

Abstract
Although there is a strong association between cigarette smoking exposure (CSE) and vascular endothelial dysfunction (VED), the underlying mechanisms by which CSE triggers VED remain unclear. Therefore, studies were performed to define these mechanisms using a chronic mouse model of cigarette smoking (CS)-induced cardiovascular disease mirroring that in humans. C57BL/6 male mice were subjected to CSE for up to 48 wk. CSE impaired acetylcholine (ACh)-induced relaxation of aortic and mesenteric segments and triggered hypertension, with mean arterial blood pressure at 32 and 48 wk of exposure of 122 ± 6 and 135 ± 5 mmHg compared with 99 ± 4 and 102 ± 6 mmHg, respectively, in air-exposed mice. CSE led to monocyte activation with superoxide generation in blood exiting the pulmonary circulation. Macrophage infiltration with concomitant increase in NADPH oxidase subunits p22phox and gp91phox was seen in aortas of CS-exposed mice at 16 wk, with further increase out to 48 wk. Associated with this, increased superoxide production was detected that decreased with Nox inhibition. Tetrahydrobiopterin was progressively depleted in CS-exposed mice but not in air-exposed controls, resulting in endothelial nitric oxide synthase (eNOS) uncoupling and secondary superoxide generation. CSE led to a time-dependent decrease in eNOS and Akt expression and phosphorylation. Overall, CSE induces vascular monocyte infiltration with increased NADPH oxidase-mediated reactive oxygen species generation and depletes the eNOS cofactor tetrahydrobiopterin, uncoupling eNOS and triggering a vicious cycle of oxidative stress with VED and hypertension. Our study provides important insights toward understanding the process by which smoking contributes to the genesis of cardiovascular disease and identifies biomarkers predictive of disease.
NEW & NOTEWORTHY In a chronic model of smoking-induced cardiovascular disease, we define underlying mechanisms of smoking-induced vascular endothelial dysfunction (VED). Smoking exposure triggered VED and hypertension and led to vascular macrophage infiltration with concomitant increase in superoxide and NADPH oxidase levels as early as 16 wk of exposure. This oxidative stress was accompanied by tetrahydrobiopterin depletion, resulting in endothelial nitric oxide synthase uncoupling with further superoxide generation triggering a vicious cycle of oxidative stress and VED.
Keywords: cigarette smoking, endothelial dysfunction, hypertension, inflammation, nitric oxide synthase uncoupling, reactive oxygen species, superoxide
INTRODUCTION
The endothelium lines the interior surface of blood vessels of the entire circulatory system, forming an interface between circulating blood in the lumen and the rest of the vessel wall. Healthy endothelium plays a crucial role in the modulation of vascular activity, inhibition of platelet aggregation, proliferation of vascular smooth muscle cells (VSMCs), angiogenesis, and the vascular inflammatory process. It senses mechanical stimuli, such as pressure and shear stress, and hormonal stimuli, such as vasoactive substances. One of the most important regulatory and vasoactive mediators produced by the endothelial cells is nitric oxide (NO). Physiologically produced NO inhibits inflammation, VSMC proliferation, and platelet adhesion and plays a pivotal role in the maintenance of vascular tone and reactivity. Additionally, it opposes the actions of potent endothelium-derived contracting factors such as angiotensin II and endothelin-1 (66).
Vascular abnormalities due to vascular endothelial dysfunction (VED) have been detected in many cardiovascular and metabolic disorders such as hypertension, coronary heart disease, dyslipidemia, chronic heart failure, peripheral artery disease, chronic renal failure, and diabetes (81). Inflammation and oxidative stress mediated by reactive oxygen species (ROS), including superoxide and secondary oxidants, have been shown to be critical mediators of VED (15). Endothelial damage is characterized by decreased vessel dilation and increased contraction, prothrombotic and proinflammatory states, and VSMC proliferation (29). Decreased NO has often been reported in the presence of VED due to reduced activity or expression of endothelial NO synthase (eNOS) and/or decreased bioavailability of NO. The oxygen free radical, superoxide, readily reacts with NO to form peroxynitrite, a cytotoxic oxidant that affects protein function and therefore endothelial function (83, 86). Moreover, superoxide or peroxynitrite can degrade the eNOS cofactor tetrahydrobiopterin (BH4), causing uncoupling (36, 69). Oxidative stress is linked to a proinflammatory state of the vessel wall, which in turn decreases NO bioavailability, further contributing to VED. This has been reported in hypertensive (24) and diabetic (42) animal models, hypertensive patients (74), and patients with chronic renal failure and diabetes (5). In these conditions, NADPH oxidase is reported as a main source for oxidative stress in the vasculature (46, 79). Other sources, including xanthine oxidase (53), the mitochondria (35), and uncoupled NOS (22, 69), have also been reported.
A strong epidemiological association between cigarette smoking exposure (CSE) and cardiovascular disorders (CVD) has been shown in several studies (34). Because cigarette smoke is known to contain a large number of oxidants, it has been hypothesized that many of the damaging effects of cigarette smoking (CS) may result from oxidative damage to cells and macromolecules. Such deleterious effects may result both from oxidants present in cigarette smoke and from cellular pathways that generate ROS (25, 57). CS has been shown to be associated with impaired endothelium-dependent vasodilatation (EDV) of different vessels. These studies suggest that CSE may impair the NO biosynthetic pathway or enhance the rate of NO degradation, leading to VED (8). However, the exact mechanism of CSE-induced VED is not fully understood, and existing data are often ambiguous and contradictory (8, 84, 90).
We have reported in a chronic model of exposure that CS induces alterations in vascular and cardiac function, structure, and physiology with cellular oxidative stress and decreased NO bioavailability (75). In the chronic mouse model, it has also been demonstrated that CSE induces changes in the plasma protein expression profile with alterations in a number of inflammation sensitive proteins involved in vascular function, metabolism, and immune function, and these changes closely correlated with the initial onset of physiological changes of cardiovascular and pulmonary disease (78). Recently, in vitro cellular studies have shown that CS induces eNOS dysfunction in vascular endothelial cells through depletion of the essential eNOS cofactor tetrahydrobiopterin (BH4), as well as total biopterin levels, and decreased the expression level of guanosine triphosphate cyclohydrolase (GTPCH), the rate-limiting enzyme in BH4 biosynthesis (2). This depletion of BH4 was seen to trigger eNOS uncoupling with loss of NO production and concomitant increase in superoxide generation. ROS-induced stimulation of the ubiquitin-proteasome system led to loss of eNOS and GTPCH (2).
The present study investigates the cellular and molecular mechanisms by which CSE alters endothelial function in a well-defined chronic mouse model of CS-induced cardiovascular disease, which mirrors that in humans. We aim to better define these mechanisms and their temporal onset and through this knowledge identify key initiating processes and alterations that could serve as early biomarkers to identify CS-induced vascular disease as well as ways to ameliorate it. Based on prior studies, we hypothesize that oxidative stress and vascular inflammation are key triggers of endothelial dysfunction and secondary cardiovascular disease. However, the precise alterations that occur with in vivo chronic exposure similar to that in human smokers and their temporal progression remain unclear. Therefore, the current study was performed to investigate the effects of chronic CSE on the processes of vascular inflammation and NOS function and regulation. eNOS protein expression and its phosphorylation, plasma BH4 level, and the production and source of ROS were measured. We observed that CSE activates ROS production from monocytic leukocytes, followed by their vascular adhesion and infiltration. Concurrently, CSE depletes BH4, leading to eNOS uncoupling with further ROS formation and impaired Akt-mediated eNOS activation, followed by loss of eNOS expression, all of which contribute to VED and hypertension. Overall, our study provides important insights toward understanding how CS contributes to the genesis of cardiovascular disease and identifies biomarkers that may enable early detection of this process.
MATERIALS AND METHODS
Materials.
Chemicals, reagents, and other materials were obtained from Sigma-Aldrich (St. Louis, MO) unless noted otherwise.
Animals and exposure.
Male C57BL/6 mice, purchased at 11–12 wk of age (Charles River Laboratories International, Inc., Shrewsbury, MA), were housed at 23 ± 2°C at 55% relative humidity with 12:12-h light-dark cycle and maintained on a standard rodent chow and water ad libitum. After acclimatization for ~1 wk, mice were either exposed to air (control animals) or whole body mainstream and side stream CS (exposed animals) generated from 3R4F reference research cigarettes (University of Kentucky, Center for Tobacco Reference Products) that deliver 9.4 mg tar/0.726 mg nicotine per cigarette under the standard Cambridge filter smoking condition. We used the following standard parameters (ISO 1991) for puffing: one 35-mL puff of 2-s duration taken once a minute and followed by 58 s of fresh air at a rate of 6 mL/s. The smoke was directed in the exposure chamber at a smoke-to-air ratio of 1:10. This exposure protocol gave an average concentration of 300 mg/m3 of total particulate matter (TPM) in air. The smoking machine (TE-10; Teague Enterprises, Woodland, CA) was programmed to generate a smoke/air mix in three cycles, 24 min each. Each exposure cycle was followed by a break of fresh air for ~20 min. The total exposure time was ~72 min/day, 6 days/wk for 16, 32, and 48 wk. Carboxyhemoglobin (CO-Hb) levels in whole blood were measured spectrophotometrically using an Agilent 8453 Diode Array UV/VIS spectrophotometer (Agilent Technologies). The CO-Hb level immediately after the exposure was ∼12% in CS-exposed mice and ∼1% in air-exposed control mice. The body weight and vital signs were continuously monitored during the exposure. The use of animals and the animal protocol were approved by The Institutional Animal Care and Use Committee at The Ohio State University.
Noninvasive blood pressure monitoring.
Blood pressure was measured in lightly anesthetized animals by the tail-cuff method using the CODA-8 system (Kent Scientific). After a week of daily training, blood pressure measurements were made three times per week. The equipment and software parameters used were according to the manufacturer’s recommendations as previously described (27, 76).
Vascular reactivity.
The vessel reactivity was done, with some modifications, according to Talukder et al. (76). Briefly, the thoracic aorta and the main mesenteric artery were rapidly excised, cleaned, and placed in an ice-cold buffer (pH 7.4) containing the following: 118 mM NaCl, 24 mM NaHCO3, 4.6 mM KCl, 1.2 mM NaH2PO4, 1.2 mM CaCl2, 4.6 mM HEPES, and 18 mM glucose. Rings of ~2 mm length were mounted in a wire Myograph System-61M (Danish Myo Technology, Denmark), equilibrated in the buffer purged with 95% O2-5% CO2 for 1 h at 37°C under a resting initial tension of 1 g. The buffer in the organ bath was changed at 15-min intervals. After equilibration, the responsiveness and stability of each individual ring were checked by the administration of 1 μM L-phenylephrine hydrochloride (PE). The integrity of the vascular endothelium was assessed by acetylcholine (ACh)-induced relaxation on PE-preconstricted rings. Vessel rings were then washed three times with buffer and allowed to relax for 30 min before the experimental protocol began. To determine the vasodilator responses to ACh, the rings were preconstricted with PE and dose-response relaxation to cumulative addition of ACh in the organ bath was recorded through changes in isometric tension using a PowerLab/8sp multichannel data acquisition system (ADInstruments) and the ADI Chart software (version 5.3). The concentration of agonist in the organ bath was increased in steps of 1-log units. ACh was added to yield the next higher concentration only when the response to the lower dose reached a steady level. A dose-response curve for ACh was constructed for each ring. The vasodilator (relaxant) responses were expressed as percent decreases of PE-induced precontraction, where the contraction produced by 1 μM PE in each ring from its initial resting tension (1 g) was considered as 100%.
Measurement of superoxide in blood leukocytes.
Whole blood was obtained from the mice through cardiac puncture of the left ventricle. Blood cells were separated from the plasma by centrifugation at 1,000 rpm for 2 min. Buffy coat containing blood was carefully aspirated from the remaining red blood cells and the sample was resuspended in Hanks’ balanced salt solution (HBSS), pH 7.4. ROS generation in leukocytes was determined using dihydroethidium (DHE; 50 µM) along with nuclear dye Hoechst-33342 (1 µM) as previously reported (22). DHE enters cells freely and reacts with superoxide to form 2-hydroxy ethidium and ethidium with red fluorescence. DHE was added simultaneously with Hoechst-33342 dye to the cell suspensions and incubated for 30 min at 37°C. Initial control assays in the presence of pre-added superoxide dismutase mimetic (SODm) Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP; 100 μM) demonstrated quenching of the observed fluorescence. After an incubation, cells were washed three times and then mounted on a glass slide using mounting media. The fluorescence derived from DHE was detected at excitation/emission wavelengths of 490 and 555 nm, respectively, and images were acquired by a Nikon Fluorescent microscope using a ×40 objective. Monocytes were identified by nuclear morphology. Averaged individual intensities of the cells were analyzed using Metamorph software.
In situ superoxide detection in vessels.
DHE-derived fluorescence staining was used to determine superoxide generation in aorta, with or without preincubation with the superoxide dismutase mimetic (SODm) MnTBAP, the NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 1 mM), or diphenyleneiodonium (DPI; 100 µM), which functions as a flavin-modifying NADPH oxidase inhibitor, and the more recently developed specific pan-NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870; 10 μM) (50, 87). Cleaned aortas were frozen in optimal cutting temperature compound (OCT; Tissue-Tek, Sakura Finetek, Inc., Torrance, CA), cryosectioned to 5-µm sections, and then stained with DHE (Molecular Probes, Inc., Eugene, OR) in the dark. The slides were rinsed extensively with PBS, mounted in antifade mounting media Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL), coverslipped, and digitally imaged with confocal microscopy (FluoView 1000; Olympus America, Inc., Center Valley, PA). The DHE-derived fluorescence was detected at excitation/emission wavelengths of 490 and 555 nm. The mean fluorescence intensity per image was calculated using the software provided by the manufacturer.
Immunofluorescence and hematoxylin eosin (H&E) staining.
Cleaned mouse aortas were isolated and placed in a block holder containing OCT embedding compound, snap frozen in dry ice, and cut at 4-µm thickness on chilled slides using a cryotome. Aortic sections were blocked with 1% BSA in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBST), permeabilized with 0.25% Triton X100 in TBS and washed three times with PBS. Slides were incubated overnight with the primary antibodies at 4°C, followed by 1 h incubation with the corresponding secondary antibodies according to the manufacturer’s recommended concentrations. The antibodies used for the immunofluorescence experiments were rabbit polyclonal anti-eNOS, rabbit polyclonal anti-p22phox, mouse monoclonal anti-gp91phox, and rat monoclonal monocyte/macrophage marker (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Secondary antibodies conjugated with Alexa Fluor 488 goat anti-mouse, Alexa Fluor 594 goat anti-mouse, Alexa Fluor 594 goat anti-rat, or Alexa Fluor 488 goat anti-rabbit (Invitrogen, Carlsbad, CA) were used, and 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) was used as a counterstain. The sections were digitally imaged with confocal microscopy, and the mean fluorescence intensity per image was calculated. H&E staining was performed on formalin-fixed sections.
Western blot analysis.
Whole cell protein extracts were obtained after homogenization of aortas in ice-cold lysis buffer containing 62 mM Tris (pH 6.8), 10% glycerol, and 2% sodium dodecyl sulfate. The protein concentration was determined using the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA). Proteins were separated in a graded (4–20%) polyacrylamide gel (Invitrogen, Carlsbad, CA) and electroblotted on PVDF membranes (Bio-Rad, Hercules, CA). The following antibodies were used according to the manufacturer’s recommended concentration: rabbit polyclonal anti-eNOS, rabbit polyclonal anti-p-eNOS ser1177, rabbit polyclonal anti-p22phox, mouse monoclonal anti-gp91phox (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit polyclonal anti-p-Akt Ser437, or rabbit polyclonal anti-GAPDH (1:500; Cell Signaling Technology, Danvers, MA). Anti-mouse and anti-rabbit linked to horseradish peroxidase were used as secondary antibodies. Because of the limited amount of proteins extracted from mouse aorta and for the sake of having internal controls for each run, we used a gradient gel to resolve multiple proteins, along with size markers. We transferred the whole gel, located the proteins of interest based on the colored size marker, and cut the membrane to different pieces accordingly. We aligned the bands on the film with the colored size makers on the membrane for accurate size identification. Protein expression was quantified by a high-resolution Pharos FX Plus Molecular Imager (Bio-Rad, Hercules, CA).
High-performance liquid chromatography analysis of BH4.
BH4 content of plasma samples or cells was performed with some modifications according to the method previously described (11). Ascorbate (1 mg/mL) was added to the plasma to prevent BH4 autoxidation. Plasma samples were loaded into microcentricon tubes with a molecular mass cutoff of 4 kDa and centrifuged for 30 min at 10,000 g at 4°C. The filtrate was removed and subjected to HPLC analysis. The chromatographic system and method was as previously described (11).
Statistical analysis.
GraphPad InStat software, version 3.06 (GraphPad, San Diego, CA), was used to compute statistical data. Data are expressed as means ± SE of 3–10 independent experiments. Statistical comparisons were performed using the unpaired Student’s t test. Multiple comparisons were done by two-way repeated-measures ANOVA followed by a Bonferroni test. The 0.05 level of probability was used as the criterion of significance.
RESULTS
Cigarette smoke exposure increases blood pressure.
Endothelial dysfunction has been reported as an early marker of many cardiovascular disorders and is associated with hypertension. We tested whether CSE induces functional changes that impact the blood pressure (BP) of CS-exposed mice. Time-dependent alterations in systolic, diastolic, and mean BP and pulse pressure (PP) were observed following CSE (Fig. 1). With exposure duration up to 16 wk, no significant changes in BP values were seen. However, after 32 wk of exposure, clear significant elevations in BP were observed. At 32 wk of exposure, CS-exposed mice showed elevation in systolic pressure with values of 140 ± 6 mmHg, compared with 127 ± 5 mmHg in the matched air-exposed controls (Fig. 1A), and an even more marked elevation in diastolic pressure with values of 115 ± 6 mmHg versus 85 ± 6 mmHg in controls (Fig. 1C). Mean arterial BP of CS-exposed mice significantly increased after 32 wk, reaching 122 ± 6 mmHg compared with 99 ± 4 mmHg in air-exposed controls (Fig. 1B). This difference in mean BP was further increased after 48 wk, reaching 135 ± 5 mmHg compared to 102 ± 6 mmHg in controls. With 48 wk of exposure, further increases in systolic, diastolic, and mean BP occurred. As the systemic PP can be conceptualized as being proportional to stroke volume and inversely proportional to the compliance of the aorta, we calculated the PP following CSE. Figure 1D shows that CSE decreased PP in CS-exposed mice compared with air-exposed controls. At 32 wk of exposure, PP values were calculated to be 43 ± 2 mmHg and 25 ± 3 mmHg for control and CS-exposed mice, respectively. PP values were similarly altered after 48 wk of exposure, with values of 39 ± 3 mmHg and 22 ± 2 mmHg for control and CS-exposed mice, respectively. These data indicate that CSE induced hypertension by 32 wk, which progressively increased with further smoking exposure.
Fig. 1.

Blood pressure (BP) in control and smoking-exposed mice. BP was measured using a noninvasive tail-cuff method. Cigarette smoking exposure (CSE) significantly elevated systolic (A), diastolic (C), and mean arterial (B) BP and lowered pulse pressure (D) in 32- and 48-wk CS-exposed mice compared with air-exposed controls. Data are presented as means ± SE of 10 independent measurements in each group at each exposure time. *Significance from controls at P ≤ 0.05.
Cigarette smoke exposure induces vascular endothelial dysfunction.
The relaxation of aortic rings in response to endothelium-dependent vasodilator, ACh, was evaluated with studies of the aorta of 32- and 48-wk CS- or air-exposed mice. CSE significantly impaired ACh-induced endothelium-dependent relaxation in mouse aortic segments after 32 wk (Fig. 2A), with marked shift to the right in the dose-response curve and similar impairment after 48 wk (Fig. 2C). Sixteen weeks of CSE did not affect endothelium-dependent vasorelaxation induced by ACh (data not shown). Overall maximum response to ACh was markedly attenuated in the CSE group after 32 wk and even more blunted after 48 wk, with a rightward and upward shift in the ACh-concentration endothelium-dependent relaxation response curve. In contrast to this, CSE did not significantly alter the endothelium-independent relaxation in these aortic segments (Fig. 2, B and D).
Fig. 2.

Relaxation response of aorta and mesenteric artery to acetylcholine and nitroprusside. Mice were exposed to cigarette smoke (smoking) or fresh air (control). Aortic and mesenteric artery rings were mounted in a wire myograph and constricted with phenylephrine (PE; 1 µM), and the concentration-response to acetylcholine and sodium nitroprusside was measured. Endothelial-dependent and independent relaxation in aortic rings from mice exposed to cigarette smoke for 32 wk is shown in A and B, respectively, while C and D show similar data following 48 wk of exposure. E and F: endothelial-dependent and independent relaxation, respectively, of mesenteric artery following 48 wk of exposure. Vessels of CS-exposed mice exhibited significant endothelial dysfunction compared with air-exposed controls. Data are presented as means ± SE of 6 independent experiments. *Significance from controls at P ≤ 0.05.
To determine if resistance vessels were similarly affected, we evaluated the relaxation response of mesenteric artery segments from CS- or air-exposed animals. Similar changes occurred with this long-term CSE, with a rightward shift in the ACh dose-dependent relaxation and decreased maximum relaxation compared with the responses in the control vessels (Fig. 2 E). Again, no significant change in endothelium-independent relaxation was seen compared with matched control vessels (Fig. 2F).
Cigarette smoke exposure increases superoxide radical production in mouse aorta.
DHE staining was used to image superoxide generation in mice aortic rings. Superoxide production was markedly increased in aortic sections of CS-exposed mice compared with air-exposed controls with clear increase in DHE-derived fluorescence (Fig. 3A). As observed, DHE reacts with superoxide to form its red fluorescent product. To confirm the specificity of the observed fluorescence for superoxide, control measurements were performed in aortic sections preincubated with the SODm (MnTBAP) before the addition of DHE. Overall, this technique enables qualitative detection of superoxide production in the tissue and the relative changes with inhibition of this process. Pretreatment with the SODm largely quenched the observed DHE fluorescence. The flavin-modifying enzyme inhibitor, DPI, that has been commonly used to inhibit NADPH oxidases, greatly decreased the DHE-derived fluorescence in tissue sections with decrease to the low levels seen in the controls (Fig. 3, A and B). Additionally, the more recently developed specific pan-NADPH oxidase inhibitor VAS2870 (50, 87) largely decreased the DHE fluorescence by ~65%. Furthermore, the NOS inhibitor (l-NAME) also markedly decreased the superoxide generation in aortic sections with ~50% decrease, indicating that CSE induces NOS-dependent generation of superoxide.
Fig. 3.

Superoxide radical generation in mouse aorta. Optimal cutting temperature-preserved aortic sections from 48-wk CS (smoking)- or air (control)-exposed mice were incubated with dihydroethidium (DHE) alone or together with 100 µM superoxide dismutase mimetic (SODm) Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP), NADPH oxidase inhibitors diphenyleneiodonium (DPI; 100 µM) and 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870; 10 µM), or NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 1 mM). Sections were visualized with confocal fluorescence microscopy. Red fluorescence arises from superoxide-mediated oxidation of DHE (A). B: quantitation of the fluorescence in A. Data represent means ± SE of 3 independent experiments. *Significance from the corresponding control at P ≤ 0.05. #Significance from the untreated smoking sections (DHE) at P ≤ 0.05.
Cigarette smoke exposure increases the levels of NADPH oxidase in the vasculature.
We assessed the relative protein levels of NADPH oxidase subunits p22phox and gp91phox (NOX2) in vessels of mice exposed to CS or air. Western blot analysis for both p22phox and gp91phox subunits showed a prominent increase in the levels of both of these NADPH oxidase subunits in the homogenate from aorta of CS-exposed animals compared with air-exposed controls (Fig. 4). The levels of these subunits increased progressively with the duration of exposure. Immunofluorescence measurements were performed to confirm the immunoblotting data and to visualize the localization of these NADPH oxidase subunits within the vessel. From the observed fluorescence, both p22phox and gp91phox were seen to be present in CS-exposed animals, with clear increases in their levels compared with those in air-exposed controls (Fig. 5B). For both NADPH oxidase subunits, maximum fluorescence is seen adjacent to the endothelium and in the subendothelial layer, with weaker fluorescence present throughout the vessel wall. p22phox and gp91phox were largely colocalized, as can be seen from the yellow color on the merged image (Fig. 5B). Immunoblotting showed that NADPH oxidase subunits were observed as early as after 16 wk of CSE, with further increase out to 48 wk of exposure. This indicates the early contribution of NADPH oxidase in the process of CS-induced superoxide generation. Thus, presence of the superoxide-generating NADPH oxidase would contribute to impaired eNOS activity with decreased NO production and bioavailability leading to the vascular endothelial dysfunction that occurs.
Fig. 4.

Expression of NADPH oxidase protein subunits p22phox, gp91phox in mouse aorta with variable duration of cigarette smoke exposure. A: immunoblot of NADPH oxidase subunits in aortic homogenate from cigarette smoke (CS) or air (C)-exposed mice. B: quantitation of p22phox, gp91phox band intensities from a series of immunoblots as in A. GAPDH is shown as a loading control. Data show time-dependent increase of NADPH oxidase subunit expression following smoke exposure. Data are presented as means ± SE of 3 independent experiments. *Significance from controls at P ≤ 0.05.
Fig. 5.

Macrophage activation and expression of NADPH oxidase protein subunits p22phox and gp91phox in mouse aorta. A: hematoxylin-eosin staining of aorta following 48 wk of cigarette smoke (smoking) or air (control) exposure. B: optimal cutting temperature-preserved aortic sections from mice exposed to 48 wk of cigarette smoke or air were incubated with antibodies against p22phox (red) and gp91phox (green). Blue fluorescence shown in the merged image (B) is due to nuclear staining with DAPI. Graph to the right shows the quantitative analysis of the fluorescence intensity of gp91phox and p22phox. C: aortic sections from mice exposed to 48 wk of cigarette smoke or air were incubated with antibodies against gp91phox (red) and monocyte/macrophage marker (MOMA-2; green). In the merged image, yellow shows colocalization of gp91phox and MOMA-2. Graph to the right shows the quantitative analysis of the fluorescence intensity of gp91phox and MOMA-2. Data are presented as means ± SE of 3 independent experiments. *Significance from control at P ≤ 0.05.
Cigarette smoke exposure induces monocytic leukocyte endothelial adhesion and infiltration into the vessel wall.
From tissue histopathology, inflammatory cell infiltration was observed within the aorta of CS-exposed mice but not in vessels from air-exposed controls. In the vessels of CS-exposed mice, marked monocytic leukocyte infiltration was present just below or adjacent to the endothelium, with leukocytes seen throughout the vessel wall (Fig. 5A). To further assess leukocyte infiltration in the aorta of CS-exposed mice, immunohistology was performed in frozen sections using the macrophage/monocyte-specific marker MOMA-2. Immunofluorescence staining shows infiltrated macrophages in the aorta of CS-exposed animals, and these macrophages largely colocalized with gp91phox and appear to be most prominent adjacent to or just below the endothelial layer (Fig. 5C). Thus, this macrophage infiltration provides a source of the measured NADPH oxidase and CS-induced ROS generation.
Cigarette smoke exposure decreases eNOS and p-eNOS Ser1177 expression in aorta.
ROS generation has been shown to downregulate eNOS expression through activation of the Rho/ROCK pathway and through proteosomal degradation (1, 73). Therefore, we investigated the effect of CSE on eNOS expression in the aorta. Western blot analysis of aortic tissue homogenate revealed that CSE downregulated eNOS expression (Fig. 6, A and B). From the Western blot data, eNOS levels remain unchanged after 16 wk exposure; however, this was followed by a significant loss of eNOS after 32 or 48 wk of CSE, with 47% and 50% declines, respectively. Immunohistology demonstrated that eNOS expression in the endothelial cell layer of aortas was clearly decreased after 48 wk of exposure in CS-exposed mice (Fig. 6C). We further examined the effect of CSE on eNOS phosphorylation at Ser1177 (p-eNOS). Western blot analysis of aortic tissue homogenate demonstrated a significant decrease in eNOS phosphorylation at 32 wk of CSE, and this effect became more prominent after 48 wk of the exposure (Fig. 6, A and B). The decrease in the levels of p-eNOS were greater than that for total eNOS. With calculation of the ratio of p-eNOS to eNOS in CS-exposed samples, the values were 0.52 and 0.43 at 32 and 48 wk of the exposure, respectively. Because Akt is the main kinase involved in this phosphorylation, the low expression levels of p-eNOS at Ser1177 detected indicate that Akt activity may be decreased as a result of CSE. Therefore, we measured the Akt kinase-mediated phosphorylation that activates Akt. As shown in Fig. 6, A and B, CSE significantly reduced the phosphorylation of Akt at Ser473 at 32 and 48 wk of exposure compared with air-exposed controls.
Fig. 6.
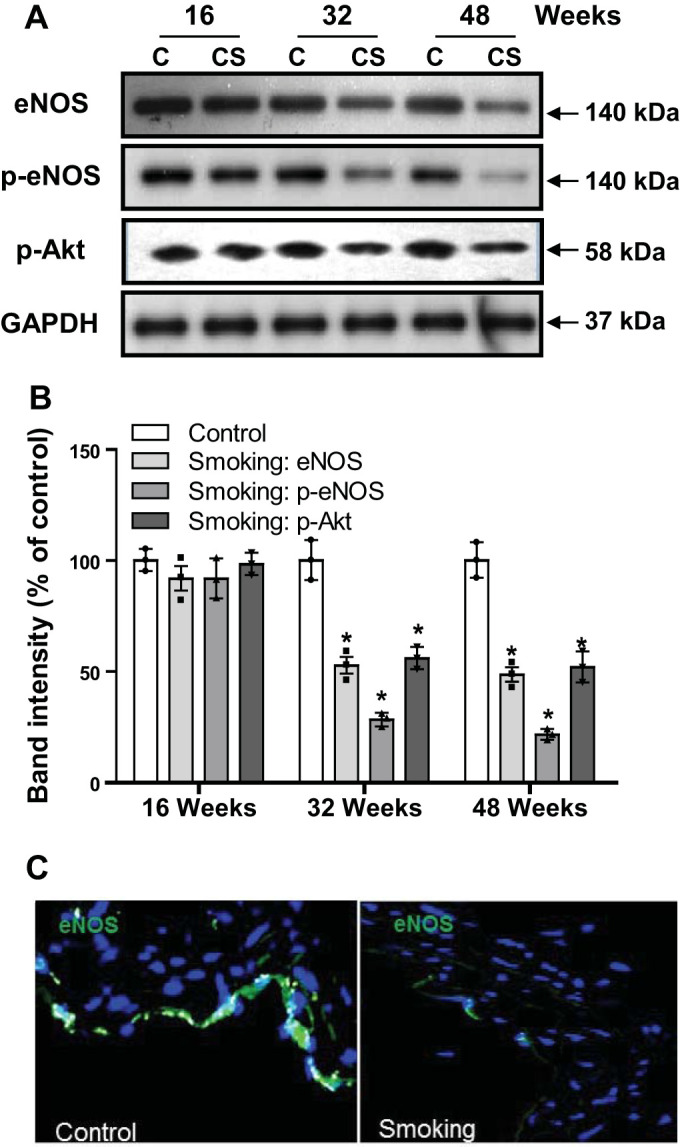
Measurement of endothelial nitric oxide synthase (eNOS), p-eNOS Ser1177, p-Akt, and GAPDH with variable duration of cigarette smoking exposure. A: immunoblotting for eNOS, p-eNOS Ser1177, and p-Akt in aortic homogenates from cigarette smoke (CS) or air-exposed (C) mice with exposure times of 16, 32, or 48 wk. B: quantitation of band intensities from a series of experiments as in A. C: aortic sections from mice exposed to 48 wk of cigarette smoking or air (control) with sections incubated with antibody against eNOS (green) and the nuclear stain DAPI (blue). Data are presented as means ± SE of 3 independent experiments. *Significance from controls at P ≤ 0.05.
Cigarette smoke exposure decreases BH4 levels.
Superoxide generation and other ROS produce highly reactive intermediates that may lead to oxidation and depletion of BH4 (11, 23). Because BH4 is a key cofactor required for eNOS function and its depletion results in eNOS uncoupling with superoxide generation from the enzyme, alterations in BH4 levels could serve as a key biomarker of endothelial dysfunction. Plasma BH4 levels are thought to reflect tissue levels and have been used in prior clinical studies of vascular disease (5a). Therefore, we measured the plasma levels of BH4 at different time points following either CS or air exposure. Figure 7 shows the time-dependent decrease in BH4 levels of CS-exposed mice compared with air-exposed controls. A significant modest decrease in BH4 levels was seen as early as 16 wk of CSE compared with controls, and this was followed by much larger declines after 32 or 48 wk CSE, with 50% and 75% decreases in BH4 levels, respectively.
Fig. 7.
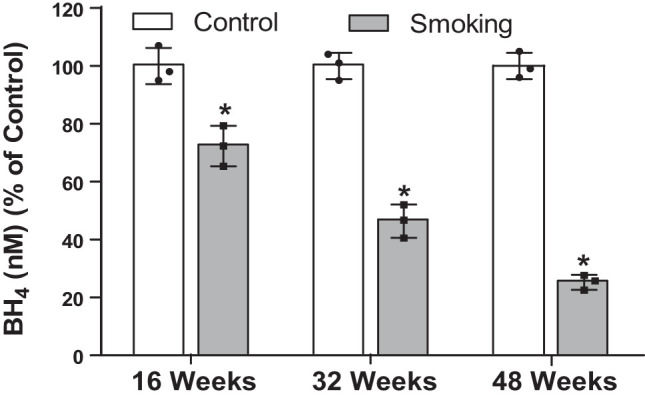
HPLC quantitation of plasma tetrahydrobiopterin (BH4) at different times following cigarette smoke exposure. Data are presented as means ± SE of 3 independent experiments. *Significance from air-exposed controls at P ≤ 0.05.
Cigarette smoke exposure induces monocytic leukocyte activation in the circulation.
One can hypothesize that leukocytes may be activated in the pulmonary circulation through exposure to CS-derived toxicants and oxidants that are inhaled into the lungs and traverse the pulmonary epithelium and endothelium. Once activated, these leukocytes would generate additional oxidants and adhere to vascular endothelium, followed by transmigration leading to tissue inflammation and vascular stress. Detection of activated monocytic leukocytes and their oxidant formation could be an early biomarker of CS-induced oxidant stress leading to vascular and other disease. Therefore, we performed measurements of oxidant formation from monocytic leukocytes obtained from freshly collected blood exiting the pulmonary circulation. In CS-exposed mice, mononuclear cells were activated to produce superoxide, with prominent DHE-derived red fluorescence seen in these cells (Fig. 8). In contrast, the cells from air-exposed mice exhibited little if any red fluorescence. Thus, CSE caused increased activation and ROS generation in monocytes exiting the pulmonary circulation. This, in turn, may lead to monocyte adhesion to the endothelium, with transmigration accounting for the monocyte/macrophage infiltration seen within the vessels of the CS-exposed mice.
Fig. 8.

Superoxide radical generation in mononuclear leukocytes following cigarette smoke exposure. Measurements are shown from blood obtained at 16 wk of exposure to cigarette smoke or air (control) (A). Lower panels show higher magnification images of representative mononuclear cells from control and smoke-exposed mice. In these images, the fluorescence derived from dihydroethidium in red is overlaid with Hoechst stain of nuclei in blue. B: mean fluorescence intensities in mononuclear cells from control and cigarette smoke-exposed mice. Smoking induced clear activation with enhanced reactive oxygen species generation. Data are presented as means ± SE of 8 independent experiments. *Significance from air-exposed control cells at P ≤ 0.05.
DISCUSSION
Several in vitro and short-term in vivo studies have suggested that CSE alters NO homeostasis, resulting in impairment of NO vasodilatory function; however, the precise underlying mechanisms have remained unclear. To elucidate the mechanisms underlying tobacco cigarette smoking-induced VED, we performed studies in a chronic mouse model of CSE that mimics human cardiovascular and pulmonary disease. Our studies possess several strengths, including: 1) the use of a well-characterized mouse model of chronic smoking-induced CVD in otherwise healthy animals, which enables delineation of the effects of CSE alone without superimposed alterations that might be caused by other overlying cardiovascular disease; 2) the use of a chronic moderate CSE regimen that induces gradual disease onset, as is the case with human smoking-related disease, rather than acute toxicity as occurs with higher dosing (75); and 3) our exposure was designed to deliver an estimated dose of nicotine that is within the relevant limits of nicotine exposure in humans, considering the differences in lung volume, body weight, and body surface area of mice and humans (19). Consequently, the cotinine plasma level in our CS-exposed animals, 80 ng/mL, is within the plasma levels found in human smokers (9, 49, 60).
In our chronic exposure model, the onset of hypertension was observed after 32 wk, with progressive increase thereafter. The lack of hypertension at 16 wk and the onset at 32 wk is consistent with our prior report (75). As reported previously, we observed systolic and diastolic hypertension, with elevations in mean blood pressure while the pulse pressure declined. In the current study, we observed a 23% rise in mean blood pressure at 32 wk and a 33% increase after 48 wk. In this chronic mouse CSE model after 32 wk, cardiac hypertrophy occurred with ~25% increase in left ventricular mass and ~20% decrease in cardiac output (75). The increase in blood pressure, despite the decrease in cardiac output, indicates that systemic vascular resistance is greatly increased by ~54% at 32 wk and ~65% at 48 wk. Although vascular tone is regulated by multiple vasoconstricting and vasodilating factors, alterations in endothelial-derived NO with resulting VED are of key importance in the onset of a variety of cardiovascular diseases. Alterations in NO production from eNOS, or in the metabolism of this NO, have been demonstrated to produce large alterations in blood pressure and vascular resistance (22, 58). Oxidative stress and inflammation are well-established triggers of VED (15). Our study examined the underlying mechanisms of the oxidant stress that triggers CS-induced VED.
With 16 wk of CSE, we observed from blood exiting the pulmonary circulation that monocyte activation with superoxide generation occurs. With this exposure duration, macrophage infiltration with concomitant increase in NADPH oxidase subunits p22phox and gp91phox was first seen in vessels of CS-exposed mice, and this was further increased with 32 and 48 wk of exposure. No such changes were seen in matched air-exposed control mice. As early as 16 wk of exposure, significant decreases in plasma BH4 levels were also detected, with near total deletion by 48 wk of exposure. Following these processes, alterations in eNOS function with uncoupling, as well as partial loss of the enzyme, were observed after 32 wk CSE, resulting in impairment of vascular endothelial function and systemic hypertension. Increased superoxide formation was detected in vessels and was abolished by a combination of NADPH oxidase inhibition and NOS inhibition. From these results, we can infer that oxidant-induced depletion of the critical eNOS cofactor BH4 triggered a switch of eNOS from NO production to superoxide generation. Overall, we observed that chronic CSE triggers a vicious cycle whereby monocyte/macrophage-mediated oxidant stress and eNOS uncoupling cause loss of NO production with further ROS production, which can lead to further monocyte/macrophage activation and adhesion to the endothelium with transmigration into the vessel wall (Fig. 9). It has been well documented that oxidative stress can lead to eNOS uncoupling due to cofactor depletion or oxidative modification of the enzyme (21, 22, 88). Additionally, oxidants trigger leukocyte activation with increased adhesion molecule expression (41, 70).
Fig. 9.

Mechanisms of oxidant production that trigger vascular endothelial dysfunction and cardiovascular disease. With chronic smoking exposure, oxidants in cigarette smoke activate monocytic leukocytes, traversing the pulmonary circulation and triggering their vascular adhesion and uptake. In parallel to this, oxidants in cigarette smoke deplete tetrahydrobiopterin (BH4) and antioxidants, leading to endothelial nitric oxide synthase (eNOS) uncoupling and oxidative modification and dysfunction with loss of nitric oxide (NO) synthesis with switch to superoxide (O2·−) generation, which leads to formation of the potent oxidant peroxynitrite (ONOO−). Both NADPH oxidase and eNOS-derived reactive oxygen species (ROS) formation can feedback on each other and themselves, resulting in a vicious cycle of ROS-induced ROS release and injury.
Although CS-induced impairment of endothelial relaxation has been previously reported, there has been conflicting data and controversy regarding the mechanisms involved. Several investigators have reported that CS is associated with impairment of endothelium-dependent relaxation in both macrovascular and microvascular beds of brachial and coronary artery circulation (8, 18, 61). In vitro studies have also found that CS extract decreased NO bioavailability, although the mechanisms involved were not clear (82, 91). The fact that CSE influences the eNOS pathway differently, depending on exposure time, dose, or experimental model used, has contributed to the controversy regarding the mechanisms of VED. It has been reported that treatment of endothelial progenitor cells with CS extract downregulated eNOS expression (47). Incubation of endothelial cells with sera from cigarette smokers for 12 h upregulated eNOS expression with decreased NO production and eNOS activity (8). Zhang et al. (91) did not see changes in eNOS expression following 1 h of CS extract exposure; however, lower eNOS activity and NO production were observed. With 4-h exposure of endothelial cells to CS extract, eNOS uncoupling has been observed due to loss of the critical cofactor BH4, and this was followed by proteosomal degradation of the BH4-synthesizing enzyme GTP cyclohydrolase (2). Inhibition of proteosomal degradation restored BH4 levels and eNOS-mediated NO production. This loss of NOS function was highly dependent on both the dose of CS extract and the duration of exposure. There are limited, yet conflicting, data on the influence of long-term CSE on the eNOS pathway in in vivo models (45, 61).
Impaired endothelium-dependent relaxation is typically caused by alterations in the NO-mediated smooth muscle relaxation cascade pathway (8, 18, 61). Potential sites for these alterations include a decrease in eNOS activity due to decreased levels of eNOS protein; reversible inhibition of eNOS; depletion of important eNOS cofactors; scavenging of NO by increased ROS production; interaction between ROS and the NO target soluble guanylate cyclase (sGC), thus modifying its cellular response to NO; and the inability or insufficiency of NO to counteract the overproduction of vasoconstrictors (65).
In the current study, chronic in vivo exposure to CS induces VED. Both eNOS uncoupling, as well as loss of eNOS protein, occur as a function of exposure duration, as demonstrated by Western blots and immunofluorescence (Fig. 6). CS-induced superoxide generation was partially inhibited by the eNOS inhibition, indicating the role of uncoupled eNOS as a critical source (Fig. 3). The loss of eNOS with prolonged CS exposure may be due to protein degradation that could result from CS-induced protein modifications such as nitration or oxidation (2). On the other hand, a prior study has suggested that CSE also reduces eNOS mRNA expression level (39).
The loss of ACh-induced relaxation with CS exposure was not mirrored when the NO donor nitroprusside was used. This suggests that the observed vascular dysfunction in CS-exposed mice is primarily due to loss of endothelium-derived NO production from eNOS, while smooth muscle function is not affected. In view of the elevated superoxide production observed in these vessels, it is surprising that endothelial-independent relaxation to the NO donor remained unchanged. This may be the case because the donor is added to the vessel bath, which is primarily in contact with the adventitial surface of the vessel, requiring diffusion to reach the vessel lumen. Because the presence of macrophage infiltration was seen to be primarily at endothelial and subendothelial locations and eNOS-derived superoxide would also be endothelial, the NO released by nitroprusside in contact with the adventitia of the vessel would still reach its sGC target in the smooth muscle wall of the aorta. Furthermore, amounts of exogenous NO available from the NO donor as added to the bath may well exceed the levels of superoxide in the smooth muscle. It is also possible that there may be some upregulation of sGC and the downstream cGMP-mediated pathways of relaxation that occur.
Phosphorylation of eNOS at Ser1177 results in an increase in electron flux through the reductase domain of eNOS, with increased eNOS activity due to increased Ca2+/CaM binding (20, 33, 40). We observed that CSE decreased this eNOS phosphorylation in an exposure duration-dependent manner. Although we also observed a loss of eNOS expression, it is clear that a much larger decrease in the levels of p-eNOS occurred that cannot be explained simply by the loss of total eNOS (Fig. 6). This loss of eNOS and p-eNOS at Ser1177 further explains our observation, and that of others, that CSE impairs NO production causing VED (8, 1).
Akt is known to mediate many downstream events controlled by phosphatidylinositol 3-kinase (PI3K) (20, 55) and to directly phosphorylate eNOS at Ser1177 (33, 62). Conflicting data on the effect of CS-induced Akt-dependent phosphorylation of eNOS have been reported (82). In our studies, we further investigated the Akt kinase-mediated Akt phosphorylation in vessels of CS-exposed mice. CSE reduced the phosphorylation of Akt at Ser473 at 32 wk of exposure. A further decrease in p-Akt expression was observed at 48 wk in CS-exposed mice compared with air-exposed controls. Thus, both a profound loss in eNOS expression and its Akt-mediated phosphorylation occurs.
From our observations, CS-exposed animals showed a decrease of the critical cofactor BH4 and subsequent partial loss of eNOS protein. A modest decrease in plasma BH4 levels was seen as early as 16 wk of CSE, and this was followed by much larger declines after 32 or 48 wk. Plasma levels of the redox sensitive eNOS cofactor BH4 could serve as a biomarker of both oxidative stress and a predictor of VED. Although we did not assay BH4 in the mouse aorta due to lack of sufficient tissue, the plasma levels would be expected to serve as an indicator of the BH4 status in tissues (5a). In prior assays of hearts isolated from CS-exposed mice after 32 wk, as reported previously (75), BH4 depletion has been observed. Circulating BH4 levels depend on the balance between its de novo synthesis and its oxidation/degradation. Oxidation of BH4 by superoxide or other ROS could occur because of CS-derived oxidants or secondary activation of leukocyte or endothelial ROS production, leading to depletion of BH4 with eNOS uncoupling, with loss of NO production and switch to superoxide generation. The combination of early depletion of BH4, along with l-NAME-mediated inhibition of superoxide generation (Fig. 3), suggests that CSE leads to eNOS uncoupling. This is consistent with the studies in isolated endothelial cells exposed to cigarette smoke extract that have shown that exposure triggers BH4 depletion, which triggers eNOS uncoupling with oxidative damage of the protein, leading to its ubiquitination and proteasomal degradation (2, 37). From these prior studies, the loss of BH4 was observed to arise because of oxidative degradation, as well as loss of the key BH4 synthesizing enzyme GTP cyclohydrolase. Oxidative protein modification was shown to lead to protein ubiquitination and proteosomal degradation of GTP cyclohydrolase and eNOS (2, 37).
The increased level of superoxide generation that we observe would also serve to scavenge NO, limiting its bioavailability and leading to the formation of the potent oxidant, peroxynitrite (64, 83). The interaction between NO and superoxide occurs at a nearly diffusion-limited rate and ~6 times faster than the rate of superoxide removal by superoxide dismutase. This results in generation of highly reactive peroxynitrite and loss of NO. CS-induced superoxide and peroxynitrite have been reported to decrease eNOS activity. We have previously shown that peroxynitrtie oxidizes BH4, causing eNOS uncoupling, and, at high concentrations, irreversibly destroys the heme center in eNOS (22, 23). We have also observed that physiological levels of NO in the presence of activated leukocytes are converted to peroxynitrite, and in turn enhance superoxide generation from leukocyte NADPH oxidase through activation of the ERK MAPK pathway (56).
The excessive generation of ROS that accompanies various cardiovascular risk factors has been implicated as a common pathway for the development of VED. There is a general consensus that CSE induces oxidative stress, which plays a major role in the pathogenesis of different cardiovascular disorders (3, 10, 26). It is known that cigarette smoke contains a high amount of free radicals and reactive aldehydes that are exogenously introduced to the body of smokers, which may activate endogenous sources of ROS, such as the leukocyte NADPH oxidase, uncoupled endothelial NO synthase, xanthine oxidase, and leak from the mitochondrial electron transport chain (10, 88, 92).
Our results clearly demonstrate that NADPH oxidase has a major role in the increased superoxide generation that occurs in the aorta of CS-exposed mice, since this increase was largely blocked by the specific NADPH oxidase inhibitor VAS2870 (Fig. 3). With leukocyte activation, adhesion molecule expression is increased, which triggers adhesion to the endothelium. Superoxide is known to induce further leukocyte adhesion molecule expression and adhesion to the endothelium, and this adhesion can be followed by subendothelial infiltration (41, 70). Of note, the levels of p22phox and gp91phox protein, as seen in the current study, were much higher in the aorta of CS-exposed mice than air-exposed controls (Figs. 4 and 5). This increase in the levels of NADPH oxidase subunits was observed as early as 16 wk of exposure. Several studies have shown that the NADPH oxidase Nox2 can be a major source of superoxide and other ROS in endothelial cells and can be activated after exposure to CS extract (7). It is also well known that the catalytic subunit gp91phox, Nox2, is highly expressed in phagocytic leukocytes (38, 54). Leukocyte NADPH oxidase consists of a membrane-localized cytochrome b558 composed of p22phox and gp91phox and the cytosolic components p47phox and p67phox. On stimulation, a multimeric protein complex (also including Rac1) is formed, leading to the production of superoxide (17, 44). Tissue histopathology showed leukocyte infiltration most marked adjacent to and beneath the endothelium of the vessel (Fig. 5). Because of the structural homology of leukocyte and vascular NADPH oxidase, we performed studies to discern the cellular source of the NADPH oxidase detected. Immunofluorescence staining showed colocalization of both p22phox and gp91phox and also the macrophage marker MOMA-2 with gp91phox, suggesting that infiltrating macrophages are a major source of the observed NADPH oxidase (Fig. 5). It is also possible, however, that some of the Nox-2 subunits detected could be present in endothelial cells as well.
It is important to note that under our experimental conditions, elevated BP or VED were first seen at 32 wk of CSE. This observation is at odds with the concept that NO in the body is directly and immediately affected by inhaled oxidants in CS. Although this direct effect would likely occur with regard to NO levels in the lungs, most of the CS-inhaled oxidants would not reach the systemic circulation. The levels reaching the circulation would be highly dependent on the doses inhaled. Acute models of CS-induced disease have been developed and report much earlier effects of CS following 4 days or 2 wk of exposure (31, 67). In general, these models utilize higher dose exposure with less admixing of the smoke with air. Higher levels of total particulate matter are present, and higher levels of CO and nicotine and its metabolites occur. There are several major differences between our study and those acute studies. Mainly, the exposure protocols, types of cigarettes used, and the smoke intensity are different. Unlike our study, those studies (31, 67) use nose-only exposure that results in much higher concentrations of total particulate matter, TPM, reaching to 420 mg/m3.
The delayed response reported in our model and the current study can be explained through understanding different compensatory mechanisms that animals may develop to protect the vascular wall from initial oxidative stress induced by CS-derived oxidants or their secondary activation of inflammation with additional oxidant stress. Indeed, in human smokers disease onset requires chronic exposure and is delayed by many years. Systemic antioxidants and antioxidative enzymes may largely neutralize CS-induced oxidant injury up to a given dose threshold. Such antioxidant defense mechanisms are basally present but can be further induced and activated to prevent oxidative stress and injury. The ability to detoxify the CS-induced oxidant stress delays the onset of tissue injury and disease for some time, but eventually this can be overcome depending on the dose and duration of exposure. The loss of protection may also be age associated, as it has been observed that oxidative stress increases with age due to dysregulation of Nrf-2-mediated antioxidant response (80). Other early compensatory processes with respect to delaying the onset of VED could include increased eNOS phosphorylation, higher levels of calcium/calmodulin, or lower levels of caveolin in endothelial cells or elevated levels of sGC in vascular smooth muscle cells. Additionally, there could be downregulation of the NO dioxygenase systems, such as cytoglobin and cytochrome b5/cytochrome b5 reductase, which have a major role in the degradation of NO in the smooth muscle wall of vessels (58).
In view of the critical role of oxidant stress and inflammation in the onset of VED and secondary cardiovascular disease, therapeutic interventions can be considered that may prevent, ameliorate, or even reverse this CS-induced vascular dysfunction. In this regard, a number of studies have targeted CS-induced oxidant stress through enhancing the level of natural antioxidants such as ascorbate, α-tocopherol, and carotenoids, as well as antioxidant enzymes such as SOD and catalase (85). Alternatively, inhibition of the endogenous sources of ROS formation, such as macrophage or endothelial-derived Nox, uncoupled eNOS, and the mitochondrial electron transport chain, has been considered (43). Ascorbate, α-tocopherol, and carotenoids are depleted by CS, and therapeutic restoration has been proposed to ameliorate the cardiovascular effects of CSE (48, 51, 59). Although cellular and animal models showed potential benefit, no benefit has been established in clinical application (28). Additionally, concerns have arisen about possible harm, as higher doses of these supplements could become pro-oxidants and induce toxicity (51). There are reports that SOD is depleted in tobacco smokers, and some SOD mimetics have been used to protect from smoking-induced toxicity in cellular and animal models (57, 71, 72). Recently, high-activity Mn-based SOD mimetics have been developed and advanced to clinical testing in cancer radiotherapy to protect surrounding normal tissues (4). It has been reported that there is crosstalk between mitochondria and NADPH oxidases, and this can also provide a vicious cycle of ROS production, which can be pharmacologically targeted (30). In an acute model of CSE, mitochondrial-targeted catalase blocked the increase in superoxide production in the aorta of CS-exposed mice (32). In the future, it will be important to compare the relative protective efficacy of targeted versus nontargeted antioxidants in chronic models of CSE.
Based on our study results, we propose that specific Nox2 inhibitors and therapeutics aimed at reversing eNOS uncoupling would hold promise in ameliorating CS-induced VED. Several prior studies have also suggested that targeting Nox using genetic tools or chemical inhibitors can protect against CS-induced toxicity (52, 63). Thus, there is much data to suggest that specific inhibition of Nox would serve to ameliorate CS-induced vascular disease. In recent years, new specific inhibitors for the Nox isoforms have been developed that may be translatable to humans (14). Based on the present studies as well as many prior studies, it is clear that eNOS dysfunction and uncoupling is a central cause of CS-induced VED. The eNOS dysfunction that was observed could be targeted by BH4 repletion strategies, as have been shown to be successful in models of cardiac ischemia-reperfusion injury and human trials (5a, 6, 68, 89). Additionally, therapeutic strategies to supplement and preserve NOS substrates, including l-arginine and NADPH, may be of value (12, 13, 16).
In conclusion, the present study in a well-defined chronic in vivo mouse model of CS-induced cardiovascular disease that mimics human disease demonstrates that CSE induces VED through macrophage infiltration with increased levels of NADPH oxidase and generation of ROS in vessels that is accompanied by eNOS dysfunction and uncoupling, triggering further ROS generation and leading to loss of NO production and increased superoxide generation. The resulting VED leads to increased vascular tone and the onset of hypertension. Thus, chronic CSE triggers VED through a vicious cycle of leukocyte-mediated oxidant stress that, in turn, causes eNOS uncoupling with further oxidant stress, which can lead to further leukocyte activation and adhesion to the endothelium with transmigration into the vessel wall. Overall, our study provides important insights toward understanding how smoking contributes to the genesis of VED, an early marker preceding the clinical manifestations of cardiovascular disease. It also points the way toward the development of biomarker assays for the early detection of this disease as well as therapeutic approaches to prevent or reverse it.
GRANTS
This work is supported by National Heart, Lung, and Blood Institute Grants R01-HL-135648 and R01-HL-131941.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.A.E.-M. and J.L.Z. conceived and designed research; M.A.E.-M., T.M.A., C.H., M.G.E., E.M.M., M.S.E., M.T.S., and Y.A.A. performed experiments; M.A.E.-M., T.M.A., C.H., M.G.E., E.M.M., M.S.E., M.T.S., Y.A.A., and J.L.Z. analyzed data; M.A.E.-M., T.M.A., M.G.E., M.S.E., M.T.S., Y.A.A., and J.L.Z. interpreted results of experiments; M.A.E.-M., T.M.A., C.H., M.G.E., E.M.M., M.S.E., and Y.A.A. prepared figures; M.A.E.-M., T.M.A., C.H., M.G.E., E.M.M., M.S.E., Y.A.A., and J.L.Z. drafted manuscript; M.A.E.-M., C.H., Y.A.A., and J.L.Z. edited and revised manuscript; M.A.E.-M. and J.L.Z. approved final version of manuscript.
REFERENCES
- 1.Abdelghany TM, El-Sherbiny GA, El-Mahdy MA, Zweier JL. Chronic cigarette smoke exposure impairs vascular endothelial function through a tetrahydrobiopterin-dependent mechanism (Abstract). 64th Tobacco Science Research Conference Hilton Head Island, SC, October 3-6, 2010, p. 48. [Google Scholar]
- 2.Abdelghany TM, Ismail RS, Mansoor FA, Zweier JR, Lowe F, Zweier JL. Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of GTP cyclohydrolase. Nitric Oxide 76: 113–121, 2018. doi: 10.1016/j.niox.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol 43: 1731–1737, 2004. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 4.Anderson CM, Lee CM, Saunders DP, Curtis A, Dunlap N, Nangia C, Lee AS, Gordon SM, Kovoor P, Arevalo-Araujo R, Bar-Ad V, Peddada A, Colvett K, Miller D, Jain AK, Wheeler J, Blakaj D, Bonomi M, Agarwala SS, Garg M, Worden F, Holmlund J, Brill JM, Downs M, Sonis ST, Katz S, Buatti JM. Phase IIb, randomized, double-blind trial of GC4419 versus placebo to reduce severe oral mucositis due to concurrent radiotherapy and cisplatin for head and neck cancer. J Clin Oncol 37: 3256–3265, 2019. doi: 10.1200/JCO.19.01507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annuk M, Zilmer M, Lind L, Linde T, Fellström B. Oxidative stress and endothelial function in chronic renal failure. J Am Soc Nephrol 12: 2747–2752, 2001. [DOI] [PubMed] [Google Scholar]
- 5a.Antoniades C, Shirodaria C, Crabtree M, Rinze R, Alp N, Cunnington C, Diesch J, Tousoulis D, Stefanadis C, Leeson P, Ratnatunga C, Pillai R, Channon KM. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 116: 2851–2859, 2007. doi: 10.1161/CIRCULATIONAHA.107.704155. [DOI] [PubMed] [Google Scholar]
- 6.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 114: 1193–1201, 2006. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 7.Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life 50: 267–269, 2000. doi: 10.1080/15216540051080976. [DOI] [PubMed] [Google Scholar]
- 8.Barua RS, Ambrose JA, Eales-Reynolds LJ, DeVoe MC, Zervas JG, Saha DC. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation 104: 1905–1910, 2001. doi: 10.1161/hc4101.097525. [DOI] [PubMed] [Google Scholar]
- 9.Benowitz NL, Hall SM, Herning RI, Jacob P III, Jones RT, Osman AL. Smokers of low-yield cigarettes do not consume less nicotine. N Engl J Med 309: 139–142, 1983. doi: 10.1056/NEJM198307213090303. [DOI] [PubMed] [Google Scholar]
- 10.Bernhard D, Moser C, Backovic A, Wick G. Cigarette smoke—an aging accelerator? Exp Gerontol 42: 160–165, 2007. doi: 10.1016/j.exger.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 11.Biondi R, Ambrosio G, De Pascali F, Tritto I, Capodicasa E, Druhan LJ, Hemann C, Zweier JL. HPLC analysis of tetrahydrobiopterin and its pteridine derivatives using sequential electrochemical and fluorimetric detection: application to tetrahydrobiopterin autoxidation and chemical oxidation. Arch Biochem Biophys 520: 7–16, 2012. doi: 10.1016/j.abb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD(P)(H). J Pharmacol Exp Ther 361: 99–108, 2017. doi: 10.1124/jpet.116.239459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boslett J, Reddy N, Alzarie YA, Zweier JL. Inhibition of CD38 with the thiazoloquin(az)olin(on)e 78c protects the heart against postischemic injury. J Pharmacol Exp Ther 369: 55–64, 2019. doi: 10.1124/jpet.118.254557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 76: 208–226, 2014. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 15.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 16.Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JL. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem 282: 879–887, 2007. doi: 10.1074/jbc.M603606200. [DOI] [PubMed] [Google Scholar]
- 17.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 8: 691–728, 2006. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 18.Celermajer DS, Sorensen KE, Georgakopoulos D, Bull C, Thomas O, Robinson J, Deanfield JE. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation 88: 2149–2155, 1993. doi: 10.1161/01.CIR.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 19.Center for Drug Evaluation and Research, U.S. Department of Health and Human Services Food and Drug Administration . Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers. 2005. [last updated 2005].
- 20.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem 283: 27038–27047, 2008. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CA, Lin CH, Druhan LJ, Wang TY, Chen YR, Zweier JL. Superoxide induces endothelial nitric-oxide synthase protein thiyl radical formation, a novel mechanism regulating eNOS function and coupling. J Biol Chem 286: 29098–29107, 2011. doi: 10.1074/jbc.M111.240127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, Tsai AL, Zweier JL. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry 49: 3129–3137, 2010. doi: 10.1021/bi9016632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 38: 606–611, 2001. doi: 10.1161/hy09t1.094005. [DOI] [PubMed] [Google Scholar]
- 25.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 64: 111–126, 1985. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-κB. J Appl Physiol (1985) 105: 1333–1341, 2008. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daugherty A, Rateri D, Hong L, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. J Vis Exp 27: 1291, 2009. doi: 10.3791/1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.M Davies A, G Holt A. Why antioxidant therapies have failed in clinical trials. J Theor Biol 457: 1–5, 2018. doi: 10.1016/j.jtbi.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation 109, Suppl 1: III27–III32, 2004. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 30.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301, 2011. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dikalov S, Itani H, Richmond B, Vergeade A, Rahman SM, Boutaud O, Blackwell T, Massion PP, Harrison DG, Dikalova A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol 316: H639–H646, 2019. doi: 10.1152/ajpheart.00595.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 305: H1417–H1427, 2013. doi: 10.1152/ajpheart.00089.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimmeler S, Zeiher AM. Exercise and cardiovascular health: get active to “AKTivate” your endothelial nitric oxide synthase. Circulation 107: 3118–3120, 2003. doi: 10.1161/01.CIR.0000074244.82874.A0. [DOI] [PubMed] [Google Scholar]
- 34.Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ 328: 1519, 2004. doi: 10.1136/bmj.38142.554479.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabó C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112: 1049–1057, 2003. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci USA 104: 15081–15086, 2007. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Mahdy MA, Abdelghany TM, Hemann C, De Pascali F, Esmat A, Zweier JL. Cigarette smoke extract causes endothelial nitric oxide synthase dysfunction through stimulation of ubiquitin proteasome system. FASEB J 27 Suppl 1: 654.12, 2013. [Google Scholar]
- 38.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res 65: 495–504, 2005. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 39.Ferrer E, Peinado VI, Díez M, Carrasco JL, Musri MM, Martínez A, Rodríguez-Roisin R, Barberà JA. Effects of cigarette smoke on endothelial function of pulmonary arteries in the guinea pig. Respir Res 10: 76, 2009. doi: 10.1186/1465-9921-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 88: E68–E75, 2001. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 41.Fraticelli A, Serrano CV Jr, Bochner BS, Capogrossi MC, Zweier JL. Hydrogen peroxide and superoxide modulate leukocyte adhesion molecule expression and leukocyte endothelial adhesion. Biochim Biophys Acta 1310: 251–259, 1996. doi: 10.1016/0167-4889(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 42.Frisbee JC, Stepp DW. Impaired NO-dependent dilation of skeletal muscle arterioles in hypertensive diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol 281: H1304–H1311, 2001. doi: 10.1152/ajpheart.2001.281.3.H1304. [DOI] [PubMed] [Google Scholar]
- 43.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108: 1912–1916, 2003. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 44.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86: 494–501, 2000. doi: 10.1161/01.RES.86.5.494. [DOI] [PubMed] [Google Scholar]
- 45.Guo X, Oldham MJ, Kleinman MT, Phalen RF, Kassab GS. Effect of cigarette smoking on nitric oxide, structural, and mechanical properties of mouse arteries. Am J Physiol Heart Circ Physiol 291: H2354–H2361, 2006. doi: 10.1152/ajpheart.00376.2006. [DOI] [PubMed] [Google Scholar]
- 46.Hamilton CA, Brosnan MJ, Al-Benna S, Berg G, Dominiczak AF. NAD(P)H oxidase inhibition improves endothelial function in rat and human blood vessels. Hypertension 40: 755–762, 2002. doi: 10.1161/01.HYP.0000037063.90643.0B. [DOI] [PubMed] [Google Scholar]
- 47.He Z, Chen Y, Hou C, He W, Chen P. Cigarette smoke extract changes expression of endothelial nitric oxide synthase (eNOS) and p16(INK4a) and is related to endothelial progenitor cell dysfunction. Med Sci Monit 23: 3224–3231, 2017. doi: 10.12659/MSM.902746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heitzer T, Just H, Münzel T. Antioxidant vitamin C improves endothelial dysfunction in chronic smokers. Circulation 94: 6–9, 1996. doi: 10.1161/01.CIR.94.1.6. [DOI] [PubMed] [Google Scholar]
- 49.Hukkanen J, Jacob P 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev 57: 79–115, 2005. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 50.Kahles T, Brandes RP. NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci 69: 2345–2363, 2012. doi: 10.1007/s00018-012-1011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelly G. The interaction of cigarette smoking and antioxidants. Part III: ascorbic acid. Altern Med Rev 8: 43–54, 2003. [PubMed] [Google Scholar]
- 52.Kim M, Han CH, Lee MY. NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicol Res 30: 149–157, 2014. doi: 10.5487/TR.2014.30.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, Harrison DG, Hornig B, Drexler H. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation 106: 3073–3078, 2002. doi: 10.1161/01.CIR.0000041431.57222.AF. [DOI] [PubMed] [Google Scholar]
- 54.Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91phox homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88: 888–894, 2001. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 55.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114: 2903–2910, 2001. [DOI] [PubMed] [Google Scholar]
- 56.Lee C, Miura K, Liu X, Zweier JL. Biphasic regulation of leukocyte superoxide generation by nitric oxide and peroxynitrite. J Biol Chem 275: 38965–38972, 2000. doi: 10.1074/jbc.M006341200. [DOI] [PubMed] [Google Scholar]
- 57.Lehr HA, Kress E, Menger MD, Friedl HP, Hübner C, Arfors KE, Messmer K. Cigarette smoke elicits leukocyte adhesion to endothelium in hamsters: inhibition by CuZn-SOD. Free Radic Biol Med 14: 573–581, 1993. doi: 10.1016/0891-5849(93)90138-K. [DOI] [PubMed] [Google Scholar]
- 58.Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT, Kawada N, Zweier JL. Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall. Nat Commun 8: 14807, 2017. doi: 10.1038/ncomms14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr 71: 530–536, 2000. doi: 10.1093/ajcn/71.2.530. [DOI] [PubMed] [Google Scholar]
- 60.Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 190: 269–319, 2007. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 61.McVeigh GE, Lemay L, Morgan D, Cohn JN. Effects of long-term cigarette smoking on endothelium-dependent responses in humans. Am J Cardiol 78: 668–672, 1996. doi: 10.1016/S0002-9149(96)00391-8. [DOI] [PubMed] [Google Scholar]
- 62.Ndiaye M, Chataigneau T, Chataigneau M, Schini-Kerth VB. Red wine polyphenols induce EDHF-mediated relaxations in porcine coronary arteries through the redox-sensitive activation of the PI3-kinase/Akt pathway. Br J Pharmacol 142: 1131–1136, 2004. doi: 10.1038/sj.bjp.0705774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol 292: H130–H139, 2007. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 64.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem 266: 4244–4250, 1991. [PubMed] [Google Scholar]
- 65.Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens 19: 891–897, 2001. doi: 10.1097/00004872-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 66.Rapoport RM. Nitric oxide inhibition of endothelin-1 release in the vasculature: in vivo relevance of in vitro findings. Hypertension 64: 908–914, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03837. [DOI] [PubMed] [Google Scholar]
- 67.Raza H, John A, Nemmar A. Short-term effects of nose-only cigarette smoke exposure on glutathione redox homeostasis, cytochrome P450 1A1/2 and respiratory enzyme activities in mice tissues. Cell Physiol Biochem 31: 683–692, 2013. doi: 10.1159/000350087. [DOI] [PubMed] [Google Scholar]
- 68.Reyes LA, Boslett J, Varadharaj S, De Pascali F, Hemann C, Druhan LJ, Ambrosio G, El-Mahdy M, Zweier JL. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc Natl Acad Sci USA 112: 11648–11653, 2015. doi: 10.1073/pnas.1505556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulz E, Jansen T, Wenzel P, Daiber A, Münzel T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal 10: 1115–1126, 2008. doi: 10.1089/ars.2007.1989. [DOI] [PubMed] [Google Scholar]
- 70.Serrano CV Jr, Ramires JAF, Venturinelli M, Arie S, D’Amico E, Zweier JL, Pileggi F, da Luz PL. Coronary angioplasty results in leukocyte and platelet activation with adhesion molecule expression. Evidence of inflammatory responses in coronary angioplasty. J Am Coll Cardiol 29: 1276–1283, 1997. doi: 10.1016/S0735-1097(97)00070-3. [DOI] [PubMed] [Google Scholar]
- 71.Naga Sirisha CV, Manohar RM. Study of antioxidant enzymes superoxide dismutase and glutathione peroxidase levels in tobacco chewers and smokers: a pilot study. J Cancer Res Ther 9: 210–214, 2013. doi: 10.4103/0973-1482.113352. [DOI] [PubMed] [Google Scholar]
- 72.St. Clair DK, Jordan JA, Wan XS, Gairola CG. Protective role of manganese superoxide dismutase against cigarette smoke-induced cytotoxicity. J Toxicol Environ Health 43: 239–249, 1994. doi: 10.1080/15287399409531918. [DOI] [PubMed] [Google Scholar]
- 73.Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang JS, Zweier JL, Chen LC, Rajagopalan S. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol 28: 1760–1766, 2008. doi: 10.1161/ATVBAHA.108.166967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taddei S, Virdis A, Ghiadoni L, Magagna A, Salvetti A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 97: 2222–2229, 1998. doi: 10.1161/01.CIR.97.22.2222. [DOI] [PubMed] [Google Scholar]
- 75.Talukder MA, Johnson WM, Varadharaj S, Lian J, Kearns PN, El-Mahdy MA, Liu X, Zweier JL. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am J Physiol Heart Circ Physiol 300: H388–H396, 2011. doi: 10.1152/ajpheart.00868.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Talukder MA, Morrison RR, Mustafa SJ. Comparison of the vascular effects of adenosine in isolated mouse heart and aorta. Am J Physiol Heart Circ Physiol 282: H49–H57, 2002. doi: 10.1152/ajpheart.2002.282.1.H49. [DOI] [PubMed] [Google Scholar]
- 78.Tewari AK, Popova-Butler A, El-Mahdy MA, Zweier JL. Identification of differentially expressed proteins in blood plasma of control and cigarette smoke-exposed mice by 2-D DIGE/MS. Proteomics 11: 2051–2062, 2011. doi: 10.1002/pmic.201000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 90: 1205–1213, 2002. doi: 10.1161/01.RES.0000020404.01971.2F. [DOI] [PubMed] [Google Scholar]
- 80.Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, Ballabh P, de Cabo R, Sonntag WE, Csiszar A. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am J Physiol Heart Circ Physiol 301: H363–H372, 2011. doi: 10.1152/ajpheart.01134.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vita JA. Endothelial function. Circulation 124: e906–e912, 2011. doi: 10.1161/CIRCULATIONAHA.111.078824. [DOI] [PubMed] [Google Scholar]
- 82.Wagner L, Laczy B, Tamaskó M, Mazák I, Markó L, Molnár GA, Wagner Z, Mohás M, Cseh J, Fekete A, Wittmann I. Cigarette smoke-induced alterations in endothelial nitric oxide synthase phosphorylation: role of protein kinase C. Endothelium 14: 245–255, 2007. doi: 10.1080/10623320701606707. [DOI] [PubMed] [Google Scholar]
- 83.Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem 271: 29223–29230, 1996. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- 84.Wang XL, Sim AS, Wang MX, Murrell GA, Trudinger B, Wang J. Genotype dependent and cigarette specific effects on endothelial nitric oxide synthase gene expression and enzyme activity. FEBS Lett 471: 45–50, 2000. doi: 10.1016/S0014-5793(00)01356-9. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Chun OK, Song WO. Plasma and dietary antioxidant status as cardiovascular disease risk factors: a review of human studies. Nutrients 5: 2969–3004, 2013. doi: 10.3390/nu5082969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.White CR, Brock TA, Chang LY, Crapo J, Briscoe P, Ku D, Bradley WA, Gianturco SH, Gore J, Freeman BA. Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA 91: 1044–1048, 1994. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wingler K, Altenhoefer SA, Kleikers PWM, Radermacher KA, Kleinschnitz C, Schmidt HH. VAS2870 is a pan-NADPH oxidase inhibitor. Cell Mol Life Sci 69: 3159–3160, 2012. doi: 10.1007/s00018-012-1107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 273: 25804–25808, 1998. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 89.Xie L, Talukder MA, Sun J, Varadharaj S, Zweier JL. Liposomal tetrahydrobiopterin preserves eNOS coupling in the post-ischemic heart conferring in vivo cardioprotection. J Mol Cell Cardiol 86: 14–22, 2015. doi: 10.1016/j.yjmcc.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang S, Day I, Ye S. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis 154: 277–283, 2001. doi: 10.1016/S0021-9150(00)00475-5. [DOI] [PubMed] [Google Scholar]
- 91.Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension 48: 278–285, 2006. doi: 10.1161/01.HYP.0000231509.27406.42. [DOI] [PubMed] [Google Scholar]
- 92.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70: 181–190, 2006. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]