

Abstract
Regulatory T cells (Tregs) are a promising therapy for several immune-mediated conditions but manufacturing a homogeneous and consistent product, especially one that includes cryopreservation, has been challenging. Discarded pediatric thymuses are an excellent source of therapeutic Tregs with advantages including cell quantity, homogeneity and stability. Here we report systematic testing of activation reagents, cell culture media, restimulation timing, and cryopreservation to develop a good-manufacturing-practice (GMP)-compatible method to expand and cryopreserve Tregs. By comparing activation reagents, including soluble antibody tetramers, antibody-conjugated beads and artificial antigen-presenting cells (aAPCs), and different media, we found that the combination of Dynabeads Treg Xpander and ImmunoCult-XF medium preserved FOXP3 expression and suppressive function, and resulted in expansion that was comparable to a single stimulation with aAPCs. Cryopreservation tests revealed a critical timing effect: only cells cryopreserved 1–3 days, but not >3 days, after restimulation maintained high viability and FOXP3 expression upon thawing. Restimulation timing was a less critical process parameter than the time between restimulation and cryopreservation. This systematic testing of key variables provides increased certainty regarding methods for in vitro expansion and cryopreservation of Tregs. The ability to cryopreserve expanded Tregs will have broad-ranging applications including enabling centralized manufacturing and long-term storage of cell products.
Keywords: Regulatory T cell, cryopreservation, T cell manufacturing, GMP, cell therapy, tolerance
Introduction
Regulatory T cell (Treg) therapy is a promising approach to prevent or treat graft-versus-host disease (GVHD) following hematopoietic stem cell transplantation, graft rejection following solid organ transplantation or autoimmune conditions (1). Several Phase I clinical trials have been completed (2–11) and Phase I/II clinical trials of Treg therapy are underway (reviewed in (12)). However, significant obstacles remain to the broad use of Treg-based therapies, including developing and optimizing manufacturing protocols to generate large numbers of cells with the desired phenotype and function, while limiting manufacturing complexity and cost (13).
We have recently shown that discarded pediatric thymuses are a feasible source of clinically-applicable Tregs, with numerous advantages over blood- or cord blood-derived cells. Specifically, thymus-derived Tregs are abundant (14), and because in the thymus CD25 expression on CD4+CD8− T cells exclusively marks Tregs (15), a homogeneous population of Tregs can be recovered by magnetic bead-based separation of CD25+ and CD8− cells. Since thymus-derived Tregs are naive and homogenous, they are also resistant to inflammatory cytokine-induced destabilization (14, 16), making them a promising autologous cell therapy in children undergoing heart transplantation, or an allogeneic cell therapy in the setting of other diseases (13). Since ~ 1% of children are born with congenital heart defects, many of whom require surgery including removal of the thymus (17), thymuses are routinely discarded. This material could be handled using procedures similar to those used to collect pancreases for islet transplantation and then used as a source of material from which to isolate Tregs.
Multiple protocols have been developed to manufacture Tregs for clinical trials, with variations in almost all key process parameters, including media, activation reagents, IL-2 concentration, restimulation timing and culture duration (2, 4, 5, 8–11, 18–24). Recently, a variety of new good-manufacturing-practice (GMP)-compatible reagents to activate T cells have been released but there are no reported comparisons of the performance of these now commercially available products. The lack of systematic studies to define critical process parameters and optimal reagents leads to uncertainty about the best methods to isolate and expand Tregs for clinical use (13).
Another uncertainty in the Treg manufacturing process is the feasibility of cryopreserving expanded cells. Some studies reported that cryopreservation reduces Treg quality, resulting in decreased FOXP3 expression and impaired suppressive capacity (25, 26). Consequently, to date only a limited number of clinical trials have used cryopreserved Tregs (21, 22, 27). Using fresh cells as a clinical treatment creates many logistical hurdles: release testing must be done in a shorter time and there is limited notice of manufacturing failure; there is less infusion time flexibility to accommodate changes in patient health status; and it is difficult to quickly produce cells for rapidly progressing diseases such as acute GVHD. Finding ways to cryopreserve clinical-grade Tregs to be used as an “off-the-shelf” product would overcome these hurdles, enable centralized manufacturing and allow a single product to be used to treat multiple patients (28, 29). Indeed, the concept of using “off-the-shelf” allogeneic cells is seen as the future of cell therapy products.
In order to develop a protocol to manufacture clinical-grade thymus-derived Tregs, we have comprehensively compared multiple process parameters using GMP-compatible reagents. We also report on a new protocol to cryopreserve expanded thymus-derived Tregs to enable their use as an “off-the-shelf” cell therapy product.
Results
Development of clinical-grade thymic Treg isolation protocol
We previously showed that CD25+CD8− Tregs can be isolated from pediatric thymus tissue using manual dissociation with scissors, complement-mediated lysis to deplete CD8-expressing cells, and magnetic-bead-based positive selection of CD25+ cells (14). To develop a protocol that would be appropriate for GMP manufacturing, we first tested alternative methods for thymus tissue processing, seeking a method that minimized manual steps, ideally in a closed system. Thymuses were collected and processed either using manual dissociation (with scissors, razor blades or a tissue chopper), or with the gentleMACS dissociator. The number of live thymocytes per gram thymus released by each method was compared, revealing that manual dissociation resulted in a significantly higher thymocyte yield and viability (Figure 1A&B). No differences in the frequency of Tregs (%CD4+CD8−CD25+ cells) within the isolated thymocytes were observed (Figure 1C).
Figure 1: Isolation of thymic Treg using a two-step magnetic-bead-based selection process.
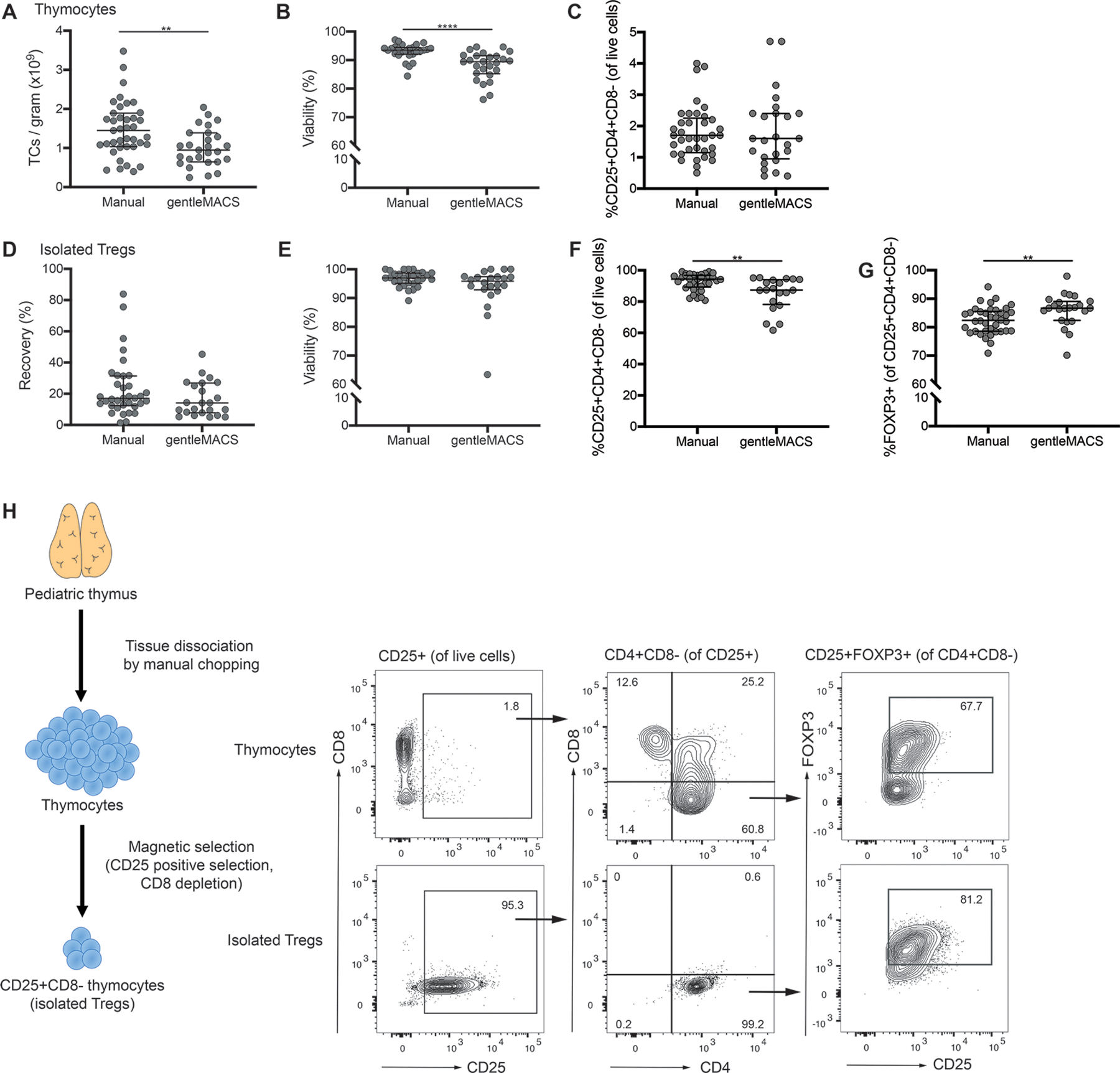
Pediatric thymuses discarded from cardiac surgery were collected and processed within 24 hours into a single cell suspension using either manual dissociation or the gentleMACS tissue dissociator. (A) Yield of thymocytes (TCs) per gram of tissue, (B) viability of isolated thymocytes and (C) frequency of CD25+CD4+CD8− cells within total live thymocytes. A two-step magnetic cell selection process with positive selection for CD25 followed by negative selection for CD8 was used to isolate Tregs from thymocytes. (D) Recovery of Tregs, defined as the number of CD25+CD4+CD8− cells isolated relative to the number of CD25+CD4+CD8− cells within the starting population of thymocytes. (E) Viability and (F) purity, defined as %CD25+CD4+CD8− cells, of isolated cells. (G) Proportion of FOXP3+ cells within the %CD25+CD4+CD8− population. (H) Schematic diagram of cell isolation process with representative flow cytometry data for thymocytes and isolated Tregs. Each symbol represents an individual subject, bars indicate median ± interquartile range; n=27–39 for manual and n=20–26 for gentleMACS tissue dissociator processing. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as determined by a Mann-Whitney test.
To eliminate the need for complement-mediated lysis of CD8+ cells, we next tested a magnetic bead-based separation protocol with two steps: positive selection of CD25+ cells using Releasable RapidSpheres (reagents not currently produced in a GMP format but which could be validated for use in a clinical setting), followed by negative selection of CD8+ cells. With this method, the median Treg recovery from thymocytes using manual dissociation was 17.0% (range: 1.4–83.9%, n=35) and using the gentleMACS was 14.1% (range: 5.0–45.3%, n=23) (Figure 1D) with no significant difference in viability (Figure 1E) or yield (data not shown).
Flow cytometry was used to characterize the purity of the resulting cells, revealing that the Treg purity (defined as CD25+CD4+CD8− cells) was significantly higher when they were isolated from manually-dissociated (median: 94.4%, range: 80.8–99.1%, n=37) versus gentleMACS-dissociated thymocytes (median: 87.4%, range: 61.7–95.3% n=20) (Figure 1F); however, within the CD25+CD4+CD8− cell population, FOXP3 expression was slightly higher in samples processed using the gentleMACS (Figure 1G). These data show that a two-step, GMP-compatible process for thymic Treg isolation by magnetic selection is feasible (Figure 1H) and that the advantage of the gentleMACS closed system is outweighed by the higher yield and viability of thymocytes obtained with manual dissociation.
Comparison of activation reagents for thymic Treg expansion
We previously developed a method to expand thymic Tregs by activation with an artificial antigen-presenting cell (aAPC)-based system using L cells, a mouse fibroblast cell line expressing CD58, CD80 and CD32, and loaded with anti-CD3 monoclonal antibodies (mAbs) (14, 16). Although aAPCs have been used to expand Tregs in a GMP-setting (9, 30), the increased cost and complexity for a manufacturing process with aAPCs led us to seek a cell-free alternative. We compared our original aAPC-based protocol with four cell-free activation reagents (ImmunoCult CD3/CD28 T Cell Activator, ImmunoCult CD3/CD28/CD2 T Cell Activator, Dynabeads Treg Xpander or T Cell TransAct) to determine their ability to effectively expand thymic Tregs without loss of FOXP3 expression. We used ImmunoCult-XF medium for CD3/CD28 and CD3/CD28/CD2 T Cell Activators; X-Vivo 15 with 5% CTS Immune Cell Serum Replacement (SR) for Treg Xpander; and TexMACS with 5% human serum for T Cell TransAct. ImmunoCult-XF medium was used for the aAPC conditions. On the basis of protocols used in previous studies (14), thymic Tregs were activated on day 0, restimulated on day 7 then counted and analyzed on day 12 and 15 (Figure 2A). Rapamycin was included in the culture medium from day 0–7 to prevent the outgrowth of effector T cells during Treg expansion (31, 32).
Figure 2: Comparison of cell-free activation reagents for thymic Treg expansion.
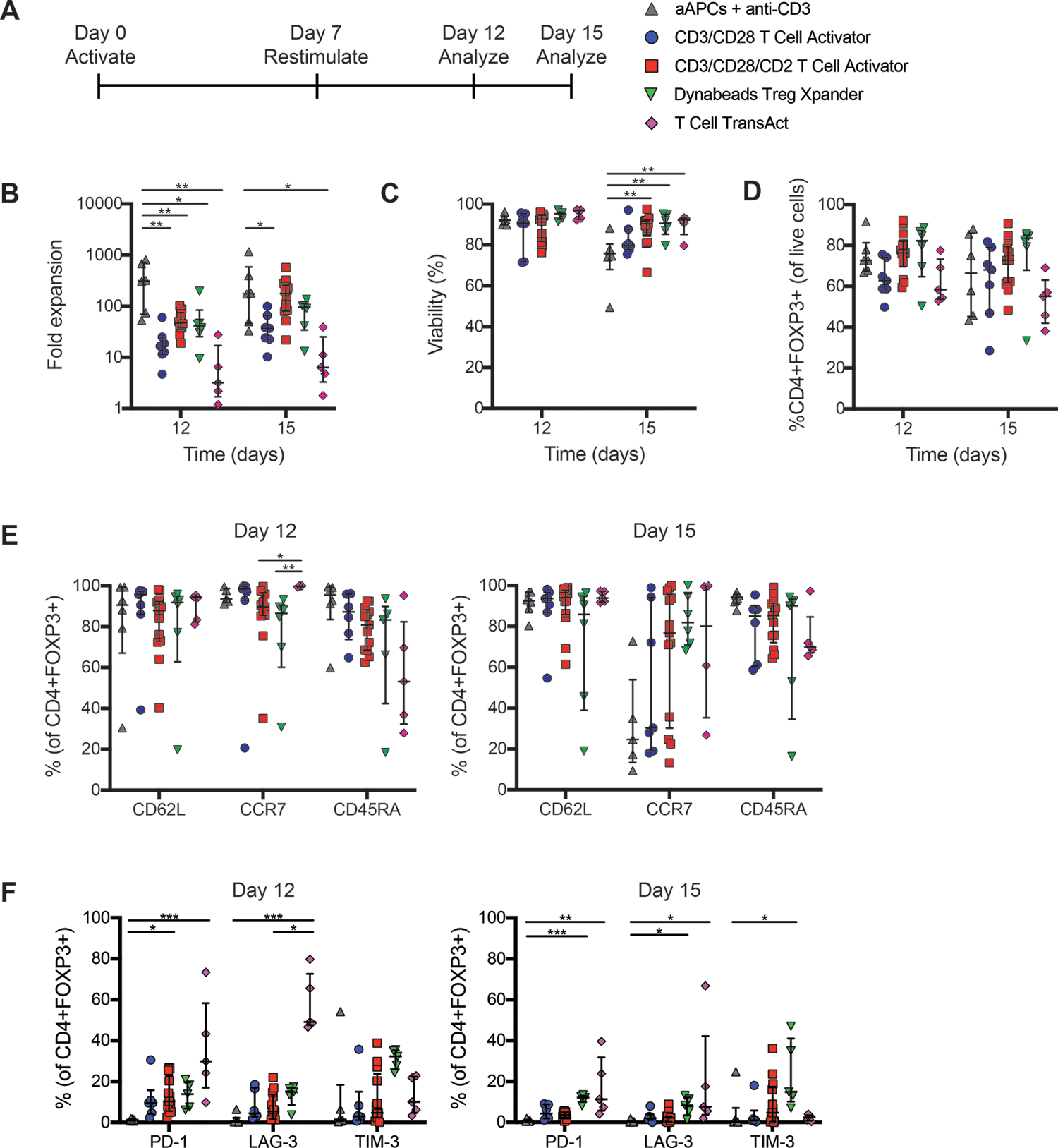
Isolated thymic Tregs were expanded and restimulated with the indicated type of activation reagent as indicated in (A). After 12 or 15 days in culture, (B) fold expansion, (C) viability and (D) FOXP3 expression, measured using flow cytometry, were determined. Flow cytometry was also used to quantify expression of markers of (E) T cell differentiation (F) or activation. Within each group, each symbol represents cells from a different subject and bars indicate median ± interquartile range. n=5–6 for L cell-based aAPCs + anti-CD3 mAbs, n=6–7 for CD3/CD28 T Cell Activator, n=13–14 for CD3/CD28/CD2 T Cell Activator, n=5–6 for Dynabeads Treg Xpander, and n=4–5 for T Cell TransAct, all tested in 9–10 experiments. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by a (B-C) two-way ANOVA with Tukey’s multiple comparisons test to compare expansion or viability between conditions on each day or (E-F) Kruskal-Wallis test with Dunn’s multiple comparisons test to compare expression for each protein among conditions.
We found that at both day 12 and 15, the aAPC-based method stimulated the highest fold expansion, and that among the cell-free reagents, activation with T Cell TransAct resulted in the lowest expansion(Figure 2B). Whereas thymic Treg viability remained high throughout culture with the cell-free activation reagents, by day 15 it declined in cultures activated with aAPCs (Figure 2C). There was a trend towards higher FOXP3 expression in cultures expanded with Treg Xpander compared to those with the other activation reagents; however, this did not reach statistical significance (Figure 2D).
We next compared the phenotypes of the expanded cells, focussing on markers of T cell differentiation and activation/exhaustion. After 12 days of culture, expression of markers associated with naive T cells remained high, with the majority of cells expressing CD62L, CCR7, and CD45RA regardless of the activation condition (Figure 2E). However, after 15 days the naive phenotype became more variable, particularly for CCR7, suggesting that prolonged culture may affect lymph node homing capacity. In CD8+ T cells, expression of PD-1, LAG-3 and TIM-3 is associated with exhaustion (33) whereas in Tregs this is associated with suppressive function (34–36). We found that Tregs expanded with cell-free activation reagents had higher expression of PD-1 and LAG-3 than those expanded with aAPCs at day 12, with the highest expression on cells expanded with T Cell TransAct (Figure 2F). Expression of these markers was generally reduced at day 15, suggesting that in Tregs these markers were likely associated with cell activation rather than exhaustion.
Effect of activation reagents on thymic Treg function
We next compared selected activation reagents for their ability to promote favourable cytokine production profiles and preserve thymic Treg suppressive function. Cultures stimulated with T Cell TransAct were excluded due to their low expansion (Figure 2B). Thymic Tregs expanded as in Figure 2A were stimulated with PMA and ionomycin and intracellular cytokine expression was analyzed by flow cytometry. As expected, a relatively low proportion of cells expressed inflammatory cytokines (14, 16), with those activated with Treg Xpander containing the lowest proportion of cells producing IL-2 (Figure 3A). Consistent with previous observations, very few cells expressed IL-10 (14, 16).
Figure 3: Function of thymic Tregs expanded with cell-free activation reagents.
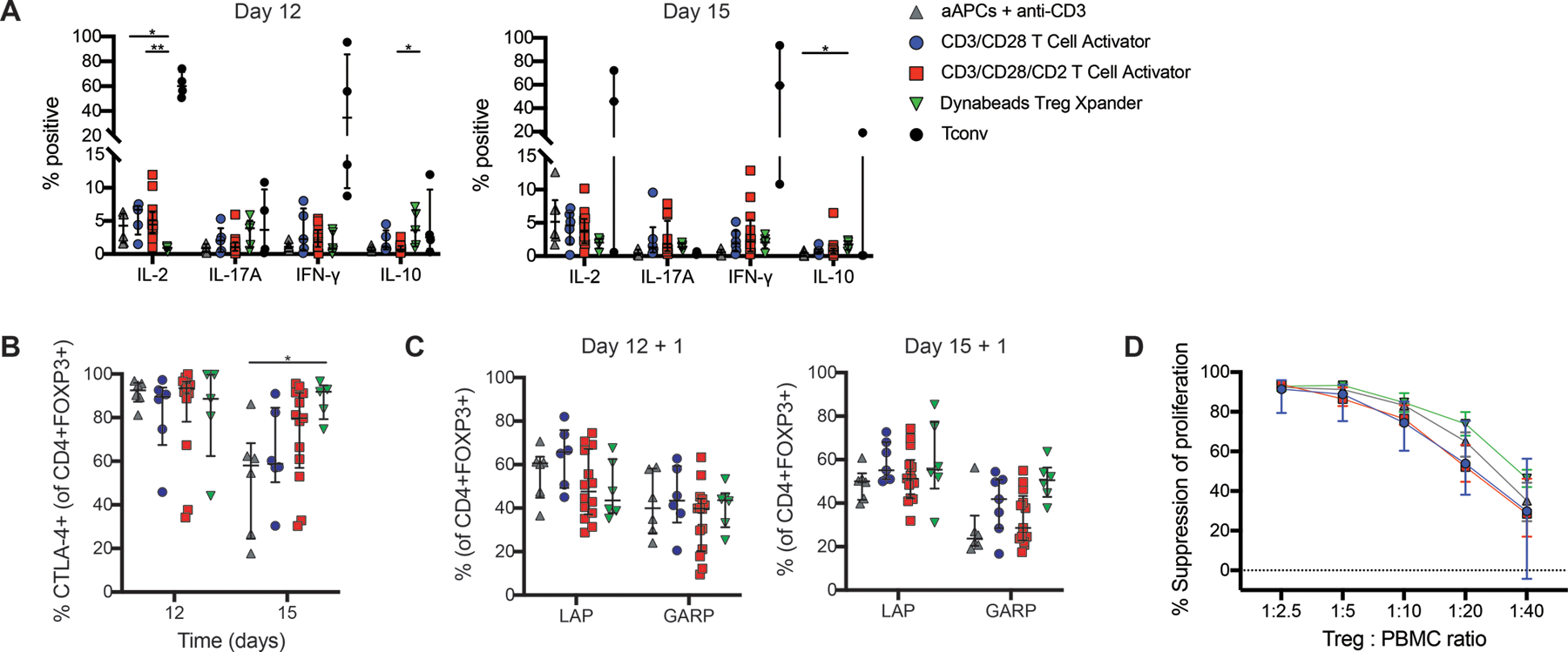
Isolated thymic Tregs were expanded with the indicated type of activation reagent and analyzed by flow cytometry after 12 or 15 days. Expression of (A) intracellular cytokines in Treg (CD4+FOXP3+) and Tconv after 4 hours of activation with PMA, ionomycin and brefeldin A, (B) CTLA-4 or (C) LAP and GARP after 24 hours of activation with anti-CD3/CD28 beads at a 1:16 bead to cell ratio. (D) After 15 days of expansion, thymic Tregs were cocultured with cell proliferation dye (CPD)-labeled PBMC at the indicated ratios and stimulated with a 1:16 ratio of anti-CD3/CD28 beads for 4 days. Suppression of CD8+ T cells within PBMC was determined by division index. For A-C, within each group, each symbol represents cells from a different subject and bars indicate median ± interquartile range. For D, median ± interquartile range is shown. n=4–6 for L cell aAPCs + anti-CD3 mAbs, n=4–6 for CD3/CD28 T Cell Activator, n=4–14 for CD3/CD28/CD2 T Cell Activator, n=2–6 for Dynabeads Treg Xpander, and n=3–4 for Tconv all tested in 4–9 individual experiments. *P < 0.05, **P < 0.01 as determined by a (A) Kruskal-Wallis test with Dunn’s multiple comparisons test to compare expression for each cytokine among activation reagents or (B) two-way ANOVA with Tukey’s multiple comparisons test to compare CTLA-4 expression between conditions on each day.
To assess the suppressive function of expanded thymic Tregs, we measured expression of proteins known to be major mediators of human Treg suppression, specifically cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (37), latency-associated peptide (LAP, the inactive form of TGF-β) and glycoprotein A repetitions predominant (GARP) (38). Thymic Tregs expanded with Treg Xpander had higher expression of CTLA-4 than those expanded with aAPCs (Figure 3B), but expression of LAP and GARP (Figure 3C) and suppressive function (Figure 3D) were similar among all conditions tested.
Effect of cell culture media on thymic Treg expansion
In Figures 2&3, the activation reagents were tested in different cell culture media, raising the possibility that some of the observed differences could be due to the media. We selected the two best-performing cell-free activation reagents (as defined by the highest fold expansion and FOXP3 expression): Treg Xpander and CD3/CD28/CD2 T Cell Activator, and compared their effects in four different media. Specifically, cells were expanded in ImmunoCult-XF, X-Vivo 15 and OpTmizer with serum replacement and ImmunoCult-XF without serum replacement. As before, the cells were activated at day 0, restimulated on day 7, and then analyzed on days 12 and 15 (Figure 4A). We found that cells cultured in ImmunoCult-XF had the highest fold expansion on day 15, and that this was not significantly affected by the presence or absence of serum replacement (Figure 4B). All media conditions preserved high cell viability (Figure 4C). Notably, expansion in OpTmizer resulted in significantly lower expression of FOXP3 with either activation reagent (Figure 4D).
Figure 4: Effect of cell culture media on thymic Treg expansion.
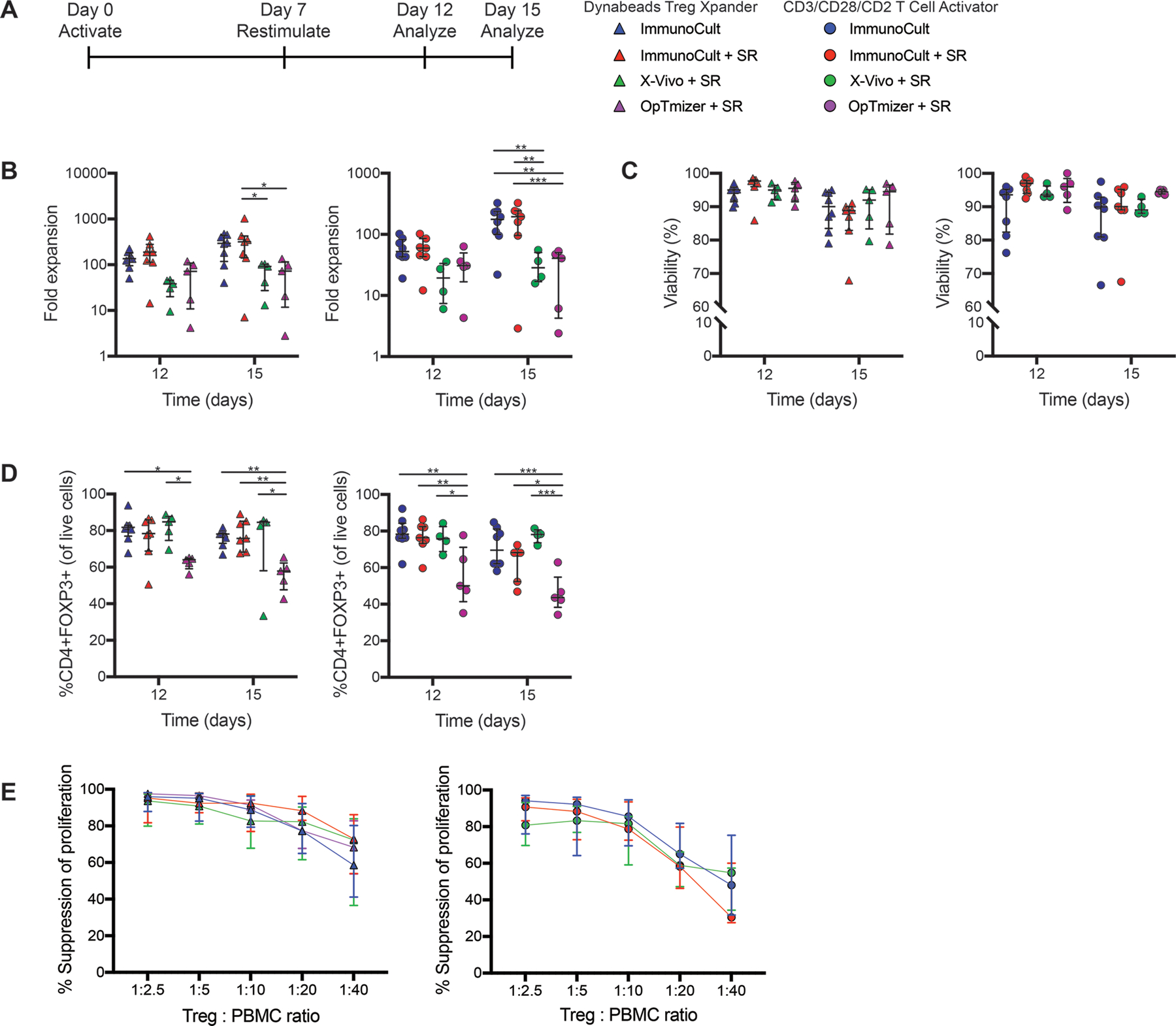
Isolated thymic Tregs were expanded and restimulated with Dynabeads Treg Xpander or CD3/CD28/CD2 T Cell Activator in different types of media as indicated in (A). After 12 or 15 days, cells were analyzed for (B) fold expansion, (C) viability and (D) FOXP3 expression. (E) After 15 days of expansion, thymic Tregs were cocultured with CPD-labeled PBMC at the indicated ratios and stimulated 1:16 with anti-CD3/CD28 beads for 4 days. Suppression of CD8+ T cells within PBMC was determined by division index. For B-D, within each group, each symbol represents cells from a different subject and bars indicate median ± interquartile range. For E, median ± interquartile range is shown. n=6–8 for ImmunoCult, n=4–7 for ImmunoCult + serum replacement (SR), n=4–5 for X-Vivo + SR, and n=2–5 for OpTmizer + SR tested in 4–5 experiments. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by a two-way ANOVA with Tukey’s multiple comparisons test to compare expansion or FOXP3 expression between conditions on each day.
Analysis of the effects of media on the thymic Treg phenotype revealed no significant differences in expression of naive cell markers (CD62L, CCR7, CD45RA) at day 12 (Figure S1B) or 15 (data not shown), with the exception of cells expanded with CD3/CD28/CD2 T Cell Activator in ImmunoCult-XF which showed higher levels of CD45RA expression but no difference in CD62L or CCR7. The media conditions also had little effect on markers of activation on day 12 (Figure S1C) or 15 (data not shown), with the exception of a higher proportion of cells expressing TIM3 when cultured in X-Vivo 15 with serum replacement. Similarly, varying the type of medium had little effect on intracellular cytokine production (Figure S1D), expression of CTLA-4, LAP and GARP (Figure S1E&F) or suppressive function. (Figure 4E).
Table 1 summarizes the results of media comparisons using the Treg Xpander or CD3/CD28/CD2 T Cell Activator. Expanding thymic Tregs in ImmunoCult-XF using Treg Xpander gave the best combination of fold expansion, viability and FOXP3 expression.
Table 1: Effects of media on fold expansion, viability and phenotype of thymic Tregs.
Cells were activated with Dynabeads Treg Xpander or CD3/CD28/CD2 T Cell Activator in ImmunoCult-XF, ImmunoCult-XF with serum replacement (SR), X-Vivo + SR or OpTmizer + SR. After 7 days the cells were restimulated with the same activators and after a total of 15 days of expansion fold expansion, viability and expression of FOXP3 was determined. All values are listed as median (range).
| Dynabeads Treg Xpander | CD3/CD28/CD2 T Cell Activator | |||||||
|---|---|---|---|---|---|---|---|---|
| ImmunoCult-XF (n=8) | ImmunoCult-XF + SR (n=7) | X-Vivo + SR (n=5) | OpTmizer + SR (n=5) | ImmunoCult-XF (n=8) | ImmunoCult-XF + SR (n=7) | X-Vivo + SR (n=4) | OpTmizer + SR (n=5) | |
| Fold expansion (day 15) | 295 (40–475) | 317 (7–1024) | 92 (13–103) | 73 (3–135) | 177 (22–326) | 194 (3–326) | 28 (16–55) | 41 (2–53) |
| Viability | 90% (79–95%) | 89% (68–91%) | 92% (80–95%) | 95% (79–97%) | 90% (67–98%) | 90% (68–96%) | 89% (88–93%) | 94% (94–95%) |
| %FOXP3+ | 78% (67–82%) | 76% (68–89%) | 85% (33–86%) | 58% (43–65%) | 70% (58–85%) | 68% (47–72%) | 78% (72–81%) | 44% (34–63%) |
| % Suppression of proliferation (1:20 Treg to PBMC) | 77% (43–95%) | 89% (83–97%) | 82% (46–96%) | 78% (68–87%) | 65% (51–92%) | 58% (43–86%) | 59% (45–67%) | --- |
Comparison of thymic Treg expansion with Treg Xpander versus GMP-compatible aAPCs
The selected combination of reagents tested above stimulated ~300 fold expansion of thymic Tregs over two weeks (Table 1). This fold expansion is substantially lower than the reported fold expansion for cord blood-derived Tregs using a GMP-validated aAPC system involving activation with K562 cells expressing CD64 (loaded with anti-CD3 mAbs) and CD86 (these cells hereafter referred to as KT64/86 aAPCs) (9, 30). We therefore tested if the fold expansion of thymic Tregs could be increased through activation with KT64/86 aAPCs. Replicate aliquots of CD25+CD8− thymic Tregs were expanded and restimulated using the optimized Treg Xpander-based system in ImmunoCult-XF as described above, or expanded with KT64/86 aAPCs in a previously-selected medium (X-Vivo 15 with 10% human AB serum) (30) without restimulation (Figure 5A).
Figure 5: Thymic Treg expansion with KT64/86 aAPCs.
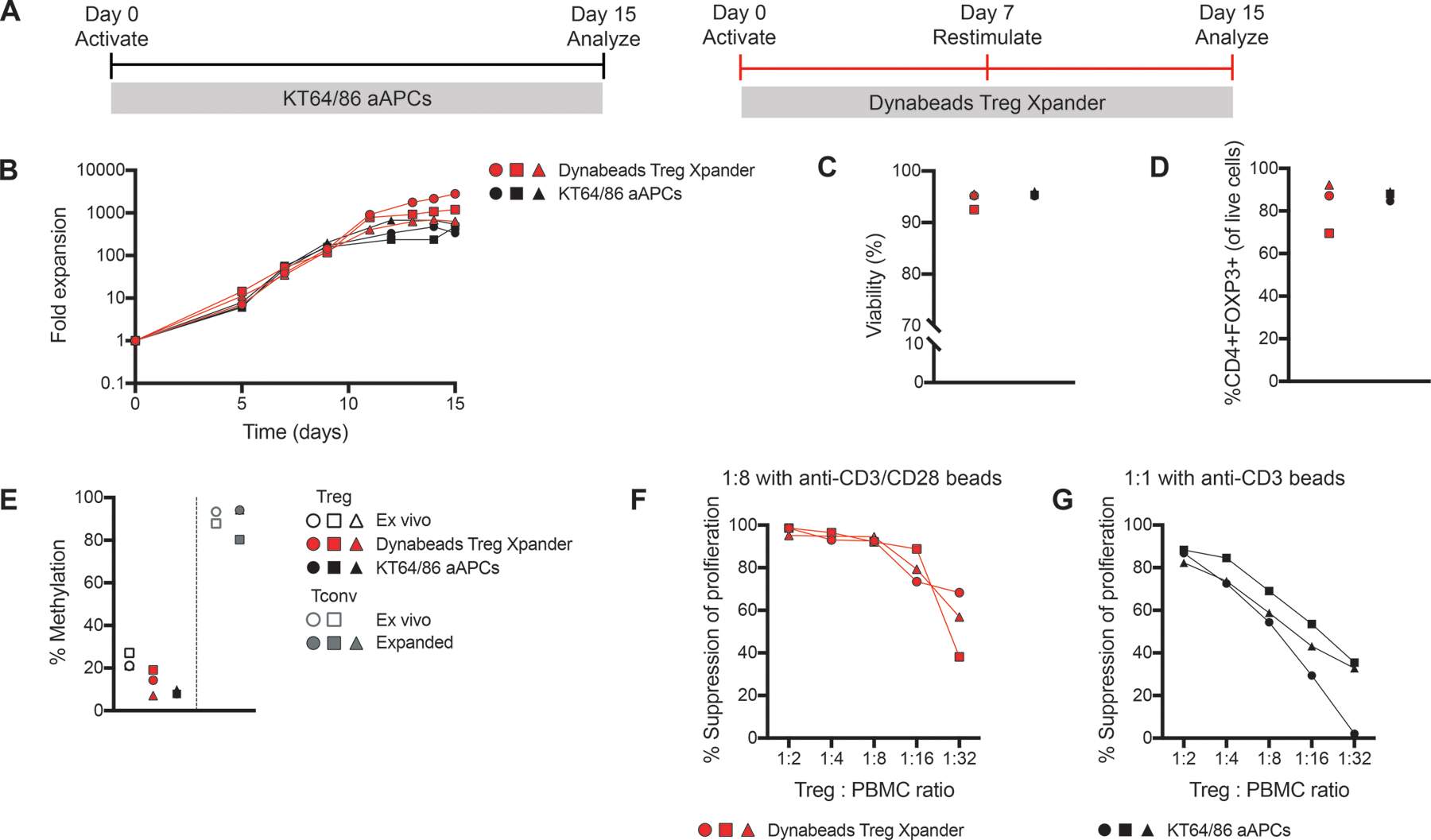
Replicate aliquots of thymic Tregs were expanded using KT64/86 aAPCs or Treg Xpander as indicated in (A). (B) Cells were counted to determine fold expansion. After 15 days, cells were analyzed for (C) viability and (D) FOXP3 expression. (E) TSDR analysis of ex vivo and expanded thymic Tregs and Tconv; all data are from males and the average methylation for 7 CpGs within the TSDR is shown. (F-G) Expanded thymic Tregs were cocultured with CPD-labeled (F) or CFSE-labeled (G) PBMC at the indicated ratios and stimulated at 1:8 with anti-CD3/CD28 beads or (F) 1:1 with anti-CD3 beads (G) for 4 days. Suppression of CD8+ T cells within PBMC was determined by division index. Within each group, each symbol represents cells from a different subject and matched subjects are shown with the same symbol. n=3 from 1–3 experiments.
We found that expansion of thymic Tregs after a single activation with KT64/86 aAPCs median 474-fold; range 338–557, n=3) resulted in a similar expansion as that previously reported for cord blood-derived Tregs (30). The median expansion after two stimulations with the Treg Xpander-based protocol was 1199-fold (range: 637–2806, n=3) (Figure 5B), and both protocols demonstrated similar viability (Figure 5C). Both protocols resulted in similar FOXP3 expression (Figure 5D), methylation at the Treg-specific demethylation region (TSDR)(Figure 5E) and suppressive function (Figure 5F&G). Comparison of Tregs expanded with KT64/86 aAPCs or Treg Xpander revealed similar patterns of differentiation and homing marker expression, as well as cytokine production(Figure S2A–C). As such, the remainder of the experiments were performed with Treg Xpander.
Cryopreservation 1–3 days post-restimulation preserves Treg viability, phenotype and function
In order to facilitate their use as an “off-the-shelf” product, we next sought to optimize the cryopreservation of thymic Tregs. As we hypothesized that activation state may affect recovery after cryopreservation, we examined the effect of cryopreserving Tregs at different time points following restimulation with Treg Xpander (Figure S3A), with and without a second restimulation before harvest and cryopreservation. We found that after a second restimulation at day 14, the cells continued to expand and maintained high viability but FOXP3 expression began to decline after day 16 (Figure S3B–D). Cell diameter increased after restimulation at day 14, reaching its peak at day 16 (Figure S3E).
We next tested how the day of cryopreservation relative to the last restimulation affected Treg recovery post-thaw. Expanded Tregs were cryopreserved at day 14 or 15 (7 or 8 days post-day 7 restimulation), or restimulated a second time on day 14 and then cryopreserved on days 15–19 (1–5 days post-restimulation) (Figure S3F). Upon thawing, cells were either immediately analyzed for recovery, viability and FOXP3 expression, or cultured overnight in ImmunoCult-XF with IL-2 (without additional TCR stimulation). We found that cell recovery was significantly higher for cells that were cryopreserved 1 or 2 days following their last stimulation than at 4 days (i.e. days 15–16 vs. day 18 in Figure S3G). Viability and FOXP3 expression had a similar trend, decreasing with longer times between restimulation and cryopreservation. After overnight culture, the effect of the day of cryopreservation was even more striking: if cells were cryopreserved >3 days following restimulation there was a dramatic drop in viability, live apoptosis-negative cells and FOXP3 expression (Figure S3H).
Increased expansion when Tregs are restimulated after returning to resting size
In light of the dramatic increase in post-thaw Treg health when cryopreserved shortly after restimulation, we altered our expansion protocol to allow cryopreservation within 1–3 days after restimulation. As a second restimulation increases the risk of Treg instability and/or outgrowth of contaminating non-Tregs (39, 40), and cryopreserving cells 2 days following restimulation at day 7 would curtail cell yields, we tested extending the length of the first activation period from 7 to 9 days. Cells restimulated at day 7 were analyzed after 2, 3 or 7 days and those restimulated at day 9 were analyzed after 2, 3 or 5 days (Figure 6A). Cells restimulated at day 9 reached a significantly higher level of overall expansion (Figure 6B&C) and sustained a higher growth rate following restimulation(Figure 6D) compared to those restimulated at day 7. It has been reported that allowing T cells to return to resting size (~8.5μm) prior to restimulation maximizes their subsequent expansion (39, 41, 42), and that if cells are restimulated too early, they likely undergo activation-induced cell death (42, 43). We found that thymic Tregs returned to resting size by day 9 (Figure 6E), providing an explanation for the superior expansion of cells restimulated on day 9.
Figure 6: Effect of restimulation time on thymic Treg expansion.
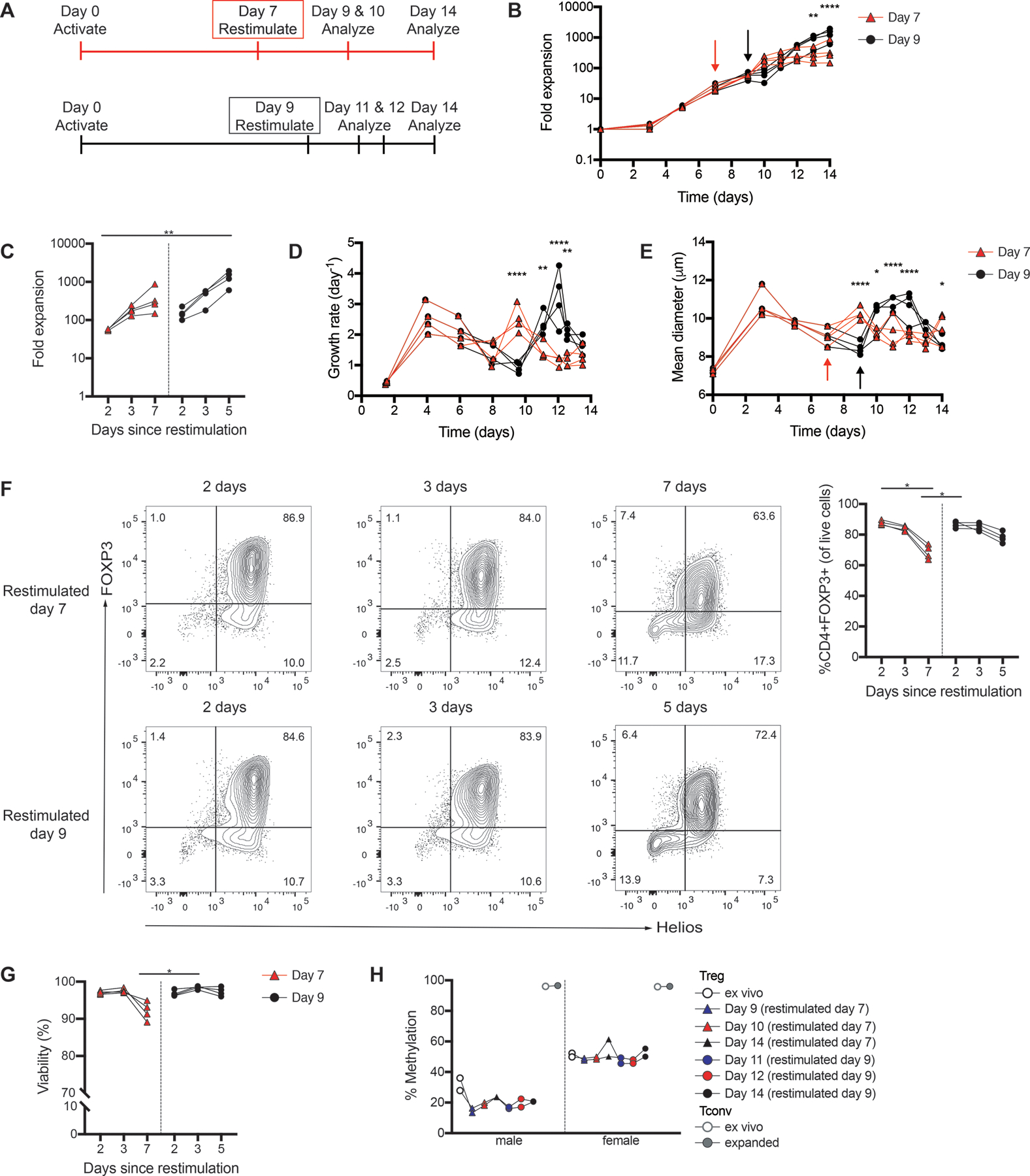
Thymic Tregs were activated and restimulated with Dynabeads Treg Xpander as indicated (A). (B) Fold expansion of thymic Tregs restimulated on day 7 or day 9. Arrows indicate the day cells were restimulated. (C) Fold expansion was measured 2, 3 or 7 days following restimulation at day 7 (day 9, 10 or 14), or 2, 3 or 5 days following restimulation at day 9 (day 11, 12 or 14). (D) Growth rate of thymic Tregs restimulated on day 7 or day 9, calculated from cell concentration over the course of culture. (E) Mean diameter of thymic Tregs restimulated on day 7 or day 9 over the course of culture. Arrows indicate the day cells were restimulated. (F) FOXP3 expression and (G) viability were measured 2, 3 or 7 days following restimulation at day 7, or 2, 3 or 5 days following restimulation at day 9. Flow cytometry plots are shown for a representative donor. (H) TSDR analysis of ex vivo and expanded thymic Tregs and Tconv. Average data from male and female donors shown is the average methylation for 7 CpGs within the TSDR. Each symbol represents cells from a different subject; data points from the same subject are linked. n=4 from 2 experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as determined by a (B,D,E) repeated measures two-way ANOVA with Sidak’s multiple comparisons test to compare between cells restimulated on day 7 or day 9 on each day, or (C,F,G) Friedman test with Dunn’s multiple comparison test.
Although cell viability and FOXP3 levels began to decrease 5 days after restimulation (Figure 6F&G), TSDR methylation levels remained similar (Figure 6H), suggesting that lower FOXP3 expression is likely the consequence of a decreased activation state (44) rather than Treg instability.
Thymic Treg cryopreservation timing is a more critical process parameter than restimulation day
We next investigated the impact of cryopreservation timing on cells restimulated at day 9. Cells were restimulated at day 9, then cryopreserved after 2, 3 or 5 days. Consistent with Figure S3, we found that higher Treg viability and FOXP3 expression were obtained when cells were cryopreserved shortly after restimulation. If they were cryopreserved >3 days after restimulation, there was a significant drop in recovery, viability and FOXP3 expression (Figure 7A&B). These declines were even greater after the thawed cells were cultured overnight, with viability and FOXP3 expression dropping well below what would be acceptable for a therapeutic product (Figure 7C&D). Consistent with their low viability and FOXP3 expression, thymic Tregs cryopreserved >3 days post-restimulation were also significantly less potent at suppressing the proliferation of CD8+ T cells (Figure 7E). In summary, we observed that cells cryopreserved 2 days following restimulation had the highest viability and FOXP3 expression upon thawing.
Figure 7: Cryopreservation of thymic Tregs following restimulation on day 9.
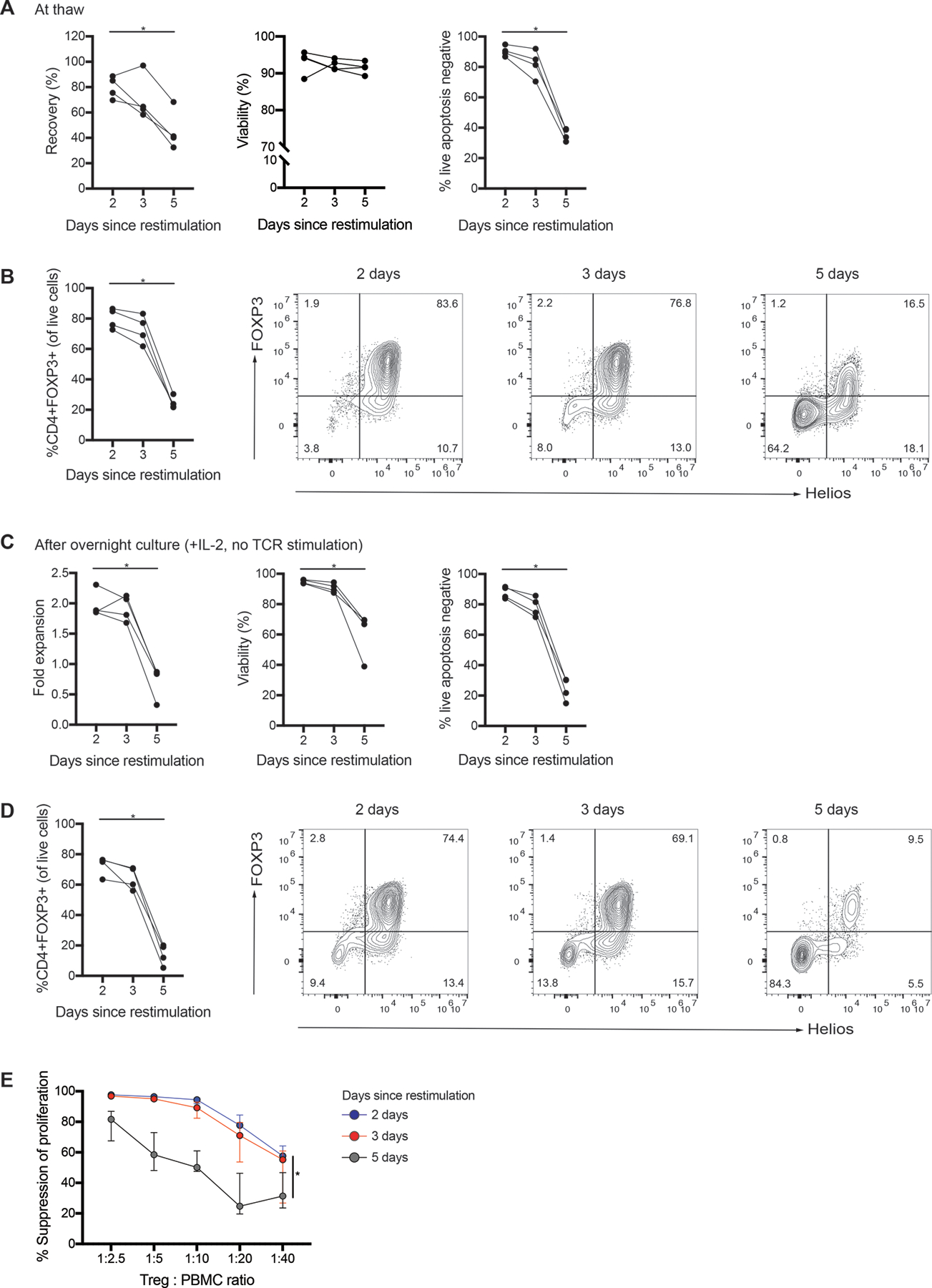
Isolated thymic Tregs were expanded with Dynabeads Treg Xpander and cryopreserved 2, 3 or 5 days following restimulation on day 9. (A) Recovery (defined as the number of live apoptosis negative cells thawed relative to the number of live cells cryopreserved), viability (measured by acridine orange/propidium iodide staining and apoptosis assay) and (B) FOXP3 expression for cells upon thawing. (C) Fold expansion, viability (measured by acridine orange/propidium iodide staining and apoptosis assay) and (D) FOXP3 expression for cryopreserved cells after thawing and overnight culture with IL-2. (E) After thawing and overnight culture, thymic Tregs were cocultured with CPD-labeled PBMC at the indicated ratios and stimulated 1:16 with anti-CD3/CD28 beads for 4 days. Suppression of CD8+ T cells within PBMC was determined by division index. Each symbol represents the mean of 3 technical replicates of cryopreserved cells from a different subject; data points from the same subject are linked. For E, median ± interquartile range of subjects is shown. n=4 from 2 experiments. *P < 0.05 as determined by a (A-D) Friedman test with Dunn’s multiple comparison test or (E) Friedman test with Dunn’s multiple comparison test to compare total area under each curve (AUC). Statistics on graph are for AUC of 2 days since restimulation relative to 5 days since restimulation.
To investigate if the effect of cryopreservation timing was specific to thymic Tregs or more broadly applicable, the experiments were repeated with naïve peripheral blood-derived Tregs (CD4+CD25+CD127−CD45RA+) and Tconv (CD4+CD25−CD127+CD45RA+). Cells were activated and restimulated at day 9 with Treg Xpander (for Tregs) or Dynabeads Human T-Expander (for Tconvs), then cryopreserved 2, 3 or 5 days following restimulation (Figure S4A–D). Analysis of cells post thawing revealed results that were consistent with Figure 7. Specifically, naïve peripheral blood-derived Tregs cryopreserved 2 or 3 days after restimulation had higher viability and FOXP3 expression than those cryopreserved at day 5 (Figure S4E). These findings were even more pronounced after culturing the thawed cells overnight (Figure S4F). Notably, we did not observe a large increase in TSDR methylation post cryopreservation (Figure S4G), suggesting that loss of FOXP3 expression was likely more related to cell viability than lineage instability. This conclusion is further supported by the finding that inflammatory cytokine production did not change significantly between cells cryopreserved at different days, although there was a slight increase in the production of IL-2 by FOXP3− cells in thymic Tregs cryopreserved 5 days following restimulation (Figure S4H).
To ask if the decrease in viability and FOXP3 expression was functionally relevant, the suppressive function of the thawed cells was tested, revealing that for both thymic and naive peripheral blood Tregs, cells cryopreserved at day 5 were substantially less able to suppress the proliferation of CD8+ T cells in comparison to those cryopreserved 2–3 days following restimulation (Figure S5A). Consistent with their lower suppressive function, Tregs cryopreseved 5 days following restimulation also had a lower proportion of FOXP3+ cells expressing CTLA-4, LAP and/or GARP (Figure S5B).
Finally, we tested whether the length of the first activation period could be extended longer than 9 days by measuring the expansion, viability and phenotype of cells restimulated at days 9–14 (Figure 8A). In each case, cells were analyzed 2 days following restimulation. For each restimulation day tested, thymic Treg numbers doubled 2 days following restimulation (Figure 8B). Cells restimulated on days 11–14 reached higher levels of overall expansion than those restimulated on day 9; however, this difference was only reached significance for the cultures restimulated on day 14(Figure 8C). Viability, FOXP3 expression and TSDR methylation were similar for the restimulation days tested (Figures 8D–F). FOXP3 expression decreased from day 7 to the day of restimulation, but increased following restimulation on all days (shown in Figure S6 for cells restimulated on day 11).
Figure 8: Effect of restimulation on day 9–14.
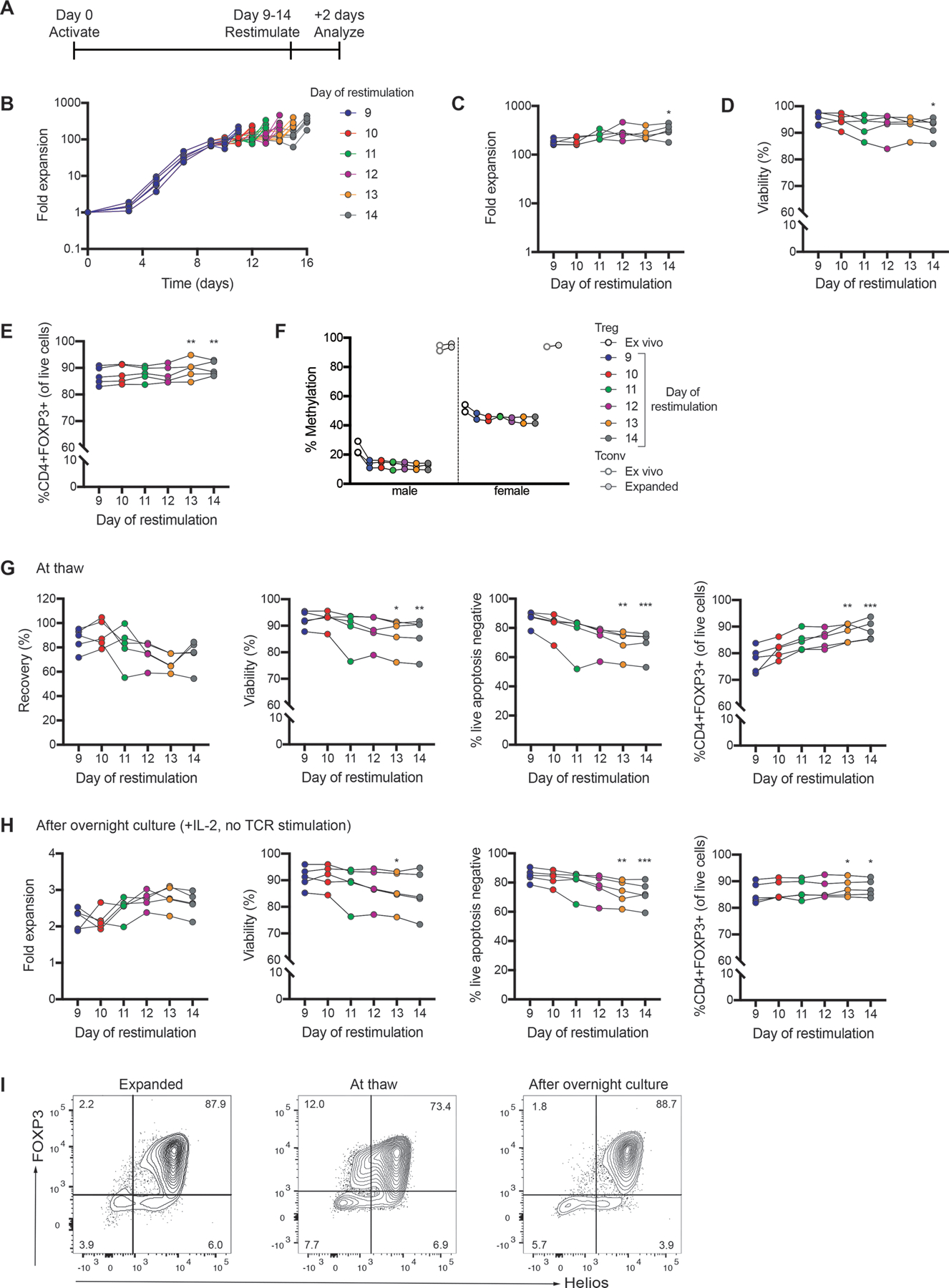
Isolated thymic Tregs were expanded with Treg Xpander and restimulated on day 9–14 as indicated in (A). In each case, cells were analyzed and cryopreserved 2 days following restimulation. (B) Fold expansion of thymic Tregs restimulated on day 9–14. The day in the legend refers to the day cells were restimulated. (C) Fold expansion, (D) viability and (E) FOXP3 expression measured 2 days following restimulation. (F) TSDR analysis of ex vivo and expanded thymic Tregs and Tconv. Average data from male and female donors shown is the average methylation for 7 CpGs within the TSDR. (G) Recovery (defined as the number of live apoptosis negative cells thawed relative to the number of live cells cryopreserved), viability (measured by acridine orange/propidium iodide staining and apoptosis assay) and FOXP3 expression for cryopreserved thymic Tregs shown at thaw. (H) Fold expansion, viability (measured by acridine orange/propidium iodide staining and apoptosis assay) and FOXP3 expression for cryopreserved thymic Tregs after thawing and overnight culture with IL-2. (I) Flow cytometry plots are shown for a representative donor. For B-F, each symbol represents cells from a different subject and matched subjects are linked. For G-H, each symbol represents the mean of 3 technical replicates of cryopreserved cells from a different subject and matched subjects are linked. n=5 from 3 experiments. *P < 0.05, **P < 0.01, ***P < 0.001 as determined by a Friedman test with Dunn’s multiple comparison test to compare each restimulation day to day 9.
In parallel, cells were cryopreserved 2 days following each day of restimulation. These cells were thawed and tested immediately or after overnight culture. There was a trend towards lower viability for cells restimulated on days 13–14 compared to day 9(Figures 8G&H). At thaw, there was a trend towards higher FOXP3 expression for cells restimulated on days 13–14 compared to day 9; however, FOXP3 expression was similar between conditions after overnight culture (Figures 8H&I). Overall, the differences between cells restimulated on different days were far less than those seen in Figure 7. Thus, the cryopreservation timing relative to the restimulation day is a much more critical process parameter than the day of restimulation.
Discussion
Here we report the first comprehensive testing of activation reagents, media, restimulation timing and cryopreservation conditions to optimize a GMP-compatible manufacturing protocol for Tregs isolated from discarded pediatric thymuses. We found large differences between the types of activation reagents and media, with the combination of Dynabeads Treg Xpander and ImmunoCult-XF medium resulting in the most efficient expansion of cells which retained high FOXP3 expression and suppressive capacity. Allowing cells to return to resting size (~8.5μm) prior to restimulation resulted in greater expansion following restimulation and the timing of cryopreservation had a major effect on cell viability, FOXP3 expression and suppressive function. Only cells cryopreserved 1–3 days following restimulation retained phenotypic and functional properties that would be acceptable for a clinical grade cell product upon thawing. These data have significant implications for the development of more feasible and cost-effective approaches to manufacture Tregs for clinical use.
Manual tissue dissociation and magnetic selection resulted in a Treg product with high (median of 94%) purity, as judged by CD25 and CD8 expression. Notably, as CD25 expression precedes that of FOXP3 in human Treg development (45), ~20% of the isolated CD25+ cells did not express FOXP3 directly after isolation; however, its expression was strongly upregulated after TCR activation. An advantage of thymic Tregs, is that magnetic-bead-based selection alone was sufficient to achieve a high purity cell product, without the additional need for GMP-grade flow cytometric sorting.
We found that the maximum fold expansion for thymic Tregs was less than that which has been previously reported for Tregs isolated from peripheral or cord blood (22, 30, 39). In comparison to Tregs from peripheral blood, the difference in expansion likely relates to the relative purity of the cultures and/or the state of T cell maturation. Unless peripheral blood Tregs are sorted as CD45RA+ cells, they contain a significant proportion of activated, non-regulatory T cells (14, 47, 48), which have a higher expansion potential. Indeed, as mentioned above, some of the isolated CD25+ cells were still undergoing differentiation and did not yet express FOXP3. This immaturity of the thymus-derived Tregs may underlie their lower expansion potential (16).
The type of activation reagent and media used had surprisingly large effects on Treg expansion potential and phenotype. Similar findings have been reported in other studies; for example, activation with KT64/86 aAPCs promotes higher expansion than antibody-conjugated beads (30, 39), and differences in expansion have been observed for beads from different suppliers (42). These differences are likely related to the strength of stimulation/costimulation provided by each reagent and the format in which the signals are provided (soluble antibodies, beads, cells, etc.). Many commercially available activation reagents are designed for Tconv rather than Tregs, and since Tregs are thought to experience stronger TCR stimulation than Tconv during development (49), the two cell types may require a different strength of stimulation for optimal expansion in vitro. As both activation reagents and media composition are proprietary, it is difficult to know the exact mechanism driving the differences observed here. However, these data clearly show that testing of activation reagents and media from a variety of manufacturers is warranted.
Similar to other reports, we found that expression of FOXP3 waxed and waned over the course of cell expansion (39, 50). These data raise the question of whether lower FOXP3 expression is the result of activation state, impending cell death and/or loss of Treg stability. FOXP3 expression in Tregs is known to vary with activation state, with higher expression shortly following activation/restimulation (50). FOXP3 expression is also known to decline in cell populations with low viability, such as after cryopreservation (25). We have previously shown that thymus-derived Tregs are stable, even when exposed to inflammatory cytokines (16), consistent with the findings reported here that despite variation in FOXP3 expression during expansion, expanded thymic Tregs maintained a demethylated TSDR and did not produce IL-2. Although there was a trend to a higher proportion of IL-2+ cells in Tregs cryopreserved 5 days following restimulation, the effect on viability was much more pronounced, suggesting that the low FOXP3 expression and suppressive function of these cells is likely mostly related to the high proportion of dead/dying cells.
We found that expanded Tregs cryopreserved 1–3 days following restimulation survived cryopreservation much better than those cryopreserved >3 days following restimulation. Tregs are known to be sensitive to cryopreservation, with many cells in the early stages of apoptosis at the time of thaw (25), although their phenotype and suppressive function can be rescued by restimulation (25, 26). The impact of cryopreservation on different cell types is influenced by many variables including metabolic state, membrane permeability, and sensitivity to stresses such as osmotic shock, toxicity and cold shock (51), all of which make cells susceptible to apoptosis and necrosis. Activation state has also been shown to affect the ability of T cells to recover from cryopreservation (52), but to the best of our knowledge, the critical effect of timing post-T cell stimulation on the success of cryopreservation has not been previously described. Further studies are required to determine the underlying mechanism for this effect which may be influenced by factors such as metabolism, speed of ice crystallization, cell composition and/or cell size. A limitation of our study is that cells were cryopreserved using Mr. Frosty freezing containers for all cryopreservation experiments. While these containers provide a rate of cooling near 1°C/min, the cooling rate is less controlled than with controlled rate freezers.
In summary, we comprehensively tested protocols to isolate, expand and cryopreserve thymus-derived Tregs. This work will enable large numbers of Tregs to be produced and cryopreserved for cell therapy applications. The ability to effectively cryopreserve expanded Tregs is step towards the development of these cells as an “off-the-shelf” cell therapy product.
Methods
Study approval.
Human research was approved by the University of British Columbia (UBC) Research Ethics Board (H17–01490 and H18–02553) and the University of Alberta Human Research Ethics Board (Pro00001408).
Cell isolation.
Thymus tissue was collected during infant cardiac surgery at British Columbia Children’s Hospital or University of Alberta Stollery Children’s Hospital. Tissue was dissociated in RPMI medium (Thermo Fisher Scientific) with 10% heat-inactivated fetal bovine serum (FBS) (VWR), 1% GlutaMAX (Thermo Fisher Scientific) and 1% penicillin/streptomycin (P/S) (Thermo Fisher Scientific) or ImmunoCult-XF T cell Expansion Medium (ImmunoCult-XF) (STEMCELL Technologies) with 1% P/S by manual dissociation using scissors, razor blades or McIlwain tissue chopper (Campden Instruments Ltd.) or by using the gentleMACS Dissociator (Miltenyi Biotec). CD25+CD8− thymic Tregs were isolated by CD25 positive selection using Releasable RapidSpheres, followed by CD8 depletion according to manufacturer’s instructions (STEMCELL Technologies). CD25−CD8− thymic Tconvs were isolated from the CD25− fraction by CD8 depletion followed by CD3 positive selection using EasySep Human CD3 Positive Selection Kit (STEMCELL Technologies). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Lymphoprep (STEMCELL Technologies).
Isolated thymic Tregs and Tconv were cryopreserved prior to expansion. Cells expanded to generate data for Figures 2–4 were cryopreserved in FBS with 10% DMSO. Cells expanded to generate data for Figure 5 were cryopreserved in human serum (HS) (Wisent Bioproducts) with 10% DMSO. Cells expanded to generate data for Figures 6–8 were cryopreserved in either FBS with 10% DMSO or CryoStor 10 (CS10) (STEMCELL Technologies). In data not shown, viability and recovery did not differ between these two types of freezing media.
For naïve peripheral blood Tregs, CD4+CD25+ cells were pre-enriched using RosetteSep Human CD4+ T Cell Enrichment Cocktail (STEMCELL Technologies) and CD25 Microbeads II (Miltenyi Biotec), then sorted as CD4+CD25+CD127−CD45RAhi Tregs (BD FACSAriaII). Naïve peripheral blood Tconv were sorted as CD4+CD25−CD127+CD45RAhi from the CD25− fraction.
Cell expansion.
For cell-free expansion, thawed thymic Tregs were activated with ImmunoCult Human CD3/CD28 T Cell Activator, ImmunoCult Human CD3/CD28/CD2 T Cell Activator (both STEMCELL Technologies), CTS Dynabeads Treg Xpander (Treg Xpander) (Thermo Fisher Scientific) at a 4:1 bead to cell ratio, or T cell TransAct (Miltenyi Biotec). Tregs were cultured as indicated in ImmunoCult-XF with 1% P/S, with or without 5% CTS Immune Cell Serum Replacement, X-Vivo 15 (Lonza) with 5% CTS Immune Cell Serum Replacement (Thermo Fisher Scientific), 1% P/S and phenol red, TexMACS Medium (Miltenyi Biotec) with 5% human serum (Wisent Bioproducts) and 1% P/S, or OpTmizer T Cell Expansion Media (Thermo Fisher Scientific) with 5% CTS Immune Cell Serum Replacement, 1% GlutaMAX and 1% P/S. Thymic Tconv were activated with ImmunoCult Human CD3/CD28/CD2 T Cell Activator and cultured in ImmunoCult-XF with 1% P/S. Naïve peripheral blood Tregs were activated with Treg Xpander at a 4:1 bead to cell ratio and cultured in ImmunoCult-XF with 1% P/S. Naïve peripheral blood Tconv were activated with Dynabeads Human T-Expander at a 3:1 bead to cell ratio and cultured in ImmunoCult-XF with 1% P/S. 100ng/mL rapamycin (Sigma Aldrich) was added to Treg cultures from day 0–7. Recombinant human (rh) IL-2 (Proleukin) (Tregs: 1000IU/mL, Tconv: 100IU/mL) was added from day 0. Cells were seeded at 0.5×106/mL in 96-well or 24-well plates with the concentration maintained at 0.5×106 cells/mL and media/additives refreshed every 2–3 days. Cells were restimulated as indicated. For cultures with Treg Xpander or Dynabeads Human T-Expander, a 1:1 bead to cell ratio was used for restimulation.
aAPC-based stimulation.
In some cases, cells were activated with irradiated (75 Gy) L cells preloaded with 100ng/mL anti-CD3 mAb (OKT3, UBC AbLab) at a 1:1 ratio (aAPCs : Tregs) (14) and cultured as indicated in ImmunoCult-XF with 1% P/S. 100ng/mL rapamycin was added from day 0–7 and 1000IU/mL IL-2 was added from day 0. Cell concentration was maintained at 0.5×106 cells/mL with media/additives refreshed every 2–3 days. Cells were restimulated as indicated. Alternatively, cells were activated with irradiated (100 Gy) K562 cells transduced to express CD64 and CD86 (KT64/86) that had been pre-loaded with 1μg/mL anti-CD3 (OKT3, Miltenyi Biotec) at a 1:1 ratio. Tregs were cultured in X-Vivo 15 with 10% heat inactivated human AB serum (Valley Biomedical), L-glutamine (Invitrogen) and N-acetylcysteine (American Regent). 300IU/mL recombinant human IL-2 (Proleukin) was added at day 2. Cell concentration was maintained at 0.25×106/mL from day 0–11, 0.5×106/mL from days 12–15 with media/additives refreshed every 2–3 days (30).
Cryopreservation of expanded thymic Tregs:
Expanded cells were resuspended at 4×106/mL in CS10. Cells were aliquoted into cryotubes (0.5–1mL per tube) which were transferred to a Mr. Frosty Freezing Container (Thermo Fisher Scientific) and placed at −80°C overnight. Once frozen, cryopreserved cells were stored in liquid nitrogen for 1–4 weeks. Cells were thawed by placing cryovials in a 37°C water bath until just melted. RPMI medium with 10% heat inactivated FCS, 1% GlutaMAX and 1% P/S was used as thawing medium. 1mL of warmed thawing medium was added dropwise to each cryovial, then the cell suspension was transferred to a Falcon tube containing an additional 9mL of warmed thawing medium. Thawed cells were washed once with PBS, then resuspended in ImmunoCult-XF with 1% P/S. For overnight culture after thawing, cells were seeded at 1×106/mL and cultures were supplemented with IL-2 (Tregs: 1000IU/mL, Tconv: 100IU/mL).
Flow cytometry and cytokine analysis.
Surface markers and fixable viability dye (FVD, Thermo Fisher Scientific) were stained in PBS or Brilliant Stain Buffer (BD Biosciences). Cells were fixed and intracellular staining was done using the FOXP3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific). For intracellular cytokine staining, expanded Tregs were activated for 4 hours using phorbol myristate acetate (PMA, 10 or 2 ng/mL), Ionomycin (500 or 750 ng/mL) and 10 μg/mL brefeldin A prior to staining. Data were acquired on a BD LSRII, BD LSRFortessa X-20 or Beckman Coulter CytoFLEX. Analysis was done using FlowJo version 10.
DNA isolation and Treg-specific demethylation region (TSDR) analysis.
The EZ DNA Methylation-Direct Kit (Zymo Research) was used to isolate DNA from frozen cell pellets and to perform disulfite conversion according to manufacturer’s instructions. The TSDR region was PCR amplified using AllTaq PCR core kit (QIAGEN). PCR products were analyzed with the PyroMark Q96 MD pyrosequencing system (QIAGEN) and results were calculated with Pyro-CpG software (Biotage).
In vitro suppression assays.
PBMCs and Tregs were labeled with cell proliferation dye or CFSE (Thermo Fisher Scientific). Cells were mixed at indicated ratios, stimulated with anti-CD3/CD28 beads (Dynabeads, Thermo Fisher Scientific) at a 1:8 or 1:16 bead to cell ratio or anti-CD3 beads (Invitrogen) at 1:1 ratio and cultured in X-Vivo 15 supplemented with 5 or 10% human serum, 1% GlutaMAX, 1% P/S, and/or L-glutamine and N-acetylcysteine at 37°C for 4 days. Percentage of suppression was calculated using division index (DI) as 100-[DI(sample)/DI(positive control)]*100. PBMCs stimulated with anti-CD3/CD28 beads without Tregs served as positive controls.
Apoptosis assay:
Cell viability was determined using Abcam’s blue, green, red Apoptosis/Necrosis Assay Kit according to the manufacturer’s instructions. Staining was performed in Assay Buffer for 30 min at room temperature. The volume was topped up using Assay Buffer after staining. Live apoptosis negative cells were defined as CytoCalcein Violet 450 positive, Apopxin Green negative.
Statistical analyses.
Statistical tests used are indicated in the Figure legends and were performed using Graphpad Prism 8. P values: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary Material
Acknowledgements
The authors thank volunteers, patients and their parents for contribution of samples, as well as the surgical and cardiac clinic staff at the British Columbia Children’s Hospital and the University of Alberta Stollery Children’s Hospital who made this study possible; special thanks to Melanie Ganshorn, Allison Jamieson, Lyn Nguyen and Colleen Ring. We also thank ThermoFisher Inc for the provision of in-kind reagents. This work was supported by grants from the Canadian Institutes of Health Research (CIHR) through the Canadian National Transplant Research Program (TFU 127880 to MKL and LJW), the Stem Cell Network and BioCanRx Network Centres of Excellence (to MKL and LJW), a donation from STEMCELL Technologies (to MKL) and from the National Institutes of Health (R01 HL11879, R01 HL56067, R37 AI 34495 and P01 CA065493 to BRB). MKL, KNM and REH receive salary awards from the BC Children’s Hospital Research Institute. KNM is also supported by a CIHR doctoral award. MGH receives a BioCanRx Summer Studentship salary award.
Abbreviations
- APC
antigen presenting cell
- CFSE
Carboxyfluorescein succinimidyl ester
- CPD
cell proliferation dye
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- GARP
glycoprotein A repetitions predominant
- GMP
good manufacturing practice
- GVHD
graft-versus-host disease
- LAP
latency-associated peptide
- PMA
phorbol myristate acetate
- SR
serum replacement
- Tconv
conventional T cell
- TCR
T cell receptor
- TSDR
Treg-specific demethylation region
- Treg
regulatory T cell
Footnotes
Conflict of Interest Statement: ThermoFisher provided in-kind reagents and STEMCELL Technology provided donations to support this project. MKL has received research funding from Bristol Myers Squibb, Takeda, Pfizer, CRISPR Therapeutics and Sangamo for work not related to this project. JMP has received research funding from Amgen, Bayer and Pfizer; has patents pending with AbCellera and SonoSep Technologies, as well as ownership of SonoSep, all for work not related to this project. BRB receives remuneration as an advisor to Kamon Pharmaceuticals, Five Prime Therapeutics, Regeneron Pharmaceuticals, Magenta Therapeutics and BlueRock Therapeutics; research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Abbvie, Leukemia and Lymphoma Society, Childrens’ Cancer Research Fund, KidsFirst Fund and is a co-founder of Tmunity. LJW has received research support from Astellas Canada, Novartis Canada, Roche, BD Biosciences, Canadian Glycomics Network (GlycoNet NCE), Women and Children’s Health Research Institute for work unrelated to this project.
References
- 1.Gliwiński M, Iwaszkiewicz-Grześ D, and Trzonkowski P. Cell-Based Therapies with T Regulatory Cells. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2017;31(4):335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clinical Immunology. 2009;133(1):22–6. [DOI] [PubMed] [Google Scholar]
- 3.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–8. [DOI] [PubMed] [Google Scholar]
- 4.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Techmańska I, Juścińska J, et al. Administration of CD4+CD25highCD127- Regulatory T Cells Preserves β-Cell Function in Type 1 Diabetes in Children. Diabetes Care. 2012;35(9):1817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. 2014;124(4):638–44. [DOI] [PubMed] [Google Scholar]
- 7.Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — Results of one year follow-up. Clinical Immunology. 2014;153(1):23–30. [DOI] [PubMed] [Google Scholar]
- 8.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science translational medicine. 2015;7(315):315ra189–315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. American Journal of Transplantation. 2017;17(11):2945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathew JM, H.-Voss J, LeFever A, Konieczna I, Stratton C, He J, et al. A Phase I Clinical Trial with Ex Vivo Expanded Recipient Regulatory T cells in Living Donor Kidney Transplants. Scientific Reports. 2018;8(1):7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duggleby R, Danby RD, Madrigal JA, and Saudemont A. Clinical Grade Regulatory CD4(+) T Cells (Tregs): Moving Toward Cellular-Based Immunomodulatory Therapies. Frontiers in immunology. 2018;9:252-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacDonald KN, Piret JM, and Levings MK. Methods to manufacture regulatory T cells for cell therapy. Clinical & Experimental Immunology. 2019;0(0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy AN, et al. Discarded Human Thymus Is a Novel Source of Stable and Long-Lived Therapeutic Regulatory T Cells. American Journal of Transplantation. 2016;16(1):58–71. [DOI] [PubMed] [Google Scholar]
- 15.Wing K, Ekmark A, Karlsson H, Rudin A, and Suri-Payer E. Characterization of human CD25+ CD4+ T cells in thymus, cord and adult blood. Immunology. 2002;106(2):190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. American Journal of Transplantation. 2019;19(1):62–76. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Chen S, Zühlke L, Black GC, Choy M-K, Li N, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48(2):455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parmar S, Liu X, Tung SS, Robinson SN, Rodriguez G, Cooper LJN, et al. Third-party umbilical cord blood–derived regulatory T cells prevent xenogenic graft-versus-host disease. Cytotherapy. 2014;16(1):90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parmar S, Liu X, Najjar A, Shah N, Yang H, Yvon E, et al. Ex vivo fucosylation of third-party human regulatory T cells enhances anti–graft-versus-host disease potency in vivo. Blood. 2015;125(9):1502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canavan JB, Scottà C, Vossenkämper A, Goldberg R, Elder MJ, Shoval I, et al. Developing in vitro expanded CD45RA(+) regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut. 2016;65(4):584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. 2016;7(7):7563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A Rapamycin-Based GMP-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Molecular therapy Methods & clinical development. 2018;8:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurology-Neuroimmunology Neuroinflammation. 2018;5(4):e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsuliman A, Appel SH, Beers DR, Basar R, Shaim H, Kaur I, et al. A robust, good manufacturing practice–compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy. 2016;18(10):1312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gołąb K, Grose R, Placencia V, Wickrema A, Solomina J, Tibudan M, et al. Cell banking for regulatory T cell-based therapy: strategies to overcome the impact of cryopreservation on the Treg viability and phenotype. Oncotarget. 2018;9(11):9728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJPM, and Joosten I. Clinical grade Treg: GMP isolation, improvement of purity by CD127 Depletion, Treg expansion, and Treg cryopreservation. PloS one. 2008;3(9):e3161–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer EH, Laport G, Xie BJ, MacDonald K, Heydari K, Sahaf B, et al. Transplantation of donor grafts with defined ratio of conventional and regulatory T cells in HLA-matched recipients. JCI Insight. 2019;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torikai H, and Cooper LJN. Translational Implications for Off-the-shelf Immune Cells Expressing Chimeric Antigen Receptors. Molecular Therapy. 2016;24(7):1178–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aijaz A, Li M, Smith D, Khong D, LeBlon C, Fenton OS, et al. Biomanufacturing for clinically advanced cell therapies. Nature Biomedical Engineering. 2018;2(6):362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna DH Jr., Sumstad D, Kadidlo DM, Batdorf B, Lord CJ, Merkel SC, et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy. 2017;19(2):250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss L, Czystowska M, Szajnik M, Mandapathil M, and Whiteside TL. Differential Responses of Human Regulatory T Cells (Treg) and Effector T Cells to Rapamycin. PLOS ONE. 2009;4(6):e5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111(1):453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wherry EJ, and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology. 2015;15:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2(4):e23849–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in Regulatory T Cells. Immunity. 2004;21(4):503–13. [DOI] [PubMed] [Google Scholar]
- 36.Ha S-J, Park HJ, Park JS, Jeong YH, Son J, Ban YH, et al. Role of PD-1 in regulatory T cells during chronic virus infection. The Journal of Immunology. 2016;196(1 Supplement):79.1–.1. [Google Scholar]
- 37.Hou TZ, Qureshi OS, and Sansom DM. In: Boyd AS ed. Immunological Tolerance: Methods and Protocols. New York, NY: Springer New York; 2019:87–101. [Google Scholar]
- 38.Liénart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, et al. Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science. 2018;362(6417):952–6. [DOI] [PubMed] [Google Scholar]
- 39.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Science translational medicine. 2011;3(83):83ra41–83ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. European Journal of Immunology. 2009;39(4):1088–97. [DOI] [PubMed] [Google Scholar]
- 41.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. The Journal of Immunology. 1997;159(12):5921–30. [PubMed] [Google Scholar]
- 42.Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical Grade Manufacturing of Human Alloantigen-Reactive Regulatory T Cells for Use in Transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013;13(11):3010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, et al. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol. 2008;181(4):2855–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. International Immunology. 2007;19(4):345–54. [DOI] [PubMed] [Google Scholar]
- 45.Caramalho I, Nunes-Silva V, Pires AR, Mota C, Pinto AI, Nunes-Cabaço H, et al. Human regulatory T-cell development is dictated by Interleukin-2 and −15 expressed in a non-overlapping pattern in the thymus. Journal of Autoimmunity. 2015;56:98–110. [DOI] [PubMed] [Google Scholar]
- 46.Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, et al. Active Demethylation of the Foxp3 Locus Leads to the Generation of Stable Regulatory T Cells within the Thymus. The Journal of Immunology. 2013;190(7):3180–8. [DOI] [PubMed] [Google Scholar]
- 47.Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108(13):4260–7. [DOI] [PubMed] [Google Scholar]
- 48.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity. 2009;30(6):899–911. [DOI] [PubMed] [Google Scholar]
- 49.Li MO, and Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nature Reviews Immunology. 2016;16:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes. 2009;58(3):652–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woods EJ, Thirumala S, Badhe-Buchanan SS, Clarke D, and Mathew AJ. Off the shelf cellular therapeutics: Factors to consider during cryopreservation and storage of human cells for clinical use. Cytotherapy. 2016;18(6):697–711. [DOI] [PubMed] [Google Scholar]
- 52.Luo Y, Wang P, Liu H, Zhu Z, Li C, and Gao Y. The state of T cells before cryopreservation: Effects on post-thaw proliferation and function. Cryobiology. 2017;79:65–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.