

Abstract
Hybridization between invasive and native species has raised global concern, given the dramatic increase in species range shifts and pest outbreaks due to anthropogenic dispersal. Nevertheless, secondary contact between sister lineages of local and invasive species provides a natural laboratory to understand the factors that determine introgression and the maintenance or loss of species barriers. Here, we characterize the early evolutionary outcomes following secondary contact between invasive Helicoverpa armigera and native H. zea in Brazil. We carried out whole-genome resequencing of Helicoverpa moths from Brazil in two temporal samples: during the outbreak of H. armigera in 2013 and 2017. There is evidence for a burst of hybridization and widespread introgression from local H. zea into invasive H. armigera coinciding with H. armigera expansion in 2013. However, in H. armigera, the admixture proportion and the length of introgressed blocks were significantly reduced between 2013 and 2017, suggesting selection against admixture. In contrast to the genome-wide pattern, there was striking evidence for adaptive introgression of a single region from the invasive H. armigera into local H. zea, including an insecticide resistance allele that increased in frequency over time. In summary, despite extensive gene flow after secondary contact, the species boundaries are largely maintained except for the single introgressed region containing the insecticide-resistant locus. We document the worst-case scenario for an invasive species, in which there are now two pest species instead of one, and the native species has acquired resistance to pyrethroid insecticides through introgression.
Keywords: hybridization, secondary contact, insecticide resistance evolution, gene flow, invasive species
Introduction
Hybridization between local and introduced species is an underestimated threat to global agriculture (Mooney and Cleland 2001; Blair and Hufbauer 2010; Paini et al. 2016). Although hybridization is often maladaptive, introgressive hybridization can occasionally introduce adaptive variants and potentially contribute to the establishment and invasiveness of the newcomer species (Currat et al. 2008; Saarman and Pogson 2015; Hall 2016; Mesgaran et al. 2016). This makes invasive hybridization a major global concern impacting natural and agricultural systems, since species introductions are predicted to continue at an accelerating rate following trade, human movement, and climate change (Saarman and Pogson 2015; Paini et al. 2016). Simultaneously, invasive species offer a unique opportunity to study evolutionary outcomes following secondary contact. Although secondary contact zones have played a major role in the study of reproductive isolation and speciation, it remains a challenge to disentangle the contribution of ancient and recent gene flow to the contemporary patterns of introgression (Barton and Hewitt 1985; Abbott et al. 2013). This is primarily because ancestral range reconstruction is challenging, and so it is hard to confidently estimate the extent of historical contact between species (Barton and Hewitt 1985; Abbott et al. 2013; Saarman and Pogson 2015). However, in the case of invasive species, the geographical reunion of divergent species can be observed in “real time.” This allows us to systematically test hypotheses regarding the possible outcomes of secondary contact including fusion, reinforcement, hybrid speciation, and adaptive introgression.
The recent invasion by the Old World cotton bollworm Helicoverpa armigera of South America (Czepak et al. 2013), where it hybridizes with its sister species H. zea (Tay et al. 2013; Anderson et al. 2018), represents an excellent scenario in which to study the evolutionary trajectories following secondary contact. Helicoverpa armigera comprises two subspecies: H. armigera conferta in Oceania (Australia, New Zealand) and H. armigera armigera in its Old World native range, subsequently introduced to South America (Tay et al. 2013; Anderson et al. 2018) (hereafter H. armigera refers to H. armigera armigera unless otherwise specified). Helicoverpa zea is present throughout the temperate and tropical regions of North and South America. Helicoverpa armigera and H. zea are estimated to have diverged around 1.5 Ma (Behere et al. 2007; Pearce et al. 2017) as a result of a transoceanic dispersion of H. armigera into the New World, and thus represent a classic case of divergence due to allopatric distributions (Pearce et al. 2017; Anderson et al. 2018). There is no evidence of introgression between the species during this period of divergence until the recent incursions of H. armigera into Brazil (Pearce et al. 2017; Anderson et al. 2018). The invasion is thought to have resulted from international trade (Pearce et al. 2017; Anderson et al. 2018; Mallet 2018; Tay and Gordon 2019) and there is evidence for initial independent incursions, centered on numerous geographic locations (Tay et al. 2017; Arnemann et al. 2019). Multiple and likely ongoing introductions of H. armigera from the Old World mean that the genetic diversity of the New World populations is very high and possibly comparable to that in the species’ ancestral range (Tay et al. 2017; Arnemann et al. 2019; Gonçalves et al. 2019).
Although the two species are very similar morphologically and their genomes share high levels of orthology, H. armigera has twice the genome-wide genetic diversity and a much greater (∼10×) effective population size compared with H. zea (Mallet et al. 1993; Pearce et al. 2017). In addition, H. zea shows a relatively narrow host-plant range and is mainly known as a pest of corn and cotton, although it is also found on over 100 plants belonging to 29 families (Pearce et al. 2017; Anderson et al. 2018; Mallet 2018). Helicoverpa armigera is more devastating to agriculture and feeds on over 300 species belonging to 68 families (Pearce et al. 2017; Anderson et al. 2018; Mallet 2018). These differences in host range are associated with fewer gustatory receptor and detoxification genes in the H. zea genome, which showed no evidence for gain of any genes after the species diverged (Pearce et al. 2017).
Another substantial phenotypic difference between H. armigera and H. zea lies in their susceptibility to insecticides widely used for pest control (Anderson et al. 2018; Walsh, Joussen, et al. 2018). Helicoverpa armigera shows by far the highest number of reported cases of insecticide resistance worldwide, having evolved resistance against pyrethroids, organophosphates, carbamates, organochlorines, and recently to macrocyclic lactone spinosad, and several Bacillus thuringiensis toxins (McCaffery 1998; Xu et al. 2005; Mahon et al. 2007; Joußen et al. 2012; Tay et al. 2015; Durigan et al. 2017; Pearce et al. 2017; Walsh, Joussen, et al. 2018). Importantly, the resistance toward pyrethroids in H. armigera is due to a unique P450 enzyme, CYP337B3, that is capable of metabolizing these pesticides into nontoxic hydropyrethroids (Joußen et al. 2012). Helicoverpa zea, in contrast, remains susceptible to many major insecticides and tends to cause less damage than its Old World relative, although there are recent reports of resistance to Bt toxins (Moar et al. 2010; Brévault et al. 2015; Reisig and Kurtz 2018). Therefore, one of the major concerns following the invasion of H. armigera is the formation of novel hybrid ecotypes or the introduction of genes associated with insecticide resistance and host-range expansion.
Anderson et al. (2018) unambiguously identified nine genetically intermediate individuals using whole-genome sequences between the two species from 2013, at least 6–8 years after its possible initial introduction into Brazil, when significant crop damages were reported (Tay et al. 2013, 2017; Sosa-Gómez et al. 2016). Of the limited individuals analyzed, eight were genetically closer to H. armigera (between 91% and 98% H. armigera-like) with some introgressed haplotypes from H. zea scattered throughout the genome. The ninth individual resembled an F2 hybrid with stretches of homozygosity for each parental species, and all H. armigera surveyed individuals contained the CYP337B3 gene involved in pesticide resistance. The apparent rarity of backcrossing to H. zea in these individuals (Anderson et al. 2018) initially suggested that hybridization would not introduce novel insecticide resistance to H. zea as had been feared (Mallet 2018). However, intensive pesticide-based control programs in Brazil aimed at controlling the invasive H. armigera provide a scenario in which there is strong selection for adaptive introgression of insecticide resistance genes into H. zea (Leite et al. 2014; Durigan et al. 2017; Pinto et al. 2017). These efforts to control H. armigera may also have had the converse effect of facilitating the establishment of H. armigera due to differential survival enabled by its resistance alleles (Norris et al. 2015; Dermauw et al. 2018).
Here, we use whole-genome resequencing of individuals in two temporal samples, soon after the outbreak of H. armigera in Brazil (2012–2013) and more recent populations (2016–2017) from a region with large areas cultivating cotton, soybean, and corn to examine the evolutionary outcomes following secondary contact between the Helicoverpa sister species. We first identify genome-wide patterns of species distinctiveness and then explore signatures of introgression. We find that the species boundaries are strongly maintained in recent Brazilian populations. Then, we show that introgression is heterogeneous throughout the genome and that admixture has decreased over time in the Brazilian H. armigera population. Conversely, for H. zea, there is a single strong signature of introgression around a region containing a novel, chimeric gene implicated in insecticide resistance (CYP337B3 gene) which was previously only documented in H. armigera and identified in the invasive population in Brazil (Anderson et al. 2018; Walsh, Joussen, et al. 2018).
Results
Sympatric H. zea and H. armigera Postincursion Persist as Two Well-Distinct Species
We analyzed whole-genome sequence data from 137 moths from across the world: 90 H. armigera armigera (hereafter H. armigera), 39 H. zea, 2 hybrid individuals (hybrids considered as F1–F3), and 6 H. punctigera. The Brazilian individuals were collected in two temporal samples: soon after the H. armigera expansion in 2012–2013 (Sosa-Gómez et al. 2016) (59 individuals hereafter 2013) and more recent populations from 2016 to 2017 (37 individuals hereafter 2017). The Brazilian sampling included three regions of sympatry in Brazil that resulted from the invasion of H. armigera into local H. zea territory. We also included, as a baseline comparison, four preinvasion Brazilian H. zea individuals sampled in 2006 (Behere et al. 2007), as well as eight individuals of H. zea from allopatric (USA) populations. Additionally, nine individuals of H. armigera from the Old World which are all considered to be geographically or temporally isolated from the Brazilian invasion (fig. 1A and supplementary tables S1–S3, Supplementary Material online), as well as 14 H. a. conferta individuals from Australia, and six individuals from an outgroup Australian endemic species H. punctigera (fig. 1A).
Fig. 1.

Sampling locations and PCA. (A) Sampling locations of Helicoverpa individuals. (B). Genetic PC1 and PC2 of ∼50,000 genome-wide SNPs. Color codes indicate different samples and species based on mitochondrial sequences and the groups used for the analyses. Helicoverpa armigera (Australia) refers to the subspecies H. armigera conferta and H. armigera (Old World) to the subspecies H. armigera armigera in the rest of the Old World. Helicoverpa zea (New World) comprises Brazilian preinvasion and United States, and finally, H. armigera 2013/2017 and H. zea 2013/2017 refer to the postinvasion sympatric populations in Brazil.
Principal component analysis (PCA) based on ∼50,000 whole-genome single-nucleotide polymorphism (SNP) data separated H. zea from H. armigera on principal component 1, which explained 39.41% of the variation in the data (fig. 1B and supplementary fig. S1A, Supplementary Material online). Across the wide geographic range covered by our sampling, there was no evidence of population substructure. The reported separation between H. armigera conferta from Australia and H. armigera armigera from the rest of the Old World (Anderson et al. 2016, 2018) was evident when resuming at least the first 25 principal component axes (supplementary fig. S1B, Supplementary Material online). Two individuals fell between H. armigera and H. zea, likely representing early generation hybrids. The remaining 97 individuals from Brazil clearly have a closer genetic affinity to one or other of the parental species. Consistent with PC1, ADMIXTURE (Alexander and Lange 2011) analyses showed strong differentiation between the two species, which is supported by the lowest cross-validation error of any assessed when the number of populations is K = 2 (supplementary fig. S2, Supplementary Material online).
To classify parental and hybrid genotypes, we calculated the proportion of ancestry and heterozygosity based on SNPs that strongly segregated between allopatric populations of the species. In accordance with the PCA and ADMIXTURE results, the classification of Brazilian individuals identified only two early generation hybrids (fig. 2). One individual resembled an F2 hybrid, whereas the other individual resembled an F3 hybrid, both from 2013. All remaining individuals grouped with one of the parental species, although with various fractions of their genome coming from the other species. (Proportions of ancestry are listed in supplementary table S4, Supplementary Material online). A total of 66 individuals were closer to H. armigera, with ∼ 0–9.2% of alleles derived from H. zea. Meanwhile, the 27 individuals that grouped with H. zea samples had ∼ 0–4.1% of H. armigera ancestry (supplementary table S4, Supplementary Material online). The tight within-species clustering and low prevalence of intermediate individuals indicated that almost none of the recently sampled individuals resulted from contemporary hybridization, suggesting that hybridization is rare on a per-individual basis in recent populations.
Fig. 2.

Classification of hybrid and parental genotypes. Classification is based on the hybrid index and interspecific heterozygosity for Helicoverpa zea and H. armigera individuals. The hybrid index and the interspecific heterozygosity were calculated based on ∼1,000,000 SNPs that are fixed differences between allopatric populations of the parental species.
Reduced Interspecific Differentiation in Sympatry around a Single Genomic Region
We characterized patterns of divergence between sympatric and allopatric pairs of H. zea and H. armigera populations using the fixation index FST. We found similar high genome-wide differentiation between H. zea and H. armigera for allopatric and recent sympatric populations with a mean autosome-wide FST of 0.474 ± 0.001 (mean ± SE) and 0.496 ± 0.001, respectively (supplementary fig. S3 and table S1, Supplementary Material online). By contrast, FST values for intraspecific comparisons were low, consistent with a clear genetic identity within species (supplementary fig. S3, Supplementary Material online). Indeed, the FST between the invasive Brazilian and Old World H. armigera was 0.0062 ± 0.0001 (supplementary fig. S3A, Supplementary Material online), and the pairwise FST between Brazilian H. zea and North American H. zea averaged 0.0118 ± 0.0002 (supplementary fig. S3A, Supplementary Material online).
To compare differentiation across the genome, we calculated FST values in 100-kb nonoverlapping windows. Interspecific FST values for allopatric (0.474 ± 0.0012) and recent sympatric (0.489 ± 0.0068) populations were consistently high throughout the genome except for a single region on chromosome 15 (fig. 3C–F). This region included the insecticide resistance gene CYP337B3, whereby FST significantly dropped up to 0.14 (−log10[P value] = 13.96) for the recent sympatric Brazilian populations of H. armigera and H. zea (fig. 3C and E). Intraspecific FST values across the genome between Brazilian and Old World H. armigera populations were also consistently low (2013: 0.0107 ± 0.001; 2017: 0.0162 ± 0.0016), although there was a slight peak at the CYP337B3 locus (2013: FST = 0.11, −log10(P value) ≫ 30; 2017: FST = 0.19, −log10(P value) ≫ 30) (supplementary fig. S3B, Supplementary Material online). Between H. zea from United States and Brazil, there was also only one peak of differentiation (−log10[P value] ≫ 30), corresponding to the same region on chromosome 15 which contains the insecticide-resistant locus and that showed low differentiation for the interspecific comparison between Brazilian H. zea and H. armigera (fig. 3D). Although both FST estimates at a genome-wide scale and across windows showed that differentiation between species is high between recent sympatric (0.489 ± 0.0068) and allopatric (0.474 ± 0.0012) populations, these patterns of reduced between-species and elevated within-species differentiation around the same region hint at recent localized introgression.
Fig. 3.

FST scans across the genome. FST across 100-kb genomic windows between sympatric and allopatric populations of Helicoverpa armigera and H. zea. Inter-specific comparisons between allopatric populations (A) and sympatric populations during the outbreak in 2013 (B) and more recent populations in 2017 (C). Intra-specific comparison between H. zea 2013 and 2017 populations (D). Squares on (C) and (D) highlight chromosome 15 and (E) and (F) zoom in on FST values on chromosome 15, showing the location of the CYP337B3 gene.
Widespread Phylogenetic Discordance Is Consistent with Recent Gene Flow
We explored species phylogenies across the genome using Twisst (Martin and Van Belleghem 2017), which quantifies the frequency of alternative topological relationships among all individuals in sliding windows, in this case, of 50 SNPs. These phylogenetic comparisons across the genome indicated large-scale introgression among recent samples of Brazilian H. armigera and H. zea (fig. 4). The three possible unrooted topologies that describe the relationships between allopatric and sympatric populations were represented across the genome (supplementary fig. S4, Supplementary Material online). Around 60% of the windows had completely sorted genealogies (i.e., weighting of 1), most of which matched the expected species branching order (blue in fig. 4 and supplementary fig. S4, Supplementary Material online). Around 90% of the topologies are concordant with the species tree (supplementary fig. S4, Supplementary Material online). The two discordant topologies are likely explained by incomplete lineage sorting of the nonstructured populations of H. zea and H. armigera (green and orange in supplementary fig. S4, Supplementary Material online). However, the topology that groups recent H. zea and H. armigera together from Brazil is more common and is consistent with a recent burst of gene flow between sympatric species and introgression (green in fig. 4 and supplementary fig. S4, Supplementary Material online). In addition, there is a correlation between the weights of the topology grouping the Brazilian populations (green in fig. 4 and supplementary fig. S4, Supplementary Material online) and the tree grouping H. zea 2017 and H. armigera from the Old World, which is expected given that there is not population substructure within H. armigera. Notably, from these regions of phylogenetic incongruence, the longest continuous interval with a discordant topology was on chromosome 15, around the CYP337B3 locus (fig. 4). Therefore, given that introgression seems to be restricted to a single region, the similar frequencies of the nonspecies phylogenies suggest that introgression in recent populations of H. zea is not widespread.
Fig. 4.

Topology weightings across the Z-chromosome and chromosome 15. Twisst analysis on 50 SNPs windows for a species topology with the relations: ((Old World Helicoverpa armigera, Brazilian H. armigera 2017) (New World H. zea, Brazilian H. zea 2017)). Blue represents the weight of the species topology ((Old World H. armigera, Brazilian H. armigera 2017) (New World H. zea, Brazilian H. zea 2017)). Green ((Brazilian H. zea 2017, Brazilian H. armigera 2017) (New World H. zea, Old World H. armigera)) and orange ((Brazilian H. zea 2017, Old World H. armigera) (New World H. zea, Brazilian H. armigera 2017)) are alternative topologies to the species tree. See supplementary figure S4, Supplementary Material online, for explanations of the topologies.
Introgression Is Biased toward Specific Genomic Regions and Has Decreased over Time in H. armigera and Increased in H. zea
To test for genetic admixture, we calculated Patterson’s D-statistics, which test for an imbalance of discordant SNPs of derived alleles that is indicative of introgression (Green et al. 2010; Patterson et al. 2012). We found strong signals of gene flow between Brazilian H. zea and H. armigera for 2013 and 2017 samples (table 1). We then examined f, the proportion of the genome that has been shared between species (Green et al. 2010), and observed a decreased f over time in H. armigera (fig. 5 and supplementary table S2, Supplementary Material online). The fraction of ancestry derived from their sibling species was higher in H. armigera populations than for H. zea in 2013, suggesting asymmetric introgression early after the invasion (fig. 5).
Table 1.
Results of D-Statistic.
| Target Population | D-Statistic | Z-Score | P Value |
|---|---|---|---|
| H. armigera 2013a | 0.1203 ± 0.005015 | 23.986 | <0.0001 |
| H. armigera 2017a | 0.0259 ± 0.004774 | 5.427 | <0.0001 |
| H. zea 2013b | 0.0068 ± 0.004373 | 1.551 | 0.1209 |
| H. zea 2017b | 0.0554 ± 0.010163 | 5.455 | <0.0001 |
Note.—Admixture test for Brazilian populations of Helicoverpa in 2013 and 2017. Model used for H. armigera populations (((A, B)C)D): (((x, H. armigera Old World) H. zea New World) H. punctigera). Model used for H. zea populations (((A, B)C)D): (((x, H. zea New World) H. armigera Old World) H. punctigera). A significantly positive Z-score implies gene flow between A and C. A significantly negative Z-score implies gene flow between B and C. P values <0.05 indicates D-statistic significantly different from 0.
Fig. 5.
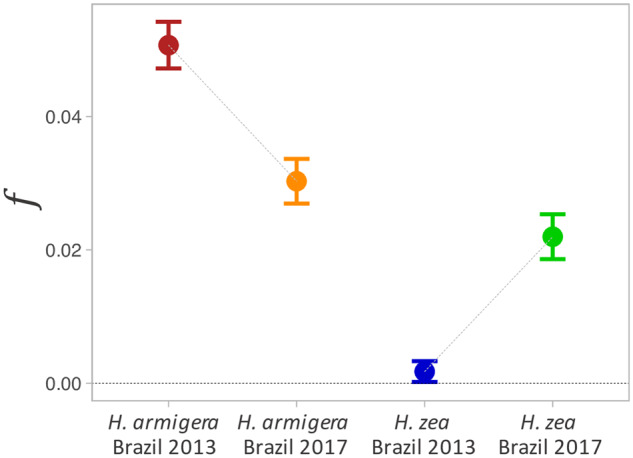
Genome-wide proportion of admixture. Proportion of admixture (f) estimated over time for Helicoverpa armigera and H. zea in Brazil estimated as an F4 ratio for the genealogy ((Australian H. armigera conferta, Old World H. armigera; X; New World H. zea).
To locate introgressed loci, we used the summary statistic fd, since it is better suited than f to quantify admixture in small genomic regions and which is roughly proportional to the effective migration rate (Martin et al. 2015). As expected from the genome-wide f, fd values also decreased over time in H. armigera populations (fig. 6). However, the proportion of admixture estimated in sliding windows showed considerable heterogeneity in the extent of introgression across the genome (fig. 6). In both temporal samples and species, admixture was minimal on the Z-chromosome (P value < 1 × 10−4), indicating a strong barrier to introgression (fig. 6B and D). For Brazilian H. armigera, we found high heterogeneity in admixture proportion in the autosomes, with peaks scattered throughout different chromosomes (fig. 6D). Some regions, including the entire sequence of chromosome 15, exhibited fd values below the genome-wide average (P value < 1 × 10−4), implying localized barriers to gene flow. Conversely, in local H. zea populations, fd values were lower throughout the genome compared with the invasive H. armigera in 2013 (P value < 2.2 × 10−6). Yet, for recent H. zea populations, we found a peak showing a higher proportion of admixture (fig. 6B), than any peak in H. armigera and reflecting almost complete replacement. This peak was located across the region on chromosome 15 containing the CYP337B3 gene responsible for pyrethroid resistance, and that initially was only present in the invasive species (fig. 6). Additionally, gene scans of H. zea revealed that CYP337B3 was present in 14 out of 18 H. zea individuals from 2017 (frequency ∼0.70) (supplementary table S4, Supplementary Material online). Therefore, the insecticide resistance locus has not swapped to fixation yet, although fd suggested a maximal admixture proportion, which may be explained by a large excess of ABBA patterns due to the recent gene flow. The introgressed sequence was similar to that found in European H. armigera (window 11,435,001–11,440,000, dXY = 0.0064, −log10(P value) = 2.14 genome-wide dXY = 0.040 ± 0.29) compared with African (dXY = 0.025, −log10(P value) = 2.15, genome-wide dXY = 0.040 ± 0.029, Z = 86.918, P value < 2.2 × 10−6) and Asian H. armigera (dXY = 0.021, −log10(P value) = 1.85, genome-wide dXY = 0.053 ± 0.031, Z = 49.094, P value < 2.2 × 10−6).
Fig. 6.

Proportion of admixture across the genome. (fd) and introgressed tracks per individual. (A) Admixture tracks displaying Helicoverpa zea ancestry in 100-kb windows for individuals of H. zea 2013, H. zea 2017, and H. zea (New World). (B) Proportion of admixture fd across 100-kb windows, for populations of H. zea 2013 (blue) and H. zea 2017 (green). (C) Admixture tracks displaying H. zea ancestry in 100-kb windows for individuals of H. armigera 2013, H. armigera 2017, and H. armigera (Old World). (D) Proportion of admixture fd across 100-kb windows, for populations of H. armigera 2013 (dark red) and H. armigera 2017 (orange).
Increased FST values in both within-species comparisons around the CYP337B3 locus might also be a result of within-species population dynamics. However, the joint estimation of FST and fd allowed us to disentangle linked selection in H. armigera from differentiation due to admixture in H. zea. FST is expected to be highly heterogeneous even in the absence of gene flow, with elevated values in regions of the genome that experience strong linked selection (Jakobsson et al. 2013; Martin et al. 2015). Therefore, the weak FST peak within H. armigera is likely an artifact of the recent selective sweep in this region in chromosome 15 (Walsh, Joussen, et al. 2018) (supplementary fig. S3, Supplementary Material online). In contrast, the FST peak on chromosome 15 in the within-species comparison of H. zea populations pre- and post-H. armigera invasion coincided with the fd peak in recent H. zea populations, implying that differentiation was caused by introgression.
We then calculated the proportion of introgressed ancestry across the genome of all individuals using a data set of diagnostic SNPs that differentiate the two species and estimated a hybrid index in 100-kb sliding windows (fig. 6A). To disentangle the role of neutral recombination and selection shaping the size of the admixed blocks over time, we characterized the extent of linkage disequilibrium and estimated a genome-wide recombination rate. Then, we used these parameters to simulate the expected distribution of block sizes under neutral recombination following (Racimo et al. 2015). Across individuals, there was evidence for long regions acquired through introgression (fig. 6A). We found that temporal samples showing a higher proportion of admixture (fig. 5) also exhibited stronger linkage disequilibrium (P value < 2.2 × 10−6) (supplementary fig. S6, Supplementary Material online) In the recent H. zea population, the mean introgressed block size was higher in 2017 (509 kb) compared with 2013 (Z = 5.23, P value < 1 × 10−4) (supplementary fig. S7, Supplementary Material online) and the admixed haplotypes are restricted to chromosome 15 (fig. 6A and supplementary fig. S6, Supplementary Material online). Therefore, consistent with the fd analyses, recent individuals of Brazilian H. zea exhibited a single clearly introgressed block around the CYP337B3 locus.
By contrast, in early H. armigera, long introgressed regions were pervasive, indicating introgression of almost whole chromosomes. Particularly long admixture tracks derived from H. zea on chromosome 18 seemed to be persistent across most H. armigera individuals in 2013 and 2017. Nevertheless, we observed shorter introgressed blocks (Z = 7.79, P value < 1 × 10−4) and a lower effective migration rate (Z = 289.18, P value < 0.001) in recent populations of H. armigera. Similarly, the extent of Linkage Disequilibrium (LD) was greater in the 2013 population (P value = 0.002) (supplementary fig. S6, Supplementary Material online). Moreover, the mean introgressed block size in the recent H. armigera population was around 227 kb, significantly shorter (Z = 7.88, P value < 1 × 10−4) than the theoretically predicted mean length of 470 kb after 36 generations (4 years between 2013 and 2017) of neutral recombination (supplementary fig. S7, Supplementary Material online). Thus, suggesting that the admixed regions have been removed faster than expected just from recombination over time, thus suggesting strong purifying selection against introgression in H. armigera.
To characterize recent selection around the CYP337B3 locus, we calculated Tajima’s D on 10-kb windows across the region on chromosome 15 containing this gene. There was a trough in Tajima’s D consistent with a selective sweep at the location of CYP337B3 from invasive H. armigera in 2013 (fig. 7A and supplementary table S3, Supplementary Material online) that is absent in the nonintrogressed H. zea from the same period (fig. 7B and supplementary fig. S5, Supplementary Material online), suggesting strong selection on this locus. In recent populations of Brazilian H. armigera, Tajima’s D is negative in the vicinity of the insecticide resistance allele (Tajima’s D = −0.60) although not lower compared with the genome-wide average (Tajima’s D ± SD: −0.98 ± 0.36) (fig. 7A). In contrast, recent populations of H. zea harboring the CYP337B3 haplotype exhibited significantly lower Tajima’s D along this region (Tajima’s D = −0.72; genome-wide Tajima’s D ± SD: 0.57 ± 0.59) suggesting recent positive selection on this introgressed locus (fig. 7B and supplementary table S3, Supplementary Material online).
Fig. 7.

Tajima’s D in 10-kb windows along the region in chromosome 15 containing CYP337B3 gene for populations of Helicoverpa armigera and H. zea in Brazil. (A) Tajima’s D for H. armigera 2013 (dark red) and for H. armigera 2017 (orange). (B) Tajima’s D for H. zea 2013 (blue) and for H. zea 2017 (green) containing CYP337B3 gene. Gray shading indicates the location of CYP337B3.
Discussion
We have used whole-genome sequencing of 137 individuals to document the interaction between native and invasive crop pest species. This has demonstrated that these species are maintaining their differentiation in the face of hybridization, showing evidence for a reduction in admixture over time in the invasive species and localized admixture in the native. Indeed, a single locus associated with insecticide resistance shows dramatic evidence for an introgressed selective sweep from H. armigera into H. zea. Whole-genome sequencing, therefore, offers an opportunity to track the fate of invasive species in the face of hybridization. In this particular case, we are witnessing the emergence of a new paradigm where insecticide resistance or other adaptive traits can be passed rapidly between species in the native range, potentially leading to the formation of a “megapest” species. This has serious implications for pest control and agricultural crop production both in South America and globally, as the movement of pests is not unidirectional.
Sympatric H. zea and H. armigera Persist as Two Well-Differentiated Species
In secondary contact after strict allopatric speciation, there are a number of possible outcomes, with barriers to gene flow potentially being either strengthened or broken down and resulting in the fusion of the divergent species. Despite pervasive introgression following invasion and secondary contact between H. armigera and H. zea, we found that the species boundaries are strongly maintained. Most individuals showed some proportion of introgression, but early hybrids and backcrosses were scarce, suggesting low levels of ongoing hybridization. Differentiation between species is roughly the same between allopatric and sympatric populations except for a single “pocket” of introgression driven by strong selection. Therefore, from the myriad of possible outcomes after secondary contact, our results do not lend support to H. armigera and H. zea fusing back into one species, nor formation of a new hybrid species, at least not in the time frame examined here. On the contrary, the predominance of parental forms, the rarity of hybrids, particularly in the recent samples, and the distribution of introgressed block sizes suggest selection against most of the introgressed variation.
The Helicoverpa sibling species exhibit both prezygotic and postzygotic barriers that can account for the low rates of natural hybridization (Hardwick 1965; Laster and Sheng 1995; Berg et al. 2014; Mallet 2018). Despite experiments having demonstrated no instances of sterility in H. armigera × H. zea hybrids in reciprocal crosses inbred for two generations or in lines backcrossed for four, they exhibit strong assortative mating (Hardwick 1965; Laster and Sheng 1995). Mating is particularly difficult to obtain between an F1 hybrid and H. zea (AZ × Z), which can account for the lower admixture proportion we observed in H. zea populations in 2013. Traits linked to prezygotic isolation include morphological differences in the male and female genitalia as well as divergent pheromonal bouquets (Hardwick 1965; Laster and Sheng 1995; Berg et al. 2014). A possible outcome of mating between H. zea and H. armigera is to “lock up,” leading to death without reproduction of the individuals involved (Hardwick 1965). This is apparently due to differences in the length of an elaborate inflating appendix in the genitalia, which makes the male unable to successfully withdraw his aedeagus extension from the female (Hardwick 1965; Laster and Sheng 1995; Mallet 2018). Although this can also occur in intraspecific mates, this clogging is more common in interspecific matings (Hardwick 1965; Mallet 2018). Furthermore, behavioral tests in Helicoverpa moths showed that female pheromones and their ratios are used as critical signals to avoid interspecific matings (Fadamiro and Baker 1997; Quero et al. 2001; Berg et al. 2014). Even though H. zea and H. armigera both use Z-11-hexadecenal and Z-9-hexadecenal as the primary sex pheromones emitted by females and detected by males, the ratio of the latter to the former is about 3-fold higher in H. armigera (Fadamiro and Baker 1997; Quero and Baker 1999; Quero et al. 2001; Berg et al. 2014). Therefore, both mechanical and chemical reproductive isolations likely play a role in assortative mating and the reduction of successful interspecific mating.
In addition to prezygotic isolation, selection against hybridization could be the result of intrinsic postzygotic barriers, including Bateson–Dobzhansky–Müller incompatibilities (BDMIs) or alternatively “pathway” incompatibilities. BDMIs cause reduced fitness in hybrids but can be broken down by recombination in backcross progeny (Bomblies et al. 2007; Lee et al. 2008; Schumer et al. 2018). Conversely, “pathway” incompatibilities reduce fitness in recombinant hybrids in which coadapted alleles become separated (Lindtke and Buerkle 2015). Simulations and empirical data show that BDMIs usually have an even, genome-wide effect, whereas “pathway” incompatibilities produce distinct localized barriers to introgression (Lindtke and Buerkle 2015; Martin et al. 2019). Our results show a genome-wide reduction of admixture over time in H. armigera and strongly localized adaptive introgression in H. zea. Furthermore, the mean length of introgressed blocks was shorter than expected due to neutral recombination across generations, suggesting pervasive selection against hybridization likely as a result of widespread BDMIs throughout the genome. Moreover, patterns of differentiation captured by FST also suggested highly polygenic barriers that maintain H. armigera and H. zea as distinctive species despite the recent burst in gene flow. BDMIs are more likely to act as polygenic barriers and are a central mechanism underlying reproductive isolation of species formed through allopatric divergence, as in the case of H. armigera and H. zea (Bomblies et al. 2007; Lee et al. 2008). Therefore, we hypothesize that barrier loci like BDMIs accumulated rapidly during the 1.5 My of allopatric divergence, contributing to selection against admixture across the genome upon secondary contact. In summary, our findings highlight that species can be unambiguously different but coupled through the interchange of adaptive genes.
Rapid Adaptive Introgression of an Insecticide Resistance Gene
Several lines of evidence suggested rapid adaptive introgression between the Helicoverpa sibling species after the H. armigera invasion in South America. In order to provide evidence for adaptive introgression, the introgressed haplotype needs to be shown to be responsible for a phenotype with a beneficial impact on fitness and to have increased in frequency under the action of selection. The candidate region containing CYP337B3 appears to satisfy both requirements. First, pesticide resistance toward pyrethroids is due to the unique P450 enzyme encoded by this gene (Joußen et al. 2012; Walsh, Joussen, et al. 2018). This chimeric enzyme arose independently, at least eight times, from unequal crossover events between two parental P450 genes (Walsh, Joussen, et al. 2018), and is capable of metabolizing pyrethroids to nontoxic compounds in larvae and adults (Joußen et al. 2012). Indeed, the exclusive presence of CYP337B3 has been shown to confer a 42-fold resistance to fenvalerate in H. armigera (Joußen et al. 2012). Furthermore, Durigan et al. (2017) showed that CYP337B3 is associated with the very low mortality of larvae when sprayed with the pyrethroids deltamethrin and fenvalerate in invasive H. armigera armigera in Brazil.
Second, the CYP337B3 introgression into H. zea and subsequent increase in frequency coincided with an intensification in pesticide use in Brazil directed to control outbreaks generated by the invasive H. armigera (Leite et al. 2014; Durigan et al. 2017; Pinto et al. 2017). In the 2012/2013 growing season, cotton, soybean, and maize crops were impacted by Helicoverpa species at unusually high infestations resulting in billion dollar losses to Brazilian agriculture (Durigan et al. 2017). Initially, the species was presumed to be H. zea, but during this season, growers reported a reduced efficacy of insecticides despite the increase in sprays and the known susceptibility of H. zea to pesticides (Durigan et al. 2017). The resistant individuals were subsequently confirmed as H. armigera, confirming its successful spread and establishment after being introduced into Brazil from ∼2007 onward (Tay et al. 2013, 2015; Lopes-da-Silva et al. 2014; Sosa-Gómez et al. 2016). Furthermore, the survival and establishment of H. armigera in the studied region during the 2012/2013 season may have been facilitated by a decline in the use of Bt cotton cultivars during this season due to problems with the quality of fibers produced. Since H. armigera was detected in 2013, the frequency and dosages of insecticide sprayings, including pyrethroids, dramatically increased (Durigan et al. 2017). Thus, the survival of early hybrids and backcrosses enabling introgression is likely related to the rapid scale-up of insecticide-based control measures in Brazil after the outbreak generated by the expansion of H. armigera. The observation of early generation hybrids mostly during the outbreak in 2013 and not in recent samples, as well as the selective sweep in early H. armigera around CYP337B3, further supports this hypothesis. Furthermore, our population genomic analyses demonstrate a signature of recent selection at the introgressed allele in H. zea that is not present in the background of the ancestral allele. Meanwhile, H. zea populations, which were susceptible to pyrethroids, seem to have responded to insecticide selection pressure by co-opting resistance from H. armigera through adaptive introgression.
The introgression of a gene involved in insecticide resistance from H. armigera into H. zea joins a series of animal examples of rapid adaptive introgression triggered by human-mediated alteration of fitness landscapes. These examples include the European house mouse, Simulium black flies, and Anopheles mosquitos whereby the increase in pesticide exposure acted as a selective force sufficient to drive introgression across species boundaries (Djogbénou et al. 2008; Adler et al. 2010; Song et al. 2011; Clarkson et al. 2014; Norris et al. 2015). Remarkably, in all these cases, adaptive introgression occurred from the species with a higher effective population size into the species with lower Ne and lower genetic diversity. This pattern has also been found in natural populations in which the selective agent is not human mediated. For instance, adaptive introgression of color pattern genes from Heliconius melpomene into H. timareta because of natural selection for mimicry (Pardo-Diaz et al. 2012; Edelman et al. 2019), tidal marsh adaptations from Ammospiza caudacauta into A. nelson sparrow (Walsh, Kovach, et al. 2018), and female-competitive traits from Jacana spinose into J. jacana (Lipshutz et al. 2019). This pattern is expected given that species with higher Ne harbor more standing variation—the raw material for adaptation—so introgression is less likely to add beneficial variants (Barton 2010; Jensen and Bachtrog 2011). Moreover, small populations have a higher mutational load because they are more prone to accumulate deleterious mutations, whereas large populations are less subjected to drift (Kimura and Ohta 1969; Barton 2010). However, the most famous example of adaptive introgression from Neanderthals and Denisovans into out-of-Africa human populations seems to be in the reverse direction, undermining the generality of this pattern and highlighting the bidirectional nature of introgression (Green et al. 2010; Patterson et al. 2012; Gazave et al. 2014; Huerta-Sánchez et al. 2014; Mafessoni and Prüfer 2017). Of note, Neanderthals were established in Eurasia when humans were expanding into Europe, they suffered from inbreeding depression (Harris and Nielsen 2016), and although less is known about introgression from humans to Neanderthals and Denisovans, there is evidence of gene flow in this direction (Green et al. 2010; Patterson et al. 2012; Huerta-Sánchez et al. 2014; Prüfer et al. 2014).
The most comprehensive genomic study of rapid adaptive introgression resulting from pesticide pressure is between Anopheles gambiae and An. coluzzi, the major malaria vectors in Africa (Clarkson et al. 2014; Fontaine et al. 2015; Norris et al. 2015). Several studies have shown introgression of mutations conferring resistance to pyrethroids from An. gambiae into An. coluzzi (Djogbénou et al. 2008; Clarkson et al. 2014; Norris et al. 2015). The CYP337B3 trickle from H. armigera into H. zea coincided with an increase in pesticide spraying, similar to introgression of insecticide resistance alleles in Anopheles overlapping with the start of a significant insecticide-treated bed net distribution campaign in Mali (Norris et al. 2015). After introgression of the insecticide-resistant genes, these alleles almost reached fixation in 4 years in An. coluzzi populations (Norris et al. 2015), which is a temporal scale that strikingly resembles introgression and near fixation of the CYP337B3 loci in H. zea in Brazil.
Despite the similitudes between Anopheles and Helicoverpa examples of rapid adaptive introgression, a critical difference relies on the speciation mechanism underlying the divergence between the sibling species. Anopheles gambiae and An. coluzzi diversified in sympatry only about 400,000 years ago (Clarkson et al. 2014; Fontaine et al. 2015), whereas H. armigera and H. zea differentiated in strict allopatry for 1.5 Ma and came into contact only in the recent 2013 invasion (Pearce et al. 2017; Anderson et al. 2018). Accordingly, the genomes of An. gambiae and An. coluzzi exhibit very low differentiation except for two large regions that show exceptional divergence known as “genomic islands” of diversification (Clarkson et al. 2014). The insecticide-resistant alleles are located within one of these major diverged regions, and its introgression resulted in the homogenization of the entire island, though no apparent impact on reproductive isolation (Clarkson et al. 2014). Conversely, in H. armigera and H. zea, the differentiation is not restricted to “islands” but rather, we observed consistently high FST values throughout the genome. Furthermore, incompatibilities are more likely to accumulate in allopatry, and therefore are expected to be more pervasive in Helicoverpa than in Anopheles sibling species (Bank et al. 2012). This suggests that in the face of intense selective pressure, adaptive variation can rapidly move across species boundaries even when differentiation and incompatibilities are pervasive across the genome, and not only between slightly differentiated species as in Anopheles mosquitos.
Asymmetric Introgression at the Front of an Invasion
We have primarily focused on the adaptive introgression of CYP337B3 from the invasive H. armigera into local H. zea even though we observed a higher effective migration of alleles from H. zea into H. armigera. This pattern is in agreement with theoretical models of secondary contact after invasions that account for demographic processes, which predict greater introgression of neutral genes from the local into the foreign species (Currat et al. 2008; Mesgaran et al. 2016; Quilodrán et al. 2019). This asymmetry is partly explained by a demographic imbalance between the two species at the wavefront, where the invading species is at lower densities (Currat et al. 2008; Hall 2016; Mesgaran et al. 2016). Therefore, it is likely that early invading H. armigera individuals had few available conspecific mates and thus were involved in more heterospecific crosses.
Most of the admixed alleles in the invasive H. armigera are likely to be neutral (Currat et al. 2008). However, introgression can also provide variants that have already been tested by natural selection and in precisely the geographic area in which they are already adapted (Martin and Jiggins 2017). Both models and empirical data show that genes from the resident species associated with local adaptation are easily introgressed into the invading species (Currat et al. 2008; Saarman and Pogson 2015). Contrastingly, genes from the invading species introgress into the local species only if they are under very strong positive selection in the local environment (Currat et al. 2008), as appears to be the case for the CYP337B3 gene. For genes with a large effect on fitness and a clear signal of admixture localized into a single region, it is reasonable to suggest that the increased frequency of the introgressed haplotypes was driven by strong positive selection. However, if introgression is pervasive, it is challenging to distinguish introgressed alleles that are beneficial from neutral genes that have also cross species boundaries. Although H. zea has developed resistance to two-toxin Bt maize and cotton (Cry1A and Cry2a) in North America (Reisig and Kurtz 2018), no single specific genes or mutations have yet been identified. Therefore, there are currently no plausible candidates for adaptive introgression from H. zea into H. armigera, nor has any Bt resistance been observed in H. zea in South America. Nonetheless, studies on gene flow between H. zea populations across North, Central, and South America would be valuable to understand the possible movement of resistance genes such as the CYP337B3 gene introgressed from H. armigera. Further temporal monitoring of the invasive H. armigera populations in Brazil will permit the detection of fine-scale selection on any introgressed alleles that may be identified.
Practical Implications of Rapid Evolution Triggered by Invasive Hybridization
The pervasive introgression between local H. zea and invasive H. armigera demonstrates the risks of human-mediated dispersion and selection. Invasive species are a major cause of crop loss and a global threat to food security (Blair and Hufbauer 2010; Paini et al. 2016). With increased geographic connectedness via trade, the threat of invasive species arriving in countries where they were previously absent is expected to increase (Saarman and Pogson 2015; Paini et al. 2016). In pest invasions, special attention has been given to newcomer species that can rapidly proliferate as they no longer face their natural enemies (Sakai et al. 2001; Clavero and García-Berthou 2005; Reisig and Kurtz 2018). However, given the evidence of bidirectional introgression following invasion, the importance of the local species should not be underestimated when developing effective biosecurity programs. Through introgression, local species can provide the invasive species with genes associated with local adaptation, thus contributing to its settlement. In addition, hybridization with the local species can reduce allee effects in the introduced species (Mesgaran et al. 2016). Indeed, there is compelling evidence that species that hybridize have a higher likelihood of successful establishment (Currat et al. 2008; Saarman and Pogson 2015; Mesgaran et al. 2016).
In addition to the human-mediated introduction of species, there is growing evidence that anthropogenically altered selection regimes drive exceptionally rapid adaptation (Song et al. 2011; Norris et al. 2015). Agricultural pests are subjected to massive insecticidal pressure, which drives selection for rapid development of resistance (Clarkson et al. 2014; Norris et al. 2015). Our observations suggest that increased insecticide spraying in Brazil to control H. armigera in the early stages of invasion acted as a selective force driving introgression of CYP337B3, by temporarily elevating the fitness of hybrids over that of the insecticide-susceptible H. zea. Furthermore, the selection regimes are rapidly changing in Brazil, and the widespread use of Bt-crops has considerably increased following the expansion of H. armigera. Although there is field-evolved resistance to some Bt toxins for H. zea in North America, this appears to result from a downregulation of midgut protease activity that reduces toxin activation and is associated with fitness costs (Moar et al. 2010; Brévault et al. 2015; Reisig and Kurtz 2018), so that, as noted above, it appears unlikely there will be any adaptive introgression of specific Bt-resistance-related genes into H. armigera, whose far greater genetic diversity already includes many specific resistance alleles with little or no fitness costs. In any case, the fate of the genes that cross species barriers will also be strongly affected by the demographic dynamics of these species which have substantially declined since 2012 along with other species of Noctuid moths (dos Santos et al. 2017; Piovesan et al. 2018; Fonseca-Medrano et al. 2019). These population declines seem to be driven by a large-scale selection regime linked to “El Niño-Southern Oscillation Effect” and other components of global change (dos Santos et al. 2017; Piovesan et al. 2018; Fonseca-Medrano et al. 2019). On a finer scale, invasive species substantially change native communities and ecosystems, and their expansion is often correlated with serious declines of closely related species (Bradley et al. 2019; Brzeziński et al. 2019; Larson et al. 2019). Understanding the fine and large-scale factors affecting these demographic changes may contribute to explain the variance in hybridization rates and the idiosyncratic responses to selection observed in each species. Thus, neglecting the interplay between selection due to local environmental heterogeneity in pesticide use and selection imposed by different components of global change can obscure the prediction of the species’ evolutionary responses and the long-term dynamics of hybridization in the sympatric populations of these Helicoverpa moths.
Hybridization and introgression represent serious risks associated with invasive species and understanding the mechanisms that determine their impact is necessary for the effective management of biological invasions. Recent advances in whole-genome sequencing give us a chance to monitor the rapid evolution of invasive and local species with important practical implications to design agricultural management programs. As whole-genome data accumulate for biological invasions, the consequences of invasive species in adaptive evolution can be systematically characterized.
Materials and Methods
Sample Collection and DNA Extraction
Helicoverpa armigera and H. zea moths were collected between 2012 and 2017 from three different localities in Brazil. The samples from 2012 and 2013 are referred to as “2013,” and the samples collected between 2016 and 2017 were treated as “2017” or recent populations. The Brazilian samples included 67 H. armigera, 27 H. zea, and 2 early hybrids closer to H. zea, from Bahia, Planaltina, and Mato Grosso states. As complementary groups for the admixture analyses, we included 4 H. zea preinvasion from 2006 in Brazil, 8 H. zea from the United States, 9 H. armigera armigera from Asia, Europe, and Africa, and 14 H. armigera conferta and 6 H. punctigera from Australia which are all considered to be unrelated to the current H. armigera invasion in Brazil. Sample collection data are listed in supplementary tables S1–S4, Supplementary Material online. All samples were preserved in ethanol and RNAlater or stored at −20 °C following collection. DNA was extracted using DNeasy blood and tissue kits (Qiagen) before quantification with a Qubit 2.0 fluorometer (Thermo Fisher Scientific).
Whole-Genome Resequencing and SNP Genotyping
Illumina libraries were produced following the manufacturer’s instructions, and 100-bp paired-end reads were generated (Illumina HiSeq. 2000, and Novogene, Biological Resources Facility, Australian National University, Australia). For BR samples, libraries were prepared an adapted tn5 transposase protocol (Picelli et al. 2014), and 150-bp paired-end sequences were generated (Illumina HiSeq Xten, BGI, China) (Picelli et al. 2014). Raw sequence reads were aligned to the H. armigera genome using BWA v 0.7.12. Reads were trimmed when quality in at least two bases fell below Q30 using VCFTOOLS v. 0.1.15, and only uniquely aligning reads were included in the analysis. Resulting BAM files were sorted before duplicate reads were annotated using Picard v. 1.138 and indexed with SAMTOOLS v. 1.3.1 (Li et al. 2009). HaplotypeCaller in GATK v. 3.7 (McKenna et al. 2010) was used to call SNPs in individual samples, and we used GenotypeGVCFs in GATK (McKenna et al. 2010) to estimate genotypes across all individuals simultaneously, implementing a heterozygosity value of 0.01. We used VCFTOOLS v. 0.1.15 (Danecek et al. 2011) to calculate mean coverage statistics for each sample. We further filtered our SNP data set to include a minor allele count 2, mean site depth 5 and 200, missing data per site 0.50, and Phred-scale mapping quality 30, resulting in 17,257,450 genome-wide biallelic SNPs for the 150 individuals.
Classification of Parental and Hybrids Individuals
We conducted a genetic PCA and discriminant analysis of principal components to visualize clustering of parental and hybrid genotypes using ∼50,000 SNP markers randomly sampled across all chromosomes with the packages “adegenet” (Jombart 2008), “Poppr” (Kamvar et al. 2014), and “vcfR” (Knaus and Grünwald 2017) in R v.3.3.2.
We further classified individuals as parental H. zea and H. armigera, hybrids or backcrosses based on the interspecific heterozygosity and the proportion of ancestry from parental species. We estimated the proportion of the genome that was ancestrally derived from H. zea using SNPs that strongly segregated between that species and H. armigera armigera. These diagnostic SNPs were determined via an association test in PLINK v1.90b (Purcell et al. 2007), taking only variants where P value < 3.83 × 10−14, thereby limiting the maximum number of alleles to fixed alleles from either H. zea or H. armigera. We used only preincursion allopatric populations of H. zea from Brazil and the United States and H. armigera armigera from the Old World for the association test resulting in ∼1,000,000 ancestry-informative SNPs. We calculated the proportion of ancestry and interspecific heterozygosity for the individuals using custom python scripts considering the sum of H. zea alleles divided by all alleles and the proportion of heterozygous sites (https://github.com/wvalencia-montoya/helicoverpa-project, last accessed April 29, 2020).
Genomic Differentiation between and within Species
To estimate genome-wide differentiation, we calculated FST using the popgenWindows.py script from the repository (https://github.com/simonhmartin/genomics_general, last accessed April 29, 2020) in 100-kb nonoverlapping windows. We calculated FST between allopatric populations of H. zea and H. armigera and sympatric postincursion populations for the two temporal samples 2013 and 2017. To assess outliers, we calculated Z-scores and −log10P values. In order to test for differences between populations, we used a permutation-based independence test implemented in the R package, coin (Hothorn et al. 2008).
Phylogenetic Weighting
We used Twisst (Martin and Van Belleghem 2017) to explore evolutionary relationships across the genome of allopatric and sympatric recent populations of H. zea and H. armigera. We performed phasing and imputation of missing bases using Beagle (Browning and Browning 2007) with 10,000-bp step size and 100-kb overlapping sliding windows. Maximum likelihood trees for Twisst were generated with the phyml_sliding_window.py script (Martin and Van Belleghem 2017) with the GTR model in 50 SNPs sliding windows in PhyML (Guindon et al. 2010).
Introgression across the Genome
To test for the genome-wide extent of admixture resulting from hybridization between H. armigera and H. zea, we performed the D-statistic test implemented in AdmixTools v. 5.1 with a block jackknife size of 61 bp (Patterson et al. 2012). We considered that D-statistic estimates were indicative of significant introgression when Z-scores 2.5, and thus with a P value of ≤0.05. To estimate the genome-wide proportion of admixture, we conducted an F4 ratio incorporating H. armigera conferta from Australia as the fifth population (description of the model in supplementary table S2, Supplementary Material online) and using AdmixTools v. 5.1 with a block jackknife size of 61 bp (Patterson et al. 2012).
For H. armigera-like individuals, we implemented a model phylogenetic model with the following branching: (((Old World H. armigera, Brazilian H. armigera Date 1 or Date 2) North American H. zea) H. punctigera) and (((Old World H. armigera, Australian H. armigera conferta) North American H. zea) H. punctigera) as a control (supplementary fig. S1, Supplementary Material online). For H. zea populations, we considered the model (((North American H. zea, Brazilian H. zea Date 1 or Date 2) Old World H. armigera), H. punctigera). To detect introgression in specific regions across the genome, we calculated fd in 100-kb sliding windows with the ABBA-BABA.py scripts as in Martin et al. (2015) and implementing the same topology models used for the D-statistic.
To further characterize introgressed regions from either parental species across the genome of individuals, we calculated the proportion of ancestry in sliding windows, using custom python scripts. The calculations incorporated the set of SNPs ancestry-informative SNPs with hybrid index averages across 100-kb nonoverlapping windows. Following Anderson et al. (2018), we considered a window as significantly introgressed with a P value threshold <5 × 10−7. Differences in block length per date were evaluated using permutation tests. Sliding windows averages of the proportion of ancestry were plotted using custom R scripts, and using “ggplot2” (Wickham 2009), “Hmisc” (Harrell and Dupont 2019), and “Rmisc” (Hope 2013) packages.
To test if the change over time of the block length is faster than would be expected due to neutral recombination, we calculated the expected ancestry block size using a formula from (Racimo et al. 2015). Because of recent introgression, the lengths of admixed tracts may be highly correlated, violating the assumption of exponentially distributed block sizes (Liang and Nielsen 2014). However, because there is no certainty about the time of the invasion, we used this framework as an approximation to a null expectation under neutrality. We assumed nine generations per year (Walsh TK, personal communication), 4 years since admixture (from 2013 to 2017), and the admixture proportions calculated from the F4-ratio tests (fig. 5 and supplementary table S2, Supplementary Material online). To estimate local recombination rate, we generated a consensus haplotype based on the phased data set of the nonadmixed H. armigera individuals, using SAMTOOLS v. 1.3.1 (Li et al. 2009) and FastaAlternateReferenceMaker within GATK v. 3.7 (McKenna et al. 2010). We inferred the population recombination parameter using LDhlemet (Chan et al. 2012). By incorporating the recombination rate, the initial admixture proportion, and the generation time, we performed 10,000 simulations of the distribution of the admixture tract length using the parameters obtained from (Racimo et al. 2015) equation and compared with the observed distribution using independence permutation tests (Hothorn et al. 2008). Furthermore, we run two additional sets of simulations varying generation time to account for realistic variation in this parameter in warmer (up to 12 generations) and cooler climates (3 generations including diapause). We did not infer a population recombination parameter for H. zea because of the lack of a high-quality, chromosome-assemblage reference genome.
To characterize linkage disequilibrium, we included parsimony SNPs, thinning the phased data to about 25% of its original size using the software package MapThin (http://www.staff.ncl.ac.uk/richard.howey/mapthin/, last accessed April 29, 2020). We used PLINK v1.90b (Purcell et al. 2007) to calculate r2 within 1-kb windows for H. armigera and within 2-kb windows for H. zea because disequilibrium decayed at a slower rate (Seymour et al. 2016). To test for differences in the extent of LD between temporal samples, we applied a two-sided Wilcoxon signed-rank test using the mean differences across 10-bp windows.
To confirm introgression of CYP337B3, all individuals of H. zea 2017 were screened for the presence of this gene as in Joußen et al. (2012). Additionally, we mapped reads of recent individuals of H. zea to the complete coding sequence of CYP337B3 and confirmed presence with the Integrative Genome Viewer IGV_2.4.14 (Robinson et al. 2011).
Tajima’s D
Tajima’s D was calculated in sliding windows of 10 kb in VCFTOOLS v. 0.1.15 (Danecek et al. 2011). We computed mean and standard deviation of Tajima’s D values for the Brazilian populations: H. armigera (2013), H. armigera (2017), H. zea (2013), and H. zea (2017) including individuals with the CYP337B3 haplotype and H. zea (2017), following Norris et al. (2015). We plotted Tajima’s D using ggplot and ggalt along a region in chromosome 15, whereby CYP337B3 gene extends from ∼11,436,000 to 11,440,000 bp.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Simon H. Martin, who provided key theoretical and practical advice on the early stages of this study and Hilde Schneemman, Alejandra Duque, Federico Tamayo, and Bregje Wertheim, who made insightful comments on the manuscript. Gabriela Montejo–Kovacevich provided useful introductory scripts for efficient mapping. We are also grateful to the cotton farmers in Western Bahia for their support during sampling collections and ICMBio and MMA for the Authorizations for Scientific Activities (SISBIO Nos. 48218-3 and 38547/6). This work was supported by the Royal Society Exchange (Grant No. IES\R3\170415) awarded to C.D.J. and S.E. and the EMBO short-term fellowship (ASTF-6889) awarded to S.E. T.K.W., W.T.T., K.H.J.G., and S.E. were supported by the CSIRO H&B Genes of Biosecurity Importance Fund (R-8681-1). W.A.V-M. and H.L.N. were supported by the European Union through the Erasmus Mundus Master in Evolutionary Biology (MEME 2017-2019). We are grateful to the Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq for A.S. fellowship (process number 306601/2016-8) and research fund (process number 403376/2013-0) and Empresa Brasileira de Pesquisa Agropecuária—Embrapa (SEG MP2 No. 02.13.14.006.00.00). We are also grateful to Juanita Valencia for her support.
References
- Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R, et al. 2013. Hybridization and speciation. J Evol Biol. 26(2):229–246. [DOI] [PubMed] [Google Scholar]
- Adler PH, Cheke RA, Post RJ.. 2010. Evolution, epidemiology, and population genetics of black flies (Diptera: Simuliidae). Infect Genet Evol. 10(7):846–865. [DOI] [PubMed] [Google Scholar]
- Alexander DH, Lange K.. 2011. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics 12(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Oakeshott JG, Tay WT, Gordon KHJ, Zwick A, Walsh TK.. 2018. Hybridization and gene flow in the mega-pest lineage of moth, Helicoverpa. Proc Natl Acad Sci U S A. 115(19):5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Tay WT, McGaughran A, Gordon K, Walsh TK.. 2016. Population structure and gene flow in the global pest, Helicoverpa armigera. Mol Ecol. 25(21):5296–5311. [DOI] [PubMed] [Google Scholar]
- Arnemann JA, Roxburgh SH, Walsh T, Guedes JVC, Gordon KHJ, Smagghe G, Tay WT.. 2019. Multiple incursion pathways for Helicoverpa armigera in Brazil show its genetic diversity spreading in a connected world. Sci Rep. 9(1):19380. [DOI] [PMC free article] [PubMed]
- Bank C, Bürger R, Hermisson J.. 2012. The limits to parapatric speciation: Dobzhansky–Muller incompatibilities in a continent–island model. Genetics 191(3):845–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton N. 2010. Understanding adaptation in large populations. PLoS Genet. 6: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, Hewitt GM.. 1985. Analysis of hybrid zones. Annu Rev Ecol Syst. 16(1):113–148. [Google Scholar]
- Behere GT, Tay WT, Russell DA, Heckel DG, Appleton BR, Kranthi KR, Batterham P.. 2007. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. zea. BMC Evol Biol. 7(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg B, Zhao X-C, Wang G.. 2014. Processing of pheromone information in related species of heliothine moths. Insects 5(4):742–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair AC, Hufbauer RA.. 2010. Hybridization and invasion: one of North America’s most devastating invasive plants shows evidence for a history of interspecific hybridization. Evol Appl. 3(1):40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Lempe J, Epple P, Warthmann N, Lanz C, Dangl JL, Weigel D.. 2007. Autoimmune response as a mechanism for a Dobzhansky–Muller-type incompatibility syndrome in plants. PLoS Biol. 5(9):e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley BA, Laginhas BB, Whitlock R, Allen JM, Bates AE, Bernatchez G, Diez JM, Early R, Lenoir J, Vilà M, et al. 2019. Disentangling the abundance–impact relationship for invasive species. Proc Natl Acad Sci U S A. 116(20):9919–9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brévault T, Tabashnik BE, Carrière Y.. 2015. A seed mixture increases dominance of resistance to Bt cotton in Helicoverpa zea. Sci Rep. 5(1):9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL.. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 81(5):1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzeziński M, Żmihorski M, Zarzycka A, Zalewski A.. 2019. Expansion and population dynamics of a non-native invasive species: the 40-year history of American mink colonisation of Poland. Biol Invasions 21(2):531–545. [Google Scholar]
- Chan AH, Jenkins PA, Song YS.. 2012. Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLoS Genet. 8(12):e1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson CS, Weetman D, Essandoh J, Yawson AE, Maslen G, Manske M, Field SG, Webster M, Antão T, MacInnis B, et al. 2014. Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nat Commun. 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavero M, García-Berthou E.. 2005. Invasive species are a leading cause of animal extinctions. Trends Ecol Evol. 20(3):110. [DOI] [PubMed] [Google Scholar]
- Currat M, Ruedi M, Petit RJ, Excoffier L.. 2008. The hidden side of invasions: massive introgression by local genes. Evolution 62:1908–1920. [DOI] [PubMed] [Google Scholar]
- Czepak C, Albernaz KC, Vivan LM, Guimarães HO, Carvalhais T.. 2013. Primeiro registro de ocorrência de Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) no Brasil. Pesqui Agropecu Trop. 43(1):110–113. [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermauw W, Pym A, Bass C, Van TL, Feyereisen R.. 2018. Does host plant adaptation lead to pesticide resistance in generalist herbivores? Curr Opin Insect Sci. 26:25–33. [DOI] [PubMed] [Google Scholar]
- Djogbénou L, Chandre F, Berthomieu A, Dabiré R, Koffi A, Alout H, Weill M.. 2008. Evidence of introgression of the ace-1R mutation and of the ace-1 duplication in West African Anopheles gambiae s. s. PLoS One 3(5):e2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos SR, Specht A, Carneiro E, Paula-Moraes S. V D, Casagrande MM.. 2017. Interseasonal variation of Chrysodeixis includens (Walker, [1858]) (Lepidoptera: Noctuidae) populations in the Brazilian Savanna. Rev Bras Entomol. 61(4):294–299. [Google Scholar]
- Durigan MR, Corrêa AS, Pereira RM, Leite NA, Amado D, de Sousa DR, Omoto C.. 2017. High frequency of CYP337B3 gene associated with control failures of Helicoverpa armigera with pyrethroid insecticides in Brazil. Pestic Biochem Physiol. 143:73–80. [DOI] [PubMed] [Google Scholar]
- Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, García-Accinelli G, Belleghem SMV, Patterson N, Neafsey DE, et al. 2019. Genomic architecture and introgression shape a butterfly radiation. Science 366(6465):594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadamiro HY, Baker TC.. 1997. Helicoverpa zea males (Lepidoptera: Noctuidae) respond to the intermittent fine structure of their sex pheromone plume and an antagonist in a flight tunnel. Physiol Entomol. 22(4):316–324. [Google Scholar]
- Fonseca-Medrano M, Specht A, Silva FAM, Otanásio PN, Malaquias JV.. 2019. The population dynamics of three polyphagous owlet moths (Lepidoptera: Noctuidae) and the influence of meteorological factors and ENSO on them. Rev Bras Entomol.63(4):308–315. [Google Scholar]
- Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, Jiang X, Hall AB, Catteruccia F, Kakani E, et al. 2015. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347(6217):1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazave E, Ma L, Chang D, Coventry A, Gao F, Muzny D, Boerwinkle E, Gibbs RA, Sing CF, Clark AG, et al. 2014. Neutral genomic regions refine models of recent rapid human population growth. Proc Natl Acad Sci U S A. 111(2):757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves RM, Mastrangelo T, Rodrigues JCV, Paulo DF, Omoto C, Corrêa AS, Azeredo-Espin AML.. 2019. Invasion origin, rapid population expansion, and the lack of genetic structure of cotton bollworm (Helicoverpa armigera) in the Americas. Ecol Evol. 9(13):7378–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MHY, et al. 2010. A draft sequence of the Neandertal genome. Science 328(5979):710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O.. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321. [DOI] [PubMed] [Google Scholar]
- Hall RJ. 2016. Hybridization helps colonizers become conquerors. Proc Natl Acad Sci U S A. 113(36):9963–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick DF. 1965. The corn earworm complex. Mem Entomol Soc Can. 97(40):1–247. [Google Scholar]
- Harrell FE, Jr, Dupont C. 2019. Hmisc: Harrell miscellaneous. Available from: https://CRAN.R-project.org/package=Hmisc.
- Harris K, Nielsen R.. 2016. The genetic cost of Neanderthal introgression. Genetics 203(2):881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope RM. 2013. Rmisc: Ryan miscellaneous. Available from: https://CRAN.R-project.org/package=Rmisc.
- Hothorn T, Hornik K, Wiel MA, van de Zeileis A.. 2008. Implementing a class of permutation tests: the coin package. J Stat Softw. 28(8):1–23.27774042 [Google Scholar]
- Huerta-Sánchez E, Jin X, Asan Bianba Z, Peter BM, Vinckenbosch N, Liang Y, Yi X, He M, Somel M, et al. 2014. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512:194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Edge MD, Rosenberg NA.. 2013. The relationship between FST and the frequency of the most frequent allele. Genetics 193(2):515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Bachtrog D.. 2011. Characterizing the influence of effective population size on the rate of adaptation: Gillespie’s Darwin domain. Genome Biol Evol. 3:687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405. [DOI] [PubMed] [Google Scholar]
- Joußen N, Agnolet S, Lorenz S, Schöne SE, Ellinger R, Schneider B, Heckel DG.. 2012. Resistance of Australian Helicoverpa armigera to fenvalerate is due to the chimeric P450 enzyme CYP337B3 Proc Natl Acad Sci U S A. 109(38):15206–15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamvar ZN, Tabima JF, Grünwald NJ.. 2014. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ohta T.. 1969. The average number of generations until fixation of a mutant gene in a finite population. Genetics 61:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus BJ, Grünwald NJ.. 2017. vcfr: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour. 17(1):44–53. [DOI] [PubMed] [Google Scholar]
- Larson ER, Kreps TA, Peters B, Peters JA, Lodge DM.. 2019. Habitat explains patterns of population decline for an invasive crayfish. Ecology 100(5):e02659. [DOI] [PubMed] [Google Scholar]
- Laster ML, Sheng CF.. 1995. Search for hybrid sterility for Helicoverpa zea in crosses between the North American H. zea and H. armigera (Lepidoptera: Noctuidae) from China. J Econ Entomol. 88(5):1288–1291. [Google Scholar]
- Lee H-Y, Chou J-Y, Cheong L, Chang N-H, Yang S-Y, Leu J-Y.. 2008. Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 135(6):1065–1073. [DOI] [PubMed] [Google Scholar]
- Leite NA, Alves-Pereira A, Corrêa AS, Zucchi MI, Omoto C.. 2014. Demographics and genetic variability of the new world bollworm (Helicoverpa zea) and the Old World bollworm (Helicoverpa armigera) in Brazil. PLoS One 9(11):e113286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Nielsen R.. 2014. The lengths of admixture tracts. Genetics 197(3):953–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindtke D, Buerkle CA.. 2015. The genetic architecture of hybrid incompatibilities and their effect on barriers to introgression in secondary contact. Evolution 69(8):1987–2004. [DOI] [PubMed] [Google Scholar]
- Lipshutz SE, Meier JI, Derryberry GE, Miller MJ, Seehausen O, Derryberry EP.. 2019. Differential introgression of a female competitive trait in a hybrid zone between sex‐role reversed species. Evolution 73(2):188–201. [DOI] [PubMed] [Google Scholar]
- Lopes-da-Silva M, Sanches MM, Stancioli AR, Alves G, Sugayama R.. 2014. The role of natural and human-mediated pathways for invasive agricultural pests: a historical analysis of cases from Brazil. Agric Sci. 5(7):634–646. [Google Scholar]
- Mafessoni F, Prüfer K.. 2017. Better support for a small effective population size of Neandertals and a long shared history of Neandertals and Denisovans. Proc Natl Acad Sci U S A. 114(48):E10256–E10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon RJ, Olsen KM, Garsia KA, Young SR.. 2007. Resistance to Bacillus thuringiensis toxin Cry2Ab in a strain of Helicoverpa armigera (Lepidoptera: Noctuidae) in Australia. J Econ Entomol. 100(3):894–902. [DOI] [PubMed] [Google Scholar]
- Mallet J. 2018. Invasive insect hybridizes with local pests. Proc Natl Acad Sci U S A. 115(19):4819–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, Korman A, Heckel DG, King P.. 1993. Biochemical genetics of Heliothis and Helicoverpa (Lepidoptera: Noctuidae) and evidence for a founder event in Helicoverpa zea. Ann Entomol Soc Am. 86(2):189–197. [Google Scholar]
- Martin SH, Davey JW, Jiggins CD.. 2015. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol Biol Evol. 32(1):244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Davey JW, Salazar C, Jiggins CD.. 2019. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLoS Biol. 17(2):e2006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Jiggins CD.. 2017. Interpreting the genomic landscape of introgression. Curr Opin Genet Dev. 47:69–74. [DOI] [PubMed] [Google Scholar]
- Martin SH, Van Belleghem SM.. 2017. Exploring evolutionary relationships across the genome using topology weighting. Genetics 206(1):429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffery AR. 1998. Resistance to insecticides in Heliothine Lepidoptera: a global view. Philos Trans R Soc Lond Ser B Biol Sci. 353(1376):1735–1750. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesgaran MB, Lewis MA, Ades PK, Donohue K, Ohadi S, Li C, Cousens RD.. 2016. Hybridization can facilitate species invasions, even without enhancing local adaptation. Proc Natl Acad Sci U S A. 113(36):10210–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moar W, Dennehy T, Anilkumar K, Head G.. 2010. Bt resistance in Helicoverpa zea (Boddie): from biology to monitoring. Southwest Entomol. 35(3):395–398. [Google Scholar]
- Mooney HA, Cleland EE.. 2001. The evolutionary impact of invasive species. Proc Natl Acad Sci U S A. 98(10):5446–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ, Lanzaro GC.. 2015. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc Natl Acad Sci U S A. 112(3):815–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paini DR, Sheppard AW, Cook DC, De Barro PJ, Worner SP, Thomas MB.. 2016. Global threat to agriculture from invasive species. Proc Natl Acad Sci U S A. 113(27):7575–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, McMillan WO, Jiggins CD.. 2012. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 8(6):e1002752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D.. 2012. Ancient admixture in human history. Genetics 192(3):1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce SL, Clarke DF, East PD, Elfekih S, Gordon KHJ, Jermiin LS, McGaughran A, Oakeshott JG, Papanikolaou A, Perera OP, et al. 2017. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biol. 15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Reinius B, Sagasser S, Winberg G, Sandberg R.. 2014. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24(12):2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto FA, Mattos MVV, Silva FWS, Rocha SL, Elliot SL.. 2017. The spread of Helicoverpa armigera (Lepidoptera: Noctuidae) and coexistence with Helicoverpa zea in southeastern Brazil. Insects 8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan M, Specht A, Carneiro E, Paula-Moraes SV, Casagrande MM.. 2018. Phenological patterns of Spodoptera Guenée, 1852 (Lepidoptera: Noctuidae) is more affected by ENSO than seasonal factors and host plant availability in a Brazilian Savanna. Int J Biometeorol. 62(3):413–422. [DOI] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505(7481):43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quero C, Baker TC.. 1999. Antagonistic effect of (Z)-11-hexadecen-1-ol on the pheromone-mediated flight of Helicoverpa zea (Boddie) (Lepidoptera: Noctuidae). J Insect Behav. 12(5):701–710. [Google Scholar]
- Quero C, Fadamiro HY, Baker TC.. 2001. Responses of male Helicoverpa zea to single pulses of sex pheromone and behavioural antagonist. Physiol Entomol. 26(2):106–115. [Google Scholar]
- Quilodrán CS, Nussberger B, Montoya‐Burgos JI, Currat M.. 2019. Hybridization and introgression during density-dependent range expansion: European wildcats as a case study. Evolution 73(4):750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E.. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 16(6):359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisig DD, Kurtz R.. 2018. Bt resistance implications for Helicoverpa zea (Lepidoptera: Noctuidae) insecticide resistance management in the United States. Environ Entomol. 47(6):1357–1364. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP.. 2011. Integrative genomics viewer. Nat Biotechnol. 29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarman NP, Pogson GH.. 2015. Introgression between invasive and native blue mussels (genus Mytilus) in the central California hybrid zone. Mol Ecol. 24(18):4723–4738. [DOI] [PubMed] [Google Scholar]
- Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, Baughman S, Cabin RJ, Cohen JE, Ellstrand NC, et al. 2001. The population biology of invasive species. Annu Rev Ecol Syst. 32(1):305–332. [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG, et al. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360(6389):656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour M, Perera OP, Fescemyer HW, Jackson RE, Fleischer SJ, Abel CA.. 2016. Peripheral genetic structure of Helicoverpa zea indicates asymmetrical panmixia. Ecol Evol. 6(10):3198–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Endepols S, Klemann N, Richter D, Matuschka F-R, Shih C-H, Nachman MW, Kohn MH.. 2011. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between Old World mice. Curr Biol. 21(15):1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Gómez DR, Specht A, Paula-Moraes SV, Lopes-Lima A, Yano SAC, Micheli A, Morais EGF, Gallo P, Pereira P, Salvadori JR, et al. 2016. Timeline and geographical distribution of Helicoverpa armigera (Hübner) (Lepidoptera, Noctuidae: Heliothinae) in Brazil. Rev Bras Entomol. 60(1):101–104. [Google Scholar]
- Tay WT, Gordon K.. 2019. Going global—genomic insights into insect invasions. Curr Opin Insect Sci. 31:123–130. [DOI] [PubMed] [Google Scholar]
- Tay WT, Mahon RJ, Heckel DG, Walsh TK, Downes S, James WJ, Lee S-F, Reineke A, Williams AK, Gordon K.. 2015. Insect resistance to Bacillus thuringiensis toxin Cry2Ab is conferred by mutations in an ABC transporter subfamily A protein. PLoS Genet. 11(11):e1005534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay WT, Soria MF, Walsh T, Thomazoni D, Silvie P, Behere GT, Anderson C, Downes S.. 2013. A brave new world for an Old World pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil PLoS One 8: e80134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay WT, Walsh TK, Downes S, Anderson C, Jermiin LS, Wong TKF, Piper MC, Chang ES, Macedo IB, Czepak C, et al. 2017. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Sci Rep. 7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J, Kovach AI, Olsen BJ, Shriver WG, Lovette IJ.. 2018. Bidirectional adaptive introgression between two ecologically divergent sparrow species. Evolution 72(10):2076–2089. [DOI] [PubMed] [Google Scholar]
- Walsh JN, Joussen N, Tian K, McGaughran A, Anderson CJ, Qiu X, Ahn S-J, Bird L, Pavlidi N, Vontas J, et al. 2018. Multiple recombination events between two cytochrome P450 loci contribute to global pyrethroid resistance in Helicoverpa armigera. PLoS One 13:e0197760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag. [Google Scholar]
- Xu X, Yu L, Wu Y.. 2005. Disruption of a cadherin gene associated with resistance to Cry1Ac {delta}-endotoxin of Bacillus thuringiensis in Helicoverpa armigera. Appl Environ Microbiol. 71(2):948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.