

Abstract
Cystic Fibrosis (CF), an inherited multi‐system disease, is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) that disrupt its ability to secrete anions from epithelia. Recovery of functional anion secretion may be curative for CF, so different components of the ion transport machinery have become attractive therapeutic targets. Several members of the SLC26 ion transporter family have been linked to epithelial ion flux, some through putative functional interactions with CFTR. Using a small‐scale qPCR screen, we confirmed that the anion transporter SLC26A4 (pendrin) is downregulated in CF. Upregulation of pendrin using interleukins IL‐4 or IL‐13 increased Cl− secretion through CFTR in human bronchial epithelial cell (HBEC) derived epithelia differentiated in vitro and measured in the Ussing Chamber. Inhibition or knockdown of pendrin prevented this increased secretion. Increased CFTR activity was not driven by increases in CFTR protein or upstream regulatory pathway components. When basolateral Cl− absorption through NKCC1 was inhibited, a pendrin‐dependent Cl− absorption pathway allowing CFTR to continue secreting Cl− from the epithelium was revealed. Although CFTR is often considered the bottleneck in the transepithelial Cl− transport pathway, these studies indicate that basolateral Cl− permeability becomes limiting as CFTR activity increases. Therefore, an increase of epithelial Cl− absorption via pendrin might have additional therapeutic benefit in combination with CFTR modulators.
Keywords: electrophysiology, epithelium, HBE
1. INTRODUCTION
Cystic Fibrosis (CF) is an inherited multi‐system disease with approximately 70,000 patients worldwide. 1 CF is characterized by cyst formation and eventual destruction of the pancreas, obstruction and malabsorption in the intestines, salty sweat, and chronic bacterial lung infections. Following advances in the treatment of gastrointestinal pathology, a decrease in lung function—driven by lung damage sustained due to infection and inflammation in the lung—is the primary cause of mortality. 2 The function of the lung epithelium is defective in CF, allowing opportunistic bacteria to colonize the airways. 3 This leads to chronic infection and inflammation, scarring and damage to the lung tissue, and a decline in lung function. The immune cells in the lung may also be impacted by the disease. 4
Mutations in the cystic fibrosis transepithelial conductance regulator (CFTR) cause CF. 5 CFTR is a member of the ABCC transporter family but encodes an anion permeant ion channel gated by ATP and phosphorylation. 6 , 7 , 8 Functional studies have demonstrated that chloride (Cl−) and bicarbonate (HCO3 −) are the primary anions which permeate the CFTR channel. 6 , 9 In cells, CFTR couples the binding of ATP with coincident phosphorylation by protein kinase A (PKA) at multiple amino acid residues to open its channel gate and permit ion permeation. 10 , 11 , 12 Since the cloning of CFTR, nearly 2000 different mutations in CFTR have been identified in CF patients. 13 However, most patients have at least one allele containing the ΔF508 mutation, a deletion of 3 nucleotide bases which results in the loss of phenylalanine at position 508. This mutation causes protein folding, interdomain assembly, stability, and functional defects leaving homozygous patients with less than 2% of normal CFTR function. 14 , 15 , 16 , 17
When studied both in vivo and in vitro, loss of CFTR virtually eliminates epithelial secretion of Cl− and HCO3 − anions, even when residual CFTR function is maximally stimulated via PKA activation. Differentiated airway epithelial cells from CF donors exhibit reduced apical anion secretion, substantially dehydrated airway surface liquid (ASL), reduced ciliary beat frequency (CBF), and highly viscous mucus. 18 , 19 The loss of apical Cl− secretion and concomitant increase in Na+ absorption (likely due to changes in apical transmembrane voltage) dehydrate the ASL. In support of this hypothesis, hypertonic saline transiently improves lung function in CF patients. 19 , 20 Additionally, HCO3 − may play a role in maintaining proper mucus viscosity, and exogenous addition of HCO3 − modifies ciliary beating in vitro. 21 Therefore, an increase in CFTR function should increase anion secretion, providing substantial therapeutic benefit to CF patients. In fact, the approved CFTR‐directed therapeutics lumacaftor and ivacaftor improve CFTR trafficking and function, respectively, providing significant improvement in lung function in CF patients. 22 , 23 The newer generation of CFTR trafficking correctors tezacaftor and elexacaftor produce even large impacts on lung function, especially when elexacaftor is combined with the first generation modulators. 24 Additional therapeutic strategies target other mutant forms of CFTR 25 and studies underline the importance of CFTR interactors 26 and other apical Cl− secretion pathways. 27 Given these observations, apical Cl− secretion is usually considered to be the bottleneck in transepithelial Cl− transport, while the higher capacity NKCC1‐based basolateral absorption of Cl− is not thought to be limiting.
CFTR may regulate or functionally interact with other ion transporters. 28 , 29 , 30 Pendrin (aka. SLC26A4) is an anion exchanger found in multiple tissues whose expression is upregulated by interleukin signaling pathways. 31 , 32 Mutations in pendrin cause Pendred syndrome, a disease characterized by deafness and thyroid goiter, and are associated with enlarged vestibular aqueducts. 33 , 34 In the pancreas and thyroid, pendrin's primary function is to exchange bicarbonate for iodide. However, pendrin is also capable of exchanging bicarbonate for chloride. 35 A recent study identified pendrin as a potential modifier of CFTR function. 36 We hypothesized that pendrin expressed in the airway might increase epithelium Cl− secretion through CFTR by increasing basolateral Cl− absorption. As such, pendrin activation alone or in combination with CFTR‐directed modulators could be a therapeutic strategy for the treatment of CF.
2. MATERIALS AND METHODS
2.1. Reagents
Unless otherwise indicated, all reagents (including IL‐4, IL‐13, IL‐17a, benzamil, bumetanide, forskolin, cftr(inh)‐172, niflumic acid, and components of solutions) were purchased from Sigma. Lumacaftor, ivacaftor, and elexacaftor were purchased from Selleck Chemicals (Houston, TX, USA).
2.2. Cell culture
Human bronchial epithelial cells from ΔF508/ΔF508 Cystic Fibrosis patients (CF HBECs) and non‐CF donors (normal HBECs) were purchased from The University of North Carolina Cystic Fibrosis Tissue Procurement and Cell Culture Core. HBECs were thawed into T75 Collagen I coated flasks (Corning, Corning, NY, USA) at 2.5 × 105 cells/flask in BEGM growth medium (Lonza, Walkersville, MD, USA) supplemented with 100 U Antibiotic/Antimycotic (Invitrogen/ThermoFisher, Waltham, MA, USA), 10 nmol/L Retinoic Acid, and 10 µmol/L Y‐27632 Rock inhibitor (Roche, Rotkreuz, Switzerland) and grown for 7 days at 37OC in a 5% CO2 incubator (Eppendorf, Hauppage, NY, USA). HBECs were then expanded into T150 Collagen I coated flasks (Corning) at 1 × 106 cells/flask for 7 days. HBECs were collected and plated onto Snapwell 6‐well inserts (Corning) at 5 x 105 cells per well in VALI differentiation medium (AthenaES, Bethesda, MD, USA) consisting of DMEM, 2% Hyclone Fetal clone II, 0.25% Bovine brain extract (Lonza), 10 nmol/L Retinoic acid, 2.5 µg/mL Insulin, 1.5 µmol/L Epinephrine, 250 nmol/L Phosphoethanolamine, 20 nmol/L Hydrocortisone, 2.5 µg/mL Human Transferrin, 500 nmol/L Triiodothyronine, 250 nmol/L Ethanolamine, 0.5% Ultorser G (Pall, Saint‐Germain‐en‐Laye, France), and 100 U Antibiotic/Antimycotic (Invitrogen) and cultured for 4 to 5 weeks at 37°C in a 5% CO2 incubator prior to use in all experiments, with the exception of siRNA experiments as indicated. Fisher Rat Thyroid (FRT) cells expressing CFTRΔF508 were cultured in Coon's F12 media (Biochrom GmbH, Berlin, Germany) with 10% FBS (Invitrogen) as previously described. 37 FRTs were collected and plated onto Snapwell 6‐well inserts (Corning) at 5 × 105 cells per well in Coon's F12 and cultured under media for 7 to 10 days at 37°C in a 5% CO2 incubator.
2.3. RT‐PCR
HBECs from ΔF508/ΔF508 CF patients and non‐CF normal donors were grown as indicated above. The monolayers on the apical side of the inserts were quickly rinsed with warm calcium‐magnesium free DPBS (Invitrogen). After aspirating the DPBS, cells were placed in fresh DPBS on both the apical and basolateral sides of the insert and incubated for 5 minutes to disrupt the tight junctions. The DPBS on the apical side was aspirated and 0.25% Trypsin with EDTA (Invitrogen) was added and incubated for 15 minutes at 37°C. Extraction of the cells was performed by vigorously pipetting up and down. The cells were pelleted and RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacture's protocol. 500 ng of RNA was synthesized into cDNA using the RT2 First Strand Kit (Qiagen) according to the manufacturer's instructions. A custom RT2 PCR array (Qiagen) containing CFTR, ANO1, SCNN1A, SCNN1B, SCNN1G, SLC4A1, SLC4A2, SLC4A3, SLC4A8, SLC4A10, SLC12A1, SLC12A2, SLC26A3, SLC26A4, SLC26A6, SLC26A7, SLC26A9, KCNQ1, KCNQ2, KCNQ3, KCNQ4, KCNQ5, KCNN4, KCNMA1, and ATP1A1 along with the 2x RT2 SYBR Green mastermix kit (Qiagen) was used for Quantitative Real‐time PCR performed by the Quant Studio 7 Flex Real‐time PCR system (Applied Biosystems, Beverly, MA, USA). Data was analyzed using the web‐based Array Data Analysis software available at www.SABiosciences.com/pcrarrydataanalysis.php.
2.4. siRNA knockdown
The following siRNA sequences were used: for SLC26A3, GGACAACAAUCAGAUAGAAtt (sense) and UUCUAUCUGAUUGUUGUCCag (antisense); for SLC26A4/Pendrin, CCCAGUCCCUAUUCCUAUAtt (sense) and UAUAGGAAUAGGGACUGGGat (antisense); for SLC26A6, UGAAGAAUAUUUUCCAUGAtt (sense) and UCAUGGAAAAUAUUCUUCAgg (antisense). CF HBECs were plated onto transwell inserts (Corning Costar 3470) at 1 x 105 cells/well in BEGM (Lonza). Cells were incubated at 37OC in a 5% CO2 incubator overnight, then transfected with Opti‐MEM (Invitrogen) containing SLC26A4, SLC26A3, or SLC26A6 siRNAs (Invitrogen) and Lipofectamine RNAi Max (Invitrogen) on both apical and basolateral sides for 3 days. A non‐targeting siRNA was used as a negative control (Invitrogen). The cells were transfected for 3 days before the media was changed. The cells were differentiated for 21 days in VALI medium (AthenaES) prior to use.
2.5. Using chamber recordings
Well‐differentiated normal and CF HBECs grown on transwell inserts were mounted in Ussing Chambers (Physiologic Instruments, San Diego, CA, USA), bathed in symmetrical physiological saline (140 mmol/L NaCl, 5 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L HEPES, 10 mmol/L Glucose, pH 7.4 with NaOH) and aerated. Benzamil (10 µmol/L apically), forskolin (5 µmol/L, both sides), bumetanide (20 µmol/L basolaterally), and CFTR(inh)‐172 (20 µmol/L, both sides) were added to the chambers. Transepithelial current (Isc) and conductance (Gt) were measured using a multichannel voltage/current clamp VCCM8 system (Physiological Instruments) and recorded using the Acquire and Analyze 2.0 software (Physiological Instruments).
2.6. Western blots
Warm PBS (Invitrogen) was added to the apical side of 6‐well transwell inserts containing HBECs to wash away mucus, and then aspirated. 100 µL of ice cold IP Lysis buffer (Pierce/ThermoFisher, Waltham, MA, USA) containing protease inhibitors (Roche) was then added to the apical side of the inserts. Cells were scraped off with a cell scraper and pipetted up and down to collect the cells in a centrifuge tube. Cells were incubated on ice for 10 minutes then centrifuged. The supernatant/protein lysate was transferred to a new tube and stored at −80°C. Protein from HBECs was quantified using a BCA protein assay kit (Pierce). Cell lysate was added to Nupage LDS Sample buffer (Invitrogen) and separated on a Nupage 4‐12% Bis‐Tris gel (Invitrogen) incubated on ice. Proteins were transferred to a membrane using the IBlot 2 transfer device and Nitrocellulose stacks (Invitrogen). The membrane was blocked with 5% nonfat milk in Tris‐buffered saline containing 0.05% Tween 20 (TTBS) for 2 hours at room temperature and then incubated overnight with anti‐SLC26A4 antibody (abcam #ab98091, Cambridge, MA, USA) at 1 µg/mL, anti‐CFTR antibody (UNC 596) at 1:1000, and/or anti‐glyceraldehyde‐3‐phosphate dehydrogenase (abcam #ab9485) at 1:100 in blocking buffer. UNC 596 antibody to CFTR was provided by Tim Jensen through the University of North Carolina and Cystic Fibrosis Foundation antibody distribution program (GENTZS18XX0). The membrane was washed three times with TTBS for 10 minutes each and incubated for 2 hours at room temperature with horseradish peroxidase‐conjugated goat anti‐rabbit antibody (abcam #ab205718) or donkey anti‐mouse antibody (The Jackson Laboratory #711‐035‐152, Bar Harbor, ME, USA) diluted 1:10,000. After washing, the blot was developed with Supersignal West Pico Chemiluminescence Substrate (Thermo Scientific, Waltham, MA, USA) and digitized with a Chemidoc Touch imaging System (BioRad, Hercules, CA, USA). No adjustments were made to Western Blot digital images after acquisition.
2.7. Ciliary beat frequency (CBF)
Monolayer images of CF HBECs were visualized using a 10x lens on an inverted microscope (Olympus, Tokyo, Japan). Data were captured using the high speed scan monochromatic digital video camera model AcA604‐120uM (Basler AG, Ahrensburg, Germany) at a sampling rate of 120 frames per second and a resolution of 640 x 480 pixels. Video images were analyzed using the Sisson‐Ammons Video Analysis system (SAVA), version 2.1.6 (Ammons Engineering, Clio, MI, USA).
2.8. Statistical analysis
Values are mean +SEM. Statistical significance was analyzed with Prism 6 (Graphpad Software, San Diego, CA, USA), using one‐way or two‐way ANOVA with Sidak's multiple comparisons test. Significance was defined as p < 0.05, with * = p < 0.05, ** = p < 0.01, *** = p < 0.001 for all tests.
3. RESULTS
To identify potential genetic modulators of CFTR anion secretion correlating with CF disease, we conducted RT‐qPCR studies of 25 ion transport related genes in both normal and ΔF508 homozygous CF primary Human Bronchial Epithelial Cells (HBECs) from the lungs of CF donors. Cells were well differentiated at the air liquid interface for 4 weeks, and mRNA levels were quantified using the RT2profiler system (Figure 1A). We found that SLC26A4 (pendrin) was substantially downregulated in CF HBECs compared to normal controls. This was confirmed using a TaqMan assay for pendrin mRNA (Figure 1B). CF‐dependent downregulation of pendrin has previously been described in CF animals 38 ; additionally, several interleukin cytokines have been shown to upregulate pendrin expression on the apical membrane of HBECs 39 . To confirm that cytokines could exert such an effect in our differentiated primary HBEC cultures, we performed Taqman qPCR analysis after 48 hours of treatment with 10 ng/mL IL‐13. We observed a large increase in pendrin mRNA in response to IL‐13 treatment in both normal and ΔF508 homozygous CF HBECs (Figure 1C). To confirm that protein levels followed changes in mRNA, we performed a western blot on lysates from normal HBECs incubated with IL‐4, IL‐13, and IL‐17a at 10 ng/mL for 48 hours and probed with an anti‐pendrin antibody (Figure 1D). Though IL‐4, IL‐13, and IL‐17a all increased pendrin protein expression, IL‐17a showed a lesser effect than IL‐4 or IL‐13. Therefore, for subsequent studies we utilized either IL‐4 or IL‐13. We then confirmed that IL‐4 could also increase pendrin protein in CF HBECs by Western Blot (Figure 1E). Finally, qPCR experiments using the RT2profiler screen again confirmed that IL‐13 dramatically increased pendrin (SLC26A4) mRNA levels in ΔF508 homozygous CF HBECs (Figure 1F), while also causing smaller increases in ANO1 (TMEM16A calcium‐activated chloride channel) and ATPA1 (Na+/K+ ATPase alpha subunit) mRNA levels.
FIGURE 1.
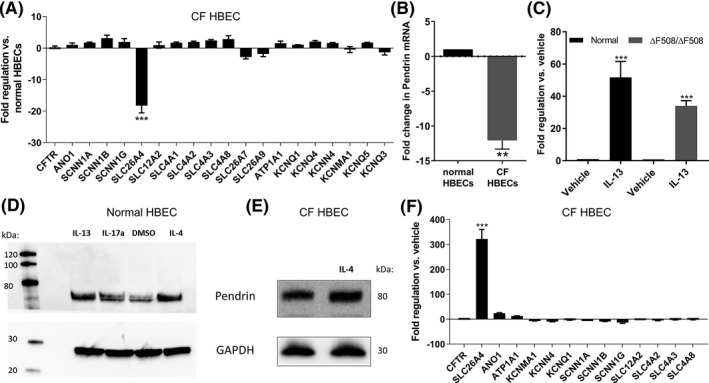
Pendrin is downregulated in CF HBECs but can be upregulated by interleukin treatment. A, RT‐PCR screen (RT2Profiler) comparing ion transport gene transcripts from well‐differentiated normal HBECs versus ΔF508/ΔF508 CF HBECs identified SLC26A4 (pendrin) as significantly downregulated in CF. B and C, Taqman assay confirmed pendrin mRNA differences between normal and CF HBECs (B), and 48 hours of treatment with 10 ng/mL IL‐13 increased pendrin mRNA in both normal and CF HBECs (C). D, Western blot against pendrin in whole cell lysates from normal HBECs treated with 10 ng/mL IL‐4, IL‐13, or IL‐17a for 48 hours showed that interleukin treatment increased pendrin protein levels. E, Same experiment as in D but using CF HBECs treated with IL‐4. F, RT‐PCR (RT2profiler) of CF HBECs after 48 h of IL‐13 treatment (10 ng/mL) showed that IL‐13 greatly increased pendrin mRNA, and modestly increased ANO1 (TMEM16A) and ATP1A1 transcripts
Given the reduced pendrin expression we observed in CF HBECs, we asked whether pendrin might impact CFTR function. CFTR‐dependent Cl− secretion can be measured in epithelia by assessing the magnitude of the short‐circuit current (Isc) response to forskolin in the Ussing Chamber. Once apical Na+ absorption has been eliminated through the addition of benzamil and resulting blockage of the epithelial sodium channel ENaC, a well‐differentiated HBEC epithelium displayed minimal ion transport (Figure 2A). Subsequent application of forskolin led to a large increase in Isc. Forskolin stimulates cAMP production via adenylate cyclase; cAMP activates protein kinase A (PKA) which phosphorylates CFTR, leading to increased CFTR channel openings and anion secretion 6 . At the end of the experiment, the increased transepithelial current resulting from forskolin addition can be confirmed as CFTR‐dependent by eliminating it with the CFTR‐specific inhibitor CFTR(inh)‐172. 40 Incubation of normal HBECs with 10 nmol/L IL‐4, IL‐13, and IL‐17a for 48 hours led to a significantly increased Isc peak response to forskolin (Figure 2A,B), a direct measure of CFTR‐dependent Cl‐ secretion. In all cases, the forskolin response was completely inhibited by addition of CFTR(inh)‐172, indicating CFTR dependence. Cells were bathed in symmetrical physiological saline without HCO3 −and maintained at physiological temperature throughout Ussing Chamber experiments (see Methods for composition).
FIGURE 2.
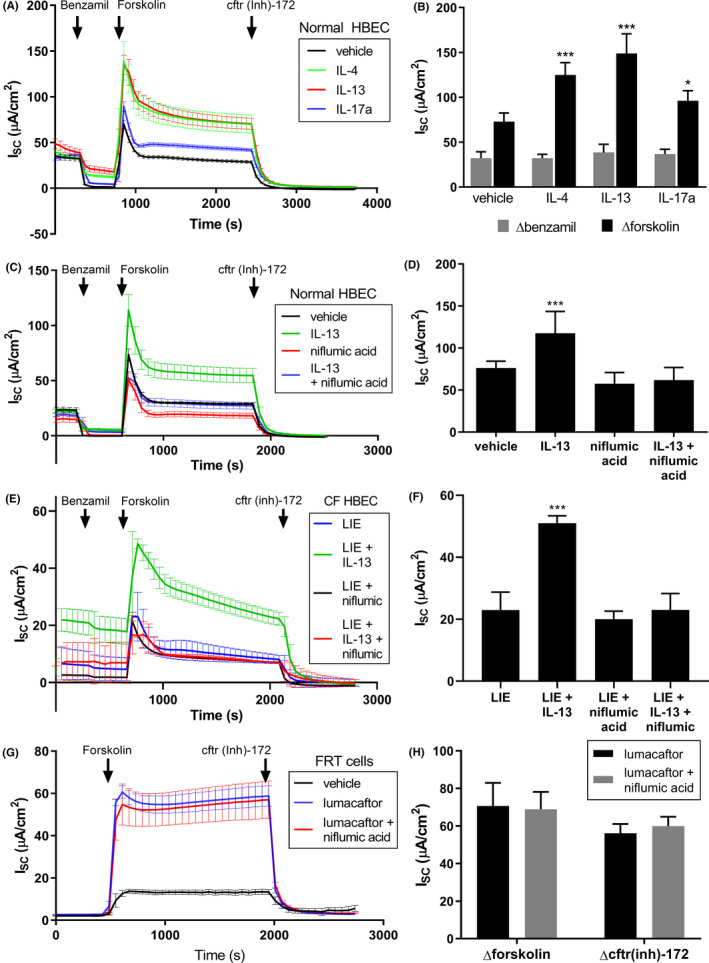
Interleukins increased CFTR current in HBECs, and pendrin inhibitor niflumic acid blocked this increase. A, Ussing Chamber recordings from well‐differentiated normal HBEC epithelia treated with 10 ng/mL IL‐4, IL‐13, or IL‐17a for 48 hours prior to recording showed significant CFTR current increases in response to interleukins. The benzamil responses and peak forskolin responses are quantified in (B). C, Ussing Chamber recordings from normal HBEC epithelia treated with 10 ng/mL IL‐13 for 48 hours and/or 10 µmol/L niflumic acid for 30 min showed that niflumic acid blocked the IL‐13 mediated increase in CFTR current. The peak forskolin responses are quantified in (D). E, Ussing Chamber recordings from ΔF508/ΔF508 CF HBEC epithelia treated with the LIE triple combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) for 48 h plus and minus 10 ng/mL IL‐13 and/or 10 µmol/L niflumic acid (for 30 minutes) again showed that niflumic acid blocked the IL‐13 mediated increase in CFTR current. The peak forskolin responses are quantified in (F). G, Ussing Chamber recordings from FRT cells overexpressing CFTRΔF508 treated with 5 µmol/L lumacaftor and/or niflumic acid showed that niflumic acid did not inhibit CFTR directly. Peak forskolin response and cftr(inh)‐172 response are quantified in H. For all graphs, n = 4
To confirm that pendrin was functionally responsible for the increased CFTR current in response to interleukins shown in Figure 2A, we tested the non‐specific pendrin inhibitor niflumic acid 41 on normal HBE cells. Niflumic acid caused a small decrease in CFTR current (Figure 2C‐D). As previously observed, IL‐13 caused a significant increase in current. However, co‐incubation of HBECs with IL‐13 and niflumic acid completely blocked the IL‐13‐dependent increase in Isc peak response to forskolin, indicating that pendrin function might be responsible for the increased CFTR current in response to IL‐13. These findings were also replicated in CF HBECs treated with a triple combination of lumacaftor + ivacaftor + elexacaftor (LIE), which generated a very large CFTR current in CF HBECs that was further additive with IL‐13 treatment (Figure 2E‐F). As in normal HBECs, the IL‐13 effect was blocked by niflumic acid. This indicated that pendrin's effects were additive with CFTR modulators. We confirmed that niflumic acid does not block CFTR directly by measuring CFTR current size in the presence and absence of niflumic acid in Fischer Rat Thyroid (FRT) cells heterologously expressing human CFTRΔF508 (Figure 2G,H). We utilized lumacaftor treatment to correct CFTRΔF508, increasing the current size in our FRT cell recordings to ensure we did not miss a subtle effect of niflumic acid. However, there was no difference in peak or inhibitable CFTR current size in response to forskolin when cells were exposed to niflumic acid, indicating that it does not block CFTR directly.
To further confirm that pendrin is required for the interleukin‐dependent increase in CFTR current, we performed siRNA knockdown experiments. We again treated CF HBECs with the LIE triple combination for these experiments. Using siRNAs targeting either pendrin (SLC26A4) or the closely related transporters SLC26A3 and SLC26A6, we transfected well‐differentiated ΔF508/ΔF508 CF HBECs with siRNAs at the time of plating and measured the effect on LIE‐evoked CFTR currents 3 weeks later. In CF HBECs transfected with siRNA targeting SLC26A4, IL‐4 incubation failed to increase forskolin‐stimulated CFTR current (Figure 3A). However, in cells transfected with siRNAs targeting either SLC26A3 or SLC26A6, IL‐4 treatment still caused a significant increase in CFTR current that was inhibited by niflumic acid. Due to the challenges of introducing and maintaining siRNA in primary cells over several weeks, we achieved only 25 – 30% knockdown of mRNA (Figure 3B). Western Blot against pendrin in the siRNA‐treated epithelia confirmed a similar reduction in pendrin protein, and a comparable inhibition of the effect of IL‐4 (Figure 3C,D). This level of pendrin knockdown was sufficient for a significant effect on CFTR current, but perhaps more knockdown of SLC26A3 or SLC26A6 might reveal further effects.
FIGURE 3.

siRNA knockdown demonstrated that pendrin, but not SLC26A3 or SLC26A6, is required for the interleukin‐dependent increase in CFTR current. A, Left, Ussing Chamber recordings from ΔF508/ΔF508 CF HBECs transfected with pendrin (SLC26A4) siRNA and treated with the LIE combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) and 10 ng/mL IL‐4 for 48 hours showed that pendrin knockdown prevented the IL‐4 dependent increase in CFTR current (n = 4). Center and right, Ussing Chamber recordings from CF HBECs transfected with either SLC26A3 or SLC26A6 siRNAs showed no significant reduction in the IL‐4 dependent increase in CFTR current (n = 4). B, Taqman RT‐PCR results for SLC26A4 (left), SLC26A3 (center), and SLC26A6 (right) from the same cells as in (A) showed approximately 30% knockdown of target mRNAs. Niflumic acid also reduced SLC26A4 expression, but did not affect SLC26A3 or SLC26A6 (n = 4). C, Western Blot of pendrin in whole cell lysates of epithelia from (A), treated with control or pendrin (SLC26A4) siRNAs, the LIE combination and other agents as indicated. D, Quantitation of the results in C showing concomitant decreases in pendrin protein (normalized to GAPDH) in the SLC26A4 siRNA‐treated epithelia
Given pendrin's primary function as an anion exchanger, it is possible that pendrin exchanges intracellular anions (eg, HCO3 −) for extracellular Cl− in HBECs. This could directly or indirectly lead to an increase in HBEC Cl− absorption, supporting increased efflux through CFTR. Although CFTR is usually considered the bottleneck to epithelial Cl− transport, at higher levels of CFTR expression or function (such as in normal HBECs, or CF HBECs treated with LIE), basolateral Cl− absorption might instead become the limiting factor. To test this hypothesis, we performed Ussing Chamber experiments in which we first inhibited both ENaC and NKCC1, the primary basolateral Cl− absorption pathway in HBECs, using benzamil and bumetanide respectively (Figure 4A,B). Cells were bathed in symmetrical saline containing Cl− but no HCO3 −. We then measured HBEC responses to forskolin. In LIE‐treated CF HBECs, forskolin evoked only a small transepithelial current, while addition of IL‐4 caused a much larger response which was inhibited by niflumic acid. The addition of CFTR(inh)‐172 along with benzamil and bumetanide completely abrogated the effect of IL‐4, again indicating that currents were mediated by CFTR, and revealing the non‐CFTR current baseline in the experiment (indicated by the dashed line in Figure 4B). When considering the size of the CFTR responses relative to this background current baseline, IL‐4 increased CFTR current >5‐fold. Such a large IL‐4 induced, CFTR‐dependent current in the absence of NKCC1 activity suggested that a previously undescribed NKCC1‐independent, pendrin‐dependent Cl− absorption pathway had been activated.
FIGURE 4.
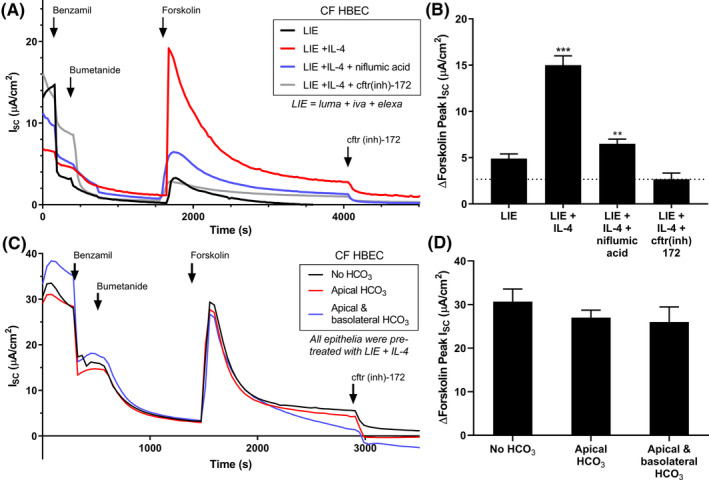
Interleukin treatment increased CFTR current via a NKCC1‐independent, pendrin‐dependent mechanism. A, Ussing Chamber recordings from ΔF508/ΔF508 CF HBECs treated with the LIE combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) and 10 ng/mL IL‐4 for 48 h. Some chambers were also pre‐incubated with 10 µmol/L niflumic acid or 20 µmol/L cftr(inh)‐172 for 30 min prior to recording, as indicated. ENaC and NKCC1 were both blocked with benzamil and bumetanide respectively prior to forskolin addition. IL‐4 treatment preserved a large CFTR current even in the absence of NKCC1 activity which was inhibited by niflumic acid. B, Quantitation of peak Isc forskolin responses from (A), n = 4. C, Ussing Chamber recordings from CF HBECs treated with the LIE combination and 10 ng/mL IL‐4 for 48 h. 10 mmol/L NaHCO3 was added to the apical chamber or both chambers as indicated, but did not alter pendrin‐dependent CFTR currents. D, Quantitation of peak Isc forskolin responses from C, n = 4
We also investigated whether bicarbonate (HCO3 −) might impact the pendrin‐dependent component of CFTR current. If cellular HCO3 − was secreted through CFTR as a result of pendrin activity, application of high apical HCO3 − should blunt the driving force and reduce the current. As in Figure 4A, we compared the forskolin responses of CF HBECs treated with LIE and IL‐4 after the addition of both benzamil and bumetanide. In this experiment however, we varied the Ussing Chamber solutions (Figure 4C,D). When 10 mmol/L NaHCO3 was added to the apical chamber, we observed no changes in CFTR current. Further addition of 10 mmol/L NaHCO3 to the basolateral chamber (symmetrical HCO3 −) also did not change CFTR current. Therefore, Cl− appears responsible for the increased current through CFTR resulting from pendrin activity.
There are additional mechanisms by which interleukins or pendrin might increase CFTR activity in HBECs independent of a change in basolateral Cl− absorption. To rule out some of these, we first blocked basolateral K+ channels with 5 mmol/L BaCl2 to ensure that the effect of IL‐4/13 or pendrin on CFTR was not due to membrane potential changes (Figure 5A,B). BaCl2 slightly reduced the IL‐4 dependent increase in CFTR current, but only when NKCC1 was active. Inhibiting NKCC1 with bumetanide abrogated the effects of BaCl2, indicating that any effect of K+ channels likely impacted NKCC1 but not a pendrin‐dependent Cl− absorption pathway. Pendrin might also increase the efficacy of forskolin by increasing intracellular ATP or cAMP concentrations, or PKA function. However, measurements of cellular ATP showed no changes upon IL‐13 treatment (Figure 5D), and surprisingly there were decreases in both cAMP (Figure 5E) and PKA function (Figure 5F), perhaps due to higher CFTR activity. Finally, we performed a Western blot against CFTR to ensure that protein levels and maturation were not altered. IL‐13 did not significantly increase CFTR protein abundance; rather, there was a trend toward a decrease (Figure 5G). Taken together, these data suggest that possibility that pendrin increases CFTR current by increasing basolateral Cl− absorption.
FIGURE 5.
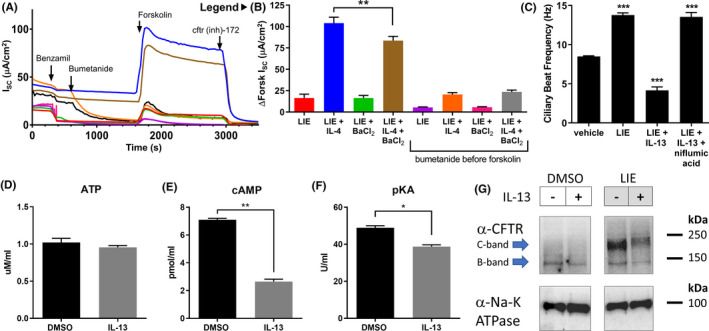
The effects of interleukins and pendrin are not due to K+ channels, increased CFTR levels, or increases in endogenous CFTR agonists. A and B, Ussing Chamber recordings from CF HBECs treated with the LIE combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) and/or 10 ng/mL IL‐4 for 48 h showed that blocking basolateral K+ channels with 5 mmol/L BaCl2 slightly reduced the IL‐4 dependent increase in CFTR current, but only when NKCC1 was active. Inhibiting NKCC1 by adding bumetanide to the indicated chambers abrogated the effects of BaCl2 on forskolin‐evoked CFTR current. Peak forskolin responses are quantified in (B) (n = 4). Colors & labels in (B) serve as the legend for (A). C, Ciliary beat frequency (CBF) measurements from CF HBECs treated with the LIE combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) and/or 10 ng/mL IL‐13 for 48 hours with or without a 30 minute pre‐treatment with 10 µmol/L niflumic acid confirmed that IL‐13 negatively impacted CBF. However, IL‐13’s effect was blocked by niflumic acid (n = 4). D, E, and F, 48 h treatment of normal HBECs with IL‐13 did not alter cellular ATP levels (D), but did significantly reduce cellular cAMP levels (E), and slightly reduced PKA function (F), all n = 4. G, Western blot against CFTR in whole‐cell lysates from CF HBECs treated with the LIE combination (1 µmol/L Lumacaftor +0.02 µmol/L Ivacaftor +3 µmol/L Elexacaftor) and/or 10 ng/mL IL‐13 for 48 hours showed that IL‐13 treatment did not increase CFTR protein levels. Na‐K ATPase was used as a control
Pendrin has been reported to contribute to the regulation of ciliary beating. We examined the impact of IL‐13 treatment and pendrin inhibition on ciliary beat frequency in ΔF508/ΔF508 CF HBECs (Figure 5C). Treatment with LIE increased ciliary beat frequency (CBF) by nearly 2‐fold above baseline, indicating that increased CFTR activity improves ciliary function. However, the addition of IL‐13 reduced CBF below baseline. Co‐incubation of IL‐13 and the pendrin inhibitor niflumic acid restored CBF to the level observed with LIE treatment alone, indicating that pendrin activity was likely responsible for the IL‐13 mediated drop in CBF. Therefore, pendrin's effects on CFTR and CBF may be independent. Although we have demonstrated that pendrin increases CFTR current, this increase appears insufficient to overcome the negative effect of pendrin on CBF in cultured HBECs.
4. DISCUSSION
Ion secretion and absorption are key determinants of airway epithelium function that are disrupted in CF. CFTR works in concert with other ion channels and ion transporters to maintain epithelial ion flux and homeostasis of critical ions such as Cl− and HCO3 −. There is substantial evidence for the functional interaction of CFTR with members of the SLC26 family of anion exchangers such as SLC26A3, SLC26A6, and SLC26A9. 28 , 29 , 30 Here we have demonstrated that SLC26A4 (pendrin), is downregulated at the mRNA level in human bronchial epithelial cells (HBECs) from CF patients. Pendrin expression was increased by exogenous application of the interleukin cytokines IL‐4, IL‐13, or IL‐17a. Upregulation of pendrin led to increased CFTR‐dependent transepithelial current in the Ussing Chamber, a measure of Cl− secretion. Importantly, this pendrin‐dependent increase in CFTR current was additive with the CFTR modulators lumacaftor, ivacaftor, and elexacaftor. siRNA knockdown of pendrin mRNA or inhibition of pendrin activity with niflumic acid blocked this interleukin‐stimulated increase in CFTR current. These observations suggested functional coupling between pendrin and CFTR; this coupling has also been suggested by another recent publication. 36
We sought to investigate the mechanism of this putative functional connection between pendrin and CFTR. Inhibition of basolateral Cl− absorption with bumetanide abolished CFTR‐dependent transepithelial Cl− transport, but interleukin treatment rescued transport. The rescue could be reduced by a pendrin inhibitor, suggesting that pendrin might activate an NKCC1‐independent basolateral Cl− absorption pathway. Several experiments ruled out alternative mechanisms of action, such as changes in HCO3 − permeability, K+ channel‐dependent membrane potential, ATP or cAMP levels, or PKA activity. Also, CFTR protein levels were slightly decreased by interleukin treatment, indicating that pendrin function does not increase CFTR expression. These results suggest that pendrin's primary mode of action may be to elevate intracellular Cl−, perhaps as the result of increased basolateral Cl− absorption. Whether pendrin itself exchanges intracellular anions for extracellular Cl− to directly generate this increased Cl− absorption, or whether it indirectly stimulates such activity via another protein is not yet clear. Importantly, these data suggest that at higher levels of CFTR functional expression in HBECs, basolateral Cl− absorption becomes the bottleneck to transepithelial Cl− secretion, rather than CFTR.
This work demonstrates that pendrin plays a role in determining the CFTR current size observed in epithelial electrophysiological recordings from HBECs. When measuring changes in CFTR current resulting from drugs, signaling molecules or other proteins, alterations in pendrin could cause unintended changes in CFTR current. CFTR conductance seems less sensitive to pendrin‐dependent changes than current and could prove a more reliable metric for the assessment of CFTR‐directed therapies. Alternately, IL‐4 or IL‐13 could be added to experiments to eliminate the potential bottleneck of basolateral Cl− absorption. This may be necessary as therapies achieve higher levels of CFTR functional restoration, and in fact we have shown that the effect of pendrin is additive with CFTR modulators, supporting increase epithelial Cl− secretion. The contribution of pendrin to CFTR recordings from HBECs should be carefully considered, especially in the context of therapeutic development.
It has been suggested that pendrin inhibitors could be useful for the treatment of CF, based on the observation that interleukin treatment decreases ciliary beat frequency (CBF) and other epithelial parameters in CF HBECs. 39 We did reproduce the CBF findings, and additionally demonstrated that pendrin inhibition prevents the IL‐13 effect on CBF. However, other data presented here indicate that inhibition of pendrin decreases transepithelial Cl− secretion, which would likely be undesirable. This apparent contradiction of increased Cl− secretion but decreased CBF resulting from pendrin activity could occur due to competing effects on chloride and bicarbonate ions, via competing functional interactions with different proteins, or as the result of studying these effects in vitro. To the latter point, both sets of experiments were conducted at the extremes of pendrin function (maximal or absent). In vivo, it may be that pendrin activity is modulated in response to the immediate needs of the epithelium; perhaps a balanced level of pendrin activity—one that maximizes CFTR current while avoiding levels that might have a negative impact on CBF—is ideal for proper lung function.
An interesting intersect with the concept of properly balancing pendrin function relates to our finding of pendrin downregulation in CF epithelia. This observation differs from a recent publication which instead reported an upregulation of pendrin. 36 The discrepancy could arise from differences in culture conditions, but might also arise from tissue donor differences. One intriguing possibility is that some CF patients, perhaps those with lower levels of immune cell infiltration into the lung or without a recent acute infection, upregulate pendrin as a compensatory mechanism to improve anion efflux through CFTR. Other patients however, perhaps those who are suffering from acute infection with high levels of interleukin release, might downregulate pendrin to prevent potentially deleterious effects of overactivation of this protein. Again, this might support the concept that a balanced level of pendrin activity is most beneficial. The details of the donors used in our studies was blinded due to privacy concerns, but studies in collaboration with donors able to share more details about their health and immune status might help to shed light on this question. Therefore, modulation of pendrin activity as a therapeutic strategy requires further validation, as the state of pendrin regulation in vivo in CF lung is not well described. Nevertheless, the observation that basolateral Cl− absorption becomes limiting at higher levels of CFTR activity suggests that increasing Cl− absorption via pendrin might provide additive or synergistic benefit with CFTR‐directed therapeutics.
CONFLICTS OF INTEREST
J. S. Kaczmarek, G. H. Hurlbut, and S. Altmann were previously employed by Sanofi. Sanofi has received research funding, reagents, and intellectual support from the Cystic Fibrosis Foundation. The authors have no further conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
J. S. Kaczmarek, J. Bajko, and G. D. Hurlbut designed research; J. Bajko, M. Duguid, and S. Altmann performed research; J. S. Kaczmarek, J. Bajko, M. Duguid, and S. Altmann analyzed data; J. S. Kaczmarek and J. Bajko wrote the paper.
ACKNOWLEDGEMENTS
The authors wish to thank Scott Bercury and Kailene Simon for their expertise with CFTR westerns; Aliza Majewski for help with culturing HBECs for the Ussing experiments; Qiuming Chu for providing reagents and suggestions; Dr. Seng H. Cheng for support and mentorship. FRT cells expressing CFTRΔF508 were kindly provided by Dr. Phil Karp.
Bajko J, Duguid M, Altmann S, et al. Pendrin stimulates a chloride absorption pathway to increase CFTR-mediated chloride secretion from Cystic Fibrosis airway epitheliaPendrin stimulates a chloride absorption pathway to increase CFTR-mediated chloride secretion from Cystic Fibrosis airway epithelia. FASEB BioAdvances. 2020;2:526–537. 10.1096/fba.2020-00012
REFERENCES
- 1. Rafeeq M, Murad H. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017;15:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reis FJ, Damaceno N. Cystic fibrosis. J Pediatr. 1998;74:S76–S79. [DOI] [PubMed] [Google Scholar]
- 3. Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261:5–16. [DOI] [PubMed] [Google Scholar]
- 4. Ratner D, Mueller C. Immune responses in cystic fibrosis: are they intrinsically defective? Am J Respir Cell Mol Biol. 2012;46:715–722. [DOI] [PubMed] [Google Scholar]
- 5. Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. [DOI] [PubMed] [Google Scholar]
- 6. Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Generation of cAMP‐activated chloride currents by expression of CFTR. Science. 1991;251:679–682. [DOI] [PubMed] [Google Scholar]
- 7. Rich DP, Anderson MP, Gregory RJ, et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347:358–363. [DOI] [PubMed] [Google Scholar]
- 8. Venglarik CJ, Schultz BD, Frizzell RA, Bridges RJ. ATP alters current fluctuations of cystic fibrosis transmembrane conductance regulator: evidence for a three‐state activation mechanism. J Gen Physiol. 1994;104:123–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest. 1992;89:1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson MP, Berger HA, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 1991;67:775–784. [DOI] [PubMed] [Google Scholar]
- 11. Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, Nairn AC. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- 12. Bozoky Z, Krzeminski M, Muhandiram R, et al. Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho‐dependent intra‐ and intermolecular interactions. Proc Natl Acad Sci U S A. 2013;110:E4427–E4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. US CF Foundation, J. HUTHfSC . (2019). The Clinical and Functional Translation of CFTR (CFTR2).
- 14. Cheng SH, Gregory RJ, Marshall J, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. [DOI] [PubMed] [Google Scholar]
- 15. Thibodeau PH, Richardson J, Wang W, et al. The cystic fibrosis‐causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem. 2010;285:35825–35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mendoza JL, Schmidt A, Li Q, et al. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rabeh W, Bossard F, Xu H, et al. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cholon DM, Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros. 2018;17:S52–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goralski JL, Wu D, Thelin WR, Boucher RC, Button B. The in vitro effect of nebulised hypertonic saline on human bronchial epithelium. Eur Respir J. 2018;51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wark PA, McDonald V. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2000;2:CD001506. [DOI] [PubMed] [Google Scholar]
- 21. Schmid A, Sutto Z, Schmid N, et al. Decreased soluble adenylyl cyclase activity in cystic fibrosis is related to defective apical bicarbonate exchange and affects ciliary beat frequency regulation. J Biol Chem. 2010;285(39):29998–30007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del‐CFTR protein processing defect in vitro by the investigational drug VX‐809. Proc Natl Acad Sci USA. 2011;108:18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX‐770. Proc Natl Acad Sci USA. 2009;106:18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double‐blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang F, Shang H, Jordan NJ, et al. High‐throughput screening for readthrough modulators of CFTR PTC mutations. SLAS Technol. 2017;22:315–324. [DOI] [PubMed] [Google Scholar]
- 26. Benedetto R, Ousingsawat J, Cabrita I, et al. Plasma membrane‐localized TMEM16 proteins are indispensable for expression of CFTR. J Mol Med (Berl). 2019;97:711–722. [DOI] [PubMed] [Google Scholar]
- 27. Rock JR, O'Neal WK, Gabriel SE, et al. Transmembrane protein 16A (TMEM16A) is a Ca2+‐regulated Cl‐ secretory channel in mouse airways. J Biol Chem. 2009;284:14875–14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bertrand CA, Mitra S, Mishra SK, et al. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am J Physiol Lung Cell Mol Physiol. 2017;312:L912–L925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. El Khouri E, Touré A. Functional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): physiological and pathophysiological relevance. Int J Biochem Cell Biol. 2014;52:58–67. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Soyombo AA, Shcheynikov N, et al. Slc26a6 regulates CFTR activity in vivo to determine pancreatic duct HCO3‐ secretion: relevance to cystic fibrosis. EMBO J. 2006;25:5049–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pedemonte N, Caci E, Sondo E, et al. Thiocyanate transport in resting and IL‐4‐stimulated human bronchial epithelial cells: role of pendrin and anion channels. J Immunol. 2007;178:5144–5153. [DOI] [PubMed] [Google Scholar]
- 32. Nofziger C, Dossena S, Suzuki S, Izuhara K, Paulmichl M. Pendrin function in airway epithelia. Cell Physiol Biochem. 2011;28:571–578. [DOI] [PubMed] [Google Scholar]
- 33. Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. 1997;17:411–422. [DOI] [PubMed] [Google Scholar]
- 34. Reardon W, OMahoney CF, Trembath R, Jan H, Phelps PD. Enlarged vestibular aqueduct: a radiological marker of pendred syndrome, and mutation of the PDS gene. QJM. 2000;93:99–104. [DOI] [PubMed] [Google Scholar]
- 35. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride‐iodide transport protein. Nat Genet. 1999;21:440–443. [DOI] [PubMed] [Google Scholar]
- 36. Kim D, Huang J, Billet A, et al. Pendrin mediates bicarbonate secretion and enhances cftr function in airway surface epithelia. Am J Respir Cell Mol Biol. 2019;60. [DOI] [PubMed] [Google Scholar]
- 37. Sheppard DN, Carson MR, Ostedgaard LS, Denning GM, Welsh MJ. Expression of cystic fibrosis transmembrane conductance regulator in a model epithelium. Am J Physiol. 1994;266:L405–L413. [DOI] [PubMed] [Google Scholar]
- 38. Varasteh Kia M, Barone S, McDonough AA, Zahedi K, Xu J, Soleimani M. Downregulation of the Cl‐/HCO3‐exchanger pendrin in kidneys of mice with cystic fibrosis: role in the pathogenesis of metabolic alkalosis. Cell Physiol Biochem. 2018;45:1551–1565. [DOI] [PubMed] [Google Scholar]
- 39. Adams KM, Abraham V, Spielman D, et al. IL‐17A induces Pendrin expression and chloride‐bicarbonate exchange in human bronchial epithelial cells. PLoS One. 2014;9:e103263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma T, Thiagarajah JR, Yang H, et al. Thiazolidinone CFTR inhibitor identified by high‐throughput screening blocks cholera toxin‐induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bernardinelli E, Costa R, Nofziger C, Paulmichl M, Dossena S. Effect of known inhibitors of ion transport on pendrin (SLC26A4) activity in a human kidney cell line. Cell Physiol Biochem. 2016;38:1984–1998. [DOI] [PubMed] [Google Scholar]