). Therefore, we propose that the fast migrating cytosolic G6PD515 is involved in regulating the levels of NADPH cofactor for enzymes like reductases (49) that are crucial for healthy heart and blood vessels.
In conclusion, we have shown that VSMCs expresses two G6PD isoforms in different subcellular locations. This is the first report to demonstrate that a novel slow migrating isoform of G6PD (G6PD545) interacted with Cav1.2 in caveolae to downregulate intrinsic voltage-dependent inactivation of the channel to sustain large VSMC Ca2+ influx through L-type Ca2+ channels. EPI-induced disruption of the G6PD-Cav1.2 interaction shifted Cav1.2 to an intrinsically inactivated state, which reduced ICa,L, and attenuated blood pressure in metabolic syndrome rats. Consequently, we propose that the two G6PD isoforms (caveolae and cytosolic) in VSMC play a distinct but pivotal signaling role in regulating vascular function. Increased expression of G6PD in the caveolae of vasculature activates maladaptive L-type Ca2+ channel-dependent cell signaling and contributes to the pathogenesis of hypertension and potentially to other forms of vascular diseases associated with obesity and diabetes.
Limitations
Although EPI-elicited relaxation of coronary artery depends on G6PD, this 17-ketosteroid may have other nonspecific action in the cell that potentially could contribute to decrease blood pressure in JCR rat. Therefore, these results should be cautiously interpreted before more studies confirm these findings with different G6PD inhibitors or genetic approaches.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants R01-HL-085352 and R0-1HL-132574 (to S.A.G).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.G., R.O., and S.A.G. conceived and designed research; R.G., V.D., R.O., and S.A.G. performed experiments; R.G., V.D., R.O., and S.A.G. analyzed data; R.G., R.O., and S.A.G. interpreted results of experiments; R.G., V.D., R.O., and S.A.G. prepared figures; R.G., R.O., and S.A.G. drafted manuscript; R.G., V.D., P.R., R.O., and S.A.G. edited and revised manuscript; R.G., V.D., P.R., R.O., and S.A.G. approved final version of manuscript.
REFERENCES
- 1.Ata H, Rawat DK, Lincoln T, Gupte SA. Mechanism of glucose-6-phosphate dehydrogenase-mediated regulation of coronary artery contractility. Am J Physiol Heart Circ Physiol 300: H2054–H2063, 2011. doi: 10.1152/ajpheart.01155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ball WJ, Schwartz A, Lessard JL. Isolation and characterization of monoclonal antibodies to (Na+ + K+)-ATPase. Biochim Biophys Acta 719: 413–423, 1982. doi: 10.1016/0304-4165(82)90228-8. [DOI] [PubMed] [Google Scholar]
- 3.Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res 92: 237–246, 2011. doi: 10.1093/cvr/cvr230. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet S, Dumas-de-La-Roque E, Bégueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA 100: 9488–9493, 2003. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bublitz C, Steavenson S. The pentose phosphate pathway in the endoplasmic reticulum. J Biol Chem 263: 12849–12853, 1988. [PubMed] [Google Scholar]
- 6.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. doi: 10.1152/ajplung.00229.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89: 411–452, 2009. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolezelová E, Zurovec M, Böhmová M, Sehnal F. Use of two transcription starts in the G6PD gene of the bark beetle Ips typographus. Insect Mol Biol 15: 25–32, 2006. doi: 10.1111/j.1365-2583.2006.00604.x. [DOI] [PubMed] [Google Scholar]
- 9.El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, Friedman JE, Eltzschig HK, Sokol RJ, Stenmark KR. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol 193: 597–609, 2014. doi: 10.4049/jimmunol.1303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francis M, Waldrup JR, Qian X, Solodushko V, Meriwether J, Taylor MS. Functional tuning of intrinsic endothelial Ca2+ dynamics in swine coronary arteries. Circ Res 118: 1078–1090, 2016. doi: 10.1161/CIRCRESAHA.115.308141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frederiks WM, Vreeling-Sindelárová H. Localization of glucose-6-phosphate dehydrogenase activity on ribosomes of granular endoplasmic reticulum, in peroxisomes and peripheral cytoplasm of rat liver parenchymal cells. Histochem J 33: 345–353, 2001. doi: 10.1023/A:1012427224822. [DOI] [PubMed] [Google Scholar]
- 12.Gordon G, Mackow MC, Levy HR. On the mechanism of interaction of steroids with human glucose 6-phosphate dehydrogenase. Arch Biochem Biophys 318: 25–29, 1995. doi: 10.1006/abbi.1995.1199. [DOI] [PubMed] [Google Scholar]
- 13.Gupte RS, Ata H, Rawat D, Abe M, Taylor MS, Ochi R, Gupte SA. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 14: 543–558, 2011. doi: 10.1089/ars.2010.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, Wolin MS, Gupte SA. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 47: 219–228, 2009. doi: 10.1016/j.freeradbiomed.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupte RS, Pozarowski P, Grabarek J, Traganos F, Darzynkiewicz Z, Lee MY. RIalpha influences cellular proliferation in cancer cells by transporting RFC40 into the nucleus. Cancer Biol Ther 4: 429–437, 2005. doi: 10.4161/cbt.4.4.1621. [DOI] [PubMed] [Google Scholar]
- 16.Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010. doi: 10.1074/jbc.M109.092916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail 13: 497–506, 2007. doi: 10.1016/j.cardfail.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Gupte SA. Glucose-6-phosphate dehydrogenase: a novel therapeutic target in cardiovascular diseases. Curr Opin Investig Drugs 9: 993–1000, 2008. [PubMed] [Google Scholar]
- 19.Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, Koller A, Wolin MS. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol 285: H2316–H2326, 2003. doi: 10.1152/ajpheart.00229.2003. [DOI] [PubMed] [Google Scholar]
- 20.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 21.Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 301: 299–305, 2002. doi: 10.1124/jpet.301.1.299. [DOI] [PubMed] [Google Scholar]
- 22.Gupte SA, Rupawalla T, Phillibert D Jr, Wolin MS. NADPH and heme redox modulate pulmonary artery relaxation and guanylate cyclase activation by NO. Am J Physiol Lung Cell Mol Physiol 277: L1124–L1132, 1999. doi: 10.1152/ajplung.1999.277.6.L1124. [DOI] [PubMed] [Google Scholar]
- 23.Gupte SA, Zias EA, Sarabu MR, Wolin MS. Role of prostaglandins in mediating differences in human internal mammary and radial artery relaxation elicited by hypoxia. J Pharmacol Exp Ther 311: 510–518, 2004. doi: 10.1124/jpet.104.070995. [DOI] [PubMed] [Google Scholar]
- 24.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, Kim YH, Ryu JW, Yoon KH, Lee CH, Kim JB. Glucose-6-phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes 65: 2624–2638, 2016. doi: 10.2337/db16-0060. [DOI] [PubMed] [Google Scholar]
- 26.Hicks S, Labinskyy N, Piteo B, Laurent D, Mathew JE, Gupte SA, Edwards JG. Type II diabetes increases mitochondrial DNA mutations in the left ventricle of the Goto-Kakizaki diabetic rat. Am J Physiol Heart Circ Physiol 304: H903–H915, 2013. doi: 10.1152/ajpheart.00567.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horecker BL. The pentose phosphate pathway. J Biol Chem 277: 47965–47971, 2002. doi: 10.1074/jbc.X200007200. [DOI] [PubMed] [Google Scholar]
- 28.Horton P, Nakai K. Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc Int Conf Intell Syst Mol Biol 5: 147–152, 1997. [PubMed] [Google Scholar]
- 29.Joshi SR, Dhagia V, Gairhe S, Edwards JG, McMurtry IF, Gupte SA. MicroRNA-140 is elevated and mitofusin-1 is downregulated in the right ventricle of the Sugen5416/hypoxia/normoxia model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 311: H689–H698, 2016. doi: 10.1152/ajpheart.00264.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanno H, Kondoh T, Yoshida A. 5′ structure and expression of human glucose-6-phosphate dehydrogenase mRNA. DNA Cell Biol 12: 209–215, 1993. doi: 10.1089/dna.1993.12.209. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Devalaraja-Narashimha K, Padanilam BJ. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am J Physiol Renal Physiol 308: F298–F308, 2015. doi: 10.1152/ajprenal.00459.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kletzien RF, Harris PK, Foellmi LA. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 8: 174–181, 1994. doi: 10.1096/fasebj.8.2.8119488. [DOI] [PubMed] [Google Scholar]
- 33.Koziel A, Jarmuszkiewicz W. Hypoxia and aerobic metabolism adaptations of human endothelial cells. Pflugers Arch 469: 815–827, 2017. doi: 10.1007/s00424-017-1935-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labrie F, Bélanger A, Cusan L, Gomez JL, Candas B. Marked decline in serum concentrations of adrenal C19 sex steroid precursors and conjugated androgen metabolites during aging. J Clin Endocrinol Metab 82: 2396–2402, 1997. doi: 10.1210/jcem.82.8.4160. [DOI] [PubMed] [Google Scholar]
- 35.Lakhkar A, Dhagia V, Joshi SR, Gotlinger K, Patel D, Sun D, Wolin MS, Schwartzman ML, Gupte SA. 20-HETE-induced mitochondrial superoxide production and inflammatory phenotype in vascular smooth muscle is prevented by glucose-6-phosphate dehydrogenase inhibition. Am J Physiol Heart Circ Physiol 310: H1107–H1117, 2016. doi: 10.1152/ajpheart.00961.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation 112: 257–263, 2005. doi: 10.1161/CIRCULATIONAHA.104.499095. [DOI] [PubMed] [Google Scholar]
- 37.Matsui R, Xu S, Maitland KA, Mastroianni R, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6-phosphate dehydrogenase deficiency decreases vascular superoxide and atherosclerotic lesions in apolipoprotein E(-/-) mice. Arterioscler Thromb Vasc Biol 26: 910–916, 2006. doi: 10.1161/01.ATV.0000205850.49390.3b. [DOI] [PubMed] [Google Scholar]
- 38.Mohazzab KM, Wolin MS. Sites of superoxide anion production detected by lucigenin in calf pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol 267: L815–L822, 1994. doi: 10.1152/ajplung.1994.267.6.L815. [DOI] [PubMed] [Google Scholar]
- 39.Morton ME, Froehner SC. Monoclonal antibody identifies a 200-kDa subunit of the dihydropyridine-sensitive calcium channel. J Biol Chem 262: 11904–11907, 1987. [PubMed] [Google Scholar]
- 40.Nóbrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Viña J, Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun 7: 10894, 2016. doi: 10.1038/ncomms10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochi R, Chettimada S, Gupte SA. Poly(ethylene glycol)-cholesterol inhibits L-type Ca2+ channel currents and augments voltage-dependent inactivation in A7r5 cells. PLoS One 9: e107049, 2014. doi: 10.1371/journal.pone.0107049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, Limbird J, Imamura M, Gebb SA, Fagan KA, McMurtry IF. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res 74: 377–387, 2007. doi: 10.1016/j.cardiores.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan S, World CJ, Kovacs CJ, Berk BC. Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler Thromb Vasc Biol 29: 895–901, 2009. doi: 10.1161/ATVBAHA.109.184812. [DOI] [PubMed] [Google Scholar]
- 44.Pearson TA, Kramer EC, Solez K, Heptinstall RH. The human atherosclerotic plaque. Am J Pathol 86: 657–664, 1977. [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders JL, Boudreau RM, Cappola AR, Arnold AM, Robbins J, Cushman M, Newman AB. Cardiovascular disease is associated with greater incident dehydroepiandrosterone sulfate decline in the oldest old: the Cardiovascular Health Study All Stars Study. J Am Geriatr Soc 58: 421–426, 2010. doi: 10.1111/j.1532-5415.2010.02724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider AM, Rawat D, Weinstein LS, Gupte SA, Richards WO. Effects of laparoscopic Roux-en-Y gastric bypass on glucose-6 phosphate dehydrogenase activity in obese type 2 diabetics. Surg Endosc 26: 823–830, 2012. doi: 10.1007/s00464-011-1959-8. [DOI] [PubMed] [Google Scholar]
- 47.Schönrogge M, Kerndl H, Zhang X, Kumstel S, Vollmar B, Zechner D. α-Cyano-4-hydroxycinnamate impairs pancreatic cancer cells by stimulating the p38 signaling pathway. Cell Signal 47: 101–108, 2018. doi: 10.1016/j.cellsig.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 48.Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H162, 2009. doi: 10.1152/ajpheart.01142.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 64: 362–369, 2012. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90: 927–963, 2015. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strijbis K, van den Burg J, Visser WF, van den Berg M, Distel B. Alternative splicing directs dual localization of Candida albicans 6-phosphogluconate dehydrogenase to cytosol and peroxisomes. FEMS Yeast Res 12: 61–68, 2012. doi: 10.1111/j.1567-1364.2011.00761.x. [DOI] [PubMed] [Google Scholar]
- 52.Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 273: 10609–10617, 1998. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 53.Yin Y, Ashihar H. Expression of glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase isoform genes in suspension-cultured Arabidopsis thaliana cells. Z Natforsch C J Biosci 63: 713–720, 2008. doi: 10.1515/znc-2008-9-1017. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z, Yang Z, Zhu B, Hu J, Liew CW, Zhang Y, Leopold JA, Handy DE, Loscalzo J, Stanton RC. Increasing glucose 6-phosphate dehydrogenase activity restores redox balance in vascular endothelial cells exposed to high glucose. PLoS One 7: e49128, 2012. doi: 10.1371/journal.pone.0049128. [DOI] [PMC free article] [PubMed] [Google Scholar]