

Abstract
Waardenburg syndrome (WS), also known as auditory-pigmentary syndrome, is the most common cause of syndromic hearing loss. It is responsible for 2–5% of congenital deafness. WS is classified into four types depending on the clinical phenotypes. Currently, pathogenic mutation of PAX3, MITF, EDNRB, EDN3, SNAI2, or SOX10 can cause corresponding types of WS. Among them, SOX10 mutation is responsible for approximately 15% of type II WS or 50% of type IV WS. We report the case of a proband in a Chinese family who was diagnosed with WS type II. Whole exome sequencing (WES) of the proband detected a novel heterozygous spontaneous mutation: SOX10 c.246delC. According to analysis based on nucleic acid and amino acid sequences, this mutation may produce a truncated protein, with loss of the HMG structure domain. Therefore, this truncated protein may fail to activate the expression of the MITF gene, which regulates melanocytic development and plays a key role in WS. Our finding expands the database of SOX10 mutations associated with WS and provides more information regarding the molecular mechanism of WS.
1. Introduction
Waardenburg syndrome (WS), also known as auditory-pigmentary syndrome, is one of the most common causes of syndromic deafness, associated with 2–5% of congenital deafness cases [1]. According to correlational research, sporadic cases of WS have been reported worldwide; the highest reported incidence is among Kenyan Africans, who have an incidence of 1 in 20,000 [2]. WS accounts for approximately 1% of the deaf population in China. Basic clinical symptoms of WS include dystopia of the canthus; abnormal pigmentation of the skin, hair, and eyes; different degrees of unilateral or bilateral sensorineural deafness; and a high and wide nasal base. The disease is divided into four types; type I exhibits only these basic symptoms. The clinical manifestations of type II are basically the same as type I, but without dystopia of the canthus. The clinical manifestations of type III are the same as type I, but combined with upper limb deformity, while type IV exhibits basically the same symptoms as type II, but combined with Hirschsprung disease (gastrointestinal malformation) [3–6]. Mutations of the PAX3, SOX10, MITF, SNAI2, EDNRB, and EDN3 genes have been found in cases of the different types of WS [7, 8]. According to previous reports, SOX10 mutations cause approximately 15% of type II WS and 45–55% of type IV WS [9, 10]. To date, the total number of reported SOX10 pathogenic mutations related to WS is 82, according to the Leiden Open Variation Database (LOVD) (https://grenada.lumc.nl/LOVD2/WS/home).
The SOX10 gene (SRY- (sex-determining region Y-) box 10) is located on chromosome 22q13.1. It contains five exons and encodes a protein which consists of 466 amino acids, with a relative molecular weight of approximately 51,000 [11]. SOX10 belongs to the SOX (SRY-related HMG-box) family, and it is first expressed in the dorsal neural tube at the early stage of neural crest cell (NCC) migration. With differentiation of the NCC, SOX10 begins to be widely expressed throughout the adult body, such as in the hair follicles, inner ear, iris, and gastrointestinal tract. SOX10 acts as a transcription factor of the microphthalmia-associated transcription factor (MITF) gene, and MITF plays a key role in the development of melanocytes [12]. Currently, it is believed that the aetiology of WS is caused by the abnormal development of NCC [7, 13, 14]. Additionally, the hearing loss of WS may relate to the abnormal proliferation, survival, differentiation, or migration of NCC-derived melanocytes (a type of intermedia cells of the stria vascularis) [15]. In the mouse inner ear, the SOX10 gene starts to express in the otic vesicle of the cochlea at embryonic day 9.5-12.5 (E9.5-E12.5), and the expression is restricted to supporting cells (SCs) of the cochlear epithelium, glial cells, and marginal and intermedia layers of the stria vascularis after birth [16–18]. Knockout of the SOX10 gene leads to the death of NCC-derived Schwann cells in a mouse model [19]. However, the role that it played in supporting cells is still unknown.
As WS has a lot of genetic heterogeneity, the molecular mechanism of WS needs better understanding, and more cases of gene mutation associated with WS need to be collected. Here, we report a novel heterozygous SOX10 mutation in a Chinese family, which provides more information about the molecular diagnosis of WS.
2. Materials and Methods
2.1. Family Description
Family member II-1: a seven-year-old boy, who failed to pass hearing screening and who had white hair at the front of his forehead and bilateral blue irises, was diagnosed with type II WS. Neither parent of the boy exhibited similar symptoms (Figure 1(a) and Table 1).
Figure 1.
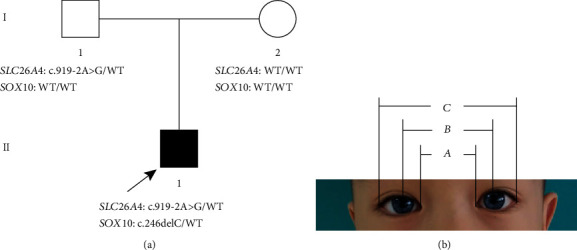
Family pedigree and clinical features of the proband. The (a) pedigree indicates that Family II-1 had a spontaneous heterozygous mutation (SOX10 c.246delC), which is marked black. Family I-1 only carried the mutation of the SLC26A4 gene. (b) The iris heterochromia in both eyes of the proband, which are blue. a, b, and c indicate the distance between the inner canthi, pupils, and outer canthi. WT: wild type.
Table 1.
Genetic variants found in this family.
| Gene | Variant | Protein level | Type | Father | Mother | Proband |
|---|---|---|---|---|---|---|
| SOX10 | c.246delC | p.Giy82GlyfsX27 | Heterozygous | Normal | Normal | Heterozygous |
| SLC26A4 | c.919-2A>G | Splicing | Heterozygous | Heterozygous | Normal | Heterozygous |
2.2. Clinical Examination
The proband underwent audiological examination including otoscopic examination, pure tone audiometry, auditory brainstem response (ABR), distortion product otoacoustic emissions (DPOAE), and auditory immittance and auditory steady-state evoked responses (ASSR). A computed tomography (CT) scan of the proband was also performed. The distance between the inner canthi (a) of the p5roband was measured at 29 mm, the distance between the pupils (b) was 54 mm, and the distance between the outer canthi (c) was 89 mm. The W exponent was obtained according to the formulae X = (2a − 0.2119c − 3.909)/c; Y = (2a − 0.2479b − 3.909)/b; and W = X + Y + a/b (Figure 1(b)) [20].
2.3. Mutation Detection and Analysis
Targeted sequencing and Sanger sequencing were performed by BGI Genomics (Wuhan, China). After obtaining informed consent, we collected 3–5 mL of venous peripheral blood from the proband and his parents to prepare DNA. Genomic DNA from all the family members was extracted according to the manufacturer's standard procedure using the QIAamp DNA Blood Midi Kit (Qiagen Inc., Hilden, Germany). The genomic DNA of the family was fragmented using a Covaris LE220 ultrasonicator (Covaris Inc., Woburn, Massachusetts, USA) to generate a paired-end library (200–250 bp). Following array hybridization, elution, and postcapture amplification, the library was enriched. An Agilent 2100 Bioanalyzer and ABI StepOne were used to estimate the magnitude enrichment of the products. Subsequently, the amplified libraries were used for circularization and sequencing on the BGISEQ-500 platform. For circularization, PCR products with different barcodes were pooled together at equimolar concentrations to yield a final amount of 80 ng. Each pool was subsequently heat-denatured, and the single-strand DNA was mixed with an MGIEasy™ DNA Library Prep Kit V1 (PN: 85–05533-00, BGI, Shenzhen, China) to form a 60 μL reaction system, which was subsequently incubated at 37°C for 30 minutes. Finally, 20 μL of each single-circle-library pool was used as input to prepare the DNB. Each pool was then sequenced on one lane, using 100SR chemistry with a BGISEQ-500RS high-throughput sequencing kit (PN: 85–05238-01, BGI). After sequencing, the data were automatically demultiplexed by index. The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference (hg19) using the BWA (Burrows Wheeler Aligner) Multi-Vision software package. After alignment, the output files were used to perform sequencing coverage and depth analysis of the target region, single-nucleotide variants (SNVs), and INDEL calling. We detected SNVs and indels using GATK software. All SNVs and indels were referenced and compared with multiple databases, including the National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/nuccore/), the Database of Single Nucleotide Polymorphisms (dbSNP) (http://www.ncbi.nlm.nih.gov/projects/SNP/), and the 1000 Genomes Database (https://www.internationalgenome.org). According to the high-throughput sequencing results, Sanger sequencing was performed to confirm whether his parents had the same mutations.
3. Results
3.1. Clinical Data
The proband had white hair at the front of his forehead and bilateral blue irises. His parents had no pigmentary abnormalities in the skin, the hair, and the eye or any other WS-associated phenotypes. The audiology examination of the proband showed failed bilateral otoacoustic emissions; all bilateral ABR thresholds were over 105 dB nHL; ASSR showed the thresholds of the left ear were 105 dB nHL at 1 kHz, 105 dB nHL at 2 kHz, and 90 dB nHL at 4 kHz, while the thresholds of the right ear were 90 dB nHL at 500 Hz, 105 dB nHL at 1 kHz, and 100 dB nHL at 4 kHz (Figures 2(a) and 2(b)). The temporal bone CT scan suggested that the shape and size of the bilateral cochleae were not obviously abnormal; however, the middle and top circles of the cochleae were obscurely decomposed, the vestibule was slightly enlarged on both sides, all the right semicircular canals were fused with the vestibule, the left posterior and superior semicircular canals were short, and the horizontal semicircular canal was fused with the vestibule (Figures 2(c) and 2(d)). We substituted the measured distances of inner canthi (a), pupil (b), and outer canthi (c) into the W exponent formula (as introduced in Section 2.2) and obtained that the W exponent was 1.687, less than 1.95 (Figure 1(b)).
Figure 2.
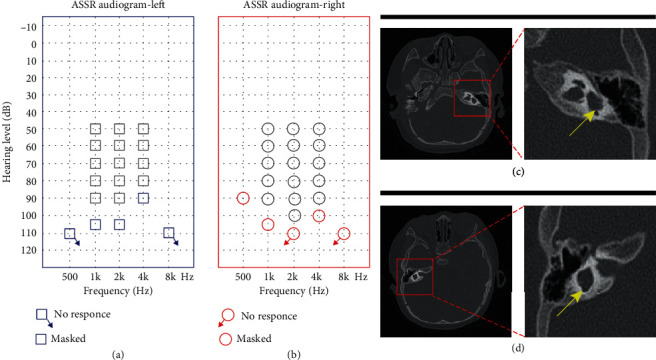
Clinical audiology examination and temporal bone CT scan of the proband. (a) ASSR (auditory steady-state responses) of the left ear: 105 dB, 105 dB, and 90 dB at 1, 2, and 4 kHz, respectively; the remaining frequencies showed no response. (b) ASSR of the right ear: 90 dB, 105 dB, and 100 dB at 500 Hz, 1, and 4 kHz; the remaining frequencies showed no response. (c) Semicircular canal abnormalities shown on high-resolution axial CT in the red square (left ear). (d) Semicircular canal abnormalities shown on high-resolution axial CT in the red square (right ear).
3.2. Mutation Identification Data
The NGS results were compared with the human reference genome (GRCh37/hg19). The proband carried two heterozygous mutations: the SOX10 c.246delC and the SLC26A4 c.919-2A>G; the c.246delC mutation was a truncation with deletion of the no. 246 nucleotide cytosine, occurring in EX2/CDS1 of SOX10. No report related to the c.246delC mutation in SOX10 was found in the HGMD (Figure 3(b)). The c.919-2A>G mutation was a splice mutation of no. 919-2 nucleotide from adenine to guanine, occurring in intron7 of SLC26A4 (Figure 3(a)). The father of the proband only carried the mutation of the SLC26A4 gene, at the same mutation site as that of the proband, while the mother of the proband had the wild-type SOX10 and SLC26A4 genes.
Figure 3.
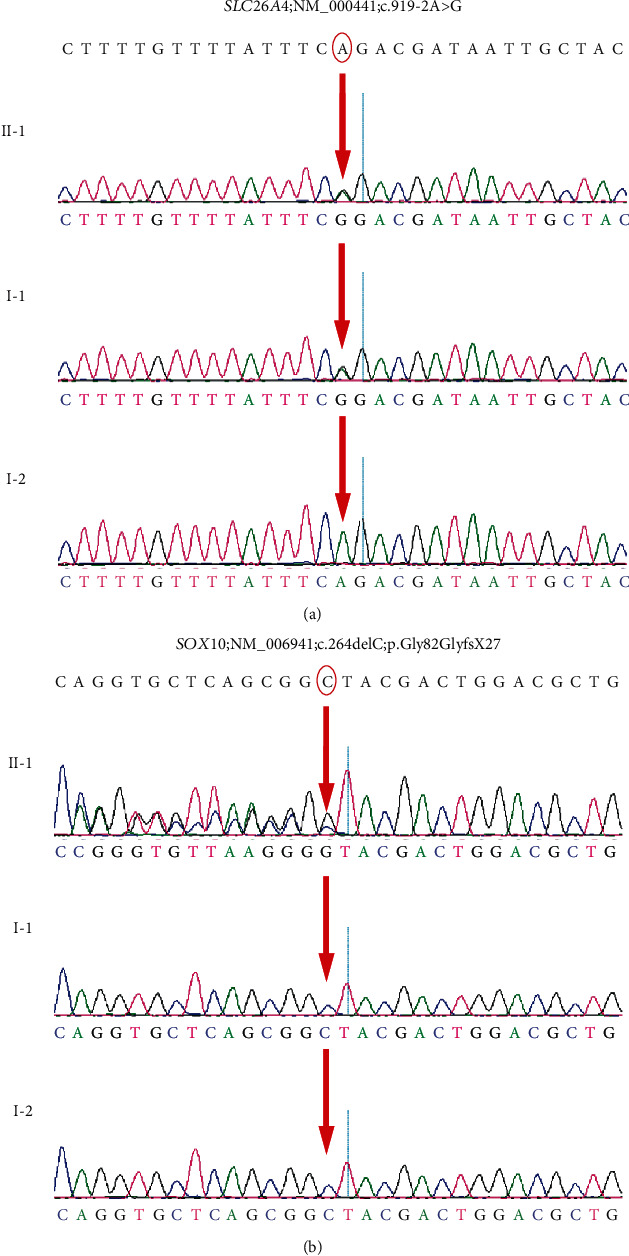
Genetic sequencing results of the proband and his parents. The red arrow indicates the site of the base deletion or substitution.
3.3. Functional Analysis of the Mutant Protein
The SOX10 gene contains three main functional domains: a SOX Group E domain which is highly conserved, the carboxy terminal (C-terminal) transactivation (TA) domain, and a highly conserved and highly active component domain: the highly mobility group (HMG) (102–181 amino acids). The mutation identified in the proband occurred in amino acid no. 82, with deletion of nucleotide C in position no. 246 causing a frameshift mutation. Consequently, the original glycine encoded by GGC at nucleotide nos. 244–246 was changed into glycine encoded by GGG. Following the change at no. 82 glycine, the no. 108 amino acid was converted into a termination codon (Figure 4).
Figure 4.
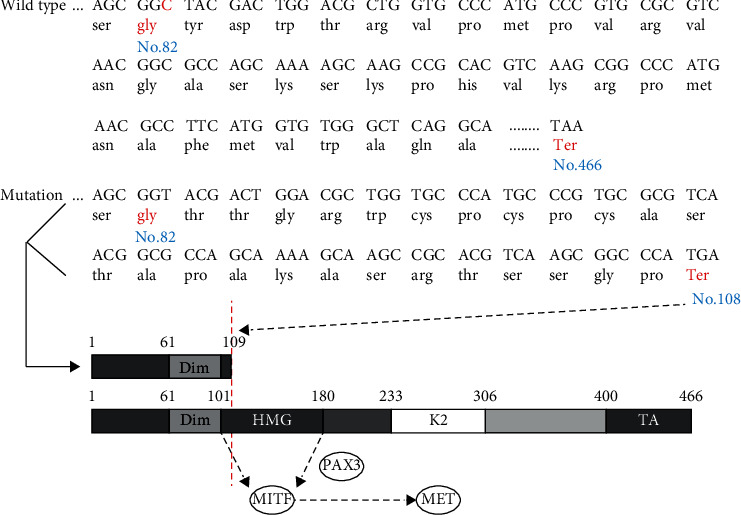
Amino acid coding diagram for the proband and schematic diagram of the SOX10 gene domain. The red letters indicate the changed amino acids and the site of the stop codon. The mutation caused early termination of the coding sequence. If a deletion mutation occurs in front of the HMG domain, the HMG domain cannot combine with the promoter of the target gene (MITF).
4. Discussion
A diagnosis of type II WS was established in the proband. In terms of clinical diagnosis, audiology testing showed that the proband suffered from profound bilateral congenital sensorineural deafness; we calculated a W index of 1.687, less than 1.95, indicating that the proband had no dystopia of the canthus; the proband had white hair on his head and had bilateral blue irises at birth, but no digestive tract abnormalities [21]. In terms of the genetic test results, we found two genetic mutations in the proband; the SLC26A4 gene related to the syndrome type deafness with vestibular pipe expansion showed autosomal recessive inheritance. According to a previous report, in the appropriate clinical context, bilateral agenesis or hypoplasia of the semicircular canals or both, associated with an enlarged vestibule and cochlear deformity, strongly suggests a diagnosis of WS linked to a SOX10 mutation, so we speculate that the spontaneous SOX10 c.246delC mutation may be the cause of the type II WS of the proband [9]. Previous studies have suggested that type II WS patients with SOX10 mutations have a very high spontaneous mutant rate [22].
Sensorineural hearing loss is a common clinical phenotype in WS patients with SOX10 mutations. In the inner ear, most of the hearing loss induced by gene mutations, ototoxic drugs, and aging is caused by the hair cell malfunction [23–27]. A previous study showed that SOX10 mutation will cause both hair cell and SC loss in a heterozygous Dom mouse model [15]. In pigs, shorter cochlear conduct was induced by SOX10 p.Arg109Trp missense mutation [28]. There are complex regulatory networks between SCs and hair cells [29, 30]. Although SOX10 is only expressed in SCs, it may affect the survival of hair cells by regulating the function of SCs. Therefore, more detailed observations should be made to explore the effect of the SOX10 gene on SC function. Moreover, SC-targeted gene therapy can be tried in a murine model [31]. SOX10 is a key transcription factor in the migration and differentiation of NCC, and its mutations lead to abnormal differentiation of NCC-derived melanocytes, which results in abnormal pigment distribution and deafness and is the main cause of WS [32]. SOX10 can exert its function by binding to the promoter or enhancer of the target gene alone or together with other transcription factors. MITF, TYR, TYRP1, DCT, MPZ, GJB1, RET, DCT, and EDNRB are the downstream target genes directly regulated by SOX10. These target genes are directly or indirectly involved in melanin synthesis, among which MITF is a key regulatory gene for melanocyte development and melanin synthesis. SOX10 can act alone or directly with PAX3 to generate a coeffect to stimulate and upregulate the expression of MITF [12, 33]. The SOX10 c.246delC mutation resulted in early termination of the coding protein sequence at amino acid position 108, and consequently, the mutant protein did not contain the HMG domain and the TA domain. The main function of the HMG domain is to identify and bind the promoter of the target gene [34] (Figure 4), so the heterozygous SOX10 c.246delC mutant protein could not effectively activate the MITF promoter, causing a decrease in effective MITF protein expression, leading to an insufficient dose effect, and resulting in disordered melanocyte development and abnormal melanin synthesis. The main function of melanocytes is to produce melanin to ensure the pigmentation of hair and skin. Melanocytes developed from NCC are widely expressed in the dermis, epidermis, vascular striae of the inner ear, and choroid of the eye, and their developmental disorder will lead to WS characterized by hearing loss and abnormal distribution of pigment in the skin and hair [12, 35]. The mutation SOX10 c.246delC (exon 2 in NM_006941) has not been reported according to the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php) [7].
In addition, through analysis of the reported cases and the literature, we found that when the SOX10 mutation site occurred behind the 180th amino acid, or you could say after the HMG domain, it caused more severe symptoms of type IV WS [5, 6, 36–39] (Table 2). We speculate that the reason may be due to the truncated mutation which means that the HMG domain loses the normal function of the protein, but can combine with the target gene promoter. It then competes with the normal protein for binding sites and restrains the influence of the normal protein, leading to more serious consequences. The exact molecular mechanism remains to be confirmed.
Table 2.
The mutation of SOX10 in WS4 probands in the literature.
| Gene | Nucleotide changes | Amino acid changes | Exon | WS subtype | Reference |
|---|---|---|---|---|---|
| SOX10 | c.1333deIT | p.Ser445Glnfs∗57 | 5 | WS4 | 6 |
| SOX10 | c.1107ins19 | p.Thr370Serfs∗38 | 5 | WS4 | 40 |
| SOX10 | c.752_753ins7 | p.Gly252Alafs∗31 | 5 | WS4 | 41 |
| SOX10 | c.895delC | p.Gln299Serfs∗12 | 5 | WS4 | 5 |
5. Conclusion
We identified a new mutation site in the SOX10 gene, explored the possible pathological mechanism of the clinical phenotype caused by this mutation, expanded the database of WS pathogenic gene mutations, and deepened the association between the mutation site and the clinical phenotype, so as to further explore the molecular pathogenic mechanism of WS.
Acknowledgments
This work was supported by a grant from the National Nature Science Foundation of China (81771003, 81570923, 81470696, and 81500793). The authors thank the family for their participation in this study.
Contributor Information
Wei-Jia Kong, Email: entwjkong@hust.edu.cn.
Yu Sun, Email: sunyu@hust.edu.cn.
Data Availability
The data used to support the findings of this study are included within the article.
Ethical Approval
This study was reviewed and approved by the responsible committee on human experimentation (Tongji Medical College, Huazhong University of Science and Technology). No animals were used for the studies. All human procedures were conducted in accordance with the Declaration of Helsinki (1964).
Consent
Written informed consent was obtained from all participants or their guardians.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
Sen Chen and Yuan Jin contributed equally to this work.
References
- 1.Read A. P., Newton V. E. Waardenburg syndrome. Journal of Medical Genetics. 1997;34(8):656–665. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nayak C. S., Isaacson G. Worldwide distribution of Waardenburg syndrome. Annals of Otology, Rhinology & Laryngology. 2016;112(9):817–820. doi: 10.1177/000348940311200913. [DOI] [PubMed] [Google Scholar]
- 3.Pingault V., Bondurand N., Kuhlbrodt K., et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nature Genetics. 1998;18(2):171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 4.Ma J., Lin K., Jiang H. C., et al. A novel mutation of the PAX3 gene in a Chinese family with Waardenburg syndrome type I. Molecular Genetics & Genomic Medicine. 2019;7(7, article e00798) doi: 10.1002/mgg3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogan A. R., Rao K. A., Thorson W. L., Neville H. L., Sola J. E., Perez E. A. Waardenburg syndrome type IV de novo SOX10 variant causing chronic intestinal pseudo-obstruction. Pediatr Gastroenterol Hepatol Nutr. 2019;22(5):487–492. doi: 10.5223/pghn.2019.22.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X., Zhu Y., Shen N., et al. A de novo deletion mutation in SOX10 in a Chinese family with Waardenburg syndrome type 4. Scientific Reports. 2017;7(1) doi: 10.1038/srep41513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pingault V., Ente D., Dastot-Le Moal F., Goossens M., Marlin S., Bondurand N. Review and update of mutations causing Waardenburg syndrome. Human Mutation. 2010;31(4):391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 8.Issa S., Bondurand N., Faubert E., et al. EDNRB mutations cause Waardenburg syndrome type II in the heterozygous state. Human Mutation. 2017;38(5):581–593. doi: 10.1002/humu.23206. [DOI] [PubMed] [Google Scholar]
- 9.Elmaleh-Berges M., Baumann C., Noel-Petroff N., et al. Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations. AJNR. American Journal of Neuroradiology. 2013;34(6):1257–1263. doi: 10.3174/ajnr.A3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki N., Mutai H., Miya F., et al. A case report of reversible generalized seizures in a patient with Waardenburg syndrome associated with a novel nonsense mutation in the penultimate exon of SOX10. BMC Pediatrics. 2018;18(1):p. 171. doi: 10.1186/s12887-018-1139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X. P., Hao Z. Q., Liu Y. L., et al. Functional analysis of a SOX10 gene mutation associated with Waardenburg syndrome II. Biochemical and Biophysical Research Communications. 2017;493(1):258–262. doi: 10.1016/j.bbrc.2017.09.034. [DOI] [PubMed] [Google Scholar]
- 12.Saleem M. D. Biology of human melanocyte development, Piebaldism, and Waardenburg syndrome. Pediatric Dermatology. 2019;36(1):72–84. doi: 10.1111/pde.13713. [DOI] [PubMed] [Google Scholar]
- 13.Mollaaghababa R., Pavan W. J. The importance of having your SOX on: role of SOX10? in the development of neural crest-derived melanocytes and glia. Oncogene. 2003;22(20):3024–3034. doi: 10.1038/sj.onc.1206442. [DOI] [PubMed] [Google Scholar]
- 14.Spritz R. A., Chiang P. W., Oiso N., Alkhateeb A. Human and mouse disorders of pigmentation. Current Opinion in Genetics & Development. 2003;13(3):284–289. doi: 10.1016/s0959-437x(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 15.Tachibana M., Kobayashi Y., Matsushima Y. Mouse models for four types of Waardenburg syndrome. Pigment Cell Research. 2003;16(5):448–454. doi: 10.1034/j.1600-0749.2003.00066.x. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe K.-i., Takeda K., Katori Y., et al. Expression of the Sox10 gene during mouse inner ear development. Molecular Brain Research. 2000;84(1-2):141–145. doi: 10.1016/s0169-328x(00)00236-9. [DOI] [PubMed] [Google Scholar]
- 17.Wakaoka T., Motohashi T., Hayashi H., et al. Tracing Sox10-expressing cells elucidates the dynamic development of the mouse inner ear. Hearing Research. 2013;302:17–25. doi: 10.1016/j.heares.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Hao X., Xing Y., Moore M. W., et al. Sox10 expressing cells in the lateral wall of the aged mouse and human cochlea. Plos One. 2014;9(6, article e97389) doi: 10.1371/journal.pone.0097389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mao Y. Y., Reiprich S., Wegner M., Fritzsch B. Targeted deletion of Sox10 by Wnt1-cre defects neuronal migration and projection in the mouse inner ear. Plos One. 2014;9(4, article e94580) doi: 10.1371/journal.pone.0094580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reynolds J. E., Marazita M. L., Meyer J. M., et al. Major-locus contributions to variability of the craniofacial feature dystopia canthorum in Waardenburg syndrome. American Journal of Human Genetics. 1996;58(2):384–392. [PMC free article] [PubMed] [Google Scholar]
- 21.Rosa Junior M., Santana L. M., Ramos B. F., Ramos H. F. Teaching NeuroImages: Waardenburg syndrome type 2. Neurology. 2019;92(16):e1935–e1936. doi: 10.1212/WNL.0000000000007318. [DOI] [PubMed] [Google Scholar]
- 22.Sun L., Li X., Shi J., et al. Molecular etiology and genotype-phenotype correlation of Chinese Han deaf patients with type I and type II Waardenburg syndrome. Scientific Reports. 2016;6(1) doi: 10.1038/srep35498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Z. H., Zou S. Y., Li M., et al. The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biology. 2020;28:p. 101364. doi: 10.1016/j.redox.2019.101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu W., Xu X., Fan Z., et al. Wnt signaling activates TIGAR and protects against cisplatin-induced spiral ganglion neuron damage in the mouse cochlea. Antioxidants & Redox Signaling. 2019;30(11):1389–1410. doi: 10.1089/ars.2017.7288. [DOI] [PubMed] [Google Scholar]
- 25.He Z., Guo L., Shu Y., et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy. 2017;13(11):1884–1904. doi: 10.1080/15548627.2017.1359449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y., Liu L., Zhang Y., Chai R., Li H. Wnt activation protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Death & Disease. 2016;130(S3):p. S105. doi: 10.1017/s0022215116003923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y., Qi J., Chen X., et al. Critical role of spectrin in hearing development and deafness. Science Advances. 2019;5(4):p. eaav7803. doi: 10.1126/sciadv.aav7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao Q. Q., Li L., Chen W., et al. Key genes and pathways associated with inner ear malformation in SOX10?p.R109W mutation pigs. Frontiers in Molecular Neuroscience. 2018;11:p. 181. doi: 10.3389/fnmol.2018.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan G., Corfas G., Stone J. S. Inner ear supporting cells: rethinking the silent majority. Seminars in Cell & Developmental Biology. 2013;24(5):448–459. doi: 10.1016/j.semcdb.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S., Zhang Y., Dong Y., et al. Knockdown of Foxg1 in supporting cells increases the trans-differentiation of supporting cells into hair cells in the neonatal mouse cochlea. Cellular and Molecular Life Sciences. 2020;77(7):1401–1419. doi: 10.1007/s00018-019-03291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan F., Chu C., Qi J., et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nature Communications. 2019;10(1):p. 3733. doi: 10.1038/s41467-019-11687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bondurand N., Sham M. H. The role of SOX10 during enteric nervous system development. Developmental Biology. 2013;382(1):330–343. doi: 10.1016/j.ydbio.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H., Chen H., Luo H., et al. Functional analysis of Waardenburg syndrome-associated PAX3 and SOX10 mutations: report of a dominant-negative SOX10 mutation in Waardenburg syndrome type II. Human Genetics. 2012;131(3):491–503. doi: 10.1007/s00439-011-1098-2. [DOI] [PubMed] [Google Scholar]
- 34.Wissmuller S., Kosian T., Wolf M., Finzsch M., Wegner M. The high-mobility-group domain of Sox proteins interacts with DNA-binding domains of many transcription factors. Nucleic Acids Research. 2006;34(6):1735–1744. doi: 10.1093/nar/gkl105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma J., Zhang Z., Jiang H. C., et al. A novel dominant mutation in the SOX10 gene in a Chinese family with Waardenburg syndrome type II. Molecular Medicine Reports. 2019;19(3):1775–1780. doi: 10.3892/mmr.2019.9815. [DOI] [PubMed] [Google Scholar]
- 36.Ma J., Zhang T. S., Lin K., et al. Waardenburg syndrome type II in a Chinese patient caused by a novel nonsense mutation in the SOX10 gene. International Journal of Pediatric Otorhinolaryngology. 2016;85:56–61. doi: 10.1016/j.ijporl.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 37.Chen K., Zong L., Liu M., et al. De novo dominant mutation of SOX10 gene in a Chinese family with Waardenburg syndrome type II. International Journal of Pediatric Otorhinolaryngology. 2014;78(6):926–929. doi: 10.1016/j.ijporl.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez R. M., Nunez-Ramos R., Enguix-Riego M. V., et al. Waardenburg syndrome type 4: report of two new cases caused by SOX10 mutations in Spain. American Journal of Medical Genetics. Part A. 2014;164A(2):542–547. doi: 10.1002/ajmg.a.36302. [DOI] [PubMed] [Google Scholar]
- 39.Liang F., Zhao M., Fan L., et al. Identification of a de novo mutation of SOX10 in a Chinese patient with Waardenburg syndrome type IV. International Journal of Pediatric Otorhinolaryngology. 2016;91:67–71. doi: 10.1016/j.ijporl.2016.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.