

Abstract
For many years, major differences in morphology, motility, and mechanical characteristics have been observed between transformed cancer and normal cells. In this review, we consider these differences as linked to different states of normal and transformed cells that involve distinct mechanosensing and motility pathways. There is a strong correlation between repeated tissue healing and/or inflammation and the probability of cancer, both of which involve growth in adult tissues. Many factors are likely needed to enable growth, including the loss of rigidity sensing, but recent evidence indicates that microRNAs have important roles in causing the depletion of growth-suppressing proteins. One microRNA, miR-21, is overexpressed in many different tissues during both healing and cancer. Normal cells can become transformed by the depletion of cytoskeletal proteins that results in the loss of mechanosensing, particularly rigidity sensing. Conversely, the transformed state can be reversed by the expression of cytoskeletal proteins—without direct alteration of hormone receptor levels. In this review, we consider the different stereotypical forms of motility and mechanosensory systems. A major difference between normal and transformed cells involves a sensitivity of transformed cells to mechanical perturbations. Thus, understanding the different mechanical characteristics of transformed cells may enable new approaches to treating wound healing and cancer.
Keywords: transformation, regeneration, apoptosis, rigidity sensing, cancer
INTRODUCTION
A long history of correlation between repeated trauma or inflammation and the frequency of cancers supports the hypothesis that cancer may start when wound healing goes awry. In particular, the proliferative phase of wound healing should cease when cell numbers are restored, but when proliferation continues, it becomes a cancerous growth. There are many other aspects needed for large tumor growth, such as telomere elongation and vascularization, but enabling cell growth is required. Early studies of cancer cells showed that such cells grew on soft surfaces, whereas normal cells required rigid matrices to grow. Furthermore, the metastasis of cancers to other tissues, where growth depends upon distinct activation pathways, implies that there is an alteration in cancer cells that enables growth via multiple local signaling pathways in different tissues. In other words, a general block to growth is missing in cancer cells such that local growth activation pathways will cause inappropriate growth. We suggest that the loss of growth control can result from the loss of mechanosensors that normally respond to insults such as obesity and wasting in adults to maintain proper tissue size. In line with that hypothesis, cytoskeletal proteins involved in mechanosensing and motility are major tumor suppressors (Wolfenson et al. 2019). Moreover, the mechanosensory module that senses matrix rigidity appears to be depleted in transformed cancer cells (Wolfenson et al. 2016, Yang et al. 2019). The loss of a major mechanosensing pathway has many implications for cell behavior since mechanosensors impact the stereotypical motility cycles that drive cell movements and affect cell shape. In this review, we explore the mechanosensory pathways linked to stereotypical motility cycles that are activated by endogenous mechanosensory, or hormonal signals. The motility and sensing cycles can be described as elements of robust cellular processes that appear in many eukaryotic cells, in which the basic engineering principles of robust devices apply.
MECHANOBIOLOGICAL BEHAVIORS OF CELLS AS ROBUST MACHINES
The shaping of cells and tissues involves the coordination of biochemical, motile, and mechanosensing functions. To understand these processes, it is useful to consider cells as robust devices, and their functions follow the principles of robust devices. In support of this hypothesis, life forms have been evolving over billions of years in response to changing environmental stresses and are robust to many factors. Basic engineering principles employed in the design of robust machines, such as computers, cars, or phones, do seem to apply to complex functions like cell mechanosensing and shape determination (Thomas et al. 2004). An important task of biologists and physicians is to reverse-engineer normal and diseased cells to understand how the desired functions are performed. Significantly, biochemical and mechanical functions occur on the timescale of seconds to minutes, and after hours or days, the phenotype of a cell is the integrated outcome of hundreds to thousands of motility and enzymatic cycles. Genetics and genomics can be informative of the potential proteins that are involved in a given function, but they do not provide information as to the sequence of events needed to complete the function. Novel microscopy methods can provide real-time measurements to localize components in important processes and to visualize their dynamics. This information can enable us to develop step-by-step models of events as they occur and to ascertain where specific proteins are in individual steps. In developing models of cellular motile functions, it is useful to consider them as robust functions (see Sheetz & Yu 2018), which are characterized by the features outlined in the below list of engineering principles for robust functions (adapted from Thomas et al. 2004):
compartmentalization of functions (localized)
modular components
largely digital (not analog) functions
term limits (functions stop after relatively short periods and have to be restarted)
cyclic in nature
several variations of important functions
phases (linked functions) with sharp transitions
An additional feature of robust cellular systems is that they use a limited number of standard motile processes to shape themselves and their environment. These motile processes are activated as needed to perform the proper mechanical behavior.
BASIC MOTILITY PROCESSES OF CELLS
Although there are some semantic issues involved in describing the common motility processes exhibited by eukaryotic cells, most cell motile behaviors result from the basic processes discussed below. In terms of how these types of motility relate to robust functions, motility processes are localized, modular, digital, and cyclic and have term limits. Localization involves the proper placement of actin assembly factors and the recruitment of myosin or other motor systems. Modularity lies in the filament assembly complexes and in most of the other enzymatic complexes that sense mechanical parameters and regulate motility. Because motile processes are either on or off, they appear digital, as reported by us and by others (Dubin-Thaler et al. 2008, Tyson & Novak 2014). Although the rates of actin polymerization or myosin contraction can change, they are not continuously adjusted when motility is active. Motile processes usually cease after roughly a minute, which is what we mean by term limits (Dubin-Thaler et al. 2008, Vasquez et al. 2014). This observation implies that the default state of motile functions is off. The cyclic nature of functions refers to the fact that important aspects of a given function are active for only one step of the cycle, whether it is myosin movement on actin or actin filament assembly and disassembly. Thus, the consideration of motile functions as robust functions can help us understand some of the important elements involved.
COORDINATION OF FUNCTIONS IN CELLULAR STATES AND INFLAMMATION/REPAIR
Just as a complex device such as a phone or car can switch between modes in which different functions are active (e.g., forward or reverse in a car), cells can also exhibit different sets of coordinated functions that are part of more complex functions such as fibroblastic migration, epithelial formation, and mitosis. Extensive descriptions of functions involved in cell spreading, cell migration, and endocytosis have been published (van Helvert et al. 2018, Weinberg & Drubin 2012, Wolfenson et al. 2019). In each of those complex functions, there is a stereotypical sequence of events in which one set of motile functions is followed by another to give rise to an emergent behavior. In fibroblasts, the cycles of cell edge extension and retraction are linked to the sequential activation of small G proteins (Machacek et al. 2009). This process can be explained as a stereotypical process that requires initial activation but, once activated, will normally be followed by a programmed set of motile events. Similarly, filopodial extensions are cyclic and require the binding of myosin X to cargo for multiple cycles of extension by a single filopodium (Watanabe et al. 2010). In the sequence of motile events, local changes in mechanical properties will be sensed and may alter the process. Thus, mechanosensing is an integral part of the motility cycles, as illustrated in Figure 1. It is important to remember that motility-sensing cycles typically take only 1–2 min and involve only a limited region of a cell. They are often repeated many times within limited regions to obtain an averaged mechanical signal. Thus, to understand how mechanosensing is involved in the final shape of cells requires that we understand the signal from individual motility-sensing cycles as well as how signals are integrated over time.
Figure 1.

Diagram of the cyclic nature of motility and mechanosensing that emphasizes the integration of motility and mechanosensing functions. In many cases in which the details are known, motility, which can include both membrane extension and contraction, is activated for only a brief period before it stops. Motility is often integrated with a mechanosensing function. Subsequently, the cell can go into stasis, during which there can be processing events that consolidate the effects of the motility events. The mechanosensing function will generate signals that will either activate further motile events or cause reversion to stasis.
FUNCTIONAL STATES RELATED TO GROWTH
Higher-order states such as growth and senescence influence not only the types of motility functions that can be activated, but also how the cell reacts to various signals. We discuss transformation in the context of a high-level state that enables cell growth in response to a variety of signals. Certain motility functions are missing or are altered in the transformed state (Wolfenson et al. 2019). Thus, transformed cells will exhibit altered behavior due to changes in motility and/or mechanosensing systems. In particular, such cells are able to grow outside of a normal tissue environment and ignore cues that would typically block growth. Because of the link between healing and cancer, we suggest that a better understanding of the transformed state can provide new approaches to treating cancer and enhancing the healing process.
Somewhat surprisingly, the literature indicates that a common feature of regeneration in many tissues and species is the upregulation of a specific microRNA, miR-21. In brain, skin, and the proliferative phase of liver regeneration, there are increased levels of miR-21 (Ge et al. 2014, Han et al. 2017, Holman et al. 2012, John et al. 2014, Kennedy et al. 2016, Li et al. 2018). Furthermore, in salamanders and fish, regeneration of limbs and kidneys involves an increase in miR-21 levels (Hoppe et al. 2015, King & Yin 2016). Also, there appear to be negative effects of downregulation of miR-21 on regenerative processes such as those following spinal cord injury (Zhang et al. 2018). At the risk of oversimplifying the control of growth in adult tissues, a working hypothesis based upon these findings is that there are two levels of growth control for tissue regeneration: First, there is a master switch that either blocks or allows growth, and then a tissue-specific growth activation process is needed to drive growth under local controls. Thus, both the master switch and local growth need to be activated for growth in an adult tissue. In terms of molecular mechanisms, miR-21 is apparently part of the general growth pathway, although knockout and knockdown experiments indicate that other factors are important. Activation by receptor tyrosine kinases, Src-integrin signaling, or other cytokines locally is also required for growth.
The case for increased miR-21 levels playing a major role in cancer is very strong. A screen of all major microRNAs identified miR-21 as the strongest activator of basal cell carcinoma tumor formation by far (Ge et al. 2016). For cancers originating from many different tissues, miR-21 levels are increased, and increased miR-21 levels correlate with poor survival (Adhami et al. 2018, Jiang et al. 2016, Lubov et al. 2017, Lv et al. 2013, Masoudi et al. 2018, Sekar et al. 2016, Zhao et al. 2018). In addition, depression of miR-21 by agents such as curcumin has anticancer effects (Momtazi et al. 2016, Taverna et al. 2016, Wang et al. 2017, Zhang et al. 2014). Even exosomal miR-21 from cancer-associated fibroblasts correlates with greater metastasis in a colorectal cancer model (Bhome et al. 2017). Although miR-21 is only one part of the problem in cancer, it is strongly linked to a general growth state.
For the purpose of this review, we want to consider how mechanical signaling differs between normal and transformed cells, since such information will also be informative regarding regeneration. Although externally applied forces can stimulate cell activity, cells have many mechanosensing processes that often involve directed motility to test the local environment of the cell. Mechanosensing events involve a cycle of the following events: activation of motility, movement for a limited period, sensing, and response; however, cells can exit the cycle for periods of stasis. These cycles are repeated many times over the course of hours (Saxena et al. 2017b) (see Figure 2). When cells are observed after days in culture, their ultimate shape is the end product of many mechanosensing events. We therefore discuss the types of motility in terms of the mechanical tests that are associated with motile events and then, where there is experimental evidence, relate these motile events to the long-term integrated behavior of the cell, which is manifest in its shape.
Figure 2.

Experimental examples of cyclic motility processes that are tied to mechanosensing. In panels a–c, the cyclic apical contractions of Drosophila epithelial cells are shown as adapted from Martin et al. (2009). In panel a, the rates of change in the apical areas of 40 cells are color coded and show seemingly stochastic contractions. The rate of change in the area of one cell (marked by the red arrow in panel a) is plotted versus time to show that the contractions are brief and are followed by periods of stasis during which time mechanosensing as well as consolidation of the contraction occurs (panel b). The outlines of the cells also show that the contraction of one cell is not correlated with the motility of its neighbors (panel c). Since the transient contractions continue until the epithelium has achieved the proper curvature, there must be a sensory process that will signal the cell to activate further contractions (C) or stop (S) contractions and to go on to the next phase in development. In panels d and e, the spreading of a fibroblast on glass is documented. In panel d, the rate of edge movement is noted by the color code for the whole edge of a mouse embryo fibroblast spreading on fibronectin (FN)-coated glass. The kymographs in panel e show that the designated area of the edge moves in an oscillatory fashion while undergoing net spreading, shown in the bottom-left and bottom-right images of panel e, along with the boxed area that was used for the kymograph. Panel e adapted from Giannone et al. (2007).
CELL EXTENSIONS ENABLE MECHANOSENSING
There are four basic types of cell extensions: filopodia, lamellipodia, blebs, and podosomes (invadopodia). In all cases, actin polymerization is the important element. However, the primary mode of actin polymerization is different in each case. For example, filopodial extensions are driven primarily by formins, whereas podosome extension is driven primarily by Arp2/3. In contrast to extensions driven by actin polymerization, blebs form by fluid pressure pushing out unsupported membranes that then recruit actin filaments to become stabilized. We consider the mechanosensing aspects of these extension processes and how they feed back onto other cellular activities. There are two basic aspects of extension in mechanosensing. (a) At the simplest level, the extension pushes on the immediate microenvironment, and the microenvironment pushes back. This pushback provides a stimulus for a retraction signal that can cause a change in direction. (b) Receptors in the extending membrane can bind to new ligands, which initiates mechanosensing through adhesive contacts that include rigidity sensing and/or force-dependent signals.
Activation of extension is under complex control and is needed for the subsequent sensing events to occur. A good example of activation is activation through the addition of hormones such as epidermal growth factor (EGF) or platelet-derived growth factor that can cause chemotaxis up a hormone concentration gradient (Biswenger et al. 2018, Saxena et al. 2018). Mechanical activation seems to occur either by adding detergent to decrease membrane tension (Raucher & Sheetz 2000) or by physical stretch followed by relaxation (Pontes et al. 2017). Similarly, fluid flow forces or poking of cells will activate membrane extension. Once activated, motility persists for only a short period before it turns off, and then in many cases further mechanosensing processes start. If the signal from the mechanosensing process is positive, there can be another round of extension, as in the periodic sensing of rigidity during lamellipodial extensions (Giannone et al. 2007). If the signal is negative, other motile processes can be activated, or motility can cease for a period.
FORCE GENERATION BY ACTIN FILAMENT ASSEMBLY
Numerous studies of actin polymerization have shown that the assembly of actin monomers into filaments can generate force through either a simple Brownian ratchet or directed polymerization by formins or nucleation-promoting factors. Recent studies indicate that the FMNL formins are required for high force generation by lamellipodia and that, without them, the ARP2/3 complex will still cause actin filament assembly at the normal rate, but with a much lower force threshold (Kage et al. 2017). The measurements of the force-velocity dependence of lamellipodial extension provide insights into the important parameters driving extension and indicate that the actin polymerization and cytoplasmic pressure forces are approximately equal (Manoussaki et al. 2015). The force-velocity dependence of the extension has been measured by atomic force microscopy, and mathematical models provide a reasonable fit of the data (Ryan et al. 2017, Schreiber et al. 2010). However, the process of actin filament assembly and turnover in lamellipodia is complicated, as might be expected of a robust system that can adapt to different microenvironments. For example, a complete accounting of the actin filament dynamics in lamellipodia revealed that turnover of filaments occurs throughout the lamellipodium and that nearly two-thirds of the actin diffuses, often as small filaments (Raz–Ben Aroush et al. 2017). In addition, two distinct filament populations, one branched and the other unbranched and containing tropomyosin, are required to work in concert to achieve efficient, persistent forward movement (Brayford et al. 2016, Hsiao et al. 2015). Thus, there appear to be complex relationships between actin filament assembly, membrane tension, myosin contractility, and substrate adhesions.
In terms of signaling from the extension forces, the presence of a barrier to lamellipodial extension will cause the cell to change direction and extend lamellipodia in other directions. This is indeed a form of signaling since the extension is an intermittent process, and barriers could simply prevent extension, rather than redirecting it. There may be other signals from the mechanical inhibition of extension, but there is insufficient evidence at the present.
FILOPODIA BINDING TO LIGANDS TO DIRECT MOTILITY
The cycles of extension and consolidation are different for each extension process, and they result in different tests of the environment. In the case of filopodia, a stepwise extension of the filopodia is dependent upon the myosin X FERM domain. In this example, a unit of extension is followed by consolidation, followed by another extension event and ultimately retraction (Watanabe et al. 2010). Since filopodia are to randomly probe the microenvironment, they will eventually retract unless they encounter a positive or a negative stimulus. In the case of growth cone pathfinding, the mechanical contact with a guidance molecule that is attached to a substrate enables the filopodium to generate force on the ligand-receptor complex (Boyer & Gupton 2018). The force-dependent signal directs the path of growth cone movement through steps of initial attachment and pulling, recruitment of other filopodia, contraction to bring the growth cone forward, and then further extension to find the next target. These steps have been shown for netrin-dependent guidance, in which force on the netrin receptor [DCC (deleted in colorectal cancer)] activates phosphorylation of focal adhesion kinase (FAK) in filopodia (Moore & Sheetz 2011; Moore et al. 2009, 2012). An analogous mechanism of directing fibroblast movement has been reported for guidance by patterned silk films (You et al. 2015). There is still debate about the mechanisms of neuronal pathfinding (Boyer & Gupton 2018), but mechanosensing is essential for developing stable neuronal patterns.
LAMELLIPODIAL EXTENSION AND MEMBRANE TENSION CONTROL DIRECTION OF MIGRATION
Migration of cells can also be directed by hormone signals that stimulate directed actin polymerization by seemingly changing membrane tension (Saha et al. 2018). Such stimulation can be mimicked by detergents that simply decrease membrane tension by expanding the membrane (Raucher & Sheetz 2000). Recent studies describe a link between membrane tension control of cell polarization and the enzymes PLD2 and mTORC2 (Diz-Muñoz et al. 2016). In addition to the role of membrane tension, there is likely a role for myosin contractility in controlling cell polarity, since myosin activation is linked to membrane signaling processes (Lou et al. 2015). Thus, the enzymatic pathways involved in controlling directional migration may be sensitive to local changes in membrane properties, including tension.
MODES OF CONTRACTION
The major forces generated by cells are typically contractile forces that require myosin II bipolar filament assembly. When myosin II activity is inhibited, the network of actin filaments in the cytoplasm disassembles, causing dramatic distortions in cell shape (Rossier et al. 2010). Formins polymerize actin and form nodes that are cross-linked by filamin A to create the basis of a network in which tension is provided by myosin IIA (Luo et al. 2013). In epithelial cells, a similar contractile network of actomyosin is responsible for the intermittent changes in apical area during epithelial shape changes (Vasquez et al. 2014) (Figure 2). The coherence of cell cytoplasm is initially dominated by the actin meshwork (Vasquez & Martin 2016); however, at later times, a strong intermediate filament network in epithelia inhibits overstretching and maintains some epithelial tension without continued myosin contraction (Harris et al. 2014). Mechanical coherence from one side of the cell to the other is required for different functions, not only for transport of materials on microtubules but also for generation of force to keep the tissue together. Since both tissue shape and cell shape depend on the interplay of extensions and contractions, as well as adhesion formation, understanding the major types of contractile systems is critical.
LAMELLIPODIAL CONTRACTION AND FORCE GENERATION
Similar to filopodia, lamellipodia extend into, attach to, and can mechanically test the new environment. There are basically two different modes of mechanical testing: radial pulling and local contractions. If the lamellipodium contacts new matrix sites, it will assemble nascent adhesions and test rigidity before forming mature adhesions for growth or motility. Over longer times, the greater stability of adhesions on rigid surfaces relative to soft surfaces results in durotaxis of cells. Traction forces are higher for cancer cells than for control cells. Higher traction forces correlate with increased metastasis and reduced survival (Indra et al. 2011, Kim et al. 2013, Koch et al. 2012, Kraning-Rush et al. 2012). An RNAi screen of adhesosome proteins identified 64 microRNAs that affect breast cancer cell migration and adhesion lifetime. Four of these proteins were validated for reduced migration after knockdown, while PPP1R12B, HIPK3, and RAC2 knockdown increased traction forces (Fokkelman et al. 2016). The increased contractility of cancer cells has been linked to TGFβ1-induced expression of caldesmon (Nalluri et al. 2018), possibly through an increase in miR-21 expression. Furthermore, as assessed by traction force microscopy, TGFβ activation of RalB, but not RalA, was required for matrix deformation and cell dissemination. RalB acts via the RhoGEF GEF-H1 complex, which associates with the exocyst complex, a major Ral effector (Biondini et al. 2015). Thus, contractions that originate from lamellipodial extensions play major roles in organizing the cytoplasm and in controlling the downstream processes of migration and traction force generation.
RADIAL CONTRACTIONS
The general flow of actin from the periphery to the nuclear region is a very common mode of contraction, and this contraction maintains general tension on the extracellular matrix. Upon inhibition of myosin contraction, the matrix adhesions disassemble. Therefore, continued contraction is critical for the preservation of cell shape. Complicating our understanding of how forces are maintained is the observation that actin filament assembly is dynamic throughout the lamellipodium (Raz–Ben Aroush et al. 2017), as is myosin activity (Baird et al. 2017). After adhesion to rigid substrates, there are three basic contractile networks involved in radial flow: radial actin bundles, circumferential cables with bipolar myosin, and nodal networks. In all cases, actin filaments appear to move from the periphery to the cell center, which belies the rapid dynamics of actin subunits in the filaments, as measured by photobleaching recovery and protein dynamics studies (mentioned above). Thus, although radial flow appears in many different cells, particularly on 2D surfaces, the many variations in the dynamics of actin filaments, adhesions, and their interactions have important implications for cell growth and motility through mechanosensory signaling processes.
STRESS FIBER CONTRACTIONS
Because of the prominence of stress fibers in F-actin stained cells and their anchorage at matrix adhesion sites, much attention has been paid to stress fiber formation and contractility. It is important to note that early spreading cells generate high radial forces without stress fibers, indicating that contractile networks other than stress fibers generate significant traction forces (Dubin-Thaler et al. 2008). With adhesion maturation, the movements of circumferential actin bundles can give rise to stress fibers that contain alternating stripes of myosin and α-actinin (Hotulainen & Lappalainen 2006). Adhesions anchor the ends of stress fibers, and the force generated on adhesions is intermediate between the values observed for actin flow forces and the values observed for local contraction forces (Ghassemi et al. 2012). Within longer adhesions, local oscillations in traction forces could belie dynamics in adhesion coupling to actin or myosin dynamics (Baird et al. 2017). There are two types of stress fibers: Ventral stress fibers affect nuclear expression patterns, and dorsal stress fibers are involved in organizing perinuclear actin fibers (Maninova & Vomastek 2016). Although stress fibers are not commonly found in tissues, they are likely to be used by cells for repeatedly developing high forces in recurring tissue contractions. Assembly and function of stress fibers require five different actin filament populations, each composed of a different tropomyosin and actin, which contribute different capabilities to the stress fibers (Tojkander et al. 2015). Again, turnover of the majority of actin in stress fibers occurs in minutes; inhibition of actin filament assembly by latrunculin A causes actin to release from adhesions and to rapidly contract (Rossier et al. 2010). When force is lost, adhesions disassemble, and there is an increase in the turnover rate of actin. Such turnover raises questions regarding how dynamic actomyosin networks maintain stable tension.
EPITHELIAL CONTRACTIONS AND SORTING
Numerous distinct contractions in epithelia shape the epithelium and result in segregation or cell extrusion. A critical part of epithelial contractions is the control of actin dynamics and integrity (Jodoin et al. 2015, Wu et al. 2014). An emerging property of mature epithelia is line tension along the cell boundaries that is regulated by control of the actomyosin network (e.g., Ect2 control of RhoA) (Ratheesh et al. 2012). In early development, epithelial monolayers undergo dramatic bending movements and convergent extension to create the proper form of the embryo. The bending movements are driven by dynamic actomyosin networks that span the apical portion of the cells. Similar to other types of motility, the contraction cycles are short lived and involve a series of steps with different biochemical and motile components (Martin et al. 2009) (see Figure 2). There are steps of activation, contraction, consolidation, and mechanosensing (Figure 1), and genetic studies in Drosophila showed that contraction requires the snail protein, whereas consolidation requires the twist transcription factor (Martin et al. 2009). Furthermore, the contraction cycles involve the cyclic activation of myosin through cycles of light chain phosphorylation and dephosphorylation (Vasquez et al. 2014). Additional proteins and signaling systems are linked to the process of epithelial contraction, but a detailed description of the steps and the sensing mechanisms is not available.
Another mechanical activity in epithelia is the sorting of dissimilar cells from one another through lateral rearrangements of the cells. This was observed early in the demixing of sponge cells from different species to form separate aggregates of a single species. Demixing was considered to be a simple matter of differential affinity between cell adhesion molecules (Steinberg & Takeichi 1994). However, physical considerations of the diffusional motility of cells make that scenario impossible, and demixing must involve active movements of cells past one another by myosin contraction. Again, we suggest that these movements are incremental and involve cycles of activation, movement, cessation, and sensing (Yang et al. 2018).
LOCAL CONTRACTIONS
There are many observations of single myosin filaments in the periphery of spreading cells. However, only through the nanofabrication of submicrometer pillars were researchers able to observe localized (1–3-μm) matrix site contractions that were correlated with rigidity sensing (Ghassemi et al. 2012). These 1–2-μm sarcomeric units are common in many different cell types and can assemble on both matrix adhesions (Meacci et al. 2016; Saxena et al. 2017b; Wolfenson et al. 2016; Yang et al. 2016, 2019) and cadherin adhesions (Yang et al. 2018). Models of the two local contraction modules are shown in Figure 3 and highlight that nonmuscle myosin IIA powers matrix contractions, whereas myosin IIB powers cadherin contractions. In general, the sarcomeric units assemble and contract to a fixed distance, and they relax and disassemble in approximately a minute (Wolfenson et al. 2019). They are sensory modules that develop different signals, depending upon the maximum force of the contractions. In the case of matrix adhesions, signals can control cell growth, death, or differentiation, whereas in the case of cadherin adhesions, signals control the organization of the epithelium. In the case of normal fibroblasts, matrix rigidity is a major factor in determining whether cells grow. In the case of stem cells, matrix rigidity affects the differentiation process (Engler et al. 2006). Because these local contraction modules require a variety of different proteins to function (see Figure 3), they can be inactivated as the result of depletion or mutation of any one of those important components of the modular complex. When sensory modules are absent in cells, the cells change state and behave dramatically differently than when modules are present. The change in cells from a matrix rigidity–dependent to a matrix rigidity–independent (transformed) growth state results from the loss of local matrix rigidity–sensing contractions. No exceptions to this link have been discovered to date.
Figure 3.
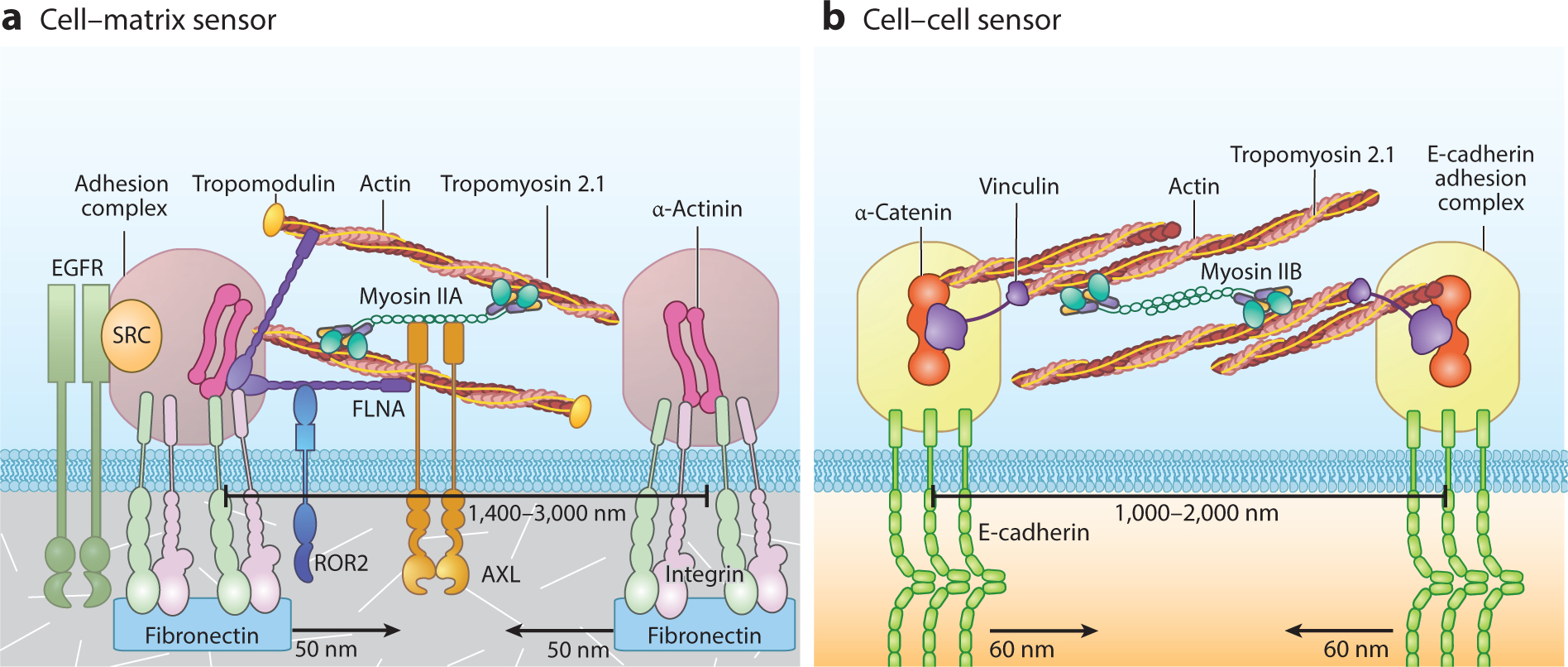
Diagram of the local contraction modules that are involved in measuring (a) the rigidity of matrix adhesions and (b) the rigidity of E-cadherin adhesions. Components of the matrix rigidity sensors (reviewed in Wolfenson et al. 2019) are derived from Meacci et al. (2016), Saxena et al. (2017b), Wolfenson et al. (2016), and Yang et al. (2016). The components of the E-cadherin rigidity sensors were defined in a study of MDCK and COS-7 cells (Yang et al. 2018). Abbreviations: EGFR, epidermal growth factor receptor; FLNA, filamin A; ROR2, receptor tyrosine kinase–like orphan receptor 2.
COMPONENTS OF MATRIX RIGIDITY–SENSING MODULES
The criteria for components of the rigidity-sensing modules are that the proteins associate with the local contraction units and that the depletion or alteration of the proteins alters local contraction activity. The density of local contractions was greatly diminished in cells that were depleted of one of the following module-associated cytoskeletal proteins: tropomyosin 2.1 (Wolfenson et al. 2016, Yang et al. 2019), α-actinin 4 (Meacci et al. 2016), myosin IIA (Saxena et al. 2017b), talin1 (Zhang et al. 2008), or filamin A (B. Yang, unpublished results). In addition, overexpression of tropomyosin 3.1/3.2 that competed with tropomyosin 2.1 caused the loss of rigidity sensing (Yang et al. 2019). Furthermore, local contractions are greatly depleted without the activity of the EGF receptor (EGFR) and HER2 (Saxena et al. 2017b). An RNAi screen of all the receptor tyrosine kinases in the human genome reported approximately 15 tyrosine kinases that altered rigidity sensing and cell (adhesion) morphology on soft or rigid surfaces (Prager-Khoutorsky et al. 2011). Further analysis of the receptor tyrosine kinases AXL and ROR2 (receptor tyrosine kinase–like orphan receptor 2) showed that they bound to cytoskeletal components in the modules and regulated the length and duration of the contractions, respectively (Yang et al. 2016). Thus, the rigidity sensors are a complex mixture of components, which, when depleted or aberrant, can result in the loss of rigidity sensing.
LOCAL E-CADHERIN CONTRACTIONS
Recent studies have reported local contractions of E-cadherin adhesions that are similar to local matrix contractions, but with major differences (Figure 3). As with matrix contractions, local contractions of cadherin adhesions are fixed in length (~120 nm), are independent of the rigidity of cadherin-coated pillars, and last for less than a minute (~20 s for half-maximal displacement) (Yang et al. 2018). Myosin bipolar filaments are located between the contracted pillars. Spreading of MDCK cells on E-cadherin surfaces is greater on stiff surfaces when local contractions occur, but depletion of the contractions causes cells to spread equally on rigid and soft surfaces. Unlike matrix contractions, local E-cadherin contractions require both α-catenin and vinculin and are powered by myosin IIB (see Figure 3). The latter contractions also involve tropomyosin 2.1, whose role in local contractions may be related to its roles in collective cell migration and aggregation (Shin et al. 2017). In terms of mechanosensing, local E-cadherin contractions sense the rigidity of the neighboring cells, which is related to myosin IIA activity. In monolayers, cells deficient in myosin IIA will be segregated from cells that have myosin IIA and that have local contractions. If the local contraction units are depleted, then sorting does not occur, and the myosin-depleted cells mix freely with myosin-containing cells. Thus, local contractions of E-cadherin provide important mechanosensing of neighboring cells.
CELL GROWTH ON RIGID MATRICES
Normal cells require rigid matrices for growth, whereas cancer cells grow equally well on rigid and soft matrices. The basic rigidity-sensing process (reviewed in Wolfenson et al. 2019) involves assembly of antiparallel actin filaments from nearby (within 1–3-μm) adhesion sites that are mechanically joined by a bipolar myosin filament (diagrammed in Figure 3). Recent studies indicate that rigidity sensing is linked to many different signaling pathways and receptor tyrosine kinases in unexpected ways (Saxena et al. 2017a, Yang et al. 2016). Moreover, lamellipodial extensions are directly tied to rigidity sensing in that cells activate rigidity sensing when contacting new regions of matrix (Saxena et al. 2017b), and not in areas with stable adhesions.
We presently have an incomplete understanding of the relationships between the component interactions in the rigidity-sensing modules and the activation of cell growth on stiff surfaces. The talin rod rescued epithelial cell growth in talin1-depleted cells through increased FAK phosphorylation and suppression of the cell cycle inhibitor p21 (Wang et al. 2011). However, the rod needs to be cleaved from the talin head, since recent studies find that full-length, noncleavable talin will not rescue growth in talin1 knockout cells but that the talin rod will. Furthermore, when cleavage of talin1 is blocked by either a point mutation in talin1 or calpain inhibition, the cell is not responsive to EGF, as is also the case on soft surfaces, indicating that talin1 cleavage and rigidity are important for EGFR function (Saxena et al. 2017a). Because calpain cleavage of talin1 normally involves calcium entry, the mechanosensitive channel Piezo1, which interacts with integrins, may be involved in providing local calcium for the cleavage process (McHugh et al. 2012). All these early events occur in the initial 5–7 min of the cell binding to matrix. Calpain cleavage renders the process irreversible until new proteins are synthesized. Thus, the details of mechanical signaling and motility are intimately linked to the signaling pathways involved in growth.
INCREASED ADHESION DYNAMICS AND APOPTOSIS ON SOFT SURFACES
If the matrix is soft, cells rapidly disassemble adhesions and, after further attempts to form adhesions, activate apoptosis processes termed anoikis. Death-associated protein kinase 1 (DAPK1) is activated on soft surfaces and causes apoptosis through activation of caspases (Ivanovska et al. 2014). Surprisingly, DAPK1 phosphorylates tropomyosin 1 on serine residues that are critical for stress fiber and adhesion assembly in endothelial cells (Houle et al. 2007). Conversely, in the absence of DAPK1 activity, the adhesions do not assemble properly, and local matrix contractions are prolonged for ~200 s instead of the normal 30 s (Qin et al. 2018). In terms of the mechanism of activation of DAPK1 on soft surfaces, the tyrosine phosphatase PTPN12 is needed (Qin et al. 2018) and this role may relate to the function of PTPN12 in disassembling adhesions (Angers-Loustau et al. 1999). Thus, DAPK1 is involved both in the proper assembly of contractile units and adhesions on rigid surfaces and in activating apoptosis on soft surfaces. DAPK1 is a major tumor suppressor (Maslikowski et al. 2017, Rose et al. 2017), as is its activator on soft surfaces, PTPN12 (Li et al. 2015, Sun et al. 2011). This strong interlinking of matrix sensing and motility occurs at a variety of levels, which may help to explain how robust cell functions are managed. However, the activation of apoptosis on soft surfaces by the rigidity-sensing module is clearly important for tumor suppression.
TRANSFORMED CELL STATE
The growth of cancer cells occurs in various environments; the ability of metastasized cancers to adapt to different growth stimulation pathways has made effective treatment of cancer very difficult. However, the ability of cancer cells to grow on soft surfaces has been a hallmark of cancer cells. One possible explanation for the transformed growth phenotype is that cancer cells lack a high-level block to growth and, as in wound healing in different tissues, can grow in response to a variety of different stimuli. The block to growth can be a critical mechanosensing pathway such as rigidity sensing. This hypothesis is consistent with a common pathway of growth stimulation by miR-21 and other common factors in wound healing and in cancer. To avoid oversimplification, it is important to mention that in the miR-21 knockout mouse, wound healing still occurs, indicating that additional factors are involved. However, there is evidence that the transformed state shares many properties with the growth phase of wound healing. Thus, we suggest that a major mechanosensor is removed in wound healing and cancer, enabling cells to grow in different environments with local growth promotors.
Transformation by oncogenes has provided many insights into the cancer cell state. In the case of v-Src transformation, cell motility and transcription patterns are altered by v-Src phosphorylation (Frame 2004). In the early characterization of v-Src transformed cells, the increased formation of podosomes/invadopodia was noted (Gelman & Gao 2006, Tanji et al. 2010). In terms of molecular mechanisms, v-Src causes specific morphological changes, podosome formation, and activation of the Akt pathway downstream of the phosphatase Shp-2 (Hakak et al. 2000). Furthermore, matrigel invasion of v-Src transformed fibroblasts, formation of large podosome structures, and tyrosine phosphorylation of Src substrates, including FAK, paxillin, and cortactin, were strictly dependent on the YxxP tyrosines in Crk-associated substrate (Brabek et al. 2005). In terms of the signaling pathways, the activation of AP-1 is common in many cancers, and inhibition by depletion of D-Jun will result in apoptosis if DAPK1 is active (Maslikowski et al. 2017). Thus, there are major changes in both motility and signaling pathways upon v-Src transformation, indicative of a change in cellular state resulting from oncogene activation. However, relatively little literature exists on the mechanical signaling changes in oncogene-transformed cells.
There appears to be an important role for the cytoskeleton in tumor cells since the expression of cytoskeletal proteins in transformed tumor cells can restore normal behavior. Of particular note is the expression of tropomyosins, which form copolymers with actin and are one of the earliest indicators of transformation (Helfman et al. 2008). Expression of tropomyosin restores normal behaviors in viral transformed cells (Gimona et al. 1996) and anoikis in tumor cells (Bharadwaj et al. 2005). Moreover, tropomyosins have major roles in TGFβ signaling, which has a complex role in inflammation and in cancer cells (Zheng et al. 2008). Since increased expression of tropomyosin 2.1 may restore rigidity sensing and rigidity-dependent growth in cancer cells, it is perhaps not surprising that the depletion of tropomyosin 2.1 in normal cells causes loss of rigidity sensing and transformed growth (Wolfenson et al. 2016). Furthermore, depletion of other components of the rigidity-sensing complex such as myosin IIA and α-actinin 4 also blocks rigidity sensing and causes transformed growth (Meacci et al. 2016, Yang et al. 2019). Without mechanosensing modules, cells adopt totally different phenotypes that are under different growth control mechanisms. In recent RNA-seq experiments, we identified nearly 1,000 altered mRNA levels after restoration of tropomyosin 2.1 to cancer cells or after depletion of tropomyosin 2.1 from normal cells (B. Yang & M. Sheetz, unpublished results). Thus, rigidity-sensing modules rely upon specific cytoskeletal proteins to form, and depletion of rigidity-sensing modules causes a major change in cell composition and cell state.
Although signaling pathways activated in tumor cells differ among various tissues, some changes in the motility properties of cancer cells appear to be common. Additionally, cytoskeletal changes can be diagnostic beyond growth on soft agar (Pawlak & Helfman 2001). More recent studies find that cancer cells are less stiff than normal cells from the same tissue (Plodinec et al. 2012, Runge et al. 2014, Weder et al. 2014), and cancer cells in vitro produce larger traction forces on matrices (Indra et al. 2011, Kraning-Rush et al. 2012). The somewhat paradoxical softness and increase in traction force can be explained by a decrease in the overall density of contractile elements with greater local activation of contraction. In the case of fibroblasts on very soft surfaces with occasional rigid barriers, a single cell will produce podosomes in low-force regions and normal adhesions at the force-bearing sites (Yu et al. 2013), indicating that different regions of the cytoplasm can exhibit different types of motility. In the case of cancer, there has been an emphasis on changes in signaling pathways; however, approximately 40% of the major tumor-associated mutations are in cytoskeletal proteins that likely affect motility (Wolfenson et al. 2019). Thus, transformed tumor cells appear to have altered mechanical properties, which may explain their altered mechanosensing as well as motility properties.
LOSS OF RIGIDITY-SENSING MODULES IN TRANSFORMED CANCER CELLS ALTERS SIGNALING PATHWAYS
Transformed cancer cells can be missing rigidity-sensing contractions for a variety of reasons, but growth on soft surfaces is the common feature. Restoring components that are depleted in rigidity sensors typically restores rigidity sensing and blocks growth on soft surfaces, independently of the tissue of origin (Yang et al. 2019). Different signaling pathways for growth are necessarily activated in the transformed cells that may relate to the altered activation of AP-1 from matrix adhesion signals. Furthermore, AP-1 activation causes a decrease in DAPK1 expression that is further inhibited by Src phosphorylation (Maslikowski et al. 2017). These and other factors can enable cells to directly activate growth pathways and avoid DAPK1 apoptosis. The multiple pathways of ERK activation in cancer cells that enable metastatic growth have caused considerable confusion about how to treat cancer. Because there is a common thread of increased miR-21 and changes in cytoskeleton-associated protein levels in many cancer cells, there may be some common features of the adhesion signaling in transformed cancer cells that could be exploited.
INCREASED CALPAIN ACTIVITY IN TRANSFORMED CELLS
There is an extensive literature on the correlation between increased calpain levels in cancer cells and the severity of disease (Carragher et al. 2002, 2004; Liu et al. 2017; Ma et al. 2017; Storr et al. 2015). In terms of the mechanism of growth activation in cancer cells, calpain1/2-CAST is associated with ERK1/2 activation, which is important for disease progression (Starska et al. 2016). Another possible relationship is the calpain-dependent cleavage of talin that produces the talin rod needed for cell growth (Saxena et al. 2017a, Wang et al. 2011). Mechanical unfolding of talin was required for cleavage, making the process mechanosensitive. Many additional targets of calpain may contribute to increased growth of cancer cells when calpain levels are high.
The high level of calpain in cancer cells appears to produce a sensitivity to calcium-dependent apoptosis. Treatments with natural products that increase cancer cell apoptosis correlate with increased levels of calcium and calpain cleavage (Chan et al. 2015, Hou et al. 2018, Su et al. 2015). In addition, cancer cells may be susceptible to increased calcium entry due to higher levels of the mechanosensitive Piezo1 channel observed in lung and bladder cancers (Etem et al. 2018, Pappu et al. 2016).
TRANSFORMATION AND SENSITIVITY TO MECHANICAL DAMAGE
Several reports indicate that cancer cell growth is vulnerable to mechanical forces. Studies reporting inhibition of tumor growth after stretching or exercise in mice models could be explained through mechanical force–dependent growth inhibition (Berrueta et al. 2018, Betof et al. 2015). Fluid shear–induced killing of circulating tumor cells and adhesive cancer cells could also be explained by increased sensitivity to mechanical forces (Lien et al. 2013, Regmi et al. 2017). In addition, ultrasonic and shock-wave therapies have demonstrated that cancer cells are particularly sensitive to mechanical distortion, although concerns were raised that these treatments might drive increased metastasis and healthy tissue damage (Lin et al. 2012, Marano et al. 2017, Nicolai et al. 1994, Zequi et al. 2018). In very recent studies, mechanical stretching of cancer cells and normal cells that were transformed inhibited growth and activated cell apoptosis. However, when rigidity sensing was restored in cancer cells, they grew better with mechanical stretch, like normal cells (Cui et al. 2015, Tijore et al. 2018). Thus, the mechanical differences in transformed cells may make them more sensitive to mechanical damage, which could be exploited in the future as a mode of cancer treatment.
MECHANOSENSORY MODULES ARE ALTERED IN TISSUE REPAIR AND CANCER
Modular mechanosensing systems cause major changes in cell state, whether they are active in a fibroblast or in an epithelial cell background. With the loss of specific mechanosensors such as rigidity sensors, cells adopt a different phenotypic behavior that is clearly linked to changes in the levels of cytoskeletal proteins. In transformed cancer cells, the ability to grow on soft surfaces correlates with loss of the ability to sense rigidity, i.e., loss of rigidity-sensing modules. Because mechanosensors have many protein components that are needed for their rapid assembly, action, and disassembly, mechanosensory pathways can be disrupted by the depletion or mutation of a large repertoire of components. When there is repeated injury in a tissue requiring activation of growth for wound healing, there is an increased likelihood that one or more of the components in the mechanosensory module required for cell growth control will be stably depleted, resulting in cancerous growth in that tissue. The observation that mechanosensory modules rely on cytoskeletal proteins is consistent with the observation that cytoskeletal protein expression levels are altered in transformed growth relative to normal phenotypes. This relationship can provide new potential targets for treatment of cancer and of wound healing.
CONCLUSION
Because cells are robust devices, they have many of the engineering features of robust devices in modular components that drive standard cyclic functions. The standard motile functions of cells are linked with mechanosensory functions in cycles of motility and sensing that incrementally drive cell and tissue shape changes. Different cell types or states are linked to changes in motility functions and in the sensory modules that are active. In the cases of wound healing and cancerous growth, there appears to be a common general activation of growth that is in part related to the overexpression of miR-21 to inhibit mechanosensing. Without inhibition from mechanosensors such as a rigidity sensor, local growth can be activated. The transformed growth of cancer cells is linked to altered expression of cytoskeletal proteins, resulting in the loss of rigidity-sensing modules that are evident in local matrix contractions. Although many other factors contribute to tumor growth, cancer cell lines generally lack rigidity-sensing activity. Other, perhaps related changes in transformed cell motility and behavior such as decreased cell rigidity, increased calpain levels, and altered calcium homeostasis provide potential avenues for intervention in cancer and wound healing. In particular, there is mounting evidence that transformed cells are sensitive to mechanically induced apoptosis through calcium-activated calpain cleavage.
ACKNOWLEDGMENTS
I wish to thank the Singapore Ministry of Education for supporting my work through the funding of the Mechanobiology Institute and tier 2 and tier 3 grants. In addition, there was important support from the National University of Singapore. This work grew out of studies funded by the NIH (R01GM113022).
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Adhami M, Haghdoost AA, Sadeghi B, Malekpour Afshar R. 2018. Candidate miRNAs in human breast cancer biomarkers: a systematic review. Breast Cancer 25:198–205 [DOI] [PubMed] [Google Scholar]
- Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, et al. 1999. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J. Cell Biol 144:1019–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird MA, Billington N, Wang A, Adelstein RS, Sellers JR, et al. 2017. Local pulsatile contractions are an intrinsic property of the myosin 2A motor in the cortical cytoskeleton of adherent cells. Mol. Biol. Cell 28:240–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrueta L, Bergholz J, Munoz D, Muskaj I, Badger GJ, et al. 2018. Stretching reduces tumor growth in a mouse breast cancer model. Sci. Rep 8:7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betof AS, Lascola CD, Weitzel D, Landon C, Scarbrough PM, et al. 2015. Modulation of murine breast tumor vascularity, hypoxia and chemotherapeutic response by exercise. J. Natl. Cancer Inst 107:djv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. 2005. Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase–dependent cytoskeleton and adhesion. Oncogene 24:8291–303 [DOI] [PubMed] [Google Scholar]
- Bhome R, Goh RW, Bullock MD, Pillar N, Thirdborough SM, et al. 2017. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: role in driving cancer progression. Aging 9:2666–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondini M, Duclos G, Meyer-Schaller N, Silberzan P, Camonis J, Parrini MC. 2015. RalB regulates contractility-driven cancer dissemination upon TGFβ stimulation via the RhoGEF GEF-H1. Sci. Rep 5:11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswenger V, Baumann N, Jurschick J, Hackl M, Battle C, et al. 2018. Characterization of EGF-guided MDAMB-231 cell chemotaxis in vitro using a physiological and highly sensitive assay system. PLOS ONE 13:e0203040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer NP, Gupton SL. 2018. Revisiting Netrin-1: one who guides (axons). Front. Cell. Neurosci 12:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabek J, Constancio SS, Siesser PF, Shin NY, Pozzi A, Hanks SK. 2005. Crk-associated substrate tyrosine phosphorylation sites are critical for invasion and metastasis of SRC-transformed cells. Mol. Cancer Res 3:307–15 [DOI] [PubMed] [Google Scholar]
- Brayford S, Bryce NS, Schevzov G, Haynes EM, Bear JE, et al. 2016. Tropomyosin promotes lamellipodial persistence by collaborating with Arp2/3 at the leading edge. Curr. Biol 26:1312–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher NO, Fonseca BD, Frame MC. 2004. Calpain activity is generally elevated during transformation but has oncogene-specific biological functions. Neoplasia 6:53–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher NO, Westhoff MA, Riley D, Potter DA, Dutt P, et al. 2002. v-Src-induced modulation of the calpain-calpastatin proteolytic system regulates transformation. Mol. Cell. Biol 22:257–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan ML, Liang JW, Hsu LC, Chang WL, Lee SS, Guh JH. 2015. Zerumbone, a ginger sesquiterpene, induces apoptosis and autophagy in human hormone-refractory prostate cancers through tubulin binding and crosstalk between endoplasmic reticulum stress and mitochondrial insult. Naunyn Schmiedebergs Arch. Pharmacol 388:1223–36 [DOI] [PubMed] [Google Scholar]
- Cui Y, Hameed FM, Yang B, Lee K, Pan CQ, et al. 2015. Cyclic stretching of soft substrates induces spreading and growth. Nat. Commun 6:6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz-Muñoz A, Thurley K, Chintamen S, Altschuler SJ, Wu LF, et al. 2016. Membrane tension acts through PLD2 and mTORC2 to limit actin network assembly during neutrophil migration. PLOS Biol. 14:e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin-Thaler BJ, Hofman JM, Cai Y, Xenias H, Spielman I, et al. 2008. Quantification of cell edge velocities and traction forces reveals distinct motility modules during cell spreading. PLOS ONE 3:e3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. 2006. Matrix elasticity directs stem cell lineage specification. Cell 126(4):677–89 [DOI] [PubMed] [Google Scholar]
- Etem EO, Ceylan GG, Ozaydin S, Ceylan C, Ozercan I, Kuloglu T. 2018. The increased expression of Piezo1 and Piezo2 ion channels in human and mouse bladder carcinoma. Adv. Clin. Exp. Med 27:1025–31 [DOI] [PubMed] [Google Scholar]
- Fokkelman M, Balcioglu HE, Klip JE, Yan K, Verbeek FJ, et al. 2016. Cellular adhesome screen identifies critical modulators of focal adhesion dynamics, cellular traction forces and cell migration behaviour. Sci. Rep 6:31707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame MC. 2004. Newest findings on the oldest oncogene; how activated Src does it. J. Cell Sci 117:989–98 [DOI] [PubMed] [Google Scholar]
- Ge XT, Lei P, Wang HC, Zhang AL, Han ZL, et al. 2014. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci. Rep 4:6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Zhang L, Nikolova M, Reva B, Fuchs E. 2016. Strand-specific in vivo screen of cancer-associated miRNAs unveils a role for miR-21* in SCC progression. Nat. Cell Biol 18:111–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman IH, Gao L. 2006. SSeCKS/Gravin/AKAP12 metastasis suppressor inhibits podosome formation via RhoA- and Cdc42-dependent pathways. Mol. Cancer Res 4:151–58 [DOI] [PubMed] [Google Scholar]
- Ghassemi S, Meacci G, Liu S, Gondarenko AA, Mathur A, et al. 2012. Cells test substrate rigidity by local contractions on submicrometer pillars. PNAS 109:5328–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, Dubin-Thaler BJ, Rossier O, Cai Y, Chaga O, et al. 2007. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128:561–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M, Kazzaz JA, Helfman DM. 1996. Forced expression of tropomyosin 2 or 3 in v-Ki-ras-transformed fibroblasts results in distinct phenotypic effects. PNAS 93:9618–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Hsu YS, Martin GS. 2000. Shp-2 mediates v-Src-induced morphological changes and activation of the anti-apoptotic protein kinase Akt. Oncogene 19:3164–71 [DOI] [PubMed] [Google Scholar]
- Han Z, Chen Y, Zhang Y, Wei A, Zhou J, et al. 2017. MiR-21/PTEN axis promotes skin wound healing by dendritic cells enhancement. J. Cell. Biochem 118:3511–19 [DOI] [PubMed] [Google Scholar]
- Harris AR, Daeden A, Charras GT. 2014. Formation of adherens junctions leads to the emergence of a tissue-level tension in epithelial monolayers. J. Cell Sci 127:2507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfman DM, Flynn P, Khan P, Saeed A. 2008. Tropomyosin as a regulator of cancer cell transformation. Adv. Exp. Med. Biol 644:124–31 [DOI] [PubMed] [Google Scholar]
- Holman EC, Campbell LJ, Hines J, Crews CM. 2012. Microarray analysis of microRNA expression during axolotl limb regeneration. PLOS ONE 7:e41804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe B, Pietsch S, Franke M, Engel S, Groth M, et al. 2015. MiR-21 is required for efficient kidney regeneration in fish. BMC Dev. Biol 15:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. 2006. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol 173:383–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou D, Che Z, Chen P, Zhang W, Chu Y, et al. 2018. Suppression of AURKA alleviates p27 inhibition on Bax cleavage and induces more intensive apoptosis in gastric cancer. Cell Death Dis. 9:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle F, Poirier A, Dumaresq J, Huot J. 2007. DAP kinase mediates the phosphorylation of tropomyosin-1 downstream of the ERK pathway, which regulates the formation of stress fibers in response to oxidative stress. J. Cell Sci 120:3666–77 [DOI] [PubMed] [Google Scholar]
- Hsiao JY, Goins LM, Petek NA, Mullins RD. 2015. Arp2/3 complex and cofilin modulate binding of tropomyosin to branched actin networks. Curr. Biol 25:1573–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra I, Undyala V, Kandow C, Thirumurthi U, Dembo M, Beningo KA. 2011. An in vitro correlation of mechanical forces and metastatic capacity. Phys. Biol 8:015015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovska J, Mahadevan V, Schneider-Stock R. 2014. DAPK and cytoskeleton-associated functions. Apoptosis 19:329–38 [DOI] [PubMed] [Google Scholar]
- Jiang J, Yang P, Guo Z, Yang R, Yang H, et al. 2016. Overexpression of microRNA-21 strengthens stem cell–like characteristics in a hepatocellular carcinoma cell line. World J. Surg. Oncol 14:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodoin JN, Coravos JS, Chanet S, Vasquez CG, Tworoger M, et al. 2015. Stable force balance between epithelial cells arises from F-actin turnover. Dev. Cell 35:685–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John K, Hadem J, Krech T, Wahl K, Manns MP, et al. 2014. MicroRNAs play a role in spontaneous recovery from acute liver failure. Hepatology 60:1346–55 [DOI] [PubMed] [Google Scholar]
- Kage F, Winterhoff M, Dimchev V, Mueller J, Thalheim T, et al. 2017. FMNL formins boost lamellipodial force generation. Nat. Commun 8:14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy LL, Meng F, Venter JK, Zhou T, Karstens WA, et al. 2016. Knockout of microRNA-21 reduces biliary hyperplasia and liver fibrosis in cholestatic bile duct ligated mice. Lab. Investig 96:1256–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Yin B, Christudass CS, Terada N, Rajagopalan K, et al. 2013. Acquisition of paclitaxel resistance is associated with a more aggressive and invasive phenotype in prostate cancer. J. Cell. Biochem 114:1286–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BL, Yin VP. 2016. A conserved microRNA regulatory circuit is differentially controlled during limb/ appendage regeneration. PLOS ONE 11:e0157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch TM, Munster S, Bonakdar N, Butler JP, Fabry B. 2012. 3D traction forces in cancer cell invasion. PLOS ONE 7:e33476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraning-Rush CM, Califano JP, Reinhart-King CA. 2012. Cellular traction stresses increase with increasing metastatic potential. PLOS ONE 7:e32572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Davidson D, Martins Souza C, Zhong MC, Wu N, et al. 2015. Loss of PTPN12 stimulates progression of ErbB2-dependent breast cancer by enhancing cell survival, migration, and epithelial-to-mesenchymal transition. Mol. Cell. Biol 35:4069–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Guo L, Liu Y, Su Y, Xie Y, et al. 2018. MicroRNA-21 promotes wound healing via the Smad7-Smad2/3-Elastin pathway. Exp. Cell Res 362:245–51 [DOI] [PubMed] [Google Scholar]
- Lien SC, Chang SF, Lee PL, Wei SY, Chang MD, et al. 2013. Mechanical regulation of cancer cell apoptosis and autophagy: roles of bone morphogenetic protein receptor, Smad1/5, and p38 MAPK. Biochim. Biophys. Acta Mol. Cell Res 1833:3124–33 [DOI] [PubMed] [Google Scholar]
- Lin CY, Tseng HC, Shiu HR, Wu MF, Chou CY, Lin WL. 2012. Ultrasound sonication with microbubbles disrupts blood vessels and enhances tumor treatments of anticancer nanodrug. Int. J. Nanomed 7:2143–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zhou Y, Lu D, Liu Y, Zhang SQ, et al. 2017. Comparison of the protein expression of calpain-1, calpain-2, calpastatin and calmodulin between gastric cancer and normal gastric mucosa. Oncol. Lett 14:3705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou SS, Diz-Muñoz A, Weiner OD, Fletcher DA, Theriot JA. 2015. Myosin light chain kinase regulates cell polarization independently of membrane tension or Rho kinase. J. Cell Biol 209:275–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubov J, Maschietto M, Ibrahim I, Mlynarek A, Hier M, et al. 2017. Meta-analysis of microRNAs expression in head and neck cancer: uncovering association with outcome and mechanisms. Oncotarget 8:55511–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Yu CH, Lieu ZZ, Allard J, Mogilner A, et al. 2013. Analysis of the local organization and dynamics of cellular actin networks. J. Cell Biol 202:1057–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv L, Huang F, Mao H, Li M, Li X, et al. 2013. MicroRNA-21 is overexpressed in renal cell carcinoma. Int. J. Biol. Markers 28:201–7 [DOI] [PubMed] [Google Scholar]
- Ma D, Fang J, Liu Y, Song JJ, Wang YQ, et al. 2017. High level of calpain1 promotes cancer cell invasion and migration in oral squamous cell carcinoma. Oncol. Lett 13:4017–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, et al. 2009. Coordination of Rho GTPase activities during cell protrusion. Nature 461:99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maninova M, Vomastek T. 2016. Dorsal stress fibers, transverse actin arcs, and perinuclear actin fibers form an interconnected network that induces nuclear movement in polarizing fibroblasts. FEBS J. 283:3676–93 [DOI] [PubMed] [Google Scholar]
- Manoussaki D, Shin WD, Waterman CM, Chadwick RS. 2015. Cytosolic pressure provides a propulsive force comparable to actin polymerization during lamellipod protrusion. Sci. Rep 5:12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marano F, Frairia R, Rinella L, Argenziano M, Bussolati B, et al. 2017. Combining doxorubicin-nanobubbles and shockwaves for anaplastic thyroid cancer treatment: preclinical study in a xenograft mouse model. Endocr. Relat. Cancer 24:275–86 [DOI] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF. 2009. Pulsed contractions of an actin-myosin network drive apical constriction. Nature 457:495–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslikowski BM, Wang L, Wu Y, Fielding B, Bedard PA. 2017. JunD/AP-1 antagonizes the induction of DAPK1 to promote the survival of v-Src-transformed cells. J. Virol 91:e01925–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoudi MS, Mehrabian E, Mirzaei H. 2018. MiR-21: a key player in glioblastoma pathogenesis. J. Cell. Biochem 119:1285–90 [DOI] [PubMed] [Google Scholar]
- McHugh BJ, Murdoch A, Haslett C, Sethi T. 2012. Loss of the integrin-activating transmembrane protein Fam38A (Piezo1) promotes a switch to a reduced integrin-dependent mode of cell migration. PLOS ONE 7:e40346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacci G, Wolfenson H, Liu S, Stachowiak MR, Iskratsch T, et al. 2016. α-Actinin links extracellular matrix rigidity–sensing contractile units with periodic cell-edge retractions. Mol. Biol. Cell 27:3471–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momtazi AA, Shahabipour F, Khatibi S, Johnston TP, Pirro M, Sahebkar A. 2016. Curcumin as a MicroRNA regulator in cancer: a review. Rev. Physiol. Biochem. Pharmacol 171:1–38 [DOI] [PubMed] [Google Scholar]
- Moore SW, Biais N, Sheetz MP. 2009. Traction on immobilized netrin-1 is sufficient to reorient axons. Science 325:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Sheetz MP. 2011. Biophysics of substrate interaction: influence on neural motility, differentiation, and repair. Dev. Neurobiol 71:1090–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Zhang X, Lynch CD, Sheetz MP. 2012. Netrin-1 attracts axons through FAK-dependent mechanotransduction. J. Neurosci 32:11574–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalluri SM, O’Connor JW, Virgi GA, Stewart SE, Ye D, Gomez EW. 2018. TGFβ1-induced expression of caldesmon mediates epithelial-mesenchymal transition. Cytoskeleton 75:201–12 [DOI] [PubMed] [Google Scholar]
- Nicolai H, Steinbach P, Knuechel-Clarke R, Grimm D, Roessler W, et al. 1994. Proliferation of tumor spheroids after shock-wave treatment. J. Cancer Res. Clin. Oncol 120:438–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappu P, Madduru D, Chandrasekharan M, Modhukur V, Nallapeta S, Suravajhala P. 2016. Next generation sequencing analysis of lung cancer datasets: a functional genomics perspective. Indian J. Cancer 53:1–7 [DOI] [PubMed] [Google Scholar]
- Pawlak G, Helfman DM. 2001. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev 11:41–47 [DOI] [PubMed] [Google Scholar]
- Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, et al. 2012. The nanomechanical signature of breast cancer. Nat. Nanotechnol 7(11):757–65 [DOI] [PubMed] [Google Scholar]
- Pontes B, Monzo P, Gole L, Le Roux AL, Kosmalska AJ, et al. 2017. Membrane tension controls adhesion positioning at the leading edge of cells. J. Cell Biol 216:2959–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, et al. 2011. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechanosensing. Nat. Cell Biol 13:1457–65 [DOI] [PubMed] [Google Scholar]
- Qin R, Wolfenson H, Saxena M, Sheetz M. 2018. Tumor suppressor DAPK1 catalyzes adhesion assembly on rigid but anoikis on soft matrices. bioRxiv 320739. 10.1101/320739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, et al. 2012. Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nat. Cell Biol 14:818–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher D, Sheetz MP. 2000. Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol 148:127–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz–Ben Aroush D, Ofer N, Abu-Shah E, Allard J, Krichevsky O, et al. 2017. Actin turnover in lamellipodial fragments. Curr. Biol 27:2963–73.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regmi S, Fu A, Luo KQ. 2017. High shear stresses under exercise condition destroy circulating tumor cells in a microfluidic system. Sci. Rep 7:39975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M, Kloten V, Noetzel E, Gola L, Ehling J, et al. 2017. ITIH5 mediates epigenetic reprogramming of breast cancer cells. Mol. Cancer 16:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier OM, Gauthier N, Biais N, Vonnegut W, Fardin MA, et al. 2010. Force generated by actomyosin contraction builds bridges between adhesive contacts. EMBO J. 29:1055–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge J, Reichert TE, Fritsch A, Käs J, Bertolini J, Remmerbach TW. 2014. Evaluation of single-cell biomechanics as potential marker for oral squamous cell carcinomas: a pilot study. Oral Dis. 20(3):e120–27 [DOI] [PubMed] [Google Scholar]
- Ryan GL, Holz D, Yamashiro S, Taniguchi D, Watanabe N, Vavylonis D. 2017. Cell protrusion and retraction driven by fluctuations in actin polymerization: a two-dimensional model. Cytoskeleton 74:490–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Nagy TL, Weiner OD. 2018. Joining forces: crosstalk between biochemical signalling and physical forces orchestrates cellular polarity and dynamics. Philos. Trans. R. Soc. B Biol. Sci 373:20170145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Changede R, Hone J, Wolfenson H, Sheetz MP. 2017a. Force-induced calpain cleavage of talin is critical for growth, adhesion development, and rigidity sensing. Nano Lett. 17:7242–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Liu S, Yang B, Hajal C, Changede R, et al. 2017b. EGFR and HER2 activate rigidity sensing only on rigid matrices. Nat. Mater 16:775–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena N, Mogha P, Dash S, Majumder A, Jadhav S, Sen S. 2018. Matrix elasticity regulates mesenchymal stem cell chemotaxis. J. Cell Sci 131:jcs211391. [DOI] [PubMed] [Google Scholar]
- Schreiber CH, Stewart M, Duke T. 2010. Simulation of cell motility that reproduces the force-velocity relationship. PNAS 107:9141–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar D, Krishnan R, Thirugnanasambantham K, Rajasekaran B, Islam VI, Sekar P. 2016. Significance of microRNA 21 in gastric cancer. Clin. Res. Hepatol. Gastroenterol 40:538–45 [DOI] [PubMed] [Google Scholar]
- Sheetz MP, Yu H. 2018. The Cell as a Machine. Cambridge, UK: Cambridge Univ. Press [Google Scholar]
- Shin H, Kim D, Helfman DM. 2017. Tropomyosin isoform Tpm2.1 regulates collective and amoeboid cell migration and cell aggregation in breast epithelial cells. Oncotarget 8:95192–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starska K, Forma E, Jozwiak P, Lewy-Trenda I, Danilewicz M, et al. 2016. Gene/protein expression of CAPN1/2-CAST system members is associated with ERK1/2 kinases activity as well as progression and clinical outcome in human laryngeal cancer. Tumour Biol. 37:13185–203 [DOI] [PubMed] [Google Scholar]
- Steinberg MS, Takeichi M. 1994. Experimental specification of cell sorting, tissue spreading, and specific spatial patterning by quantitative differences in cadherin expression. PNAS 91:206–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storr SJ, Thompson N, Pu X, Zhang Y, Martin SG. 2015. Calpain in breast cancer: role in disease progression and treatment response. Pathobiology 82:133–41 [DOI] [PubMed] [Google Scholar]
- Su CC, Liu SH, Lee KI, Huang KT, Lu TH, et al. 2015. Cantharidin induces apoptosis through the calcium/PKC-regulated endoplasmic reticulum stress pathway in human bladder cancer cells. Am. J. Chin. Med 43:581–600 [DOI] [PubMed] [Google Scholar]
- Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, et al. 2011. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 144:703–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji M, Ishizaki T, Ebrahimi S, Tsuboguchi Y, Sukezane T, et al. 2010. mDia1 targets v-Src to the cell periphery and facilitates cell transformation, tumorigenesis, and invasion. Mol. Cell. Biol 30:4604–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Fontana S, Monteleone F, Pucci M, Saieva L, et al. 2016. Curcumin modulates chronic myelogenous leukemia exosomes composition and affects angiogenic phenotype via exosomal miR-21. Oncotarget 7:30420–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Lee T, Suh NP. 2004. A function-based framework for understanding biological systems. Annu. Rev. Biophys. Biomol. Struct 33:75–93 [DOI] [PubMed] [Google Scholar]
- Tijore A, Yao M, Wang YH, Nematbakhsh Y, Hariharan A, Lim CT, Sheetz M. 2018. Mechanical stretch kills transformed cancer cells. bioRxiv 491746. 10.1101/491746 [DOI] [PubMed] [Google Scholar]
- Tojkander S, Gateva G, Husain A, Krishnan R, Lappalainen P. 2015. Generation of contractile actomyosin bundles depends on mechanosensitive actin filament assembly and disassembly. eLife 4:e06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Novak B. 2014. Control of cell growth, division and death: information processing in living cells. Interface Focus 4:20130070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helvert S, Storm C, Friedl P. 2018. Mechanoreciprocity in cell migration. Nat. Cell Biol 20:8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez CG, Martin AC. 2016. Force transmission in epithelial tissues. Dev. Dyn 245:361–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez CG, Tworoger M, Martin AC. 2014. Dynamic myosin phosphorylation regulates contractile pulses and tissue integrity during epithelial morphogenesis. J. Cell Biol 206:435–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Ballestrem C, Streuli CH. 2011. The C terminus of talin links integrins to cell cycle progression. J. Cell Biol 195:499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hang Y, Liu J, Hou Y, Wang N, Wang M. 2017. Anticancer effect of curcumin inhibits cell growth through miR-21/PTEN/Akt pathway in breast cancer cell. Oncol. Lett 13:4825–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe TM, Tokuo H, Gonda K, Higuchi H, Ikebe M. 2010. Myosin-X induces filopodia by multiple elongation mechanism. J. Biol. Chem 285:19605–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weder G, Hendriks-Balk M, Smajda R, Rimoldt D, Liley M, et al. 2014. Increased plasticity of the stiffness of melanoma cells correlates with their acquisition of metastatic properties. Nanomedicine 10(1):141–48 [DOI] [PubMed] [Google Scholar]
- Weinberg J, Drubin DG. 2012. Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 22:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenson H, Meacci G, Liu S, Stachowiak MR, Iskratsch T, et al. 2016. Tropomyosin controls sarcomere-like contractions for rigidity sensing and suppressing growth on soft matrices. Nat. Cell Biol 18:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfenson H, Yang B, Sheetz MP. 2019. Steps in mechanotransduction pathways that control cell morphology. Annu. Rev. Physiol 81:585–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SK, Budnar S, Yap AS, Gomez GA. 2014. Pulsatile contractility of actomyosin networks organizes the cellular cortex at lateral cadherin junctions. Eur. J. Cell Biol 93:396–404 [DOI] [PubMed] [Google Scholar]
- Yang B, Lieu ZZ, Wolfenson H, Hameed FM, Bershadsky AD, Sheetz MP. 2016. Mechanosensing controlled directly by tyrosine kinases. Nano Lett. 16:5951–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Wolfenson H, Nakazawa N, Liu S, Hu J, Sheetz MP. 2019. Stopping transformed growth by rigidity sensing. Nature Mater. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Nguyen E, Mege R-M, Ladoux B, Sheetz MP. 2018. Local contractions test rigidity of E-cadherin adhesions. bioRxiv 318642. 10.1101/318642 [DOI] [Google Scholar]
- You R, Li X, Luo Z, Qu J, Li M. 2015. Directional cell elongation through filopodia-steered lamellipodial extension on patterned silk fibroin films. Biointerphases 10:011005. [DOI] [PubMed] [Google Scholar]
- Yu CH, Rafiq NB, Krishinasamy A, Hartman KL, Jones GE, et al. 2013. Integrin-matrix clusters form podosome-like adhesions in the absence of traction forces. Cell Rep. 5(5):1456–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zequi SC, Mourão TC, Guimarães GC. 2018. Prostate cancer—local treatment after radiorecurrence: HIFU—high-intensity focused ultrasound. Int. Braz. J. Urol 44:429–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou Y, Huang T, Wu F, Liu L, et al. 2018. PIEZO1 functions as a potential oncogene by promoting cell proliferation and migration in gastric carcinogenesis. Mol. Carcinog 57:1144–55 [DOI] [PubMed] [Google Scholar]
- Zhang W, Bai W, Zhang W. 2014. MiR-21 suppresses the anticancer activities of curcumin by targeting PTEN gene in human non–small cell lung cancer A549 cells. Clin. Transl. Oncol 16:708–13 [DOI] [PubMed] [Google Scholar]
- Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP. 2008. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol 10:1062–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Chen S, Zhu Z, Yu L, Ren Y, et al. 2018. miR-21 promotes EGF-induced pancreatic cancer cell proliferation by targeting Spry2. Cell Death Dis. 9:1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Safina A, Bakin AV. 2008. Role of high-molecular weight tropomyosins in TGF-β-mediated control of cell motility. Int. J. Cancer 122:78–90 [DOI] [PubMed] [Google Scholar]