

Abstract
SARS-CoV-2 is an enveloped virus responsible for the COVID-19 pandemic. Despite recent advances in the structural elucidation of SARS-CoV-2 proteins, the detailed architecture of the intact virus remains to be unveiled. Here we report the molecular assembly of the authentic SARS-CoV-2 virus using cryoelectron tomography (cryo-ET) and subtomogram averaging (STA). Native structures of the S proteins in pre- and postfusion conformations were determined to average resolutions of 8.7–11 Å. Compositions of the N-linked glycans from the native spikes were analyzed by mass spectrometry, which revealed overall processing states of the native glycans highly similar to that of the recombinant glycoprotein glycans. The native conformation of the ribonucleoproteins (RNPs) and their higher-order assemblies were revealed. Overall, these characterizations revealed the architecture of the SARS-CoV-2 virus in exceptional detail and shed light on how the virus packs its ∼30-kb-long single-segmented RNA in the ∼80-nm-diameter lumen.
Keywords: coronavirus, SARS-CoV-2, virus structure, virus assembly, spike glycoprotein, ribonucleoprotein, cryo-electron tomography, subtomogram averaging, cryo-EM
Graphical Abstract
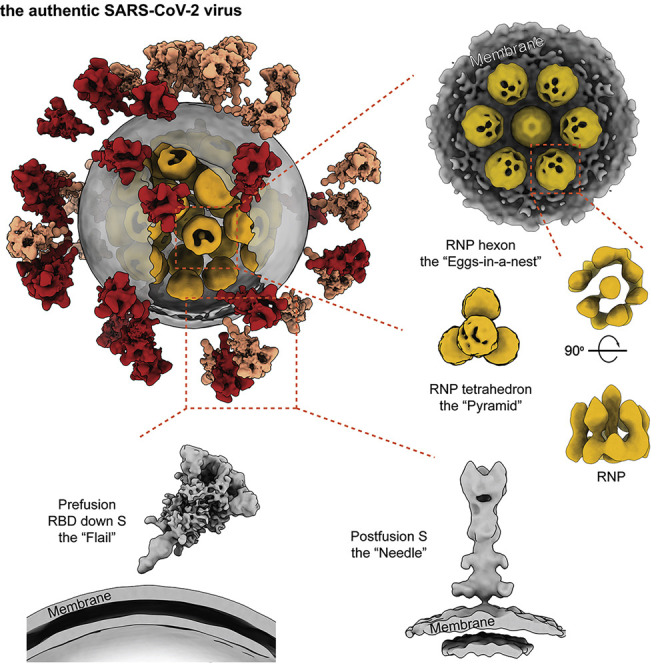
Combined imaging analyses of 2,294 intact virions from the authentic SARS-CoV-2 virus resolve the S protein in pre- and postfusion conformations and characterize the molecular architecture of SARS-CoV-2 at high resolution.
Introduction
As of September 21, 2020, over 31 million cases of COVID-19 have been reported, and more than 960,000 lives were claimed globally (https://covid19.who.int). The causative pathogen, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel β-coronavirus (Lu et al., 2020; Wu et al., 2020; Zhou et al., 2020). SARS-CoV-2 encodes at least 29 proteins in its (+) RNA genome, four of which are structural proteins: the spike (S), membrane (M), envelope (E) and nucleocapsid (N) proteins (Kim et al., 2020).
The ∼600-kDa trimeric S protein, one of the largest known class I fusion proteins, is heavily glycosylated with 66 N-linked glycans (Walls et al., 2020; Watanabe et al., 2020a; Wrapp et al., 2020). Each S protomer comprises the S1 and S2 subunits and a single transmembrane (TM) anchor (Wrapp et al., 2020). The S protein binds to the cellular surface receptor angiotensin-converting enzyme 2 (ACE2) through the receptor binding domain (RBD), an essential step for membrane fusion (Hoffmann et al., 2020; Lan et al., 2020; Shang et al., 2020; Wang et al., 2020; Yan et al., 2020; Zhou et al., 2020). Activation of S requires cleavage of S1/S2 by furin-like protease and undergoes a conformational change from prefusion to postfusion (Belouzard et al., 2009; Kirchdoerfer et al., 2018; Simmons et al., 2004, 2013; Song et al., 2018). Several prefusion conformations have been resolved for the S protein, where the three RBDs display distinct orientations: up or down (Walls et al., 2020; Wrapp et al., 2020). The receptor binding sites are exposed only when the RBDs adopt an up conformation. The “RBD down,” “one RBD up,” and “two-RBD up” conformations have been observed in recombinantly expressed S proteins of SARS-CoV-2 (Henderson et al., 2020; Walls et al., 2020; Wrapp et al., 2020). Upon activation, S follows a classic pathway among class I fusion proteins (Rey and Lok, 2018): it undergoes dramatic structural rearrangements involving shedding its S1 subunit and inserting the fusion peptide (FP) into the target cell membrane (Cai et al., 2020). Following membrane fusion, S transforms to a needle-shaped postfusion form, having three helixes entwining coaxially (Cai et al., 2020). Despite efforts to elucidate the SARS-CoV-2 virus host recognition and entry mechanism at near-atomic resolution using recombinant proteins, high-resolution information regarding the in situ structures and landscape of the authentic virus is in demand.
Coronaviruses have the largest genome among all RNA viruses. It is enigmatic how the N protein oligomerizes, organizes, and packs the ∼30-kb-long single-stranded RNA into the viral lumen. Early negative-staining electron microscopy of coronaviruses showed single-strand helical RNPs with a diameter of ∼15 nm (Caul and Egglestone, 1979). Cryo-electron tomography (cryo-ET) of SARS-CoV revealed that RNPs organized into lattices under the envelope at ∼4- to 5-nm resolution (Neuman et al., 2006). However, such an ultrastructure is not observed in the mouse hepatitis virus (MHV), the prototypic β-coronavirus (Bárcena et al., 2009). So far, no molecular model exists for the coronavirus RNP, and little is known about the architecture, assembly, and RNA packaging of RNPs of other (+) RNA viruses.
To address these questions, we combined cryo-ET and subtomogram averaging (STA) for imaging analysis of 2,294 intact virions propagated from an early virus strain (Yao et al., 2020). To our knowledge, this is the largest cryo-ET dataset of the SARS-CoV-2 virus to date. Here we report the architecture and assembly of the authentic SARS-CoV-2 virus.
Results and Discussion
The Molecular Landscape of the SARS-CoV-2 Virus
SARS-CoV-2 virions (ID: ZJU_5) were collected on January 22, 2020 from a patient with severe symptoms and were propagated in Vero cells. The patient was infected during a conference with attendees from Wuhan (Yao et al., 2020). For cryo-electron microscopy (cryo-EM) analysis, the virus sample was fixed with paraformaldehyde, which has minor effects on protein structure at 7- 20-Å resolution (Li et al., 2016; Wan et al., 2017). Intact and unconcentrated virions were directly visualized from the supernatant by cryo-EM, showing ellipsoidal and spherical enveloped particles (Figure S1 A), consistent with the observation of virions concentrated by ultracentrifugation through a sucrose cushion (Figure S1B). We modeled 2,294 virions as ellipsoids by meshing their lipid envelopes, measuring average diameters of 64.8 ± 11.8, 85.9 ± 9.4, and 96.6 ± 11.8 nm (average ± SD) for the short, medium, and long axis of the envelope, respectively. S proteins and RNPs (Figures 1 A, S1B, and S5A) represented the most distinctive features of the virus. A comprehensive data analysis has been carried. In total, 56,832 spikes were manually identified from the virions, approximately 97% of which were in the prefusion conformation and 3% in the postfusion conformation (Method Details). An average of 26 ± 15 prefusion Ss were found to be randomly distributed on each virion (Figures 1B and 1C). The spike copy number per virion is comparable with human immunodeficiency virus (HIV) (Liu et al., 2008), but ∼5 times less than that of the Lassa virus (LASV) (Li et al., 2016) and ∼10 times less than that of the influenza virus (Harris et al., 2006). 18,500 RNPs were manually identified in the viral lumen (Table S1), giving an average of 26 ± 11 RNPs per virion. However, because the viral lumen is tightly packed with RNPs and electron opaque, the actual number of RNPs per virion was estimated to be 20%–30% more; i.e., 30–35 RNPs per virion. Regularly ordered RNP ultrastructures were occasionally observed (Figure S5 A), indicating that the RNPs could form local assemblies.
Figure S1.

Comparison of Unconcentrated and Concentrated SARS-CoV-2 Virions, Related to Figure 1
Cryo-electron microscopy of virions present in the supernatant of infected Vero cells (A), and virions concentrated by ultracentrifugation (B), showing both spherical and ellipsoidal particles. The long/short axis ratio, as well as the average diameters of the viral envelope measured from the micrographs, are similar between the concentrated (157 virions) (C) and unconcentrated (113 virions) virions (D).
Figure 1.

The Molecular Architecture of the SARS-CoV-2 Virus
(A) A representative tomogram slice (5 Å thick) showing pleomorphic SARS-CoV-2 virions.
(B) A representative virus (boxed in A) is reconstructed by projecting the prefusion S in the “RBD down” conformation (salmon) and “one RBD up” conformation (red), the lipid envelope (gray), and RNPs (yellow) onto their refined coordinates. RNPs away from the envelope are hidden for clarity. The unsolved stem regions of the spikes are sketched from a predicted model (https://zhanglab.ccmb.med.umich.edu/COVID-19).
(C) Summary of structural observations. Top: number of prefusion Ss and RNPs per virion. An average of 26 prefusion Ss and RNPs are found in a virion. Bottom left: ratio of the S proteins between the “RBD down” and “one RBD up” conformations. An average of 54% prefusion Ss are in the “RBD down” conformation. Bottom right: statistics of the dimension of SARS-CoV-2 viral envelopes. The average diameters for the short, medium, and long axis of the envelope are 64.8, 85.9, and 96.6 nm, respectively. A boxplot shows the outliers, minimum, first quartile, medium, third quartile, and maximum of the data.
(D) Distribution of the spike tilt angle reveals a prevailing tilt of 40° relative to the normal axis of the envelope. Shown is a representative “RBD down” spike in authentic orientation to the envelope (gray). The spike is colored by local resolution, and the predicted model of the stem is fitted for illustration purpose.
See also Figure S1 and Video S1.
Figure S5.

Ultrastructure of the RNPs, Related to Figure 4
(A) Example tomogram slices (lowpassed to 80 Å) of in situ RNP assembly. Thickness of the slice is 5 Å. (B) RNP tetrahedrons are projected onto their coordinates to show their characteristic higher-order organization. (C) An RNP tetrahedron is overlaid with hexon, showing identical spacing between two neighboring RNPs on either of the assembly. (D) Lipid bilayer density appears, when all RNPs are aligned and averaged using a large spherical mask. (E-G) Topview and sideview of the RNP tetrahedron and hexon reconstructions, showing RNPs are tightly packed in the viral lumen.
Two conformations of the prefusion S, “RBD down” and “one RBD up”, from inactivated SARS-CoV-2 virions were classified and reconstructed to 8.7- and 10.9-Å resolution by STA, with local resolution reaching 7.8 Å (Figures S3 A–S3C). The heptad repeat 1 (HR1) and central helix (CH) domains of the S2 subunit are the best-resolved domains (Figure S3D). The proportion of the “RBD down” conformation among all prefusion Ss was estimated to be 54% per virion (Figure 1C). The membrane-proximal stem of S was the poorest-resolved region, with a local resolution of ∼20 Å, showing no trace of the TM or membrane in the structure (Figure 1D). When the tomogram slices were scrutinized, spike populations that stood perpendicular to or leaned toward the envelope were observed, suggesting that the spike ectodomain can rotate around the stem.
Figure S3.

Local Resolution and Fourier Shell Correlation (FSC) Curves, Related to Figure 2
(A, B) Maps of prefusion S in RBD down or one RBD up conformations are colored by their local resolution ranging between 7.8-19.8 Å. Among all domains, the central helical (CH) region is the best resolved, as evidenced by tubular densities of the alpha-helix bundles. (C) Resolution of the spikes in RBD down, one RBD up, the postfusion conformations, and the RNP was estimated from the FSC curves, using a criterion 0.143. (D) The best solved domains of the RBD down S, HR1 and CH from the S2 subunit, are highlighted with the fitted PDB: 6XR8. The domains are colored as in the schematic. NTD, N-terminal domain; RBD, receptor binding domain; S1/S2, S1/S2 cleavage site; S2′, S2′ cleavage site; FP, fusion peptide; HR1, heptad repeat 1; CH, central helix; CD, connector domain; HR2, heptad repeat 2; TM, transmembrane anchor; CT, cytoplasmic tail. (E) Comparison of the rigidly and flexibly fitted PDB: 6XR8 to the RBD down S. The NTD has shifted by 9 Å away from the CH (centroid distance).
Refined orientations of the prefusion Ss showed that they rotate around their stalks almost freely outside of the envelope, leaning at an average angle of 40° ± 20° relative to the normal axis of the envelope (Figures 1B and 1D). The rotational freedom of spikes is allowed by its low population density, which is prominently distinct from other enveloped viruses possessing class I fusion proteins (Harris et al., 2006; Li et al., 2016; Liu et al., 2008). Interestingly, a minor population of Y-shaped spike pairs having two heads and one combined stem were observed (Figures S2 A and S2C), which possibly represent spikes intertwined with their stems. These observations suggest that the SARS-CoV-2 spikes have unusual freedom on the viral envelope. Such unique features may facilitate the virus exploring the surrounding environment and better engaging with the cellular receptor ACE2, allowing multiple spikes to bind with one ACE2 or one spike with multiple ACE2s simultaneously. However, the sparsely packed spikes on the viral envelope are also more vulnerable to neutralizing antibodies that bind the otherwise less accessible domains (Chi et al., 2020) or glycan holes (Walls et al., 2019). Our observations of the structures and landscape of the intact SARS-CoV-2 are consistent with two other cryo-ET studies that were published at the same time (Turoňová et al., 2020; Ke et al., 2020).
Figure S2.

Example Tomogram Slices of Spikes, Related to Figures 1 and 3
S in (A) prefusion (5 Å thick) and (B) postfusion (5 nm thick) conformations. (C) A representative pair of Y-shaped spikes in RBD down conformation (marked with ∗ in A) is illustrated by projecting the refined structures onto their coordinates and overlaying with the raw tomogram (lowpassed to 80 Å resolution).
Native Structures of S in the Prefusion Conformation
The native structures of S in the “RBD down” and “one RBD up” conformations were similar to the rigidly fitted recombinant protein structures (PDB: 6XR8, 6VYB; Cai et al., 2020; Walls et al., 2020), except for the N terminus domain (NTD). Comparison of the rigidly and flexibly fitted PDB: 6XR8 suggested that the NTD on the native spike structure shifted 9 Å (centroid distance) away from S2 (Figure S3E). The slight dilation and lower local resolution (Figures S3A and S3B) of the NTD on the native spike against recombinant spike structures was also observed on the other cryo-ET structures (Ke et al., 2020). It is known that the NTD exhibits a certain mobility as a rigid body (Cai et al., 2020; Walls et al., 2020; Wrapp et al., 2020; Xiong et al., 2020). Through large date set and classification, the near-atomic resolution cryo-EM reconstruction of the spike possibly represents its metastable conformation, whereas the cryo-ET reconstructions represent an average of various dynamic states of the NTD.
Ten N-linked glycans are visible in the “RBD down” and seven in the “one RBD up” conformations, of which N61, N282, N801, N1098, and N1134 were best resolved. Interestingly, densities for glycans N1158 and N1173/N1194 are visible on the stem of the spike (Figures 2 B and S4 C). In general, the glycan densities observed on the native spike fit well with the full-length recombinant structure (Cai et al., 2020); however, they are bulkier than those observed in the TM truncated recombinant spikes (Walls et al., 2020; Wrapp et al., 2020). Similar observations have been reported by two studies of the native spike structures that were published at the same time (Turoňová et al., 2020; Ke et al., 2020).
Figure 2.

The Native Structure of the S Protein in the Prefusion Conformation
(A) S in the “RBD down” (resolution, 8.7 Å; threshold, 1.2) and “one RBD up” (resolution, 10.9 Å; threshold, 1.5) conformations. Shown are a side view (top) and top view (bottom) of the maps fitted with PDB: 6XR8, 6VYB.
(B) The “RBD down” S maps fitted with PDB: 6XR8. Densities of ten glycans are highlighted in the insets.
(C) Compositional analysis of the virus sample. The treated viruses, designated ZJU_5, were resolved by SDS-PAGE and visualized by Coomassie blue staining. Lane 1, protein ladder; lane 2, purified ZJU_5; lane 3, ZJU_5 treated with PNGase F; lane 4, PNGase F as control. After PNGase F treatment, the molecular weight of the S1 subunit is reduced by ~30 kDa and that of S2 by ~20 kDa.
(D) Compositional analysis of the surface glycans. The identity and proportion of 22 N-linked glycans from the native S glycans were analyzed by MS and are presented in pie charts.
See also Figures S3 and S4.
Figure S4.

N-linked Glycans of the Native S Proteins, Related to Figure 2
(A) Glycan composition of recombinant S, as reported by Watanabe et al. (2020a). (B) Glycan composition of the native full-length S (Oligomannose: M12-M5; Hybrid: Hybrid, Fhybrid; Complex: A0, FA0, A1, FA1, A2/A1B, FA2/FA1B, A3/A2B, FA3/FA2B, A4/A3B, FA4/FA3B). Please check the attached excel data form for the detailed glycan composition. (C) The RBD down S colored by its oligomer subunits. Densities of ten glycans are visible on the map (gray).
We further determined the native glycan identity by analyzing the virus sample using mass spectrometry (MS). The viral particles, with or without Peptide -N-Glycosidase F (PNGase F) digestion, were resolved on SDS-PAGE. After PNGase F treatment, the S1 and S2 subunits were reduced by ∼30 kDa and ∼20 kDa in weight, respectively (Figure 2C). The bands corresponding to S1 and S2 before PNGase F treatment were analyzed by MS to reveal the glycan compositions at each of the 22 glycosylation sites (Figures 2D and S4B). The overall processing states of the native glycans are highly similar to that of the recombinant glycoprotein glycans (Figures S4A and S4B; Watanabe et al., 2020a), a feature shared with Middle East Respiratory Syndrome (MERS) and SARS-CoV (Walls et al., 2019). Populations of under-processed oligomannose-type glycans are found at the same sites as seen in the recombinant material, including at N234, where the glycan is suggested to have a structural role (Casalino et al., 2020). However, as observed at many sites in HIV (Cao et al., 2018; Struwe et al., 2018), the virus exhibits somewhat lower levels of oligomannose-type glycosylation compared with the recombinant, soluble mimetic. Overall, the presence of substantial complex-type glycosylation suggests that the budding route of SARS-CoV-2 into the lumen of endoplasmic reticulum-Golgi apparatus intermediate compartments (ERGICs) is not an impediment to glycan maturation and is consistent with analysis of SARS-CoV glycans (Ritchie et al., 2010) and identification of neutralizing antibodies targeting the fucose at the N343 glycan on SARS-CoV-2 (Pinto et al., 2020). Furthermore, the lower levels of oligomannose-type glycans compared with HIV and LASV are also consistent with lower glycan density (Watanabe et al., 2018, 2020b). Comparing the structures between our native prefusion S with the recombinant ones, we conclude that (1) the N-linked glycans present on the native spike are bulkier and contain elevated levels of complex-type glycans and (2) the recapitulation of the main features of native viral glycosylation by soluble, trimeric recombinant S glycoprotein is encouraging for vaccine strategies utilizing recombinant S protein.
The Native Structure of S in the Postfusion Conformation
Apart from the triangular prefusion S, needle-like densities were occasionally observed on the viral envelope (Figure S2B). A 15.3-Å-resolution structure was solved and well fitted with a postfusion form of S (PDB: 6XRA), suggesting a postfusion conformation of the S protein. The ectodomain measures 22.5 nm in length and 6 nm in width and stands perpendicular to the viral envelope. Densities of five N-linked glycans (N1098, N1074, N1158, N1173, and N1194) are displayed on the spike (Figure 3 D), reminiscent of the recombinant postfusion structures (Cai et al., 2020; Fan et al., 2020). In comparison with the prefusion conformation, the fixation in orientation to the envelope suggests dramatic conformational reordering of the stem region to achieve the postfusion conformation. The postfusion Ss were found on only a small fraction of the viruses (18% of all viruses, each carrying an average of 5 postfusion Ss), hindering us from reaching higher resolutions. Interestingly, among postfusion S-carrying virions, virions possessing fewer spikes in total tended to have a higher proportion of postfusion Ss (Figure 3C). Statistics for the refined coordinates (Figures 3A–3C) suggested a tendency of the postfusion Ss to bundle on the virus surface. One such bundle was reconstructed, showing four postfusion Ss spaced by ∼10 nm on the virus surface (Figure 3B) compared with the ∼15-nm average distance between the nearest prefusion Ss.
Figure 3.

The Native Structure of S2 in the Postfusion Conformation
(A) A representative tomogram slice (5 nm thick), showing a cluster of spikes in the postfusion conformation on a SARS-CoV-2 virion.
(B) 3D reconstructions of S2 in the postfusion state. The boxed region in (A) is reconstructed by projecting the solved postfusion S structure onto the refined coordinates. Resolution, 15.3 Å; threshold, 1.0.
(C) Distribution of the postfusion Ss. Left: statistics indicate that the postfusion Ss are closer to each other on the viral surface compared with the prefusion Ss. Right: viruses possessing less spikes in total tend to have more postfusion Ss (only virions possessing postfusion Ss were counted).
(D) The postfusion S structure fitted with PDB: 6XRA, displaying densities of N1098, N1074, N1158, N1173, and N1194. The density of N1134 is not visible on the map and is colored gray.
Distinguished from the SARS-CoV virus, which was estimated to possess an average of ∼50–100 spikes per virion (Neuman et al., 2011), the SARS-CoV-2 virus possesses approximately half of the prefusion Ss and occasionally some postfusion Ss. The postfusion Ss observed on the SARS-CoV-2 virus may come from (1) products of occasional, spontaneous dissociation of S1 (Cai et al., 2020), which was cleaved by host proteinases; (2) syncytium naturally formed on infected cells (Xia et al., 2020) when budding progeny virions carried a few residual postfusion Ss from the cell surface; (3) the sample preparation procedure because cryo-EM images of β-propiolactone-fixed viruses show that most spikes present on the virus are postfusion like (Liu et al., 2020; Gao et al., 2020). Such instability of the prefusion Ss has been reported on the other β-coronaviruses (Pallesen et al., 2017). In addition, the distribution graph (Figure 3C) implies that the kinetically trapped prefusion S is more fragile than the postfusion S and could even dissociate from the virus. The speculation is based on the fact that intracellular virions, on average, possess more spikes (Klein et al., 2020) than the extracellular virions reported by us and others (Turoňová et al., 2020; Ke et al., 2020) and the occasional observation of the spike-less “bald” virus in our data.
In summary, we conclude that SARS-CoV-2 prefusion Ss are unstable, indicating that the distribution of solvent-exposed epitopes on virions is more complicated than observations of the recombinant proteins. Our observation has implications for efficient vaccine design and neutralizing antibody development, which prefer a sufficient number of stable antigens.
Architecture and Assembly of RNPs in Intact Virions
It remains enigmatic how coronaviruses pack the ∼30-kb RNA into the ∼80-nm-diameter viral lumen. Are the RNPs ordered relative to each other to avoid RNA entangling, knotting, or even damage or are they involved in virus assembly? When raw tomogram slices were inspected, densely packed, bucket-like densities were discernible throughout the virus lumen, some of which appeared to be locally ordered (Figure S5A). Combining previous cryo-ET observations of coronaviruses (Bárcena et al., 2009) and SDS-PAGE/MS analysis (Figure 2C), the densities most likely represent RNPs.
In total, 18,500 RNPs were picked in the viral lumen and initially aligned using a sphere as the template and a large spherical mask. A bucket-like conformation with few structural features emerged adjacent to the density for the lipid bilayer (Figure S5D), suggesting that a significant number of RNPs were membrane proximal. Alignment using a small spherical mask revealed a 13.1-Å-resolution reverse G-shaped architecture of the RNP, measuring 15 nm in diameter and 16 nm in height (Figure 4 B). Its shape is comparable with a recently reported SARS-CoV-2 RNP conformation (Klein et al., 2020) as well as the in situ conformation of the chikungunya viral RNP, which is also positive stranded (Jin et al., 2018), but different from the MHV RNP released using detergent (Gui et al., 2017).
Figure 4.

Native Assembly of the Ribonucleoproteins (RNPs)
(A) 2D class averages of RNPs reveal two distinct types of RNP ultrastructure: hexameric (boxed in orange) and tetrahedral (boxed in red) assembly.
(B) 3D reconstruction and 2D slices of the RNP. Resolution, 13.1 Å; threshold, 1.0.
(C) Ultrastructure of the RNP hexon and tetrahedron assemblies. Seven RNPs are packed against the viral envelope (gray), forming an “eggs in a nest”-shaped hexagonal assembly (top). Four RNPs are packed as a membrane-free tetrahedral assembly (bottom), most of which were found in the virus away from the envelope. The structural features of the RNPs on the assembly are smeared because of the symmetry mismatch between individual RNPs and the assembly.
(D) Representative projection of RNP hexons assembling into a spherical virus and tetrahedrons into an ellipsoidal virus.
(E) Statistics of the ratio of tetrahedron/hexon assembly reveals that spherical and ellipsoidal virions are more likely to be packed with hexons and tetrahedrons, respectively. The ratio is estimated by sorting 382 virions that have over 5 RNP assemblies by their ratio of long/short axis and counting their ratio of tetrahedron to hexon RNPs. A boxplot shows the minimum, first quartile, medium, third quartile, and maximum of the data.
The map was segmented into five head-to-tail, reverse L-shaped densities, each fitted with a pair of N proteins ([N_NTD], PDB: 6WKP; C terminus domain [N_CTD], PDB: 6WJI) dimerized by the N_CTD (Chen et al., 2007; Figures S6 A and S6C). We analyzed the electrostatic potential distribution on the surface of the decamer and suggested a tentative structural model of RNA-winded RNP (Figures S6B and S6D). Interestingly, an early observation of the MHV RNPs showed ∼15-nm-diameter helices with five subunits per turn (Caul and Egglestone, 1979). Because of the limited resolution and little previous structural knowledge about the (+) RNA virus RNPs, our model should be interpreted with caution.
Figure S6.

A Tentative Model of the SARS-CoV-2 RNP, Related to Figure 4
(A) Alignment of all single RNPs in regardless of their assembly types using a tight spherical mask revealed a 13.1 Å resolution reverse G-shaped architecture of the RNP, measuring 15 nm in diameter and 16 nm in height. (C) The map was segmented into five head-to-tail reverse L-shaped densities, each fitted with a pair of N (N_NTD: 6WKP, N_CTD: 6WJI) dimerized by the N-CTD, leaving two upper segments unoccupied. Based on the electrostatic potential distribution on the surface of the decamer (B), we propose a tentative model of RNP winded with RNA (D).
Further 2D classification of the RNPs revealed three classes: (1) closely packed against the envelope and (2) hexagonally and (3) triangularly packed RNPs (Figure 4A). Following 3D refinement, a membrane-proximal, “eggs-in-a-nest”-shaped RNP assembly (referred to as the “hexon”) and a membrane-free, “pyramid”-shaped RNP assembly (referred to as the “tetrahedron”) emerged (Figures 4C and S5E–S5G). Projection of the two class averages back onto their refined coordinates revealed that the majority of hexons came from spherical virions, whereas more tetrahedrons came from ellipsoidal virions (Figure 4D). This was quantified by statistics; ellipsoidal virions tended to pack more RNP tetrahedrons (Figure 4E). Furthermore, the spacing between two neighboring RNPs (∼18 nm) was the same for tetrahedrons and hexons, and some tetrahedrons could assemble into hexons when projected onto their in situ coordinates (Figure S5B), suggesting that the RNP triangle is a key and basic packing unit throughout the virus. We further propose that the RNPs are involved in coronavirus assembly and help strengthen the virus against environmental and physical challenges because purified virions remained intact after five cycles of freezing and thawing (Figure S7 ). Such involvement of RNPs in viral assembly has also been reported by Neuman et al. (2006), who showed that RNPs form a lattice under the envelope, and has also been seen in intracellular virions (Klein et al., 2020). However, it remains unanswered whether the ultrastructures of RNPs are assembled by RNA, the M or E proteins, the RNP itself, or multiples of these.
Figure S7.

Environmental Challenges for SARS-CoV-2 Virions, Related to Figure 4
(A-C) SARS-CoV-2 virions remained intact after five cycles of freeze-and-thaw treatment, as shown by negative staining microscopy.
Solving RNPs to subnanometer resolution was hindered by the crowding of RNPs against each other (Figures S5E–S5G). Furthermore, the structural features of the RNPs on higher-order assemblies smeared, possibly because of the symmetry mismatch between individual RNPs and the assembly (Figures 4C and 4D). No virus with strictly ordered RNPs throughout the lumen was found by projections. We conclude that the native RNPs are highly heterogeneous and densely packed but locally ordered in the virus, possibly interacting with the RNA in a “beads on a string” stoichiometry.
From ∼2,300 intact virions and over 300 tilt series, we provide molecular insights into the structures of spikes in the pre- and postfusion conformations, the RNPs, and how they assemble on the authentic virus. We also analyzed detailed glycan compositions of native spikes. The reconstructed virus map (Figure 1B), consisting of the “RBD down” S, “one RBD up” S, lipid envelope, and RNP components of the authentic SARS-CoV-2, has been deposited in the Electron Microscopy Data Bank under accession code EMD-30430, which provides a model for full virus molecular dynamic simulation, 3D printing, education, or public media.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| SARS-CoV-2 (ID: ZJU_5) | Isolated from a patient (Yao et al.,2020) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PNGase F | New England Biolabs | Cat#P0704S |
| Trypsin | Promega | Cat#V5280 |
| Chymotrypsin | Promega | Cat#V1062 |
| Alpha lytic protease | Sigma-Aldrich | Cat#A6362 |
| Critical Commercial Assays | ||
| SARS-CoV-2 qRT-PCR Kit(targeting ORF1ab, E, and N genes) | Liferiver Biotech | Cat#Z-RR-0479-02 |
| Deposited Data | ||
| Prefusion RBD down S cryo-ET map | This paper | EMD-30426 |
| Prefusion one RBD up S cryo-ET map | This paper | EMD-30427 |
| Postfusion S cryo-ET map | This paper | EMD-30428 |
| RNP cryo-ET map | This paper | EMD-30429 |
| SARS-CoV-2 virus cryo-ET map | This paper | EMD-30430 |
| Experimental Models: Cell Lines | ||
| Vero cells | ATCC | Cat#CCL-81 |
| Software and Algorithms | ||
| MotionCor | Li et al.,2013 | https://emcore.ucsf.edu/ucsf-software |
| MotionCor2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-software |
| Gctf | Zhang, 2016 | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf |
| Novactf | Turoňová et al., 2017 | https://github.com/turonova/novaCTF/wiki |
| IMOD | Kremer et al., 1996 | https://bio3d.colorado.edu/imod/ |
| Dynamo | Castaño-Díez et al., 2012 | https://wiki.dynamo.biozentrum.unibas.ch |
| Gold standard plugin for Dynamo | This paper | https://github.com/SaiLi-Lab/dynamo_gold_standard |
| Relion | Bharat and Scheres, 2016 | https://scicomp.ethz.ch/wiki/RELION |
| Jsubtomo | Huiskonen et al., 2014 | https://www.opic.ox.ac.uk/jsubtomo |
| UCSF Chimera | Pettersen et al., 2004 | https://www.rbvi.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 2018 | https://www.rbvi.ucsf.edu/chimerax/ |
| VMD | Humphrey et al., 1996 | http://www.ks.uiuc.edu/Research/vmd/ |
| NAMD 2.12 | Phillips et al., 2005 | http://www.ks.uiuc.edu/Research/namd/ |
| CHARMM36 force field | MacKerell et al., 1998 | https://www.charmm.org/charmm/resources/charmm-force-fields/ |
| Byonic (Version 2.8.2) | PROTEIN METRICS | https://www.proteinmetrics.com/products/byonic/ |
| Xcalibur 4.3.73.11 | Thermo Fisher | https://www.thermofisher.com/order/catalog/product/OPTON-30967?SID=srch-srp-OPTON-30967 |
| Proteome Discoverer (Version 1.4) | Thermo Fisher | https://thermo.flexnetoperations.com/control/thmo/login?nextURL=%2Fcontrol%2Fthmo%2Fproduct%3FplneID%3D820497 |
| Other | ||
| Gold grids coated with holey carbon, R 2/2 200 mesh | Quantifoil | Cat#Q34567 |
| Copper grids coated with holey carbon, R 2/2 300mesh | Quantifoil | Cat#Q36516 |
| Gold fiducial beads (10 nm diameter) | Aurion | Cat#25486 |
| Copper grids | Zhongjingkeyi Technology | Cat#BZ11024a |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sai Li (sai@tsinghua.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Cryo-ET maps of the reported structures have been deposited in the Electron Microscopy Data Bank under accession codes EMD-30426, EMD-30427, EMD-30428, EMD-30429 and EMD-30430. The Gold standard plugin for subtomogram averaging in Dynamo used in this work is available at: https://github.com/SaiLi-Lab/dynamo_gold_standard.
Experimental Model and Subject Details
Cell lines
The Vero cells (African green monkey kidney, ATCC CCL-81, sex unknown) for virus propagation were cultured at 37°C and 5% CO2 in Modified Eagle Medium (MEM, Corning) supplemented with 10% fetal bovine serum (FBS, GIBCO) and 1% Penicillin-Streptomycin (GIBCO) in T75 culture flask (Grenier). When the cells confluence reached 90%, the cells were harvested with 0.25% Trypsin-EDTA (GIBCO) and passaged at a split ratio of 1:4.
Method Details
Sample preparation
SARS-CoV-2 virions isolated from the patient’s sputum (ID: ZJU_5) (Yao et al., 2020) were propagated in Vero cells (ATCC CCL-81). Sputum was diluted by 5 volumes of Modified Eagle Medium (MEM) complete medium supplemented with 2% fetal bovine serum (FBS), Amphotericin B (100 ng/ml), Penicillin G (200 units/ml), Streptomycin (200 μg/ml) and centrifuged to remove impurities at 3000 rpm for 10 min in room temperature. Finally, the supernatant was collected and filtered through a 0.45 μm filter. 3 mL of filtered supernatant was added to Vero cells in a T25 culture flask. After incubation at 35°C for 2 hours to allow binding, the inoculum was removed and replaced with fresh culture medium. The cells were incubated at 35°C and observed daily to evaluate cytopathic effects (CPE). The SARS-CoV-2 was tested by qRT-PCR and sequencing. For the preparation of enough virus samples, viruses were proliferated using Vero cells in T75 culture flasks. On four days post-infection, 100 mL cell supernatant was cleared from cell debris at 4,000 g centrifugation for 30 min and inactivated with paraformaldehyde (PFA; final concentration 3%) for 48 hours at 4°C. The supernatant was kept at 4°C afterward. All experiments involving infectious virus were conducted in approved biosafety level (BSL)-3 laboratory.
Purification, concentration, biochemical analysis and sample preparation for electron microscopy of inactivated virions were carried out in a BSL-2 lab. For cryo-ET, the fixed virions were pelleted through 30% sucrose cushion by ultracentrifugation (Beckman, IN) at 100,000 g for 3 hours at 4°C and resuspended in 60 μl HEPES-saline buffer containing 10 mM HEPES, pH 7.3, 150 mM NaCl at 4°C overnight (Neuman et al., 2008).
Deglycosylation and SDS-PAGE
To deglycosylate S protein of virions, 20 μg purified virion sample was treated with 500 units PNGase F (New England Biolabs, Ipswich, MA) at 37°C for 1 hour. 20 μg of treated and untreated samples were kept at 100°C for 15 min. Deglycosylation of S protein was characterized by 4 to 12% NuPAGE Bis-Tris gel (Invitrogen,Carlsbad, CA). The protein band was stained by One-Step Blue Protein Gel Stain (Biotium, Fremont, CA).
Mass spectrometric analysis
For protein identification, the gel bands were excised from the gel, reduced with 5 mM of DTT and alkylated with 11 mM iodoacetamide which was followed by in-gel digestion with sequencing grade modified trypsin (Promega, Madison, WI) in 50 mM ammonium bicarbonate at 37°C overnight. The sample was quenched by adding 10% trifluoroacetic acid (TFA) to adjust the pH to below 2. The peptides were extracted twice with 0.1% TFA in 50% acetonitrile aqueous solution for 1 hour and then dried in a speedVac. Peptides were dissolved in 25 μL 0.1% TFA 6 μL of the extracted peptides was analyzed by Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
MS/MS spectra from each LC-MS/MS run were searched against the SARS-CoV-2 database using Proteome Discoverer (Version 1.4) searching algorithm. The search criteria were as follows: full tryptic specificity was required; two missed cleavages were allowed; carbamidomethylation was set as fixed modification; oxidation (M) were set as variable modifications; precursor ion mass tolerance was 20 ppm for all MS acquired in the Orbitrap mass analyzer; and fragment ion mass tolerance was 0.02 Da for all MS2 spectra acquired in the Orbitrap. High confidence score filter (FDR < 1%) was used to select the “hit” peptides and their corresponding MS/MS spectra were manually inspected
For glycan analysis, the gel bands corresponding to S protein were treated with trypsin (Promega, Madison, WI), chymotrypsin (Promega, Madison, WI) and alpha lytic protease (Sigma-Aldrich, St. Louis, MO) similarly to the above procedures.The extracted peptides was analyzed by Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
For LC-MS/MS analysis, the peptides were separated by a 40 min gradient elution at a flow rate 0.30 μl/min with a Thermo-Dionex Ultimate 3000 HPLC system, which was directly interfaced with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The analytical column was a home-made fused silica capillary column (75 μm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C-18 resin (300 Å, 5 μm, Varian, Lexington, MA). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. An LTQ-Orbitrap mass spectrometer was operated in the data-dependent acquisition mode using Xcalibur 4.3.73.11 software and there was a single full-scan mass spectrum in the Orbitrap (300–1500 m/z, 120,000 resolution) followed by 3 s data-dependent MS/MS scans in an Ion Routing Multipole at stepped 27, 30, 33 normalized collision energy (HCD).
Glycopeptide fragmentation data were extracted from the raw file using Byonic (Version 2.8.2). The MS data was searched using the Protein Metrics 309 N-glycan library. The search criteria were as follows: Non-specificity; carbamidomethylation (C) was set as the fixed modifications; the oxidation (M) was set as the variable modification; precursor ion mass tolerances were set at 20 ppm for all MS acquired in an orbitrap mass analyzer; and the fragment ion mass tolerances were set at 0.02 Da for all MS2 spectra acquired.
The intensities of each glycan type in identical site were combined and analyzed for proportion. Data with score less than 30 were discarded. The glycans were classfied into oligomannose, hybrid and complex type based on composition. Hybrid and complex type glycan were subdivided according to fucose component and antenna.
Cryo-electron tomography and electron microscopy
7 μl virus sample was applied onto a glow discharged copper grid coated with holey carbon (R 2/2; Quantifoil, Jena, Germany), and subsequently dipped onto 500 μl HEPES-saline buffer for 1 s to clear the sucrose. A drop of 3 μl gold fiducial beads (10 nm diameter; Aurion, the Netherlands) was applied and the grid was blotted for 4.5 s, vitrified by plunge-freezing into liquid ethane using a Cryo-plunger 3 (Gatan, CA). Fixed cells cultured on grids were applied with 2 μl fiducial beads (10 nm diameter; Aurion, the Netherlands) prior to single-sided plunge-frozen.
The grids were imaged on a Titan Krios microscope (Thermo Fisher Scientific, Hillsboro, OR) operated at a voltage of 300 kV equipped with an energy filter (slit width 20 eV; GIF Quantum LS, Gatan, CA) and K3 direct electron detector (Gatan, CA). Virions were recorded in super-resolution mode at a nominal magnification of 64,000 × , resulting in a calibrated pixel size of 0.68 Å. 361 sets of tilt-series data were collected using the dose-symmetric scheme (Hagen et al., 2017) from −60° to 60° at 3° steps and at various defocus between −1.7 and −5 μm in SerialEM (Mastronarde, 2005). At each tilt, a movie consisting of 8 frames was recorded with 0.0265 s/frame exposure, giving a total dose of 131.2 e-/Å2 per tilt series.
For the freeze-and-thaw test, 1.5 μl purified virions were diluted in 6 μl HEPES-saline buffer at 4°C, then subjected to five cycles of freezing in liquid nitrogen and thawing in water bath at 37°C. For the negative staining microscopy, 4 μl freeze-and-thaw sample was applied on copper grids (Zhongjingkeyi Technology, Beijing, China), stained using 2% Uranium acetate and imaged using a Tecnai Spirit TEM (Thermo Fisher Scientific, Hillsboro, OR).
Data processing
Tilt series data was analyzed in a high-throughput pre-processing suite developed in our lab. The electron beam induced motion was corrected using a combination of MotionCor (Li et al., 2013) and MotionCor2 (Zheng et al., 2017) by averaging eight frames for each tilt. Defocuses of the tilt series were measured using Gctf (Zhang, 2016). The tilt series were contrast transfer function corrected using Novactf (Turoňová et al., 2017), 319 tilt-series with good fiducial alignment and relative thin ice thickness were reconstructed to tomograms by weighted back projection in IMOD (Kremer et al., 1996), resulting in a final pixel size of 1.36 Å/pixel. The tomograms were 2 × and 4 × binned for subsequent processing. 2,294 virions, 54,878 prefusion S, 2,010 postfusion S and 18,500 RNPs were manually picked. The viral envelopes were manually selected and modeled as ellipsoids using the mesh methods in Dynamo. On average, 425 equally spaced vectors normal to the viral envelope were established per virus. Initial orientations of all spikes were applied using these vectors. The metadata containing all viral parts was organized with Dynamo catalog (Castaño-Díez et al., 2017) for further analysis.
Subtomogram averaging was done using Dynamo (Castaño-Díez et al., 2012). For the prefusion S reconstruction, 54,878 subtomograms were extracted from 4 × binned tomograms into boxes of 96 × 96 × 96 voxels and EMD-21452 (Walls et al., 2020) was used as the template for their alignment. The resolution was restricted to 40 Å and C3 symmetry was applied at this stage. 8,562 spikes present at the edges of the tomograms were removed to minimize the impact of air-water interface effect and incomplete signal on the structure. The remaining particles were subjected to multi-reference alignment imposing C1 symmetry using EMD-21452 and EMD-21457 lowpassed to 30 Å resolution as the templates, resulting in 25,236 spikes (54.5%) classified into RBD down conformation and 21,080 spikes (45.5%) into one RBD up conformation. Coordinates of the two spike conformations were used to extract boxes of 160 × 160 × 160 voxels from the 2 × binned tomograms for further alignment. To prevent overfitting, a customized ‘gold-standard adaptive bandpass filter’ method was used for the alignment at this stage, and a criterion of 0.143 for the Fourier shell correlation were used to estimate the resolution. The 2 × binned spikes in the RBD down and one RBD up conformations were independently further aligned imposing C3 or C1 symmetry respectively, to 9.5 and 10.9 Å resolution. Finally, the RBD down spike subtomograms were extracted from unbined tomograms into boxes of 256 × 256 × 256 voxels and aligned to 8.7 Å resolution. The prefusion S maps were lowpassed according to the estimated local resolutions of the reconstructed subunits. Universal empirical B-factors of −1200 and −2000 were applied to sharpen the RBD down and one RBD up spikes, respectively (Wan et al., 2017).
For the postfusion S reconstruction, 2,010 subtomograms were extracted from 4 × binned tomograms into boxes of 96 × 96 × 96 voxels, which were averaged to give an initial template for their alignment. The resolution was restricted to 30 Å and C3 symmetry was applied at this stage. Next, the refined coordinates were used to extract 1,954 postfusion S from the 2 × binned tomograms into boxes of 160 × 160 × 160 voxels for gold-standard alignment. Subsequent alignment achieved 15.3 Å resolution.
For the RNP reconstruction, 18,500 manually picked RNPs were extracted into subtomograms of 80 × 80 × 80 voxels from 4 × binned tomograms and globally aligned using a large sphere (radius 36 pixels) as the template. The resolution was restricted to 40 Å and no symmetry was applied at this stage. Lipid bilayers were visible in the aligned maps, suggesting part of the RNPs are relatively packed with the membrane. The alignment was repeated using a small spherical mask (radius 18 pixels). A reverse “G”-shaped structure appeared after this stage and the refined coordinates were used to extract particles from the 2 × binned tomograms into boxes of 128 × 128 × 128 voxels. Gold-standard was applied to align the RNP to a final resolution at 13.1 Å.
To analyze the local pattern of the RNP assembly, the picked RNPs’ coordinates were imported into the Relion subtomogram averaging pipeline (Bharat and Scheres, 2016). The RNP particles were extracted and projected into 2D images. Three characteristic patterns of the 2D classification are selected and subjected to 3D initial model generation and 3D classification: 1) closely packed toward the envelope, 2) hexagonally packed and 3) triangularly packed RNPs. Following 3D refinement, the first class converged only on the membrane. The second class aligned into a hexagonally packed, membrane proximal RNP assembly, and the third class aligned into a tetrahedrally packed, membrane-free assembly. Refined coordinates and orientations of the hexagonal particles (2,270 hexons) and tetrahedral particles (3,659 tetrahedrons) were converted and exported for further alignment in Dynamo. For the RNP hexons, 2,270 particles were extracted into subtomograms of 128 × 128 × 128 voxels from 4 × binned tomograms. A shell-shaped mask and C6 symmetry were applied during the alignment. For the RNP tetrahedrons, 3,659 particles were extracted into subtomograms of 90 × 90 × 90 voxels from 4 × binned tomograms. A spherical mask and C3 symmetry were applied during the alignment. Both assemblies were only aligned using non-gold standard.
Assembly structure reconstruction
Three representative SARS-CoV-2 virus (Figures 1B and 4D) and a bundle of postfusion S (Figure 3B) were reconstructed by projecting all spikes and RNPs onto their refined coordinates and merging the structures using Jsubtomo (Huiskonen et al., 2014). For other map-projection to coordinates (Figures 1D, S2C, and S5B), the ‘dtplot’ function in Dynamo was used. UCSF Chimera (Pettersen et al., 2004) and ChimeraX (Goddard et al., 2018) were used for rendering the graphics.
Fitting
Atomic models (PDB accession code 6XR8, 6VYB, 6XRA) of the pre- and postfusion S were rigidly fitted to the corresponding densities using the Fit in Map tool (Pettersen et al., 2004).
The RNP map was segmented into five reverse L-shaped units, which can be further ungrouped into 7 segments above and 10 segments on the base. According to the previous Small-angle X-ray scattering (SAXS) (Chang et al., 2009) and cryo-EM (Gui et al., 2017) reports, the N_NTD and N_CTD possibly form a reverse L-shaped unit; the N_CTD dimer was suggested to be an assembly unit of the RNP (Chen et al., 2007). One segment above and two on the base forming a reverse “L” from the best solved region were selected, and were fitted with a N_CTD dimer (6WJI) using the ‘fit to segments’ tool (Pintilie et al., 2010) in UCSF Chimera. The segment with the best fitting score (0.78 against 0.74 and 0.72) were adopted as the N_CTD. The other two segments were fitted with the N_NTD monomer (6WKP, score 0.92 and 0.90). With the reverse “L”-shaped N_NTD-CTD pair formed, we fitted the rest of RNP with four such units, leaving two upper segments unoccupied. Together, the map was interpreted as a decamer of N.
Molecular dynamic flexible fitting (MDFF) (Trabuco et al., 2009) was applied to improve the fitting of the atomic model to the S in RBD down conformation. PDB: 6XR8 was prepared in VMD (Humphrey et al., 1996) for the MDFF, which was performed in vacuum until convergence using NAMD 2.12 (Phillips et al., 2005) and the CHARMM36 force field (MacKerell et al., 1998). A scaling factor ζ = 0.3 kcal/mol; the secondary structure, domain and symmetry restraints were applied for the simulation.
Quantification and Statistical Analysis
Morphological description of virions (Figures 1C and S1)
1,959 ellipsoidal models for the envelopes were used for virus diameter estimation statistics; For the statistics in Figure S1, membranes of virion were fitted by ellipse, and 113 virions without high-speed centrifugation and 157 virions with high-speed centrifugation were used.
Distribution of spikes in different conformation (Figures 1C and 3C)
1,743 virions were used for calculating the proportion of RBD down/one RBD up conformations per virus; the tilt angle statistics was performed on 39,112 4 × binned spikes (CC > 0.1); and 742 virions were used for RNP statistics. 422 virions were used for the postfusion S statistics. 139 virions which had both prefusion and postfusion S were used for the statistics of conformation proportion. 91 virions which have more than 3 postfusion S and prefusion S were used for spike average distance statistics. And Paired t test showed average distance among postfusion S and prefusion S is significantly different (p value < 0.01). Statistics was performed using Python package Scipy.
Distribution of spikes’ tilt angles (Figure 1D)
The aligned location and orientation were used for the statistics of the spike tilt angles. For a given spike on a given virus, the nearest envelope mesh to the spike stem end was found. The angle between vector A (the spike’s Z axis) and vector B (normal to the envelope mesh) was calculated. For the statistics in Figure S1C, envelopes of 113 virions before ultracentrifugation and 157 after ultracentrifugation were fitted by ellipse to estimate their diameters.
382 virions with more than 5 tetrahedron/hexon RNP assemblies were included for the statistics shown in Figure 4E. They were sorted into three classes according to the ratio of the longest to the shortest axis: spherical (long/short axis: 1-1.24, 127 virions), intermediate (long/short axis: 1.24-1.43, 127 virions), and ellipsoidal (long/short axis: 1.43-2.45, 128 virions). For each class, the ratio of tetrahedrons to hexons was calculated. Wilcoxon test was applied and showed that the frequency of hexons and tetrahedrons is significantly different between these three classes (p < 0.01).
Acknowledgments
S.L. thanks Tsinghua University for providing a start-up fund; the Tsinghua University Branch of China National Center for Protein Sciences (Beijing) for the cryo-EM facility and computational facility support; and Dr. Fan Yang, Jie Wen, and Danyang Li for technical support. We thank Dr. Haiteng Deng and Chongchong Zhao in the Proteinomics Facility at the Technology Center for Protein Sciences, Tsinghua University, for protein MS analysis. We thank Tao Yang, Yakun Wang, and Anbao Jia at the cluster of Bio-Computing Platform (Tsinghua University Branch of China National Center for Protein Sciences Beijing) and Nvidia for providing computational facility support. We are in debt to Dr. Hongwei Wang, Dr. Nieng Yan, Dr. Xinquan Wang, Dr. Lingqi Zhang, Dr. Qiang Ding, and Dr. Xueming Li for critical advice. This work was supported in part by the Major Project of Zhejiang Provincial Science and Technology Department (2020C03123-1) and National Science and Technology Major Project for the Control and Prevention of Major Infectious Diseases in China (2018ZX10711001 and 2018ZX10102001).
Author Contributions
S.L. conceived and supervised the project. H.Y., N.W., T.W., Z.W., L.C., D.S., and X.L. propagated, fixed, and verified fixation of the virus sample. Y. Song and S.L. isolated the viruses, performed biochemical analyses, and prepared the cryosample for cryo-EM. Y. Song, Y.C., S.L., J.Z., and J.L. collected the EM data. Y. Song, Y.C., J.Z., J.X., C.S., Z.Z., and S.L. processed the EM data. S.L. and J.X. performed subtomogram averaging and classification. Y.C., J.X., S.L., and Y. Song analyzed the structures. C.S. and J.X. performed the statistical analysis. M.C., Y. Song, and Z.Z. analyzed the glycan data. S.L., Y. Shi, M.C., and L.L. wrote the manuscript. All authors critically revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 6, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.09.018.
Supplemental Information
References
- Bárcena M., Oostergetel G.T., Bartelink W., Faas F.G., Verkleij A., Rottier P.J., Koster A.J., Bosch B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA. 2009;106:582–587. doi: 10.1073/pnas.0805270106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Chu V.C., Whittaker G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA. 2009;106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat T.A.M., Scheres S.H.W. Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION. Nat. Protoc. 2016;11:2054–2065. doi: 10.1038/nprot.2016.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Jr., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020:eabd4251. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Pauthner M., Andrabi R., Rantalainen K., Berndsen Z., Diedrich J.K., Menis S., Sok D., Bastidas R., Park S.R. Differential processing of HIV envelope glycans on the virus and soluble recombinant trimer. Nat. Commun. 2018;9:3693. doi: 10.1038/s41467-018-06121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalino L., Gaieb Z., Dommer A.C., Harbison A.M., Fogarty C.A., Barros E.P., Taylor B.C., Fadda E., Amaro R.E. Shielding and Beyond: The Roles of Glycans in SARS-CoV-2 Spike Protein. bioRxiv. 2020 doi: 10.1101/2020.1106.1111.146522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño-Díez D., Kudryashev M., Arheit M., Stahlberg H. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J. Struct. Biol. 2012;178:139–151. doi: 10.1016/j.jsb.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Castaño-Díez D., Kudryashev M., Stahlberg H. Dynamo Catalogue: Geometrical tools and data management for particle picking in subtomogram averaging of cryo-electron tomograms. J. Struct. Biol. 2017;197:135–144. doi: 10.1016/j.jsb.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Caul E.O., Egglestone S.I. Coronavirus-like particles present in simian faeces. Vet. Rec. 1979;104:168–169. doi: 10.1136/vr.104.8.168. [DOI] [PubMed] [Google Scholar]
- Chang C.K., Hsu Y.L., Chang Y.H., Chao F.A., Wu M.C., Huang Y.S., Hu C.K., Huang T.H. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J. Virol. 2009;83:2255–2264. doi: 10.1128/JVI.02001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Chang C.K., Chang Y.W., Sue S.C., Bai H.I., Riang L., Hsiao C.D., Huang T.H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007;368:1075–1086. doi: 10.1016/j.jmb.2007.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Cao D., Kong L., Zhang X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020;11:3618. doi: 10.1038/s41467-020-17371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Bao L., Mao H., Wang L., Xu K., Yang M., Li Y., Zhu L., Wang N., Lv Z. Development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369:77–81. doi: 10.1126/science.abc1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M., Liu X., Guo D., Zhang Z., Yin C.C., Chen Y., Xiang Y. Electron microscopy studies of the coronavirus ribonucleoprotein complex. Protein Cell. 2017;8:219–224. doi: 10.1007/s13238-016-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen W.J.H., Wan W., Briggs J.A.G. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J. Struct. Biol. 2017;197:191–198. doi: 10.1016/j.jsb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A., Cardone G., Winkler D.C., Heymann J.B., Brecher M., White J.M., Steven A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R., Edwards R.J., Mansouri K., Janowska K., Stalls V., Gobeil S.M.C., Kopp M., Li D., Parks R., Hsu A.L. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020 doi: 10.1038/s41594-020-0479-4. Published online July 22, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huiskonen J.T., Parsy M.L., Li S., Bitto D., Renner M., Bowden T.A. Averaging of viral envelope glycoprotein spikes from electron cryotomography reconstructions using Jsubtomo. J. Vis. Exp. 2014;(92):e51714. doi: 10.3791/51714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- Jin J., Galaz-Montoya J.G., Sherman M.B., Sun S.Y., Goldsmith C.S., O’Toole E.T., Ackerman L., Carlson L.A., Weaver S.C., Chiu W., Simmons G. Neutralizing Antibodies Inhibit Chikungunya Virus Budding at the Plasma Membrane. Cell Host Microbe. 2018;24:417–428.e5. doi: 10.1016/j.chom.2018.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C.J., Cerikan B. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020 doi: 10.1038/s41586-020-2665-2. Published online August 17, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Wang N., Pallesen J., Wrapp D., Turner H.L., Cottrell C.A., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018;8:15701. doi: 10.1038/s41598-018-34171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B., Stanifer M.L., Boulant S., Bartenschlager R., Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. bioRxiv. 2020 doi: 10.1101/2020.06.23.167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J.R., Mastronarde D.N., McIntosh J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Sun Z., Pryce R., Parsy M.L., Fehling S.K., Schlie K., Siebert C.A., Garten W., Bowden T.A., Strecker T., Huiskonen J.T. Acidic pH-Induced Conformations and LAMP1 Binding of the Lassa Virus Glycoprotein Spike. PLoS Pathog. 2016;12:e1005418. doi: 10.1371/journal.ppat.1005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Bartesaghi A., Borgnia M.J., Sapiro G., Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Yang Y., Gao Y., Shen C., Ju B., Liu C., Tang X., Wei J., Ma X., Liu W. Viral Architecture of SARS-CoV-2 with Post-Fusion Spike Revealed by Cryo-EM. bioRxiv. 2020 doi: 10.1101/2020.1103.1102.972927. [DOI] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A.D., Bashford D., Bellott M., Dunbrack R.L., Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Neuman B.W., Adair B.D., Yoshioka C., Quispe J.D., Orca G., Kuhn P., Milligan R.A., Yeager M., Buchmeier M.J. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 2006;80:7918–7928. doi: 10.1128/JVI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Adair B.D., Yeager M., Buchmeier M.J. Purification and electron cryomicroscopy of coronavirus particles. Methods Mol. Biol. 2008;454:129–136. doi: 10.1007/978-1-59745-181-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman B.W., Kiss G., Kunding A.H., Bhella D., Baksh M.F., Connelly S., Droese B., Klaus J.P., Makino S., Sawicki S.G. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011;174:11–22. doi: 10.1016/j.jsb.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J., Wang N., Corbett K.S., Wrapp D., Kirchdoerfer R.N., Turner H.L., Cottrell C.A., Becker M.M., Wang L., Shi W. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA. 2017;114:E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie G.D., Zhang J., Goddard T.D., Chiu W., Gossard D.C. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A. Structural and functional analysis of a potent sarbecovirus neutralizing antibody. bioRxiv. 2020 doi: 10.1101/2020.1104.1107.023903. [DOI] [Google Scholar]
- Rey F.A., Lok S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell. 2018;172:1319–1334. doi: 10.1016/j.cell.2018.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie G., Harvey D.J., Feldmann F., Stroeher U., Feldmann H., Royle L., Dwek R.A., Rudd P.M. Identification of N-linked carbohydrates from severe acute respiratory syndrome (SARS) spike glycoprotein. Virology. 2010;399:257–269. doi: 10.1016/j.virol.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Zmora P., Gierer S., Heurich A., Pöhlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 2013;100:605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Gui M., Wang X., Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14:e1007236. doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struwe W.B., Chertova E., Allen J.D., Seabright G.E., Watanabe Y., Harvey D.J., Medina-Ramirez M., Roser J.D., Smith R., Westcott D. Site-Specific Glycosylation of Virion-Derived HIV-1 Env Is Mimicked by a Soluble Trimeric Immunogen. Cell Rep. 2018;24:1958–1966.e5. doi: 10.1016/j.celrep.2018.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco L.G., Villa E., Schreiner E., Harrison C.B., Schulten K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Schur F.K.M., Wan W., Briggs J.A.G. Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4Å. J. Struct. Biol. 2017;199:187–195. doi: 10.1016/j.jsb.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Sikora M., Schürmann C., Hagen W.J.H., Welsch S., Blanc F.E.C., von Bülow S., Gecht M., Bagola K., Hörner C. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020:eabd5223. doi: 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Xiong X., Park Y.J., Tortorici M.A., Snijder J., Quispe J., Cameroni E., Gopal R., Dai M., Lanzavecchia A. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell. 2019;176:1026–1039.e15. doi: 10.1016/j.cell.2018.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W., Kolesnikova L., Clarke M., Koehler A., Noda T., Becker S., Briggs J.A.G. Structure and assembly of the Ebola virus nucleocapsid. Nature. 2017;551:394–397. doi: 10.1038/nature24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.Y. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020;181:894–904.e9. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Raghwani J., Allen J.D., Seabright G.E., Li S., Moser F., Huiskonen J.T., Strecker T., Bowden T.A., Crispin M. Structure of the Lassa virus glycan shield provides a model for immunological resistance. Proc. Natl. Acad. Sci. USA. 2018;115:7320–7325. doi: 10.1073/pnas.1803990115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020:eabb9983. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Berndsen Z.T., Raghwani J., Seabright G.E., Allen J.D., Pybus O.G., McLellan J.S., Wilson I.A., Bowden T.A., Ward A.B., Crispin M. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020;11:2688. doi: 10.1038/s41467-020-16567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Qu K., Ciazynska K.A., Hosmillo M., Carter A.P., Ebrahimi S., Ke Z., Scheres S.H.W., Bergamaschi L., Grice G.L., CITIID-NIHR COVID-19 BioResource Collaboration A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020 doi: 10.1038/s41594-020-0478-5. Published online July 31, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H., Lu X., Chen Q., Xu K., Chen Y., Cheng L., Liu F., Wu Z., Wu H., Jin C. Patient-derived mutations impact pathogenicity of SARS-CoV-2. medRxiv. 2020 doi: 10.1101/2020.1104.1114.20060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-ET maps of the reported structures have been deposited in the Electron Microscopy Data Bank under accession codes EMD-30426, EMD-30427, EMD-30428, EMD-30429 and EMD-30430. The Gold standard plugin for subtomogram averaging in Dynamo used in this work is available at: https://github.com/SaiLi-Lab/dynamo_gold_standard.